

Supplemental Digital Content is available in the text.
Keywords: hypertension; hypertrophy; myocyte, cardiac; renin-angiotensin system; transgenes
Abstract
Activation of AT1 (type 1 Ang) receptors stimulates cardiomyocyte hypertrophy in vitro. Accordingly, it has been suggested that regression of cardiac hypertrophy associated with renin-Ang system blockade is due to inhibition of cellular actions of Ang II in the heart, above and beyond their effects to reduce pressure overload. We generated 2 distinct mouse lines with cell-specific deletion of AT1A receptors, from cardiomyocytes. In the first line (C-SMKO), elimination of AT1A receptors was achieved using a heterologous Cre recombinase transgene under control of the Sm22 promoter, which expresses in cells of smooth muscle lineage including cardiomyocytes and vascular smooth muscle cells of conduit but not resistance vessels. The second line (R-SMKO) utilized a Cre transgene knocked-in to the Sm22 locus, which drives expression in cardiac myocytes and vascular smooth muscle cells in both conduit and resistance arteries. Thus, although both groups lack AT1 receptors in the cardiomyocytes, they are distinguished by presence (C-SMKO) or absence (R-SMKO) of peripheral vascular responses to Ang II. Similar to wild-types, chronic Ang II infusion caused hypertension and cardiac hypertrophy in C-SMKO mice, whereas both hypertension and cardiac hypertrophy were reduced in R-SMKOs. Thus, despite the absence of AT1A receptors in cardiomyocytes, C-SMKOs develop robust cardiac hypertrophy. By contrast, R-SMKOs developed identical levels of hypertrophy in response to pressure overload–induced by transverse aortic banding. Our findings suggest that direct activation of AT1 receptors in cardiac myocytes has minimal influence on cardiac hypertrophy induced by renin-Ang system activation or pressure overload.
Hypertension is a major public health problem causing significant risk of end-organ damage including stroke and cardiovascular disease.1 Left ventricular hypertrophy (LVH) is a frequent complication of chronic hypertension, increasing risk for heart failure and cardiovascular mortality.2 The renin-Ang (angiotensin) system (RAS) plays a prominent role in blood pressure (BP) regulation and hypertension pathogenesis.3 Components of the RAS are expressed in organ systems and cell lineages contributing to BP regulation. In addition to its effects to cause hypertension, Ang receptors may directly promote end-organ damage in target tissues. In this regard, it has been suggested that actions of Ang II mediated by its receptors in the kidney, heart, and central nervous system may contribute to morbidity and mortality above and beyond increases BP.
The classical effects of the RAS are mediated by Ang II acting via AT1 (type 1 angiotensin) receptors. This activation triggers a cascade of classic physiological responses, including vasoconstriction, activation of sympathetic nervous system, and stimulation of sodium reabsorption by the kidney, which all contribute to elevated BP in hypertension.4 AT1 receptor blockers and ACE (angiotensin-converting enzyme) inhibitors are effective and widely used antihypertensive agents.5 RAS blockade using Ang receptor blockers and ACE inhibitors are not only effective at lowering BP but also attenuating end-organ damage, such as reducing LVH, and decreasing cardiovascular mortality.6,7
The clinical evidence indicating a key contribution of RAS activation to development of LVH in hypertension is clear-cut.8–10 However, the mechanism of this effect is potentially complex as AT1 receptor activation triggers an array of cellular, vascular, and neuroendocrine responses that each might distinctly promote the development of hypertrophy. For example, activation of AT1 receptors triggers signaling pathways that have been linked to LVH,11 and indeed, Ang II induces hypertrophy of cultured cardiac myocytes.12,13 Furthermore, models of forced overexpression of wild-type (WT) and constitutively activated AT1 receptors in cardiomyocytes have consistently been shown to induce hypertrophy in vivo.14,15 In contrast, several studies using mice completely deficient in AT1A receptors, the major murine AT1 receptor, suggest that BP elevation and after-load have the predominant role to drive Ang II–dependent LVH.16,17 However, it is difficult to specifically isolate cardiac and vascular actions in systems where AT1 receptors are absent in all tissues.
In this study, we reexamine this issue, taking advantage of 2 mouse models lacking AT1 receptors in cardiac myocytes, but with divergent expression of AT1 receptors in resistance vessels. Thus, although AT1 receptor signaling is abrogated in cardiac myocytes in both groups, pressor responses to Ang II are preserved in one group (conduit vessel smooth muscle AT1A receptor knockout [C-SMKO]) but are absent in the other (resistance vessel smooth muscle AT1A receptor knockout [R-SMKO]). We find no evidence for a significant impact of direct effects of AT1 receptor signaling in cardiomyocytes to promote hypertrophy. Instead, the extent of hypertrophy seems to be predominantly driven by pressure.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Details of Mice
A mouse line with a conditional Agtr1aflox allele was generated using homologous recombination in embryonic stem cells, as described previously.18,19 To delete the AT1A receptor from vascular smooth muscle cells, we crossed inbred C57BL/6 transgenic mouse lines expressing Cre recombinase specifically in smooth muscle cells under the control of the sm22α promoter (KIsm22α-Cre, Strain Name B6.129S6-Taglntm2(cre)Yec/J, Stock Number 006878, The Jackson Laboratory, Bar Harbor, ME) or the (Tgsm22α-Cre, Strain Name B6Tg[Tagln-cre]1Her, Stock Number 004746, The Jackson Laboratory, Bar Harbor, ME). Therefore, we generated 2 separate mouse models, KIsm22α -Cre+Agtr1aflox/flox (R-SMKO) mice and Tgsm22α -Cre+Agtr1aflox/flox (C-SMKO), both on inbred C57BL/6 background. All experiments were performed on 8- to 24-week old male mice. In all experimental models, mice were randomized and researchers were blinded to genotype. Membrane-targeted tdTomato (mT)/membrane–targeted EGFP (mG) mice with loxP sites flanking the membrane-targeted tdTomato cassette followed by an N-terminal membrane-tagged version of EGFP were purchased from The Jackson Laboratory (Strain Name B6.129[Cg]-Gt[ROSA]26Sortm4(ACTB-tdTomato,-EGFP)Luo/J, Stock Number 007676) and crossed with either the constitutively expressed Tagln-Cre or KIsm22α-Cre recombinase transgenic lines as above. Mice possessing the mTmG transgene normally express red fluorescence protein (mT) in all tissues. However, when Cre recombinase is present, the mT cassette is deleted, triggering expression of the membrane-targeted EGFP (mG).20 Assessment of red and green fluorescence were conducted in male mice at 8 weeks of age.
All experimental mice were bred in an Association for Assessment and Accreditation of Laboratory Animal Care international accredited animal facility at the Duke University and Durham Veterans Affairs medical centers under National Institutes of Health guidelines for care and use of laboratory animals and housed with free access to standard rodent chow and water unless otherwise specified.
BP Measurements in Conscious Mice
BPs were measured in conscious, unrestrained 12- to 20-week-old male C-SMKO, R-SMKO, and WT mice using radiotelemetry (PA-C10), as described previously.16 Arterial BP was collected, stored, and analyzed using Dataquest A.R.T software (version 4.0; Data Sciences International, Saint Paul, MN). Mice were allowed to recover for 7 days after telemetry implantation to reestablished normal circadian rhythms.21 After recovery, telemetry data were collected continuously with sampling every 5 minutes for 10-s intervals. Baseline measurements were recorded for 7 consecutive days while mice ingested a normal sodium diet. On day 8, an osmotic minipump (Alzet, Cupertino, CA) infusing Ang II (Sigma Aldrich, St. Louis, MO) at a rate of 1000 ng/(kg·min) were implanted subcutaneously and BP measurements continued for at least 18 days in all 3 groups of mice (WT, C-SMKO, R-SMKO).
Real-Time Reverse Transcription Polymerase Chain Reaction of B-Type Natriuretic Peptide and AT1A mRNA Levels in Left Ventricle
RNA was extracted from the left ventricle of mice at baseline (uninfused) and after 4 weeks of Ang II infusion (1000 ng/kg/min) using an RNeasy Fibrous mini kit (Qiagen), cDNA was made using the High-Capacity cDNA Reverse Transcription Kit (4368814, ThermoFisher Scientific). Real-time quantitative polymerase chain reaction (PCR) was performed using PerfeCTa quantitative PCR FastMix II (Quanta) using the ΔCT method. Primers were from Taqman (ThermoFisher Scientific)- endogenous control 18s- 4352930E, Mm01255770_g1 BNP (B-type natriuretic peptide; Nppb). As previously described, relative levels of mRNA for the AT1A receptor were measured from the heart in WT, C-SMKO, and R-SMKO in male mice averaging 15±2 weeks of age, and there was no difference in age between groups.22 RNA was isolated and reverse transcription performed using qScript cDNA Supermix (Quanta Biosciences, Gaithersburg, MD). SYBR Green-based quantitative PCR was carried out using the SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, CA). For AT1A receptor expression, the amount of target gene relative to endogenous control was determined by the change in threshold cycle method. The following primer sequences were used: type 1 angiotensin Forward-5′-ACTCACAGCAACCCTCCAAG-3′, type 1 angiotensin Reverse-5′-ATCACCACCAAGCTGTTTCC-3′ (Amplicon size: 236bp), GAPDH Forward-5′-TCACCACCATGGAGAAGGC-3′, GAPDH Reverse-5′-GCTAAGCAGTTGGTGGTGCA-3′ (Amplicon size: 168bp). Control tubes lacking cDNA and containing RNA were included in each run.
Analysis of Oxidized Amino Acids in Heart
After 28 days of Ang II infusion, the ventricles were dissected from WT and C-SMKO mice and immediately placed in antioxidant buffer (100 μmol/L diethylene tetramino pentaacetic acid (metal chelator), 50 μmol/L butylated hydroxytoluene (lipid-soluble antioxidant), 10 μL/mL protease inhibitor (Halt Protease Inhibitor Cocktail; Pierce) in 50 mmol/L sodium phosphate buffer, pH 7.4) at −80 °C. Amino acids were isolated from the acid hydrolysate using a solid-phase column (Supelclean ENVI ChromP column; Supelco, Inc) as described.23 Oxidized amino acids were quantified by liquid chromatography–electrospray ionization tandem mass spectrometry using multiple reaction monitoring mode. Under these chromatography conditions, authentic compounds and isotopically labeled standards were baseline separated and exhibited retention times identical to those of analytes derived from tissue samples. 3-nitrotyrosine and dityrosine were detected by characteristic liquid chromatography retention time and specific ion transitions in the multiple reaction monitoring mode. The ratio of the peak areas of the analyte with corresponding 13C internal standard was used to quantify levels of oxidized amino acids in tissue. Results are normalized to protein content of tyrosine, the precursor of 3-nitrotyrosine, and dityrosine.
Pressure Overload
Animal studies were carried out according to approved protocols and animal welfare regulations of Duke University Medical Center’s Institutional Review Boards. Mice were anesthetized with a mixture of ketamine (100 mg/kg) and xylazine (2.5 mg/kg). Chronic pressure overload was induced by transverse aortic constriction (TAC) in R-SMKO and WT mice for 8 weeks, whereas in sham-operated mice the suture was passed around the aorta but not ligated as previously described.24 TAC gradients (Figure S1 in the Data Supplement) and mortality (40%) were not different between WT controls and R-SMKO mice (Figure S3). Mice with pressure gradients of ≤15 mm Hg were excluded from analysis as previously described.25
Histopathologic Analysis
After 28 days of Ang II infusion, C-SMKO hearts were harvested, weighed, fixed in formalin, sectioned, and stained with Masson trichrome. The severity of cardiac pathology was determined by a pathologist (P. Ruiz) without knowledge of genotypes based on the presence and severity of component abnormalities, including cellular infiltrate, myocardial cell injury, vessel wall thickening, and fibrosis. Grading for each component was performed by using a semiquantitative scale where 0 was normal and 1–3+ represented mild through severe abnormalities. The total cardiac injury score for each heart was a summation of the component injury scores across 5 domains scored 0 to 3 (interstitial Infiltrate, myocyte injury, vascular medial/fibrointimal changes, interstitial fibrosis, chronic vascular injury grade, total score: 0–15). For quantitation of fibrosis, 6 to 9 images were randomly captured for each sample. Percentage of fibrosis area (blue color on Masson trichrome stained sections) were measured by using a computer image analysis system (National Institutes of Health image J, 1.8.0) and averaged for each animal and then across each group by F. Rianto without knowledge of the genotype or treatment. Cardiomyocyte diameters were obtained by measuring the shortest perpendicular axis of a transverse section of a single cardiomyocyte using a computer image analysis system (National Institutes of Health image J, 1.8.0). Analysis of hematoxylin and eosin–stained sections was performed by F. Rianto without knowledge of genotype or treatment condition. Diameters of between 100-250 cardiomyocytes were measured for each sample from different areas at ×40 magnification.
Statistical Analysis
The values for each parameter within a group are expressed as the mean±SEM. For comparisons between groups with normally distributed data, statistical significance was assessed using 2-way ANOVA followed by Sidak adjusted for multiple comparisons. For comparisons within groups, normally distributed variables were analyzed by a paired t test, whereas non-normally distributed variables were analyzed by the Wilcoxon signed-rank test. Normality was determined using the Shapiro-Wilk W test. A P value of <0.05 was considered statistically significant.
Results
Mice With Cell-Specific Deletion of AT1 Receptors in Heart and Vascular Smooth Muscle Cells
Generation of mice with cell-specific deletion of the AT1A receptor from smooth muscle and myocyte cell lineages using the KISm22α-Cre (R-SMKO) and (transgene) Sm22α-Cre (C-SMKO) has been described previously.19,26 We examined cardiac expression of Cre recombinase in both lines using the mTmG dual-reporter mouse line20 and found equivalent, robust GFP (green fluorescent protein) fluorescence in hearts of both KIsm22α-Cre-mTmG and TgSm22α-Cre-mTmG mice, which was absent in WT Cre-negative-mTmG mice (Figure 1A, 1B, and 1C), indicating robust Cre recombinase activity in cardiomyocytes. Successful excision of Agtraflox allele from heart was confirmed in reverse transcription PCR analysis of AT1A receptor mRNA expression shown in Figure 1D and 1E. Although AT1A receptor mRNA was easily detected in the hearts of WT mice, levels of AT1A mRNA were substantially reduced by >95% in hearts of both C-SMKO (P=0.02) and R-SMKO mice (P<0.0001). Thus, cardiac expression of AT1A receptors is similarly and dramatically reduced in both C- and R-SMKOs.
Figure 1.
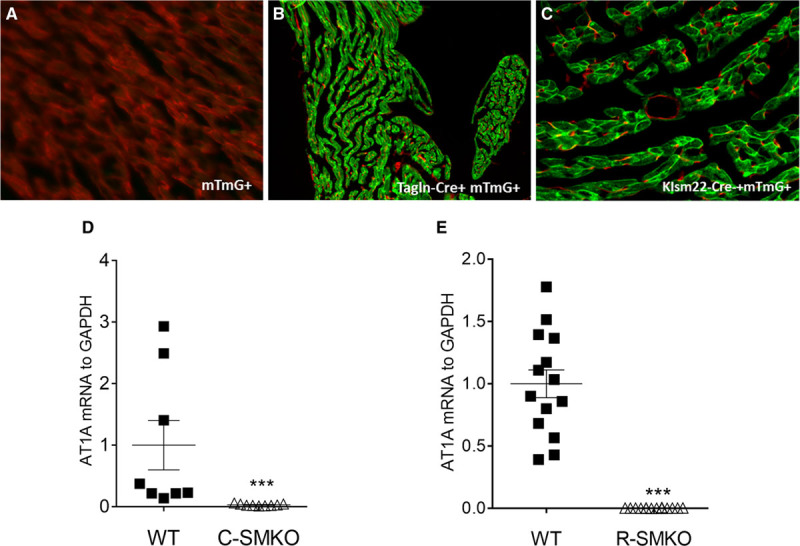
Verification of cardiomyocyte-specific Cre recombinase expression and AT1A deletion. Cre expression patterns of both the Tagln-Cre and KIsm22-Cre mouse lines were verified by intercrossing with the mTmG reporter mouse line, which contains a Cre-activated GFP (green fluorescent protein) transgene. A, GFP expression indicated by green fluorescence was not seen in cardiac myocytes from Crenegative mTmG+ mice. B, Robust GFP expression reflecting Cre activity was observed in cardiomyocytes from Tagln-Cre+ mTmG+ and (C) in KIsm22-Cre+ mTmG+ mice. D and E, AT1A receptor is efficiently deleted from the hearts of both C-SMKO and R-SMKO mice. Quantitative reverse transcription polymerase chain reaction performed on whole left ventricle in C-SMKO, R-SMKO, and wild-type (WT) mice. D, mRNA for the AT1A receptor was easily detected from the hearts of WT mice (n=8) but not hearts of C-SMKO mice (n=10; WT1±0.4 vs C-SMKO 0.03±0.0005 AT1A to GAPDH mRNA, arbitrary units, ***P=0.02). E, Similarly, AT1A receptor mRNA expression was virtually absent in the hearts of R-SMKO mice (control; n=14, R-SMKO n=14; control 1±0.11 vs R-SMKO 0.003±0.0005 AT1A to GAPDH mRNA, arbitrary units, ***P<0.0001, unpaired t test). Data are expressed as mean±SEM.
Using mTmG mice, we have previously demonstrated robust Cre expression and coincident deletion of AT1AR mRNA in large arteries of both C-SMKO19 and R-SMKO26 mice, whereas Cre expression in small resistance vessels such as preglomerular arterioles was only observed in R-SMKO mice.26 In this regard, expression of AT1A receptors in preglomerular vessels of C-SMKOs was not different from WT controls.19 Furthermore, acute vasoconstrictor responses to Ang II are dramatically attenuated in R-SMKO26 mice but are relatively preserved in C-SMKOs,19 indicating functional absence of AT1A receptors from resistance vessels in R-SMKOs. These differing patterns of vascular Cre expression have been seen by others in KIsm22α- and Tgsm22α-Cre mouse lines.27,28 Taken together, our findings indicate virtually complete deletion of AT1A receptors from myocardium and larger, conduit arteries in both groups, whereas efficient deletion from resistance vessels is only seen in R-SMKOs.
Cardiac Hypertrophy With Chronic Ang II Infusion Follows Elevated BP
To induce cardiac hypertrophy, Ang II 1000 ng/(kg·min) was infused subcutaneously in 12- to 20-week old male WT, C-SMKO, and R-SMKO mice for 28 days using osmotic minipumps. Radiotelemetry units were also implanted to measure intraarterial pressure continuously in the conscious, unrestrained state. Heart-to-body weight ratios were determined in groups of WT, C-SMKO, and R-SMKO mice without Ang II to establish baseline values for each group. No differences in heart weight to body weight ratio were detected between groups that did not receive Ang II infusion (Figure 2A). As shown in Figure 2A, after 4 weeks of Ang II infusion, heart-to-body weight ratios were significantly increased to a similar extent in WT (44%; versus uninfused) and C-SMKO (51% versus Uninfused) mice. By contrast, Ang II caused only a modest increase of 23% in Heart-to-body weight ratio in the R-SMKO mice versus uninfused, and this was significantly less than the extent of cardiac hypertrophy in the other 2 groups WT, 44% and C-SMKO, 51%). We did not detect any differences in cardiac injury or cardiomyocyte diameter between WT and C-SMKO mice after 4 weeks of Ang II infusion (Figure 6A and 6B). Thus, although both C- and R-SMKOs lack AT1A receptors in the heart, C-SMKOs developed robust cardiac hypertrophy with Ang II infusion that was not different from WTs, whereas cardiac hypertrophy was significantly attenuated in the R-SMKO group.
Figure 2.

Reduction in cardiac hypertrophy and systolic blood pressure (SBP) observed in mice lacking AT1A receptors in resistance vessels. A, Heart-to-body weight ratios were calculated at baseline in wild-type (WT; n=17), C-SMKO (n=12), and R-SMKO (n=6) mice and separate groups after 28 d of Ang (angiotensin) II infusion; WT (n=18), C-SMKO (n=13), and R-SMKO (n=9). No differences in heart-to-body weight ratio were detected between the 3 groups at baseline, and Ang II infusion caused significant increases in heart-to-body weight ratio in all groups: WT: 5.0±0.1–7.1±0.2 mg/g *P<0.0001; C-SMKO: 4.7±0.2–7.1±0.1 mg/g #P<0.0001; and R-SMKO: 4.7±0.2–5.8±0.2 mg/g ^P=0.01, analyzed by 2-way ANOVA with Sidak test for multiple comparisons. After Ang II, heart-to-body weight ratios were significantly lower in R-SMKOs compared with both WT and C-SMKO (R-SMKO: 5.8±0.2 vs WT: 7.1±0.2, ϕP<0.0001 vs ΔC-SMKO: 7.1±0.1 mg/g, P<0.0001). B, Systolic BP was measured via radiotelemetry in WT (n=14), C-SMKO (n=8), and R-SMKO (n=6) mice. At baseline, R-SMKO have had lower systolic BP than WT and C-SMKO mice (123±5 vs137±3 and 143±3 mm Hg, *P=0.01, **P=0.003). Compared with baseline, Ang II infusion caused significant increases in systolic BP in WT (137±3–167±3 mm Hg; #P<0.0001) and C-SMKO (143±3 to 179±7 mm Hg; ϕP<0.0001) but not R-SMKO (123±5 to 136±2 mm Hg; P=nonsignificant) mice (daily averaged). C, Correlation between average systolic BP during Ang II–induced hypertension and cardiac hypertrophy. The degree of cardiac hypertrophy as measured by heart-to-body weight ratio (mg/g). Across the experimental groups, cardiac hypertrophy was significantly correlated with average SBP during the Ang II infusion period. Combined, solid line: r=0.55, R2=0.31, P=0.005. Pearson correlation. D, Twenty-four–hour heart rates, (E) SBP, and (F) diastolic BP (DBP) of WT, C-SMKO, and R-SMKO at baseline and after Ang II–induced hypertension. Daily heart rates are higher during days 1 and 2 of Ang II–induced hypertension in R-SMKO compared with C-SMKO and WT mice (589±51 vs 535±24 vs 536±54 bpm, P=0.005 R-SMKO to C-SMKO, P=0.002 R-SMKO to WT). SBP and DBP were significantly decreased at baseline and throughout the entire Ang II period, similar to averages in B.
Figure 6.

Assessment of cardiac histology of C-SMKO mice after Ang (angiotensin) II–induced hypertension and in R-SMKO mice after transverse aortic constriction (TAC). A, Histolopathological analysis of hearts from wild-type (WT; n=5) and C-SMKO (n=5) mice was performed after 28 of Ang II–induced hypertension. Masson trichrome staining showed only minimal to mild cardiac injury in WT and C-SMKO with no difference between groups (WT: 0.9±0.78 vs C-SMKO 2.6±1.2 cumulative cardiac injury score). Representative ×10 images of WT and C-SMKO hearts. B, Cardiomyocyte diameter was assessed in hematoxylin and eosin–stained hearts from Ang II–infused WT (n=5) and C-SMKO (n=5) mice. There was no difference in cardiomyocyte diameter between Ang II infused WT and C-SMKO mice (WT 20±0.9 vs C-SMKO 21±1.3 μm; P=nonsignificant [NS]). C, The extent of cardiac fibrosis in hearts WT and R-SMKO mice from sham surgery and TAC groups stained with Masson trichrome were determined using a computer image analysis system. Scale bar=50 μm. Scale bar=50 μm. More fibrosis was seen after TAC, but these differences did not achieve statistical significance in any of the groups (WT:sham: 4.4±0.9 vs TAC 6.7±1.2% fibrosis; R-SMKO:sham: 3.9±0.4 vs TAC 10.6±5.4% fibrosis). D, Cardiomyocyte diameter was assessed in hematoxylin and eosin stained hearts from WT and R-SMKO mice 8 wk after sham surgery or TAC. Scale bar=50μm. There was no difference in cardiomyocyte diameter in sham mice (WT 14±0.3 vs R-SMKO 14±0.3 μm; P=NS). Cardiomyocyte diameter increased in both WT and R-SMKO mice after 8 wk of TAC (WT 17±0.4 μm; P=0.06 vs WT sham; R-SMKO 21±0.7 μm; ***P=0.02 vs R-SMKO sham). After TAC, cardiomyocyte diameter was significantly greater in R-SMKO compared with WT mice (**P=0.002). Error bars represent SEM. Statistics performed with 2-way ANOVA with Sidak multiple comparisons.
We also measured systolic BP at baseline and with Ang II infusion (Figure 2B). R-SMKO mice had lower systolic BP at baseline (123±5 mm Hg) compared with both WT (137±3 mm Hg) and C-SMKO (143±3 mm Hg) mice (Figure 2B). Compared with baseline, Ang II caused significant increases in mean systolic BP in WT of 30±4 mm Hg and C-SMKO of 37±7 mm Hg. The magnitude of systolic BP elevation was similar between these 2 groups. In contrast, mean systolic BP only increased by 14±2 mm Hg in R-SMKO mice with Ang II infusion, which was significantly less than WTs or C-SMKOs, reflecting the key role of AT1A receptors in resistance vessels in Ang II–dependent hypertension. Moreover, there was a correlation between achieved average systolic BP and heart/body weight ratio (Figure 2C). Thus, in Ang II hypertension, the relative extent of cardiac hypertrophy in the groups mirrored the mean increases in BP and not presence or absence of AT1A receptors in the heart.
Cardiac BNP mRNA Expression
As transcription of the BNP gene is characteristically enhanced with cardiac hypertrophy,29,30 we measured mRNA levels of BNP in the various experimental groups using real-time reverse transcription quantitative PCR. Levels of BNP mRNA were measured in ventricular tissues from mice in each of the experimental groups after 4 weeks of Ang II infusion, compared with respective WT controls that did not receive Ang II infusion. As shown in Figure 3, there were no differences in mRNA levels of BNP at baseline between the 3 experimental groups, while chronic infusion of Ang II caused significant increases in BNP expression in WT and C-SMKO but not R-SMKO hearts compared with their respective uninfused control hearts. However, after Ang II infusion, BNP mRNA levels in R-SMKO mice were significantly lower than WT and C-SMKO mice.
Figure 3.
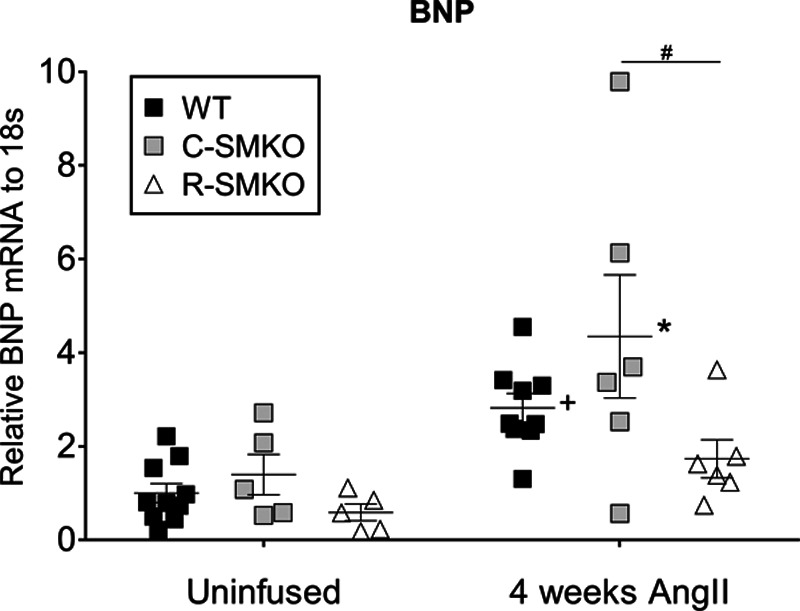
Levels of heart BNP (B-type natriuretic peptide) before and after chronic Ang (angiotensin) II. Left ventricular (LV) BNP mRNA expression was measured using real-time reverse transcription–quantitative polymerase chain reaction at baseline and after 4 wk of Ang II infusion in wild-type (WT), C-SMKO, and R-SMKO mice and normalized to 18s mRNA levels within each sample. For comparisons between groups, all values were normalized to the uninfused WT group. In uninfused mice, there were no differences in BNP levels between WT (n=10), C-SMKO (n=5), or R-SMKO (n=5) mice. After 4 wk of chronic Ang II infusion, BNP mRNA levels in LV increased significantly in all groups (WT [n=9], C-SMKO [n=6], and R-SMKO [n=6]; +P=0.0001 for Ang II effect, F[1, 35]=18.2). However, after Ang II infusion, BNP values in R-SMKO were significantly lower than C-SMKO mice (#P=0.04). Error bars represent SEM. Statistics were performed using a 2-way ANOVA with Sidak post-test for multiple comparisons.
Diminished Oxidative Stress in Hearts Lacking AT1A Receptors in Cardiomyocytes
Activation of AT1 receptors in cardiomyocytes stimulates free radical generation, which may contribute to end-organ damage in hypertension.31,32 To examine levels of oxidative stress in the heart, we assessed oxidation of proteins in WT and C-SMKO mice to assess if downstream AT1A receptor effects are altered in the hearts even with a similar degree of hypertension after Ang II infusion. We determined the content of the 2 oxidized amino acids, 3-nitrotyrosine, and dityrosine, as molecular signatures of peroxynitrite-mediated oxidation using isotope dilution liquid chromatography–tandem mass spectrometry.19,23 As shown in Figure 4, levels of both dityrosine (162±29 versus 68±22 μmol/mol tyrosine) and 3-nitrotyrosine (4899±2022 versus 569±125 μmol/mol tyrosine) were significantly reduced in the hearts from C-SMKO compared with WT mice (Figure 4B). This indicates a significant role for AT1 receptors in cardiomyocytes to promote oxidative stress in the heart, yet, these actions do not seem to affect the development of cardiac hypertrophy, since heart weights and BNP levels in Ang II–infused C-SMKOs were similar to WTs (Figures 2 and 3).
Figure 4.

C-SMKO mice have reduced oxidative stress in cardiac myocytes after 4 wk of Ang (angiotensin) II infusion. Oxidized amino acids in hearts were quantified by liquid chromatography–electrospray ionization tandem mass spectrometry using multiple reaction monitoring mode in wild-type (WT; n=5) and C-SMKO (n=5) mice after 4 wk of Ang II. Compared with WT mice, markedly diminished levels of (A) dityrosine (162±29 vs 68±22 μmol/mol tyrosine; ***P=0.03) and (B) 3-nitrotyrosine (4899±2022 vs 569±125 μmol/mol tyrosine; **P=0.008) were detected in the hearts of C-SMKO mice after Ang II infusion. Error bars represent SEM.
Absence of Cardiomyocyte AT1A Receptors Does Not Affect Responses to TAC
To examine cardiac hypertrophy responses in R-SMKO mice in a model where pressure load could be directly controlled, we utilized TAC. As shown in Figure 5A, significant increases in LV weight/tibia length were observed in both WT (6.0±0.6 versus 8.6±0.4) and R-SMKO (5.6±0.1 versus 9.2±0.8) mice after TAC compared with matched sham WTs. There was no difference in LV weight/tibia length between WT and R-SMKO after 8 weeks of TAC (8.6±0.4 versus 9.2±0.8). Thus, in contrast to the Ang II–infusion model, the absence of AT1A receptors in cardiomyocytes in R-SMKO mice does not diminish ventricular weight after TAC. We measured fractional shortening, as a reflection of ventricular function, at baseline and 8 weeks after TAC. As depicted in Figure 5B, TAC was associated with significant reductions of fractional shortening in R-SMKO mice (55±1.2% versus 37±6.3%). However, WT mice had reduced fractional shortening but this was not significant (53±1.3% versus 47±3.1%). Although fractional shortening after TAC was numerically lower in R-SMKO than WTs (37±6.3% versus 47±3.1%), this difference between genotypes at 8 weeks did not achieve statistical significance. In these studies, we also used echocardiography to assess posterior and septal wall thicknesses at baseline and after TAC. As shown in Figure 5C, WT mice (n=7) had significant increases in ventricular wall thickness by echocardiography after TAC (2.3±0.08 versus 2.7±0.09 mm). Likewise, ventricular wall thickness also tended to be increased in R-SMKO mice (n=7) 8 weeks after TAC (2.5±0.09 versus 2.7±0.15 mm) but this did not reach statistical significance. There was no significant difference in ventricular wall thickness between the WT and R-SMKO groups at 8 weeks after TAC. We measured the end-diastolic dimension (EDD) at baseline and after 8 weeks of TAC in both WT and R-SMKO mice (Figure 5D). At baseline, we did not detect a difference in EDD between WT and R-SMKO (3.7±0.1 versus 3.5±0.2 mm). After 8 weeks of TAC, the EDD did not change in WT mice (3.7±0.1–3.5±0.1 mm). However, the EDD increased in R-SMKO mice pre to 8 weeks (3.5±0.2–4.3±0.3 mm). Thus, EDD was greater after 8 weeks of TAC in R-SMKOs compared with WT (4.3±0.3 versus 3.5±0.1 mm). We also examined end-systolic diameter (ESD) in WT and R-SMKO mice (Figure 5E). At baseline, we did not detect a difference in ESD between WT and R-SMKO (1.6±0.07 versus 1.5±0.09 mm). After 8 weeks of TAC, the ESD did not change in WT mice (1.7±0.1–1.9±0.1 mm). However, the ESD increased in R-SMKO mice pre to 8 weeks of TAC (1.6±0.1–2.8±0.5 mm). Thus, ESD was greater after 8 weeks of TAC in R-SMKOs compared with WT (2.8±0.4 versus 1.9±0.1 mm). With regard to histopathology, no difference in severity of fibrosis was observed between WT and R-SMKO mice. However, R-SMKO mice had larger cardiomyocyte diameter compared with controls (Figure 6C/6D). These findings further indicate that while the absence of AT1A receptors in cardiomyocytes does not prevent development of hypertrophy and in this TAC model, severity of cardiomyopathy may actually be enhanced. WT mice develop concentric hypertrophy after acute pressure overload, whereas R-SMKO mice develop ventricular dilation and eccentric hypertrophy. Thus, AT1A receptors in cardiomyocytes may provide protection against adverse remodeling in this acute pressure overload model.
Figure 5.

Transverse aortic banding (TAC) causes hypertrophy and reduced cardiac function in R-SMKO mice. TAC was performed in wild-type (WT; n=7) and R-SMKO (n=7) mice. As controls, sham surgeries were carried out in separate groups of WT (n=5) and R-SMKO (n=5). A, TAC induced similar levels of cardiac hypertrophy assessed as left ventricular (LV) weight-to-tibia length in WT and R-SMKO mice. **P=0.02; ***P=0.004. B, Fractional shortening (FS) fell in both WT and R-SMKO mice 8 wk after TAC but this reduction only achieved statistical significance in R-SMKOs (**P=0.02). There was no significant difference in FS between WT and R-SMKO mice after 8 wk exposure to TAC. C, Cardiac hypertrophy was also assessed by echocardiography as the sum of posterior wall (PW) and septal wall (SW) thicknesses. There were numerical increases in this parameter in both groups, but this only achieved statistical significance in WT mice (**P<0.01). There was no significant difference in ventricular wall thickness between the WT and R-SMKO groups at 8 wk after TAC. D, End-diastolic dimension (EDD) was measured at baseline and after 8 wk of TAC in both WT and R-SMKO mice. A baseline, there was no difference in EDD between WT and R-SMKO (P=nonsignificant [NS]) and 8 wk of TAC had no effect on EDD in the WT group. However, EDD increased significantly in R-SMKO after TAC (*P=0.04) and EDD post-TAC was significantly greater in R-SMKOs than WT mice (#P=0.02). E, At baseline, we did not detect differences in end-systolic diameter (ESD) between WT and R-SMKO (P=NS). TAC did not affect ESD WT mice (P=NS) but caused a significant increase in ESD in R-SMKO mice (**P=0.01) such that ESD was significantly greater after 8 wk of TAC in R-SMKOs compared with WT (#P=0.03). Error bars represent SEM. Statistics performed with 2-way ANOVA (repeated measures) with Sidak multiple comparisons.
Discussion
Activation of the RAS potently induces hypertension and its associated end-organ damage.3,33 The major pathological consequences of RAS activation are mediated by AT1 receptors. Along with their actions to increase BP, AT1 receptors may contribute to complications of hypertension through direct actions in target tissues to promote injury.34 For example, AT1 receptors are expressed in cardiac myocytes where they can have potentially maladaptive actions, such as induction of cellular hypertrophy35 and activation of pathways associated with fibrosis and inflammation.36,37
LVH is one of the most common complications of hypertension and is associated with significant increase in cardiovascular risk.38,39 In humans, a role for AT1 receptors in development of LVH has been demonstrated by clinical trials showing better efficacy of Ang receptor blockers in preventing or attenuating LVH relative to other classes of antihypertensive agents.40,41 Moreover, findings of enhanced regression of LVH with AT1 receptor blockade at comparable levels of BP control seem to support the importance of direct AT1 receptor actions to promote hypertrophy independent of BP.10,41 However, studies from our laboratory and others utilizing AT1A receptor-null mice16,42 have suggested that AT1 receptors drive the development of cardiac hypertrophy primarily by increasing BP rather than through direct cellular actions.43
In this study, we reexamined the relative impact of hemodynamic and cellular effects of the AT1 receptor in cardiac hypertrophy using newly developed mouse models.19,26 We generated 2 mouse lines, both lacking AT1A receptors in the heart but with divergent vascular responses to Ang II due to the presence (C-SMKO) or absence (R-SMKO) of AT1 receptors in vessels mediating Ang II–dependent control of peripheral vascular resistance.19,26 These models provide a unique opportunity to precisely assess the contributions of AT1 receptors actions in cardiac myocytes compared with their effects to increase vascular resistance and pressure load.
We found that robust cardiac hypertrophy developed in C-SMKOs, lacking AT1AR in cardiac myocytes but with preserved expression in resistance vessels. By contrast, hypertrophy was attenuated in R-SMKOs, which fail to develop full-fledged hypertension because of impaired vascular responses to Ang II. Thus, eliminating AT1A receptors from myocardium is not sufficient to protect from cardiac hypertrophy when BP is elevated. Moreover, by increasing pressure load directly with TAC, hypertrophy could easily be induced in the R-SMKOs that had been protected in the Ang II infusion model. R-SMKO mice also had evidence of eccentric rather than concentric hypertrophy, along with increased cardiomyocyte diameters, when subjected to pressure overload. One explanation for this might be loss of AT1 receptor–dependent, noncanonical β-arrestin signaling in cardiomyocytes perhaps leading to cytotoxicity and worsening heart function. β-arrestin signaling through the AT1 receptor is cytoprotective and promotes cardiac contractility.44,45
The dominant role of pressure to drive hypertrophy was further corroborated by the strong positive correlation between BP and heart weight across all groups (Figure 2C). Our findings also highlight the key contribution of AT1 receptor-induced peripheral vasoconstriction to cardiac hypertrophy.
The development of LVH is associated with a well-characterized molecular responses including enhanced transcription of the BNP gene.46 Moreover, elevated BNP level is a useful clinical biomarker in LVH and heart failure.47,48 We compared BNP expression in WT, C- and R-SMKO mice at baseline and after chronic infusion of Ang II. BNP levels increased with Ang II infusion and were highest in WT and C-SMKO mice, but diminished in R-SMKOs (Figure 3). Furthermore, we found significant correlations between BNP expression, BPs, and extent of cardiac hypertrophy across the various mouse lines. Therefore, BNP mRNA levels in Ang II–dependent hypertension are driven by pressure load, with no evidence for an appreciable influence of AT1 receptors actions in cardiac myocytes on BNP gene expression.
Activation of AT1 receptors stimulates generation of reactive oxygen species.49 Although it has been suggested that these free radicals may participate in physiological AT1 receptor signaling,4 Ang II–dependent oxidative stress also contributes to tissue damage and end-organ injury in hypertension.32 Thus, we were interested in understanding the relationship between AT1 receptor–mediated oxidative stress in the heart and development of cardiac hypertrophy in our models. To this end, we measured 3-nitrotyrosine and dityrosine in heart tissue from WT and C-SMKO mice after Ang II infusion as both of these lines develop marked and equivalent cardiac hypertrophy with Ang II (Figure 2), but AT1A receptor expression is absent in the C-SMKOs. These oxidized amino acids provide a molecular signature of reactive oxygen species generated in tissues.50 Although there were prominent levels of 3-nitrotyrosine and dityrosine levels in WT hearts, these were dramatically reduced in C-SMKOs indicating a critical role for AT1 receptors in cardiomyocytes to promote cardiac oxidative stress in Ang II hypertension (Figure 4). Nonetheless, despite the marked attenuation of free radical generation in C-SMKOs, cardiac hypertrophy occurred unabated consistent with dominant effects of pressure load in this process.
There is ample clinical evidence indicating superiority of RAS blockade in prevention and regression of LVH compared with other therapies for hypertension. Our studies suggest that this enhanced efficacy cannot be explained by blockade of AT1 receptor actions in the heart, including attenuation of oxidative stress. Instead, they highlight the importance of pressure load contributed in part by AT1 receptor–dependent increases in peripheral vascular resistance. It follows that the beneficial actions of RAS antagonists in LVH may relate to the specific characteristics of their BP lowering, such as enhanced reduction of central arterial pressures or sustained duration of antihypertensive effects. We acknowledge that with extended RAS activation over many years in humans with hypertension, pathological consequences of direct cardiac actions of AT1 receptors might become more important. Nonetheless, the lack of any detectable impact of cardiac AT1 receptors on development of hypertrophy in animal models with sustained elevation of Ang II levels is striking and suggests a limited capacity to promote LVH.
Perspectives
Cardiac hypertrophy is one of the most common complications of hypertension and is associated with increased cardiovascular risk. Clinical studies show that blockade of the renin-Ang system promotes regression of cardiac hypertrophy, and it has been suggested that these effects are above and beyond the impact of BP lowering. In addition, Ang II consistently causes hypertrophy of cultured cardiomyocytes. Using novel genetically modified mice, we show here that cardiac hypertrophy is primarily driven by increased pressure and not direct activation of AT1 receptors in cardiomyocytes.
Acknowledgments
The Duke Cardiovascular Physiology Core and the University of Michigan George M. O`Brien Kidney Research Core Center (P30-DK081943). Thanks to Caitlyn Vlasschaert for making the graphical abstract.
Sources of Funding
This work was supported by the Edna and Fred L. Mandel Center for Hypertension and Atherosclerosis Research, National Institutes of Health grants HL056122 and P30DK096493 (to T.M. Coffman), a Career Development Award IK2BX002240 from the Biomedical Laboratory Research and Development Service of the Department of Veterans Affairs Office of Research and Development (to M.A. Sparks), a Scientist Development Award 13SDG13990017 from the American Heart Association (to M.A. Sparks), a Chair’s Research Award from the Duke Department of Medicine (to M.A. Sparks), and a grant from the Institute for Medical Research at the Durham VAMC (to M.A. Sparks). The views expressed in this article are those of the authors and do not necessarily represent the policy or position of the United States Department of Veteran Affairs or the United States government.
Disclosures
None.
Supplementary Material
{kind=link}
Nonstandard Abbreviation and Acronyms
- ACE
- angiotensin-converting enzyme
- Ang
- angiotensin
- AT1
- type 1 angiotensin
- BP
- blood pressure
- LVH
- left ventricular hypertrophy
- RAS
- renin-angiotensin system
- TAC
- transverse aortic constriction
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/HYPERTENSIONAHA.119.14079.
This article is part of the Null Hypothesis Collection, a collaborative effort between CBMRT, AHA Journals, and Wolters Kluwer, and has been made freely available through funds provided by the CBMRT. For more information, visit https://www.ahajournals.org/null-hypothesis.
For Sources of Funding and Disclosures, see page 403.
Contributor Information
Fitra Rianto, Email: fitra.rianto@duke.edu.
Edward Diaz, Email: edward_diaz@med.unc.edu.
Ritika Revoori, Email: ritika.revoori@duke.edu.
Thien Hoang, Email: thihoang@utmb.edu.
Lucas Bouknight, Email: lucas@med.unc.edu.
Johannes Stegbauer, Email: johannes.stegbauer@googlemail.com.
Anuradha Vivekanandan-Giri, Email: anuvivek15@gmail.com.
Phillip Ruiz, Email: PRuiz@med.miami.edu.
Subramaniam Pennathur, Email: spennath@med.umich.edu.
Dennis M. Abraham, Email: dennis.abraham@duke.edu.
Susan B. Gurley, Email: gurley@ohsu.edu.
Steven D. Crowley, Email: steven.d.crowley@duke.edu.
Thomas M. Coffman, Email: tcoffman@duke.edu.
Novelty and Significance
What Is New?
The absence of AT1 (type 1 angiotensin) receptors in cardiomyocytes does not impair the development of cardiac hypertrophy in 2 distinct experimental models.
Increases in blood pressure driven by changes in peripheral vascular resistance are a critical determinant of cardiac hypertrophy in hypertension.
What Is Relevant?
Blood pressure control is paramount to reducing the development of cardiac hypertrophy in hypertension.
Summary
We generated 2 novel mouse models in which the AT1A receptor was knocked out specifically in cardiomyocytes and vascular smooth muscle. Each mouse had differential hypertensive responses to Ang II–induced hypertension. The mice that developed cardiac hypertrophy, had blood pressure responses indistinguishable to wild-type controls. Whereas, mice with an attenuated blood pressure response to Ang II–induced hypertension had similarly reduced cardiac hypertrophy development. In the mice with protection of cardiac hypertrophy with Ang II infusion, cardiac hypertrophy was induced using a pressure overload model. Thus, demonstrating the primacy of pressure increases in the development of heart hypertrophy.
References
- 1.Muntner P, Carey RM, Gidding S, Jones DW, Taler SJ, Wright JT, Jr, Whelton PK. Potential US population impact of the 2017 ACC/AHA high blood pressure guideline. Circulation. 2018;137:109–118. doi: 10.1161/CIRCULATIONAHA.117.032582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frohlich ED. Cardiac hypertrophy in hypertension. N Engl J Med. 1987;317:831–833. doi: 10.1056/NEJM198709243171310 [DOI] [PubMed] [Google Scholar]
- 3.Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM. Classical Renin-Angiotensin system in kidney physiology. Compr Physiol. 2014;4:1201–1228. doi: 10.1002/cphy.c130040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, Scalia R, Eguchi S. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev. 2018;98:1627–1738. doi: 10.1152/physrev.00038.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matchar DB, McCrory DC, Orlando LA, Patel MR, Patel UD, Patwardhan MB, Powers B, Samsa GP, Gray RN. Systematic review: comparative effectiveness of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers for treating essential hypertension. Ann Intern Med. 2008;148:16–29. doi: 10.7326/0003-4819-148-1-200801010-00189 [DOI] [PubMed] [Google Scholar]
- 6.CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the cooperative north scandinavian enalapril survival study (consensus). N Engl J Med. 1987;316:1429–1435. doi: 10.1056/NEJM198706043162301 [DOI] [PubMed] [Google Scholar]
- 7.Yusuf S, Pitt B, Davis CE, Hood WB, Jr, Cohn JN; SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med. 1992;327:685–691. doi: 10.1056/NEJM199209033271003 [DOI] [PubMed] [Google Scholar]
- 8.Mathew J, Sleight P, Lonn E, Johnstone D, Pogue J, Yi Q, Bosch J, Sussex B, Probstfield J, Yusuf S; Heart Outcomes Prevention Evaluation (HOPE) Investigators. Reduction of cardiovascular risk by regression of electrocardiographic markers of left ventricular hypertrophy by the angiotensin-converting enzyme inhibitor ramipril. Circulation. 2001;104:1615–1621. doi: 10.1161/hc3901.096700 [DOI] [PubMed] [Google Scholar]
- 9.Klingbeil AU, Schneider M, Martus P, Messerli FH, Schmieder RE. A meta-analysis of the effects of treatment on left ventricular mass in essential hypertension. Am J Med. 2003;115:41–46. doi: 10.1016/s0002-9343(03)00158-x [DOI] [PubMed] [Google Scholar]
- 10.Dahlöf B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, et al. ; LIFE Study Group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:995–1003. doi: 10.1016/S0140-6736(02)08089-3 [DOI] [PubMed] [Google Scholar]
- 11.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37–45. doi: 10.1172/JCI62839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watkins SJ, Borthwick GM, Oakenfull R, Robson A, Arthur HM. Angiotensin II-induced cardiomyocyte hypertrophy in vitro is TAK1-dependent and Smad2/3-independent. Hypertens Res. 2012;35:393–398. doi: 10.1038/hr.2011.196 [DOI] [PubMed] [Google Scholar]
- 13.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006 [DOI] [PubMed] [Google Scholar]
- 14.Hein L, Stevens ME, Barsh GS, Pratt RE, Kobilka BK, Dzau VJ. Overexpression of angiotensin AT1 receptor transgene in the mouse myocardium produces a lethal phenotype associated with myocyte hyperplasia and heart block. Proc Natl Acad Sci U S A. 1997;94:6391–6396. doi: 10.1073/pnas.94.12.6391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paradis P, Dali-Youcef N, Paradis FW, Thibault G, Nemer M. Overexpression of angiotensin II type I receptor in cardiomyocytes induces cardiac hypertrophy and remodeling. Proc Natl Acad Sci U S A. 2000;97:931–936. doi: 10.1073/pnas.97.2.931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103:17985–17990. doi: 10.1073/pnas.0605545103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harada K, Komuro I, Zou Y, Kudoh S, Kijima K, Matsubara H, Sugaya T, Murakami K, Yazaki Y. Acute pressure overload could induce hypertrophic responses in the heart of angiotensin II type 1a knockout mice. Circ Res. 1998;82:779–785. doi: 10.1161/01.res.82.7.779 [DOI] [PubMed] [Google Scholar]
- 18.Gurley SB, Riquier-Brison ADM, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, et al. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. 2011;13:469–475. doi: 10.1016/j.cmet.2011.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sparks MA, Parsons KK, Stegbauer J, Gurley SB, Vivekanandan-Giri A, Fortner CN, Snouwaert J, Raasch EW, Griffiths RC, Haystead TA, et al. Angiotensin II type 1A receptors in vascular smooth muscle cells do not influence aortic remodeling in hypertension. Hypertension. 2011;57:577–585. doi: 10.1161/HYPERTENSIONAHA.110.165274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335 [DOI] [PubMed] [Google Scholar]
- 21.Butz GM, Davisson RL. Long-term telemetric measurement of cardiovascular parameters in awake mice: a physiological genomics tool. Physiol Genomics. 2001;5:89–97. doi: 10.1152/physiolgenomics.2001.5.2.89 [DOI] [PubMed] [Google Scholar]
- 22.Herrera M, Sparks MA, Alfonso-Pecchio AR, Harrison-Bernard LM, Coffman TM. Lack of specificity of commercial antibodies leads to misidentification of angiotensin type 1 receptor protein. Hypertension. 2013;61:253–258. doi: 10.1161/HYPERTENSIONAHA.112.203679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pennathur S, Bergt C, Shao B, Byun J, Kassim SY, Singh P, Green PS, McDonald TO, Brunzell J, Chait A, et al. Human atherosclerotic intima and blood of patients with established coronary artery disease contain high density lipoprotein damaged by reactive nitrogen species. J Biol Chem. 2004;279:42977–42983. doi: 10.1074/jbc.M406762200 [DOI] [PubMed] [Google Scholar]
- 24.Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, Ross J, Jr, Chien KR. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci U S A. 1991;88:8277–8281. doi: 10.1073/pnas.88.18.8277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abraham DM, Lee TE, Watson LJ, Mao L, Chandok G, Wang HG, Frangakis S, Pitt GS, Shah SH, Wolf MJ, et al. The two-pore domain potassium channel TREK-1 mediates cardiac fibrosis and diastolic dysfunction. J Clin Invest. 2018;128:4843–4855. doi: 10.1172/JCI95945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sparks MA, Stegbauer J, Chen D, Gomez JA, Griffiths RC, Azad HA, Herrera M, Gurley SB, Coffman TM. Vascular type 1A angiotensin II receptors control BP by regulating renal blood flow and urinary sodium excretion. J Am Soc Nephrol. 2015;26:2953–2962. doi: 10.1681/ASN.2014080816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang L, Villacorta L, Zhang J, Garcia-Barrio MT, Yang K, Hamblin M, Whitesall SE, D’Alecy LG, Chen YE. Vascular smooth muscle cell-selective peroxisome proliferator-activated receptor-gamma deletion leads to hypotension. Circulation. 2009;119:2161–2169. doi: 10.1161/CIRCULATIONAHA.108.815803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang N, Yang G, Jia Z, Zhang H, Aoyagi T, Soodvilai S, Symons JD, Schnermann JB, Gonzalez FJ, Litwin SE, et al. Vascular PPARgamma controls circadian variation in blood pressure and heart rate through Bmal1. Cell Metab. 2008;8:482–491. doi: 10.1016/j.cmet.2008.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakagawa O, Ogawa Y, Itoh H, Suga S, Komatsu Y, Kishimoto I, Nishino K, Yoshimasa T, Nakao K. Rapid transcriptional activation and early mRNA turnover of brain natriuretic peptide in cardiocyte hypertrophy. Evidence for brain natriuretic peptide as an “emergency” cardiac hormone against ventricular overload. J Clin Invest. 1995;96:1280–1287. doi: 10.1172/JCI118162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maeda K, Tsutamoto T, Wada A, Hisanaga T, Kinoshita M. Plasma brain natriuretic peptide as a biochemical marker of high left ventricular end-diastolic pressure in patients with symptomatic left ventricular dysfunction. Am Heart J. 1998;1355 Pt 1825–832. doi: 10.1016/s0002-8703(98)70041-9 [DOI] [PubMed] [Google Scholar]
- 31.Palomeque J, Rueda OV, Sapia L, Valverde CA, Salas M, Petroff MV, Mattiazzi A. Angiotensin II-induced oxidative stress resets the Ca2+ dependence of Ca2+-calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ Res. 2009;105:1204–1212. doi: 10.1161/CIRCRESAHA.109.204172 [DOI] [PubMed] [Google Scholar]
- 32.Briones AM, Touyz RM. Oxidative stress and hypertension: current concepts. Curr Hypertens Rep. 2010;12:135–142. doi: 10.1007/s11906-010-0100-z [DOI] [PubMed] [Google Scholar]
- 33.Dahlöf B. Left ventricular hypertrophy and angiotensin II antagonists. Am J Hypertens. 2001;14:174–182. doi: 10.1016/s0895-7061(00)01257-7 [DOI] [PubMed] [Google Scholar]
- 34.Weir MR. Effects of renin-angiotensin system inhibition on end-organ protection: can we do better? Clin Ther. 2007;29:1803–1824. doi: 10.1016/j.clinthera.2007.09.019 [DOI] [PubMed] [Google Scholar]
- 35.Dostal DE, Baker KM. Angiotensin II stimulation of left ventricular hypertrophy in adult rat heart. Mediation by the AT1 receptor. Am J Hypertens. 1992;55 Pt 1276–280. doi: 10.1093/ajh/5.5.276 [DOI] [PubMed] [Google Scholar]
- 36.Rogg H, de Gasparo M, Graedel E, Stulz P, Burkart F, Eberhard M, Erne P. Angiotensin II-receptor subtypes in human atria and evidence for alterations in patients with cardiac dysfunction. Eur Heart J. 1996;17:1112–1120. doi: 10.1093/oxfordjournals.eurheartj.a015008 [DOI] [PubMed] [Google Scholar]
- 37.Chen W, Frangogiannis NG. The role of inflammatory and fibrogenic pathways in heart failure associated with aging. Heart Fail Rev. 2010;15:415–422. doi: 10.1007/s10741-010-9161-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown DW, Giles WH, Croft JB. Left ventricular hypertrophy as a predictor of coronary heart disease mortality and the effect of hypertension. Am Heart J. 2000;140:848–856. doi: 10.1067/mhj.2000.111112 [DOI] [PubMed] [Google Scholar]
- 39.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203 [DOI] [PubMed] [Google Scholar]
- 40.Fagard RH, Celis H, Thijs L, Wouters S. Regression of left ventricular mass by antihypertensive treatment: a meta-analysis of randomized comparative studies. Hypertension. 2009;54:1084–1091. doi: 10.1161/HYPERTENSIONAHA.109.136655 [DOI] [PubMed] [Google Scholar]
- 41.Yasunari K, Maeda K, Watanabe T, Nakamura M, Yoshikawa J, Asada A. Comparative effects of valsartan versus amlodipine on left ventricular mass and reactive oxygen species formation by monocytes in hypertensive patients with left ventricular hypertrophy. J Am Coll Cardiol. 2004;43:2116–2123. doi: 10.1016/j.jacc.2003.12.051 [DOI] [PubMed] [Google Scholar]
- 42.Harada K, Komuro I, Shiojima I, Hayashi D, Kudoh S, Mizuno T, Kijima K, Matsubara H, Sugaya T, Murakami K, et al. Pressure overload induces cardiac hypertrophy in angiotensin II type 1A receptor knockout mice. Circulation. 1998;97:1952–1959. doi: 10.1161/01.cir.97.19.1952 [DOI] [PubMed] [Google Scholar]
- 43.Ahmad S, Cesana F, Lamperti E, Gavras H, Yu J. Attenuation of angiotensin II-induced hypertension and cardiac hypertrophy in transgenic mice overexpressing a type 1 receptor mutant. Am J Hypertens. 2009;22:1320–1325. doi: 10.1038/ajh.2009.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abraham DM, Davis RT, III, Warren CM, Mao L, Wolska BM, Solaro RJ, Rockman HA. β-Arrestin mediates the Frank-Starling mechanism of cardiac contractility. Proc Natl Acad Sci U S A. 2016;113:14426–14431. doi: 10.1073/pnas.1609308113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim KS, Abraham D, Williams B, Violin JD, Mao L, Rockman HA. β-Arrestin-biased AT1R stimulation promotes cell survival during acute cardiac injury. Am J Physiol Heart Circ Physiol. 2012;303:H1001–H1010. doi: 10.1152/ajpheart.00475.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dries DL. Natriuretic peptides and the genomics of left-ventricular hypertrophy. Heart Fail Clin. 2010;6:55–64. doi: 10.1016/j.hfc.2009.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maisel AS, Krishnaswamy P, Nowak RM, McCord J, Hollander JE, Duc P, Omland T, Storrow AB, Abraham WT, Wu AH, et al. ; Breathing Not Properly Multinational Study Investigators. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med. 2002;347:161–167. doi: 10.1056/NEJMoa020233 [DOI] [PubMed] [Google Scholar]
- 48.Orlowska-Baranowska E, Baranowski R, Greszata L, Stepinska J. Brain natriuretic peptide as a marker of left ventricular hypertrophy in patients with aortic stenosis. J Heart Valve Dis. 2008;17:598–605 [PubMed] [Google Scholar]
- 49.Pendergrass KD, Gwathmey TM, Michalek RD, Grayson JM, Chappell MC. The angiotensin II-AT1 receptor stimulates reactive oxygen species within the cell nucleus. Biochem Biophys Res Commun. 2009;384:149–154. doi: 10.1016/j.bbrc.2009.04.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pennathur S, Wagner JD, Leeuwenburgh C, Litwak KN, Heinecke JW. A hydroxyl radical-like species oxidizes cynomolgus monkey artery wall proteins in early diabetic vascular disease. J Clin Invest. 2001;107:853–860. doi: 10.1172/JCI11194 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.