

Abstract
Intracellular delivery of oligonucleotides is important for their use as therapeutic drugs. The conjugation of molecules interacting with cell membrane proteins to enhance their internalization into cells is an effective strategy for delivering oligonucleotides. In the present study, we focused on creating aptamers, which are single-stranded oligonucleotides that bind target molecules with high affinity and specificity, as membrane protein-binding molecules. With an evolutionary selection approach using a random DNA library containing a uracil derivative with a hydrophobic functional group at the 5 position, we successfully obtained aptamers that are efficiently internalized into A549 cells. The efficacies of the aptamers were tested by further conjugation with MALAT1-targeting antisense oligonucleotides (ASOs), and the expression levels of MALAT1 RNA were examined. The aptamer-ASO conjugates were taken up by A549 cells, although there was no observable reduction in MALAT1 RNA levels. In contrast, the activity of the aptamer-ASO conjugate was potentiated when endosomal/lysosomal escape was enhanced by the addition of chloroquine. Thus, we showed that the hydrophobic modification of the nucleobase moiety is useful for developing highly internalizing aptamers and that endosomal/lysosomal escape is important for the intracellular delivery of ASOs by aptamers.
Keywords: artificial nucleic acid, aptamer, SELEX, MALAT1, antisense oligonucleotide
Graphical Abstract
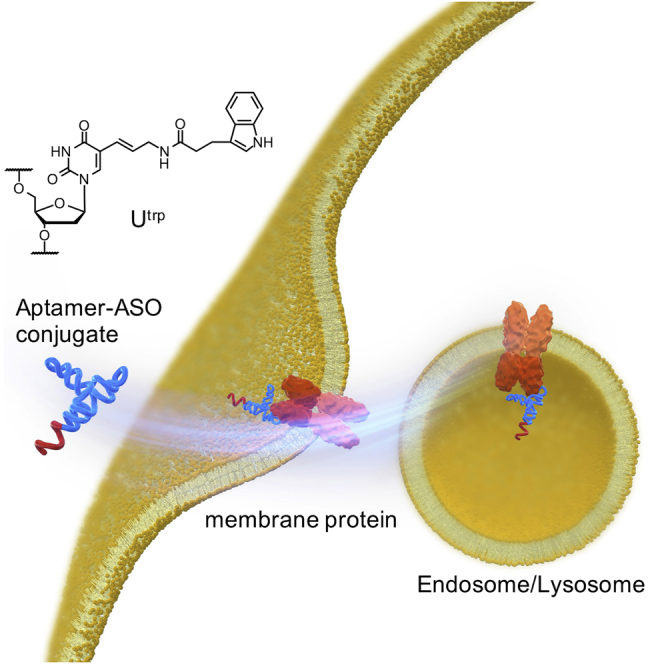
With the hydrophobic modification of nucleobase, we successfully developed cell-internalizing aptamers. The aptamers had higher cell-internalizing ability than did those developed without the modification. Although the aptamers could deliver antisense oligonucleotide into cells, endosomal/lysosomal escape was a bottleneck to increase target RNA degradation by the delivered antisense oligonucleotide.
Introduction
Drug delivery systems are used to deliver the drugs to desired tissues, organs, cells, and subcellular organs through a variety of drug carriers, and the use of appropriate drug delivery systems has been shown to improve drug potency.1 The carriers increase the specificity of small-molecule drugs in target organs for easy cellular uptake (e.g., antibody drug conjugates),2 and they also enhance the efficacy of macromolecular drugs by improving the intracellular translocations.3 For instance, lipid nanoparticles and conjugation with N-acetylgalactosamine have greatly improved the efficacy of oligonucleotide therapeutics in the liver.4,5 Lipid nanoparticles can protect oligonucleotides from degradation and enhance their uptake into cells via endocytosis. Therefore, the delivery of small interfering RNAs (siRNAs) and antisense oligonucleotides (ASOs) using nanoparticles has been widely studied.6 Ligands that interact with cell membrane proteins and are taken up into the cell can be covalently linked to oligonucleotides to enhance delivery. Various types of ligands, such as peptides,7 glycans,8 antibodies,9 and aptamers,10 have been studied for efficient intracellular delivery of siRNAs and ASOs.
Aptamers are single-stranded oligonucleotides that specifically bind target molecules, including small molecules or recombinant proteins.11 Aptamers that bind cellular membrane proteins can be internalized into cells and have been studied as drug carriers.12 As drug carriers, aptamers have the advantages of low immunogenicity, smaller size than antibodies, and adaptability to chemical production. ASOs/siRNAs and aptamers can be synthesized consecutively with a DNA synthesizer. For example, a sense strand fused to an aptamer is synthesized, and then an antisense strand is annealed to the sense strand.10,13,14 Such aptamer-siRNA conjugates can be cleaved by Dicer in the target cell and the siRNA incorporated into the silencing complex.10 ASO-aptamer conjugates have been less studied than the siRNA/microRNA (miRNA)-aptamer conjugates, and their performance has not been fully discussed.15 There is limited knowledge about the aptamers suitable for ASO or siRNA delivery. Therefore, more cell-internalizing aptamers need to be generated, and their functions as ASO/siRNA carriers need to be studied.
Aptamers are developed from a random library via systematic evolution of ligands by exponential enrichment (SELEX).16,17 In this method, oligonucleotides that bind to the target molecule are separated from those that do not bind and subsequently amplified. There are two methods for obtaining cell-internalizing aptamers. One method is to generate aptamers for specific cellular membrane proteins, in which caseecombinant proteins or cells that express high levels of target proteins are used as targets for SELEX.18,19 The other method is to generate aptamers against the target cells.20 After treatment of the cells with a library, the oligonucleotides in the cells are recovered. In this method, called cell-internalization SELEX, it is not necessary to determine the target protein prior to SELEX. The advantage of cell-internalization SELEX is that aptamers against unknown membrane proteins can be generated within their native environment. In addition, by adjusting the SELEX selection method, aptamers can be developed that exhibit unique intracellular behaviors. Recently, aptamers that localize to the nucleus were developed using an endogenous ligase.21 Beyond their development, it is important to investigate the intracellular behaviors of these highly internalized carriers.
One of the features of aptamers is the ease of introducing artificial nucleic acids. SELEX using artificial nucleic acids has been extensively studied for several recombinant proteins, and various modifications that enhance binding affinities have been identified.22 One of the most successful modifications is the aromatic ring modification of the 5 position of uracil.23 The aromatic ring gives the aptamers a high binding affinity owing to hydrophobic interactions.24 Although natural RNA and DNA aptamers have been mainly used with a few exceptions,25,26 modifications to nucleobases have not been frequently applied in cell-internalization SELEX. Considering the previous results of nucleobase-modified aptamers,23 aromatic ring modification is expected to enhance the cell-internalization ability of aptamers.
Herein, we introduced artificial nucleic acids into a SELEX library and carried out cell-internalization SELEX to develop aptamers with high cell-internalizing ability. The intracellular behaviors of the developed aptamers were investigated using real-time PCR and confocal laser microscopy. Furthermore, the aptamers were conjugated with an ASO, and the behavior of the resulting conjugate was evaluated. Our findings will accelerate the use of aptamers as drug carriers.
Results
Selection of aptamers
We prepared two SELEX libraries, a natural DNA library and an artificial DNA library. For the artificial DNA library, we introduced 5-((3-indolyl)propionamide-N-allyl)-2′-deoxyuridine (Utrp) synthesized from 5-aminoallyl-2′-deoxyuridine triphosphate (N-1062; TriLink BioTechnologies, CA, USA) instead of deoxythymidine (Figure 1A).27 Our library consisted of a forward primer region (20 bases), a random region (30 bases), and a reverse primer region (20 bases). Utrps were introduced in the random and reverse primer regions with KOD Dash DNA polymerase. KOD DNA polymerase could incorporate 5 position-modified deoxyuridine triphosphates (dUTPs) into the SELEX library by primer extension.28 Cell-internalization SELEX was carried out against a human lung cancer cell line (A549) with each library. Cells were incubated with each SELEX library for 30 min, and the oligonucleotides that penetrated the cells were collected by lysing the cells after washing (Figure 1B). Washing conditions were made more stringent as SELEX progressed to obtain aptamers with high cell-internalizing ability. The collected oligonucleotides were amplified by PCR. The sense strands of the PCR products were purified and used as the library of the next SELEX round with the natural DNA library. With the artificial DNA library, antisense strands of the PCR products were purified. The sense strands containing Utrp were synthesized by single-primer PCR with deoxyadenosine triphosphate (dATP), deoxycytidine triphosphate (dCTP), deoxyguanosine triphosphate (dGTP), and Utrp triphosphate (UtrpTP) using the purified antisense strands. The obtained sense strands containing Utrp were purified and used as the library for the next SELEX round. After eight rounds of SELEX, the sequences of each library round were investigated using next-generation sequencing (NGS). The results of NGS were analyzed by FASTAptamer29 or AptaSUITE,30 and similar sequences were clustered. In the artificial DNA library, the total number of sequences in the top 10 clusters accounted for more than 75% of the eighth round (Figure S1). Representative sequences were selected from the top 5 clusters (DNA-1 to DNA-5) in the natural DNA library and the top 10 clusters (Apt-1 to Apt-10) in the artificial DNA library for further studies. The sequences and the number of Utrps of the aptamers are listed in Table S2. The number of Utrps in the random region (30 bases) of the aptamers ranged from 5 to 10, with an average of 7, suggesting that a higher number of Utrps does not necessarily correlate to higher uptake of the aptamer into the cell.
Figure 1.

Selection of cell-internalizing aptamers
(A) Chemical structure of Utrp. (B) Schematic representation of cell-internalization SELEX.
Cell-internalizing ability of the aptamers
To determine the cell-internalizing abilities of the selected aptamers, DNA-1 to DNA-5 and Apt-1 to Apt-10, we quantified the aptamers that penetrated the cells by using real-time PCR. The comparison of the natural DNA library and the artificial DNA library revealed that the artificial DNA library internalized into the cells about 3- to 5-fold higher than did the natural DNA library (Figures S2 and S4B). In A549 cells incubated with the aptamers for 30 min, DNA-1 to DNA-5 showed low cell-internalizing ability (less than 3-fold that of the DNA random library) (Figure S3). On the contrary, all Utrp-containing aptamers penetrated the cells more easily than did the natural DNA random library (Figure 2A). Notably, Apt-2 had more than 20-fold higher cell-internalizing ability than that of the DNA random library. With an incubation time of 4 h, Apt-1 to Apt-10 were internalized almost 10-fold more than the DNA random library (Figure 2B), with Apt-5 demonstrating the maximum cell internalization. The substitution of Utrps in the aptamers with deoxythymidine reduced their internalization abilities (Figures S4A and S4B). This suggests that Utrp plays an important role in the function of aptamers.
Figure 2.

Cell-internalizing ability of generated aptamers
(A and B) Quantification of the aptamers internalized into cells. The natural DNA library and Apt-1 to Apt-10 were incubated with A549 cells for 30 min (A) and 4 h (B) at 37°C. The amounts of internalized aptamers were evaluated using real-time PCR and were normalized to the amount of internalized DNA library. Error bars show the mean ± SD values of three culture wells. The experiments were repeated three times independently, with similar results. (C and D) Fluorescence images were taken every 20 min, while the Alexa 594-labeled natural DNA library, Apt-2, Apt-5, Apt-6, Apt-9, and Apt-10 were incubated with A549 cells at 37°C. (C) Images were taken after 4 h of incubation. Cell nuclei were stained with Hoechst 33342. Scale bars represent 30 μm. (D) The average number of observed red fluorescence dots in a cell was counted using CellPathfinder. With live-cell confocal imaging, the medium, which contained aptamers, showed weak fluorescence. The contrast of the images was adjusted to the levels so that the fluorescence of the medium was not visible using CellPathfinder. (E) Alexa 594-labeled natural DNA library, Apt-2, and Apt-10 were incubated with HepG2, MCF7, and PC-9 cells at 37°C for 4 h. After incubation, cell nuclei were stained with Hoechst 33342. The staining solution was replaced with a growth medium, and the fluorescence images were taken. Scale bars represent 20 μm. The contrast was adjusted using CellPathfinder.
Using real-time PCR, we were able to relatively quantify the amounts of aptamers taken up into cells, even small amounts that could not be detected by fluorescence microscopy. However, we cannot exclude the possibility that the aptamers quantified by real-time PCR may not be internalized even if they bind to the cellular membrane. In addition, since only aptamers with primer sequences were amplified using real-time PCR, it was not possible to quantify aptamers rapidly degraded by cells, even if they had easily entered. Therefore, to confirm the internalization of aptamers from another perspective, live-cell imaging of Alexa Fluor 594 (Alexa 594)-labeled aptamers was performed using confocal laser microscopy. Imaging of A549 cells incubated with Alexa 594-labeled aptamers every 20 min for 6 h revealed that the aptamers did not remain in the cellular membrane, with Apt-2 and Apt-10 displaying high cell-internalization capacity (Figure 2C). Intense red fluorescence was observed as a dot-like structure, and the number of red fluorescence dots increased with incubation time. The change in the average number of dots in a cell over time is shown in Figure 2D. For Apt-2 and Ap2-10, the number of fluorescence dots was saturated at approximately 4–5 h of incubation. In contrast, no colored dots were observed with Apt-5, for which the real-time PCR demonstrated efficient cellular uptake.
Subsequently, we investigated the internalization pathway of Apt-2 and Apt-10 by incubating the aptamer-treated cells on ice or chlorpromazine, an inhibitor of clathrin- and caveolae-mediated endocytosis.31 It was observed that the treatments decreased the internalization of the aptamers (Figure S5A). Moreover, the aptamers bound to the cells detached by EDTA and cell scraper, but not to the cells detached by trypsin/EDTA (Figure S5B). These results suggested that the aptamers bound cellular membrane proteins and internalized the cells via the endocytosis pathway. Furthermore, we determined the dissociation constants (KDs) of aptamers against A549 cells using flow cytometry. The KDs of Apt-2, Apt-5, and Apt-10 were 198, 295, and 301 nM, respectively (Figure S5C); however, no correlation between the affinity for A549 cells and the internalization activity of the aptamers was observed.
Next, we tested the internalization of Apt-2 and Apt-10, which were shown to have high cell-internalizing imaging ability into other cell lines. PC-9 (lung cancer cell line), MCF-7 (breast cancer cell line), and HepG2 (liver cancer cell line) cells were incubated with Alexa 594-labeled aptamers. Fluorescence images at 4 h of incubation are shown in Figure 2E. The internalization of Apt-2 and Apt-10 was observed for HepG2, MCF-7, and PC-9 cells. At this time point, the DNA library was not internalized into these cells. This result suggests that Apt-2 and Apt-10 were internalized into cells via interaction with the proteins that are also expressed in HepG2, MCF-7, and PC-9 cells as well as A549 cells.
Localization of aptamers
The dot-like fluorescence observed by fluorescence microscopy suggests that the aptamers localized in endosomes or lysosomes. To confirm this, the co-localization of the internalized aptamers and early endosome/lysosome markers was evaluated by live-cell imaging using confocal laser microscopy (Figure 3). GFP-Rab5 and GFP-LAMP1 were expressed using their respective baculoviruses. Rab5 is an early endosome marker, and LAMP1 is a lysosome marker. Alexa 594-labeled Apt-2 and Apt-10 were incubated with cells expressing Rab5 or LAMP1, and the cells were imaged every 20 min for 6 h. Fluorescence images taken after a 6-h incubation are shown in Figure 3. The aptamers co-localized slightly with GFP-Rab5, but mainly co-localized with GFP-LAMP1. These findings suggest that the internalized aptamers accumulated in lysosomes.
Figure 3.

Co-localization of aptamers and endosome/lysosome marker
Fluorescence images were taken every 20 min, while Alexa 594-labeled Apt-2 and Apt-10 were incubated with GFP-Rab5- or GFP-LAMP1-expressing A549 cells. Images that were taken after 6 h of incubation are shown. (A) GFP-Rab5 was expressed. (B) GFP-LAMP1 was expressed. Cell nuclei were stained with Hoechst 33342. Scale bars represent 10 μm. The contrast of green channels is different in (A) and (B). The contrast of the images was adjusted to the levels so that the fluorescence of the medium was not visible using CellPathfinder. See also Figure S6.
ASO delivery by aptamers
To investigate the delivery of ASOs by the aptamers, we prepared ASO-aptamer conjugates by primer extension using primers with an ASO sequence at the 5′ end (Figure 4A). In this study, we used a 15-mer phosphorothioate gapmer ASO modified with locked nucleic acids to target MALAT1. The internalization of the ASO-aptamer conjugates into cells was checked using confocal laser microscopy (Figure 4B). Attachment of the ASO to the 5′ end of aptamers did not reduce the ability of Apt-2, Apt-5, or Apt-10 to internalize into cells. Moreover, the ASO-aptamer conjugates internalized into cells more quickly than did the ASO itself. In cells transfected with ASO or ASO-aptamer conjugates using Lipofectamine 3000, real-time PCR showed that ASO efficiently degraded the target MALAT1 RNA (Figure S7A). Furthermore, we also found that with Lipofectamine 3000, the ASO-aptamer conjugates degraded MALAT1 RNA more efficiently than did the ASO alone. However, the ASO-library conjugates also degraded the target RNA with similar efficiency (Figure S7B), suggesting that the increased efficiency of RNA degradation by the ASO-aptamer conjugates was not dependent on the aptamer sequence. Furthermore, we investigated the transfection efficiency of FAM-labeled ASO and ASO-library conjugates (Figure S7C), which indicated that the length of oligonucleotides also affects the rate of internalization and the intracellular behavior of oligonucleotides.
Figure 4.

ASO delivery by aptamers
(A) Construction of ASO-aptamer and ASO-primer conjugates. ASO-primer conjugates were ASOs with the forward primer sequences of aptamers. (B) FAM-labeled ASO, aptamers, and ASO-aptamer conjugates were incubated with A549 cells at 37°C. After 1 h of incubation, the cells were fixed and permeabilized, and fluorescence images were subsequently taken with a CV7000 screening system. Cell nuclei were stained with Hoechst 33342. Scale bars represent 20 μm. The contrast was adjusted using CellPathfinder. (C) ASO and ASO-aptamer conjugates were incubated with A549 cells for 8 h, the oligonucleotides were removed, and the cells were further incubated for 16 h. MALAT1 expression was evaluated by qRT-PCR. GAPDH expression was used as a control, and MALAT1 expression was normalized to control cells treated with PBS alone. NEG, non-targeting antisense oligonucleotide. Error bars show the mean ± SD values of three independent experiments. See also Figure S8A. (D) ASO and ASO-aptamer conjugates were incubated with A549 cells in the presence of 100 μM chloroquine for 8 h, after which the oligonucleotides and chloroquine were removed. The cells were incubated without the oligonucleotides and chloroquine for 16 h. MALAT1 expression was evaluated by qRT-PCR. GAPDH expression was used as a control, and MALAT1 expression was normalized to control cells treated with chloroquine alone. Error bars show the mean ± SD values of five independent experiments. Statistical significance was assessed using Student’s t test. ∗p < 0.0005 (ASO-apt-2 [100 nM] versus ASO [100 nM] or ASO-primer [100 nM] and ASO-apt-10 [100 nM] versus ASO [100 nM]), ∗∗p < 0.005 (ASO-apt-10 [100 nM] versus ASO-primer [100 nM]). See also Figure S8B.
Next, we examined the target RNA degradation activities of the ASO and the ASO-aptamer conjugates in the absence of lipofection reagents to evaluate how aptamer conjugation affects ASO activity. Cells were incubated with the ASO-aptamer conjugates for 8 h. The medium was replaced with the culture medium, and cells were incubated for an additional 16 h. The inhibition of gene expression by ASO was measured using real-time PCR (Figure 4C). Contrary to expectations, conjugation of Apt-2, Apt-5, and Apt-10 did not increase MALAT1 RNA degradation, indicating that the ASOs delivered into the cell by conjugation with aptamers do not reach the target RNA.
Endosomal escape by chloroquine
To promote endosomal/lysosomal escape of ASO-aptamer conjugates, we used a small molecule, chloroquine, which becomes protonated in acidic environments (e.g., those in the late endosome and lysosome) and disrupts the membranes of late endosomes and lysosomes.32,33 As shown by the real-time PCR data in Figure 4D, the ASO did not reduce the amount of MALAT1 RNA in the same way as the non-targeting ASO (NEG) in A549 cells treated with chloroquine. However, ASO-Apt-2 and ASO-Apt-10 conjugates degraded the target RNA in a dose-dependent manner. In the absence of chloroquine, 400 nM ASO-Apt-2 did not affect the RNA level of MALAT1, whereas in the presence of chloroquine, the RNA level was reduced to less than 40% with 100 nM ASO-Apt-2. Moreover, with chloroquine, we observed high cellular accumulation and nuclear translocation of ASO-aptamer conjugates (Figure S9). This high accumulation might be the result of lysosomal dysfunction by chloroquine. Chloroquine could increase the pH of endosome/lysosome and inhibit aptamer degradation and exocytosis. There was a possibility that chloroquine could increase the internalization activity of aptamers. However, this increase might not be the main reason for the increase in RNA degradation activity, because even a higher concentration of ASO-aptamer conjugates did not degrade RNA in the absence of chloroquine. This suggests that the lack of activity of ASO-aptamer conjugates in the absence of chloroquine was not due to low intracellular uptake of ASO-aptamer conjugates, but it was rather due to insufficient escape from endosomes or lysosomes.
Discussion
In this study, we successfully generated cell-internalizing aptamers by cell-internalization SELEX. The aptamers selected from the artificial DNA library showed higher cell-internalizing ability than did those from the natural DNA library. Aptamers with hydrophobic genetic alphabets, artificial extra base pairs, have a high cell-internalizing ability.26 This report and our results suggest that the incorporation of hydrophobic groups, such as aromatic rings, into nucleobases improves hydrophobic interactions between aptamers and membrane proteins to enhance the cell-internalizing ability of aptamers. In addition to the hydrophobic genetic alphabet,26 introduction of a non-hydrophobic genetic alphabet expands the diversity of libraries and results in successful cell-SELEX.34 In the present study, we showed that introduction of hydrophobic modification resulted in high cell-internalizing ability without expanding the diversity of libraries. However, the high affinity against membrane proteins does not ensure a high cell-internalizing ability. The nature of the target proteins (e.g., expression levels and recycling) influences the internalization of the aptamers. Although Apt-2, Apt-5, and Apt-10 did not show a very high KD, Apt-2 and Apt-10 had a high cell-internalizing ability. This difference between Apt-2/Apt-10 and Apt-5 could be due to the nature of their target proteins, indicating that the high cell-internalizing ability of the aptamers is dependent on the binding of the aptamers to proper targets. The introduction of hydrophobic modification may enable aptamers to get high affinity and bind to membrane proteins to which natural DNA aptamers cannot bind. Therefore, both the hydrophobic modification and the nature of target proteins are important for high cell-internalizing ability.
We investigated aptamer internalization by two methods: real-time PCR and confocal laser microscopy. Microscopic images showed that Apt-2 and Apt-10 had the highest internalization capacity, while the results of real-time PCR after 4 h of incubation showed that the amount of Apt-2 and Apt-10 in the cells was similar to or less than that of Apt-5. This difference could be due to differences in the stability of the aptamers inside the cells. This consideration is consistent with the localization of Apt-2 and Apt-10. Accumulation in lysosomes may induce the degradation of aptamers. The amount of internalized Apt-10 was much lower than that of Apt-2 in the real-time PCR experiment at 30 min of incubation; however, by 4 h of incubation, real-time PCR and imaging for both aptamers were similar. Thus, the difference at the 30-min incubation point might be caused by variations between the internalization and/or degradation speeds of Apt-2 and Apt-10.
Our aptamers did not increase RNA degradation with aptamer-delivered ASO, possibly because of lysosomal accumulation. The deliveries of ASO,15 siRNA,10 RNA,35 and DNAzyme18 using aptamers have been shown to increase their activities. However, some differences were observed between the current study and previous studies that showed increased activity of aptamer-delivered oligonucleotide therapeutics. Although in some of the above reports, the internalization of the aptamers was mediated by endocytosis, as in the present study, our aptamers did not increase ASO activity, which indicated that different proportions of aptamers could have escaped the endosome. In the present study, we first developed aptamers by cell-SELEX without specifying target membrane proteins; therefore, the behavior of aptamers after endocytosis could have differed depending on the kinds of membrane proteins with which they interact. Earlier studies have suggested that the discovery of membrane proteins that prefer the endosomal escape pathway could identify promising aptamer targets. For example, Wang et al.18 reported a low-density lipoprotein (LDL) receptor-targeting aptamer that internalized into Huh-7 cells and enhanced DNAzyme activity. Nucleolin was also reported as a good target.15 Thus, it is important to establish a selection method for aptamers that prefer endosomal escape to lysosomal accumulation while performing SELEX without specifying target proteins. In this study, we collected all of the sequences that internalized into cells, but it would be useful to specifically recover the sequences according to distinct cellular compartments.21
Second, we introduced an artificial nucleic acid with a hydrophobic moiety, which might have affected the endosomal escape of aptamers. The pathway after endocytosis depends on the type of protein,36 and with high binding affinity by the artificial nucleic acid,24 the intracellular behavior of aptamers is more affected by the nature of the proteins to which the aptamers bind.
In this study, we enhanced endosomal/lysosomal escape by using chloroquine. With chloroquine, 100 nM ASO-Apt-2 decreased MALAT1 RNA to lower than 40%. However, even with chloroquine, 400 nM ASO decreased MALAT1 RNA only to 70% (data not shown). Based on these results, the amount of ASO-Apt-2 in endosomes could have been at least 8-fold higher than that of ASO. Despite higher endosomal accumulation, ASO-aptamer conjugates did not increase RNA degradation without chloroquine. Thus, there is a possibility that attachment to aptamers reduced the rate of endosomal escape by ASO. In a previous study, phosphorothioate ASOs internalized into cells via the endocytosis pathway37 and were released from endosomes by interactions with various proteins (e.g., STX538 and M6PR39). This suggests that the attachment of the aptamers altered the interactions between ASO and intracellular proteins, and if the attachment to aptamers hindered endosomal escape by ASO, the release of ASO from aptamers in the endosome might improve mRNA degradation. Moreover, the attachment of ASO to aptamers by hybridization with RNA or a biodegradable linker could be a useful strategy.
This study has a potential limitation. We used only one ASO, so there is a concern that the sequence or target of other ASOs could affect the delivery efficiency of our aptamers. When aptamers have a complementary strand of ASOs, ASOs can hybridize with aptamers intramolecularly. This hybridization disrupts the aptamers’ structure and function. Therefore, while conjugating another ASO with aptamers, the aptamers’ function should be checked. Furthermore, if attachment of an ASO disrupts aptamers’ function, suitable linkers and position (5′ or 3′) should be investigated.
In summary, we successfully generated cell-internalizing aptamers with hydrophobic base modification, and the aptamers could deliver ASO inside A549 cells. Lysosomal accumulation hindered RNA degradation with aptamer-delivered ASO. With chloroquine, endosomal/lysosomal escape was enhanced, and aptamer-delivered ASO increased RNA degradation. Thus, endosomal/lysosomal escape is a bottleneck of ASO delivery with our aptamers. The development of aptamers that efficiently escape from endosomes will accelerate the use of aptamers as ASO/siRNA carriers.
Materials and methods
Cell culture
A549 (JCRB0076), HepG2 (JCRB1054), and MCF-7 (JCRB0134) cells were obtained from the JCRB Cell Bank (Osaka, Japan). PC-9 (RCB4455) cells were obtained from the RIKEN BioResource Center (Ibaraki, Japan) through the National Bio-Resource Project of the MEXT/AMED, Japan. A549, HepG2, and MCF-7 cells were cultured in DMEM (10% [v/v] fetal bovine serum and 1% penicillin-streptomycin). PC-9 cells were cultured in RPMI 1640 (10% [v/v] fetal bovine serum and 1% penicillin-streptomycin). Cells were cultured at 37°C and 5% CO2.
Cell-internalization SELEX
The natural DNA library (antisense strand and sense strand) was procured from Japan Bio Service (Saitama, Japan). The sequences of oligonucleotides used are listed in Table S1. Modified libraries were prepared by primer extension (14 cycles at 94°C for 30 s, 54°C for 30 s, 74°C for 1 min) in the presence of forward primer_FAM (0.4 μM), KOD Dash DNA polymerase (0.025 U/μL) (Toyobo, Osaka, Japan), dATP (0.1 mM), dGTP (0.1 mM), dCTP (0.1 mM), UtrpTP (0.1 mM), and antisense strand DNA library (80 nM). The scheme of UtrpTP synthesis is shown in Scheme S1 (UtrpTP can also be purchased from TriLink [N-2065]). The modified libraries were purified with denaturing polyacrylamide gels. One day before selection, cells were plated at a density of 1.2 × 106 cells per 6-cm dish. Natural and modified DNA libraries were initially dissolved in PBS (with 0.5 mM magnesium chloride) and further diluted with binding buffer (DMEM + 0.1 mg/mL yeast tRNA [Invitrogen, Carlsbad, CA, USA]) to final concentrations of 100 nM (round 1) or 50 nM (rounds 2–8) and 50 nM (rounds 1 and 2) or 25 nM (rounds 3–8), respectively. For selection, the seeded cells were incubated with binding buffer for 5 min. The binding buffer was discarded, and the diluted library was added. After a 30-min incubation period, the cells were washed with 0.5 M NaCl and PBS. The washing conditions of each round are described in detail in Table S3. Cells were collected by trypsinization and centrifugation at 300 × g for 5 min. The pelleted cells were lysed with 500 μL of DNAzol Direct (Molecular Research Center, Cincinnati, OH, USA) for 30 min at room temperature or overnight at 4°C.
For natural DNA library SELEX, eluted DNA was subjected to PCR (20–22 cycles under conditions described above) in the presence of forward primer_FAM (0.4 μM), reverse primer_PHO (0.3 μM), KOD Dash DNA polymerase (0.005 U/μL), and dNTPs (0.2 mM). The antisense strand of the amplified library was degraded by a 60-min incubation with lambda exonuclease (0.025 U/μL) (New England Biolabs, Ipswich, MA, USA). The sense strand was purified with denaturing polyacrylamide gels, and the purified library was used for the next selection. The selection process was repeated eight times.
For modified-library SELEX, the eluted DNA was subjected to PCR (18–20 cycles under conditions described above) in the presence of forward primer_PHO (0.3 μM), reverse primer_HEX (0.4 μM), KOD Dash polymerase (0.005 U/μL), and dNTPs (0.2 mM). The sense strand of the amplified library was degraded by a 60-min incubation with lambda exonuclease (0.025 U/μL). The mixture was desalted using a Vivaspin instrument (Sartorius, Goettingen, Germany). The remaining antisense strand (80 nM) was subjected to one primer extension (16 cycles under conditions described above) in the presence of forward primer_FAM (0.4 μM), KOD Dash DNA polymerase (0.025 U/μL), dATP (0.1 mM), dGTP (0.1 mM), dCTP (0.1 mM), and UtrpTP (0.1 mM). The modified libraries were purified with denaturing polyacrylamide gels, and the purified library was used for the next selection. The selection process was repeated eight times.
NGS
The libraries resulting from each round were modified by an adaptor sequence attachment and purified with denaturing polyacrylamide gels. The libraries were subjected to NGS using MiSeq (Illumina, San Diego, CA, USA) according to the supplied manual, and the obtained results were analyzed using FASTAptamer29 (natural DNA library) or AptaSUITE (artificial DNA library).30
Preparation of aptamers
Antisense DNA strands of the aptamers were purchased from Invitrogen. The antisense strands (0.5 μM) were subjected to one primer extension in the presence of a forward primer (0.4 μM), KOD Dash DNA polymerase (0.025 U/μL), dATP (0.05 mM), dGTP (0.05 mM), dCTP (0.05 mM), and UtrpTP (0.05 mM). In preparing ASO-aptamer conjugates, the antisense strands with phosphate modifications at the 3′ ends were used to make purification easier. The forward primer with the ASO sequence at the 5′ end was purchased from GeneDesign (Osaka, Japan). The aptamers were purified with denaturing polyacrylamide gels. The purification and mass spectrometry (MS) of the aptamers were verified using liquid chromatography (LC)-MS (Figure S10).
Real-time PCR for aptamer quantification
One day before the experiment, cells were plated at a density of 3.0 × 104 cells per well in a 96-well plate. The aptamers and natural DNA library (100 nM) were dissolved in PBS (with 0.5 mM magnesium chloride) and diluted in binding buffer (DMEM + 0.1 mg/mL yeast tRNA) to a final concentration of 10 nM. The seeded cells were initially incubated in binding buffer for 5 min, which was replaced with the diluted aptamers for an additional 30 min or 4 h of incubation. Cells were washed twice with 0.5 M NaCl and incubated in the same solution for 5 min, then subsequently washed with PBS. Cells were collected by trypsinization and centrifugation at 800 × g for 5 min, and the cell pellets were dried under reduced pressure. Dried pellets were lysed with 20 μL of DNAzol Direct by overnight incubation at 4°C. The lysates were diluted with deionized water and subjected to real-time PCR with the forward primer (0.2 μM), reverse primers (0.2 μM), and Applied Biosystems PowerUp SYBR Green master mix (Applied Biosystems, Foster City, CA, USA) using a StepOnePlus real-time PCR system (Applied Biosystems). Data were analyzed by the standard curve method using standards prepared with the DNA library at 10, 1, 0.1, 0.01, and 0.001 pM (or 10, 1, 0.1, 0.02, 0.01 pM) diluted with deionized water.
Live-cell confocal imaging
A549 cells were plated overnight at a density of 8.0 × 103 cells per well in a 96-well plate (Screenstar 655866; Greiner Bio-One International, Kremsmünster, Austria). HepG2 and MCF-7 cells were plated at a density of 1.0 × 104 cells per well, while PC-9 cells were plated at a density of 1.2 × 104 cells per well. The aptamers and natural DNA library (1 μM) were dissolved in PBS (with 0.5 mM magnesium chloride) and diluted in DMEM (10% [v/v] fetal bovine serum) to a final concentration of 200 nM. For A549 cells, cell nuclei were stained with Hoechst 33342 (5 μg/mL in DMEM, 10% [v/v] fetal bovine serum). The staining medium was replaced with the aptamers, and fluorescence images were taken every 20 min for 6 h on a CV7000 screening system (Yokogawa, Tokyo, Japan) using a ×60 objective lens with excitation at 405 and 561 nm. During the experiment, the stage incubator was kept at 37°C and 5% CO2. For HepG2, MCF-7, and PC-9 cells, cells were incubated with aptamers for 4 h. After incubation, cell nuclei were stained with Hoechst 33342 (5 μg/mL in DMEM or RPMI 1640, 10% [v/v] fetal bovine serum). The staining medium was replaced with a growth medium, and fluorescence images were taken on the CV7000. The images were analyzed with CellPathfinder (Yokogawa). The contrast of the red channel was adjusted to the levels so that the fluorescence of the medium, which contained Alexa 594-labeled aptamers, was not visible. The contrast of the blue channel was adjusted to the levels so that only the fluorescence of the nucleus was visible.
Flow cytometry assay
Cells that were almost 90% confluent were detached using 5 mM EDTA (PBS) and cell scraper or 2.5 g/L trypsin in 1 mM EDTA. The cells were washed with PBS and suspended in binding buffer (with 1 mg/mL BSA). The cells were filtered with a cell strainer and counted. The numbers of cells were adjusted to 1.0 × 106 cells per mL, and 40 μL of cell suspension was dispensed to a 96-well plate. 10 μL of FAM-labeled aptamer solution was added to the cell suspension, and the cell was incubated on ice for 30 min. After incubation, the cells were washed twice with FCM wash buffer (PBS with 0.5 mM magnesium chloride and 1 mg/mL BSA) and fixed with 4% paraformaldehyde for 10 min. The fixed cells were again washed with FCM wash buffer and suspended in the same solution. The fluorescence intensity of the cells was monitored with LSRFortessa X-20 (BD Biosciences, CA, USA), and the mean fluorescence intensity (MFI) was analyzed with FACSDiva. To determine KD, the MFIs were plotted, and the points were fitted using ImageJ according to the following formula: Y = BMAXX/(KD + X), where X is aptamer concentration and Y is MFI.
Co-localization study
Cells were plated at a density of 8.0 × 103 cells per well in 96-well plates (Greiner Bio-One International). After 6 h, CellLight BacMam 2.0 early endosome or lysosome-GFP (Invitrogen) was added to the culture medium at a dose of 30 particles per cell. After 18 h of incubation, the addition of aptamers and imaging were carried out under the same conditions as live-cell confocal imaging, with excitation at 405, 488, and 561 nm. The images were analyzed with CellPathfinder (Yokogawa). The contrast was adjusted to the same as used for the live-cell confocal imaging.
Confocal fluorescence imaging with cell fixation
FAM-labeled ASO-aptamer conjugates and ASO at a final concentration of 100 nM were added to cells plated at a density of 1.4 × 104 cells per well in 96-well plates and incubated for 1 h at 37°C and 5% CO2. Cells were washed with PBS, fixed with 4% paraformaldehyde for 10 min, and permeabilized for 5 min with 0.2% Triton X-100 in PBS. After three washes with PBS, cell nuclei were stained with Hoechst 33342 in PBS. The staining solution was replaced, and cells were imaged using a CV7000 with a ×60 objective lens and excitation at 405 and 488 nm. The images were analyzed with CellPathfinder. The contrast of the blue channel was adjusted to the levels so that only the fluorescence of the nucleus was visible.
ASO delivery
ASO-aptamer conjugates and ASO diluted as described earlier were added to an overnight culture of cells plated at a density of 8.0 × 103 cells per well in a 96-well plate and incubated for 8 h. The treatment medium was replaced with DMEM (10% [v/v] fetal bovine serum), and cells were further incubated for 16 h. cDNA synthesis was carried out with a SuperPrep cell lysis & RT kit for qPCR (Toyobo) according to the manufacturer’s guidelines. The cDNA was subjected to real-time PCR with a forward primer (50 nM), reverse primers (50 nM), and Applied Biosystems PowerUp SYBR Green master mix (Applied Biosystems) using a StepOnePlus real-time PCR system. The primers for MALAT1 and GAPDH are listed in Table S1. Data were analyzed with the comparative ΔΔCt method. MALAT1 expression levels were normalized with GAPDH expression levels.
For the experiments with chloroquine, cell density was 1.0 × 104 cells per well, and ASOs were diluted with DMEM containing 125 μM chloroquine diphosphate and 10% (v/v) fetal bovine serum to a final chloroquine concentration of 100 μM. Subsequent operations were the same as in experiments without chloroquine.
For transfection, cells were plated at a density of 8.0 × 103 cells per well in a 96-well plate. The ASO-aptamer conjugates and ASO were dissolved in PBS (with 0.5 mM magnesium chloride) to the concentrations of 1, 2, and 4 μM. The ASOs were mixed with Lipofectamine 3000 (Invitrogen) and P3000 reagent according to the manufacturer’s guidelines. The mixtures were added at 10 μL/well into 90 μL of culture medium, and the cells were incubated for 24 h. Subsequent operations were the same as in experiments without Lipofectamine.
Acknowledgments
This research was supported by the Japan Agency for Medical Research and Development (AMED) under grant no. JP19ak0101102 and by the Nagai Memorial Research Scholarship from the Pharmaceutical Society of Japan.
Author contributions
K.T. designed and conducted the experiments and wrote the manuscript; T.O. conducted the synthesis of UtrpTP; and Y.K. and S.O. conceptualized and supervised the study and revised and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.11.016.
Supplemental information
References
- 1.Rosen H., Abribat T. The rise and rise of drug delivery. Nat. Rev. Drug Discov. 2005;4:381–385. doi: 10.1038/nrd1721. [DOI] [PubMed] [Google Scholar]
- 2.Chari R.V., Miller M.L., Widdison W.C. Antibody-drug conjugates: an emerging concept in cancer therapy. Angew. Chem. Int. Ed. Engl. 2014;53:3796–3827. doi: 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- 3.Torchilin V.P., Lukyanov A.N. Peptide and protein drug delivery to and into tumors: challenges and solutions. Drug Discov. Today. 2003;8:259–266. doi: 10.1016/s1359-6446(03)02623-0. [DOI] [PubMed] [Google Scholar]
- 4.Chen S., Tam Y.Y., Lin P.J., Leung A.K., Tam Y.K., Cullis P.R. Development of lipid nanoparticle formulations of siRNA for hepatocyte gene silencing following subcutaneous administration. J. Control. Release. 2014;196:106–112. doi: 10.1016/j.jconrel.2014.09.025. [DOI] [PubMed] [Google Scholar]
- 5.Prakash T.P., Graham M.J., Yu J., Carty R., Low A., Chappell A., Schmidt K., Zhao C., Aghajan M., Murray H.F. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014;42:8796–8807. doi: 10.1093/nar/gku531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen T.M., Cullis P.R. Liposomal drug delivery systems: from concept to clinical applications. Adv. Drug Deliv. Rev. 2013;65:36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 7.Ämmälä C., Drury W.J., 3rd, Knerr L., Ahlstedt I., Stillemark-Billton P., Wennberg-Huldt C., Andersson E.-M., Valeur E., Jansson-Löfmark R., Janzén D. Targeted delivery of antisense oligonucleotides to pancreatic β-cells. Sci. Adv. 2018;4:eaat3386. doi: 10.1126/sciadv.aat3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu L., Mahato R.I. Targeted delivery of siRNA to hepatocytes and hepatic stellate cells by bioconjugation. Bioconjug. Chem. 2010;21:2119–2127. doi: 10.1021/bc100346n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song E., Zhu P., Lee S.K., Chowdhury D., Kussman S., Dykxhoorn D.M., Feng Y., Palliser D., Weiner D.B., Shankar P. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 2005;23:709–717. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- 10.McNamara J.O., 2nd, Andrechek E.R., Wang Y., Viles K.D., Rempel R.E., Gilboa E., Sullenger B.A., Giangrande P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006;24:1005–1015. doi: 10.1038/nbt1223. [DOI] [PubMed] [Google Scholar]
- 11.Bunka D.H., Stockley P.G. Aptamers come of age—at last. Nat. Rev. Microbiol. 2006;4:588–596. doi: 10.1038/nrmicro1458. [DOI] [PubMed] [Google Scholar]
- 12.Zhu G., Niu G., Chen X. Aptamer-drug conjugates. Bioconjug. Chem. 2015;26:2186–2197. doi: 10.1021/acs.bioconjchem.5b00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Affinito A., Quintavalle C., Esposito C.L., Roscigno G., Vilardo C., Nuzzo S., Ricci-Vitiani L., De Luca G., Pallini R., Kichkailo A.S. The discovery of RNA aptamers that selectively bind glioblastoma stem cells. Mol. Ther. Nucleic Acids. 2019;18:99–109. doi: 10.1016/j.omtn.2019.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esposito C.L., Cerchia L., Catuogno S., De Vita G., Dassie J.P., Santamaria G., Swiderski P., Condorelli G., Giangrande P.H., de Franciscis V. Multifunctional aptamer-miRNA conjugates for targeted cancer therapy. Mol. Ther. 2014;22:1151–1163. doi: 10.1038/mt.2014.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong S., Sun N., Liu M., Wang J., Pei R. Building a chimera or aptamer-antisense oligonucleotide for silencing galectin-1 gene. RSC Advances. 2016;6:112445–112450. [Google Scholar]
- 16.Tuerk C., Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 17.Ellington A.D., Szostak J.W. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 18.Wang T., Rahimizadeh K., Veedu R.N. Development of a novel DNA oligonucleotide targeting low-density lipoprotein receptor. Mol. Ther. Nucleic Acids. 2020;19:190–198. doi: 10.1016/j.omtn.2019.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thiel K.W., Hernandez L.I., Dassie J.P., Thiel W.H., Liu X., Stockdale K.R., Rothman A.M., Hernandez F.J., McNamara J.O., 2nd, Giangrande P.H. Delivery of chemo-sensitizing siRNAs to HER2+-breast cancer cells using RNA aptamers. Nucleic Acids Res. 2012;40:6319–6337. doi: 10.1093/nar/gks294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thiel W.H., Bair T., Peek A.S., Liu X., Dassie J., Stockdale K.R., Behlke M.A., Miller F.J., Jr., Giangrande P.H. Rapid identification of cell-specific, internalizing RNA aptamers with bioinformatics analyses of a cell-based aptamer selection. PLoS ONE. 2012;7:e43836. doi: 10.1371/journal.pone.0043836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smestad J., Wilbanks B., Maher L.J., 3rd An in vitro selection strategy identifying naked DNA that localizes to cell nuclei. J. Am. Chem. Soc. 2019;141:18375–18379. doi: 10.1021/jacs.9b06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ni S., Yao H., Wang L., Lu J., Jiang F., Lu A., Zhang G. Chemical modifications of nucleic acid aptamers for therapeutic purposes. Int. J. Mol. Sci. 2017;18:1683. doi: 10.3390/ijms18081683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gold L., Ayers D., Bertino J., Bock C., Bock A., Brody E.N., Carter J., Dalby A.B., Eaton B.E., Fitzwater T. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE. 2010;5:e15004. doi: 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ren X., Gelinas A.D., von Carlowitz I., Janjic N., Pyle A.M. Structural basis for IL-1α recognition by a modified DNA aptamer that specifically inhibits IL-1α signaling. Nat. Commun. 2017;8:810. doi: 10.1038/s41467-017-00864-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sefah K., Yang Z., Bradley K.M., Hoshika S., Jiménez E., Zhang L., Zhu G., Shanker S., Yu F., Turek D. In vitro selection with artificial expanded genetic information systems. Proc. Natl. Acad. Sci. USA. 2014;111:1449–1454. doi: 10.1073/pnas.1311778111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Futami K., Kimoto M., Lim Y.W.S., Hirao I. Genetic alphabet expansion provides versatile specificities and activities of unnatural-base DNA aptamers targeting cancer cells. Mol. Ther. Nucleic Acids. 2019;14:158–170. doi: 10.1016/j.omtn.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dolot R., Lam C.H., Sierant M., Zhao Q., Liu F.W., Nawrot B., Egli M., Yang X. Crystal structures of thrombin in complex with chemically modified thrombin DNA aptamers reveal the origins of enhanced affinity. Nucleic Acids Res. 2018;46:4819–4830. doi: 10.1093/nar/gky268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaught J.D., Bock C., Carter J., Fitzwater T., Otis M., Schneider D., Rolando J., Waugh S., Wilcox S.K., Eaton B.E. Expanding the chemistry of DNA for in vitro selection. J. Am. Chem. Soc. 2010;132:4141–4151. doi: 10.1021/ja908035g. [DOI] [PubMed] [Google Scholar]
- 29.Alam K.K., Chang J.L., Burke D.H. FASTAptamer: a bioinformatic toolkit for high-throughput sequence analysis of combinatorial selections. Mol. Ther. Nucleic Acids. 2015;4:e230. doi: 10.1038/mtna.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoinka J., Backofen R., Przytycka T.M. AptaSUITE: a full-featured bioinformatics framework for the comprehensive analysis of aptamers from HT-SELEX experiments. Mol. Ther. Nucleic Acids. 2018;11:515–517. doi: 10.1016/j.omtn.2018.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L.H., Rothberg K.G., Anderson R.G.W. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J. Cell Biol. 1993;123:1107–1117. doi: 10.1083/jcb.123.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varkouhi A.K., Scholte M., Storm G., Haisma H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release. 2011;151:220–228. doi: 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 33.Du Rietz H., Hedlund H., Wilhelmson S., Nordenfelt P., Wittrup A. Imaging small molecule-induced endosomal escape of siRNA. Nat. Commun. 2020;11:1809. doi: 10.1038/s41467-020-15300-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang L., Yang Z., Sefah K., Bradley K.M., Hoshika S., Kim M.J., Kim H.J., Zhu G., Jiménez E., Cansiz S. Evolution of functional six-nucleotide DNA. J. Am. Chem. Soc. 2015;137:6734–6737. doi: 10.1021/jacs.5b02251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porciani D., Cardwell L.N., Tawiah K.D., Alam K.K., Lange M.J., Daniels M.A., Burke D.H. Modular cell-internalizing aptamer nanostructure enables targeted delivery of large functional RNAs in cancer cell lines. Nat. Commun. 2018;9:2283. doi: 10.1038/s41467-018-04691-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonifacino J.S., Traub L.M. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- 37.Crooke S.T., Wang S., Vickers T.A., Shen W., Liang X.H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017;35:230–237. doi: 10.1038/nbt.3779. [DOI] [PubMed] [Google Scholar]
- 38.Liang X.H., Sun H., Nichols J.G., Allen N., Wang S., Vickers T.A., Shen W., Hsu C.W., Crooke S.T. COPII vesicles can affect the activity of antisense oligonucleotides by facilitating the release of oligonucleotides from endocytic pathways. Nucleic Acids Res. 2018;46:10225–10245. doi: 10.1093/nar/gky841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang X.H., Sun H., Hsu C.W., Nichols J.G., Vickers T.A., De Hoyos C.L., Crooke S.T. Golgi-endosome transport mediated by M6PR facilitates release of antisense oligonucleotides from endosomes. Nucleic Acids Res. 2020;48:1372–1391. doi: 10.1093/nar/gkz1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.