

Abstract
N6-methyladenosine (m6A) methylation modification is the most prevalent and abundant internal modification of eukaryotic mRNAs. Increasing evidence has shown that mRNA m6A plays important roles in the development of stem cells. However, to the best of our knowledge, no reports about the roles of mRNA m6A in mouse female germline stem cells (mFGSCs) have been published. In this study, we compared the genome-wide profiles of mRNA m6A methylation and DNA methylation between FGSCs and sandosinbred mice (SIM) embryo-derived thioguanine and ouabain-resistant (STO) cells. qRT-PCR revealed that the expression levels of mRNA m6A-related genes (Mettl3, Alkbh5, Ythdf1, Ythdf2, Ythdc1, and Ythdc2) in FGSCs were significantly higher than those in STO cells. m6A RNA immunoprecipitation sequencing (MeRIP-seq) data further showed that the unique m6A-methylated mRNAs in FGSCs and STO cells were related to cell population proliferation and somatic development, respectively. Additionally, knockdown of Ythdf1 inhibited FGSC self-renewal. Comparison of methylated DNA immunoprecipitation sequencing (MeDIP-seq) results between FGSCs and STO cells identified that DNA methylation contributed to FGSC proliferation by suppressing the somatic program. These results suggested that m6A regulated FGSC self-renewal possibly through m6A binding protein YTHDF1, and DNA methylation repressed somatic programs in FGSCs to maintain FGSC characteristics.
Graphical Abstract
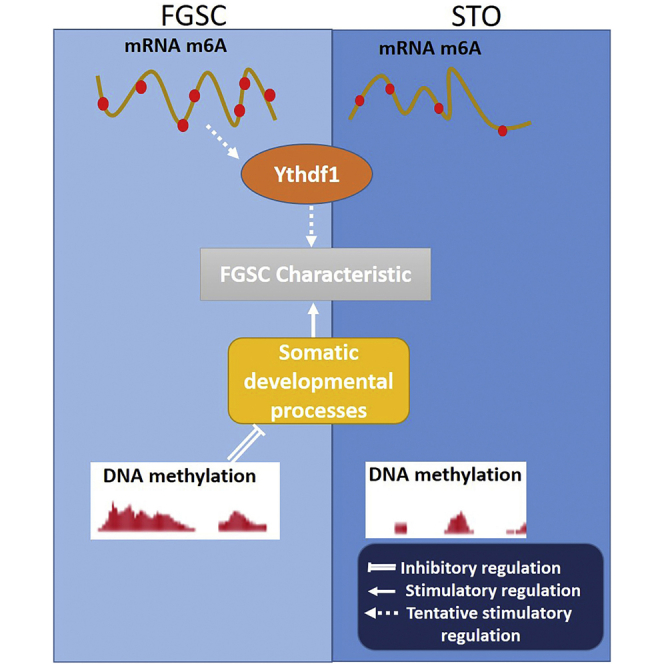
Ji Wu’s research team found that the genome-wide levels of mRNA m6A methylation and DNA methylation were higher in FGSCs compared with STO cells. Furthermore, they found that knockdown of Ythdf1 inhibited FGSC self-renewal, and DNA methylation repressed the somatic program in FGSCs to maintain FGSC characteristics.
Introduction
Germ cells are essential for passing genetic information from generation to generation and for maintaining the continuation of species.1 Female germline stem cells (FGSCs), a new class of germ cells, have been successfully isolated from postnatal mammalian and human ovarian tissue and used to establish cell lines.2, 3, 4, 5, 6, 7, 8, 9 Our previous studies demonstrated that FGSCs differentiated into functional oocytes and produced fertile offspring after transplantation into the ovaries of mice with premature ovarian failure (POF).2 By the random recombination of targeted genes in FGSCs, we generated transgenic or gene knockdown mice.10
Recently, increasing attention has been paid to the genetic and epigenetic regulation of the basic properties of stem cells and their development. Zhang et al.11 revealed the genome-wide profiles of the histone modifications H3K4me1, H3K27ac, H3K4me3, and H3K27me3; DNA methylation; and RNA polymerase II occupancy in FGSCs and found that they were involved in the unipotency of FGSCs. In addition, Sun et al.12 demonstrated that histone H1-mediated epigenetic regulation controls the self-renewal of ovary germline stem cells (OGSCs) by modulating H4K16 acetylation. Moreover, Bernstein et al.13 reported that epigenetic modifications, such as DNA modifications, play essential roles in regulating embryonic stem cells (ESCs) and their biological development. DNA methylation is one of the best studied epigenetic modifications and is important to mammalian development.14 Germline genes were acutely sensitive to loss of DNA methylation; they were derepressed in Dnmt3a/3b-knockout mouse ESCs (mESCs),15 Dnmt1-depleted human fibroblasts,16,17 and Dnmt3b mutant mouse embryos.18
N6-methyladenosine (m6A) methylation modification is the most prevalent and abundant internal modification on eukaryotic mRNAs.19 Studies involving the analysis of m6A in eukaryotes showed that hundreds of m6A sites were highly conserved in human ESCs and mESCs.20 Analogous to the epigenetic code, m6A methyltransferases (“writers”; i.e., METTL3, METTL14, WTAP, ZC3H13), demethylases (“erasers”; i.e., FTO, ALKBH5), and m6A binding proteins (“readers”; i.e., YTHDF family, YTHDC family) involved in this modification have been identified.21, 22, 23 It has been reported that METTL3, FTO, and ALKBH5 play essential roles in many biological processes, ranging from development and metabolism to fertility.24 Defects in m6A methyltransferases and demethylases might induce abnormal m6A methylation levels, leading to dysfunction of RNA and diseases. For example, the mRNA m6A level was found to be increased in Alkbh5-deficient male mice, which affected spermatocyte apoptosis at the MII (metaphase II) stage and then impaired fertility.25 The major mechanism by which m6A exerts its influences was through the recruitment of m6A binding proteins. YTHDF1 directly promoted the translation of methylated mRNAs.26 YTHDF2 had an effect on the stability of m6A-modified RNAs by localizing them to mRNA decay sites.27, 28, 29 YTHDF3 enhanced protein translation in synergy with YTHDF1 and facilitates m6A-modified RNA decay mediated via YTHDF2.30 Ythdf1 was known to regulate tumorigenicity and cancer stem cell-like activity in human colorectal carcinoma (CRC).31 The loss of Ythdf2 was shown to upregulate the level of transcription of oocyte maturation-related genes, leading to specific infertility of Ythdf2-defective female mice.32 Nevertheless, there is little information about the profile of mRNA m6A in FGSCs and SIM mouse embryo-derived thioguanine and ouabain-resistant (STO) cells belonging to somatic cells, which comprise all of the cells making up an organism besides germ cells, as well as the influence of YTHDF1 on FGSCs.
In this study, we profiled mRNA m6A modification by methylated RNA immunoprecipitation sequencing (MeRIP-seq) and DNA methylation by methylated DNA immunoprecipitation sequencing (MeDIP-seq) in FGSCs and STO cells, and we found that mRNA m6A regulated FGSC self-renewal possibly through m6A-binding protein YTHDF1, and DNA methylation repressed the somatic programs in FGSCs to maintain FGSC characteristics.
Results
Biological characterization of FGSC line and morphology of STO cells
The FGSC line used in this study had been established previously.2The cultured FGSCs formed clusters with a bead-like shape (Figure 1A). To characterize these cells, we analyzed the expression of gene markers of female germ cells: Oct4 (also called Pou5f1, POU domain, class 5, transcription factor 1),33 Stella (also known as developmental pluripotency associated factor 3, Dppa3),34 Mvh (also called Ddx4, DEAD [Asp-Glu-Ala-Asp] box polypeptide 4),35 Blimp-1 (also called Prdm1, positive regulatory [PR] domain containing 1 with zinc finger protein [ZNF] domain),36 and Fragilis (also called Ifitm3, interferon-induced transmembrane protein 3).37,38 The results of reverse-transcriptase PCR (RT-PCR) showed that the FGSCs expressed Oct4, Stella, Mvh, Fragilis, and Blimp-1 (Figure 1C). Immunofluorescence analysis also showed that the FGSCs were positive for OCT4 and MVH (Figure 1D). The morphology of STO cells was shown in Figure 1B.
Figure 1.

Characteristics of cultured FGSCs and STO cells
(A and B) Representative morphology of cultured FGSCs (A) and STO cells (B). (C) RT-PCR analysis of Oct4, Stella, Mvh, Fragilis, and Blimp-1 mRNA expression in FGSCs. M, 100 bp DNA markers. Gapdh served as a loading control. NC, negative control. (D) Immunofluorescence analysis of FGSCs with antibodies against OCT4 and MVH. Scale bars: (A and B) 50 μm; (D) 20 μm (MVH) and 25 μm (OCT4).
Comparison of mRNA m6A methylation profile between FGSCs and STO cells
To explore whether there was a difference in the m6A RNA methylation profile between FGSCs and STO cells, an mRNA dot blot assay was initially performed to determine the m6A levels in these two cell types. Compared with STO cells, we found that the global m6A level significantly increased in FGSCs (Figure 2A). We also compared the expression of m6A methyltransferases (Mettl3, Mettl14, Wtap), demethylases (Alkbh5, Fto), and binding proteins (Ythdf1, Ythdf2, Ythdf3, Ythdc1, Ythdc2) between FGSCs and STO cells using quantitative RT-PCR (qRT-PCR). The results revealed that the expression levels of Mettl3, Alkbh5, Ythdf1, Ythdf2, Ythdc1, and Ythdc2 were significantly higher in FGSCs than those in STO cells (Figure 2B). Then, we performed m6A RNA immunoprecipitation combined with deep sequencing (MeRIP-seq) to detect the m6A peaks and explored their distributions in the transcriptome of FGSCs and STO cells. We observed that the m6A peaks were principally located in the coding sequence (CDS), transcription start site (TSS), and 3′ untranslated region (3′ UTR) in both FGSCs and STO cells (Figure 3A). m6A modifications of both FGSCs and STO cells were enriched in consensus motifs (Figure 3B), but there were differences in m6A distribution patterns between these two cell types. Bioinformatic analysis revealed 2,575 and 1,265 m6A-methylated mRNAs in FGSCs and STO cells, respectively, whereas there was a total of 4,130 m6A-methylated mRNAs overlapping between these two cell types (Figure 3C). Gene Ontology (GO) analysis showed that the functional enrichment of these 4,130 overlapping m6A mRNAs was related to the mRNA metabolic processes, splicing, transport, and stabilization (Figure 3F). The unique m6A-methylated mRNAs in FGSCs (2,575) were enriched in terms related to DNA replication, the mitotic cell cycle, and cell population proliferation (Figure 3E), whereas the 1,265 unique m6A-methylated mRNAs in STO cells were enriched in GO categories related to system development (Figure 3D).
Figure 2.

The m6A level in FGSCs and STO cells
(A) Dot blot assay determined the m6A level in FGSCs and STO cells. MB, methylene blue (as loading control). (B) Mettl3, Mettl14, Alkbh5, Fto, Wtap, Ythdf1, Ythdf2, Ythdf3, Ythdc1, and Ythdc2 mRNA levels in FGSCs and STO cells were measured by qRT-PCR. The expression of these genes in STO cells was set as 1. ∗p < 0.05, ∗∗∗p < 0.001. All data are presented as the mean ± SD of three independent experiments.
Figure 3.

Dynamic m6A modification in FGSCs and STO cells
(A) Transcriptome-wide distribution of m6A peaks in FGSCs and STO cells. (B) The most common sequence motif among m6A peaks in FGSCs and STO cells. (C) Venn diagram illustrating the m6A-modified mRNAs in FGSCs and STO cells. (D and E) GO analysis of the m6A-modified mRNAs specifically identified in STO cells (D) and FGSCs (E), respectively. (F) GO analysis of the overlapping m6A-modified mRNAs in FGSCs and STO cells.
Knockdown of Ythdf1 inhibited FGSC self-renewal in vitro
To explore the function of mRNA m6A modification in regulating FGSC proliferation, we knocked down Ythdf1 using the lentiviral vectors carrying Ythdf1-specific small hairpin (sh)RNAs (shDF1) to infect FGSCs. qRT-PCR, as well as western blot analyses, showed that the mRNA and protein levels of Ythdf1 were significantly decreased in FGSCs with shDF1 lentivirus infection compared with controls (shCtrl) (Figures 4A–4D). In addition, Cell Counting Kit 8 (CCK8) assays demonstrated that the optical density values of shDF1 lentivirus-infected FGSCs were significantly lower than those of controls (Figure 4E). Furthermore, 5-ethynyl-2′-deoxyuridine (EdU) assays revealed that EdU-positive cells were significantly decreased in shDF1 lentivirus-infected FGSCs compared with controls (Figures 4F and 4G).
Figure 4.

Effects of Ythdf1 on FGSCs
(A) Fluorescence and bright-field images for FGSCs infected with lentivirus. (B) mRNA expression levels of Ythdf1 in cells infected with shYthdf1 lentivirus (shDF1) and negative control lentivirus (shCtrl) were examined by qRT-PCR. (C and D) Western blot analyses detected the protein levels of Ythdf1 in cells infected with shDF1 and shCtrl. (E and F) CCK8 assays (E) and EdU incorporation assays (F) were conducted in FGSCs infected with shDF1 and shCtrl. (G) Quantification of EdU-positive cells. Data are presented as the mean ± SD of three independent experiments. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 compared with the control group. Scale bars, 20 μm.
Comparison of genomic DNA methylation profile between FGSCs and STO cells
We next compared genomic DNA methylation between FGSCs and STO cells. Before profiling the genomic DNA methylation sequence, we compared the mRNA expression levels of DNA methyltransferases (Dnmt1, Dnmt3a, and Dnmt3b) in FGSCs and STO cells by qRT-PCR, and we found that the expression levels of Dnmt1, Dnmt3a, and Dnmt3b were significantly higher in FGSCs than those in STO cells (Figure 5A). Then, we performed clustering analysis with the datasets of the genomic DNA methylation sequence in FGSCs and STO cells (Figures 5B and 5C), and we explored the specific functions of the genes in the promoters, of which there was FGSC-specific methylation based on their genomic locations using Genomic Regions Enrichment of Annotations Tool (GREAT) analysis (Figure 5B; cluster 1).39 The results demonstrated that the FGSC-specific DNA-methylated genes of cluster 1 were significantly correlated with the GO biological process terms “DNA methylation involved in gamete generation,” “meiosis,” “chromosome organization involved in meiotic cell cycle,” “male meiosis,” and “synapsis” (Figure 5D), which were biological processes related to germ cell development. Notably, we found that the promoters of somatic development-related genes (e.g., Tbx, Fox, and Hox family transcription factors) were DNA methylated in FGSCs but fully hypomethylated in STO cells (Figure 6; Table S3). Furthermore, our previous study found that several somatic development-related genes (e.g., Hoxa1, Hoxb1, Tbx1) with DNA methylation at promoter regions were visibly upregulated after Dnmt1 knockdown in FGSCs.11
Figure 5.

Genomic DNA methylation in FGSCs and STO cells
(A) Dnmt1, Dnmt3a, and Dnmt3b mRNA levels in FGSCs and STO cells were measured by qRT-PCR. The expression of these genes in STO cells was set as 1. ∗p < 0.05, ∗∗p < 0.01, compared with STO group. All data are presented as the mean ± SD of three independent experiments. (B and C) K-means clustering of DNA methylation at promoter regions for FGSCs and STO cells. Distribution of genome in the 5-kb upstream and downstream flanking regions of the TSS in FGSCs and STO cells. (D) Functional enrichment of FGSC-specific methylated regions by GREAT analysis (p < 0.05).
Figure 6.

UCSC genome browser view of DNA methylation features of genes (Tbx1, Tbx2, Foxa1, and Foxb1) in FGSCs and STO cells
Promoter regions of genes involved in somatic development exhibit a distinct methylation pattern in FGSCs compared with those in STO cells.
Discussion
In our previous study, through comparisons with ESCs, primordial germ cells (PGCs), and male germline stem cells, we found unique epigenetic signatures (histone modification and DNA methylation) involved in the unipotency of FGSCs.11 In the present study, to obtain a better understanding of the epigenetic signatures of FGSCs, especially the relationship between mRNA m6A and the maintenance of FGSC characteristics, we compared the profiles of mRNA m6A and DNA methylation between FGSCs and STO cells.
DNA methylation, as an epigenetic mark involved in gene silencing, was of paramount importance for mammalian embryonic development.40 For example, in mESCs, depletion of Dnmt1 would result in genome-wide loss of CpG methylation.41 When lacking both Dnmt3a and Dnmt3b, mESCs could not methylate proviral genomes and repetitive elements.42 ESCs, without CpG methylation, could maintain stem cell properties and proliferation ability. Our previous study also showed that DNA methylation played a key role in maintaining the unipotency of FGSCs by suppressing somatic programs compared with the case in ESCs and embryonic day 11.5 (E11.5) PGCs.11 In this study, compared with the findings in STO cells, a type of somatic cell, we also observed that the expression levels of DNA methyltransferases (Dnmt1, Dnmt3a, and Dnmt3b) were significantly higher in FGSCs than those in STO cells. Additionally, our MeDIP-seq data also showed that there were distinct differences in DNA methylation patterns between FGSCs and STO cells, and the somatic development-related genes (e.g., Tbx1, Tbx2, Foxb1, and Foxa1) were hypermethylated specifically in FGSCs. The previous study proved that they were notably upregulated after Dnmt1 knockdown in FGSCs.11 These findings suggested that DNA methylation was actively involved in repressing the somatic program in FGSCs.
Recently, emerging data have demonstrated that m6A affects RNA transcript splicing, RNA stability, translation, processing, and nuclear export.26,29,43, 44, 45 Many studies have revealed that the modulation of the m6A level is involved in diverse processes, including the regulation of fate determination, proliferation and differentiation of stem cells, homeostasis, DNA damage response, spermatogenesis, and circadian clock processes.46, 47, 48, 49 m6A methylation is an mRNA post-transcriptional modification that is regulated by methyltransferases (METTL3, METTL14, WTAP, ZC3H13), demethylases (FTO, ALKBH5), and binding proteins (YTHDF1–3, YTHDC1 and YTHDC2). In this study, we observed that the expression levels of Mettl3, Alkbh5, Ythdf1, Ythdf2, Ythdc1, and Ythdc2 were significantly higher in FGSCs than those in STO cells, and MeRIP-seq data further showed that the unique m6A-methylated mRNAs in FGSCs and STO cells were related to cell population proliferation and somatic development, respectively. Recent studies showed that YTHDF1 interacted with translation initiation factors to directly affect translation initiation.26 Orouji et al.50 found that Ythdf1 could impact the proliferation and clonogenic capacity of Merkel cell carcinoma (MCC) cells by regulating the expression of translation eukaryotic initiation factor 3 (eIF3) in vitro. Bai et al.31 demonstrated that Ythdf1 was overexpressed in CRC and regulated the CRC cell’s tumorigenicity and cancer stem cell-like activity. Zhao et al.51 reported that Ythdf1 was a cell-cycle-related gene and involved in regulating hepatocellular carcinoma (HCC) cell-cycle progression. To understand the detailed mechanism of mRNA m6A on FGSC proliferation, we knocked down Ythdf1 in FGSCs. The results of CCK8 and EdU assays revealed that knockdown of Ythdf1 inhibited the proliferation of FGSCs. These findings suggested that mRNA m6A was involved in the maintenance of FGSC proliferation possibly through Ythdf1.
In summary, this study shown that mRNA m6A regulated FGSC self-renewal possibly through m6A binding protein YTHDF1, and the DNA methylation repressed the somatic programs in FGSC to maintain FGSC characteristics (Figure 7).
Figure 7.

Proposed model of mRNA m6A (YTHDF1) and DNA methylation in the regulation of FGSC characteristics
mRNA m6A regulated FGSC characteristics possibly through m6A binding protein YTHDF1, and genome DNA methylation repressed the somatic programs to maintain FGSC characteristics.
Materials and methods
Animals
5-day-old C57BL/6 mice were obtained from SLAC Laboratory Animal Company (Shanghai, China). All processes involving animals were approved by the Institutional Animal Care and Use Committee of Shanghai and conducted in accordance with the National Research Council Guide for the Care and Use of Laboratory Animals.
Culture of FGSCs and STO cells
The STO cell line (ATCC, Manassas, VA, USA) was cultured in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies, NY, USA), supplemented with 10% fetal bovine serum (FBS; Life Technologies, NY, USA), 100 μg/mL penicillin (Amresco, Lardner, PA, USA), and 1 mM nonessential amino acids (Invitrogen Life Sciences, MA, USA). The medium was replaced every other day. STO cells were subcultured every 3–4 days.
The FGSC line was cultured on STO feeder cells. The culture medium for FGSCs was alpha-minimum essential medium (MEM-α; Life Technologies, NY, USA), supplemented with 10% FBS (Life Technologies, NY, USA), 30 mg/mL pyruvate (Amresco, Lardner, PA, USA), 2 mM L-glutamine (Amresco, Lardner, PA, USA), 0.1 mM β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO, USA), 1 mM nonessential amino acids (Invitrogen Life Sciences, MA,USA), 20 ng/mL mouse epidermal growth factor (mEGF; PeproTech, NJ, USA), 10 ng/mL mouse basic fibroblast growth factor (mbFGF; BD Biosciences, Franklin Lakes, NJ, USA), 10 ng/mL mouse glial cell line-derived neurotrophic factor (mGDNF; R&D Systems, Minneapolis, MN, USA), 10 ng/mL mouse leukemia inhibitory factor (mLIF; Santa Cruz Biotechnology, CA, USA), and 15 mg/mL penicillin (Amresco, Lardner, PA, USA). Cells were passaged every 2–3 days. All cultures were performed in humidified air maintained at 37°C under a 5% CO2 atmosphere.
We used the differential adhesion method52 to separate feeder from FGSCs. After the cells were digested, feeder would adhere to the well more quickly than FGSCs. When the cells adhered for 30 min, the remaining unadhered cells were removed to another well without feeder and cultured for 24 h.
RT-PCR and qRT-PCR
Total RNA was extracted from FGSCs, STO cells, and ovarian tissues of neonatal mice using the TRIzol reagent (QIAGEN, Hilden, Germany) following the manufacturer’s protocol. About 1 μg of RNA was used to synthesize cDNA using a RT kit (Takara, Tokyo, Japan) in a volume of 20 μL. RT-PCR analysis was carried out with Taq DNA polymerase. qRT-PCR was conducted with SYBR Premix Ex Taq (Takara, Tokyo, Japan) in a 20-μL vol on an Applied Biosystems 7500 Real-Time PCR System. The conditions of qRT-PCR were 94°C for 5 min; 40 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s; followed by 72°C for 10 min. The 2−ΔΔCt method was used to analyze the data. The primers used were shown in Tables S1 and S2.
Immunofluorescence staining
The cells cultured in 48-well plates were washed with phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde (PFA) at room temperature for 30 min, and washed three times with PBS. Then, the cells were incubated for 30 min at 37°C in blocking buffer (PBS containing 35% goat serum). Next, the cells were incubated overnight at 4°C with primary rabbit anti-MVH antibody (1:200; Santa Cruz Biotechnology, CA, USA) and then washed three times with PBS. After that, the cells were incubated with the secondary antibody (1:200, goat anti-rabbit immunoglobulin [IgG]; Proteintech, Rosemont, IL, USA) for 30 min at 37°C. Finally, the cells were washed with PBS three times and stained with 4′,6-diamidino-2-phenylindole at room temperature for 3 min. For OCT4 staining, before incubation in blocking buffer, cells were permeabilized with 0.5% Triton X-100 for 30 min at room temperature and then washed with PBS three times. The primary antibody was anti-rabbit OCT4 (1:100; Santa Cruz Biotechnology, CA, USA). The secondary antibody was goat anti-rabbit IgG (1:200; Proteintech, Rosemont, IL, USA).
Lentiviral infection and selection
The lentiviral Ythdf1 knockdown (shDF1) and lentivirus carrying empty vector (shCtrl) were obtained from OBiO Technology (Shanghai, China). To generate stable lentivirus-infected cell lines, cells were infected with virus following the manufacturer’s instructions, and the cells were treated with 5 μg/mL of puromycin after infected 48 h to select the stable cell lines. The efficiency for Ythdf1 knockdown was determined by qRT-PCR and western blot. The shRNA primer sequences were listed as follows: primer (forward [F]): 5′-CCGGTTACCAGCACAGGTTTAATTTCTCGAGAAATTAAACCTGTGCTGGTAATTTTTTG-3′; primer (reverse [R]): 5′-AATTCAAAAAATTACCAGCACAGGTTTAATTTCTAGAAATTAAACCTGCTGGTAA-3′.
CCK8 assay
FGSCs were seeded at 5,000 cells/well in a 96-well plate and cultured for 24 h. The CCK8 (Research Science, Shanghai, China) was diluted at a 1:11 ratio to add the cell culture medium, and the cells were incubated for 2 h at 37°C. After that, the absorbance was measured at 450 nm using a microplate spectrophotometer.
EdU proliferation assay
Cells were incubated with 50 μM EdU kit for 2 h at 37°C and were fixed with 4% PFA for 30 min at room temperature. Then cells were washed with 2 mg/mL glycine for 5 min on a shaker and incubated with 0.5% Triton X-100 for 30 min before 1 × Apollo was added for 30 min at room temperature. After washing 3 times with PBS containing 0.5% Triton X-100, 1 × Hoechst 33342 was used to stain cell nuclei. Images were obtained using the Leica fluorescence microscope, and then, the number of EdU-positive cells was analyzed.
Western blotting
Cells were lysed in radio immunoprecipitation assay (RIPA) buffer (Yeasen, Shanghai, China), containing 1 × PIC (protease inhibitor cocktail), and then centrifugated at 12,000 × g for 20 min at 4°C. Proteins were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted on polyvinylidene fluoride (PVDF) membranes. Membranes were blocked with 5% non-fat milk for 2 h at room temperature and incubated with the primary antibodies: anti-YTHDF1 (1:1,000; Proteintech, Rosemont, IL, USA) and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:10,000; Santa Cruz Biotechnology, CA, USA) at 4°C overnight. The next day, membranes were incubated with secondary antibodies (1:2,000; Proteintech, Rosemont, IL, USA) for 1 h at room temperature following washing three times with Tris-buffered saline-Tween 20 (TBST). The protein bands were obtained with a Tanon 4600SF (Tanon, Shanghai, China).
MeDIP-seq
The preparation of MeDIP and input DNA libraries was performed as previously described.11 Briefly, genomic DNA of STO cells was extracted and then randomly sheared into 200-to 500-bp fragments by sonication. The fragmented DNA was precleared and then immunoprecipitated using a highly specific anti-5-methylcytosine monoclonal antibody supplied in the Magnetic Methylated DNA Immunoprecipitation (MagMeDIP) Kit (Diagenode, Denville, NJ, USA), in accordance with the manufacturer’s instructions. MagMeDIP and input DNA were used for library generation with NEBNext Ultra End Repair/dA-Tailing Module (NEB, Ipswich, MA, USA). Enriched libraries were evaluated for their size distribution and concentration using an Agilent Bioanalyzer 2100, and sequencing was carried out on Illumina HiSeq 2000 following the manufacturer’s instructions.
MeRIP-seq
Total RNA was extracted from FGSCs and STO cells using TRIzol reagent (QIAGEN, Hilden, Germany), in accordance with the manufacturer’s protocol. mRNA was further purified using the Dynabeads mRNA Purification Kit (Thermo Fisher Scientific, MA, USA). The purified mRNA was then fragmented into 100−200 nucleotides using Fragmentation Reagents (Thermo Fisher Scientific, MA, USA). A small proportion (10%) of the purified mRNA fragments was put aside to be used as an input sample, and the rest was incubated with m6A primary antibody (ab151230; Abcam, Cambridge, MA, USA) for 2 h at 4°C. The mixture was then immunoprecipitated by incubation with Protein A beads (Thermo Fisher Scientific, MA, USA) for 2 h at 4°C. Then, the beads were washed three times with immunoprecipitation buffer (150 mM NaCl, 0.1% NP-40, and 10 mM Tris-HCl, pH 7.4) and eluted competitively with m6A 5′-monophosphate sodium salt (Santa Cruz Biotechnology, CA, USA). The RNA in the eluate was used for library preparation with the TruSeq Stranded mRNA Sample Preparation Kit (Vazyme, Shanghai, China). Libraries were sequenced on Illumina HiSeq 2000, in accordance with the manufacturer’s instructions.
m6A dot blot assay
For m6A dot blot assays, mRNA was purified using Dynabeads mRNA Purification Kit (Thermo Fisher Scientific, MA, USA). Then, the serially diluted mRNA was denatured at 95°C for 3 min to disrupt secondary structures in a heat block. After that, the mRNA was dropped onto negative control (NC) membranes and UV crosslinked. Then, the membrane was stained with 0.02% methylene blue (MB) in 0.3 M sodium acetate (pH 5.3) as loading control. These membranes were blocked with 5% non-fat milk for 1 h at room temperature and then incubated with m6A primary antibody (ab151230; Abcam, Cambridge, MA, USA) overnight at 4°C. The next day, the membranes were incubated with goat anti-rabbit IgG (1:2,000; Proteintech, Rosemont, IL, USA) for 1 h at room temperature. The signal was detected using the Tanon detection system, and the signal density was quantified using ImageJ.
Data analysis
All DNA MeDIP-seq, m6A-MeRIP-seq, and control raw data reads were mapped using Bowtie (version 1.0.1) to the University of California, Santa Cruz (UCSC), mm9 reference genome.53 Two biological replicates were performed, which were well correlated (Figure S1). The raw data quality was analyzed using FastQC; poor-quality sequences and duplicate reads were removed, and the remainder was used for analysis. Data analysis for each experiment was as follows: (1) for DNA MeDIP-seq, we first divided the UCSC known gene promoter regions into 500-bp windows, calculated the methylation level in each window, and then calculated the Pearson correlation coefficient between them. To examine the methylation levels in FGSCs and STO cells, we evaluated them using the relative methylation score (rms), calculated with the R package MEDIPS. (2) For m6A MeRIP-seq, the m6A-enriched peaks in each m6A immunoprecipitation sample were identified by MACS2 peak-calling software (version 2.0.10)54 with the corresponding input sample acting as a control. MACS2 was run with the default options, except for “–nomodel, –keepdup all” to turn off fragment size estimation and to keep all uniquely mapping reads, respectively. A stringent cut-off threshold for the p value of 1 × 10−5 was used to obtain high-confidence peaks. Each peak was annotated based on Ensemble (release 79) gene annotation information by applying Bedtools’ Intersect Bed (version 2.16.2).55
The datasets supporting the results presented in this article are available in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO): GSM4411135.
GO analysis
GO analysis was performed using GREAT (version 2.0).39 Fisher’s exact test was used to identify the significant results, and the false discovery rate (FDR) was applied to correct the p values (p < 0.05; fold change > 1.5).
Motif analysis
We searched for de novo motifs enriched in each m6A peak using the HOMER tool.56 Motif length was restricted to six nucleotides. All peaks mapped to mRNAs were used as the target sequences, and background sequences were constructed by randomly shuffling peaks upon total mRNAs on the genome using Bedtools’ shuffle Bed (version 2.16.2).55
Data from other sources
Previously published MeDIP-seq data from FGSCs were downloaded from NCBI Sequence Read Archive (SRA): SRP066132.11
Statistical analysis
Results are shown as the mean ± SD of three independent experiments. Student’s t test was used to calculate differences between groups using SPSS17.0. A p value less than 0.05 was considered statistically significant.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2017YFA0504201); National Nature Science Foundation of China (81720108017); National Major Scientific Instruments and Equipment Development Project, National Natural Science Foundation of China (61827814); and Major Project of Key Research and Development Program of Ningxia Hui Autonomous Region (2019BFG02005).
Author contributions
J.W. conceived and designed the experiments. X.Z. and Q.F. conducted most of the experiments. X.Z. and G.G.T. collected and analyzed the data. Z.W. and X.P. conceived the experiments. X.Z. drafted the manuscript. G.G.T. and J.W. revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.11.020.
Contributor Information
Zhaoxia Wang, Email: zhaoxiaw@sjtu.edu.cn.
Ji Wu, Email: jiwu@sjtu.edu.cn.
Supplemental information
References
- 1.Lehmann R. Germline stem cells: origin and destiny. Cell Stem Cell. 2012;10:729–739. doi: 10.1016/j.stem.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zou K., Yuan Z., Yang Z., Luo H., Sun K., Zhou L., Xiang J., Shi L., Yu Q., Zhang Y. Production of offspring from a germline stem cell line derived from neonatal ovaries. Nat. Cell Biol. 2009;11:631–636. doi: 10.1038/ncb1869. [DOI] [PubMed] [Google Scholar]
- 3.Wu C., Xu B., Li X., Ma W., Zhang P., Chen X., Wu J. Tracing and Characterizing the Development of Transplanted Female Germline Stem Cells In Vivo. Mol. Ther. 2017;25:1408–1419. doi: 10.1016/j.ymthe.2017.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hou L., Wang J., Li X., Wang H., Liu G., Xu B., Mei X., Hua X., Wu J. Characteristics of Female Germline Stem Cells from Porcine Ovaries at Sexual Maturity. Cell Transplant. 2018;27:1195–1202. doi: 10.1177/0963689718784878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo K., Li C.H., Wang X.Y., He D.J., Zheng P. Germ stem cells are active in postnatal mouse ovary under physiological conditions. Mol. Hum. Reprod. 2016;22:316–328. doi: 10.1093/molehr/gaw015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan Z., Sun M., Liang X., Li J., Zhou F., Zhong Z., Zheng Y. The Controversy, Challenges, and Potential Benefits of Putative Female Germline Stem Cells Research in Mammals. Stem Cells Int. 2016;2016:1728278. doi: 10.1155/2016/1728278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White Y.A., Woods D.C., Takai Y., Ishihara O., Seki H., Tilly J.L. Oocyte formation by mitotically active germ cells purified from ovaries of reproductive-age women. Nat. Med. 2012;18:413–421. doi: 10.1038/nm.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bai Y., Yu M., Hu Y., Qiu P., Liu W., Zheng W., Peng S., Hua J. Location and characterization of female germline stem cells (FGSCs) in juvenile porcine ovary. Cell Prolif. 2013;46:516–528. doi: 10.1111/cpr.12058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou L., Wang L., Kang J.X., Xie W., Li X., Wu C., Xu B., Wu J. Production of fat-1 transgenic rats using a post-natal female germline stem cell line. Mol. Hum. Reprod. 2014;20:271–281. doi: 10.1093/molehr/gat081. [DOI] [PubMed] [Google Scholar]
- 10.Zhang C., Wu J. Production of offspring from a germline stem cell line derived from prepubertal ovaries of germline reporter mice. Mol. Hum. Reprod. 2016;22:457–464. doi: 10.1093/molehr/gaw030. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X.L., Wu J., Wang J., Shen T., Li H., Lu J., Gu Y., Kang Y., Wong C.H., Ngan C.Y. Integrative epigenomic analysis reveals unique epigenetic signatures involved in unipotency of mouse female germline stem cells. Genome Biol. 2016;17:162. doi: 10.1186/s13059-016-1023-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun J., Wei H.M., Xu J., Chang J.F., Yang Z., Ren X., Lv W.W., Liu L.P., Pan L.X., Wang X. Histone H1-mediated epigenetic regulation controls germline stem cell self-renewal by modulating H4K16 acetylation. Nat. Commun. 2015;6:8856. doi: 10.1038/ncomms9856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 14.Smith Z.D., Meissner A. DNA methylation: roles in mammalian development. Nat. Rev. Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 15.Karimi M.M., Goyal P., Maksakova I.A., Bilenky M., Leung D., Tang J.X., Shinkai Y., Mager D.L., Jones S., Hirst M., Lorincz M.C. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell. 2011;8:676–687. doi: 10.1016/j.stem.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Neill K.M., Irwin R.E., Mackin S.J., Thursby S.J., Thakur A., Bertens C., Masala L., Loughery J.E.P., McArt D.G., Walsh C.P. Depletion of DNMT1 in differentiated human cells highlights key classes of sensitive genes and an interplay with polycomb repression. Epigenetics Chromatin. 2018;11:12. doi: 10.1186/s13072-018-0182-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Auclair G., Borgel J., Sanz L.A., Vallet J., Guibert S., Dumas M., Cavelier P., Girardot M., Forné T., Feil R., Weber M. EHMT2 directs DNA methylation for efficient gene silencing in mouse embryos. Genome Res. 2016;26:192–202. doi: 10.1101/gr.198291.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borgel J., Guibert S., Li Y., Chiba H., Schübeler D., Sasaki H., Forné T., Weber M. Targets and dynamics of promoter DNA methylation during early mouse development. Nat. Genet. 2010;42:1093–1100. doi: 10.1038/ng.708. [DOI] [PubMed] [Google Scholar]
- 19.Perry R.P., Kelley D.E., LaTorre J. Synthesis and turnover of nuclear and cytoplasmic polyadenylic acid in mouse L cells. J. Mol. Biol. 1974;82:315–331. doi: 10.1016/0022-2836(74)90593-2. [DOI] [PubMed] [Google Scholar]
- 20.Jia G., Fu Y., He C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. 2013;29:108–115. doi: 10.1016/j.tig.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwartz S. Cracking the epitranscriptome. RNA. 2016;22:169–174. doi: 10.1261/rna.054502.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li S., Mason C.E. The pivotal regulatory landscape of RNA modifications. Annu. Rev. Genomics Hum. Genet. 2014;15:127–150. doi: 10.1146/annurev-genom-090413-025405. [DOI] [PubMed] [Google Scholar]
- 23.Liu N., Pan T. RNA epigenetics. Transl. Res. 2015;165:28–35. doi: 10.1016/j.trsl.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heck A.M., Wilusz C.J. Small changes, big implications: The impact of m6A RNA methylation on gene expression in pluripotency and development. Biochim. Biophys. Acta. Gene Regul. Mech. 2019;1862:194402. doi: 10.1016/j.bbagrm.2019.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng G., Dahl J.A., Niu Y., Fedorcsak P., Huang C.M., Li C.J., Vågbø C.B., Shi Y., Wang W.L., Song S.H. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X., Zhao B.S., Roundtree I.A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., He C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sheth U., Parker R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science. 2003;300:805–808. doi: 10.1126/science.1082320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X., He C. Dynamic RNA modifications in posttranscriptional regulation. Mol. Cell. 2014;56:5–12. doi: 10.1016/j.molcel.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi H., Wang X., Lu Z., Zhao B.S., Ma H., Hsu P.J., Liu C., He C. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017;27:315–328. doi: 10.1038/cr.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bai Y., Yang C., Wu R., Huang L., Song S., Li W., Yan P., Lin C., Li D., Zhang Y. YTHDF1 Regulates Tumorigenicity and Cancer Stem Cell-Like Activity in Human Colorectal Carcinoma. Front. Oncol. 2019;9:332. doi: 10.3389/fonc.2019.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ivanova I., Much C., Di Giacomo M., Azzi C., Morgan M., Moreira P.N., Monahan J., Carrieri C., Enright A.J., O’Carroll D. The RNA m(6)A Reader YTHDF2 Is Essential for the Post-transcriptional Regulation of the Maternal Transcriptome and Oocyte Competence. Mol. Cell. 2017;67:1059–1067.e4. doi: 10.1016/j.molcel.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dann C.T., Alvarado A.L., Molyneux L.A., Denard B.S., Garbers D.L., Porteus M.H. Spermatogonial stem cell self-renewal requires OCT4, a factor downregulated during retinoic acid-induced differentiation. Stem Cells. 2008;26:2928–2937. doi: 10.1634/stemcells.2008-0134. [DOI] [PubMed] [Google Scholar]
- 34.Bortvin A., Goodheart M., Liao M., Page D.C. Dppa3 / Pgc7 / stella is a maternal factor and is not required for germ cell specification in mice. BMC Dev. Biol. 2004;4:2. doi: 10.1186/1471-213X-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kobayashi T., Kajiura-Kobayashi H., Nagahama Y. Differential expression of vasa homologue gene in the germ cells during oogenesis and spermatogenesis in a teleost fish, tilapia, Oreochromis niloticus. Mech. Dev. 2000;99:139–142. doi: 10.1016/s0925-4773(00)00464-0. [DOI] [PubMed] [Google Scholar]
- 36.Ohinata Y., Payer B., O’Carroll D., Ancelin K., Ono Y., Sano M., Barton S.C., Obukhanych T., Nussenzweig M., Tarakhovsky A. Blimp1 is a critical determinant of the germ cell lineage in mice. Nature. 2005;436:207–213. doi: 10.1038/nature03813. [DOI] [PubMed] [Google Scholar]
- 37.Tanaka S.S., Yamaguchi Y.L., Tsoi B., Lickert H., Tam P.P. IFITM/Mil/fragilis family proteins IFITM1 and IFITM3 play distinct roles in mouse primordial germ cell homing and repulsion. Dev. Cell. 2005;9:745–756. doi: 10.1016/j.devcel.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 38.Lange U.C., Saitou M., Western P.S., Barton S.C., Surani M.A. The fragilis interferon-inducible gene family of transmembrane proteins is associated with germ cell specification in mice. BMC Dev. Biol. 2003;3:1. doi: 10.1186/1471-213X-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greenberg M.V.C., Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019;20:590–607. doi: 10.1038/s41580-019-0159-6. [DOI] [PubMed] [Google Scholar]
- 41.Li E., Bestor T.H., Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 42.Okano M., Bell D.W., Haber D.A., Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 43.Schwartz S., Mumbach M.R., Jovanovic M., Wang T., Maciag K., Bushkin G.G., Mertins P., Ter-Ovanesyan D., Habib N., Cacchiarelli D. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014;8:284–296. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geula S., Moshitch-Moshkovitz S., Dominissini D., Mansour A.A., Kol N., Salmon-Divon M., Hershkovitz V., Peer E., Mor N., Manor Y.S. Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. 2015;347:1002–1006. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- 45.Liu N., Dai Q., Zheng G., He C., Parisien M., Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang C., Chen Y., Sun B., Wang L., Yang Y., Ma D., Lv J., Heng J., Ding Y., Xue Y. m6A modulates haematopoietic stem and progenitor cell specification. Nature. 2017;549:273–276. doi: 10.1038/nature23883. [DOI] [PubMed] [Google Scholar]
- 47.Tang C., Klukovich R., Peng H., Wang Z., Yu T., Zhang Y., Zheng H., Klungland A., Yan W. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3′-UTR mRNAs in male germ cells. Proc. Natl. Acad. Sci. USA. 2018;115:E325–E333. doi: 10.1073/pnas.1717794115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang S., Zhao B.S., Zhou A., Lin K., Zheng S., Lu Z., Chen Y., Sulman E.P., Xie K., Bogler O. m(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell. 2017;31:591–606.e6. doi: 10.1016/j.ccell.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao X., Yang Y., Sun B.F., Shi Y., Yang X., Xiao W., Hao Y.J., Ping X.L., Chen Y.S., Wang W.J. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24:1403–1419. doi: 10.1038/cr.2014.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orouji E., Peitsch W.K., Orouji A., Houben R., Utikal J. Oncogenic Role of an Epigenetic Reader of m(6)A RNA Modification: YTHDF1 in Merkel Cell Carcinoma. Cancers (Basel) 2020;12:202. doi: 10.3390/cancers12010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao X., Chen Y., Mao Q., Jiang X., Jiang W., Chen J., Xu W., Zhong L., Sun X. Overexpression of YTHDF1 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Biomark. 2018;21:859–868. doi: 10.3233/CBM-170791. [DOI] [PubMed] [Google Scholar]
- 52.Steinberg M.S. Does differential adhesion govern self-assembly processes in histogenesis? Equilibrium configurations and the emergence of a hierarchy among populations of embryonic cells. J. Exp. Zool. 1970;173:395–433. doi: 10.1002/jez.1401730406. [DOI] [PubMed] [Google Scholar]
- 53.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.