

Abstract
Chronic hepatitis B virus (HBV) infection is a major public health problem. New treatment approaches are needed because current treatments do not target covalently closed circular DNA (cccDNA), the template for HBV replication, and rarely clear the virus. We harnessed adeno-associated virus (AAV) vectors and CRISPR-Staphylococcus aureus (Sa)Cas9 to edit the HBV genome in liver-humanized FRG mice chronically infected with HBV and receiving entecavir. Gene editing was detected in livers of five of eight HBV-specific AAV-SaCas9-treated mice, but not control mice, and mice with detectable HBV gene editing showed higher levels of SaCas9 delivery to HBV+ human hepatocytes than those without gene editing. HBV-specific AAV-SaCas9 therapy significantly improved survival of human hepatocytes, showed a trend toward decreasing total liver HBV DNA and cccDNA, and was well tolerated. This work provides evidence for the feasibility and safety of in vivo gene editing for chronic HBV infections, and it suggests that with further optimization, this approach may offer a plausible way to treat or even cure chronic HBV infections.
Keywords: HBV, AAV, Gene editing, Humanized mouse, CRISPR/Cas9
Graphical Abstract
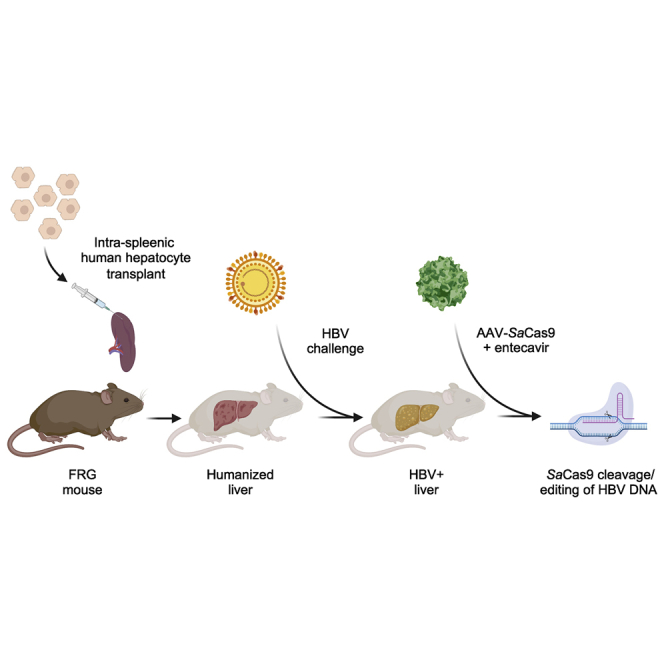
This study analyzes CRISPR-Cas9-based antiviral therapy in a model of chronic hepatitis B virus (HBV) infection. The study demonstrates that therapy is safe, and that gene editing of HBV can occur in vivo, although low levels of CRISPR-Cas9 delivery limit the current efficacy of the approach.
Introduction
An estimated 257 million people are living with chronic hepatitis B virus (HBV) infection.1 More than 850,000 die annually from complications such as cirrhosis and hepatocellular carcinoma (HCC).1 Although antiviral drugs can reduce HBV viral loads to almost undetectable levels after several months of therapy, they are only suppressive, and the virus rebounds rapidly when treatment is withdrawn. Cure of HBV will require elimination of covalently closed circular DNA (cccDNA), the long-lived viral genomic intermediate that is the template for HBV replication and persistence. For this reason, gene-editing approaches that directly target cccDNA have been proposed as potential curative therapies.2,3
Gene editing of viral DNA by the clustered regularly interspaced palindromic repeat system (CRISPR-Cas9) or other endonucleases is a promising therapy for several persistent viral infections.2, 3, 4 Gene editing can lead to mutation of essential viral genes, or even degradation and elimination of viral genomes. Gene editing of HBV has been demonstrated in experimental models using CRISPR-Cas9, zinc finger nucleases (ZFNs), and transcription activator-like effector nucleases. Most of these studies have been performed in cell culture systems, with few studies having been performed in vivo. The in vivo studies reported to date have been of limited relevance to actual human infection, consisting of transgenic mice containing integrated HBV genomes, or relying on plasmid or adeno-associated virus (AAV) vectors to introduce HBV genomes into mouse hepatocytes, which are not permissive to HBV infection. These models do not feature a complete HBV replication cycle that includes the ability to infect and re-infect hepatocytes, and importantly they do not produce cccDNA.5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 Rigorous testing of gene editing against chronic HBV infection requires an animal model that recapitulates these essential elements of HBV infection.
Recently, humanized liver mouse models have been developed that faithfully reflect human HBV infection. These models use immunodeficient mice with a genetic background lethal for mouse hepatocytes; transplanted human hepatocytes are provided to reconstitute the liver, which becomes chimeric.25 For HBV the most commonly used liver-humanized models include Alb-uPA-severe combined immunodeficiency (SCID), TK-NOG, or fumarylacetoacetate hydrolase (FAH)−/−/Rag2−/−/Il2rg−/− (FRG) mice,26, 27, 28 in which the chimeric liver provides a site for active HBV replication and cccDNA production, allowing for persistent viral infection.29, 30, 31, 32, 33, 34, 35 Although complex, costly, and lacking a functional immune system, these liver-humanized mouse models are the gold standard for validation of antiviral therapies, and they have been used to investigate new classes of small-molecule HBV inhibitors.31,36
In this study, we used the liver-humanized FRG mouse model to study the safety and antiviral efficacy of a hepatotropic AAVLK03 vector delivering Staphylococcus aureus (Sa)Cas9 and two HBV-specific single guide RNAs (sgRNAs), with concurrent entecavir treatment, in mice chronically infected with genotype C HBV for >140 days. We show that AAV-SaCas9-mediated gene editing of HBV is safe, results in mutagenesis of HBV in hepatocytes, and prolongs human hepatocyte survival. Our results support the continued development of CRISPR-Cas9-mediated gene editing as a potentially curative approach for chronic HBV infection.
Results
Selection of HBV-specific sgRNAs and in vitro efficacy
We chose to use SaCas9, which is among the smallest and most active Cas9 variants, to enable delivery of both Cas9 and sgRNAs against HBV targets via a single AAV vector. We identified all (N)20NNGRRT recognition sequences in the plus and minus strands of a consensus HBV sequence generated from 2,233 genotype B/C complete genome sequences found within an alignment of 3,847 previously described genotype A–H whole-genome sequences.7 From these, we identified highly conserved sites within overlapping HBV reading frames with the best combinations of on- and off-target specificity scores, and further validated their conservation using an alternative dataset of 2,179 genotype C complete genome sequences.37 We then selected from these six genotype C-specific sgRNA target sites for biological validation (Figure S1A; Table S1) that had no more than a single mismatch with a consensus sequence generated from an alignment of 7,108 complete genomes of all genotypes derived from the distinct dataset (Figure S1B).37
We developed an in vitro assay to analyze sgRNA activity using plasmids as a surrogate for HBV cccDNA. We built two reporter constructs, each containing tandem copies of three HBV-specific sgRNA target sites (C1-C3-C6 and C7-C14-C16) fused in-frame to the 5′ end of the green fluorescent protein (GFP) reporter gene, so that a single continuous open reading frame (ORF) is expressed (Figure S1C). We transfected each reporter into 293 cells along with a second plasmid that expresses SaCas9 in combination with an HBV-specific or control GFP-specific sgRNA. Cells were purposely transfected at low efficiency (~20%) so that the reporter plasmid copy number per cell would be low and knockdown of gene expression levels could be readily detected. For quantification purposes cells were imaged by fluorescence microscopy (Figure S1D), after which we gated GFP+ cells by flow cytometry into high-MFI cells (Hi), middle- plus high-MFI cells (Mid), and all GFP+ cells (All) using the no sgRNA control as shown (Figure S1E). Knockdown of GFP fluorescence by all HBV-specific sgRNAs in 293 cells was quantified at 24 h post-transfection in the Hi, Mid, and All groups (Figure S1F), and two previously validated and highly active SaCas9 sgRNAs targeting GFP (GFP6 and GFP7)38 were used as internal controls, allowing comparison of activity across the two reporters. For both reporters, the number of GFP+ cells was reduced in the presence of both control GFP-specific sgRNAs, with GFP7 showing higher activity than GFP6. GFP knockdown was detected in Hi, Mid, and All cells transfected with each reporter, and the relative difference in GFP knockdown between GFP6 and GFP7 was consistent across both reporters. All six HBV-specific sgRNAs reduced the number of GFP+ cells, although activity varied across the Hi, Mid, and All groups, and was more pronounced in the Hi group. The most active HBV sgRNAs, with levels of GFP knockdown comparable to the control GFP6 and GFP7 sgRNAs (C7 and C14), were selected as lead candidates for in vivo analysis. In parallel, human embryonic kidney 293 (HEK293) cells were transfected in duplicate with the same reporter and SaCas9/sgRNA-expressing plasmids, and gene editing efficiency was quantified using the T7 endonuclease I assay on PCR amplicons from genomic DNA isolated 24 h post-transfection (Figure S1G). T7 endonuclease I cleavage bands of the predicted size were seen for all GFP-specific and HBV-specific sgRNAs, demonstrating that gene editing had occurred at all target sites.
Optimization of AAV delivery to humanized FRG mouse livers
We chose to investigate AAV-SaCas9 therapy for chronic HBV in liver-humanized FRG mice, in which we previously demonstrated persistent infection with multiple HBV genotypes, including genotype C.31 Humanized FRG mice have chimeric livers that contain both human and mouse hepatocytes (Figure 1A). To determine efficiency of transgene delivery to human hepatocytes in the humanized FRG mouse, we packaged self-complementary AAV (scAAV) vector genomes expressing GFP from the strong constitutive small hybrid cytomegalovirus (CMV)/chicken β-actin (smCBA) enhancer/promoter into the human hepatocyte tropic AAVLK03 capsid.39 We delivered this scAAVLK03-smCBA-GFP vector intravenously at doses of 5 × 1010 and 2 × 1011 vector genomes/mouse and analyzed hepatocyte transduction 14 days later. By immunohistochemistry (IHC), GFP expression was detected in hepatocytes throughout the liver, predominantly around blood vessels found in hepatic lobules (Figure 1B). Direct in situ detection of GFP fluorescence in whole livers immediately after necropsy revealed a more intense fluorescent signal at the higher dose (Figure 1C). IHC staining of liver tissue for GFP and human albumin expression demonstrated that the GFP signal predominantly co-localized with human hepatocytes (Figure 1D). In contrast and for unclear reasons, identical delivery of the lower dose of this vector to liver-humanized Alb-uPA/scid mice yielded few GFP+ human hepatocytes (Figure 1E). Based on high levels of human hepatocyte transduction, we pursued AAVLK03 delivery of anti-HBV SaCas9 therapeutics in the liver-humanized FRG mouse model.
Figure 1.

Gene delivery to humanized chimeric mouse livers
(A) Livers from liver-humanized FRG mice were stained to show human (red, anti-human albumin [hAlb]) and mouse (green, anti-mouse albumin [mAlb]) hepatocytes. (B) GFP expression in humanized livers of FRG mice 14 days after retro-orbital delivery of scAAVLK03-smCBA-GFP vector at a dose of 5 × 1010 vector genomes/mouse. Livers were stained using an anti-GFP antibody. (C) In situ GFP expression in whole livers at necropsy detected using an AMG EVOS fluorescence microscope 14 days after retro-orbital administration of a low (5 × 1010 vector genomes) or high (2 × 1011 vector genomes) dose of scAAVLK03-smCBA-GFP. (D) Co-labeling with anti-GFP (green) and anti-human albumin (red) antibodies in humanized livers from a FRG mouse treated with 2 × 1011 vector genomes of scAAVLK03-smCBA-GFP. (E) Liver gene delivery in liver-humanized Alb-uPA/scid mice. Liver tissue was harvested from Alb-uPA/scid mice 14 days after intravenous retro-orbital delivery of scAAV-LK03-smCBA-GFP vector at a dose of 5 × 1010 vector genomes/mouse. Images show human albumin (red, hAlb) and GFP (green, anti-GFP).
AAV-SaCas9 construct evaluation
The ~4.8-kb packaging limit of single-stranded AAV genomes limits the genetic payload that can be used in the development of CRISPR-Cas9 therapeutics; specifically, the smCBA promoter is too large for use in combination with the SaCas9 gene plus sgRNAs within a single vector. We therefore evaluated a short EF1α promoter for expression of SaCas9 that we previously used to express anti-HBV ZFNs from AAV vectors in HCC-derived HepAD38 cells.7 To enhance SaCas9 expression we fused the short EF1α promoter to a minute virus of mice intron, which can increase promoter expression levels from AAV vectors in vivo.40,41 This hybrid EFM promoter is only 325 bp in length, which enables incorporation of two sgRNA expression cassettes in the same AAV-SaCas9 vector. We subsequently evaluated EFM-SaCas9 AAV vector constructs that utilize two different polymerase III (pol III) promoters to express two sgRNAs in order to minimize repeat sequences and potential recombination in the AAV vector backbone. AAV-SaCas9vector constructs that contain expression cassettes utilizing a combination of the hU6, hH1, or tRNAGLN pol III promoters to drive dual sgRNA expression were generated (Figure S2A). We inserted the GFP7 sgRNA sequence at each site to ensure that each promoter could produce a functional sgRNA. GFP knockdown was then analyzed in HEK293 cells and HCC-derived Huh7 cells transfected with each GFP7-expressing sgRNA AAV-SaCas9 vector plasmid plus a GFP-expressing reporter plasmid. At 24 h post-transfection each AAV vector plasmid expressing the GFP7 sgRNA from either the hU6 or hH1 promoter was able to knock down GFP expression in both HEK293 and Huh7 cells, indicating that both SaCas9 and GFP7 sgRNAs were being expressed (Figures S2B and S2C). When tRNAGLN was used to express the GFP7 sgRNA we did not see efficient GFP knockdown, in contrast to previous studies.42 These observations suggest that the EFM, hU6, and hH1 promoters can function efficiently in human hepatocyte-derived cells, and we elected to use the AAV vector construct AAV-EFM-SaCas9-SV40pA-hH1-sgRNA-hU6-sgRNA for further studies since the SV40 poly(A) signal has previously enabled higher AAV expression than a synthetic poly(A) (SPA) signal.43
AAV-SaCas9 evaluation in chronically HBV-infected FRG mice
In order to most closely model the clinical setting and enable viral gene editing with a minimum of ongoing viral replication, we analyzed AAV-SaCas9 antiviral activity in combination with RTi (entecavir) therapy (Figure 2). We used 15 humanized FRG mice chronically infected with a genotype C HBV clinical isolate.31 To ensure that the clinical isolate was susceptible to SaCas9 cleavage, HBV target sites C7 and C14 from the genotype C inoculum were sequenced by next-generation sequencing (NGS), confirming that both sites were highly conserved (>99%) at each nucleotide position (Figure S3). sgRNAs for target sites C7 and C14 were incorporated into the AAV-EFM-SaCas9-SV40pA-hH1-sgRNA-hU6-sgRNA AAV vector backbone for AAVLK03 vector production (Figure 2). In parallel, a control AAV vector backbone was generated containing the GFP-specific target sequences GFP6 and GFP7. At the initiation of the study all mice had baseline viremia, so they were treated with the RTi entecavir for 17 days to reduce viral loads and the number of intra-hepatic targets. Mice were then administered anti-HBV AAV-SaCas9 vector or control anti-GFP AAV-SaCas9 vector intravenously at a dose of 5 × 1011 vector genomes/mouse. AAV vectors were delivered 144 days after initial HBV infection, and mice remained on entecavir treatment for 27 days after AAV administration to enable CRISPR-Cas9 viral gene editing during suppression of ongoing viral replication. At this time point, a subset of anti-HBV AAV-SaCas9-treated and anti-GFP AAV-SaCas9-treated mice were sacrificed, while entecavir treatment was stopped for the remaining animals so that rebound viremia could be followed until the experimental endpoint at day 62 after AAV administration.
Figure 2.

Experimental setup for AAV-SaCas9 treatment of HBV-infected humanized FRG mice
AAV vectors were generated with capsid LK03 and express SaCas9 in combination with either two control GFP-specific sgRNAs (GFP early/late) or two HBV genotype C-specific sgRNAs (HBV early/late). Chronically HBV-infected FRG mice were given entecavir for 17 days to suppress viremia and then injected intravenously with either anti-GFP or anti-HBV AAV SaCas9 vectors, and followed for levels of viremia for 4 (early) or 9 (late) weeks, before necropsy. Mice were infected with a genotype C HBV inoculum 144 days prior to AAV administration and entecavir administration was halted at 4 weeks after AAV administration. vg, vector genomes; ITR, inverted terminal repeat; EFM, EF1α short/minute virus of mice intron hybrid promoter; pA, SV40 poly(A); sgRNA, single guide RNA; hH1, human H1 promoter; hU6, human U6 promoter.
Detection of HBV gene editing in Cas9-treated mice
HBV target site sequences in serum and livers of control and treated mice collected at necropsy were analyzed by generating PCR amplicons spanning target sites C7 and C14 and sequencing them by NGS. SaCas9 target site mean read depth was >1,000× (target site C14) and >4,500× (target site C7). In livers of treated mice, we found C7- or C14-specific target site mutations in five out of eight mice that received anti-HBV AAV treatment, presumably as a result of mutagenic DNA repair following SaCas9 target site cleavage (Figure 3A; Figure S4). Two treated mice had detectable mutations present within both target sites. Importantly, no mutations were observed at the C7 or C14 sites in any of the control mice. The frequency of indels in anti-HBV SaCas9-treated animals was low, representing less than 1% of sequencing reads at either site. We and others have previously detected endonuclease-resistant and replication-competent HIV proviruses with indels in multiples of 3 following endonuclease treatment despite altered amino acid sequences in essential genes,44, 45, 46, 47 and notably deletions of 3 and 6 nt were detected in HBV target site C14. However, site C14 contains codons within different reading frames for two essential genes (core and polymerase), and it is unlikely that altered amino acid sequences within both of these genes would produce virus retaining replication competency. At the time of death, all animals had viral loads of 104–108 IU/mL, so HBV-specific target site amplicons amplified from serum were most likely to be derived from de novo produced virus. Our analysis of both target sites in serum found no evidence of gene editing at either location in any anti-HBV SaCas9-treated animal (data not shown). This likely indicates that HBV present in blood at the time of death was derived from unmodified template cccDNA in HBV-infected hepatocytes and was thus likely to be infectious.
Figure 3.

HBV-specific and off-target gene editing
PCR amplicons spanning the C7 and C14 HBV target sites, or 20 closely related off-target sites for C7 or C14 within the human or mouse genomes (five of each) were amplified from liver genomic DNA and subjected to deep sequencing for analysis of mutations within the indicated 26-bp SaCas9 sgRNA target sequences. Mutations detected within each target site were identified using a custom script, and the mutation rate is based on mutations with multiple reads (non-singletons). (A and B) Mutation rates for the C7 and C14 target sites were determined for all mice (A), and the relative mutation rate for each off-target site was compared to the mutation rate of mice from the GFP-specific control sgRNA treatment group (B). The chromosomal location and starting nucleotide location for each off-target site are indicated along with the number of mismatches versus C17 or C14. Error bars indicate SD.
Off-target gene editing
A potential concern for gene editing therapeutics is the potential for cytotoxicity caused by off-target cleavage at similar target sites throughout the host genome. Although AAV-SaCas9 therapy was well tolerated by all mice, the potential long-term effects of off-target gene editing caused by genetic indels may not be visibly manifested within 62 days of AAV administration. We therefore analyzed a subset of off-target sites closely related to C7 and C14 found in the human and mouse genomes containing three to five sequence mismatches and determined whether gene editing was occurring at these sites. PCR amplicons spanning off-target sites were amplified and then subjected to NGS. No evidence of gene editing above background levels was seen in mice that received the C7 and C14 sgRNAs in combination with SaCas9 when compared to control animals not receiving the C7 and C14 sgRNAs (Figure 3B).
Improved human hepatocyte survival in Cas9-treated mice
Levels of human hepatocyte chimerism at the time of death were determined in each FRG mouse liver by quantifying levels of the human and mouse RPP30 (hRPP30 and mRPP30) genes via Droplet Digital PCR (ddPCR), and correcting for the presence of non-hepatocyte mouse cells in regions of both mouse and human hepatocytes, using previously determined values for the relative numbers of hepatocytes, Kupffer cells, stellate cells, and sinusoidal endothelial cells found in SCID mice or humanized uPA-SCID mouse livers (Figure 4A).48 The levels of human hepatocytes at the time of death/necropsy ranged from 3% to 44% of total hepatocytes across all animals (Figure 4B). At both early and late time points, mice receiving anti-HBV AAV-SaCas9 had significantly more human hepatocytes (7.1- and 1.7-fold more, respectively) than did control animals (anti-HBV early versus anti-GFP early, n = 2, p = 0.0045; anti-HBV late versus anti-GFP late, n = 5 versus n = 6, p < 0.0176). This observed difference in engraftment at the time of death was not due to differences in the initial levels of engraftment (Figure 4C), as human albumin levels prior to challenge with HBV were not significantly different between treatment and control groups (anti-HBV early versus anti-GFP early, n = 2, p = 0.0571; anti-HBV late versus anti-GFP late, n = 5 versus n = 6, p = 0.842). For a subset of these animals, we were able to evaluate human albumin levels in serum at the last time point before necropsy (day 56 after AAV/day 200 after HBV challenge, anti-HBV late versus anti-GFP late, n = 3 per group). Human albumin levels in two of three control anti-GFP mice were approximately one-third of those seen in anti-HBV AAV-SaCas9-treated mice at this time point (Figure 4D); this correlated with levels of human cell engraftment in these same control mice, which were significantly lower (p = 0.0431) than in anti-HBV mice by ddPCR (Figure 4E). Taken together, these observations suggest that HBV-specific AAV-SaCas9 therapy provides a selective survival advantage to human hepatocytes during chronic HBV infection in this model system.
Figure 4.

Levels of human serum albumin and human hepatocyte chimerism
The percentage of human hepatocytes at the time of death was determined by quantifying levels of human and mouse RPP30 by qPCR on total DNA extracted from livers using primer/probe sets against human and mouse RPP30. Values were corrected for the presence of non-hepatocyte mouse cells as described in Materials and Methods. (A and B) Levels of human hepatocyte chimerism are shown for each treatment group (A) and for individual mice (B). (C and D) Levels of human serum albumin were quantified by ELISA and are shown for all animals prior to initial HBV challenge (C) or for surviving animals at day 56 after AAV administration (200 days after HBV challenge) (D). (E) The percentage of human hepatocytes at death for animals surviving 56 days or longer after AAV is also shown. Asterisks indicate mice with detectable HBV gene editing. Error bars indicate SD; p values were generated using one-sided t tests. Values for the six mice that survived for 56 days or longer are color coordinated (colored dots).
Intrahepatic HBV DNA levels
Livers of all mice were flash-frozen rapidly after death, DNA was extracted, and levels of hRPP30, total HBV DNA, and HBV cccDNA in liver tissues were analyzed by qPCR or ddPCR. HBV DNA was detected in all mouse livers at necropsy, with >1 log more seen in livers of mice necropsied after entecavir withdrawal (groups anti-HBV late and anti-GFP late versus anti-HBV early and anti-GFP early) (Figure 5A), likely due to resumption of viral replication. Levels of cccDNA were also increased in mice necropsied after entecavir withdrawal, although only approximately 2- to 3-fold (Figure 5B), which was likely due to replenishment of cccDNA following resumption of viral replication and hepatocyte re-infection. When mice treated with anti-HBV AAV-SaCas9 were compared to control anti-GFP AAV-SaCas9-treated mice, a non-significant decrease in total HBV DNA was seen both before and after the withdrawal of entecavir (group anti-HBV early versus anti-GFP early, n = 2; anti-HBV late versus anti-GFP late, n = 5 versus n = 6, Figure 5A). A more dramatic reduction was observed in cccDNA levels in anti-HBV AAV-SaCas9-treated animals compared with control animals, both before and after entecavir withdrawal (Figure 5B), but again this did not reach statistical significance. Total HBV DNA levels per cccDNA increased in anti-GFP AAV-SaCas9- and anti-HBV AAV-SaCas9-treated mice by >1 log upon withdrawal of entecavir, consistent with resumption of high-level HBV replication, and no significant difference was seen between control and anti-HBV treatment groups either before or after entecavir withdrawal (Figure 5C).
Figure 5.

Liver-associated viral DNA levels at the time of death
(A–C) Levels of total HBV DNA (A), cccDNA (B), and total HBV DNA per cccDNA molecule (C) found in human hepatocytes of each liver were assayed by ddPCR using primers that detect total HBV DNA, cccDNA, and hRPP30. Grouped (left panel) and individual mouse (right panel) values are shown. Mouse livers were snap-frozen at necropsy, or as soon as possible following death. Total HBV DNA levels were analyzed using total DNA extracted from livers by a QIAGEN DNeasy blood and tissue kit. cccDNA levels were analyzed using DNA that was extracted from livers using a modified Hirt procedure, and then digested with T5 exonuclease. Error bars indicate SD; p values were generated using one-sided t tests.
Longitudinal serum viral loads
Initially, mice had baseline serum viral loads of 108–109 IU/ml, which dropped 2–2.5 logs during 17 days of entecavir treatment to ~106 IU/mL at the time of AAV delivery (Figures 6A and 6B), consistent with previous levels of entecavir suppression over a similar duration of treatment in both humanized uPA-SCID and FRG mice.31,36 After AAV administration, viral loads dropped an additional 0.5–1 logs in all experimental groups during 27 days of concurrent entecavir treatment. Upon entecavir withdrawal, viral loads in mice receiving both control and HBV-specific AAV-SaCas9 vectors rose steadily, approaching initial levels 4 weeks later at week 8 after AAV delivery. No significant difference in the rate of viral rebound was seen between the control (anti-GFP late) and treatment (anti-HBV late) groups. These data indicate that in immune-deficient mice under conditions where entecavir treatment is not fully suppressive, incomplete targeting of HBV by AAV-SaCas9 therapy has no effect on viremia once entecavir is withdrawn.
Figure 6.

Longitudinal viral loads
Chronically HBV-infected FRG mice were given entecavir to suppress viremia, then injected intravenously with either anti-GFP or anti-HBV AAV-SaCas9 vectors as shown and followed for levels of viremia for 4 (anti-GFP early, anti-HBV early) or 9 (anti-GFP late, anti-HBV late) weeks. Mice were infected with a genotype C HBV inoculum 144 days prior to AAV administration, and entecavir administration was halted at 4 weeks after AAV administration to monitor levels of viremia rebound in control or treated mice. (A and B) All mice were monitored weekly during entecavir (days −17 to 28) and AAV (days 0–62) treatment, and viral loads for grouped (A) and individual (B) mice are shown. Error bars indicate SD.
Tolerability of anti-HBV AAV-Cas9 therapy
Body weight was monitored for all mice throughout the experiment, with no apparent changes caused by treatment with either entecavir or AAV (Figure S5A). Weight loss was seen in some mice before they were either found dead or were sacrificed due to poor health, but this did not differ between treatment and control groups. Early deaths occurred in both groups and were likely due to the age of these mice, which had also received hepatotoxic injury due to nitisinone withdrawal and adenovirus-uPA administration prior to human hepatocyte transplantation and HBV infection (Figures S5B and S5C). At necropsy, 13 of 15 mice had enlarged livers, and most had liver nodules and/or hepatocellular adenomas/carcinomas, or less frequently regions of nodular hepatocellular hyperplasia (Figures S5C and S6). Such observations are common in fah−/− mice older than 7 months of age,49 and our mice were 9–13 months old at the time of death. No significant difference was seen between the day of analysis for treated and untreated groups for either the early or late time points, despite spontaneous deaths or the need for early necropsy of sick animals in all groups (Figure S5B). Tumors were negative for both human cytokeratin 18 (hCK18) and FAH, confirming that they originated from mouse and not human hepatocytes (Figures S7A and S7B), and they contained numerous Ki67+ foci indicative of active proliferation (Figure S7A). Importantly, autopsy reports for all mice suggested no findings of death that were associated with the administration of AAV or entecavir.
Liver characterization
To investigate the roles HBV infection, SaCas9 expression, cell proliferation, and cell death may have played in the prolonged hepatocyte survival seen in the anti-HBV AAV-SaCas9 treatment groups, serial liver sections were stained with an antibody specific for hCK18, in combination with antibodies against hepatitis B surface antigen (HBsAg), cleaved caspase-3, or Ki67. In parallel, RNAscope was performed on serial liver sections to detect HBV and SaCas9 RNA. Livers from all anti-GFP and anti-HBV AAV-SaCas9-treated mice showed widespread expression of HBsAg throughout humanized livers that co-localized with hCK18+ cells (Figure 7). No HBsAg staining was seen in murine tumors, where only sporadic hCK18+ cells could be found (data not shown). Cleaved caspase-3+ cells were seen at low frequency in regions of both mouse and human hepatocytes, with no obvious difference in levels seen between anti-GFP and anti-HBV AAV-SaCas9-treated mice (Figure 7). In areas of non-tumor liver parenchyma, Ki67+ cells were also seen at low frequency in regions of mouse and human hepatocytes, with no obvious difference in numbers between anti-GFP and anti-HBV AAV-SaCas9-treated mice (Figure 7). In contrast to parenchymal tissue, tumors of mouse origin showed multiple dense DAPI+ nuclei that were also Ki67+, indicative of ongoing tumor proliferation (Figure S7A). By RNAscope, HBV RNA was abundant in the livers of HBV+ humanized FRG mice, and it was found in regions containing hCK18 and HBsAg+ cells in serial sections by IHC (Figure 7). SaCas9 RNA was much less abundant than HBV RNA and was found in cells that were both positive and negative for HBV RNA. These cells were found within regions of livers also containing hCK18 and HBsAg+ cells in serial sections by IHC (Figure 7). Overall, no apparent differences in HBV replication, cell proliferation, or cell death were seen between anti-GFP and anti-HBV AAV-SaCas9-treated mice.
Figure 7.

Characterization of HBV-infected and AAV-SaCas9-treated humanized FRG mouse livers
Serial liver sections from humanized liver FRG mice were stained with hematoxylin and eosin (H&E), subjected to RNAscope for the presence of HBV and SaCas9 RNA, or co-labeled by immunohistochemistry for human cytokeratin 18 (hCK18) in combination with HBV surface antigen (HBsAg), activated caspase-3, or Ki67. Representative serial sections are shown for animal GFP-L3, which received control GFP6 and GFP7 sgRNAs. Scale bars, 1 mm.
Quantification of HBV and SaCas9 RNA
To determine how gene delivery impacted the levels of HBV gene editing seen in our mice, we quantified the levels of HBV and SaCas9 RNA by RNAscope in non-tumor tissue using sections taken from livers of three anti-GFP control and three anti-HBV AAV-SaCas9-treated mice. We used a previously described machine-learning RRScell algorithm that converts scanned fluorescent RNAscope images into a single-cell resolution profiling map of mRNA expression to identify whether HBV+ or SaCas9+ RNA foci were present in cells.50,51 For each animal, DAPI+ cells were assigned as HBV+/SaCas9+, HBV+/SaCas9−, HBV−/SaCas9+, or HBV−/SaCas9− and were quantified in non-tumor tissue (Figures 8A–8C; Figure S8). Across the six quantified mice, between 18% and 52% of all DAPI+ cells in non-tumor tissue were positive for HBV RNA (Figure 8D), and SaCas9 RNA was also present in 1.5%–13.6% of the HBV+ cells (Figure 8E). While the majority of SaCas9 RNA was detected in cells that also contained HBV RNA (58%–76%, Figure 8F), a significant proportion of SaCas9+ cells was HBV RNA−. Interestingly, mice HBV-L1 and HBV-L3, which had the highest detectable levels of HBV gene editing, had 8.6-fold and 5.6-fold higher levels of SaCas9+/HBV+ cells than mouse HBV-L5, respectively, despite having roughly equivalent levels of HBV infection per cell (19%–26%) and levels of SaCas9 RNA within HBV+ rather than HBV− cells (58%–62%). This indicates that higher levels of SaCas9 gene transfer into HBV+ cells may be the reason for the higher observed gene editing in these two mice.
Figure 8.

Quantification of HBV and SaCas9 RNA levels
(A) Liver sections from three AAV-SaCas9 control and three AAV-SaCas9-treated humanized liver FRG mice were subjected to RNAscope using custom HBV and SaCas9-specific probes. Cells in each image were assigned based on DAPI staining, and those containing foci indicative of HBV and/or SaCas9 probe hybridization were quantified using a RRScell image analysis algorithm, which was used to assign individual cell types. (B) Examples of assigned HBV+/SaCas9+ cells (red circles), HBV+/SaCas9− cells (green circles), HBV−/SaCas9+ cells (blue circles), and HBV-/SaCas9- cells (purple circles) present within the boxed region of mouse HBV-L1 shown in (A). (C) where appropriate, semi-automated image masking by RRScell was used to analyze non-tumor regions only (yellow areas). (D–F) Quantification of the percentage of HBV+ cells per DAPI+ cell (D), SaCas9+ cells per HBV+ cell (E), and HBV+ cells per SaCas9+ cell (F) is shown for individual humanized mouse livers. Tumors (T) and approximate tumor margins (dotted lines) are indicated where present.
Virus evolution
Mice were followed for up to 62 days after AAV administration, but they were infected with HBV for 144 days prior to being treated with our AAV-SaCas9 therapy, during which time they were exposed to RTi and/or RNase H small-molecule inhibitors as part of a previous study.31 To determine whether the clinical isolate used in our study evolved in any of our mice during the 155–206 days of chronic HBV infection, we obtained a 10× coverage consensus HBV sequence from the livers of mice using a probe capture method to isolate HBV-specific DNA from total liver DNA for NGS sequencing. Full consensus sequences were obtained from 12 of 15 study animals (GFP-E2, HBV-E1, HBV-E2, GFP-L1, GFP-L2, GFP-L3, GFP-L4, GFP-L5, HBV-L1, HBV-L2, HBV-L3, and HBV-L4) plus a control animal that was not part of our study but was infected with the same genotype C HBV clinical isolate at the same time (infected for > 100 days), and were compared to the consensus sequence obtained from the initial inoculum. Eleven of the 13 consensus sequences obtained from mouse livers at the time of death were 100% identical to the challenge inoculum. The consensus from two mice (GFP-L3 and HBV-L4) contained the single-nucleotide polymorphism (SNP) G782T. Both the G782 and T782 variants were found in the challenge inoculum by deep sequencing (G, 58%; T 41.8%), and the T variant is more prevalent in 7108 complete HBV sequences found in the HBVdb 2019 genome database.37 The consensus sequence from mouse GFP-L3 also contained the SNPs A287G, A1764T, and G1766A, and for each of these SNPs both variant alleles were detected in the challenge inoculum by Sanger sequencing (data not shown), indicating they were present in at least 15%–20% of the inoculum at the time of HBV challenge. Each variant allele detected at positions 287, 1764, and 1766 is also highly prevalent in the HBVdb genome database. Importantly, none of the four consensus SNPs detected in either of these two mice is found in regions of the genome that would likely be under selective pressure due to treatment with RTi inhibitors, RNase H inhibitors, or our HBV-specific AAV-SaCas9 therapy.
Discussion
In this work, we used a highly physiological liver-humanized mouse model to evaluate CRISPR-Cas9 as a therapeutic in the setting of chronic HBV infection. CRISPR-Cas9 was well tolerated, and we were able to demonstrate gene editing in five of eight treated animals. Treated animals demonstrated a highly significant improvement in human hepatocyte survival compared with control animals. Treated animals also trended toward reduced hepatic cccDNA levels at both early and late time points, although neither reached statistical significance. Taken together, these results constitute the most rigorous testing to date of anti-HBV gene editing in vivo, and they strongly support the continued development of this approach.
The liver-humanized FRG mouse represents one of the most physiologically relevant models for human HBV infection, in which the dynamics of HBV replication and responses to classical RTi therapy closely mimic those seen in infected patients. Although there have been numerous reports of gene-editing approaches to target HBV using Streptococcus pyogenes (Sp)Cas9 or other gene-editing enzymes,5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 our study utilizing SaCas9 is the first to show successful gene editing of HBV in a liver-humanized mouse model. Our work in the liver-humanized FRG mouse builds on several previous studies that also utilized AAV vectors to deliver SaCas9 as an anti-HBV therapeutic.11,14,52 In one study, Scott et al.11 showed that AAV2-SaCas9 vectors could efficiently inhibit HBV replication in vitro, and they could target cccDNA in HBV-infected HepG2-NTCP cells. In another study, Liu et al.14 saw efficient inhibition of HBV antigen production in vitro, and also in a hydrodynamic injection HBV mouse model in vivo in which SaCas9 and HBV plasmids were co-injected. In contrast, however, this group saw only limited anti-HBV efficacy from an AAV8-SaCas9 vector in AAV-HBV-transduced C3H mice. In a third study, Li et al.52 showed in HBV-transgenic mice that an AAV8-SaCas9 vector can efficiently lower HBV DNA, HBsAg, and hepatitis B e antigen (HBeAg) levels in blood or livers. As noted above, none of these models is fully permissive for HBV hepatocyte infection, and thus they do not fully mimic a natural persistent infection in which cccDNA is present and reinfection of SaCas9-treated cells might occur. Thus, the barriers for success in these models may be lower than they would be in a natural HBV infection. Our results extend these previous studies of AAV-SaCas9 therapeutics into a realistic model system in which cccDNA is present, concurrent antiviral RTi therapy can be used, and hepatocyte infection/reinfection can occur. As such, our work reveals several additional issues that must be resolved before gene editing can become a viable curative approach for HBV infection.
To achieve meaningful therapeutic benefit, the efficiency of HBV gene editing must be maximized, and it is clear that gene-editing efficacy is dependent on both efficient gene delivery of Cas9 to target cell populations, as shown by our RNAscope data, and the expression levels of Cas9 within a target cell.53,54 In previous work using a herpes simplex virus (HSV)-specific meganuclease, gene editing was more efficient when the meganuclease was expressed at higher levels from the strong ubiquitous smCBA promoter or from more transcriptionally active scAAV vectors.55,56 The EFM promoter used in the present study may be suboptimal for high-level expression in human hepatocytes in vivo, despite its demonstrated activity in HCC-derived Huh7 and HepG2.2.15 cells in vitro (Figure S2C).7 Previous studies showed that the EF1α promoter (from which our EFM promoter is derived) produced ~1 log less human factor IX than did the CMV enhancer/chicken β-actin promoter (which is highly similar to the smCBA promoter) when delivered to mouse liver by an AAV vector.57 Unfortunately, the smCBA and related promoters are too large to drive SaCas9 expression from an AAV vector, so alternative short promoters that are highly active in human hepatocytes in vivo will need to be identified.
Although high-level Cas9 expression in target cells appears to be a prerequisite for successful CRISPR-mediated gene editing in vivo, recent data suggest that pre-existing immunity to Cas9 may be a major impediment in a clinical setting. Several groups recently showed that within the human population there is a high prevalence of antibodies and T cells with specificity for SaCas9 and SpCas9, and it has been suggested that these Cas9-specific immune responses may promote elimination of Cas9-transduced target cells in vivo.58, 59, 60, 61 This is supported by data that show pre-immunization with recombinant SaCas9 or AAV-SaCas9 reduces the levels of gene editing and AAV-SaCas9-transduced hepatocytes in mouse livers following subsequent delivery of an AAV-SaCas9 vector,62,63 and that this occurs concurrently with a rise in CD8+ T cells within livers. Future studies will need to take into account how pre-existing immunity may impede the efficiency of CRISPR-mediated gene editing.
Gene-editing efficiency may also be affected by the accessibility of viral target sequences, particularly within epigenetically modified targets such as cccDNA.64,65 A detailed map of histone posttranslational modification provided by a recent chromatin immunoprecipitation sequencing (ChIP-seq) analysis of cccDNA from HBV+ liver tissue66 may prove useful in identifying the regions of HBV most amenable to gene editing. The C7 and C14 target sites used in our study are located within regions of active chromatin based on this dataset, so it is not clear that alternative targets would allow higher rates of gene editing. An attractive concept is the use of chromatin-modifying agents, which may increase target site accessibility by further opening up areas of heterochromatin within cccDNA. Previous studies have shown that histone deacetylase inhibitors such as trichostatin A and sodium butyrate can increase transcription from cccDNA,67 and we have previously shown that HSV genomes with highly organized epigenetic modifications can be edited more efficiently in the presence of histone deacetylase inhibitors including, trichostatin A and sodium butyrate.56
Our results also revealed an interesting dichotomy, in which there was a dramatic improvement in human hepatocyte survival after more than 200 days of persistent viremia following infection with a genotype (genotype C) that is known to cause more severe disease in humans,68, 69, 70, 71, 72 despite only modest indel formation in HBV DNA as measured by PCR amplification of SaCas9 target sites followed by NGS. At the same time, we observed a trend toward reduced HBV cccDNA load in hepatocytes of SaCas9-treated animals (although this did not reach statistical significance given our sample size). Since our AAV-SaCas9 vector expresses two sgRNAs targeting different parts of the HBV coding sequence, one explanation is that individual HBV genomes frequently undergo two independent cleavage events, which splits the molecule into two parts. Repair would therefore require rejoining of two independent DNA fragments, likely to represent an inefficient process, favoring degradation of the cleaved genomes. A similar phenomenon has been described by Khalili and colleagues,73, 74, 75 who have demonstrated that excision of latent HIV after gene editing in both the flanking long terminal repeats (LTRs) is inherently more efficient than mutagenesis occurring after a single cleavage event within the HIV genome. In our own work with latent HSV infection in mice, we have observed that simultaneous targeting of two sites in the viral genome leads to elimination of >90% of latent HSV, while indel frequencies in the virus remaining after therapy are typically only 5%–10%.76 In a surrogate in vitro assay in which an episomal template was cleaved by one to three independent sgRNAs, we did not see evidence of template degradation in either HepG2 or Huh7 cells (Figure S9), but these HepG2 and Huh7 models do not contain cccDNA, and 72 h may not be a sufficient period of time for degradation to occur, so the significance of this finding is unclear. It therefore remains unclear whether degradation of Cas9-linearized cccDNA is responsible for the reduction in cccDNA levels. Future studies should continue to evaluate this issue, which may also have relevance for genomic and other targets beyond persistent viruses.
The observation that anti-HBV AAV-SaCas9 treatment can promote survival of human hepatocytes was surprising, and our study does not provide a clear reason for this observation, since the levels of viremia and expression of cell death or proliferative markers did not appear to differ between groups. Of relevance to this, our ddPCR method for determining the levels of chimerism takes into account the expected levels of mouse and human cells within a humanized mouse liver but is based on assumptions made using data from “normal” liver tissue in humanized uPA-SCID mice. Unlike uPA-SCID mice, FRG mice are highly prone to develop tumors, and it is possible that the mass percentage of livers containing mouse cell-derived tumors could have been higher in anti-GFP AAV-SaCas9-treated mice, which would artificially deflate the levels of human cell chimerism found in non-tumor regions of chimeric livers. Since tumors in FRG mice are found throughout all lobes of the liver, it is challenging to determine what proportion of each liver may be made up of mouse cell-derived tumors. Further studies are needed to identify the mechanism causing the observed increase in human cell chimerism anti-HBV AAV-SaCas9-treated mice.
The unique biology of chronic HBV infection may also pose specific challenges for gene editing. Unlike cellular genomic targets, which are present at one to two copies per cell, in an HBV-infected hepatocyte multiple HBV DNA forms are present, including relaxed circular DNA (rcDNA) and cccDNA. In chronically infected humans, HBV-infected cells have been reported to contain one to five cccDNA copies per cell,77, 78, 79 but the total HBV DNA levels can range from 4 to 2,000 copies/cell.78,80, 81, 82 Furthermore, unlike in truly latent viral infections such as HSV, HBV cccDNA is slowly but continually replenished by ongoing viral replication. In our mice, the average cccDNA levels per human hepatocyte were lower (<0.1 copies/cell). The levels of total HBV DNA were two to four copies/cell in mice necropsied before entecavir withdrawal, and >60 copies/cell in mice necropsied after entecavir withdrawal. Importantly, despite entecavir therapy, viral loads in our mice remained higher during AAV-SaCas9 therapy (>105 IU/mL) than would typically be observed in human patients receiving entecavir. Thus, cells in which HBV gene editing may have occurred could retain some intact HBV, or even potentially be re-infected with HBV, thus blunting any therapeutic effect. We also note the possibility that RTi therapy might directly inhibit AAV-SaCas9 therapy. In previous work using humanized NRG mice, expression of HIV-neutralizing antibodies from AAV vectors was significantly inhibited in mice receiving an antiviral drug combination that included the RTi drugs tenofovir and emtricitabine.83 The authors suggested these nucleoside/nucleotide analogs might inhibit AAV transduction by interfering with the necessary second strand synthesis through which AAV vectors convert their single-stranded DNA genomes to double-stranded DNA.84,85 More complete suppression of HBV replication in future studies, especially using newer non-nucleoside/nucleotide compounds, may thus enable more efficacious HBV gene editing.
To summarize, our results represent the first demonstration of successful gene editing of persistent HBV infection in a liver-humanized mouse model. Despite modest levels of HBV mutagenesis at the sequence level, we observed a strong positive effect on human hepatocyte survival in treated animals, and a trend toward reduced hepatic cccDNA load. Our work reinforces the need for improved small animal models for HBV, which compared to the highly complex model used in the present study would allow increased experimental group sizes and improved statistical power. Model systems containing an intact immune system would also be beneficial, by allowing investigation of whether the immune response might successfully contain HBV after reduction of the reservoir by gene editing. Despite these challenges, our work warrants cautious optimism about the promise of gene editing as a potential cure for chronic HBV infection.
Materials and methods
Plasmids
Plasmids pscAAV-MND-GFP and pscAAV-smCBA-GFP have been described in detail before.44,56 Reporters containing HBV targets sites C1-C3-C6 and C7-C14-C16 were generated by placing three target sites in-frame with each other and then inserting them between the ATG start codon and the 5′ end of the GFP ORF within the plasmid pscAAV-MND-EGFP so that one continuous N-terminal GFP fusion ORF would be transcribed from the MND promoter (a fusion of the murine leukemia virus and myeloproliferative sarcoma virus LTRs). For each reporter, a gBlock (Integrated DNA Technologies, Coralville, IA, USA) was inserted between the HindIII and NcoI sites of pscAAV-MND-EGFP by Gibson Assembly86 (comprehensive cloning details are available upon request). The AAV vector plasmid pAAV-short CMV (sCMV)-SaCas9-SV40pA-hU6-sgRNA, which expresses SaCas9 and a single sgRNA, was generated by four-piece Gibson Assembly using the px601 XbaI/NotI linearized vector backbone, a PCR product containing the 371-bp sCMV promoter, a PCR product containing nuclear localization signal (NLS)-SaCas9-NLS-hemagglutinin (HA), and a gBlock containing the SV40 poly(A), a hU6-sgRNA expression cassette, and AarI sites for cloning sgRNA target sequences (comprehensive cloning details are available upon request). The NLS-SaCas9-NLS-HA gene was amplified from plasmid px601 (Addgene plasmid 61591), a gift from Feng Zhang.87 AAV vector plasmids were also made that express SaCas9 plus two sgRNAs using hU6, hH1, or tRNAGLN pol III promoters. Plasmid pAAV-EFM-SaCas9-SV40pA-hU6-sgRNA-hH1-sgRNA was generated by a four-piece Gibson Assembly using the pAAV-sCMV-SaCas9-SV40pA-hU6-sgRNA AscI/SpeI linearized vector backbone, a gBlock containing the 325-bp EFM promoter, a XhoI/NheI NLS-SaCas9-NLS-HA fragment from pAAV-sCMV-SaCas9-SV40pA-hU6-sgRNA, and a gBlock containing the SV40 poly(A), a hH1-sgRNA expression cassette, and BbsI sites for cloning sgRNA target sequences (comprehensive cloning details are available upon request). The EFM promoter contains a short EF1α promoter sequence7 fused to a minute virus of mice intron. We used gBlocks that contain the SV40 poly(A), a tRNAGLN-sgRNA expression cassette, and BbsI sites for cloning sgRNA target sequences, or an SPA, a hH1-sgRNA expression cassette, and BbsI sites for cloning sgRNA target sequences, to generate plasmids pAAV-EFM-SaCas9-SV40pA-hU6-sgRNA-tRNAGLN-sgRNA and pAAV-EFM-SaCas9-SPA-hU6-sgRNA-hH1-sgRNA, respectively. Sequences for all GFP- or HBV-specific sgRNA sequences are indicated in Table S1.
Cell culture
HEK293 cells88 were grown in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal calf serum, and HCC-derived Huh7 cells89 were grown in DMEM supplemented with 10% fetal calf serum (HyClone, Logan, UT, USA), 10 mmol/L l-glutamine (Thermo Fisher Scientific), and 1× non-essential amino acids (Thermo Fisher Scientific).
HBV sequence analysis and sgRNA design
Geneious Pro90 was used to generate consensus sequences and logo plots using 3,847 previously described whole-genome GenBank sequences,7 or whole-genome sequences found within an HBV database (i.e., HBVdb).37 All sgRNA design was performed using the algorithms of Doench et al.91 and Hsu et al.92 for on- and off-target specificity, respectively, with the sgRNA design tool available at https://benchling.com.
In vitro sgRNA activity assays
To test HBV-specific sgRNA activity, HEK293 cells were plated in 12-well plates at 4 × 105 cells/well and the following day transfected using polyethylenimine 25K (PEI) (Polysciences, Warrington, PA, USA) with 200 ng of C1-C3-C6 or C7-C14-C16 reporter plasmids, and 1 μg of each corresponding plasmid that expresses SaCas9 in combination with either control GFP-specific or HBV-specific sgRNAs. Gene knockdown was monitored by flow cytometry at 24 h post-transfection. For analysis of dual sgRNA AAV-SaCas9 expression constructs, 12-well plates were seeded with HEK293 or Huh7 cells at 4 × 105 cells/well or 2 × 105 cells/well, respectively. The following day, cells were transfected with 0.5 μg of pscAAV AAV-MND-GFP reporter plasmid and 0.5 μg of each expression vector using PEI (HEK293 cells) or Lipofectamine 3000 (Huh7 cells). Images demonstrating gene knockdown were taken at 24 h post-transfection. PCR amplicons spanning target sites were amplified from genomic DNA isolated after 48 h post-transfection and 200 ng of each product was digested with T7 endonuclease I (NEB) and visualized on a 2% agarose gel.
AAV vectors
AAV vectors were generated by transiently transfecting HEK293 cells using PEI according to the method of Choi et al.93 Briefly, HEK293 cells were transfected with AAV vector plasmids pscAAV-smCBA-GFP, pAAV-EFM-SaCas9-hH1-GFP6-hU6-GFP7, or AAV-EFM-SaCas9-hH1-C7-hU6-C14, in combination with a plasmid that expresses the AAV2 replication (rep) and AAV-LK03 capsid proteins, and a helper plasmid that expresses adenovirus helper proteins (pHelper). At 24 h post-transfection media were changed to serum-free DMEM, and after 72 h cells were collected and re-suspended in AAV lysis buffer (50 mM Tris, 150 mM NaCl [pH 8.5]) before freeze-thawing four times. AAV stocks were purified by iodixanol gradient separation93,94 followed by concentration into PBS using an Amicon Ultra-15 column (EMD Millipore, Burlington, MA, USA) before storage at −80°C. All AAV vector stocks were quantified by quantitative PCR using primers against the AAV inverted terminal repeat, with linearized plasmid DNA as a standard, according to the method of Aurnhammer et al.95 AAV stocks were treated with DNase I (Thermo Fisher Scientific) and Proteinase K (Thermo Fisher Scientific) prior to quantification.
In vivo studies
All mice were housed in accordance with the institutional, NIH, or Canadian Council on Animal Care guidelines on the care and use of animals in research, and all procedures were conducted with local Institutional Animal Care and Use Committee approval. For AAVLK03 gene transfer studies in the absence of HBV, liver-humanized chimeric FRG mice (Yecuris, Tualatin, OR, USA) with a human hepatocyte engraftment rate of >70%,26 or human Alb-uPA/scid28,96 mice with human albumin levels >5 mg/mL at 8 weeks after hepatocyte engraftment, were used. Alb-uPA/scid mice of both sexes were transplanted as previously described.28 For HBV studies, male FRG mice with a human hepatocyte engraftment rate of >90% were challenged with HBV at 5–6 months of age,31 following human hepatocyte transplantation at 6 weeks of age. Standard housing, diet, bedding, enrichment, and light/dark cycles were implemented, except for HBV-infected mice, which were kept in sterile microisolator housing and provided a high-energy mouse diet. For AAVLK03 gene transfer studies in the absence of HBV, AAV vectors were delivered via retro-orbital injection at the indicated doses in a volume of 50 μL, and mice were euthanized 14 days later to analyze levels of liver transduction. For HBV studies we used 15 humanized FRG mice infected with HBV genotype C (BioIVT, Westbury, NY, USA; human serum/BRH996810) at a dose of 2.6 × 108 vector genomes/25 g for 144 days, and mice were administered 5 × 1011 vector genomes of AAV vectors via tail vein injection in a volume of 100 μL. These HBV-infected mice had been used in a previous study validating small-molecule inhibitors of HBV replication.31 Prior to AAV administration, small-molecule HBV inhibitors were allowed to clear from all mice for 4 weeks and viral loads were allowed to return to post-infection baseline levels. From days −17 to 27 relative to AAV administration, mice received entecavir daily (1 mg/kg in 5 mL/kg by oral gavage). On days −17 through −15 and days 42 through 44, HBV-infected mice were given water containing 8 μg/mL nitisinone, 640 μg/mL sulfamethoxazole, 128 μg/mL trimethoprim, and 3% dextrose. Clinical observations and body weight measurements were recorded daily, and blood was drawn weekly for analysis. Human albumin levels in blood were determined using a human albumin ELISA quantitation set (Bethyl Laboratories, Montgomery, TX, USA, E80-129).
IHC
IHC was performed on 5-μm paraffin or OCT-embedded cryosections from humanized FRG mouse liver. For cryosections, sections were permeabilized in methanol for 10 min and blocked with PBS containing 13% donkey serum and 8.7% fetal bovine serum. For human and mouse albumin co-staining, sections were serially incubated with goat anti-human albumin polyclonal antibody (1:200 dilution; Bethyl Laboratories, A80-299A), Alexa Fluor 594-conjugated donkey anti-goat immunoglobulin G (IgG) secondary antibody (1:1,000 dilution; Invitrogen, Carlsbad, CA, USA, A11058), and then fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse albumin polyclonal antibody (1:200 dilution; Bethyl Laboratories, A90-234F), with three PBS wash steps after each antibody exposure. For GFP and human albumin co-staining, sections were incubated with rabbit anti-GFP polyclonal (1:100 dilution; Thermo Fisher Scientific, A11122) and goat anti-human albumin polyclonal primary antibodies followed by incubation with Alexa Fluor 594-conjugated donkey anti-goat IgG and Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:1,000 dilution; Thermo Fisher Scientific, A11008) secondary antibodies. All antibody incubations were done in PBS containing 1% bovine serum albumin (v/v), 1% donkey serum (v/v), and 0.3% Triton X-100, and sections were stained with DAPI before mounting with Vectastain fluorescence mounting medium. For some mice, 5-μm paraffin sections were treated with citrate antigen retrieval buffer after rehydration and stained using antibodies against human FAH (hFAH) (Sigma-Aldrich, AV41681), hCK18 (Agilent Technologies, DC10 #M701029-2), HBsAg (Bio-Rad, OBt0990), Ki67 (Cell Signaling Technology, D3B5 #12202), or cleaved caspase-3-Asp175 (Cell Signaling Technology, 9661). Signals were detected with secondary antibodies donkey anti-mouse Alexa Fluor 647 (Thermo Fisher Scientific, A31571) and donkey anti-rabbit Alexa Fluor 594 (Thermo Fisher Scientific, A21207), and sections were counterstained with DAPI and mounted with ProLong Gold.
RNAscope and RRScell quantification
All RNAscope was performed by the Experimental Histopathology Core at the Fred Hutchinson Cancer Research Center using a probe against SaCas9 (RNAscope 2.5 LS probe-SaCas9-C3, #501628-C3, Advanced Cell Diagnostics) and a custom probe that binds to nucleotides 2–1687 of genotype C HBV isolates (RNAscope 2.5 LS probe V-HBV-C-03-C2, #860678-C2, Advanced Cell Diagnostics). Briefly, 5-μm formalin-fixed, paraffin-embedded sections were baked for 1 h at 60°C before staining on the Leica Biosystems Bond RX automated staining platform. Sections were dewaxed, antigen retrieval was performed using Bond Epitope Retrieval 2 solution (Leica Biosystems), and enzyme digestion was performed with protease (Leica Biosystems). In situ hybridization was performed using the ACD RNAscope LS multiplex fluorescent reagent kit (Advanced Cell Diagnostics), and detection was performed using Akoya’s Opal reagents (Akoya Biosciences). Nuclear counterstaining was performed with DAPI, and coverslips were mounted with ProLong Gold mounting media (Thermo Fisher Scientific). Images were obtained from the HALO Link platform (Indica Labs) using images acquired at ×20 or ×40 using the Leica Biosystems Aperio VERSA 200 slide scanner using DAPI, Cy3, and Cy5 filters.
A previously described RRScell image analysis algorithm developed for use with RNA in situ hybridization was used to quantitate the number of cells containing HBV and SaCas9 RNA.50,51 Briefly, RRScell creates artificial membranes surrounding each individual DAPI+ nuclei, performs single-cell resolution segmentation of multicolor fluorescence images, and quantitates RNA expression levels within the cell boundaries.
Quantitative PCR
Total HBV DNA was detected in DNA extracted from serum using the MagNA Pure 96 system (Roche, Basel, Switzerland). Total HBV DNA, HBV cccDNA, AAV inverted terminal repeat, hRPP30, and mRPP30 were all detected in DNA extracted from chimeric humanized FRG mouse livers. For total HBV DNA, hRPP30, and mRPP30, ddPCR was performed using genomic DNA extracted using a DNeasy blood and tissue kit (QIAGEN, Hilden, Germany). For cccDNA, ddPCR was performed using genomic DNA extracted using a modified Hirt procedure as described below, followed by treatment with either plasmid-safe nuclease (Epicenter, Charlotte, NC, USA) or T5 exonuclease (NEB, Ipswich, MA, USA) at 37°C for 1 h. Primer/probe sets for HBV total DNA and hRPP30,7 cccDNA,97 and AAV inverted terminal repeat95 have been described previously. mRPP30 was detected using primers mRPP30F (5′-GGCGTTCGCAGATTTGGA-3′) and mRPP30R (5′-TCCCAGGTGAGCAGCAGTCT-3′) and probe musRPP30P (5′-HEX-ACCTGAAGGCTCTGCGCGGACTC-BHQ-3′).
Modified Hirt extraction
The modified Hirt extraction was performed using a previously described protocol.98 Briefly, homogenized liver tissue extracts underwent a neutral pH, three-step alkaline lysis before the supernatant was passed over a QIAGEN mini prep column to capture extra-chromosomal DNA.
HBV target site sequencing
HBV target site-specific PCR products were amplified directly from the HBV+ human serum used to infect humanized FRG mice or from serum or liver tissue isolated from HBV-infected humanized FRG mice. HBV target sites C7 and C14 were sequenced by Illumina sequencing. A 582-bp PCR product spanning target site C7 was amplified using primers HBV-534F (5′-CTGCTCAAGGAACCTCTATG-3′) and HBV-1115R (5′-GCCTTGTAAGTTGGCGAGAA-3′) and then submitted for Illumina sequencing. An 813-bp PCR product spanning target site C14 was amplified using primers HBV-2021F (5′-GCCTTAGAGTCTCCGGAACA-3′) and HBV-2833R (5′-TCCCAAGAATATGGTGACCC-3′) and then submitted for Illumina sequencing.
NGS sequencing
Target site-specific PCR products were diluted to 1 ng/μL and quarter-volume NexteraXT reactions were performed following the manufacturer’s protocol (Illumina, San Diego, CA, USA) with a sample volume of 1.25 μL. Libraries were amplified and barcoded using 12 cycles of PCR with the Nextera XT index kit (Illumina), and samples were pooled and quantitated by an Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA) such that approximately 200,000 130- to 150-bp reads were achieved per sample on a MiSeq sequencer (Illumina). Raw reads were pre-processed using tools from the Galaxy suite99 or using a previously described pipeline.100 Briefly, reads were trimmed using BBDuk (https://jgi.doe.gov/data-and-tools/bb-tools/), Trimmomatic,101 and cutadapt102 to remove adaptor contaminants and low-quality regions (Q < 30) at the 3′ and 5′ ends; any remaining reads shorter than 20 nt were discarded. Trimmed reads were mapped to the HBV sequence using Bowtie 2103 and exported for further analysis. Variant analysis was performed using a custom script that used functions from the Rsamtools, ShortRead, and Biostrings packages in R/Bioconductor.104, 105, 106 Aligned reads that completely overlapped the endonuclease target region of interest were scanned for insertions and deletions and the length and positions of each indel were recorded. Finally, the percentage of reads containing each mutation was tabulated to compute the mutation rate for each treatment.
Off-target gene-editing analysis
Genomic DNA was extracted from flash-frozen liver tissue using the DNeasy blood and tissue kit (QIAGEN, Hilden, Germany), and PCR amplicons spanning putative human or mouse off-target sites were amplified using target site-specific primers (Table S3). Each amplicon was sequenced by Illumina sequencing as described.
Quantification of liver chimerism
The percentages of human and mouse cells in chimeric livers were determined at death by quantifying hRPP30 and mRPP30 levels by ddPCR, and the results were used to calculate the percentage of human hepatocytes using values previously reported for percentages of hepatocytes, Kupffer cells, sinusoidal endothelial cells, and stellate cells in livers of SCID mice and humanized uPA-SCID mice.48 The following assumptions were made: (1) non-hepatocyte mouse cells are found in regions of mouse and human liver tissue at different levels; (2) levels of polyploid human and mouse hepatocytes are not different; (3) all hRPP30 signals are exclusively from human hepatocytes; (4) the levels of hepatocytes, Kupffer cells, sinusoidal endothelial cells, and stellate cells previously reported for SCID and uPA-SCID mice are for areas of liver that contain 100% mouse or 100% human hepatocytes;48 (5) in regions of human cells there is 0.58 non-hepatocyte mouse cell per human hepatocyte;48 and (6) 34.2% of the mRPP30 signal that is not associated with human hepatocytes is derived from mouse hepatocytes.48 Extrapolation of the percentage of total hepatocytes that are human in chimeric livers was performed as follows: (1) the mRPP30 copy number associated with human hepatocytes (non-mouse hepatocyte area derived) was determined by multiplying the total hRPP30 copy number by 0.58; (2) the levels of mRPP30 found in mouse hepatocyte areas was determined by subtracting the value from step 1 from the total mRPP30 copy number; (3) the levels of mouse hepatocyte-derived mRPP30 in mouse tissue was determined by multiplying the values from step 2 by 0.34; and (4) the percentage of human hepatocytes = total hRPP30 copy number/(total hRPP30 copy number + value from step 3) × 100.
Data and materials availability
Raw sequencing data associated with this paper are available via the NCBI sequence read archive (BioProject: PRJNA547828), and code and analysis scripts are available at https://github.com/proychou/TargetedMutagenesis.
Statistical analyses
Statistical analysis was performed using one-sided t tests.
Acknowledgments
We thank Ashley Sherrid for critical reading of this manuscript, and Robert W. Coombs and Daniel F Haile for technical assistance with the HBV cccDNA ddPCR assay. We thank Lander Foquet for useful discussions and advice on determining the levels of human liver chimerism. We thank Lisa Wilson and John Bial for technical assistance and advice for the FRG mouse AAV gene transfer study. We thank Stephanie Weaver for assistance with RNAscope analysis. This work was supported by NIH/NIAID grants 5R21AI107252-02, UM1 AI126623, and UM1AI106701, and by the Gilead Sciences HIV Cure Grant Program. Work performed within the Fred Hutchinson Cancer Research Center Comparative Medicine Shared Resources is supported by the Fred Hutch/University of Washington Cancer Consortium (P30 CA015704).
Author contributions
D.S., K.R.L., D.L.T., M.A., J.E.S., and K.R.J. conceived and planned the experiments. D.S., M.L., H.S.D.S.F., E.J.K., R.M.L., S.P., T.N., L.S., R.C.-T., L.M.K., N.D.W., C.L., J.W., E.A.G., and S.J.P. performed the experiments. D.S., K.R.L., M.L., H.S.D.S.F., P.R., K.E., A.Z.L., L.C., A.L.G., M.-L.H., S.J.P., J.E.S., and K.R.J. analyzed the data. D.S. and K.R.J. wrote the paper. All authors read and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.11.014.
Supplemental information
References
- 1.World Health Organization . 2019. Hepatitis B factsheet.https://www.who.int/en/news-room/fact-sheets/detail/hepatitis-b [Google Scholar]
- 2.Stone D., Niyonzima N., Jerome K.R. Genome editing and the next generation of antiviral therapy. Hum. Genet. 2016;135:1071–1082. doi: 10.1007/s00439-016-1686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber N.D., Aubert M., Dang C.H., Stone D., Jerome K.R. DNA cleavage enzymes for treatment of persistent viral infections: recent advances and the pathway forward. Virology. 2014;454–455:353–361. doi: 10.1016/j.virol.2013.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schiffer J.T., Aubert M., Weber N.D., Mintzer E., Stone D., Jerome K.R. Targeted DNA mutagenesis for the cure of chronic viral infections. J. Virol. 2012;86:8920–8936. doi: 10.1128/JVI.00052-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu W., Xie K., Xu Y., Wang L., Chen K., Zhang L., Fang J. CRISPR/Cas9 produces anti-hepatitis B virus effect in hepatoma cells and transgenic mouse. Virus Res. 2016;217:125–132. doi: 10.1016/j.virusres.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 6.Zhen S., Hua L., Liu Y.H., Gao L.C., Fu J., Wan D.Y., Dong L.H., Song H.F., Gao X. Harnessing the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated Cas9 system to disrupt the hepatitis B virus. Gene Ther. 2015;22:404–412. doi: 10.1038/gt.2015.2. [DOI] [PubMed] [Google Scholar]
- 7.Weber N.D., Stone D., Sedlak R.H., De Silva Feelixge H.S., Roychoudhury P., Schiffer J.T., Aubert M., Jerome K.R. AAV-mediated delivery of zinc finger nucleases targeting hepatitis B virus inhibits active replication. PLoS ONE. 2014;9:e97579. doi: 10.1371/journal.pone.0097579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J., Xu Z.W., Liu S., Zhang R.Y., Ding S.L., Xie X.M., Long L., Chen X.M., Zhuang H., Lu F.M. Dual gRNAs guided CRISPR/Cas9 system inhibits hepatitis B virus replication. World J. Gastroenterol. 2015;21:9554–9565. doi: 10.3748/wjg.v21.i32.9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seeger C., Sohn J.A. Complete spectrum of CRISPR/Cas9-induced mutations on HBV cccDNA. Mol. Ther. 2016;24:1258–1266. doi: 10.1038/mt.2016.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seeger C., Sohn J.A. Targeting hepatitis B virus with CRISPR/Cas9. Mol. Ther. Nucleic Acids. 2014;3:e216. doi: 10.1038/mtna.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott T., Moyo B., Nicholson S., Maepa M.B., Watashi K., Ely A., Weinberg M.S., Arbuthnot P. ssAAVs containing cassettes encoding SaCas9 and guides targeting hepatitis B virus inactivate replication of the virus in cultured cells. Sci. Rep. 2017;7:7401. doi: 10.1038/s41598-017-07642-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakuma T., Masaki K., Abe-Chayama H., Mochida K., Yamamoto T., Chayama K. Highly multiplexed CRISPR-Cas9-nuclease and Cas9-nickase vectors for inactivation of hepatitis B virus. Genes Cells. 2016;21:1253–1262. doi: 10.1111/gtc.12437. [DOI] [PubMed] [Google Scholar]
- 13.Ramanan V., Shlomai A., Cox D.B., Schwartz R.E., Michailidis E., Bhatta A., Scott D.A., Zhang F., Rice C.M., Bhatia S.N. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci. Rep. 2015;5:10833. doi: 10.1038/srep10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y., Zhao M., Gong M., Xu Y., Xie C., Deng H., Li X., Wu H., Wang Z. Inhibition of hepatitis B virus replication via HBV DNA cleavage by Cas9 from Staphylococcus aureus. Antiviral Res. 2018;152:58–67. doi: 10.1016/j.antiviral.2018.02.011. [DOI] [PubMed] [Google Scholar]
- 15.Liu X., Hao R., Chen S., Guo D., Chen Y. Inhibition of hepatitis B virus by the CRISPR/Cas9 system via targeting the conserved regions of the viral genome. J. Gen. Virol. 2015;96:2252–2261. doi: 10.1099/vir.0.000159. [DOI] [PubMed] [Google Scholar]
- 16.Lin S.R., Yang H.C., Kuo Y.T., Liu C.J., Yang T.Y., Sung K.C., Lin Y.Y., Wang H.Y., Wang C.C., Shen Y.C. The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Mol. Ther. Nucleic Acids. 2014;3:e186. doi: 10.1038/mtna.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H., Sheng C., Wang S., Yang L., Liang Y., Huang Y., Liu H., Li P., Yang C., Yang X. Removal of integrated hepatitis B virus DNA using CRISPR-Cas9. Front. Cell. Infect. Microbiol. 2017;7:91. doi: 10.3389/fcimb.2017.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H., Sheng C., Liu H., Liu G., Du X., Du J., Zhan L., Li P., Yang C., Qi L. An effective molecular target site in hepatitis B virus S gene for Cas9 cleavage and mutational inactivation. Int. J. Biol. Sci. 2016;12:1104–1113. doi: 10.7150/ijbs.16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kennedy E.M., Bassit L.C., Mueller H., Kornepati A.V.R., Bogerd H.P., Nie T., Chatterjee P., Javanbakht H., Schinazi R.F., Cullen B.R. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology. 2015;476:196–205. doi: 10.1016/j.virol.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karimova M., Beschorner N., Dammermann W., Chemnitz J., Indenbirken D., Bockmann J.H., Grundhoff A., Lüth S., Buchholz F., Schulze zur Wiesch J., Hauber J. CRISPR/Cas9 nickase-mediated disruption of hepatitis B virus open reading frame S and X. Sci. Rep. 2015;5:13734. doi: 10.1038/srep13734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong C., Qu L., Wang H., Wei L., Dong Y., Xiong S. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antiviral Res. 2015;118:110–117. doi: 10.1016/j.antiviral.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 22.Cradick T.J., Keck K., Bradshaw S., Jamieson A.C., McCaffrey A.P. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol. Ther. 2010;18:947–954. doi: 10.1038/mt.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J., Zhang W., Lin J., Wang F., Wu M., Chen C., Zheng Y., Peng X., Li J., Yuan Z. An efficient antiviral strategy for targeting hepatitis B virus genome using transcription activator-like effector nucleases. Mol. Ther. 2014;22:303–311. doi: 10.1038/mt.2013.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bloom K., Ely A., Mussolino C., Cathomen T., Arbuthnot P. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol. Ther. 2013;21:1889–1897. doi: 10.1038/mt.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allweiss L., Dandri M. Experimental in vitro and in vivo models for the study of human hepatitis B virus infection. J. Hepatol. 2016;64(1, Suppl):S17–S31. doi: 10.1016/j.jhep.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 26.Azuma H., Paulk N., Ranade A., Dorrell C., Al-Dhalimy M., Ellis E., Strom S., Kay M.A., Finegold M., Grompe M. Robust expansion of human hepatocytes in Fah−/−/Rag2−/−/Il2rg−/− mice. Nat. Biotechnol. 2007;25:903–910. doi: 10.1038/nbt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hasegawa M., Kawai K., Mitsui T., Taniguchi K., Monnai M., Wakui M., Ito M., Suematsu M., Peltz G., Nakamura M., Suemizu H. The reconstituted “humanized liver” in TK-NOG mice is mature and functional. Biochem. Biophys. Res. Commun. 2011;405:405–410. doi: 10.1016/j.bbrc.2011.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mercer D.F., Schiller D.E., Elliott J.F., Douglas D.N., Hao C., Rinfret A., Addison W.R., Fischer K.P., Churchill T.A., Lakey J.R. Hepatitis C virus replication in mice with chimeric human livers. Nat. Med. 2001;7:927–933. doi: 10.1038/90968. [DOI] [PubMed] [Google Scholar]
- 29.Bissig K.D., Wieland S.F., Tran P., Isogawa M., Le T.T., Chisari F.V., Verma I.M. Human liver chimeric mice provide a model for hepatitis B and C virus infection and treatment. J. Clin. Invest. 2010;120:924–930. doi: 10.1172/JCI40094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diab A., Foca A., Fusil F., Lahlali T., Jalaguier P., Amirache F., N’Guyen L., Isorce N., Cosset F.L., Zoulim F. Polo-like-kinase 1 is a proviral host factor for hepatitis B virus replication. Hepatology. 2017;66:1750–1765. doi: 10.1002/hep.29236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long K.R., Lomonosova E., Li Q., Ponzar N.L., Villa J.A., Touchette E., Rapp S., Liley R.M., Murelli R.P., Grigoryan A. Efficacy of hepatitis B virus ribonuclease H inhibitors, a new class of replication antagonists, in FRG human liver chimeric mice. Antiviral Res. 2018;149:41–47. doi: 10.1016/j.antiviral.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meuleman P., Libbrecht L., De Vos R., de Hemptinne B., Gevaert K., Vandekerckhove J., Roskams T., Leroux-Roels G. Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology. 2005;41:847–856. doi: 10.1002/hep.20657. [DOI] [PubMed] [Google Scholar]
- 33.Meuleman P., Libbrecht L., Wieland S., De Vos R., Habib N., Kramvis A., Roskams T., Leroux-Roels G. Immune suppression uncovers endogenous cytopathic effects of the hepatitis B virus. J. Virol. 2006;80:2797–2807. doi: 10.1128/JVI.80.6.2797-2807.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsuge M., Hiraga N., Takaishi H., Noguchi C., Oga H., Imamura M., Takahashi S., Iwao E., Fujimoto Y., Ochi H. Infection of human hepatocyte chimeric mouse with genetically engineered hepatitis B virus. Hepatology. 2005;42:1046–1054. doi: 10.1002/hep.20892. [DOI] [PubMed] [Google Scholar]
- 35.Kosaka K., Hiraga N., Imamura M., Yoshimi S., Murakami E., Nakahara T., Honda Y., Ono A., Kawaoka T., Tsuge M. A novel TK-NOG based humanized mouse model for the study of HBV and HCV infections. Biochem. Biophys. Res. Commun. 2013;441:230–235. doi: 10.1016/j.bbrc.2013.10.040. [DOI] [PubMed] [Google Scholar]
- 36.Klumpp K., Shimada T., Allweiss L., Volz T., Lutgehetmann M., Hartman G., Flores O.A., Lam A.M., Dandri M. Efficacy of NVR 3-778, alone and in combination with pegylated interferon, vs entecavir in uPA/SCID mice with humanized livers and HBV infection. Gastroenterology. 2018;154:652–662.e8. doi: 10.1053/j.gastro.2017.10.017. [DOI] [PubMed] [Google Scholar]
- 37.Hayer J., Jadeau F., Deléage G., Kay A., Zoulim F., Combet C. HBVdb: a knowledge database for hepatitis B virus. Nucleic Acids Res. 2013;41:D566–D570. doi: 10.1093/nar/gks1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friedland A.E., Baral R., Singhal P., Loveluck K., Shen S., Sanchez M., Marco E., Gotta G.M., Maeder M.L., Kennedy E.M. Characterization of Staphylococcus aureus Cas9: a smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol. 2015;16:257. doi: 10.1186/s13059-015-0817-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lisowski L., Dane A.P., Chu K., Zhang Y., Cunningham S.C., Wilson E.M., Nygaard S., Grompe M., Alexander I.E., Kay M.A. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2014;506:382–386. doi: 10.1038/nature12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gray S.J., Foti S.B., Schwartz J.W., Bachaboina L., Taylor-Blake B., Coleman J., Ehlers M.D., Zylka M.J., McCown T.J., Samulski R.J. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum. Gene Ther. 2011;22:1143–1153. doi: 10.1089/hum.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu Z., Sun J., Zhang T., Yin C., Yin F., Van Dyke T., Samulski R.J., Monahan P.E. Optimization of self-complementary AAV vectors for liver-directed expression results in sustained correction of hemophilia B at low vector dose. Mol. Ther. 2008;16:280–289. doi: 10.1038/sj.mt.6300355. [DOI] [PubMed] [Google Scholar]
- 42.Mefferd A.L., Kornepati A.V., Bogerd H.P., Kennedy E.M., Cullen B.R. Expression of CRISPR/Cas single guide RNAs using small tRNA promoters. RNA. 2015;21:1683–1689. doi: 10.1261/rna.051631.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choi J.H., Yu N.K., Baek G.C., Bakes J., Seo D., Nam H.J., Baek S.H., Lim C.S., Lee Y.S., Kaang B.K. Optimization of AAV expression cassettes to improve packaging capacity and transgene expression in neurons. Mol. Brain. 2014;7:17. doi: 10.1186/1756-6606-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Silva Feelixge H.S., Stone D., Pietz H.L., Roychoudhury P., Greninger A.L., Schiffer J.T., Aubert M., Jerome K.R. Detection of treatment-resistant infectious HIV after genome-directed antiviral endonuclease therapy. Antiviral Res. 2016;126:90–98. doi: 10.1016/j.antiviral.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ueda S., Ebina H., Kanemura Y., Misawa N., Koyanagi Y. Anti-HIV-1 potency of the CRISPR/Cas9 system insufficient to fully inhibit viral replication. Microbiol. Immunol. 2016;60:483–496. doi: 10.1111/1348-0421.12395. [DOI] [PubMed] [Google Scholar]
- 46.Wang G., Zhao N., Berkhout B., Das A.T. CRISPR-Cas9 can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Mol. Ther. 2016;24:522–526. doi: 10.1038/mt.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z., Pan Q., Gendron P., Zhu W., Guo F., Cen S., Wainberg M.A., Liang C. CRISPR/Cas9-derived mutations both inhibit HIV-1 replication and accelerate viral escape. Cell Rep. 2016;15:481–489. doi: 10.1016/j.celrep.2016.03.042. [DOI] [PubMed] [Google Scholar]
- 48.Tateno C., Miya F., Wake K., Kataoka M., Ishida Y., Yamasaki C., Yanagi A., Kakuni M., Wisse E., Verheyen F. Morphological and microarray analyses of human hepatocytes from xenogeneic host livers. Lab. Invest. 2013;93:54–71. doi: 10.1038/labinvest.2012.158. [DOI] [PubMed] [Google Scholar]
- 49.Grompe M., Lindstedt S., al-Dhalimy M., Kennaway N.G., Papaconstantinou J., Torres-Ramos C.A., Ou C.N., Finegold M. Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nat. Genet. 1995;10:453–460. doi: 10.1038/ng0895-453. [DOI] [PubMed] [Google Scholar]
- 50.Eichholz K., Li A.Z., Diem K., Tareen S.U., Jensen M.C., Zhu J., Corey L. A CAR RNA FISH assay reveals functional and spatial heterogeneity of chimeric antigen receptor T cells in tissue. bioRxiv. 2020 doi: 10.1101/2020.08.21.260935. [DOI] [Google Scholar]
- 51.Li A.Z., Eichholz K., Sholukh A., Corey L. RRScell method for automated learning immune cell phenotypes with immunofluorescence cancer tissue. arXiv. 2020 arXiv:2011.01002v1. [Google Scholar]
- 52.Li H., Sheng C., Liu H., Wang S., Zhao J., Yang L., Jia L., Li P., Wang L., Xie J. Inhibition of HBV expression in HBV transgenic mice using AAV-delivered CRISPR-SaCas9. Front. Immunol. 2018;9:2080. doi: 10.3389/fimmu.2018.02080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuen G., Khan F.J., Gao S., Stommel J.M., Batchelor E., Wu X., Luo J. CRISPR/Cas9-mediated gene knockout is insensitive to target copy number but is dependent on guide RNA potency and Cas9/sgRNA threshold expression level. Nucleic Acids Res. 2017;45:12039–12053. doi: 10.1093/nar/gkx843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kallimasioti-Pazi E.M., Thelakkad Chathoth K., Taylor G.C., Meynert A., Ballinger T., Kelder M.J.E., Lalevée S., Sanli I., Feil R., Wood A.J. Heterochromatin delays CRISPR-Cas9 mutagenesis but does not influence the outcome of mutagenic DNA repair. PLoS Biol. 2018;16:e2005595. doi: 10.1371/journal.pbio.2005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aubert M., Boyle N.M., Stone D., Stensland L., Huang M.L., Magaret A.S., Galetto R., Rawlings D.J., Scharenberg A.M., Jerome K.R. In vitro inactivation of latent HSV by targeted mutagenesis using an HSV-specific homing endonuclease. Mol. Ther. Nucleic Acids. 2014;3:e146. doi: 10.1038/mtna.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aubert M., Madden E.A., Loprieno M., DeSilva Feelixge H.S., Stensland L., Huang M.L., Greninger A.L., Roychoudhury P., Niyonzima N., Nguyen T. In vivo disruption of latent HSV by designer endonuclease therapy. JCI Insight. 2016;1:e88468. doi: 10.1172/jci.insight.88468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu L., Daly T., Gao C., Flotte T.R., Song S., Byrne B.J., Sands M.S., Parker Ponder K. CMV-β-actin promoter directs higher expression from an adeno-associated viral vector in the liver than the cytomegalovirus or elongation factor 1α promoter and results in therapeutic levels of human factor X in mice. Hum. Gene Ther. 2001;12:563–573. doi: 10.1089/104303401300042500. [DOI] [PubMed] [Google Scholar]
- 58.Simhadri V.L., McGill J., McMahon S., Wang J., Jiang H., Sauna Z.E. Prevalence of pre-existing antibodies to CRISPR-associated nuclease Cas9 in the USA population. Mol. Ther. Methods Clin. Dev. 2018;10:105–112. doi: 10.1016/j.omtm.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferdosi S.R., Ewaisha R., Moghadam F., Krishna S., Park J.G., Ebrahimkhani M.R., Kiani S., Anderson K.S. Multifunctional CRISPR-Cas9 with engineered immunosilenced human T cell epitopes. Nat. Commun. 2019;10:1842. doi: 10.1038/s41467-019-09693-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wagner D.L., Amini L., Wendering D.J., Burkhardt L.M., Akyüz L., Reinke P., Volk H.D., Schmueck-Henneresse M. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2019;25:242–248. doi: 10.1038/s41591-018-0204-6. [DOI] [PubMed] [Google Scholar]
- 61.Charlesworth C.T., Deshpande P.S., Dever D.P., Camarena J., Lemgart V.T., Cromer M.K., Vakulskas C.A., Collingwood M.A., Zhang L., Bode N.M. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019;25:249–254. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li A., Tanner M.R., Lee C.M., Hurley A.E., De Giorgi M., Jarrett K.E., Davis T.H., Doerfler A.M., Bao G., Beeton C., Lagor W.R. AAV-CRISPR gene editing is negated by pre-existing immunity to Cas9. Mol. Ther. 2020;28:1432–1441. doi: 10.1016/j.ymthe.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moreno A.M., Palmer N., Alemán F., Chen G., Pla A., Jiang N., Leong Chew W., Law M., Mali P. Immune-orthogonal orthologues of AAV capsids and of Cas9 circumvent the immune response to the administration of gene therapy. Nat. Biomed. Eng. 2019;3:806–816. doi: 10.1038/s41551-019-0431-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hong X., Kim E.S., Guo H. Epigenetic regulation of hepatitis B virus covalently closed circular DNA: implications for epigenetic therapy against chronic hepatitis B. Hepatology. 2017;66:2066–2077. doi: 10.1002/hep.29479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koumbi L., Karayiannis P. The epigenetic control of hepatitis B virus modulates the outcome of infection. Front. Microbiol. 2016;6:1491. doi: 10.3389/fmicb.2015.01491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tropberger P., Mercier A., Robinson M., Zhong W., Ganem D.E., Holdorf M. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc. Natl. Acad. Sci. USA. 2015;112:E5715–E5724. doi: 10.1073/pnas.1518090112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wei Z.Q., Zhang Y.H., Ke C.Z., Chen H.X., Ren P., He Y.L., Hu P., Ma D.Q., Luo J., Meng Z.J. Curcumin inhibits hepatitis B virus infection by down-regulating cccDNA-bound histone acetylation. World J. Gastroenterol. 2017;23:6252–6260. doi: 10.3748/wjg.v23.i34.6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kao J.H. Hepatitis B virus genotypes and hepatocellular carcinoma in Taiwan. Intervirology. 2003;46:400–407. doi: 10.1159/000074999. [DOI] [PubMed] [Google Scholar]
- 69.Gu X., Yang X., Wang D., Hua Z., Wu H., Chen H., Xu Y., Lu Z. Comparison and significance of specific and non-specific cellular immunity in patients with chronic hepatitis B caused by infection with genotypes B or C of hepatitis B virus. Sci. China C Life Sci. 2009;52:719–723. doi: 10.1007/s11427-009-0098-0. [DOI] [PubMed] [Google Scholar]
- 70.Chan H.L., Tsang S.W., Liew C.T., Tse C.H., Wong M.L., Ching J.Y., Leung N.W., Tam J.S., Sung J.J. Viral genotype and hepatitis B virus DNA levels are correlated with histological liver damage in HBeAg-negative chronic hepatitis B virus infection. Am. J. Gastroenterol. 2002;97:406–412. doi: 10.1111/j.1572-0241.2002.05478.x. [DOI] [PubMed] [Google Scholar]
- 71.Tsubota A., Arase Y., Ren F., Tanaka H., Ikeda K., Kumada H. Genotype may correlate with liver carcinogenesis and tumor characteristics in cirrhotic patients infected with hepatitis B virus subtype adw. J. Med. Virol. 2001;65:257–265. doi: 10.1002/jmv.2028. [DOI] [PubMed] [Google Scholar]
- 72.Kao J.H., Chen P.J., Lai M.Y., Chen D.S. Hepatitis B genotypes correlate with clinical outcomes in patients with chronic hepatitis B. Gastroenterology. 2000;118:554–559. doi: 10.1016/s0016-5085(00)70261-7. [DOI] [PubMed] [Google Scholar]
- 73.Yin C., Zhang T., Qu X., Zhang Y., Putatunda R., Xiao X., Li F., Xiao W., Zhao H., Dai S. In vivo excision of HIV-1 provirus by saCas9 and multiplex single-guide RNAs in animal models. Mol. Ther. 2017;25:1168–1186. doi: 10.1016/j.ymthe.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kaminski R., Bella R., Yin C., Otte J., Ferrante P., Gendelman H.E., Li H., Booze R., Gordon J., Hu W., Khalili K. Excision of HIV-1 DNA by gene editing: a proof-of-concept in vivo study. Gene Ther. 2016;23:690–695. doi: 10.1038/gt.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hu W., Kaminski R., Yang F., Zhang Y., Cosentino L., Li F., Luo B., Alvarez-Carbonell D., Garcia-Mesa Y., Karn J. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA. 2014;111:11461–11466. doi: 10.1073/pnas.1405186111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aubert M., Strongin D.E., Roychoudhury P., Loprieno M.A., Haick A.K., Klouser L.M., Stensland L., Huang M.L., Makhsous N., Tait A. Gene editing and elimination of latent herpes simplex virus in vivo. Nat. Commun. 2020;11:4148. doi: 10.1038/s41467-020-17936-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Laras A., Koskinas J., Dimou E., Kostamena A., Hadziyannis S.J. Intrahepatic levels and replicative activity of covalently closed circular hepatitis B virus DNA in chronically infected patients. Hepatology. 2006;44:694–702. doi: 10.1002/hep.21299. [DOI] [PubMed] [Google Scholar]
- 78.Volz T., Lutgehetmann M., Wachtler P., Jacob A., Quaas A., Murray J.M., Dandri M., Petersen J. Impaired intrahepatic hepatitis B virus productivity contributes to low viremia in most HBeAg-negative patients. Gastroenterology. 2007;133:843–852. doi: 10.1053/j.gastro.2007.06.057. [DOI] [PubMed] [Google Scholar]
- 79.Werle-Lapostolle B., Bowden S., Locarnini S., Wursthorn K., Petersen J., Lau G., Trepo C., Marcellin P., Goodman Z., Delaney W.E., 4th Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology. 2004;126:1750–1758. doi: 10.1053/j.gastro.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 80.Bayram A., Erkilic S., Ozkur A., Bayram M., Sari I. Quantification of intrahepatic total hepatitis B virus DNA in chronic hepatitis B patients and its relationship with liver histology. J. Clin. Pathol. 2008;61:338–342. doi: 10.1136/jcp.2007.050765. [DOI] [PubMed] [Google Scholar]
- 81.Shi M., Sun W.L., Hua Y.Y., Han B., Shi L. Effects of entecavir on hepatitis B virus covalently closed circular DNA in hepatitis B e antigen-positive patients with hepatitis B. PLoS ONE. 2015;10:e0117741. doi: 10.1371/journal.pone.0117741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wong D.K., Fung J., Lee C.K., Seto W.K., Leung J., Huang F.Y., Lin C.K., Lai C.L., Yuen M.F. Intrahepatic hepatitis B virus replication and liver histology in subjects with occult hepatitis B infection. Clin. Microbiol. Infect. 2016;22:290.e1-3. doi: 10.1016/j.cmi.2015.10.036. [DOI] [PubMed] [Google Scholar]
- 83.Horwitz J.A., Halper-Stromberg A., Mouquet H., Gitlin A.D., Tretiakova A., Eisenreich T.R., Malbec M., Gravemann S., Billerbeck E., Dorner M. HIV-1 suppression and durable control by combining single broadly neutralizing antibodies and antiretroviral drugs in humanized mice. Proc. Natl. Acad. Sci. USA. 2013;110:16538–16543. doi: 10.1073/pnas.1315295110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhong L., Zhou X., Li Y., Qing K., Xiao X., Samulski R.J., Srivastava A. Single-polarity recombinant adeno-associated virus 2 vector-mediated transgene expression in vitro and in vivo: mechanism of transduction. Mol. Ther. 2008;16:290–295. doi: 10.1038/sj.mt.6300376. [DOI] [PubMed] [Google Scholar]
- 85.Zhou X., Zeng X., Fan Z., Li C., McCown T., Samulski R.J., Xiao X. Adeno-associated virus of a single-polarity DNA genome is capable of transduction in vivo. Mol. Ther. 2008;16:494–499. doi: 10.1038/sj.mt.6300397. [DOI] [PubMed] [Google Scholar]
- 86.Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A., 3rd, Smith H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 87.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Graham F.L., Smiley J., Russell W.C., Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 89.Nakabayashi H., Taketa K., Miyano K., Yamane T., Sato J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 1982;42:3858–3863. [PubMed] [Google Scholar]
- 90.Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Doench J.G., Hartenian E., Graham D.B., Tothova Z., Hegde M., Smith I., Sullender M., Ebert B.L., Xavier R.J., Root D.E. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 2014;32:1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Choi V.W., Asokan A., Haberman R.A., Samulski R.J. Production of recombinant adeno-associated viral vectors for in vitro and in vivo use. Curr. Protoc. Mol. Biol. 2007;Chapter 16 doi: 10.1002/0471142727.mb1625s78. Unit 16.25. [DOI] [PubMed] [Google Scholar]
- 94.Zolotukhin S., Byrne B.J., Mason E., Zolotukhin I., Potter M., Chesnut K., Summerford C., Samulski R.J., Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 95.Aurnhammer C., Haase M., Muether N., Hausl M., Rauschhuber C., Huber I., Nitschko H., Busch U., Sing A., Ehrhardt A., Baiker A. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum. Gene Ther. Methods. 2012;23:18–28. doi: 10.1089/hgtb.2011.034. [DOI] [PubMed] [Google Scholar]
- 96.Chen R., Kobewka M., Addison W., Lachance G., Tyrrell D.L. Intrinsic viral factors are the dominant determinants of the hepatitis C virus response to interferon alpha treatment in chimeric mice. PLoS ONE. 2016;11:e0147007. doi: 10.1371/journal.pone.0147007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mu D., Yan L., Tang H., Liao Y. A sensitive and accurate quantification method for the detection of hepatitis B virus covalently closed circular DNA by the application of a droplet digital polymerase chain reaction amplification system. Biotechnol. Lett. 2015;37:2063–2073. doi: 10.1007/s10529-015-1890-5. [DOI] [PubMed] [Google Scholar]
- 98.Arad U. Modified Hirt procedure for rapid purification of extrachromosomal DNA from mammalian cells. Biotechniques. 1998;24:760–762. doi: 10.2144/98245bm14. [DOI] [PubMed] [Google Scholar]
- 99.Goecks J., Nekrutenko A., Taylor J., Galaxy Team Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Niyonzima N., Lambert A.R., Werther R., De Silva Feelixge H., Roychoudhury P., Greninger A.L., Stone D., Stoddard B.L., Jerome K.R. Tuning DNA binding affinity and cleavage specificity of an engineered gene-targeting nuclease via surface display, flow cytometry and cellular analyses. Protein Eng. Des. Sel. 2017;30:503–522. doi: 10.1093/protein/gzx037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10–12. [Google Scholar]
- 103.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.R Core Team . R Foundation for Statistical Computing; 2018. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 105.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Morgan M., Anders S., Lawrence M., Aboyoun P., Pagès H., Gentleman R. ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics. 2009;25:2607–2608. doi: 10.1093/bioinformatics/btp450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.