

Abstract
Aim: Recent studies have demonstrated that selective sodium–glucose cotransporter 2 inhibitors (SGLT2is) reduce cardiovascular events, although their mechanism remains obscure. We examined the effect of canagliflozin, an SGLT2i, on atherogenesis and investigated its underlying mechanism.
Method: Canagliflozin (30 mg/kg/day) was administered by gavage to streptozotocin-induced diabetic apolipoprotein E-deficient (ApoE−/−) mice. Sudan IV staining was performed at the aortic arch. Immunostaining, quantitative RT-PCR, and vascular reactivity assay were performed using the aorta. In vitro experiments using human umbilical vein endothelial cells (HUVECs) were also performed.
Result: Canagliflozin decreased blood glucose (P < 0.001) and total cholesterol (P < 0.05) levels. Sudan IV staining showed that 12-week canagliflozin treatment decreased atherosclerotic lesions (P < 0.05). Further, 8-week canagliflozin treatment ameliorated endothelial dysfunction, as determined by acetylcholine-induced vasodilation (P < 0.05), and significantly reduced the expressions of inflammatory molecules such as ICAM-1 and VCAM-1 in the aorta at the RNA and protein levels. Canagliflozin also reduced the expressions of NADPH oxidase subunits such as NOX2 and p22phox in the aorta and reduced urinary excretion of 8-OHdG, suggesting a reduction in oxidative stress. Methylglyoxal, a precursor of advanced glycation end products, increased the expressions of ICAM-1 and p22phox in HUVECs (P < 0.05, both). Methylglyoxal also decreased the phosphorylation of eNOSSer1177 and Akt but increased the phosphorylation of eNOSThr495 and p38 MAPK in HUVECs.
Conclusion: Canagliflozin prevents endothelial dysfunction and atherogenesis in diabetic ApoE−/− mice. Anti-inflammatory and antioxidative potential due to reduced glucose toxicity to endothelial cells might be its underlying mechanisms.
Keywords: SGLT2 inhibitor, Canagliflozin, Atherosclerosis, Endothelial dysfunction
See editorial vol. 27: 1139–1140
Introduction
The prevalence of type 2 diabetes mellitus (T2DM) in the world is increasing1–3). Even after using medical treatment and lifestyle modifications, it is still hard to achieve the goal of reducing blood glucose to an optimal level4, 5). In T2DM, atherosclerosis plays an important role in cardiovascular disease complications, and vascular inflammation due to hyperglycemia is one of the mechanisms of atherosclerosis6–10).
Recent randomized placebo-control studies found that sodium-dependent glucose transport 2 inhibitors (SGLT2is) (e.g., canagliflozin) reduced cardiovascular events in addition to their effect as antidiabetic drugs11–14). Previous studies demonstrated that SGLT2is reduce blood glucose levels, blood pressure, and body weight in T2DM patients, all of which play roles in atherogenesis11, 15–17). However, the mechanisms by which SGLT2is prevent cardiovascular events are not completely understood and need to be explored to achieve better treatment in T2DM patients with risk of cardiovascular events.
Previous preclinical studies have demonstrated that SGLT2is reduced vascular inflammation and atherogenesis in apolipoprotein e-deficient (ApoE−/−) mice18–22). Oxidative stress that is induced in a hyperglycemic state causes vascular inflammation, thus leading to the development of endothelial dysfunction, which is an initial step in atherogenesis23–25). However, few studies have investigated the effects of canagliflozin on atherogenesis, particularly on endothelial cells26, 27). Therefore, in the current study, we examined whether canagliflozin ameliorates endothelial dysfunction and prevents atherogenesis in diabetic ApoE−/− mice.
Methods
Animals and Treatment
ApoE−/− (C57BL/6J background) mice were originally purchased from The Jackson Laboratory. Male, 8-week-old ApoE−/− mice were treated with streptozotocin (STZ) (75 mg/kg/day) on three consecutive days to induce diabetes. A Western-type diet (Harlan Teklad Western diet TD.88137) was started from 9 weeks old and continued throughout the study period. From 9 weeks old, canagliflozin (30 mg/kg/day), which was supplied by Mitsubishi Tanabe Pharma, was orally administered for 12 weeks or 8 weeks to investigate its effect on atherosclerosis or endothelial function, respectively. Canagliflozin was suspended in 0.5% carboxymethyl cellulose (CMC) solution. The control group received only CMC. Mice were maintained under a 12-hour light/dark cycle. All experimental procedures conformed to the guidelines for the animal experimentation of Tokushima University.
Measurement of Metabolic Parameters
At the time of sacrifice, blood was collected from the heart into EDTA-containing tubes, and plasma was separated by centrifugation (9,000 rpm for 15 min) at 4°C. Plasma lipid levels (total cholesterol [TC], high-density lipoprotein [HDL] cholesterol, and triglycerides [TG]) were measured at LSI Medicine Corporation (Japan). Plasma levels of nonfasting insulin (FUJIFILM Wako Shibayagi Corporation) and advanced glycation end products (AGEs; Cell Biolabs, Inc.) were measured using commercially available kits. Urinary 8-hydroxy-2-deoxyguanosine (8-OHdG) concentration in urine collected for 16 hours was determined using a commercially available kit (Japan Institute for the Control of Aging, Nikken SEIL Co., Ltd.) and was corrected by creatinine level.
Analysis of Atherosclerotic Lesions
The thoracic aorta was opened longitudinally, and atherosclerotic lesions in the aortic arch were visualized using Sudan IV staining after fixation with 10% neutral buffered formalin. The luminal aspect of the stained aorta was photographed. The quantification of atherosclerotic lesions in the aortic arch was performed with Adobe Photoshop™ CS3 image analysis software. The thoracic aorta and abdominal aorta were collected and snap-frozen immediately for gene and/or protein analyses.
Histological and Immunohistochemical Analyses
Each heart was cut along a horizontal plane between the lower tips of the right and left atria. The upper portion was snap-frozen in OCT compound (TissueTeck). Sections (5 µm intervals) were stained with oil red O to detect lipid deposition. Other sections were incubated overnight with antivascular cell adhesion molecule-1 (VCAM-1) antibody, anti-intercellular adhesion molecule-1 (ICAM-1) antibody (Abcam), or antimacrophage antigen-3 (Mac-3) antibody (BD Biosciences). Thereafter, sections were incubated with biotinylated secondary antibody (VECTOR Laboratories, Inc.), followed by VECTASTAIN ABC-AP Kit (VECTOR Laboratories, Inc.) and were stained using a VectorRed AP Substrate Kit (VECTOR Laboratories, Inc.). All sections were counterstained with hematoxylin. The ratio of positive area to plaque area was then calculated28).
Vascular Reactivity Assay
The analysis of vascular reactivity was performed as described previously29). The descending thoracic aorta was cut into 2 mm rings with special care to preserve the endothelium, and the rings were mounted on a force transducer in an organ bath filled with modified Krebs–Henseleit buffer (118.4 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PH4, 1.2 mM MgSO4, 25 mM NaHCO3, and 11.1 mM glucose) aerated with 95% O2 and 5% CO2 at 37°C. Vessel rings were primed with 31.4 mM KCl and precontracted with phenylephrine to produce submaximal (60% of maximum) contraction. After the plateau was attained, the rings were exposed to increasing concentrations of acetylcholine (Ach) or sodium nitroprusside (SNP) to obtain cumulative concentration–response curves. Isometric tension was recorded on a polygraph.
Cell Culture Experiments
Human umbilical vein endothelial cells (HUVECs) were purchased from Life Technologies and cultured in EGM-2 (Lonza). HUVECs (passages 5–8) were treated with 500 µM methylglyoxal (MGO; Sigma-Aldrich) in the presence or absence of 1 mM canagliflozin in EBM-2 (Lonza) containing 2% FBS for 30 minutes or 8 hours.
Quantitative RT-PCR
Total RNA was extracted from tissues and cells by using an illustra RNAspin RNA isolation kit (GE Healthcare). Reverse transcription was performed using a QuantiTect Reverse Transcription kit (Qiagen) according to the manufacturer's instructions. Quantitative real-time PCR was performed on an Mx3000P (Agilent Technologies) by using gene-specific primers (Table 1) and Power SYBR Green PCR Master Mix (Applied Biosystems). Data are expressed in arbitrary units normalized by β-actin or GAPDH.
Table 1. Primer sequences.
| Name | Foward | Reverse |
|---|---|---|
| Mouse | ||
| ICAM-1 | TTCACACTGAATGCCAGCTC | GTCTGCTGAGACCCCTCTTG |
| VCAM-1 | CCCGTCATTGAGGATATTGG | GGTCATTGTCACAGCACCAC |
| MCP-1 | CCACTCACCTGCTGCTACTCAT | TGGTGATCCTCTTGTAGCTCTCC |
| TNF-α | ACCCTCACACTCAGATCATCTTC | TGGTGGTTTGCTACGACGT |
| F4/80 | TGCATCTAGCAATGGACAGC | GCCTTCTGGATCCATTTGAA |
| iNOS | CACCTTGGAGTTCACCCAGT | ACCACTCGTACTTGGGATGC |
| IL6 | ACAACCACGGCCTTCCCTACTT | CACGATTTCCCAGAGAACATGTG |
| NOX2 | ACTCCTTGGGTCAGCACTGG | GTTCCTGTCCAGTTGTCTTCG |
| NOX4 | TGTTGGGCCTAGGATTGTGTT | AGGGACCTTCTGTGATCCTCG |
| p22phox | CATGTGGGCCAACGAACA | CACTGTGTGAAACGTCCAGCAGTA |
| P47phox | AGACGGCTCCTATCCCTATCTCTG | TGCTAGCAATACCGGTGGAGATTA |
| RAGE | ACTACCGAGTCCGAGTCTACC | GTAGCTTCCCTCAGACACACA |
| β-actin | CCTGAGCGCAAGTACTCTGTGT | GCTGATCCACATCTGCTGGAA |
| Human | ||
| ICAM-1 | TGATGGGCAGTCAACAGCTA | GGGTAAGGTTCTTGCCCACT |
| p22phox | CCAGTGGTACTTTGGTGCCT | CCAGTGGTACTTTGGTGCCT |
| RAGE | ACCAGTTATGTAAGTCCCTGGATCA | CGTGTACGGCTGCTTGGAATAG |
| GAPDH | TGGGTGTGAACCATGAGAAG | GCTAAGCAGTTGGTGGTGC |
Western Blot Analysis
Cell lysates were prepared using RIPA buffer (Wako Pure Chemical Industries, Ltd.) containing a protease inhibitor cocktail (Takara Bio Inc.) and phosphatase inhibitors (Roche). Proteins were separated by SDS-PAGE and then transferred onto polyvinylidene difluoride membranes (Hybond-P; GE Healthcare). After blocking with 5% bovine serum albumin or 5% skimmed milk, the membranes were incubated overnight at 4°C with primary antibody against either phosphorylated-eNOSSer1177, eNOS (BD Biosciences), phosphorylated-eNOSThr495, phosphorylated-AktSer473, Akt, phosphorylated-p38 MAPK, p38 MAPK (Cell Signaling Technology), or β-actin (Sigma). Horseradish peroxidase-conjugated antimouse Ig antibody or antirabbit Ig antibody (Cell Signaling Technology) was then used as the secondary antibody. Antibody distribution was visualized with ECL-plus reagent (GE Healthcare) by using a luminescent image analyzer (LAS-1000, Fuji Film).
Statistical Analysis
All numerical values are expressed as mean ± SEM. Student's t-test was used to compare the two groups. Comparisons between multiple groups were performed with analysis of variance (ANOVA), followed by Scheffe's post hoc test for comparison between groups. The comparison of vascular response curves was performed by two-factor repeated measurement ANOVA. A value of P<0.05 was considered significant.
Results
Canagliflozin Decreased Atherosclerotic Lesions in Diabetic ApoE−/− Mice
After 12 weeks of canagliflozin administration, we examined atherosclerosis lesions in the aortic arch in diabetic ApoE−/− mice. Canagliflozin administration significantly reduced atherosclerotic lesions compared with the vehicle group (P < 0.01) (Fig. 1). The administration of canagliflozin for 12 weeks significantly reduced blood glucose level. Canagliflozin significantly reduced TC and HDL cholesterol levels and tended to reduce TG level after 12 weeks of treatment. The canagliflozin-treated group and nontreated group had no differences in blood pressure levels (Table 2).
Fig. 1.
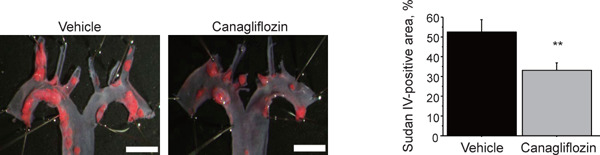
Canagliflozin attenuated atherosclerosis progression in diabetic ApoE−/− mice
En face Sudan IV staining showed that the administration of canagliflozin for 12 weeks significantly reduced atherosclerotic lesions in the aortic arch (n = 13–17 per group). Bar: 2 mm. **; P < 0.01. All values are mean ± SEM.
Table 2. Effects of canagliflozin after 12-week treatment.
| Veh (n = 13) | Cana (n = 17) | |
|---|---|---|
| Body weight, g | 27.5 ± 1.5 | 32.1 ± 1.3 |
| Blood glucose, mg/dl | 612.6 ± 43.9 | 218.9 ± 7.7*** |
| Total cholesterol, mg/dl | 2215.5 ± 338.3 | 1333.2 ± 145.5* |
| Triglyceride, mg/dl | 294.7 ± 53.9 | 186.0 ± 44.2 |
| HDL cholesterol, mg/dl | 45.1 ± 12.3 | 21.65 ± 3.8* |
| Insulin, pg/ml | 668.3 ± 162.0 | 1125.1 ± 207.7 |
| Heart rate, bpm | 652.2 ± 23.1 | 678.4 ± 13.9 |
| Systolic blood pressure, mmHg | 101.5 ± 5.7 | 92.9 ± 3.3 |
| Diastolic blood pressure, mmHg | 65.4 ± 3.3 | 59.6 ± 1.9 |
Veh; vehicle, Cana; canagliflozin, HDL; high density lipoprotein. All values are mean ± SEM.
P < 0.05
P < 0.001 vs. Veh.
Canagliflozin Ameliorated Endothelial Dysfunction in Diabetic ApoE−/− Mice
We examined the effects of canagliflozin on endothelial dysfunction, which is an initial step of atherogenesis. Canagliflozin administration for 8 weeks significantly ameliorated Ach-dependent vasodilation in diabetic ApoE−/− mice, thus suggesting the improvement of endothelial function (P < 0.05) (Fig. 2A). Our study showed that there were no significant differences in the endothelial-independent vasodilatation, as determined by the vascular response to SNP (Fig. 2B). As observed with 12-week treatment, canagliflozin treatment for 8 weeks significantly reduced blood glucose levels. The plasma AGE level in the canagliflozin-treated group was lower than that of the vehicle-treated group (vehicle vs. canagliflozin; 9.8 ± 4.0 vs. 7.9 ± 2.1 mg/ml, P = 0.12); however, it did not reach statistical significance. Canagliflozin also reduced TC level and HDL cholesterol level with this treatment protocol (Table 3). Throughout this study protocol, canagliflozin reduced food intake, water intake, and urine volume compared with the vehicle group (Table 4).
Fig. 2.
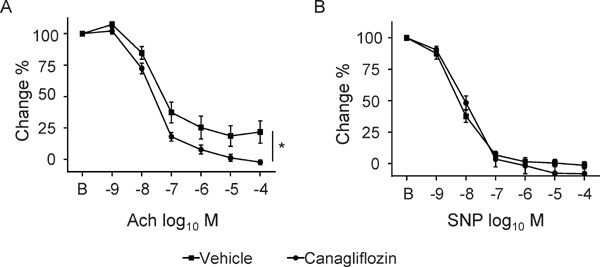
Canagliflozin ameliorated endothelium-dependent vasodilation in diabetic ApoE−/− mice
(A) After 8 weeks of treatment with canagliflozin, the vascular response to Ach was ameliorated in canagliflozin-treated diabetic ApoE−/− mice compared with the nontreated group. (B) There was no difference in vascular response to SNP between the treated group and nontreated group (n = 10–16, per group). *; P < 0.05. All values are mean ± SEM.
Table 3. Effects of canagliflozin after 8-week treatment.
| Veh (n = 10) | Cana (n = 16) | |
|---|---|---|
| Body weight, g | 21.5 ± 0.6 | 21.6 ± 0.5 |
| Blood glucose, mg/dl | 578.4 ± 51.9 | 214.8 ± 26.2*** |
| Total cholesterol, mg/dl | 3138.6 ± 356.3 | 2114.0 ± 177.3** |
| Triglyceride, mg/dl | 315.9 ± 75.5 | 187.6 ± 80.9 |
| HDL cholesterol, mg/dl | 125.0 ± 26.0 | 52.9 ± 14.5* |
| Insulin, pg/ml | 260.3 ± 97.9 | 427.6 ± 183.8 |
| Heart rate, bpm | 682.8 ± 18.3 | 696.4 ± 8.9 |
| Systolic blood pressure, mmHg | 86.0 ± 3.8 | 89.6 ± 3.0 |
| Diastolic blood pressure, mmHg | 49.0 ± 1.3 | 57.3 ± 2.4* |
Veh; vehicle, Cana; canagliflozin, HDL; high density lipoprotein.
All values are mean ± SEM.
P < 0.05
P < 0.01
P < 0.001 vs. Veh.
Table 4. Food intake, water intake, and urine output after canagliflozin treatment.
| Veh (n = 10) |
Cana (n = 16) |
|||
|---|---|---|---|---|
| 4th week | 8th week | 4th week | 8th week | |
| Food intake, g | 3.21 ± 0.27 | 3.87 ± 0.35 | 2.44 ± 0.17* | 2.7 ± 0.13** |
| Water intake, ml | 14.00 ± 1.8 | 14.05 ± 2.34 | 9.38 ± 1.09* | 9.84 ± 1.70 |
| Urine output, ml | 11.72 ± 1.83 | 14.46 ± 2.18 | 7.03 ± 1.29* | 7.12 ± 1.47** |
Veh; vehicle, Cana; canagliflozin. All values are mean ± SEM.
P < 0.05
P < 0.01 vs. Veh.
Canagliflozin Reduced Inflammatory Molecule Expression and Oxidative Stress in Diabetic ApoE−/− Mice
Canagliflozin administration for 8 weeks significantly reduced the expression of inflammatory molecules such as ICAM-1, VCAM-1, MCP-1, F4/80, and IL-6 in the aorta. Canagliflozin also decreased the expression of tumor necrosis factor-α (TNF-α) and receptor for AGEs (RAGE) in the aorta; however, it did not reach statistical significance. Furthermore, canagliflozin also reduced the expression of molecules related to oxidative stress such as iNOS and NADPH oxidase subunits (e.g., NOX2, NOX4, p22phox, and p47phox) (Fig. 3A). Canagliflozin significantly decreased the urinary excretion of 8-OHdG, which is an oxidative stress marker, compared with the vehicle group (P < 0.01) (Fig. 3B). Canagliflozin also reduced lipid deposition in atherosclerotic plaques in the aortic root (Fig. 4A). The results of immunostaining demonstrated that canagliflozin decreased the expression of inflammatory molecules such as VCAM-1 (P < 0.05) and ICAM-1 (P < 0.01) and decreased the accumulation of macrophages (P < 0.01) in atherosclerotic plaques in the aortic root (Figs. 4B–4D).
Fig. 3.
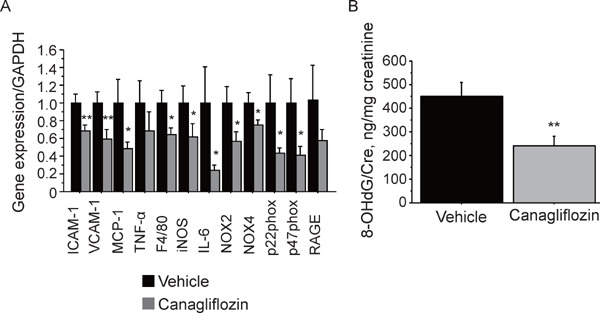
Canagliflozin reduced the expression of inflammatory molecules and oxidative stress-related molecules in diabetic ApoE−/− mice
(A) Canagliflozin treatment for 8 weeks reduced the expression of a macrophage marker, namely, F4/80, and inflammatory molecules (ICAM-1, VCAM-1, and MCP-1, and IL-6) in the aorta of diabetic ApoE−/− mice. Canagliflozin also reduced the expression of molecules that contribute to the production of oxidative stress, such as iNOS, NOX2, NOX4, p22phox, and p47phox, in the aorta. Canagliflozin tended to reduce RAGE. (B) Canagliflozin treatment for 8 weeks reduced the urinary excretion of 8-OHdG in diabetic ApoE−/− mice (n = 10–14, per group) *; P < 0.05 and **; P < 0.01. All values are mean ± SEM.
Fig. 4.
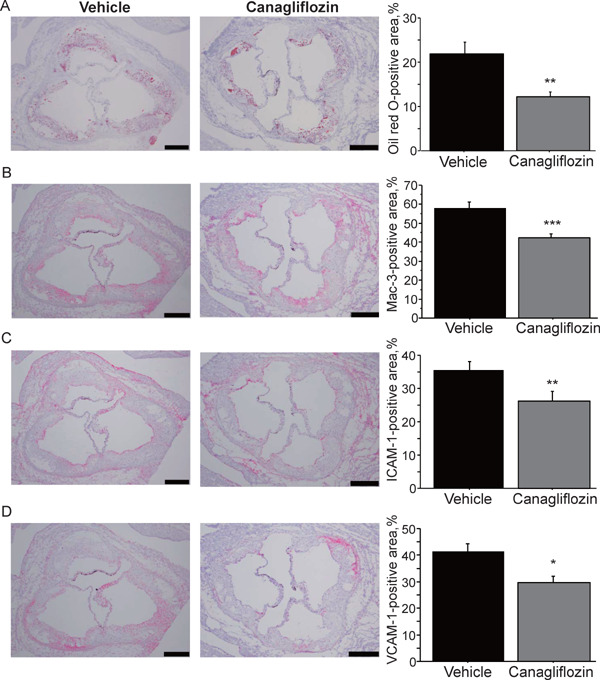
Effects of canagliflozin on the characteristics of atherosclerotic plaques
(A) Oil red O staining showed that canagliflozin treatment for 8 weeks reduced lipid deposition in atherosclerotic plaques in the aortic root compared with the nontreatment group. (B–D) Immunostaining against Mac-3 (B), ICAM-1 (C), and VCAM-1 (D). Canagliflozin treatment for 8 weeks reduced the accumulation of macrophages and the expression of adhesion molecules compared with the nontreatment group (n = 10 per group). Bar: 200 µm *; P < 0.05, **; P < 0.01, and ***; P < 0.001. All values are mean ± SEM.
Hyperglycemic Condition Contributed to Endothelial Dysfunction
Our in vivo data suggested that canagliflozin suppressed the glucose toxicity represented by the AGE–RAGE system. Thus, we treated HUVECs with MGO, which is a major precursor of AGEs, to investigate the effects of canagliflozin on endothelial cells. MGO significantly increased the expression of inflammatory molecules such as ICAM-1, RAGE, and p22phox. The presence of canagliflozin in culture medium did not affect the expressions promoted by MGO (Fig. 5A). MGO decreased the phosphorylation of eNOSSer1177 and AktSer473 (P < 0.05) (Fig. 5B). On the contrary, MGO increased the phosphorylation of eNOSThr495 and p38 MAPK in HUVECs (P < 0.05) (Fig. 5C).
Fig. 5.
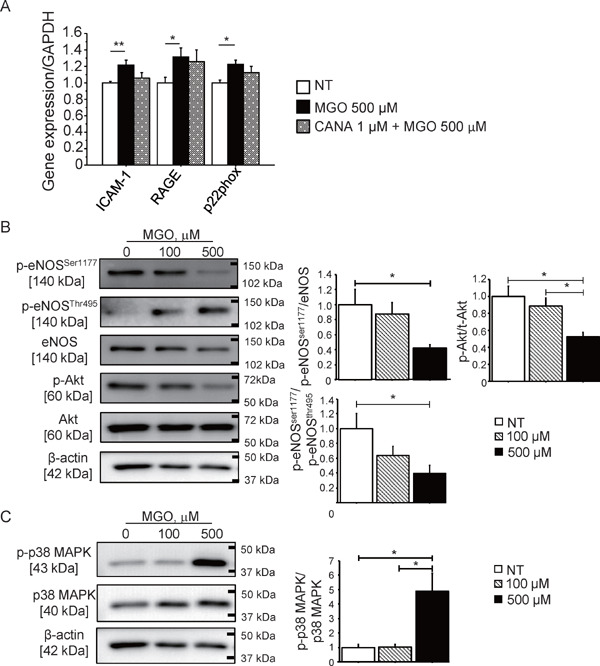
MGO induced endothelial dysfunction
(A) The incubation of HUVECs with MGO, which is one of the major precursors of AGEs, for 8 hours increased the expression of inflammatory molecules such as ICAM-1, RAGE, and p22phox, which is an NADPH oxidase subunit (n = 5, per group). The presence of canagliflozin did not affect these expressions. (B) The incubation of HUVECs with MGO (500 µM) for 30 minutes reduced the phosphorylation of eNOSSer1177 and increased the phosphorylation of the eNOS inhibitory site, namely, eNOSThr495. MGO also reduced the phosphorylation of Aktser473 (n = 8, per group). (C) MGO increased the phosphorylation of p38 MAPK in HUVECs (n = 8, per group). *; P < 0.05 and **; P < 0.01. All values are mean ± SEM.
Discussion
We demonstrated that canagliflozin attenuated the development of endothelial dysfunction and atherogenesis in diabetic ApoE−/− mice. Canagliflozin reduced the expression of inflammatory molecules and oxidative stress, thus suggesting that this drug reduced vascular inflammation. The results of in vitro experiments using HUVECs suggested that the glucose-lowering effects of canagliflozin contribute to the inhibition of inflammatory activation of endothelial cells.
Canagliflozin is a selective SGLT2i that inhibits glucose reabsorption and increases glucose excretion into urine11). Although there are numerous antidiabetic drugs, large-scale randomized trials have proved that only a few drugs decrease cardiovascular events in diabetic patients. By contrast, recent studies demonstrated that SGLT2i, including canagliflozin, reduced cardiovascular events in diabetic patients11, 12). Several studies demonstrated that canagliflozin reduced albuminuria, plasma lipid levels, blood pressure, and body weight, in addition to its effect as an antidiabetic drug, thus suggesting that these metabolic actions of canagliflozin contribute to cardiovascular protection11, 15–17). However, the mechanisms by which SGLT2is reduce cardiovascular events are not well established.
In this study, we demonstrated that the administration of canagliflozin to diabetic ApoE−/− mice for 12 weeks reduced atherosclerotic lesions in the aorta. Several previous studies also reported that SGLT2is reduced atherosclerotic lesion development in diabetic and nondiabetic mice18, 19, 26). Our results are consistent with these studies. We also showed that canagliflozin significantly reduced lipid deposition, macrophage accumulation, and expression of adhesion molecules such as ICAM-1 and VCAM-1. These results indicate that canagliflozin attenuated vascular inflammation, which is a major cause of atherosclerosis, in diabetes.
One of the pathological processes of atherogenesis is endothelial dysfunction. Endothelial dysfunction is an initial step of atherosclerosis. Our study showed that canagliflozin treatment improved endothelial dysfunction in diabetic ApoE−/− mice compared with the nontreatment group. The improvement of endothelial dysfunction by SGLT2is has been shown in other studies26, 27). In addition to these studies, our results demonstrated that canagliflozin ameliorated endothelial dysfunction in a diabetic atherosclerotic mouse model. Vascular inflammation promotes endothelial dysfunction, thus leading to the development of atherosclerosis30). In the current study, together with the amelioration of endothelial dysfunction, canagliflozin-treated mice showed lower expressions of inflammatory molecules such as VCAM-1, ICAM-1, and MCP-1 in the aorta. In addition to these inflammatory molecules, we observed that canagliflozin reduced the expression of molecules related to the elevation of oxidative stress. Metabolic disorders influence the production of oxidative stress, and a hyperglycemic state is one of them31). Numerous studies have reported that oxidative stress promotes various processes of vascular inflammation and decreases NO bioavailability, thus leading to endothelial dysfunction32). Therefore, several studies have indicated that oxidative stress is one of the therapeutic targets for vascular diseases33). In the current study, canagliflozin reduced iNOS; NOX2; NOX4; p22phox, and p47phox expression in the aorta of diabetic ApoE−/− mice. Previous studies have reported that these NADPH oxidase subunits contribute to the development of atherosclerosis34). Furthermore, we found that canagliflozin decreased the excretion of 8-OHdG, which is a marker for oxidative stress, thus suggesting that canagliflozin reduced oxidative stress in diabetic ApoE−/− mice.
We further examined the mechanism by which canagliflozin attenuated endothelial dysfunction and atherosclerosis. In this study, we focused on the role of the AGE–RAGE system. AGEs are generated in a hyperglycemic condition35). There is a growing body of evidence indicating that AGEs elicit oxidative stress via its receptor, namely, RAGE, and evoke inflammation in vascular cells, thus leading to the development of microvascular and/or macrovascular diabetic complications36, 37). In the present study, despite having no significance, canagliflozin decreased plasma AGE levels and RAGE expression. HUVECs treated with MGO, which is the main precursor of AGEs38), showed increased expression of inflammatory molecules such as ICAM-1, RAGE, and p22phox. However, canagliflozin did not reduce their expression possibly because SGLT2 is exclusively expressed on proximal tubules and because endothelial cells rarely express SGLT2 39). MGO also decreased the phosphorylation of eNOSSer1177 and Akt and increased the phosphorylation of an inhibitory site of eNOS, namely, eNOSThr495. These results suggest that a hyperglycemic condition affects endothelial function. Furthermore, MGO promoted the phosphorylation of p38 MAPK, thus suggesting activation of this pathway. The p38 MAPK pathway is associated with the promotion of inflammation40) and regulation of oxidation5). The activation of MAPK by MGO might affect endothelial function in a diabetic condition41). Our results suggest that the glucose-lowering effect of canagliflozin promotes the Akt–eNOS pathway and inhibits p38 MAPK activation, thus conferring atheroprotective effects.
Our study demonstrated that a glucose-lowering effect that subsequently suppresses the AGE–RAGE system was the one of the mechanisms by which canagliflozin attenuated the development of endothelial dysfunction and atherosclerosis. From this point of view, all antidiabetic drugs may have the same effects. However, several studies proposed the specific effects of SGLT2i. A recent study demonstrated that treatment with canagliflozin favorably affects the levels of adipokines and inflammatory molecules, which are associated with impaired adipose tissue function and cardiovascular disease, compared with glimepiride42). Furthermore, many studies demonstrated that SGLT2is decrease body weight, blood pressure, and TG. These results support the hypothesis that SGLT2is have specific cardioprotective effects16, 17). Further studies are needed to understand the cardioprotective effects of SGLT2is.
This study has several limitations. First, we used STZ-induced diabetic mice. This model does not reflect T2DM completely. Second, canagliflozin significantly reduced TC and HDL cholesterol levels and tended to reduce TG level. Several previous studies have demonstrated similar effects of SGLT2is18–21). These effects on lipid levels might have affected the development of atherogenesis and endothelial dysfunction in the treated group. Third, the glucose-lowering effect of canagliflozin contributed largely to the inhibition of atherogenesis in our mouse model. To investigate the SGLT2i-specific effects, we needed to incorporate other groups that received different classes of antidiabetic drug. Fourth, the dose of canagliflozin used in this study was higher than that for humans. In a previous study, the same dose of canagliflozin effectively increased urinary glucose excretion and decreased blood glucose levels without any harmful effects43). The difference in metabolic speed of medical substances between mice and humans may explain higher dose for mice. Finally, several studies have reported that SGLT2is have a small diuretic effect and reduce body weight; however, in the current study, the treated group did not show these effects. Furthermore, canagliflozin reduced food intake, water intake, and urine volume in this study. These might have been caused by the improvement of a hyperglycemic condition by canagliflozin. Unexpectedly, canagliflozin increased diastolic blood pressure after 8 weeks of treatment. Although the results of animal studies cannot be directly applied to humans, further research is needed to elucidate the mechanism of action of SGLT2is on both metabolic and cardiovascular systems. Despite these limitations, our research suggests that canagliflozin improves endothelial function and attenuates atherosclerosis by reducing inflammation and oxidative stress.
Conclusion
Canagliflozin ameliorates endothelial dysfunction and prevents atherogenesis in diabetic ApoE−/− mice. Anti-inflammatory and antioxidative effects that are potentially caused by the reduction of glucose toxicity might be involved in the cardioprotective effects of canagliflozin.
Acknowledgements
The authors would like to thank Shintaro Okamoto and Etsuko Uematsu (Tokushima University) for their technical assistance.
Abbreviation
- 8-OHdG
8-hydroxy-2-deoxyguanosine
- Ach
acetylcholine
- AGE
advanced glycation end products
- ApoE−/−
apolipoprotein e-deficient
- CMC
carboxymethyl cellulose
- HDL
high-density lipoprotein
- HUVEC
human umbilical vein endothelial cell
- ICAM-1
intercellular adhesion molecule-1
- IL
interleukin
- Mac-3
macrophage antigen-3
- MGO
methylglyoxal
- RAGE
receptor for AGEs
- SGLT2i
sodium-dependent glucose transport 2 inhibitor
- SNP
sodium nitroprusside
- STZ
streptozotocin
- T2DM
type 2 diabetes mellitus
- TC
total cholesterol
- TG
triglyceride
- TNF-α
tumor Necrosis Factor-α
- VCAM-1
vascular cell adhesion molecule-1
- WTD
Western-type diet
Competing Interests
The Department of Cardio-Diabetes Medicine, Tokushima University Graduate School is supported in part by unrestricted research grants from Boehringer Ingelheim. Dr. Sata received research funding and lecture fees from Mitsubishi Tanabe Pharma. Other authors declare that they have no conflicts of interest.
Sources of Funding
This work was partially supported by JSPS Kakenhi Grants (No. 19K08584 to D.F. and No. 19H03654 to M.S.), SENSHIN Medical Research Foundation (D.F.), Takeda Science Foundation (M.S.), and the Vehicle Racing Commemorative Foundation (M.S.). The funders had no role in the study design, data collection and analysis, or manuscript preparation.
References
- 1). Collaboration NCDRF. Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4.4 million participants. Lancet, 2016; 387: 1513-1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2). Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract, 2010; 87: 4-14 [DOI] [PubMed] [Google Scholar]
- 3). Whiting DR, Guariguata L, Weil C, Shaw J. IDF Diabetes Atlas: Global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res Clin Pract, 2011; 94: 311-321 [DOI] [PubMed] [Google Scholar]
- 4). Zimmet P, Alberti KGMM. Surgery or Medical Therapy for Obese Patients with Type 2 Diabetes? N Engl J Med, 2012; 366: 1635-1636 [DOI] [PubMed] [Google Scholar]
- 5). Ali MK, Bullard KM, Saaddine JB, Cowie CC, Imperatore G, Gregg EW. Achievement of Goals in U.S. Diabetes Care, 1999–2010. N Engl J Med, 2013; 368: 1613-1624 [DOI] [PubMed] [Google Scholar]
- 6). Kralova Lesna I, Tonar Z, Malek I, Maluskova J, Nedorost L, Pirk J, Pitha J, Lanska V, Poledne R. Is the amount of coronary perivascular fat related to atherosclerosis? Physiol Res, 2015; 64 Suppl 3: S435-S443 [DOI] [PubMed] [Google Scholar]
- 7). Park I, Kassiteridi C, Monaco C. Functional diversity of macrophages in vascular biology and disease. Vascul Pharmacol, 2017; 99: 13-22 [DOI] [PubMed] [Google Scholar]
- 8). Ross R. Atherosclerosis — An Inflammatory Disease. N Engl J Med, 1999; 340: 115-126 [DOI] [PubMed] [Google Scholar]
- 9). Fenyo IM, Gafencu AV. The involvement of the monocytes/macrophages in chronic inflammation associated with atherosclerosis. Immunobiology, 2013; 218: 1376-1384 [DOI] [PubMed] [Google Scholar]
- 10). Maki K, Rieko T, Koh O, Sayaka S, Hiromichi W, Hajime Y, Noriko S-A, Tatsuya M, Akira S, Yuko T, Koji H. Association between monocyte chemoattractant protein-1 and blood pressure in smokers. J Int Med Res, 2017; 46: 965-974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11). Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N Engl J Med, 2017; 377: 644-657 [DOI] [PubMed] [Google Scholar]
- 12). Wu JHY, Foote C, Blomster J, Toyama T, Perkovic V, Sundström J, Neal B. Effects of sodium-glucose cotransporter-2 inhibitors on cardiovascular events, death, and major safety outcomes in adults with type 2 diabetes: a systematic review and meta-analysis. Lancet Diabetes Endocrinol, 2016; 4: 411-419 [DOI] [PubMed] [Google Scholar]
- 13). Chao EC, Henry RR. SGLT2 inhibition--a novel strategy for diabetes treatment. Nat Rev Drug Discov, 2010; 9: 551-559 [DOI] [PubMed] [Google Scholar]
- 14). Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med, 2015; 373: 2117-2128 [DOI] [PubMed] [Google Scholar]
- 15). Budoff MJ, Wilding JPH. Effects of canagliflozin on cardiovascular risk factors in patients with type 2 diabetes mellitus. Int J Clin Pract, 2017; 71: e12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16). Davies MJ, Merton K, Vijapurkar U, Yee J, Qiu R. Efficacy and safety of canagliflozin in patients with type 2 diabetes based on history of cardiovascular disease or cardiovascular risk factors: a post hoc analysis of pooled data. Cardiovasc Diabetol, 2017; 16: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17). Stenlof K, Cefalu WT, Kim KA, Alba M, Usiskin K, Tong C, Canovatchel W, Meininger G. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab, 2013; 15: 372-382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18). Han JH, Oh TJ, Lee G, Maeng HJ, Lee DH, Kim KM, Choi SH, Jang HC, Lee HS, Park KS, Kim Y-B, Lim S. The beneficial effects of empagliflozin, an SGLT2 inhibitor, on atherosclerosis in ApoE−/− mice fed a western diet. Diabetologia, 2017; 60: 364-376 [DOI] [PubMed] [Google Scholar]
- 19). Leng W, Ouyang X, Lei X, Wu M, Chen L, Wu Q, Deng W, Liang Z. The SGLT-2 Inhibitor Dapagliflozin Has a Therapeutic Effect on Atherosclerosis in Diabetic ApoE−/− Mice. Mediators Inflamm, 2016; 2016: 6305735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20). Terasaki M, Hiromura M, Mori Y, Kohashi K, Nagashima M, Kushima H, Watanabe T, Hirano T. Amelioration of Hyperglycemia with a Sodium-Glucose Cotransporter 2 Inhibitor Prevents Macrophage-Driven Atherosclerosis through Macrophage Foam Cell Formation Suppression in Type 1 and Type 2 Diabetic Mice. PLoS One, 2015; 10: e0143396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21). Nasiri-Ansari N, Dimitriadis GK, Agrogiannis G, Perrea D, Kostakis ID, Kaltsas G, Papavassiliou AG, Randeva HS, Kassi E. Canagliflozin attenuates the progression of atherosclerosis and inflammation process in APOE knockout mice. Cardiovasc Diabetol, 2018; 17: 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22). Nakatsu Y, Kokubo H, Bumdelger B, Yoshizumi M, Yamamotoya T, Matsunaga Y, Ueda K, Inoue Y, Inoue M-K, Fujishiro M, Kushiyama A, Ono H, Sakoda H, Asano T. The SGLT2 Inhibitor Luseogliflozin Rapidly Normalizes Aortic mRNA Levels of Inflammation-Related but Not Lipid-Metabolism-Related Genes and Suppresses Atherosclerosis in Diabetic ApoE KO Mice. Int J Mol Sci, 2017; 18: 1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23). Dasu MR, Devaraj S, Jialal I. High glucose induces IL-1β expression in human monocytes: mechanistic insights. Am J Physiol Endocrinol Metab, 2007; 293: E337-E346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24). Devaraj S, Venugopal SK, Singh U, Jialal I. Hyperglycemia Induces Monocytic Release of Interleukin-6 via Induction of Protein Kinase C-α and -β. Diabetes, 2005; 54: 85-91 [DOI] [PubMed] [Google Scholar]
- 25). Piga R, Naito Y, Kokura S, Handa O, Yoshikawa T. Short-term high glucose exposure induces monocyteendothelial cells adhesion and transmigration by increasing VCAM-1 and MCP-1 expression in human aortic endothelial cells. Atherosclerosis, 2007; 193: 328-334 [DOI] [PubMed] [Google Scholar]
- 26). Oelze M, Kröller-Schön S, Welschof P, Jansen T, Hausding M, Mikhed Y, Stamm P, Mader M, Zinßius E, Agdauletova S, Gottschlich A, Steven S, Schulz E, Bottari SP, Mayoux E, Münzel T, Daiber A. The Sodium-Glucose Co-Transporter 2 Inhibitor Empagliflozin Improves Diabetes-Induced Vascular Dysfunction in the Streptozotocin Diabetes Rat Model by Interfering with Oxidative Stress and Glucotoxicity. PLoS ONE, 2014; 9: e112394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27). Salim HM, Fukuda D, Yagi S, Soeki T, Shimabukuro M, Sata M. Glycemic Control with Ipragliflozin, a Novel Selective SGLT2 Inhibitor, Ameliorated Endothelial Dysfunction in Streptozotocin-Induced Diabetic Mouse. Front Cardiovasc Med, 2016; 3: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28). Hara T, Fukuda D, Tanaka K, Higashikuni Y, Hirata Y, Nishimoto S, Yagi S, Yamada H, Soeki T, Wakatsuki T, Shimabukuro M, Sata M. Rivaroxaban, a novel oral anticoagulant, attenuates atherosclerotic plaque progression and destabilization in ApoE-deficient mice. Atherosclerosis, 2015; 242: 639-646 [DOI] [PubMed] [Google Scholar]
- 29). Gao YJ, Lu C, Su LY, Sharma AM, Lee RM. Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide. Br J Pharmacol, 2007; 151: 323-331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30). Lusis AJ. Atherosclerosis. Nature, 2000; 407: 233-241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31). Teresa Vanessa F, Annamaria P, Pengou Z, Franco F. Hyperglycemia-induced Oxidative Stress and its Role in Diabetes Mellitus Related Cardiovascular Diseases. Curr Pharm Des, 2013; 19: 5695-5703 [DOI] [PubMed] [Google Scholar]
- 32). Higashi Y, Noma K, Yoshizumi M, Kihara Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ J, 2009; 73: 411-448 [DOI] [PubMed] [Google Scholar]
- 33). Munzel T, Gori T, Bruno RM, Taddei S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur Heart J, 2010; 31: 2741-2748 [DOI] [PubMed] [Google Scholar]
- 34). Kigawa Y, Miyazaki T, Lei XF, Kim-Kaneyama JR, Miyazaki A. Functional Heterogeneity of Nadph Oxidases in Atherosclerotic and Aneurysmal Diseases. J Atheroscler Thromb, 2017; 24: 1-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35). Yamagishi SI, Nakamura N, Matsui T. Glycation and cardiovascular disease in diabetes: A perspective on the concept of metabolic memory. J Diabetes, 2017; 9: 141-148 [DOI] [PubMed] [Google Scholar]
- 36). Aronson D, Rayfield EJ. How hyperglycemia promotes atherosclerosis: molecular mechanisms. Cardiovascular Diabetology, 2002; 1: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37). Singh R, Barden A, Mori T, Beilin L. Advanced glycation end-products: a review. Diabetologia, 2001; 44: 129-146 [DOI] [PubMed] [Google Scholar]
- 38). Allaman I, Bélanger M, Magistretti PJ. Methylglyoxal, the dark side of glycolysis. Front Neurosci, 2015; 9: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39). Han Y, Cho YE, Ayon R, Guo R, Youssef KD, Pan M, Dai A, Yuan JX, Makino A. SGLT inhibitors attenuate NO-dependent vascular relaxation in the pulmonary artery but not in the coronary artery. Am J Physiol Lung Cell Mol Physiol, 2015; 309: L1027-L1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40). Pietersma A, Tilly BC, Gaestel M, de Jong N, Lee JC, Koster JF, Sluiter W. P38 Mitogen Activated Protein Kinase Regulates Endothelial VCAM-1 Expression at the Post-transcriptional Level. Biochem Biophys Res Commun, 1997; 230: 44-48 [DOI] [PubMed] [Google Scholar]
- 41). Peng X-q, Damarla M, Skirball J, Nonas S, Wang X-y, Han EJ, Hasan EJ, Cao X, Boueiz A, Damico R, Tuder RM, Sciuto AM, Anderson DR, Garcia JGN, Kass DA, Hassoun PM, Zhang J-t. Protective role of PI3-kinase/Akt/eNOS signaling in mechanical stress through inhibition of p38 mitogen-activated protein kinase in mouse lung. Acta Pharmacologica Sinica, 2010; 31: 175-183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42). Garvey WT, Van Gaal L, Leiter LA, Vijapurkar U, List J, Cuddihy R, Ren J, Davies MJ. Effects of canagliflozin versus glimepiride on adipokines and inflammatory biomarkers in type 2 diabetes. Metabolism, 2018; 85: 32-37 [DOI] [PubMed] [Google Scholar]
- 43). Liang Y, Arakawa K, Ueta K, Matsushita Y, Kuriyama C, Martin T, Du F, Liu Y, Xu J, Conway B, Conway J, Polidori D, Ways K, Demarest K. Effect of canagliflozin on renal threshold for glucose, glycemia, and body weight in normal and diabetic animal models. PLoS One, 2012; 7: e30555. [DOI] [PMC free article] [PubMed] [Google Scholar]