

Abstract
Objectives
To elucidate the motor unit response to intrathecal nusinersen in children with symptomatic spinal muscular atrophy (SMA) using a novel motor unit number estimation technique.
Methods
MScanFit MUNE studies were sequentially undertaken from the abductor pollicis brevis muscle after stimulation of the median nerve in a prospective cohort of symptomatic children with SMA, undergoing intrathecal treatment with nusinersen at a single neuromuscular centre from June 2017 to August 2019. Electrophysiological measures included compound muscle action potential (CMAP), motor unit number estimation (MUNE), motor unit number contributing to 50%–100% of CMAP (N50) and measures of collateral reinnervation including largest single motor unit potential (LSMUP) and amplitude of the smallest unit contributing to N50 (A50).
Results
Twenty children (median age 99 months, range 4–193) were followed for a median of 13.8 (4–33.5) months. Therapeutic intervention was an independent and significant contributor to an increase in CMAP (p = 0.005), MUNE (p = 0.001) and N50 (p = 0.04). The magnitude of this electrophysiological response was increased in children with shorter disease durations (p<0.05). Electrophysiological changes delineated children who were functionally stable from those who attained clinically significant gains in motor function.
Interpretation
Nusinersen therapy facilitated functional innervation in SMA through recovery of smaller motor units. Delineation of biomechanisms of therapeutic response may be the first step in identifying potential novel targets for disease modification in this and other motor neuropathies. MScanFit MUNE techniques may have a broader role in establishing biomarkers of therapeutic response in similar adult-onset diseases.
Introduction
Spinal muscular atrophy (SMA) is a rare genetic disease with a wide phenotypic spectrum.1 Disease pathophysiology centres around irreversible loss of motor neurons in the spinal cord and brainstem.2 Deficiency of survival motor neuron (SMN) protein secondary to homozygous disruption of survival motor neuron 1 gene (SMN1) forms the aetiological basis of disease.3 A paralogous gene, survival motor neuron 2 (SMN2) produces a small quantity of functional protein, varying in copy number to ameliorate phenotype in a dose-dependent manner.1
The advent of nusinersen, the first disease-modifying therapy, heralded a new treatment era for this condition acting as an SMN2 enhancer to increase SMN protein levels.4 With therapeutic intervention, improvements in survival and attainment of motor skills are noted in comparison to historical controls.5 In contrast to this well-documented clinical response, the physiological effects of SMN repletion at the motor unit level have not been elucidated in affected patients. This lack of biomechanistic knowledge is a rate limiting factor for development of therapeutic biomarkers6 and the evolution of therapeutics that may harness the motor unit’s ability to remodel, both of which impede the optimisation of clinical outcomes for affected individuals.7
Electrophysiological biomarkers of denervation were first developed to track motor neuron loss in adult-onset motor neuropathies,8 acting as indicators for treatment acceleration, modification and to define treatment limits.9 Similar potential exists in paediatric-onset conditions such as SMA. Accordingly, clinical trials for SMA have started to incorporate and track treatment-associated compound muscle action potential (CMAP) changes. Stabilisation and/or improvement of CMAP in symptomatic infants and in later-onset forms of the disease after therapeutic intervention are observed.10 11 However, this single measure is not sensitive enough to quantify subtle changes in denervation, with preserved values noted despite 50% loss of the motor neuron pool.12 Instead, electrophysiological measures such as motor unit number estimation (MUNE) and single motor unit amplitude (SMUP) more accurately reflect the health of the motor neuron pool, delineating flux in denervation (signified by MUNE) and collateral reinnervation (signified by SMUP) capacity of motor units.
Muscle Scans and the associated MUNE estimation (MScanFit) provide a novel method for motor unit number estimation, eliminating sources of error in traditional techniques.13 While this method is a sensitive and specific way of monitoring early disease progression in adult neuropathies,13 its utility in defining disease status is only now being recognised in treatment-naïve children with SMA.14 Its role in treated cohorts has not previously been investigated.
Accordingly, this is the first study to investigate the utility of five Muscle Scan parameters to define the basis, timing and extent of motor unit changes with SMN repletion therapy, in a prospectively studied cohort of children with SMA. This study uniquely investigates clinical measures that predict an electrophysiological response, exploring the interplay between denervation and reinnervation across a spectrum of clinical phenotypes, shedding light on the therapeutic window and clinical confounders that modulate response.
Methods
Study design
This was a single centre prospective cross-sectional and longitudinal study measuring clinical, functional and electrophysiological outcomes in infants, children and adolescents with SMA conducted between June 2017 and October 2019. Inclusion and exclusion criteria are detailed in table 1.
Table 1.
Study inclusion and exclusion criteria for participants in the study
| Inclusion criteria | Exclusion criteria |
| Genetically confirmed homozygous SMN1 deletions | Children who were beyond >4 months since commencement of treatment, that is, those who had already transitioned to nusinersen maintenance treatment at the start of the study |
| Clinical signs and symptoms consistent with a diagnosis of SMA | Children with comorbidities or medication use potentially associated with development of peripheral neuropathy or neuromuscular disease |
| Age 4 months–20 years | Serious illnesses/comorbidities that would affect clinical or electrophysiological assessment in the view of the researchers |
| Functional status (non-sitter, sitter, walker) | Children who did not tolerate or were unwilling to undertake sequential studies |
| Receiving intrathecal nusinersen as part of clinical management at Sydney Children’s Hospital New South Wales, Australia | Children who were treated with a disease-modifying agent other than nusinersen |
| Written informed, voluntary consent given by parent/legal guardian or young person according to the principles set out in the Declaration of Helsinki: Consent forms were signed
Non-English speakers were consented with the aid of an interpreter. |
SMA, spinal muscular atrophy.
Study measures
Clinical measures
Demographic and medical data, as detailed in table 2, were collated from electronic medical records. ‘Current’ functional motor status is a useful and pragmatic way of classifying children with SMA and has been used to functionally define children with SMA in international standard of care consensus guidelines.15 Our participants were similarly stratified as (non-sitter, sitter and walker) against WHO developmental milestones at 1 month prior to therapeutic intervention and 1 month after study completion. Classification of SMA phenotype for this study adhered to guidelines set out in the International Collaborative SMA Workshop.16
Table 2.
Clinical characteristics of participants in each SMA phenotypic subgroup and for the total cohort
| Characteristics | SMA type 1, N=6 | SMA type 2, N=10 | SMA type 3, N=4 | Total, N=20 |
| Sex | ||||
| Male | 2 (33%) | 6 (60%) | 3 (75%) | 11 (55%) |
| Female | 4 (66%) | 4 (40%) | 1 (25%) | 9 (45%) |
| SMN2 copy number | ||||
| 2 | 1 (17%) | 1 (11%) | 1 (25%) | 3 (16%) |
| 3 | 5 (83%) | 8 (89%) | 3 (75%) | 16 (84%) |
| Age at symptom onset (months) Median, (range), SD |
3.5, (2–5), 1.11 | 12, (8–18), 7.8 | 22.5, (18.5–144), 65.2 | 12, (2–144), 30.5 |
| Age at time of study (months) Median, (range), SD |
11.5, (4–178), 67.7 | 99, (13–153), 42 | 127.5, (109–193), 37.2 | 99, (4–193), 59.3 |
| Disease duration (months) Median, (range), SD |
7.2 (2–175), 68.2 | 77 (1–141), 41.7 | 104.5 (52–114), 29 | 63 (2–175), 51.6 |
| Duration of study follow-up (months) Median, (range), SD |
26.8, (21.5–33.5), 5.0 | 12.8, (4 -26), 5.9 | 12, (6–14), 3.8 | 13.8 (4–33.5), 8.6 |
Classification of SMA phenotype for this study adhered to guidelines set out in the International Collaborative SMA Workshop16; SMA type 1 (symptom onset <6 months, unable to sit independently), SMA type 2 (symptom onset 7–18 months, sits independently) and SMA type 3 (symptom onset >18 months of age, walks independently at time of diagnosis).
Age at the time of study, age at symptom onset, disease duration (interval between age of symptom onset and age at first nusinersen treatment) and study follow-up duration are expressed as median, (range), standard deviation (months).
Sex and SMN2 copy number of participants in each SMA phenotypic subgroup and for the total cohort are expressed as n (%).
*Nineteen out of 20 (95%) of children had SMN2 copy number available (missing data for a child with SMA type 2 phenotype).
SMA, spinal muscular atrophy.
Functional motor assessments
Validated assessments for use in children with SMA include the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND), for weak infants or children <2 years of age,17 and Hammersmith Functional Motor Scale/extended version (HFMS/E) for older individuals and/or in those who have demonstrated at minimum, the ability to sit.18 In both assessments, higher scores indicate better motor function. Baseline and final assessments were completed within a month of commencing therapy and completing the study, respectively. Assessments were administered and scored by a senior neuromuscular physiotherapist (KH), trained in using validated SMA motor scores. Clinically significant motor improvement or deterioration was defined as ≥4-point increase or decrease, respectively, in CHOP-INTEND scores10 or ≥3-point increase or decrease respectively in HFSM/E scores.5 Clinical stability was defined as a change in score of <4 in CHOP-INTEND and <3 change in HFSM/E.
Electrophysiological measures obtained using MScanFit MUNE
The protocol established by Jacobsen et al was used to estimate the number of motor units of the abductor pollicis brevis (APB) muscle, innervated by the median nerve.13 The same APB muscle was sequentially tested in each individual.
Each participant’s hand and forearm were cleansed with Nuprep abrasive skin prepping gel (Weaver and Company, Aurora, USA). ECG-type non-polarisable Ag/AgCl surface electrodes (4620M; Unomedical, Birkerød, Denmark) were used for recording with the active electrode placed over the APB muscle belly and the reference electrode placed distally over the tendon insertion at the metacarpophalangeal joint of the thumb. Ground electrodes were placed on the dorsum and palm of the hand. The optimal stimulation site of the median nerve was located at the wrist by identifying the largest electromyograph (EMG) response to a submaximal stimulus, using repositionable bipolar electrodes, before fixing stimulating electrodes (the same as used for recording). Movement of the thumb was limited by taping the digit to a supporting surface.
Stimulation of the median nerve was coordinated by QtracS software (H. Bostock, Institute of Neurology, University College London, UK) with the TRONDNF protocol, using a data acquisition system (PCI-6221; National Instruments, Austin, Texas, USA). The data acquisition system provided the command signals for a constant-current simulator (DS5, Digitimer, Welwyn Garden City, UK). Measurements were amplified and filtered using a purpose built low noise (amplifier gain × 250; filter 2 Hz–2 kHz)19 before removal of mains frequency noise in line with a 50 Hz noise eliminator (Humbug; Quest Scientific, Vancouver, Canada) and digitised at 10 kHz with the data acquisition system. Temperature of the forearm was maintained at or above 32°C.
The Muscle Scan protocol uses 0.2 ms wide rectangular current pulses delivered at 2 Hz and consists of three parts: a prescan, the scan itself and a postscan. The prescan and postscan are used in the muscle scan analysis to estimate CMAP variability.
The prescan records the EMG response to 20 supramaximal stimuli. Following the prescan, the CMAP scan automatically decreased the stimulus intensity in a logarithmic fashion by 0.2% of the previous value until no motor response was apparent. This subminimal stimulus intensity was used to record 20 postscan CMAPs. These cumulative data generated a detailed stimulus response (SR) curve of motor response against stimulus intensity. The MScanFit module in the Qtrac analysis software used the slope and variance of the points of the recorded SR curve to generate a preliminary model taking into consideration threshold variability and amplitude of all motor units. MScanFit then compared the recorded and model SR curves, making serial optimisations to reduce differences and improve the fit of the model to the recorded scan.20 In addition to maximal CMAP, which is a broad measure of neuromuscular health, the following Muscle Scan measures were derived:
Electrophysiological measures representative of extent of denervation
MUNE: the estimated number of functional motor units.
N50: the estimated number of larger units making up 50%–100% of the amplitude of CMAP (figure 1).
Figure 1.
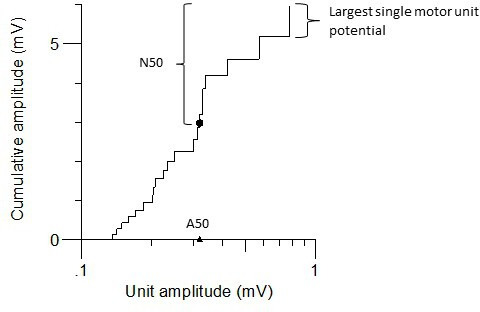
Example of a cumulative amplitude plot generated from MScanFit motor unit number estimation in a patient with spinal muscular atrophy type 1. Units are ranked in order of increasing amplitude. The filled black dot represents 50% of the cumulative amplitude and separates the largest and smallest units. The number of largest units is denoted by number of larger units making up higher 50% of CMAP amplitude (N50) (and makes up 50% of the maximal compound muscle action potential, representing these larger units). The triangle represents the amplitude of the smallest of these N50 units (A50).
Electrophysiological measures representative of collateral reinnervation
1.) A50 (mV): the amplitude of the smallest of the N50 units.
2.) Largest SMUP (mV): the amplitude of the largest unit in the motor unit pool.
Assessments were undertaken at initiation of intrathecal nusinersen therapy (0–1 months) and repeated at intervals corresponding to completion of second induction phase (2 months), first maintenance phase (6–14 months) and second maintenance phase (18–26 months) of the dosing regimen. A reliability coefficient, the interclass correlation coefficient (ICC) was used to measure reliability of all electrophysiological measures. This descriptive statistical measure reflects both degree of correlation and agreement between quantitative measurements. Test–retest pairs of data were collected within the same study visit in four children, with a 60 min break between each recording.
Statistical analysis
Statistical modelling was performed in R statistical software, V.3.6.2 (R Core Team (2019)). Throughout statistical analysis, normal quantile–quantile plots of residuals demonstrated no gross deviations from the normal distribution; residual versus fitted value plots indicated non-uniform variance, which was stabilised by log-transforming outcome variables. For cross-sectional data (at the start of therapy), one-way analysis of variance was used to ascertain differences in electrophysiological values between SMA phenotypes, WHO-derived functional motor status and SMN2 copy number at start of therapy. The Pearson’s correlation coefficient test was used to determine the relationship between baseline electrophysiological values and motor function at start of therapy as assessed on validated SMA functional motor scales. For analysis of the longitudinal data, continuous responses were modelled with a linear mixed model, as there were multiple measurements per patient. Each electrophysiological outcome variable was modelled with explanatory variables. SMA phenotype, functional motor status at start of therapy, SMN2 copy number, disease duration and age of first intervention were all included in the model. Model selections for models with and without interactions were performed using the conditional Akaike information criterion (AIC), owing to the small size of this data set. The AIC enables model selection by estimating the quality of a selected model relative to other candidate models for a given data set.21 Fitted means and 95% CI were obtained and back-transformed values reported. A p value of <0.05 was considered significant. A binary logistic regression model was used to assess change in motor function with treatment (on validated SMA motor scales) against each electrophysiological variable, and fitted probabilities with CIs were obtained.
Results
Clinical characteristics
The study population of 20 participants included 11 male and nine female (table 2). Children were Classification of SMA phenotype for this study adhered to guidelines set out in the International Collaborative SMA Workshop.16 Functionally, prior to the start of treatment, 5 children (25 %) were classified as non-sitters, 13 (65%) as sitters and 2 children (10%) walked either independently or with support.
Electrophysiological measures
Due to the nature of embedding this research into the course of clinical management, the interval between and total number of studies varied among individuals. The mean recording time for each test was 3.4 min, SD 1.4 min. All tests were well tolerated by participants with no need for extraneous sedation/anaesthetic. No side effects were observed from the technique. ICC test of reliability for MUNE and CMAP showed good reliability for all measures. ICC for MUNE was 0.95 (p=0.02) and for CMAP was 0.98 (p=0.002).
Electrophysiological measures and their association with clinical measures of disease severity prior to therapy
There were significant differences in mean CMAP (p=0.03), MUNE (p=0.01) and N50 (p=0.01) between children with different SMA phenotypes, where children with SMA type 3 had the highest number of functional motor units and CMAP values at the start of therapy (figure 2A). Children across the spectrum of SMA phenotypes exhibited large motor units as suggested by the fact that no significant differences between LSMUP (p=0.30) and A50 (p=0.6) existed between phenotypes. There were significant differences in mean values for CMAP (p=0.003), MUNE (p<0.001) and N50 (p=0.001) between functional groups (as derived by WHO motor status at start of therapy). Children who had better functional motor skills (such as ambulatory ability) prior to therapeutic intervention showed lower levels of denervation represented by higher mean CMAP, MUNE and N50 values (figure 2B). Levels of collateral reinnervation were not significantly different among functional subgroups as denoted by LSMUP (p=0.09) and A50 (p=0.3). There were no significant differences in electrophysiological measures between children with different SMN2 copy numbers (CMAP p=0.34; MUNE p=0.22; N50 p=0.24; LSMUP p=0.54 and A50 p=0.18).
Figure 2.

Comparison of electrophysiological values at the start of nusinersen therapy as a parameter of (A) SMA phenotype and (B) functional status. Black dots represent individual values, blue dots represent mean values for the group, error bars demonstrate 95% CIs. A50, amplitude of smallest unit making up N50 (mV); CMAP, compound muscle action potential (mV); LSMUP, largest single motor unit potential (mV); MUNE, motor unit number estimation; N50, number of larger units making up higher 50% of CMAP amplitude.
Longitudinal changes in electrophysiological measures with therapeutic intervention
Therapeutic intervention with nusinersen was an independent and significant contributor to an increase in CMAP (p=0.005), MUNE (p=0.001) and N50 (p=0.04). The extent of compensatory collateral reinnervation did not change with therapy (LSMUP (p=0.99) and A50 (p=0.77)). In contrast, a greater number of smaller units increasingly contributed to the overall CMAP as denoted by increasing differences between MUNE and N50 values. A representative example of electrophysiological changes with treatment from our cohort is shown in figure 3. During the first 2 months of therapy, there were no significant changes in any electrophysiological outcome measure (figure 4). MUNE was the initial electrophysiological measure to show a significant response, with increases noted after 6 months of treatment, (p=0.02), continuing without plateau for the duration of the study (figure 4). Concomitantly, a significant increase in CMAP was observed between the start and 18 months of therapy (p=0.001). The largest single motor unit potential did not change significantly over the therapeutic course (p=0.99).
Figure 3.
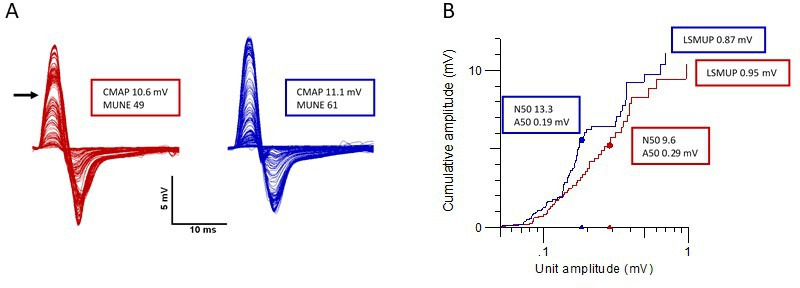
MScanFit MUNE analysis of a 10-year-old child treated with nusinersen. An example of the change in CMAP scan and its MScanFit analysis prior to treatment (red) and after 6 months of nusinersen therapy (blue). Panel A shows all the CMAP responses in response to the fine-graded stimuli delivered during a CMAP scan and panel B represents a cumulative amplitude plot of model units, ranked in order of increasing amplitude. Panel A: With treatment, CMAP and motor unit number increase. An irregular CMAP scan is noted with the horizontal arrow denoting a large motor unit secondary to collateral reinnervation at the start of treatment. This effect is ameliorated after commencement of nusinersen therapy. Panel B: With treatment, the number of units (N50 and MUNE) increases, the size of the largest single motor unit (LSMUP) stays constant and the size of the smallest of the N50 units (A50) does not significantly change. A50, amplitude of smallest unit making up N50 (mV); CMAP, compound muscle action potential (mV); LSMUP, largest single motor unit potential (mV); MUNE, motor unit number estimation; N50, number of larger units making up higher 50% of CMAP amplitude.
Figure 4.
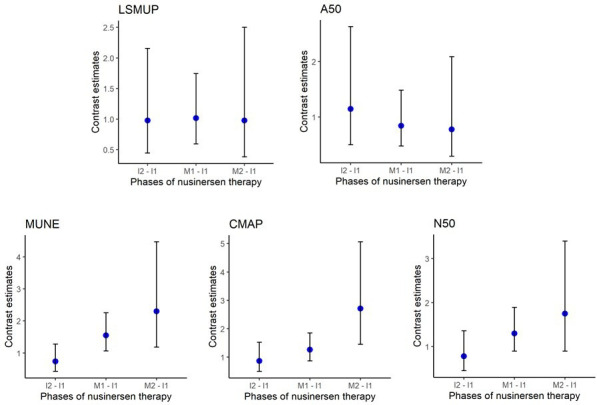
Electrophysiological changes between phases of nusinersen therapy for the cohort. Blue dots represent differences in means among later phases of nusinersen therapy in comparison to the induction phase (contrast estimates). Whiskers represent 95% CIs. Nusinersen phase equivalent to I1=first induction phase (0–1 months), I2=second induction phase (2 months), M1=first maintenance phase (6–14 months) and M2=second maintenance phase (18–26 months) of nusinersen dosing regimen. A50, amplitude of smallest unit making up N50; CMAP, compound muscle action potential; LSMUP, largest single motor unit potential; MUNE, motor unit number estimation; N50, number of larger units making up higher 50% of CMAP amplitude.
Clinical factors and their effects on the magnitude of electrophysiological response with treatment
Disease duration and therapeutic intervention in combination significantly modified MUNE (p=0.003), CMAP (p=0.04) and N50 (p=0.03) over time. Changes in MUNE secondary to therapeutic intervention, negatively correlated with disease duration. A decrease of MUNE (7%), CMAP (6%) and N50 (8%), respectively, was observed for every six additional months of disease duration. This clinical factor had no impact on LSMUP (p=0.06) or A50 (p=0.07) over time.
Age of intervention and therapeutic intervention in combination significantly modified the electrophysiological response to therapy MUNE (p=0.001) and N50 (p=0.004). MUNE and N50 values correlated positively, with a rise of 7% and 7.5%, respectively, for every additional 6 months of age at intervention.
SMA phenotype, SMN2 copy number and gross functional status (as derived by WHO assessment) at the start of therapy did not have a significant association with electrophysiological measures seen over the study period (p>0.05).
Electrophysiological measures and their association with change in motor function
Out of the cohort who underwent longitudinal electrophysiological studies, two patients, both categorised with SMA type III, did not tolerate functional assessment at the end of the study period. Of the 18 children who completed functional motor assessments, the majority remained clinically stable (n=12, 67%) or showed clinically significant improvements (as validated by change in the HFSME/CHOP-INTEND scores) in motor scores (n=6, 33%). There was a trend towards an association between increasing CMAP, MUNE and N50 values and probability of (previously defined) clinically significant improvement on validated SMA motor scales. Although none of the children in our cohort demonstrated a magnitude of electrophysiological change large enough to confirm the significance of this trend (MUNE p=0.34), (CMAP p=0.69) and (N50 p=0.35), our analysis predicted that children with an increase in CMAP≥4.5 mV or an increase in MUNE≥15 units from baseline were more likely to attain significant improvements in motor function on validated SMA scales, with treatment. Children who had an increase in N50 of ≥4 units were more likely to have a clinically significant improvement in motor function with treatment, than be classified as clinically stable.
Interpretation
This study describes the pathophysiological status of the motor unit prior to therapeutic intervention in children with symptomatic SMA, the biophysical responses to SMN repletion, detailing the limits of the ‘therapeutic window’ and characteristics of the ‘therapeutic cohort’. Our study is the first to show that commencement of antisense oligonucleotide therapy halts disease-related axonal loss, altering the established degenerative electrophysiological trajectory in untreated children.22
This study uniquely denotes that motor unit number increases soon after therapeutic induction. Biological effects continue throughout maintenance with preferential restoration of small motor units to functionality. Electrophysiological measures correlate with functional change, highlighting the capacity of MUNE measures to act as biomarkers of therapeutic response. Our findings form the foundation for the goal of precision medicine and may, in the future, have broader utility in tracking treatment response in adult-onset motor neuropathy.
Prior to treatment, children who have a higher burden of disease (characterised by SMA phenotype and/or lower functional motor status), have lower CMAP, MUNE and N50, reflecting comparatively higher levels of neuronal degeneration. Our findings mirror natural history studies showing increased levels of motor neuron loss in children with an SMA type 1 phenotype, compared with those with later and milder onset disease,22 while adding knowledge by suggesting similar associations between neuronal loss and motor function.
Our study finds that disease severity in SMA appears more closely associated with the extent of denervation, with surviving motor units across SMA phenotypes and functional groups similarly mechanistically compensated, with comparative LSMUP and A50 values. This has been demonstrated in natural history studies where a threefold increase in motor unit size appeared functionally inadequate to compensate for a 90% disease-related reduction in motor unit number.23
The present study demonstrates the differing electrophysiological trajectory in treated, symptomatic children compared with historically untreated counterparts. In the latter, early and precipitous motor unit number and corresponding CMAP decline is the main mechanism of disease.22 In contrast, at a biological level, our study suggests that nusinersen leads to cessation of disease-related denervation, remodels the motor unit pool, and is hence a true disease-modifying agent.
In contrast to other (infectious and demyelinating) motor neuropathies that show an increasing degree of collateral reinnervation as the major contributor to CMAP restoration during illness recovery,24 we observe that axonal sprouting has less of a mechanistic role to play in the therapeutic response. This is possibly due to motor unit size being maximally compensated in the process of chronic denervation, as has been electrophysiologically captured in SMA23 and other neuropathic disease processes such as poliomyelitis.25
Although small and large units simultaneously recover with intervention, smaller, units recover first and preferentially, denoted by a more significant rise in MUNE (total number of functional motor units) compared with N50 (number of larger motor units). Our study is the first to replicate in a clinical cohort, preclinical models of SMN repletion26 and increased resilience of small motor units.27
Our findings corroborate clinical trial data demonstrating increase and maintenance of CMAP in treated children with infantile onset disease (known as CMAP responders) compared with untreated patients.10 In contrast to outcomes from clinical trials in later-onset forms of SMA showing only small increments or decrements in CMAP and/or MUNE in children with SMA type 2 and 3 respectively,11 this electrophysiological response is noted across the spectrum of our heterogeneous cohort, independently of other clinical factors. Discrepancies in observations between studies may be due to the motor unit number estimation technique, measurement of median nerve pathology (ulnar nerve was tested in the clinical trial) and variation in baseline demographics.
Nusinersen exerts its biological effect early on. Motor unit number improves soon after the first 2 months of therapy, consistent with preclinical literature showing termination of motor neuron loss in the first phases of SMN restoration.28 An increase in functional motor unit number continues (>18 months) without reaching a plateau phase, mirroring outcomes from clinical trials that shows ongoing motor gains over the first 2 years of therapy.29 Our findings may indicate that premature cessation of intervention due to lack of functional benefit could interrupt an ongoing motor unit innervation process, which leads to clinical benefit in the longer term.
Our study gives rise to several hypotheses surrounding the mechanism of motor unit number restoration with treatment. Although MScan Fit MUNE takes into account threshold variability of all functional motor units30 and is a sensitive measure of motor neuron loss in adult diseases,31 challenges remain (as with all MUNE techniques) in detecting the smallest motor units with a non-invasive methodology. With treatment, these very small motor units may enlarge in size and are captured as they surpass the threshold for detection.
Axonal regeneration has been observed in acute (diabetic and chemotoxic) peripheral neuropathies when environmental insults are withdrawn32 and may represent a distinct biomechanistic pathway for motor unit number restoration. However, structural reinnervation has not previously been described in preclinical models of SMN repletion and chronic denervation leading to chronic axotomisation,(as seen in SMA) is known to especially inhibit axonal regeneration.33
A return of function to pre-existing ‘dysfunctional’ axons possibly at the cusp of degeneration is another postulated process by which chronically denervated motor units respond to SMN2 enhancing therapies.34 Our findings may support emerging biological concepts of reversible pathophysiology in affected neurones including restoration of neuromuscular junction function,35 improved synaptic and neuronal cell trafficking,36 and/or reversal of axonal conduction block,37 with commencement of therapy.
One further potential interpretation of our data is that an increase in motor unit number is secondary to a developmental phenomenon; however, prior (limited) data for healthy young subjects show that motor unit number appears stable from the neonatal period to adulthood.38 Furthermore, SMA has an established natural history of denervation, such that developmental phenomenon would not be expected in a longitudinal study in patients with SMA. Preclinical models suggest that SMA is a developmental as well as neurodegenerative pathology.39 An ‘unlocking’ and restoration of halted, disease-related neuronal development after repletion of SMN levels may underpin our electrophysiological findings. Future studies using MScan Fit MUNE methodology in age-sex matched healthy children are required to delineate this further. Excitability studies exploring biophysical properties of the motor unit in symptomatic and healthy controls may also provide a more in-depth understanding of the biomechanisms behind our results.
Clinical factors that predict treatment response on a neurophysiological level relate strongly to disease duration and age of intervention. Traditional clinical determinants of disease severity such as phenotype and SMN2 copy number do not modulate electrophysiological sequalae significantly.
Children with a longer symptomatic phase of disease have a reduced capacity to increase their functional motor unit number with treatment. The electrophysiological sequalae mirror outcomes across clinical trial and real-world settings that observe greater improvements in survival and motor outcomes in patients who are treated earlier in their disease course.10 Our findings contribute to the expanding clinical evidence base that promotes early (and potentially presymptomatic) intervention with disease-modifying agents to halt and potentially reverse rapid disease-associated motor neuron loss.
Clinical trial outcomes show clear benefit of therapy in young children with severe forms of SMA10 with less clear cut outcomes associated with other SMA phenotypes. Our study has demonstrated that the ‘therapeutic cohort’ is more heterogeneous than originally anticipated; children who are beyond the age of expected developmental gains (ie, older children/adolescents and young adults who are still ambulant) maintain a significant capacity for functional motor unit innervation with therapy and may accrue clinical benefit from nusinersen therapy. Thus, the therapeutic window is such that a broad range of patients will benefit from therapy, with patient-reported outcomes emphasising that stability is important, in the context of a neurodegenerative disease. Our findings emphasise the need to provide active intervention with allied therapy to preserve joint range, nutritional status and muscle mass as a standard of care among symptomatic children, so that the biological potential of these therapeutics can be fully harnessed.
The present study starts to explore electrophysiological parameters as biomarkers of disease severity and treatment response. Our findings propose that change in CMAP, MUNE and N50 have potential to predict individuals who are functionally stable, from those who attain clinically significant gains in motor function, with treatment. This parallels strong correlations observed between improving functional motor scales and rising CMAP values after intervention with disease-modifying agents in SMA.10 MScanFit MUNE studies may be similarly useful in categorising disease severity and therapeutic response in adult-onset motor neuropathies, including in adult-onset SMA.
The development and utility of biomarkers of disease severity and treatment response in a paediatric population are limited by concerns surrounding safety, tolerability and replicability of methods used to obtain these measures.6 Our study suggests that MScan Fit MUNE overcomes many of these barriers; the methodology is non-invasive, feasible, reproducible, tolerated (no child required extraneous sedation to complete the study) and quick to administer in children in a real-world setting. Although still considered a research tool, our study suggests that this automated technique may be adopted at the bedside to individualise treatment regimens for children with SMA. Future clinical trials incorporating MScanFit MUNE as an exploratory outcome utilising larger symptomatic cohorts and longer term follow-up are warranted to assess the wider applicability of this methodology.
Although classically thought of as a disease of proximal muscles, distal muscles are also severely affected across the phenotypic spectrum in SMA40 and have been the preferred target of investigation in clinical trials in SMA.11 We chose to study the APB muscle due to its accessibility in paediatric patients, and its importance as the most intact41 and clinically important distal muscle (for preservation of hand function) in children with SMA. The response to therapeutic intervention in other muscle groups requires further evaluation.
Our study used motor scales that have been validated as functionally meaningful for children with SMA.17 18 There is a paucity of (clinically meaningful), upper-limb-specific functional scales and strength measures that can be feasibly used and standardised across heterogeneous populations as seen in our cohort. The concurrent development of scales that are deemed functionally important and that meet the needs of a real-world population is necessary to fully interrogate the role of electrophysiological measures as clinically meaningful biomarkers of treatment response.
Footnotes
Twitter: @imichellefarrar
Contributors: DK and MAF planned the manuscript. MAF, DK, AD and CS-YL collated electrophysiological and clinical parameters during the study. DK and AD executed and prepared the first and subsequent drafts of the manuscript. KH completed all functional motor assessments. PG-S, AD and DK completed statistical analysis of the data. JH, CS-YL and MAF contributed to figures and interpretation of results. DK, MAF, AD, CS-YL, PG-S and KH contributed to manuscript revision. All authors read and approved the submitted version.
Funding: DK acknowledges funding from the RTP Scholarship, University of New South Wales and The Freedman Family Foundation Scholarship, Sydney Children’s Hospital Foundation Trust. AD acknowledges funding from the Sydney Children’s Hospital Foundation.
Competing interests: MAF has previously received compensation as a member of the scientific advisory board for Biogen, who manufactures the medication that is tested in this study. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Patient consent for publication: Parental/guardian consent obtained.
Ethics approval: The study was approved by the Sydney Children’s Hospital Network and South Eastern Sydney and Illawarra Area Health Service Human Research Ethics Committee (HREC/18/SCHN/373).
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: Data are available upon reasonable request. The data central to this study is available on request from suitably qualified researchers.
References
- 1. Awano T, Kim J-K, Monani UR. Spinal muscular atrophy: journeying from bench to bedside. Neurotherapeutics 2014;11:786–95. 10.1007/s13311-014-0293-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dubowitz V. Ramblings in the history of spinal muscular atrophy. Neuromuscul Disord 2009;19:69–73. 10.1016/j.nmd.2008.10.004 [DOI] [PubMed] [Google Scholar]
- 3. Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995;80:155–65. 10.1016/0092-8674(95)90460-3 [DOI] [PubMed] [Google Scholar]
- 4. Wurster CD, Ludolph AC. Antisense oligonucleotides in neurological disorders. Ther Adv Neurol Disord 2018;11:1756286418776932. 10.1177/1756286418776932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus sham control in Later-Onset spinal muscular atrophy. N Engl J Med 2018;378:625–35. 10.1056/NEJMoa1710504 [DOI] [PubMed] [Google Scholar]
- 6. Kariyawasam DST, D'Silva A, Lin C, et al. Biomarkers and the development of a personalized medicine approach in spinal muscular atrophy. Front Neurol 2019;10:898. 10.3389/fneur.2019.00898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kariyawasam D, Alexander IE, Kurian M, et al. Great expectations: virus-mediated gene therapy in neurological disorders. J Neurol Neurosurg Psychiatry 2020;91:849–60. 10.1136/jnnp-2019-322327 [DOI] [PubMed] [Google Scholar]
- 8. McComas AJ. 1998 ISEK Congress Keynote Lecture: motor units: how many, how large, what kind? International Society of electrophysiology and Kinesiology. J Electromyogr Kinesiol 1998;8:391–402. 10.1016/s1050-6411(98)00020-0 [DOI] [PubMed] [Google Scholar]
- 9. Vucic S, Rutkove SB. Neurophysiological biomarkers in amyotrophic lateral sclerosis. Curr Opin Neurol 2018;31:640–7. 10.1097/WCO.0000000000000593 [DOI] [PubMed] [Google Scholar]
- 10. Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med 2017;377:1723–32. 10.1056/NEJMoa1702752 [DOI] [PubMed] [Google Scholar]
- 11. Darras BT, Chiriboga CA, Iannaccone ST, et al. Nusinersen in later-onset spinal muscular atrophy: long-term results from the phase 1/2 studies. Neurology 2019;92:e2492–506. 10.1212/WNL.0000000000007527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Daube JR. Motor unit number estimates--from A to Z. J Neurol Sci 2006;242:23–35. 10.1016/j.jns.2005.11.011 [DOI] [PubMed] [Google Scholar]
- 13. Jacobsen AB, Bostock H, Tankisi H. CMAP Scan MUNE (MScan) - A Novel Motor Unit Number Estimation (MUNE) Method. J Vis Exp 2018;7:56805. 10.3791/56805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sleutjes BTHM, Wijngaarde CA, Wadman RI, et al. Assessment of motor unit loss in patients with spinal muscular atrophy. Clin Neurophysiol 2020;131:1280–6. 10.1016/j.clinph.2020.01.018 [DOI] [PubMed] [Google Scholar]
- 15. Mercuri E, Finkel RS, Muntoni F, et al. Diagnosis and management of spinal muscular atrophy: Part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord 2018;28:103–15. 10.1016/j.nmd.2017.11.005 [DOI] [PubMed] [Google Scholar]
- 16. Munsat T. Workshop report. International SMA collaboration 1991;1:81. [Google Scholar]
- 17. Glanzman AM, Mazzone E, Main M, et al. The children's Hospital of Philadelphia infant test of neuromuscular disorders (CHOP INTEND): test development and reliability. Neuromuscul Disord 2010;20:155–61. 10.1016/j.nmd.2009.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Main M, Kairon H, Mercuri E, et al. The Hammersmith functional motor scale for children with spinal muscular atrophy: a scale to test ability and monitor progress in children with limited ambulation. Eur J Paediatr Neurol 2003;7:155–9. 10.1016/S1090-3798(03)00060-6 [DOI] [PubMed] [Google Scholar]
- 19. Howells J, Bostock H, Park SB, et al. Tracking small sensory nerve action potentials in human axonal excitability studies. J Neurosci Methods 2018;298:45–53. 10.1016/j.jneumeth.2018.02.003 [DOI] [PubMed] [Google Scholar]
- 20. Bostock H. Estimating motor unit numbers from a CMAP scan. Muscle Nerve 2016;53:889–96. 10.1002/mus.24945 [DOI] [PubMed] [Google Scholar]
- 21. Akaike H. Information theory as an extension of the maximum likelihood principle : Petrov BN, Csaki F, Second International Symposium on information theory. Budapest: Akademiai Kiado, 1973: 267–81. [Google Scholar]
- 22. Swoboda KJ, Prior TW, Scott CB, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol 2005;57:704–12. 10.1002/ana.20473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Galea V, Fehlings D, Kirsch S, et al. Depletion and sizes of motor units in spinal muscular atrophy. Muscle Nerve 2001;24:1168–72. 10.1002/mus.1128 [DOI] [PubMed] [Google Scholar]
- 24. Bickerstaffe A, van Dijk JP, Beelen A, et al. Loss of motor unit size and quadriceps strength over 10 years in post-polio syndrome. Clin Neurophysiol 2014;125:1255–60. 10.1016/j.clinph.2013.11.003 [DOI] [PubMed] [Google Scholar]
- 25. Wiechers DO. Acute and latent effect of poliomyelitis on the motor unit as revealed by electromyography. Orthopedics 1985;8:870–2. [DOI] [PubMed] [Google Scholar]
- 26. McGovern VL, Iyer CC, Arnold WD, et al. SMN expression is required in motor neurons to rescue electrophysiological deficits in the SMNΔ7 mouse model of SMA. Hum Mol Genet 2015;24:5524–41. 10.1093/hmg/ddv283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sharp PS, Tyreman N, Jones KE, et al. Crush injury to motor nerves in the G93A transgenic mouse model of amyotrophic lateral sclerosis promotes muscle reinnervation and survival of functionally intact nerve-muscle contacts. Neurobiol Dis 2018;113:33–44. 10.1016/j.nbd.2018.01.019 [DOI] [PubMed] [Google Scholar]
- 28. Arnold WD, Porensky PN, McGovern VL, et al. Electrophysiological biomarkers in spinal muscular atrophy: preclinical proof of concept. Ann Clin Transl Neurol 2014;1:34–44. 10.1002/acn3.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kirschner J, Darras B, Farrar M, et al. P.352Interim report on the safety and efficacy of longer-term treatment with nusinersen in later-onset spinal muscular atrophy (SMA): results from the shine study. Neuromuscular Disorders 2019;29:S184 10.1016/j.nmd.2019.06.514 [DOI] [Google Scholar]
- 30. Jacobsen AB, Kristensen RS, Witt A, et al. The utility of motor unit number estimation methods versus quantitative motor unit potential analysis in diagnosis of ALS. Clin Neurophysiol 2018;129:646–53. 10.1016/j.clinph.2018.01.002 [DOI] [PubMed] [Google Scholar]
- 31. Kristensen RS, Bostock H, Tan SV, et al. MScanFit motor unit number estimation (MScan) and muscle velocity recovery cycle recordings in amyotrophic lateral sclerosis patients. Clin Neurophysiol 2019;130:1280–8. 10.1016/j.clinph.2019.04.713 [DOI] [PubMed] [Google Scholar]
- 32. Landowski LM, Dyck PJB, Engelstad J, et al. Axonopathy in peripheral neuropathies: mechanisms and therapeutic approaches for regeneration. J Chem Neuroanat 2016;76:19–27. 10.1016/j.jchemneu.2016.04.006 [DOI] [PubMed] [Google Scholar]
- 33. Fu SY, Gordon T. Contributing factors to poor functional recovery after delayed nerve repair: prolonged axotomy. J Neurosci 1995;15:3876–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McComas AJ, Sica RE, Campbell MJ. "Sick" motoneurones. A unifying concept of muscle disease. Lancet 1971;1:321–6. 10.1016/s0140-6736(71)91045-2 [DOI] [PubMed] [Google Scholar]
- 35. Paez-Colasante X, Seaberg B, Martinez TL, et al. Improvement of neuromuscular synaptic phenotypes without enhanced survival and motor function in severe spinal muscular atrophy mice selectively rescued in motor neurons. PLoS One 2013;8:e75866. 10.1371/journal.pone.0075866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Swoboda KJ. SMN-targeted therapeutics for spinal muscular atrophy: are we smart enough yet? J Clin Invest 2014;124:487–90. 10.1172/JCI74142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Farrar MA, Vucic S, Lin CS-Y, et al. Dysfunction of axonal membrane conductances in adolescents and young adults with spinal muscular atrophy. Brain 2011;134:3185–97. 10.1093/brain/awr229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gawel M, Kostera-Pruszczyk A, Lusakowska A, et al. Motor unit loss estimation by the multipoint incremental MUNE method in children with spinal muscular atrophy--a preliminary study. Neuromuscul Disord 2015;25:216–21. 10.1016/j.nmd.2014.11.012 [DOI] [PubMed] [Google Scholar]
- 39. Vrbová G, Sławińska U. Critical period of neuromuscular development: importance for a new treatment of SMA. Neuromuscul Disord 2018;28:385–93. 10.1016/j.nmd.2018.03.007 [DOI] [PubMed] [Google Scholar]
- 40. Bromberg MB, Swoboda KJ. Motor unit number estimation in infants and children with spinal muscular atrophy. Muscle Nerve 2002;25:445–7. 10.1002/mus.10050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Günther R, Neuwirth C, Koch JC, et al. Motor unit number index (MUNIX) of hand muscles is a disease biomarker for adult spinal muscular atrophy. Clin Neurophysiol 2019;130:315–9. 10.1016/j.clinph.2018.11.009 [DOI] [PubMed] [Google Scholar]