

Abstract
Hyriopsis cumingii is an important species for freshwater pearl cultivation in China. In terms of pearl production, males have larger pearls and better glossiness than females, but there are few reports focusing on the sex of H. cumingii. In this study, six mRNA and six microRNA (miRNA) libraries were prepared from ovaries and testes. Additionally, 28,502 differentially expressed genes (DEGs) and 32 differentially expressed miRNAs (DEMs) were identified. Compared with testis, 14,360 mRNAs and 20 miRNAs were up-regulated in ovary, 14,142 mRNAs and 12 miRNAs were down-regulated. In DEGs, the known genes related to sex determinism and/or differentiation were also identified, such as DMRT1, SOX9, SF1 for males, FOXL2 for females, and other potentially significant candidate genes. Three sex-related pathways have also been identified, which are Wnt, Notch, and TGF-beta. In 32 DEMs, the three miRNAs (miR-9-5p, miR-92, miR-184) were paid more attention, they predicted 28 target genes, which may also be candidates for sex-related miRNAs and genes. Differential miRNAs target genes analysis reveals the pathway associated with oocyte meiosis and spermatogenesis. Overall, the findings of the study provide significant insights to enhance our understanding of sex differentiation and/or sex determination mechanisms for H. cumingii.
Subject terms: RNA sequencing, Sequencing
Introduction
Hyriopsis cumingii, a kind of bivalve mollusk, its pearl is known by the smooth appearance and bright color1. It is a crucial freshwater pearl species with significantly economic in China. Studies have shown that the pearl rearing performance of H. cumingii is different between 3 and 4 years old females and males, such as the total weight, grain weight, and particle size of pearls. The average pearl weight of male mussels is 12.4–17.5% higher than that of females. Besides, The average grain weight is 13.5–17.9% higher, and the average grain size is 4.4–5.4% higher2. Obviously, the male H. cumingii has better pearl breeding performance. Hence, sex is an important factor affecting the yield and quality of the pearl. However, the present studies on sex determination or sex differentiation of bivalve shellfish are very few, which restricts the development of the pearl industry. Therefore, it is indispensable to understand the mechanism of sex determination and/or differentiation in H. cumingii.
To date, there are relatively few researches on the genetic and phenotypic basis of bivalve sex differentiation, especially in the molecular pathway of reproduction. Their reproductive system is different by the hermaphroditic, dioecism, and gender changes. For instance, as we well-known Crassostrea gigas, it is an alternative and irregular protandrous hermaphrodite: most individuals mature first as males and then change sex several times3. Some species are hermaphroditic. For example, Nodipecten subnodosus is a functional hermaphrodite in which male and female gametes mature at the same time4. Additionally, species like Placopecten magellanicus are dioecious5. In recent years, the development of high-throughput sequencing technology has accelerated the identification of transcripts. Studies on gonadal development, sex differentiation and sex determination of bivalve shellfish are increasingly in-depth at the same time. The transcriptome analysis of N. subnodosus illustrated that the genes of the sex determination/differentiation are Ns-SEX1, Ns-DMRTA2, Ns-SOX9, Ns-WNT4, Ns-DOA, Ns-OVO, and Ns-VIR4. In the differential gene analysis of the gonadal transcript of blood clam (Tegillarca granosa), sex-related genes included FOXL2, SOX, β-CATENIN, CBX, and SXL6. In yesso scallop (Patinopecten yessoensis), a hypothetical sex determination and differentiation pathway was constructed, in which PyDMRT1 may have a leading function7. The gonadal transcriptome of Pinctada Margaritifera revealed the importance of pmarg-FOXL2 and pmarg-FEM1-like for sex inversion and sex differentiation, which provided a powerful resource for the molecular mechanism of reproductive strategy in hermaphroditic mollusks8. These results may suggest that genes such as FOXL2, DMRT, SOX can play a role during sex regulation in many species, indicating that their functions are very conservative. MicroRNA is a single-stranded non-coding RNA molecule with a length of about 21–24 nucleotides. And it is a post-transcriptional regulator. miRNAs sequencing has advanced our understanding of miRNAs in biological sex differentiation and gonadal development, and it has been applied in a variety of aquatic species. The sex-biased miRNAs (such as miR-9, miR-21, miR-30a, miR-96, miR-200b, miR-212 and miR-7977) were obtained by a comprehensive analysis of miRNA and mRNA expression profiles in the gonads of tilapia at the early stage of sexual differentiation. Their target genes include FOXL2, AMH, STAR1, SF1 and DMRT1, which are key molecules involved in vertebrate sex differentiation9. Three significant miRNAs (aca-miR-30b-5p, ame-miR-263b and cfa-miR-125a) were screened from Macrobrachium nipponense. They and their predicted target genes may have a strong impact on sex differentiation/determination10. miRNAs were also identified and analyzed during the gonadal development of Macrobrachium rosenbergii and zebrafish11,12. These data will be useful for further study of miRNA mediated gonadal development and reproductive regulation mechanism. In animals, miRNAs regulate target gene expression through degradation or translation inhibition13. The relationship between miRNA and mRNA is not a one-to-one correspondence. A single miRNA can target multiple mRNAs, and one mRNA can also have several miRNAs binding sites14. During gonadal development, miRNAs are expressed differently in male and female gonads. They play a role through germ cells and gonadal cells to regulate critical proteins needed for gonadal development15. Studies have shown that miR-202-5p/3p can be expressed in a sexual dimorphism pattern as the primordial XY gonad differentiates into a testis and play an early role in mouse testis development16. The results of knockdown or overexpression of miR-124 in mouse showed that miR-124 could induce the repression of both SOX9 translation and transcription in ovarian cells17. Therefore, understanding the regulatory relationship between miRNA and sex-determining genes can help us have a better comprehending of the sex-determining mechanism.
Bioinformatics analysis of miRNA and mRNA expression profile can help us improve the reliability of prediction and understand the molecular mechanism of post-transcriptional regulation. Nowadays, there is no information about the interaction network and regulation of mRNAs and miRNAs in the gonads of H. cumingii. Therefore, our goal is to identify the essential genes and miRNAs that regulate sex determination/differentiation of H. cumingii by mRNA sequencing and miRNA sequencing. This study provides basic information for understanding sex differentiation and sex determining mRNAs and miRNAs of H. cumingii.
Results
mRNA sequencing and assembly
The samples were sequenced from the gonads of six H. cumingii, including three ovaries (F1, F2, and F3) and three testes (M1, M2, and M3). A total of 308,008,006 raw reads were produced by Illumina HiSeq X Ten sequencing. And 302,865,864 clean reads filtered by Trimomatic. The average percentage of Q30 and the G + C percentage of the six cDNA libraries were 94.75% and 41.48% (Table 1). After De novo assembly, the number of unigenes is 96,266. The length range is 301–28,485 bp. And the average length is 1101.67 bp. The annotated results of the genes in seven databases are NR 21,790 (22.64%), Swissprot 14,705 (15.28%), KEGG 9573 (9.94%), KOG 11,833 (12.29%), eggNOG 16,525 (17.17%), GO 13,429 (13.95%), Pfam 14,987 (15.57%).
Table 1.
Summary information and analysis of mRNA sequences in H. cumingii.
| Sample | Raw reads | Clean reads | Q30 (%) | GC (%) |
|---|---|---|---|---|
| F1 | 53,449,060 | 52,542,528 | 94.67 | 40.89 |
| F2 | 49,936,082 | 49,120,634 | 94.96 | 40.77 |
| F3 | 50,929,990 | 50,033,610 | 94.83 | 40.67 |
| M1 | 46,700,762 | 45,946,032 | 94.70 | 42.41 |
| M2 | 53,239,136 | 52,374,040 | 94.84 | 43.12 |
| M3 | 53,752,976 | 52,849,020 | 94.50 | 41.04 |
Raw reads: the statistics of the raw reads; Clean reads: remove the contaminated and low quality read; Q30: The ratio of bases with a value greater than 30 to the total bases in raw bases; GC: The ratio of the total number of G and C to the total number of bases in clean bases.
Screening for DEGs and functional annotation
Three female gonadal tissues (F1 ~ F3) were Group_F, and three male gonadal tissues (M1 ~ M3) were Group_M. Compared with Group_M, there were 28,502 DEGs in Group_F, including 14,360 genes up-regulated and 14,142 genes down-regulated. GO annotations consist of three categories: biological processes, cellular components, and molecular functions. DEGs were enriched to 9295 GO terms. Ten terms with P value less than 0.05 and the largest number of DEGs were selected in each of the three categories (Supplementary Fig. 1). KEGG enrichment of DEGs showed that there were 305 pathways, remove the pathways in which the number of DEGs is less than three, sort them according to the − log10 P value, and draw a bubble diagram with the first 20 pathways (Supplementary Fig. 2).
According to sex determining genes of other species18,19 and GO analysis, we screened 12 sex-related genes in H. cumingii transcriptomes (Table 2), including DMRT, TRA, SOX, FOXL genes, etc. GO analysis is to find candidate genes by searching for "sex" keywords in GO terms. The search results include nine GO terms, namely sex determination (GO:0,007,530), sex differentiation (GO:0,007,548), male sex differentiation (GO:0,046,661), male sex determination (GO:0,030,238), female somatic sex determination (GO:0,019,101), sex determination establishment of X:A ratio (GO:0,007,540), sex chromosome (GO:0,000,803) , male germ-line sex determination (GO:0,019,100), and development of primary female sexual characteristics (GO:0,046,545). The DEGs in these terms are listed in Table 2. In the KEGG enrichment results, Wnt (ko04310), Notch (ko04330), and TGF-beta (ko04350) were confirmed to be related to sex determination20. There are 49 DEGs in these three signaling pathways, which may be related to sex regulation of H. cumingii (Supplementary Table 1). Through these methods, we obtained a total of 67 sex-related candidate genes (after removing duplicates). 14 differentially expressed genes were selected for quantitative real-time PCR (qRT-PCR) detection (Supplementary Fig. 3).
Table 2.
List of important genes related to sex determination/differentiation in the H. cumingii transcriptomes.
| Gene | NR id | Name |
|---|---|---|
| SOX9 | AGI96396.1 | SRY-related HMG-domain containing transcription factor 9 |
| DMRT1 | ASV71764.1 | Double-sex and mab-3 related transcription factor 1 |
| FOXL2 | XP_022345405.1 | Forkhead box protein L2 |
| SF1 | XP_011417019.1 | Splicing factor 1 |
| TRA2 | Transformer-2 | |
| BMP2 | OWF43831.1 | Bone morphogenetic protein 2 |
| ZGLP1 | XP_011445224.2 | GATA-type zinc finger protein 1 |
| CYP17A1 | Steroid 17-alpha-hydroxylase/17,20 lyase | |
| DACH2 | XP_021340457.1 | Dachshund homolog 2 |
| DPN | AGS55441.1 | Hairy enhancer of split 7 |
| SMC5 | XP_021362293.1 | Structural maintenance of chromosomes protein 5 |
| SRD5A1 | EKC36980.1 | 3-oxo-5-alpha-steroid 4-dehydrogenase 1 |
miRNAs sequencing and assembly
We constructed six cDNA libraries of small RNAs from F1, F2, F3, M1, M2, and M3. After filtering the Rfam database, transcript sequence, and Repbase database, the reads are compared with miRBase to identify and annotate the known miRNA (Table 3). The number of miRNAs in all samples was 209, including 84 known miRNAs and 125 novel miRNAs.
Table 3.
Summary information and analysis of miRNA sequences in H. cumingii.
| Sample | Raw reads | Clean reads | Reads trimmed Q20 | Known miRNA | Novel miRNA |
|---|---|---|---|---|---|
| F1 | 30,286,657 | 18,147,307 | 18,163,822 | 65 | 81 |
| F2 | 41,577,946 | 29,989,345 | 30,009,397 | 65 | 93 |
| F3 | 36,741,132 | 19,784,177 | 19,811,678 | 66 | 92 |
| M1 | 37,370,743 | 28,974,936 | 28,988,411 | 66 | 88 |
| M2 | 29,870,390 | 18,486,304 | 18,500,540 | 70 | 74 |
| M3 | 37,527,689 | 26,717,472 | 26,755,846 | 74 | 85 |
Raw reads: the statistics of the raw reads; Clean reads: remove the contaminated and low quality read; Reads trimmed Q20: reserve the reads of the percent of Q20 > 80%.
Screening for Differentially-expressed miRNAs (DEMs) and functional annotation
Compare with miRNA libraries of Group_F vs. Group_M, 32 DEMs were obtained, including 13 known miRNAs and 19 novel miRNAs (Supplementary Table 2). Among the known miRNAs, three were ovary-biased, and ten were testis-biased. Unsupervised hierarchical clustering groups the samples in two major clusters (Fig. 1). The number of target genes of 32 DEMs are 911. The functional terms cytoplasm, ATP binding, and nucleoplasm, had a higher number of transcripts than other terms (Supplementary Fig. 4). KEGG analysis showed that the target genes were enriched in 184 signaling pathways. The largest number of target genes is found in cell cycle (Supplementary Fig. 5). To study the expression pattern of these DEMs in testis and ovary, we randomly selected three ovary-biased miRNAs (miR-184, miR-281-3p, miR-315), and five testis-biased miRNAs (miR-153, miR-29, miR-7, miR-9-5p, miR-92) to verify their expression profiles by qRT-PCR (Fig. 2). All of these miRNAs expression patterns are consistent with the results of miRNAs sequencing, indicating that the sequencing results are reliable.
Figure 1.

Heatmap plots for differentially expressed miRNAs between Group_F and Group_M. Sample names are represented in columns and significant miRNAs are represented in rows. miRNAs are clustered together based on their expression similarity. The color indicates the Z_score from high (red) to low (blue).
Figure 2.
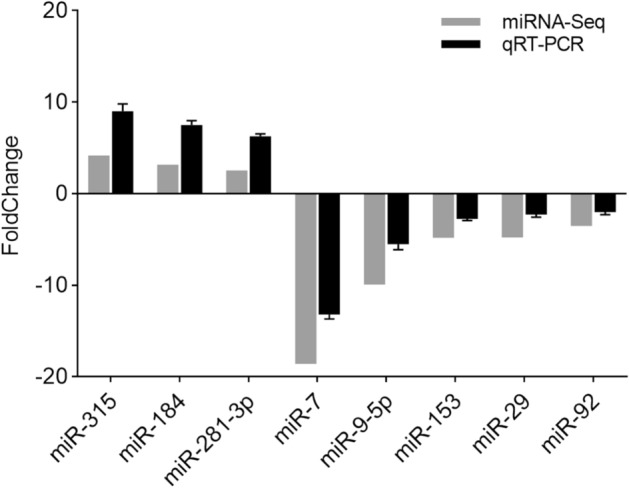
The relative expression of different miRNAs revealed by real-time quantitative PCR. Data are shown as mean ± SD (standard deviation) of tissues from three separate individuals. The figure is drawn by GraphPad Prism (version 8.0.1) software (https://www.graphpad.com/).
Integrated analysis of DEMs and DEGs
According to the results of transcriptome sequencing and miRNAs sequencing, we found three key miRNAs (miR-9-5p, miR-92, miR-184) and their target genes (Table 4). These genes may be related to the sex regulation of H. cumingii. Some of them have been reported to relates to sex. For example, REX1 is associated with X-chromosome inactivation in mice21. SMAD4 is essential factors that regulate the female fate of human germ cells22. MCM6 was identified and considered as candidate genes that may be involved in sex differentiation regulation in cucumber23. CDH5 is necessary for spermatogenesis and chromatin concentration in mice24. Besides, 361 differentially-expressed target genes were obtained from the intersection of 28,502 DEGs and 911 target genes (Fig. 3a). GO analysis showed that there were more transcripts in the cytoplasm, ATP binding, calcium ion binding, endoplasmic reticulum membrane, and microtubule (Fig. 3c). In the biological process, there is a term for spermatogenesis (GO:0,007,283), in which the genes are IFT81, LRGUK, SYNE1, TDRD1. KEGG analysis showed that 361 differentially-expressed target genes were mapped to 113 pathways. There are many genes involved in the pathways of cell cycle, viral carcinogenesis, and regulation of actin cytoskeleton (Fig. 3b). And there is an oocyte meiosis signal pathway (ko04114), which contained three genes: PPP1C, PKMYT, and CCNE. These genes were randomly selected for qRT-PCR verification, SYNE1 and TDRD1 were highly expressed in the ovary, and IFT81, PPP1C, PKMYT, and LRGUK were highly expressed in testis (Supplementary Fig. 3). The corresponding miRNAs of the seven genes (IFT81, LRGUK, SYNE1, TDRD1, PPP1C, PKMYT, and CCNE) related to spermatogenesis and oocyte meiosis are novel31, novel85, miR-71-5p, miR-750-3p, novel31, novel85, novel86, respectively.
Table 4.
The predicted target genes of miR-9-5p, miR-92 and miR-184.
| miRNA | Targeted genes | NR id |
|---|---|---|
| miR-9-5p |
BLM, GATD3A, REX1, CHIA |
|
| miR-92 |
RDH16, HEXA_B, SMAD4, LRP4, HM20A, MCM6, PNKP, ADAMTS18, CG9801, MOB1, MUT, MARK1 |
XP_014596438.1, XP_015772465.1, XP_021379469.1, XP_019918967.1, XP_011423614.1, XP_021360010.1, XP_022344800.1, OWF36531.1, OWF39070.1, XP_021349454.1, XP_019920763.1, XP_021360442.1 |
| miR-184 |
SVEP1, ZASP52, LRCH3, CHD5, SESN1, BCKDK, POL, ATP6V1FNB, MUS81, TEAD, LRCH3, PAXBP1 |
XP_021344288.1, KOF67579.1, XP_021371746.1, XP_025081588.1, XP_021352570.1, XP_014784793.1, |
Figure 3.

GO and KEGG analysis of differential target genes. (a) Integrate analysis of mRNA and miRNA. Venn diagram representing the intersection between differentially expressed genes (DEGs) and target genes of the differentially expressed miRNAs (DEMs) in the Group_F and Group_M. (b) Bubble diagramv of KEGG enrichment analysis. (c) GO enrichment analysis.
Discussion
In order to explore the mRNAs and miRNAs involved in sex differentiation and/or sex determination of H. cumingii, RNA-seq and miRNA-seq analyses were performed on ovaries and testes. To the best of our knowledge, this is the first report of miRNA and mRNA profiling of ovary and testis in H. cumingii. We analyzed miRNAs and mRNAs to provide a basis for screening candidate miRNAs and sex-related genes. This result provides an essential insight into the mechanism of sex differentiation/determination in H. cumingii.
Since there are few studies focusing on the sex of bivalve shellfish, to find candidate genes related to sex determination, we refer to other species genes that have been identified, which has important reference value for our research. After screening the transcriptome results, we obtained seven DEGs as SOX9, DMRT1, FOXL2, SF1, TRA2, BMP2 and ZGLP1. In general, SOX9 is a direct target of SRY gene in mammals and is necessary for normal testicular development25. The conservative expression of SOX9 in chicken and mouse indicates its primary role in the testicular determination of vertebrates26. SOX9 was also identified in bivalve shellfish, such as N. subnodosus, C. gigas, and Hyriopsis schlegelii4,27,28. In our results, miranda software predicted that there was an interaction site between miR-193 and SOX9. Also, SOX9 was highly expressed in testis (Supplementary Fig. 3). These results suggest the potential role of SOX9 and miR-193 in the sex of H. cumingii. Unfortunately, due to the limitation of miRNA sequencing information, other genes were not predicted by miRNAs except SOX9. Apart from that, DMRT (double sex/male-normal-3-related transcription factor) gene is a major transcription factor controlling sex determination and differentiation29. DMRT1 can not only control testicular differentiation and meiosis of male germ cells in mice30,31. But also be necessary for male sexual development in zebrafish32. In mollusks, the homologous gene of DMRT has been identified in many species, such as C. gigas, P. margaritifera, and P. yessoensis33–35. In H. cumingii, the qRT-PCR results showed that DMRT1 gene was a testis specific gene, and its expression level in the ovary was very low, and the expression level in mRNA sequencing results was not detected (Supplementary Fig. 3). It was suggested that DMRT1 was crucial for the male sexual development of H. cumingii. FOXL2, a member of the forkhead box (Fox) domain transcription factor family, plays a key role in ovarian differentiation and oogenesis in vertebrates36. FOXL2 can affect ovarian development and sex determination of mice37. In C. farreri and P. yessoensis, FOXL2 expression was a sexually dimorphic pattern, and the expression level in ovary was significantly higher than that in testis38,39. In our results, FOXL2 was highly expressed in the ovary, and the expression abundance in the testis was very low. The results of mRNA sequencing also showed that FOXL2 was not expressed in testis (Supplementary Fig. 3). It is suggested that FOXL2 is crucial for the female sexual development of H. cumingii. SF1 (Steroidogenic factor 1) is an important transcription factor involved in steroidogenesis, reproduction, and sexual differentiation40. The TRA2 gene, encoding a protein (TRA-2) which directs sex-specifically alternative splicing, has been proved to play important roles in sex differentiation and sex development of Drosophila melanogaster41. BMP2 (Bone morphogenetic protein 2) has been confirmed to be related to the fetal ovarian development and gonadal somatic cell differentiation of mice42,43. ZGLP1 (GATA-type zinc finger protein 1) is essential for the oogenic program and meiotic entry in mice44. In addition to the above genes, GO analysis also enriched some extra sex determination/differentiation genes, they are CYP17A1, DACH2, DPN, SMC5, and SRD5A1. CYP17A1 has the activity of 17α-hydroxylase and 17, 20-lyase, which is involved in the steroidogenic pathway that produces androgens and estrogens, it is crucial for male determination in amphibians45. DACH is differentially expressed in the male and female genital discs of drosophila, and plays sex-specific roles in the developmenting genitalia46. And DPN is also a primary part of sex determining signal of drosophila47. SMC5 was confirmed to be associated with spermatogenesis of mice48. In Crassostrea hongkongensis, SRD5A1 was expressed at a higher level in female gonads than in other tissues18. There is no doubt that the above role in gender regulation is vital. In the sequencing results, FOXL2, BMP2, ZGLP1, CYP17A1, DACH2, DPN, SRD5A1 were highly expressed in the ovary, and DMRT1, SF1, SMC5, SOX9 were highly expressed in the testis. This suggests that they may play a role in the formation of female or male. The results of random verification of qRT-PCR showed the accuracy of sequencing (Supplementary Fig. 3), but their position in sex regulation needs more research to explore. In KEGG results, the function of Wnt, Notch, and TGF-beta signaling pathways in sex determination and gonadal development in mammals have been relatively straightforward, including BMPs (bone morphogenetic proteins), HH (hedgehog), Hippo, nuclear receptors20,49. In addition to non-mammals, it has also been shown that the classical Wnt signaling pathway regulates gonadal differentiation in zebrafish50. The Notch signaling pathway can regulate stem cell proliferation and sex determination in the germline of Caenorhabditis elegans51. The role of AMH (anti-Müllerian hormone) in testicular differentiation of Eriocheir sinensis highlights the importance of TGF-beta pathway in sex determination of reptiles52. All three pathways exist in our results, indicating that they may be related to the sex determination/differentiation of H. cumingii. At the same time, in these three signal pathways, some gender-related genes have also been studied. WNT2, a wnt family ligand, has been demonstrated to be involved in the induction of sex-specific differences in drosophila53. HES1 is an important transcription factor in Notch signaling pathway, which is related to the mouse testis cell fate determination step from primordial stage to the differentiated stage in adulthood54. The loss of CREBBP (CREB binding protein, Notch and TGF-beta signaling pathway member) and P300 (its related paralogue) disrupts histone acetylation of mouse SRY promoter and causes XY gonadal sex reversal55. CUL-1 (CULLIN-1, TGF-beta signaling pathway member), SKR-1, and SEL-10 constitute a SCF E3 ligase complex that plays an critical role in modulating sex-determination of C. elegans56.
In this study, 32 miRNAs were identified from six miRNA libraries, among which 19 were novel miRNAs. Since novel miRNAs have not been studied in other species, we focus on 13 known miRNAs. The DEMs in our gonadal miRNA-seq results are similar to those of other species. For example, miR-7, miR-9, miR-29, miR-184 has been detected in miRNA-seq identification of other species57–59. In these miRNAs, miR-7 is a crucial factor in regulating the hypothalamus-pituitary-ovary axis of mice60. Moreover, miR-7a2 can regulate sexual maturation and reproductive function61. miR-9 is involved in spermatogenesis of spermatogonia during the natural sex change of Monopterus albus62. Furthermore, it can also regulate the critical genes in the ovarian development pathway of Mud Crab Scylla paramamosain63. miR-29 is an essential regulatory factor in the process of male meiosis of mice64. miR-184 was highly expressed in mouse testis, and down-regulated the expression of NCOR2 during spermatogenesis65. In drosophila, miR-184 can affect oogenesis, early embryonic development, and ovarian development66,67. DROSHA is essential for the coordinated development of somatic and germ cell precursors in drosophila larvae. And miR-8 and miR-184 are regulated by DROSHA67. miR-92 may play a role in early chicken gonadogenesis68. In Japanese flounder, miR-92 regulates early development and metamorphosis of Paralichthys olivaceus69. We list three miRNAs and their target genes, and there are 28 DEGs (Table 4). Based on the potential function of these miRNAs in sex, it is speculated that these DEGs may also be sex-related candidate genes of H. cumingii. However, the specific functionality needs further experimental verification. Also, qRT-PCR results showed that miR-7, miR-9-5p, miR-29, miR-92, and miR-153 were highly expressed in the testis and miR-184, miR-218, miR-315 were highly expressed in the ovary (Fig. 2). This is consistent with our sequencing results, which further indicates that these DEMs may play a specific role in the sex regulation of H. cumingii.
361 differentially-expressed target genes were obtained from the intersection analysis of 28,502 DEGs genes and 911 target genes (Fig. 3a). These genes are differentially expressed in ovaries and testes, and their associated miRNAs are also differentially expressed between the two genders. There are two terms of spermatogenesis and oocyte meiosis in GO and KEGG analysis (Fig. 3b,c). They have seven differentially expressed genes (IFT81, LRGUK, SYNE1, TDRD1, PPP1C, PKMYT, and CYCE). In mouse spermatogenesis, IFT81 plays an essential role in regulating the assembly and elongation of sperm flagellum70. And LRGUK-1 is required for basal body and manchette function during spermatogenesis and male fertility71. PPP1C is needed in the meiosis of mouse oocytes72. In P. olivaceus, PoTDRD1 is a germ line specific and sex dimorphic factor, which may play a role in the development of its reproductive system and gametogenesis73. In Xenopus, CPEB and miR-15/16 co-regulate CYCE1 and play a role in oocyte maturation74. These genes and miRNAs may be closely related to germ cell maturation and gametogenesis of H. cumingii.
Conclusion
In this study, the transcriptome and miRNA analysis were applied to explore candidate genes/miRNAs for sex determination/differentiation in H. cumingii. mRNA and miRNA expression profiles provided a rich list of genes and miRNAs expressed in testes and ovaries. In total, 95 DEGs, three DEMs, and three signaling pathways were screened, which may be closely relative with sex determination/differentiation in H. cumingii. In the intersection analysis of DEGs and DEMs target genes, two signal pathways related to gametogenesis were obtained. They include seven genes and seven miRNAs. These sex-related genes, miRNAs, and signaling pathways provided not only the basis foundation for the study of the sexual regulation mechanism in H. cumingii, but also a fundamental reference for better understanding the sex determination and/or differentiation of bivalves.
Materials and methods
Ethics statement
The samples were collected from Jinhua farm in Zhejiang province. All experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Shanghai Ocean University, Shanghai, China.
Sample collection
Specimens used in current study were obtained WeiMing aquaculture farms (ZheJiang, China). Three sexually mature females and three males H. cumingii weighing 320–338 g were collected. In general, the mussels over 2 years old are considered to be sexually mature. The samples we used in this study were 3 years old. Because the eggs of H. cumingii are very large and the sperm is very small. We took a small amount of liquid from the gonads and observed them under a microscope to judge their sex. These mussels are propagated from the same batch of parents and cultured in the same culture pond. The water temperature in the pond ranges from 12 to 32 °C throughout the year, and the pH is about 7.5. All the samples were taken back to the laboratory of Shanghai Ocean University. Keep the mussels in a tank for 3 days. The water temperature was controlled around 26 °C. The gonadal tissues of three female mussels (F1, F2, F3) were Group_F, and those of three male mussels (M1, M2, M3) were Group_M. Gonadal samples were collected with high temperature sterilized scissors and tweezers, quickly placed in liquid nitrogen, and then transferred to the − 80 °C refrigerator.
Tissue material and RNA extraction
The total RNA of the sample is extracted by TRIzol reagent (Invitrogen, Carlsbad, CA, USA). The concentration of total RNA was determined by NanoDrop 2000 ultraviolet spectrophotometer (Thermo, USA). The OD 260/280 was required to be 1.8–2.2. Then, the quality of RNAs were examined by Agilent Technologies 2100 Bioanalyze with a minimum RNA integrity number (RIN) of 8.9.
cDNA library and small RNA preparation and sequencing
Six sequencing libraries were constructed, three libraries (F1, F2, F3) from female groups and other three (M1, M2, M3) from male groups. The samples were prepared by using a mirVana miRNA Isolation Kit (Invitrogen, USA). The libraries were constructed by using TruSeq Stranded mRNA Library Preparation Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. After passing the quality test of Agilent 2100 Bioanalyzer, the constructed library was sequenced by Illumina HiSeq X Ten sequencer to produce the double-terminal data of 150 bp.
The miRNA library was prepared by using mirVana miRNA Isolation Kit (Invitrogen, USA). The main experimental steps were the ligation of 3′ and 5′ connectors. The small RNAs equipped with connectors were reversed transcript and amplified by PCR. Then 147 nt and 157 nt bands were recovered by RNA gel electrophoresis. The concentration of RNA was detected by Agilent 2100 Bioanalyzer. The platform of small RNA sequencing was Illumina HiSeq 4000. Transcriptome and small RNA sequencing were both completed by OE biotech company (Shanghai).
Assembly and annotation
A large number of mRNA‐seq raw reads were obtained from male and female gonadal tissues. Remove adaptor, low quality reads and containing ploy-N reads from the raw reads. The clean reads were assembled into expressed sequence tag clusters (contigs). The transcripts were assembled with Trinity (vesion 2.4)75. The longest transcript was selected as a unigene based the similarity and length of a sequence analysis. The transcriptome analysis was conducted by OE biotech company (Shanghai, China). The transcript annotations give functional annotations for NR (ftp://ftp.ncbi.nih.gov/blast/db), SwissProt (http://www.uniprot.org/downloads), Clusters of orthologous groups for eukaryotic complete genomes (KOG) (ftp://ftp.ncbi.nih.gov/pub/COG/KOG/kyva), Pfam, GO (Gene Ontology) classification, and KEGG (Kyoto Encyclopedia of Genes and Genomes). The Pfam database was compared with the protein family model by HMMER76 software. Other databases were annotated by Diamond77 software with a threshold E-value of 10−5. Base on the SwissProt annotation, GO classification was performed by the mapping relation between SwissProt and GO term. The unigenes were mapped to the KEGG78,79 database to annotate their potential metabolic pathways.
The small RNA sequencing reads were converted into sequence data (also called raw data/reads) by base calling. After filtering out the reads without 3′ adapter and insert tag, reading shorter than 15 nt and longer than 41nt, the clean reads were obtained. The clean reads sequences were compared with Rfam v.10.1 (http://www.sanger.ac.uk/software/Rfam) by blastn. rRNA, scRNA, Cis-reg, snRNA, tRNA and other sequences were annotated. And the transcripts and repetitive sequences were filtered. The filtered sequence was used to identify the known miRNAs by miRBase v.21 database (http://www.mirbase.org/). Unannotated small RNAs were analyzed by mirDeep280. The targets of differentially expressed miRNAs were predicted by using software miRanda81, with the parameter as follows: S ≥ 150 ΔG ≤ − 30 kcal/mol and demand strict 5′ seed pairing.
DEGs and DEMs analysis, GO and KEGG enrichment
Bowtie2 and eXpress were applied to analyze FPKM (fragments per kilobaseper million mapped reads) of each unigene. The counts number of each sample unigene was standardized by using DESeq software. The fold change was calculated. NB (negative binomial distribution test) was used to test the difference significance of reads number. P value adjusted with a false-discovery rate (FDR) correction for multiple testing by Benjamini–Hochberg method82. Finally, the DEGs were screened according to the |fold change|> 2 and P < 0.01. After getting the DEGs, GO and KEGG enrichment analysis were carried out to determine the biological functions or pathways. The number of differential mRNAs included in each GO term was counted. The significance of differential gene enrichment in each GO term was calculated by hypergeometric distribution test. The result of the calculation will return a P value of enrichment significance. And the small P indicates that the differential unigene is enriched in the GO term. Similarly, KEGG database was used to for analysis DEGs. The hypergeometric distribution test method was used to calculate the significance of differential unigene enrichment in each Pathway. At the same time, the unsupervised hierarchical clustering of DEGs is carried out. And the expression pattern of DEGs among different samples is shown in the form of heat map.
miRNA expression is calculated using TPM (transcript per million), TPM = the number of read compared to each miRNA / the total ratio of samples to the number of read × 106. The miRNAs with P value < 0.05 and fold change more than 2 times were selected as differentially expressed miRNAs. Using unsupervised hierarchical clustering analysis, a heat map is constructed based on differentially expressed miRNAs. The heat map (Fig. 1) is drawn by the pheatmap package in R83. GO enrichment and KEGG pathway enrichment analysis of different expressed miRNA-target-Gene were respectively performed using R based on the hypergeometric distribution. Figure 3a is drawn by VennDiagram package in R84. Figure 3b,c are drawn by ggplot2 R package85.
qRT-PCR of mRNA and miRNAs
We selected 12 genes and 8 miRNAs to verify the sequencing results by qRT-PCR. The primer sequences of mRNA and miRNA are shown in supplementary Table 3 and supplementary Table 4, respectively. For the verification of differentially expressed genes, the kit for RNA reverse transcription is PrimeScript RT reagent Kit (TaKaRa, Japan). The total qRT-PCR reaction of mRNA contained 2 × TB Green Premix Ex Taq II (TaKaRa, Japan) 10 μL, forward and reverse gene-specific primer (10 mM) 0.8 μL, cDNA template 1.6 μL, ddH2O 6.8 μL. The condition of the reaction is 95 °C (pre-denaturation) for 15 min, 95 °C (degeneration) for 10 s, and 60 °C (annealing) for 30 s (40 cycles), followed by dissociation curve analysis at 95 °C for 15 s, 60 °C for 1 min and 95 °C for 15 s. EF1α was used as the internal reference gene. The reverse transcription of miRNAs was performed using the Mir-X miRNA First-Strand Synthesis kit (TaKaRa, Japan). The total qRT-PCR reaction of miRNAs contained ddH2O 9 μL, TB green advantage premix 12.5 μL, Rox dye 0.5 μL, miRNA specific primer (or U6 forward primer) 0.5 μL, MRQ 3′primer (or U6 reverse primer) 0.5 μL, and cDNA template 2.0 μL. The reaction procedure was set according to the following procedure: 95 °C for 10 s; 95 °C for 5 s, 60 °C for 20 s, 39 cycles; then the PCR temperature was increased from 60 to 95 °C to generate the dissolution curve. U6 snRNA was used as internal reference gene. All qRT-PCR were performed using Bio-Rad CFX-96 (Bio-Rad, USA). The relative expression of mRNAs and miRNAs was calculated by 2−ΔΔCT method. The difference was calculated by SPSS12.0. Statistically significant differences were examined by paired t test. A value of P < 0.05 was considered to statistically significant.
Supplementary information
Acknowledgements
The authors are thankful for the samples provided by the Weiming aquaculture plant. This study was supported by the National Key R&D Program of China (grant number 2018YFD0901406), the National Natural Science Foundation of China (grant number 31772835) and the Earmarked Fund for Modern Agro-industry Technology Research System (CARS-49).
Author contributions
S.-H. D. completed the sample collection; Y.-Y. W. conceived and designed experiments, analyzed the data and interpreted results and wrote the manuscript; G.-L. W. and J.-L.L. provided feedback on discussion and results. All authors have given approval to the final version of the manuscript.
Data availability
The datasets generated and analyzed during this are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-020-80264-7.
References
- 1.Wu LM, et al. Comparative analysis of shell color variety and genetic structure among five high-quality freshwater pearl mussel populations. Genet. Mol. Res. 2016;15:2–12. doi: 10.4238/gmr15048390. [DOI] [PubMed] [Google Scholar]
- 2.Zhao Y, Bai Z, Fu L, Liu Y, Li J. Comparison of growth and pearl production in males and females of the freshwater mussel, Hyriopsis cumingii, China. Aquac. Int. 2013;21:1301–1310. doi: 10.1007/s10499-013-9632-y. [DOI] [Google Scholar]
- 3.Dheilly NM, et al. Gametogenesis in the Pacific oyster Crassostrea gigas: a microarrays-based analysis identifies sex and stage specific genes. PLoS ONE. 2012;7:e36353. doi: 10.1371/journal.pone.0036353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galindo-Torres P, et al. Sex determination and differentiation genes in a functional hermaphrodite scallop, Nodipecten subnodosus. Mar. Genom. 2018;37:161–175. doi: 10.1016/j.margen.2017.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Kenchington EL, Patwary MU, Zouros E, Bird CJ. Genetic differentiation in relation to marine landscape in a broadcast-spawning bivalve mollusc (Placopecten magellanicus) Mol. Ecol. 2006;15:1781–1796. doi: 10.1111/j.1365-294X.2006.02915.x. [DOI] [PubMed] [Google Scholar]
- 6.Chen H, et al. Transcriptome analysis of sex-related genes in the blood clam Tegillarca granosa. PLoS ONE. 2017;12:1–21. doi: 10.1371/journal.pone.0184584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou L, et al. Transcriptomics analysis revealing candidate genes and networks for sex differentiation of yesso scallop (Patinopecten yessoensis) BMC Genom. 2019;20:1–15. doi: 10.1186/s12864-018-5379-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teaniniuraitemoana V, et al. Molecular signatures discriminating the male and the female sexual pathways in the pearl oyster Pinctada margaritifera. PLoS ONE. 2015;10:1–20. doi: 10.1371/journal.pone.0122819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tao W, et al. Integrated analysis of miRNA and mRNA expression profiles in tilapia gonads at an early stage of sex differentiation. BMC Genom. 2016;17:328. doi: 10.1186/s12864-016-2636-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin S, et al. Integrated analysis of microRNA and mRNA expression profiles during the sex-differentiation sensitive period in oriental river prawn, Macrobrachium nipponense. Sci. Rep. 2017;7:16–18. doi: 10.1038/s41598-017-00039-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu X, et al. Identification and profiling of microRNAs during gonadal development in the giant freshwater prawn Macrobrachium rosenbergii. Sci. Rep. 2019;9:2406. doi: 10.1038/s41598-019-38648-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Presslauer C, Tilahun Bizuayehu T, Kopp M, Fernandes JMO, Babiak I. Dynamics of miRNA transcriptome during gonadal development of zebrafish. Sci. Rep. 2017;7:43850. doi: 10.1038/srep43850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park JH, Shin C. MicroRNA-directed cleavage of targets: mechanism and experimental approaches. BMB Rep. 2014;47:417–423. doi: 10.5483/BMBRep.2014.47.8.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:0–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 15.Grossman H, Shalgi R. A role of microRNAs in cell differentiation during gonad development. Results Probl. Cell Differ. 2016;58:309–336. doi: 10.1007/978-3-319-31973-5_12. [DOI] [PubMed] [Google Scholar]
- 16.Wainwright EN, et al. SOX9 regulates microRNA miR-202-5p/3p expression during mouse testis differentiation. Biol. Reprod. 2013;89:1–12. doi: 10.1095/biolreprod.113.110155. [DOI] [PubMed] [Google Scholar]
- 17.Real FM, et al. A microRNA (mmu-miR-124) prevents Sox9 expression in developing mouse ovarian cells. Biol. Reprod. 2013;89:1–11. doi: 10.1095/biolreprod.113.110957. [DOI] [PubMed] [Google Scholar]
- 18.Tong Y, et al. Transcriptomics analysis of crassostrea hongkongensis for the discovery of reproduction-related genes. PLoS ONE. 2015;10:e0134280. doi: 10.1371/journal.pone.0134280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Capt C, Renaut S, Stewart DT, Johnson NA, Breton S. Putative mitochondrial sex determination in the Bivalvia: insights from a hybrid transcriptome assembly in freshwater mussels. Front. Genet. 2019;10:1–13. doi: 10.3389/fgene.2019.00840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Windley SP, Wilhelm D. Signaling pathways involved in mammalian sex determination and gonad development. Sex. Dev. 2016;9:297–315. doi: 10.1159/000444065. [DOI] [PubMed] [Google Scholar]
- 21.Gontan C, et al. RNF12 initiates X-chromosome inactivation by targeting REX1 for degradation. Nature. 2012;485:386–390. doi: 10.1038/nature11070. [DOI] [PubMed] [Google Scholar]
- 22.Wu Q, et al. Sexual fate change of XX germ cells caused by the deletion of SMAD4 and STRA8 independent of somatic sex reprogramming. PLoS Biol. 2016;14:1–22. doi: 10.1371/journal.pbio.1002553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang R, et al. RNA-Seq analysis of gynoecious and weak female cucumber revealing the cell cycle pathway may regulate sex determination in cucumber. Gene. 2019;687:289–297. doi: 10.1016/j.gene.2018.11.071. [DOI] [PubMed] [Google Scholar]
- 24.Zhuang T, et al. CHD5 is required for spermiogenesis and chromatin condensation. Mech. Dev. 2014;131:35–46. doi: 10.1016/j.mod.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knower KC, et al. Failure of SOX9 regulation in 46XY disorders of sex development with SRY, SOX9 and SF1 mutations. PLoS ONE. 2011;6:e17751. doi: 10.1371/journal.pone.0017751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kent J, Wheatley SC, Andrews JE, Sinclair AH, Koopman P. A male-specific role for SOX9 in vertebrate sex determination. Development. 1996;122:2813–2822. doi: 10.1242/dev.122.9.2813. [DOI] [PubMed] [Google Scholar]
- 27.Santerre C, Sourdaine P, Adeline B, Martinez AS. Cg-SoxE and Cg-β-catenin, two new potential actors of the sex-determining pathway in a hermaphrodite lophotrochozoan, the Pacific oyster Crassostrea gigas. Comp Biochem. Physiol. Part A Mol. Integr. Physiol. 2014;167:68–76. doi: 10.1016/j.cbpa.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 28.Shi J, Hong Y, Sheng J, Peng K, Wang JD. novo transcriptome sequencing to identify the sex-determination genes in Hyriopsis schlegelii. Biosci. Biotechnol. Biochem. 2015;79:1257–1265. doi: 10.1080/09168451.2015.1025690. [DOI] [PubMed] [Google Scholar]
- 29.Picard MAL, et al. The roles of Dmrt (double sex/Male-abnormal-3 related transcription factor) genes in sex determination and differentiation mechanisms: Ubiquity and diversity across the animal kingdom. C. R. Biol. 2015;338:451–462. doi: 10.1016/j.crvi.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 30.Kim S, Bardwell VJ, Zarkower D. Cell type-autonomous and non-autonomous requirements for Dmrt1 in postnatal testis differentiation. Dev. Biol. 2007;307:314–327. doi: 10.1016/j.ydbio.2007.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matson CK, et al. The mammalian doublesex homolog DMRT1 is a transcriptional gatekeeper that controls the mitosis versus meiosis decision in male germ cells. Dev. Cell. 2010;19:612–624. doi: 10.1016/j.devcel.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Webster KA, et al. Dmrt1 is necessary for male sexual development in zebrafish. Dev. Biol. 2017;422:33–46. doi: 10.1016/j.ydbio.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naimi A, et al. Identification and expression of a factor of the DM family in the oyster Crassostrea gigas. Comp Biochem. Physiol. Part A Mol. Integr. Physiol. 2009;152:189–196. doi: 10.1016/j.cbpa.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 34.Teaniniuraitemoana V, et al. Effect of temperature, food availability, and estradiol injection on gametogenesis and gender in the pearl oyster Pinctada margaritifera. EJ Exp. Zool. Part A Ecol. Genet. Physiol. 2016;325:13–24. doi: 10.1002/jez.1992. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, et al. Transcriptome sequencing and comparative analysis of ovary and testis identifies potential key sex-related genes and pathways in scallop Patinopecten yessoensis. Mar. Biotechnol. 2016;18:453–465. doi: 10.1007/s10126-016-9706-8. [DOI] [PubMed] [Google Scholar]
- 36.Bertho S, et al. Foxl2 and its relatives are evolutionary conserved players in gonadal sex differentiation. Sex. Dev. 2016;10:111–129. doi: 10.1159/000447611. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Ortiz JE, et al. Foxl2 functions in sex determination and histogenesis throughout mouse ovary development. BMC Dev. Biol. 2009;9:36. doi: 10.1186/1471-213X-9-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu XL, Zhang ZF, Shao MY, Liu JG, Muhammad F. Sexually dimorphic expression of foxl2 during gametogenesis in scallop Chlamys farreri, conserved with vertebrates. Dev. Genes Evol. 2012;222:279–286. doi: 10.1007/s00427-012-0410-z. [DOI] [PubMed] [Google Scholar]
- 39.Wu S, et al. Identification and expression profiles of Fox transcription factors in the Yesso scallop (Patinopecten yessoensis) Gene. 2020;733:144387. doi: 10.1016/j.gene.2020.144387. [DOI] [PubMed] [Google Scholar]
- 40.Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77:481–490. doi: 10.1016/0092-8674(94)90211-9. [DOI] [PubMed] [Google Scholar]
- 41.Hoshijima K, Inoue K, Higuchi I, Sakamoto H, Shimura Y. Control of doublesex alternative splicing by transformer and transformer-2 in Drosophila. Science. 1991;252:833–836. doi: 10.1126/science.1902987. [DOI] [PubMed] [Google Scholar]
- 42.Kashimada K, et al. FOXL2 and BMP2 act cooperatively to regulate follistatin gene expression during ovarian development. Endocrinology. 2011;152:272–280. doi: 10.1210/en.2010-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cen CH, Chen M, Jiang L, Hou XH, Gao F. The regulation of gonadal somatic cell differentiation during sex determination in mice. Sheng Li Xue Bao. 2020;72:20–30. [PubMed] [Google Scholar]
- 44.Nagaoka SI, et al. ZGLP1 is a determinant for the oogenic fate in mice. Science. 2020;367:eaaw4115. doi: 10.1126/science.aaw4115. [DOI] [PubMed] [Google Scholar]
- 45.Nakamura M. Sex determination in amphibians. Semin. Cell Dev. Biol. 2009;20:271–282. doi: 10.1016/j.semcdb.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 46.Keisman EL, Baker BS. The Drosophila sex determination hierarchy modulates wingless and decapentaplegic signaling to deploy dachshund sex-specifically in the genital imaginal disc. Development. 2001;128:1643–1656. doi: 10.1242/dev.128.9.1643. [DOI] [PubMed] [Google Scholar]
- 47.Barbash DA, Cline TW. Genetic and molecular analysis of the autosomal component of the primary sex determination signal of Drosophila melanogaster. Genetics. 1995;141:1451–1471. doi: 10.1093/genetics/141.4.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hwang G, Verver DE, Handel MA, Hamer G, Jordan PW. Depletion of SMC5/6 sensitizes male germ cells to DNA damage. Mol. Biol. Cell. 2018;29:3003–3016. doi: 10.1091/mbc.E18-07-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pires-daSilva A, Sommer RJ. The evolution of signalling pathways in animal development. Nat. Rev. Genet. 2003;4:39–49. doi: 10.1038/nrg977. [DOI] [PubMed] [Google Scholar]
- 50.Sreenivasan R, et al. Gonad differentiation in zebrafish is regulated by the canonical Wnt signaling pathway. Biol. Reprod. 2014;90:1–10. doi: 10.1095/biolreprod.113.110874. [DOI] [PubMed] [Google Scholar]
- 51.Wang C, Wilson-Berry L, Schedl T, Hansen D. TEG-1 CD2BP2 regulates stem cell proliferation and sex determination in the C. elegans germ line and physically interacts with the UAF-1 U2AF65 splicing factor. Dev. Dyn. 2012;241:505–521. doi: 10.1002/dvdy.23735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou Y, et al. The role of anti-Müllerian hormone in testis differentiation reveals the significance of the TGF-b pathway in reptilian sex determination. Genetics. 2019;213:1317–1327. doi: 10.1534/genetics.119.302527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deshpande G, Nouri A, Schedl P. Wnt signaling in sexual dimorphism. Genetics. 2016;202:661–673. doi: 10.1534/genetics.115.177857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu C, Rodriguez K, Yao HHC. Mapping lineage progression of somatic progenitor cells in the mouse fetal testis. Development. 2016;143:3700–3710. doi: 10.1242/dev.135756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carré GA, et al. Loss of p300 and CBP disrupts histone acetylation at the mouse Sry promoter and causes XY gonadal sex reversal. Hum. Mol. Genet. 2018;27:190–198. doi: 10.1093/hmg/ddx398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Killian DJ, et al. SKR-1, a homolog of Skp1 and a member of the SCF(SEL-10) complex, regulates sex-determination and LIN-12/Notch signaling in C. elegans. Dev. Biol. 2008;322:322–331. doi: 10.1016/j.ydbio.2008.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li S, et al. Identification and comparison of microRNAs in the gonad of the yellowfin seabream (Acanthopagrus latus) Int. J. Mol. Sci. 2020;21:1–16. doi: 10.3390/ijms21165690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meng X, Zhang X, Li J, Liu P. Identification and comparative profiling of ovarian and testicular microRNAs in the swimming crab Portunus trituberculatus. Gene. 2018;640:6–13. doi: 10.1016/j.gene.2017.10.026. [DOI] [PubMed] [Google Scholar]
- 59.Song YN, Shi LL, Liu ZQ, Qiu GF. Global analysis of the ovarian microRNA transcriptome: Implication for miR-2 and miR-133 regulation of oocyte meiosis in the Chinese mitten crab, Eriocheir sinensis (Crustacea:Decapoda) BMC Genom. 2014;15:1–10. doi: 10.1186/1471-2164-15-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He J, et al. The role of miR-7 as a potential switch in the mouse hypothalamus-pituitary-ovary axis through regulation of gonadotropins. Mol. Cell. Endocrinol. 2020;518:110969. doi: 10.1016/j.mce.2020.110969. [DOI] [PubMed] [Google Scholar]
- 61.Ahmed K, et al. Loss of microRNA-7a2 induces hypogonadotropic hypogonadism and infertility. J. Clin. Invest. 2017;127:1061–1074. doi: 10.1172/JCI90031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao Y, Jia D, Hu Q, Li D. Foxl3, a target of MIR-9, stimulates spermatogenesis in spermatogonia during natural sex change in monopterus albus. Endocrinology. 2016;157:4388–4399. doi: 10.1210/en.2016-1256. [DOI] [PubMed] [Google Scholar]
- 63.Zhou M, et al. miR-9 and miR-263 regulate the key genes of the ERK pathway in the ovary of mud crab scylla paramamosain. Mar. Biotechnol. 2020;22:594–606. doi: 10.1007/s10126-020-09981-4. [DOI] [PubMed] [Google Scholar]
- 64.Hilz S, Fogarty EA, Modzelewski AJ, Cohen PE, Grimson A. Transcriptome profiling of the developing male germ line identifies the miR-29 family as a global regulator during meiosis. RNA Biol. 2017;14:219–235. doi: 10.1080/15476286.2016.1270002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu J, Bao J, Wang L, Hu Y, Xu C. MicroRNA-184 downregulates nuclear receptor corepressor 2 in mouse spermatogenesis. BMC Dev. Biol. 2011;11:64. doi: 10.1186/1471-213X-11-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iovino N, Pane A, Gaul U. miR-184 has multiple roles in Drosophila female germline development. Dev. Cell. 2009;17:123–133. doi: 10.1016/j.devcel.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 67.Yang H, et al. MicroRNA-dependent roles of Drosha and Pasha in the Drosophila larval ovary morphogenesis. Dev. Biol. 2016;416:312–323. doi: 10.1016/j.ydbio.2016.06.026. [DOI] [PubMed] [Google Scholar]
- 68.Miao N, Wang X, Feng Y, Gong Y. Male-biased miR-92 from early chicken embryonic gonads directly targets ATRX and DDX3X. Gene. 2017;626:326–336. doi: 10.1016/j.gene.2017.05.050. [DOI] [PubMed] [Google Scholar]
- 69.Li X, et al. The role of miR-92 in regulating early development and metamorphosis of Japanese flounder Paralichthys olivaceus. Genes Genet. Syst. 2020;95:1–10. doi: 10.1266/ggs.18-00047. [DOI] [PubMed] [Google Scholar]
- 70.Qu W, et al. The essential role of intraflagellar transport protein IFT81 in male mice spermiogenesis and fertility. Am. J. Physiol. Cell Physiol. 2020;318:C1092–C1106. doi: 10.1152/ajpcell.00450.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu Y, et al. LRGUK-1 Is required for basal body and manchette function during spermatogenesis and male fertility. PLoS Genet. 2015;11:1–20. doi: 10.1371/journal.pgen.1005090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Swain JE, Ding J, Brautigan DL, Villa-Moruzzi E, Smith GD. Proper chromatin condensation and maintenance of histone H3 phosphorylation during mouse oocyte meiosis requires protein phosphatase activity. Biol. Reprod. 2007;76:628–638. doi: 10.1095/biolreprod.106.055798. [DOI] [PubMed] [Google Scholar]
- 73.Zhao J, et al. tdrd1 is a germline-specific and sexually dimorphically expressed gene in Paralichthys olivaceus. Gene. 2018;673:61–69. doi: 10.1016/j.gene.2018.06.043. [DOI] [PubMed] [Google Scholar]
- 74.Wilczynska A, Git A, Argasinska J, Belloc E, Standart N. CPEB and miR-15/16 co-regulate translation of cyclin E1 mRNA during xenopus oocyte maturation. PLoS ONE. 2016;11:1–21. doi: 10.1371/journal.pone.0146792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grabherr MG, et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol. 2013;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mistry J, Finn RD, Eddy SR, Bateman A, Punta M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013;41:e121. doi: 10.1093/nar/gkt263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2014;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 78.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2020;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Friedländer MR, MacKowiak SD, Li N, Chen W, Rajewsky N. MiRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40:37–52. doi: 10.1093/nar/gkr688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Enright AJ, et al. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- 83.Kolde, R. pheatmap: Pretty Heatmaps. (2015). R package version1.0.8. http://CRAN.R-project.org/package=pheatmap.
- 84.Chen, H. VennDiagram: Generate High-Resolution Venn and Euler Plots. (2018). R package version 1.6.20. http://CRAN.R-project.org/package=VennDiagram.
- 85.Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York: Springer; 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during this are available from the corresponding author on reasonable request.