

Abstract
Head and neck squamous cell carcinoma (HNSCC) is a very aggressive disease with a poor prognosis for advanced-stage tumors. Recent clinical, genomic, and cellular studies have revealed the highly heterogeneous and immunosuppressive nature of HNSCC. Despite significant advances in multimodal therapeutic interventions, failure to cure and recurrence are common and account for most deaths. It is becoming increasingly apparent that tumor microenvironment (TME) plays a critical role in HNSCC tumorigenesis, promotes the evolution of aggressive tumors and resistance to therapy, and thereby adversely affects the prognosis. A complete understanding of the TME factors, together with the highly complex tumor–stromal interactions, can lead to new therapeutic interventions in HNSCC. Interestingly, different molecular and immune landscapes between HPV+ve and HPV−ve (human papillomavirus) HNSCC tumors offer new opportunities for developing individualized, targeted chemoimmunotherapy (CIT) regimen. This review highlights the current understanding of the complexity between HPV+ve and HPV−ve HNSCC TME and various tumor–stromal cross-talk modulating processes, including epithelial–mesenchymal transition (EMT), anoikis resistance, angiogenesis, immune surveillance, metastatic niche, therapeutic resistance, and development of an aggressive tumor phenotype. Furthermore, we summarize the recent developments and the rationale behind CIT strategies and their clinical applications in HPV+ve and HPV−ve HNSCC.
Subject terms: Head and neck cancer, Cancer microenvironment
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the 6th most common cancer worldwide, with an annual incidence of more than 800,000 new cases and 350,000 deaths.1–3 Clinically, pathologically, phenotypically, and biologically, HNSCC is a heterogeneous disease of the oral cavity, oropharynx, hypopharynx, larynx, and paranasal sinuses.4,5 Oral squamous cell carcinoma (OSCC) is the major subtypes of HNSCC and accounts for two-thirds of the cases in developing countries. While tobacco and alcohol consumption are responsible for ~75% of HNSCC cases, recently, a substantial increase in human papillomavirus (HPV)-associated oropharynx cancers (OPC)6 in the Western world has been observed which is expected to surpass cervical cancers by 2020 in the USA.7 In contrast to the etiological role of tobacco smoking in Western countries,8 the use of smokeless tobacco (ST) products like pan masala, gutkha, and betel quid are the major risk factors in Asian countries, including India.9–12 Other etiological factors such as exposure to radiation,13 wood dust,14 asbestos,15 salted foods,14 poor oral hygiene,16 and Epstein–Barr virus (EBV) infection17 also increase the risk of HNSCC.
HNSCCs are mostly diagnosed at an advanced stage with locally advanced (LA) or distant metastasis (DM). Despite multimodality therapeutic interventions which include surgery, radiotherapy (RT), chemotherapy (CT), and/or immunotherapy (IT), a majority (40–60%) of the LA tumors ultimately display recurrence/local progression. Treatment of metastatic and recurrent (R/M) HNSCC tumors with palliative CT also displays poor prognosis. The complete understanding of the HNSCC tumor biology might help us overcome the low therapeutic response of HNSCCs and aid in developing therapeutic strategies with minimal inherent or acquired resistance. Therefore, understanding the HNSCC biology and identifying novel therapeutic targets for effective management of this malignancy is the dire need.18
Most of the previous studies were focused on targeting only cancer cells. However, recent studies have shown that noncancerous cells surrounding the tumor and extracellular matrix (ECM) proteins, which together form the TME, play a critical role in tumorigenesis, the evolution of aggressive tumors, and the promotion of resistance to therapy.19,20 The TME is enriched with various growth factors, intermediate metabolites, nutrients, hormones, growth factors, including chemokines, cytokines, and immune modulators that are secreted by both the tumor and stromal cells. These factors promote the clonal selection of aggressive cells and the acquisition of many of the hallmarks of cancer.21,22 Thus TME provides a permissive environment for tumor progression, metastasis, and development of resistance.23
Interestingly, recently developed therapeutic strategies targeting both the cancer cells and components of the TME have shown increased efficacy and improved patient prognosis.24 In this review, we discuss the current understanding and complexity of TME in both HPV+ve and HPV−ve HNSCC, and tumor–stromal cross-talk modulating processes, including EMT, anoikis resistance, angiogenesis, immune surveillance, metastatic niche, therapeutic resistance, which together contribute to the development of aggressive tumors and resistance to therapy. We also summarize the recent developments and the rationale behind CIT strategies and their clinical applications in HNSCC.
HNSCC tumor microenvironment
Cancer was previously deemed a mass of undifferentiated tumor cells without considering the surrounding stromal cells in the microenvironment. In HNSCC, the TME represents a highly complex ecosystem of cellular and noncellular components. The cellular constituents include genetically altered stromal cells such as cancer-associated fibroblasts (CAFs), endothelial cells (EC), adipocytes, neuroendocrine cells, blood and lymphatic vascular cells,25 and infiltrating immune cells ((T cells, B cells, natural killer cells (NK cells), dendritic cells (DC), macrophages, myeloid-derived suppressor cells (MDSCs)) (Fig. 1).26–28 The noncellular components of the TME include ECM proteins such as collagen, fibronectin, elastin, laminin, and tenascin and physical and chemical parameters, such as pH, oxygen tension, interstitial pressure, and fluid flux. Recent studies have shown that stromal cells provide intermediate metabolites, nutrients, hormones, cytokines/chemokines, and growth factors to tumor cells to support their proliferation, invasion, metastasis, and survival.29–32 They also help recruit CAFs,33 tumor-promoting immune cells, inflammatory cells, and help escape immune recognition,34 thereby providing a permissive environment for tumor progression, metastasis, and development of resistance to therapy.23 These studies suggest that TME dictates aberrant tissue function and plays a critical role in the subsequent evolution of more advanced malignancies.35
Fig. 1.
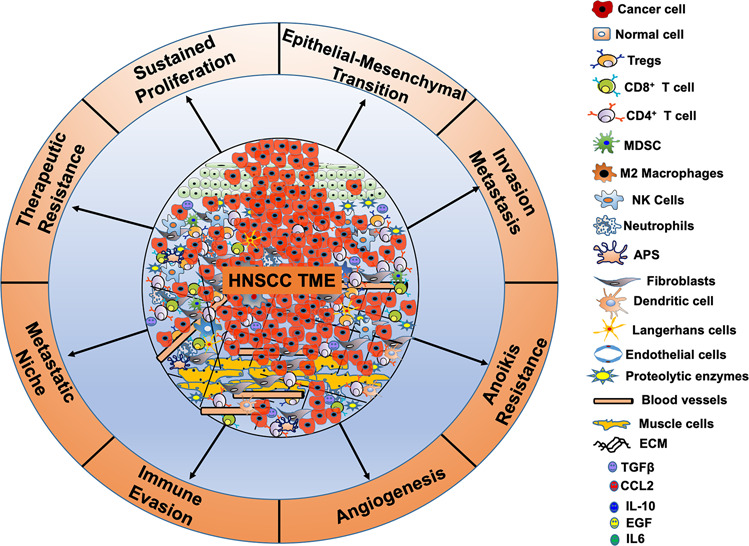
Head and neck squamous cell carcinoma (HNSCC) TME is a complex ecosystem. HNSCC TME is a complex ecosystem consisting of a fabricated network of tumor cells surrounded by non-tumor cells, including cancer-associated fibroblasts (CAFs), endothelial cells (EC), adipocytes, neuroendocrine cells, blood and lymphatic vascular cells, muscle cells, infiltrating immune cells ((T cells, B cells, natural killer cells (NK cells), neutrophils, dendritic cells (DC), Langerhans cells (LCs), macrophages, and myeloid-derived suppressor cells (MDSCs)]. In addition, stromal components, including extracellular matrix (ECM) proteins (collagen, fibronectin, elastin, laminin, and tenascin), intermediate metabolites, nutrients, hormones, growth factors, etc. are the crucial components of HNSCC TME. The complex cross talk between the tumor and the stromal components regulates cell growth, epithelial–mesenchymal transition (EMT), invasion and metastasis, anoikis resistance, angiogenesis, metastatic niche, immune surveillance, and therapeutic resistance making HNSCC tumors very aggressive
HNSCC TME is infiltrated with lymphocytes (TILs) and their subsets such as CD8+ cytotoxic T cells, CD4+ helper T cells, CD163+ and CD68+ macrophages and MDSCs, CD57+ NK cells, FOXP3+ T-regulatory (Tregs) cells36,37 attributing prognostic value to TILs. Besides, substantial infiltration of pro-inflammatory immune cells, such as tumor-associated macrophages (TAM), natural killer (NK) cells, and CD8+ T cells, HNSCC TME is immunosuppressive.34 Expression of pro-inflammatory mediators, such as IL-1α, IL-1β, IL-6, IL-8, TNF-α, TGF-β, granulocyte–macrophage colony-stimulating factor (GM-CSF), vascular endothelial growth factor (VEGF), monocyte chemoattractant protein 1 (MCP-1), RANTES (CCL5), and prostaglandin E2 (PGE2) are found to be upregulated in the premalignant lesions of HNSCC38,39 and are involved in progression and metastasis. Similarly, overexpression of alpha-smooth muscle actin (α-SMA), a marker for cancer-associated macrophages (CAFs), is observed in premalignant lesions compared to normal epithelia.40,41 A study using a 4-nitro-quinoline-2-oxide (4-NQO) induced mouse model of HNSCC found elevated levels of inflammatory Th1, TC1, Th17 cells, and increased expression of IL-17, IL-23 in premalignant lesions. Further, increased infiltration of Tregs along with the downregulation of IL-23 and upregulation of TGF-β in the tumor tissues was also observed Table 1. These results are suggestive of an immune-stimulatory microenvironment in the premalignant stages compared to an immunosuppressive TME of the established tumors.42 Similarly, increased IL-6 and TNF-α expression was observed in the saliva of HNSCC patients.43
Table 1.
Differences between HPV+ve and HPV−ve HNSCC TME
| Serial No. | Cell type | HPV status | Levels in tumor | Protein expression/factors secreted | Prognosis | Ref. |
|---|---|---|---|---|---|---|
| 1 | CD3+ | HPV+ve | Higher | IFN-γ | Better | 64 |
| 2 | CD4+ | Higher | IFN-γ, TNF-α | Better | 65 | |
| 3 | CD8+ | Higher | IFN-γ, IL-17 | Better | 64 | |
| 4 | CD4+/CD8+ ratio | Lower | Better | 74 | ||
| 5 | CD4+CD25+ Tregs | Higher | IL-10, IL-12, IL-35, TGF-β, VEGF | Better | 71 | |
| 6 | CD56+ NK cells | Higher | IFN-γ, | Better | 71 | |
| 7 | M1 macrophages | Lower | IL-12, IL-23, TNF-α, CCL5, CXCL9, CXCL5, CXCL10 | Better | 74 | |
| 8 | M2 macrophages | Higher | TGF-β, IL-13, IL-4, IL-10, MMP9, CCL2, CCL5, CXCL2, CXCL8, MIF, EGF, VEGF, ROS, arginase-1 | Poor | 72 | |
| 9 | MDSCs | Higher | iNOS, NO, ROS, MMP9, PD-L1, arginase-1 | Poor | 70 | |
| 10 | CD45+ lymphocytes | Higher | Better | 54 | ||
| 11 | CD19+/CD20+ B cells | Higher | Better | 54 | ||
| 12 | Tumor-infiltrating APCs | Higher | -- | Better | 70 | |
| 13 | Langerhans cells | Higher | -- | Better | 70 | |
| 14 | Chemokines | Higher | IL-10, CCL2, EGF | Poor | 55,56 | |
| 15 | Cytotoxic mediators | Higher | Granzyme A, granzyme B, perforin | Better | 53,76 | |
| 16 | Protein molecules | Lower | E-cadherin, MIP-3, IFN-γ, MHC1 CD1d | Poor | 79,81,85 | |
| Higher | LAG3, PD-1, TIGIT, TIM3, CD39, COX2, PD-L1, PD-L2 | Poor | 202,238 | |||
| 17 |
Metabolism Core Periphery |
HPV-ve |
Higher Higher |
Aerobic glycolysis OXPOS |
Poor | 28 |
| 18 |
Metabolic mediators Core Periphery |
Lower Higher |
GLUT1, MCT1, COX5B, and LDHB | Poor | 28 |
Tregs T regulatory cells, NK cells natural killer cells, MDSCs myeloid-derived suppressor cells, APC antigen-presenting cells, MIP-3 macrophage inflammatory protein 3, Tim-3 T-cell immunoglobulin and mucin domain-3, LAG3 lymphocyte-activation gene 3, PD-1 programmed death-1, TIGIT T-cell immune receptor with Ig and ITIM domains, GLUT1 glucose transporter-1, LDHB lactate dehydrogenase-B, MCT1 monocarboxylase transporter 1, COX2 cyclooxygenase-2, COX5B cyclooxygenase 5B, OXPHOS oxidative phosphorylation
Tumor microenvironment differs in HPV+ve and HPV−ve HNSCC
The HPV−ve tumors mostly occur in the tongue, buccal mucosa, hard palate, lips, while the HPV+ve tumors are commonly observed in the palatine and lingual tonsillar region.44,45 In addition, HPV−ve OPC and non-OPC patients are typically older when compared to HPV+ve OPC.46–48 While TP53, CCND1, CDKN2A, FGFR1, MLL2, CUL3, NSD1, PIK3CA, and NOTCH are highly mutated in HPV−ve HNSCC,49 the higher mutational incidence of DDX3X, FGFR2, FGFR3 PIK3CA, KRAS, MLL3, and NOTCH-1 is observed in HPV+ve HNSCC.50 Interestingly, increased cancer stem cells (CSC) population with higher expression of CSC markers, OCT4, SOX2, KLF4, and BIM1 were reported in HPV−ve OPCs51 and associated with a lower response to CRT and worse patient survival.52 Further comprehensive analysis of The Cancer Genome Atlas (TCGA) data has established the immunologically active nature of HNSCC tumors.53 However, further characterization of these tumors revealed HPV−ve as immunologically cold tumors as compared to their HPV+ve counterparts. Specifically, HPV+ve and HPV−ve OPC tumors have more TILs, Tregs (CD3+ and CD8+), exhausted CD4+ and CD8+ PD-1+ T cells, NK cells, and B cells54 (Fig. 2 and Table 2). Increased infiltration of these TILs is associated with increased production of CCL17, CCL21, IL-10, IL-17, IL-21, TNF-α, and IFN-γ, thereby suggesting an HPV-specific T-cell response that supports favorable OS in HPV+ve HNSCC.55–60 Other studies revealed significantly more numbers of FOXP3+ Tregs in the stromal and intraepithelial compartments of HPV+ve HNSCC tumors compared to HPV−ve tumors.61–63 While some of these studies reported an association of Tregs infiltration with better overall survival (OS) and disease-free survival (DFS),54,62 others observed inferior recurrence-free survival (RFS) and OS with Tregs infiltration.61,63 Higher CD3+ and CD8+ T-cell infiltration was also reported in HPV+ve OPCs compared to HPV−ve tumors,64 and increased CD8+ T-cell infiltration was strongly associated with improved OS and locoregional control (LRC).54,62 Similarly, higher CD4+ TILs in HPV+ve OPC were associated with better prognosis.65 The presence of HPV16 and E7-specific T-cell and circulating T lymphocytes in HPV+ve OPCs,66,67 and their correlation with survival outcome is also documented.68 Oncogenic E6 and E7 HPV proteins function as tumor-associated antigens and activate CD8+ cytotoxic T lymphocytes (CTLs) via DCs.69 However, HPV E7 has been shown to decrease the expression of Toll-like receptor-9 (TLR9), which are involved in the activation of DCs. In contrast, higher tumor intraepithelial infiltration of MDSCs was observed in HPV+ve HNSCC tumors compared to HPV−ve tumors.70 Tumor-associated macrophages (TAMs) are essential for tumorigenesis and controlling angiogenesis, invasion and migration, EMT, intravasation and extravasation, and immunosuppression.71 Furthermore, increased CD68+ macrophage infiltration in HNSCC has been reported to be associated with lymph node metastasis,72 shorter RFS and OS.73 While increased M2 macrophage infiltration in HNSCC TME has been shown to contribute to local and systemic immunosuppression,72 increased M1 macrophage levels in HPV+ve HNSCC patients showed favorable prognosis,74 possibly due to increased M1/M2 ratio. In contrast, increased macrophage recruitment in HPV+ve HNSCC tumors compared to HPV−ve tumors was reported to be associated with shorter RFS and OS.75 In addition to TAMs, significantly increased CD56+ NK cells and increased granzyme B expression were reported in HPV+ve OPCs compared to HPV−ve counterparts53,76 that correlate with improved OS.53,76 Langerhans cells (LCs) are the antigen-presenting cells (APCs) of the immune system and decreased LCs density represents compromised immune surveillance. While the progressive increase of LC infiltration was observed in the transition of normal phenotype to dysplasia and eventually to cancers,77 higher intraepithelial infiltration of LCs was observed in HPV−ve HNSCC tumors than HPV+ve tumors.77 Importantly, increased LC infiltration was associated with improved RFS and OS.77 Though the underlying mechanisms are still unclear, E6- and E7-associated decrease in E-cadherin and macrophage inflammatory protein 3 (MIP-3) has been shown to impair LC recruitment and retention.78,79
Fig. 2.

TME of HPV+ve and HPV−ve HNSCC tumors. The HPV+ve (left) and HPV−ve (right) HNSCC TME have different cell composition and differential tumor–stromal cross talk. The HPV+ve tumors show increased infiltration of CD3+, CD4+, CD8+ cells, CD56dim NK cells, APCs, MDSCs, DCs, and lower Tregs infiltration; however, the converse is true for HPV−ve tumors. HPV−ve tumors have increased the infiltration of M1 macrophages and Langerhans cells. As compared to HPV−ve tumors, HPV+ve tumors have increased secretion of various cytokines, chemokines, and growth factors, including IL-10, CCL2, IL-6, TGF-β, TNF-α, and EGF that provide a growth advantage to the tumor cells and induce immunosuppression. Increased secretion of TGF-β and CCL2 by the HPV+ve cells promotes macrophage differentiation to pro-tumorigenic M2 that stimulates Tregs. In turn, Tregs induce LT CD8+ exhaustion and apoptosis through PD-L1-PD-1 interaction. M2 secretes IL-6 and IL-10 that stimulate MDSCs, TGF-β and EGF induces epithelial-to-mesenchymal transition (EMT) on cancer cells. While the HPV+ve tumors have enhanced OXPHOS at the core and aerobic glycolysis in the tumor periphery (blue zone), the opposite is true for HPV−ve tumors and has more deposition of lactate known to suppress the immune cells
Table 2.
Clinical trials targeting HPV+ve and HPV-ve HNSCC TME
| Serial No. | Antibodies/drugs used | Target | HNSCC patients | Status | Phase | Trial ID number |
|---|---|---|---|---|---|---|
| 1 | Durvalumab (MEDI4736) monotherapy | PD-L1 | HPV+ve/−ve PR R/M | Active, not recruiting | II | NCT02207530 |
| 2 | Durvalumab (MEDI4736) + tremelimumab (anti-CTLA-4 Ab) vs Durvalumab | PD-L1 and CTLA-4 | R/M | Active, not recruiting | II | NCT02319044 |
| 3 | Durvalumab (MEDI4736) with/without tremelimumab vs SOC (docetaxel + methotrexate + 5FU + cetuximab (EAGLE) | PD-L1, CTLA-4, and EGFR | PD-L1+ve/−ve R/M | Active, not recruiting | III | NCT02369874 |
| 4 | Durvalumab with/without tremelimumab vs cetuximab + cisplatin + carboplatin + 5FU | PD-L1, CTLA-4, and EGFR | R/M | Active, not recruiting | III | NCT02551159 |
| 5 | Durvalumab + RT + cisplatin vs RT + Cisplatin + placebo (ADHERE) | PD-L1 | HPV-ve | Not yet recruiting | III | NCT03673735 |
| 6 | Avelumab + IMRT + cetuximab (anti-EGFR Ab) vs SOC (IMRT + cisplatin) (REACH) | PD-L1 and EGFR | LA | Recruiting | III | NCT02999087 |
| 7 | Avelumab + cetuximab + palbociclib | PD-L1, EGFR, and CDK4/6 | R/M HNSCC | Recruiting | I | NCT03498378 |
| 8 | Atezeolizumab + bevacizumab (anti-VEGF Ab) | PD-L1 and VEGF | HPV+ve or EBV+ve HNSCC and other solid tumors | Recruiting | II | NCT03074513 |
| 10 | Pembrolizumab (MK-3475) | PD-1 | High risk for recurrence | Recruiting | II | NCT02841748 |
| 11 | Pembrolizumab (MK-3475-012) (KEYNOTE-012) | PD-1 | HPV+ve/-ve HNSCC and others | Active, not recruiting | I | NCT01848834 |
| 12 | Pembrolizumab + RT vs RT + cisplatin | PD-1 | LA | Recruiting | II | NCT03383094 |
| 13 | Pembrolizumab (MK-3475) + platinum-based chemotherapy and 5FU with or without cetuximab (KEYNOTE-048) | PD-1 | R/M | Active, not recruiting | III | NCT02358031 |
| 14 | Pembrolizumab + cetuximab | PD-1 and EGFR | R/M | Recruiting | II | NCT03082534 |
| Pembrolizumab (MK-3475) vs SOC (methotrexate + docetaxel + cetuximab) (KEYNOTE-040) | PD-1 and EGFR | R/M | Ongoing | III | NCT02252042 | |
| 15 | Pembrolizumab (anti-PD-1 Ab, MK-3475) | PD-1 | RD Following definitive CRT | Recruiting | II | NCT02892201 |
| 17 | Pembrolizumab + cisplatin and IMRT | PD-1 | LA | Recruiting | II | NCT02777385 |
| 18 | Pembrolizumab (MK-3475) + RT + cisplatin (MK-3475-412/KEYNOTE-412) | PD-1 | LA | Active, not recruiting | III | NCT03040999 |
| 19 | Nivolumab vs cetuximab/methotrexate/docetaxel (CheckMate 141) | PD-1 | R/M HNSCC | Active, not recruiting | III | NCT02105636 |
| 20 | Nivolumab (anti-PD-1 Ab) + RT (REPORT) | PD-1 | R | Recruiting | I/II | NCT03317327 |
| 21 | Nivolumab + STBR vs nivolumab | PD-1 | Metastatic | Active, not recruiting | II | NCT02684253 |
| 22 | Nivolumab + IMRT/cisplatin vs nivolumab | PD-1 | LA | Active, not recruiting | II | NCT02764593 |
| 23 | Nivolumab (MDX1106,BMS-936558) vs therapy of investigator’s choice (methotrexate + cetuximab + docetaxel) (CheckMate 141) | PD-1 | PR R/M | Active, not recruiting | III | NCT02105636 |
| 24 | Nivolumab (BMS-936558) + RT vs cetuximab + RT vs nivolumab + cisplatin + RT vs cisplatin + RT | PD-1 and EGFR | LA | Active, not recruiting | III | NCT03349710 |
| 25 | Ipilimumab + cetuximab + IMRT | CTLA-4 and EGFR | Stage III-IVB | Active, not recruiting | Ib | NCT0186043, NCT01935921 |
| Ipilimumab + IMRT vs ipilimumab+ nivolumab | CTLA-4 and PD-1 | Stage III-IVB | Active, not recruiting | Ib | NCT01860430 | |
| 26 | Nivolumab + varlilumab (anti-CD27) | PD-1 and CD27 | LA/R | Completed | III | NCT02335918 |
| 28 | Nivolumab (BMS-936558) + ipilimumab (BMS-734016) vs SOC (cetuximab + carboplatin + platinum + 5FU) | PD-1, CTLA-4, and EGFR | R/M | Active, not recruiting | III | NCT02741570 |
| Nivolumab + ulocuplumab (BMS-936564) | PD-1 and CXCR4 | LA/M | Terminated | I/II | NCT02472977 | |
| 29 | Anti-OX40 antibody (MEDI6469) | OX40 | LA | Active, not recruiting | Ib | NCT02274155 |
| 30 | Durvalumab (MEDI4736) + agonistic anti-OX40-antibody (MEDI6383) vs MEDI6383 | PD-1 and OX40 | R/M | Completed | I | NCT02221960 |
| 31 | Nivolumab (antiPD1 Ab, BMS-936558) + lirilumab (anti-KIR Ab, BMS-986015) vs nivolumab + ipilimumab (anti-CTLA4 Ab) | PD-1, KIR, and CTLA-4 | LA/R HNSCC and solid tumors | Active, not recruiting | I/II | NCT01714739 |
| 32 | Nivolumab vs nivolumab + ipilimumab vs nivolumab + relatlimab (anti-LAG3 Ab, BMS-986016) (Checkmate 358). | PD-1, CTLA-4, and LAG3 | EBV+ve | Recruiting | I/II | NCT02488759 |
| 33 | Sunitinib (SU11248) | VEGFR, PDGFR, c-kit, RET, CSF-1R, and FLT3 | R/M | Completed | II | NCT00387335 |
| 34 | Sorafinib + cetuximab vs cetuximab | VEGFR, PDGFR, Raf, and c-kit | R/M | Completed | II | NCT00939627 |
| 35 | Dasatinib (BMS-354825) | SRC | R/M | Completed | II | NCT00507767 |
DCR disease control rate, DES dose escalation study, LA locally advanced, R recurrent, M metastatic, OS overall survival, PFS progression-free survival, PR platinum refractory, ORR objective responsive rate, SOC standard of care, IMRT intensity-modulated radiation therapy, DLT dose-limiting toxicity, EFS event-free survival, MTD maximum-tolerated dose, RD residual disease
The role of tumor suppressor p53 in promoting immune infiltration in HPV+ve HNSCC is well known; however, the underlying mechanisms of this disparate immune infiltration is yet to be established. High incidence of p53 mutations in HPV−ve HNSCCs may partially explain these observations.80 While this and other studies suggest immune-rich TME in HPV+ve tumors, E6 and E7 proteins evade immune response through downregulation of IFN-γ81 and inhibition of the NF-κB pathway,82,83 resulting in persistent HPV infection. Furthermore, increased IL-10 and TGF-β secretion by the Tregs suppressed immune cells prevent HPV clearance.79,84 Besides, HPV E5 protein leads to the downregulation of MHC1 CD1d and helps protect the recognition of infected cells from NK cell.85 In addition to the immunological differences between HPV+ve and HPV−ve tumors, recent studies have shown differential metabolic compartmentalization inside these tumors.28 While HPV+ve-associated tumors display increased oxidative phosphorylation (OXPHOS) in the tumor core and higher rate of aerobic glycolysis, the inverse is true for HPV−ve tumors.55 This differential metabolism is associated with increased centrally localized staining of glucose transporter-1 (GLUT1), lactate dehydrogenase-B (LDHB), monocarboxylase transporter 1 (MCT1), and cyclooxygenase 5B (COX5B) in HPV+ve tumors, but more peripheral staining along with the higher concentration of lactic acid in HPV−ve tumors.55 Furthermore, differential gene expression and lactate location is correlated with intratumoral abundance of cytotoxic CD8+ T-cell infiltration55 and peritumoral presence of pro-inflammatory cytokines, CD8+ IFN-γ/IL-17+ TL, and myeloid dendritic cells.86 These studies suggest that immune-metabolic cross talk and HPV−ve tumors favor more immunosuppressive TME with decreased cytotoxic tumor-infiltrating T cells and increased Tregs cells.
Though many diagnostic and prognostic biomarkers have been identified, only a few are validated for use in clinical practice for HNSCC subtypes. Concerning HPV+ve OPC, PCR-based HPV DNA/E6 or E7 mRNA detection,87 and serological markers including antibodies against HPV16 capsid protein L1, or oncoproteins E6 and E7 are routinely used.88–91 In addition, p16 overexpression is an important surrogate biomarker for HPV infection and is associated with favorable outcomes for HPV+ve OPC.92 It is interesting to mention that p16-positivity has been included in the recent WHO TNM classification for OPCs. As compared to HPV−ve tumors, HPV+ve OPCs have higher CD3+ and CD8+ TILs,64 suggesting a more antitumor immune response.54,62 Based on these studies, immunological biomarkers or immunoscore with the diagnostic and predictive outcome has been developed.93 The immunoscore quantifies CD3+ and CD8+ TILs in the tumor core and the invasive margin of resected tumors.
Furthermore, tumor mutational burden (TMB) is being currently used as a predictive marker of response to ICI’s.94 Higher TMB in patients with solid tumors was associated with better outcomes after treatment with a combination of anti-CTLA-4 and anti-PD-L1/PD-1 compared to monotherapy.95 Interestingly, in HNSCC, patients with higher TMB showed better response to anti-PD-L1/PD-1 treatment.96,97 Though differences in TMB has been reported in HPV+ve and HPV−ve HNSCC with more TMB observed in HPV−ve tumors,50 the clinical significance of these genomic alterations requires further validation in clinical settings. Similarly, the presence of EBV DNA in the plasma has a predictive and prognostic role in nasopharyngeal carcinoma (NPC). Future studies should be focused on developing biomarkers based on molecular, immunological, and TMB in combination with imaging techniques for effective diagnosis and prognosis of HNSCC subtypes.
TME promotes aggressive HNSCC tumors
The role of TME has a more profound influence on the growth and metastasis of HNSCC. Secretion of growth factors, cytokines, chemokines, hormones, and other chemical inducers by both the stromal and cancer cells activate a vast array of signaling pathways like EGFR, mitogen-activated protein kinases (MAPKs), mammalian target of rapamycin (mTOR), signal transducer and activator of transcription (STAT), Janus kinase (JAK), phosphoinositide 3-kinase (PI3K), and protein kinase C (PKC) pathways.98,99 These pathways are involved in regulating cell cycle, growth, differentiation, EMT, anoikis resistance, invasion and metastasis, angiogenesis, apoptosis, stemness in cancer cells, immune surveillance, metastatic niche, and the therapeutic response, thus making tumors more aggressive.
Role of TME in epithelial–mesenchymal transition in HNSCC
Epithelial-to-mesenchymal transition (EMT) is a crucial process whereby tumor cells lose epithelial characteristics and acquire mesenchymal properties changing cellular morphology, altered cell–cell and cell–matrix adhesion with more invasion and metastatic properties.100,101 Several proteins, including Snail, Slug, TWIST, and SMAD nuclear interacting protein 1 (SNIP1), play essential role(s) in promoting EMT by upregulating α-SMA, vimentin, fibroblast-specific protein 1 (FSP-1) and N-cadherin and downregulating E-cadherin and β-catenin expression in HNSCC.102 Many recent studies have conclusively established the involvement of TME in HNSCC EMT. Studies have shown that HNSCC cells recruit and educate monocytes into TAMs via the CCL2/CCR2 axis, which secrete migration inhibitory factor (MIF), EGF, and TGF-β, which in turn promote EMT in HNSCC.103 The TAM-secreted MIF recruit neutrophils that secrete CCL4, IL-8, and TNF-α to induce EMT and invasive behavior of HNSCC cells.27 In addition, MDSCs in HNSCC TME secrete MMP9, EGF, bFGF, IL-1β, and TGF-β, which play a critical role in inducing the EMT in HNSCC.104,105 IL-1β secreted by MDSCs reduces the expression of E-cadherin by increased binding of Zeb1 to the Ebox element on its promoter as well as through COX2-dependent upregulation of Snail.106,107 Secretion of CXCL12 by CAFs through CXCR4 signaling pathway activation108,109 induces EMT in HNSCC,110,111 and promotes tumor progression and metastasis.112 Besides, CAF-secreted molecules like endothelin-1, CCL7, TGF-β1 via the TGF-β/Smad signaling pathway, and SDF1 via activation of the PI3K-Akt/PKB signaling pathway also promote EMT in HNSCC cells.110,113–115 Furthermore, HNSCC secreted IL-1 has been shown to promote TGF-β and HGF production by CAFs known inducers of EMT. Additionally, IL-6 secreted by tumor and many stromal cells through upregulation of Snail via the STAT3 pathway, and stabilizing Twist via casein kinase 2 (CK2) promote HNSCC EMT.116 Hypoxia is the vital component of TME and a vital contributor to metastasis and has been shown to induce EMT. HIF-1α, an essential hypoxia-regulated gene, has been reported to control the expression of all the EMT regulators, including Snail, Slug, TWIST, and SNIP1.27 HIF-1α increased Twist expression by binding to its HRE proximal promoter element. Hypoxia also promotes EMT in HNSCC by NOTCH signaling activation117 and by regulating the metadherin (MTDH) loop.118 Through cooperative regulation of Twist and Bmi1, hypoxia represses E-cadherin, and p16INK4a expression to promote EMT in HNSCC.119 Interestingly, by increasing the expression of Twist, and reducing that of cyclin D1 and p16INK4, EMT promotes dormancy of disseminated tumor cells (DTCs) within the niche.
Role of HNSCC TME in anoikis resistance
Apoptosis is a programmed cell death (PCD) that occurs in response to irreparable DNA damage or induced by inflammatory cells. Anoikis is an apoptotic cell death triggered by the loss of ECM contact or inappropriate adhesion.120 Anoikis resistance is a crucial process that helps evade apoptosis of disseminating cells during metastasis and therefore promotes the development of aggressive tumors.121 Many studies have anticipated inhibitors of apoptosis (IAP) gene family as the most likely candidates conferring anoikis resistance in HNSCC.122 TIMP metallopeptidase inhibitor 1 (TIMP-1) exerts anti-apoptotic effects by activating FAK/PI3K and MAPK signaling pathways.123 However, recent studies have shown that TIMP-1 is downregulated in anoikis insensitive cells suggesting its role in regulating anoikis in HNSCC.122 Similarly, anoikis-resistant HNSCC JMAR cells grown under attached conditions show increased expression of paternally expressed gene 10 (PEG10 gene).122 This PEG10 protein has been shown to regulate apoptosis via interaction with E3 ligases, including seven in absentia homolog-1 and 2 (SIAH1 and 2) proteins.122 Further studies using these JMAR cells also showed increased expression of small proline-rich proteins (SPRK, SPRR 1A, SPRR1B, SPRR2A, and SPRR3) and S100 gene family calcium-binding secretory proteins (S100P, S100A7, and S100A9).124 Overexpression and activation of neurotrophic tyrosine kinase receptor-B (TrKB) was the first signaling pathway identified to facilitate anoikis resistance in HNSCC.125 Later, activation of integrins and their interactions with cytoplasmic kinases, small G-proteins, and scaffolding proteins mediated survival signaling pathways were found to be important for anoikis resistance.126 Tumor cells exhibiting decreased focal adhesion kinase (FAK) and increased p38 activation in the absence of adhesion undergo apoptosis.127 However, integrin-mediated activation of FAK/Src signaling has been shown to induce PI3K/AKT signaling and promote anoikis resistance in HNSCC.126 Additionally, hepatocyte growth factor (HGF) mediated activation of c-Met, PI3K/AKT/ERK, NF-κB signaling pathways have also been associated with anoikis resistance in HNSCC cells.128,129 It was recently shown that endothelins (secreted by endothelial cells) by promoting IL-6 and CCLX8 expression activates STAT3, PI3K/AKT/ERK pathways, and encourages anoikis resistance in HNSCC.130 Besides, secretion of VEGF by TME-associated endothelial cell-secreted factors also prevents anoikis via PI3K/AKT activation in HNSCC CSCs.131 In addition, EGF secretion by TAMs and endothelial cells via the ERK/c-Myc pathway induces angiopoietin-like 4 (ANGPTL4, secretory protein) expression which regulates NADPH oxidase 1 (Nox1) activation, regulates ROS levels and MMP expression. In the HNSCC cells, ANGPTL4 has been shown to activate the integrin β1 survival signaling pathway and protect the cells from anoikis.132 The formation of multicellular aggregates, also called emboli, is a crucial prerequisite to escape anoikis. Formation of a “platelet cloak” by tumor cell-induced platelet aggregation (TCIPA) shields the tumor cells from NK cell-mediated cytotoxicity and high shear forces in the bloodstream133 (Fig. 3). S100A7 or psoriasin is overexpressed in HNSCC, regulates the PKB/Akt signaling pathway, and induces anoikis resistance in HNSCC.134 Other studies reported the upregulation of S100P, KLK-related peptidase 6 (KLK6), and catenin alpha-like 1 (CTNNAL1) in the HNSCC anoikis-resistant cells compared to anoikis-sensitive cells, suggesting their role in the promotion of anoikis resistance.122 In addition to the secretory factors, many ECM protein fibronectin-mediated integrin αV-FAK activation has been shown to prevent p53-induced anoikis in HNSCC.135 Furthermore, another ECM protein collagen-I promotes tumor cell differentiation, invasion, migration and survival, and regulates anoikis resistance.136,137
Fig. 3.
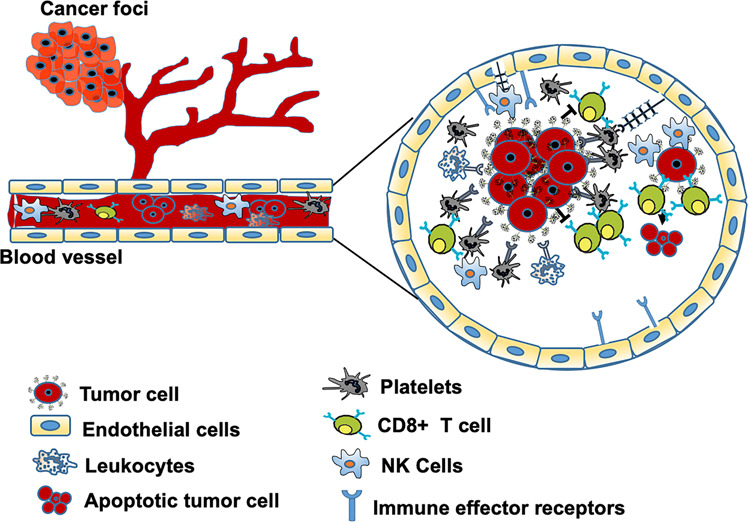
Formation of Emboli and anoikis resistance. After tumor cells leave the primary tumors and invade into the blood circulation, they activate the platelets that encase them to protect them from the shear stress and immune attack. Platelets through the secretion of VEGF, PDGF, or TGF-β promote downregulation of NKG2D receptor and induce NK cells anergy. By arresting tumor cells at the vascular wall via P-selectin and its ligands, platelets facilitate extravasation and distant metastasis
Role of HNSCC TME in angiogenesis
Tumor angiogenesis, the process of new blood vessel formation from pre-existing vessels, is critical for tumor growth and metastasis.138 Cytokines, chemokines, and growth factors, including VEGF, PDGF, and IL-8 by HNSCC cells and stromal cells, activate endothelial cells and induce angiogenesis.131 In particular, the interaction of endothelial and HNSCC cells triggers the NOTCH-1 signaling pathway and promotes capillary tubule formation. The endothelial cells secrete VEGF, which in an autocrine manner, activates VEGFR1 signaling and induces the expression of proangiogenic chemokines such as CXCL1, and CXCL8 thereby facilitating the proliferation of endothelial cells and angiogenesis.139,140 Under hypoxic conditions, TAMs by secreting TGF-β, VEGF, GM-CSF, TNF-α, IL-1, IL-6, and IL-8 stimulate angiogenesis. More specifically, IL-8, IL-6, and EGF induce phosphorylation of STAT3 and ERK in the endothelial cells resulting in their increased survival and proliferation to promote angiogenesis.130 CAF-secreted CXCL12 via CXCL12/CXCR4 signaling in endothelial cells controls angiogenesis by upregulating VEGF expression.141 TGF-β, which is present in the HNSCC TME and also secreted by Tregs, CAFs, TAMs, and MSDCs also increase angiogenesis.142 Similarly, lymphotoxin-α (LT-α), a member of the TNF superfamily, and an important pro-inflammatory cytokine secreted by B and T lymphocytes, increases angiogenesis by regulating CXCL2 expression. A recent study has shown that increased secretion of LT-α from HNSCC TILs induced angiogenesis via increasing endothelial PFKFB3 expression and enhanced glycolytic flux.143 Extracellular vesicles (EVs), secreted from many cell types, are known to mediate cell-to-cell communication by carrying signaling molecules, including VEGF, TGF-β, bFGF. Sato et al. showed that HNSCC cells secreted EVs carry ephrin type B receptor 2 (EPHB2), promoting angiogenesis by inducing the STAT3 signaling pathway in endothelial cells.144 Another study showed that tumor-derived exosomes (a form of EV) carry coagulation factor III, IGFBP-3, uPA, thrombospondin-1, endostatin, and uPA. The authors further demonstrated that through the uPA/uPAR system, exosomes promoted EC proliferation and migration and, thus, stimulate angiogenesis.145 Besides soluble factors, increased levels of nitric oxide (NO) have also shown to trigger neo-vascularization, thereby promoting tumor growth and metastasis.146
Immune surveillance of HPV+ve and HPV−ve HNSCC tumors
Frequently formed premalignant lesions are eliminated by the immune system before the development of invasive tumors.147 However, tumor cells also acquire multiple mechanisms to evade the immune system. These include downregulation of major histocompatibility complex (MHC) class I or class II molecules, loss of immune costimulatory molecules and gain of co-inhibitory signals, defects in antigen-processing machinery (APM), as well as the presentation of tumor-associated antigens (TAA), and downregulation of TAA.148 In addition, overexpression of immunosuppressive molecules, including TGF-β, PGE2, IL-6, IL-10, adenosine, and/or the expression of Fas ligand (FasL), lead to the death of TILs148 and help cancer cell to escape immune recognition. Furthermore, the recruitment of immunosuppressive and tumor-promoting Tregs, MDSCs, TAMs cells, and dysfunction of antitumor immune cells produce tumor pervasive microenvironment and aid in developing clinically evident malignancies. In HNSCC, reduced expression and function of HLA- and APM and associated poor TA processing and presentation prevent NK activation and escape from T-cell-mediated lysis.149 Although the mechanism(s) of impaired APM expression is still unclear in HNSCC, EGFR-mediated HLA downregulation is essential for evading immune recognition.150 Also, overexpression of checkpoint inhibitors (ICIs) in the HNSCC TME like CTLA-4, PD-1, LAG3, TIM3, and killer immunoglobulin-like receptor (KIR) promote T-cell anergy or apoptosis and therefore fail to induce toxicity.150 Furthermore, downregulation of the classical costimulatory molecules CD28/B7 and other ligands CD137/CD137-L, OX40/OX40-L, and CD40/CD40-L observed in HNSCC is associated with poor patient outcome.151 High expression of TGF-β1 produced by HNSCC cells is known to downregulate CD16 and the NK cell receptor NKG2D associated with decreased biological functions of NK cells.152,153 Increased expression of FasL, TRAIL by HNSCC cells, or MAGE3/6+FasL+MHC class I+ exosomes derived from serum of HNSCC patients has been shown to induce T-cells apoptosis and suppress the immune response.153,154 High expression of the tryptophan-catabolizing enzyme indoleamine 2,3-dioxygenase (IDO) arrests the clonal expansion of T cells. Increased expression of IDO by the HNSCC cells, or by MDSCs and dendritic cells has been shown to reduce tryptophan levels needed for T-cell growth and the production of Granzyme B, thereby enhancing immune evasion.155 Moreover, increased expression of HLA-E by HNSCC cells that are known to inhibit NK and CD8+ T cells via NKG2A represents another mode for immunosuppression.149 Ectonucleotidases like CD39 and CD73 expressed by Tregs convert ATP into immunosuppressive adenosine. Increased expression of CD39, CD73, and adenosine in the HNSCC patients is thought to be associated with more potent Teff cell suppression.153 HNSCC tumors show an increased number of MDSCs,156 and by producing NO and ROS catalyze the nitration of the T-cell receptor, prevent TCR-HLA interaction and subsequent activation.157 HPV-associated tumors represent a significant proportion of HNSCCs with enhanced production of TGF-β, decreased expression of IFN-α, TLR9, low viremia, infection confined to keratinocytes with no cell lysis that prevents inflammation. Therefore, no inflammatory response is employed to escape immune surveillance against HPV.153 Although specific T cells against the oncogenic HPV E7 protein have been detected, these cells can not eliminate the tumors. Though the underlying reasons still need to be clarified, it has recently been shown that NLRX1-mediated degradation of proximal nucleic acid-sensing proteins such as the interferon gene stimulator (STING) complex promotes immune escape in HPV+ve tumors.158
Role of HNSCC TME in development of metastatic niche
The development of metastatic HNSCCs remains the primary cause of morbidity and mortality. The term metastatic niche (MN) refers to the well-suited microenvironmental conditions for tumor cells to survive and colonize in and disseminate to distant organs.159 The important components of the pre-metastatic niche include secreted factors from tumor cells (TDSFs), EVs, bone marrow-derived cells (BMDCs), suppressive immune cells, and host stromal cells.159 MN formation involves deposition and remodeling of ECM components, perivascular location and vascular remodeling, regulation of growth factors, cytokines, chemokines and MMPs expression, the establishment of inflammatory milieu and hypoxia, stemness, mesenchymal-to-epithelial transition (MET), and dormancy.160 While the formation of the primary tumor is dependent on progressive genetic changes in an inflammatory stroma, formation of niche either pre-metastatically or after the settlement of DTCs serves to re-establish the stromal environment required for metastatic tumor growth.161 Integrin-mediated interaction of DTCs with the modified niche ECM induces FAK signaling, promoting their proliferation and survival.162 Endothelial cells also play other roles in the development of perivascular microenvironment (ME) and MN. By secreting pro-inflammatory cytokines, S100A8 and S100A9, endothelial cells help in the recruitment of CTC and myeloid CD11b+ cells.163 Lysyl oxidase (LOX) and LOX-like proteins (LOXL) activity at these pre-metastatic sites help cross-link collagen IV and elastin that provide the substrate for CD11b+ cell attachment.164 The expression of S100A8 and S100A9 at the pre-metastatic niche (PMN) also promotes the secretion of serum amyloid A3 (SAA3) protein that helps to recruit myeloid cells,160,163 stimulate TNF-α expression and promote inflammatory milieu. Hypoxia is also a critical TME component promoting tumor cell dormancy and its dissemination. By inducing the expression of VEGF-A, placenta growth factor (PlGF), LOX, and stromal cell-derived factor (SDF1α), hypoxia helps in the recruitment of circular hematopoietic progenitor VEGFR1+ and CD11b+ BMDCs and remodeling of ECM components during the pre-metastatic niche formation. Along with CAF-secreted SDF1, VEGFR1+ BMDC recruit CXC4+ tumor cells to pre-metastatic niche. The CD11b+ cells by stimulating the versican (ECM proteoglycan) inhibit the TGF-β/Smad2 signaling pathway, promote MET, increase proliferation, and speed up metastasis.165 Both VEGFR1+ BMDC and CD11b+ secrete large amounts of MMP9, which in turn, participate in ECM and vascular remodeling, angiogenesis, and vasculogenesis.160,166 These VEGFR1+ BMDC cells directly recruit VEGFR2+ circulating endothelial progenitor cells and thereby help to vascularize the niche and promote tumor sprouting. HNSCC cell migration and metastasis are also promoted by the expression of macrophage inflammatory protein 3α (MIP-3α).167 At the metastatic niche, TAMs provide survival signals for VCAM-1-expressing tumor cells. The MCP-1 secreted by the tumor cells help recruit MDSC and NK to the MN. Also, tumor-secreted VEGFA stimulates TAMs to produce CXCL1 thereby recruiting MDSCs to PMN. After reaching the PMN, DTCs enter a dormant state to adopt the host microenvironment or proliferate to form multicellular micrometastases. This dormancy is reversed by the activation of MET, restore epithelial features, and help tumor relapse. The CD11b+ cells by stimulating versican (ECM proteoglycan) synthesis inhibit TGF-β/Smad2 signaling pathway, stimulate MET, increase proliferation, and promote metastasis.165
TME promotes therapeutic resistance in HNSCC
Despite significant advances, resistance to therapy is still a challenge and the leading cause of poor prognosis of HNSCC patients. The mechanisms underlying resistance to CRT are very complicated and multifactorial. The genomic complexity, intratumoral genetic heterogeneity, and field cancerization result in the deregulated expression of drug target, drug transporters, pro-survival, and apoptotic pathways are considered important underlying reasons. More importantly, increased expression of transporters including ATP-binding cassette (ABC) family members, including P-glycoprotein (P-gp) drive expulsion of routinely used chemotherapeutic drugs (cisplatin, carboplatin, oxaliplatin, and 5FU) and is associated with multidrug resistance (MDR) in HNSCC. In addition to MDR, the deregulation of DNA repair signaling pathways, anti-apoptotic proteins (Bcl2, Bcl-xL, survivin, BMI), and tyrosine kinases (EGFR, cMET), also promote resistance to these DNA damaging agents.168 In addition to these acquired mechanisms, a growing body of evidence suggests that tumor-initiating cells (TICs) or CSCs are intrinsically resistant to standard CRT.169,170 All these mechanisms have been known for decades in HNSCC, but issues related to the resistance mechanism to IT have been gradually emphasized recently. These include lack of production, editing, and neo-antigen presentation, impaired immune infiltration, deregulation of immune checkpoints, T-cell exhaustion, and presence of suppressive immune cells (reviewed in ref. 171) The presence of Tregs and TAMs are significant contributors to the progression, therapeutic response, and resistance in many cancers, including HNSCC. While the recent preclinical studies showed increased Tregs population in immune ICIs and RT-treated recurrent tumors,172 the use of anti-CD25 antibody increased Teff/Treg ratio and improved ICI efficacy.173 Programmed death-ligand 1 (PD-L1), also known as cluster of differentiation 274 (CD274) or B7 homolog 1 (B7-H1) has recently been associated with resistance to immunotherapy and immune escape in multiple cancers, including HNSCC. Recent studies demonstrated increased expression of PD-L1 on CD68+ TAMs and tumor cells in the tonsillar crypts, the site of initial HPV infection. In addition, these studies also showed increased expression of PD-L1 inhibitor PD-1 on the CD8+ tumor-infiltrating lymphocytes (TILs).174 These studies suggest that tonsillar crypts are immune-privileged site with a downregulated effector function of virus-specific T cells and facilitating immune evasion of the HPV-infected cells. These findings are highly compatible with a model in which IFN-γ and potentially other cytokines associated with an immune response induce PD-L1 on tumor cells, which then downmodulates antitumor immunity to facilitate tumor survival. Thus, the PD-1: PD-L1 pathway plays a role in both persistence of HPV infection (through the expression of PD-L1) in the tonsillar crypt epithelium as well as resistance to immune elimination during malignant progression.174 In addition to inhibitory molecules, the overexpression of alternative co-inhibitory receptors like CTLA-4, TIM-3, OX40, lymphocyte-activation gene 3 (LAG3) is known to promote T-cell exhaustion and induces resistance to ICI.175 Besides, downregulation of human leukocyte antigen (HLA) class I molecules and loss of β2-microglobulin expression interfere with antigen presentation to cytotoxic T cells. Furthermore, loss of a classical tumor-suppressor gene PTEN induces expression of CCL2 and VEGF, thereby blocking T-cell infiltration and promoting resistance to the ICIs. Similarly, activation of β-catenin/WNT signaling decreases CCL4 production and inhibits the infiltration of DCs. It is essential to mention here that resistance to ICI is associated with an increased number of memory CD8 T cells (CCR7−CD45RA−), lower CD4/CD8 ratio, and increased expression of checkpoint inhibitor TIM-3 on CD4 as well as CD8 T cells.176,177
As noted earlier, hypoxia is a crucial component of HNSCC TME and plays a vital role in developing resistance to CRT and IT. Increased expression of hypoxia-inducible factors (HIF-1α and HIF-2α) has been shown to modulate both the innate and adaptive immune responses by affecting cytokine production from tumor and endothelial cells, thereby controlling the infiltration of T lymphocytes and macrophages into the tumors.178 Hypoxia induces endoplasmic reticulum (ER) stress response and promotes growth arrest and resistance (hypoxia-induced) to chemotherapy.
In addition to cytokines, chemokines, and growth factors, tumor and the stromal cells secrete many microRNAs (miRNAs) that control growth, proliferation, EMT, angiogenesis, immune surveillance, and impart CRT resistance in HNSCC. While many miRNAs are differentially expressed in HNSCC compared to normal tissues,179 many recent studies have shown the differential miRNA expression profile among HPV+ve and HPV−ve HNSCC.180,181 These studies have shown the association of core miRNAs, including miR-15a/miR-16/miR-195/miR-497 family, miR-143/miR-145, and the miR-106–363 with HPV infection in HNSCC.180 While the expression of miR-195, miR-497, miR-143, miR-145, miR-199a-3p, miR-199b-3p, miR-199b-5p, and miR-126 was significantly downregulated, miR-15a, miR-16, miR-363, miR-363, miR-20b, and miR106b~25 were found to be upregulated in HPV+ve HNSCC patients.180,182 Using the microarray analysis, miRNA-363 and miRNA-33 were found upregulated, whereas miRNA-155, miRNA-181a, miRNA-181b, miRNA-29a, miRNA-218, miRNA-222, miRNA-221, miRNA-42-5p, and miRNA-1323 were downregulated in HPV+ve SCC2 and SCC90 cells compared to HPV−ve PCI13 and PCI30 cells.183
Interestingly, significant upregulation of miR-101, miR-181d, miR-181b, and miR-195 and downregulation of miR-100, miR-130a, and miR-197 has been reported in drug-resistant HNSCC cells compared to their parent cells.184 Similar overexpression of miRNA-23a, miRNA-214, miRNA-518c, miRNA-608, let-7 family of miRNAs, and downregulation of miRNA-21 and miRNA-342 were observed in cisplatin-resistant TSCC compared to sensitive cells.185 Though the underlying reasons for imparting cisplatin resistance are entirely unknown, miRNA-214 mediated PTEN decay, thereby PI3K/Akt signaling pathway activation, is considered one of the mechanisms.185 Overexpression of miR-125a promotes cisplatin sensitivity in laryngeal CSCs by targeting hematopoietic cell-specific protein 1-associated protein X-1 (HAX-1).186 Furthermore, miR-125a-mediated HAX-1 downregulation also promoted vincristine, etoposide, and doxorubicin sensitivity in laryngeal CSCs.186 Importantly, overexpression of miR-222187 and miR-24-3p188 promoted resistance to chemotherapeutic drugs via targeting ABCG2 and CHD5, respectively.
Compared to miRNAs involved in CT resistance, many miRNAs that regulate radio-resistance (RR) have been found. For example, miR-296-5p is downregulated in LSCC and its upregulation in enhanced radiosensitivity (RS) by targeting MDR1 gene.189 Similarly, miR-125b is downregulated in OSCC, but its overexpression enhanced RS via ICAM2 downregulation and PI3K/Akt signaling pathway activation.190 Both the miR-324-3p191 and miR-185-3p192 are downregulated in the RR NPC cells, and their upregulation promoted radiosensitivity via targeting WNT2B gene. Similarly, miR-196a193 and miR-451 determine RR via targeting annexin-1 (ANXA1) and RAS related protein 14 (RAB14), respectively, in HNSCC.194 Similar to miRNA-214, miR-205195 and miR-296-5p196 via targeting PTEN promote RR in NPC. Furthermore, miR-23a197 and miR-203198 play an important role in radiosensitivity by targeting IL-8 in NPC. While the pathobiological importance of many of these miRNAs is known in HNSCC, limited studies are available showing their role in imparting CRT resistance specific to HPV+ve HNSCC.
Targeting HNSCC TME: differential response among HPV+ve and HPV−ve tumors
Tumor progression, metastasis, and response to therapy are markedly affected by the complex heterotypic multicellular interactions among tumor and other cellular and noncellular components of TME. The presence of dense stroma also limits drug availability and compromises therapeutic efficacy. HPV+ve tumors are immune-rich and more responsive to CRT with significant locoregional control199,200 than HPV−ve tumors.201 However, recent studies have shown that increased expression of PD-L1 and PD-L2 on fibroblasts and tumor cells make these tumors immunosuppressive.202 Many immunotherapies that target immune checkpoints like PD-1/PD-L1 and CTLA-4 are currently being evaluated. Several mAbs targeting these ICIs are currently being investigated to increase cancer-specific immunity in HNSCC (Table 2 and Fig. 4). However, differential mutational burden and immunologic landscape among HPV+ve and HPV−ve HNSCCs result in disparate response to therapy and prognosis. PD-L1 is a ligand for PD-1 and PD-2 receptors that limit T-cell activation during inflammation and restrict autoimmunity. PD-L1 is overexpressed in 66% of HNSCC patients and more often in HPV+ve than HPV−ve patients. Recently, fully humanized anti-PD-L1 mAb (durvalumab) was evaluated in the HAWK study on platinum-refractory (PR) R/M HNSCC patients.203 The interim results from this study showed a higher overall response rate (ORR) in HPV+ve patients than HPV−ve patients (30% vs 10%).203 Similarly, the use of FDA approved pembrolizumab (anti-PD-1)204 and nivolumab (anti-PD-1)205 mAbs in PR R/M HNSCC patients showed improved OS in HPV+ve HNSCC patients compared to HPV−ve patients. Combination therapies targeting both the inhibitory and costimulatory receptors have synergistically enhanced the immunological antitumor effects. The dual-blockade therapy combining durvalumab with tremelimumab (anti-CTLA-4 mAb) is currently being investigated for R/M HNSCC (NCT02551159, NCT02369874, and NCT02319044). Similarly, nivolumab in combination with ipilimumab (an IgG1 mAb against CTLA-4) and standard therapy cetuximab/platinum/5FU (NCT02741570) or with ulocuplumab (a fully human anti-CXCR4, NCT02472977) is also being investigated in LA/M HNSCC tumors. Cetuximab, is a chimeric murine/human IgG1 monoclonal antibody (mAb) against EGFR, and is the Food and Drug Administration (FDA) approved targeted agent for the treatment of HNSCC. Triple combination therapy involving nivolumab, ipilimumab, and anti-LAG3 mAb (BMS-986016) is also being investigated in R/M HNSCC patients (NCT02488759). Agonistic anti-OX40 and anti-CD27 agents activate OX40 and CD27, respectively, and both promote T-cell proliferation. In this direction, clinical trials to assess the safety and tolerability of durvalumab along with agonistic anti-OX40 Ab (NCT02221960), and varlilumab with agonistic anti-CD27 mAb (NCT02335918) are currently ongoing in HNSCC patients.
Fig. 4.
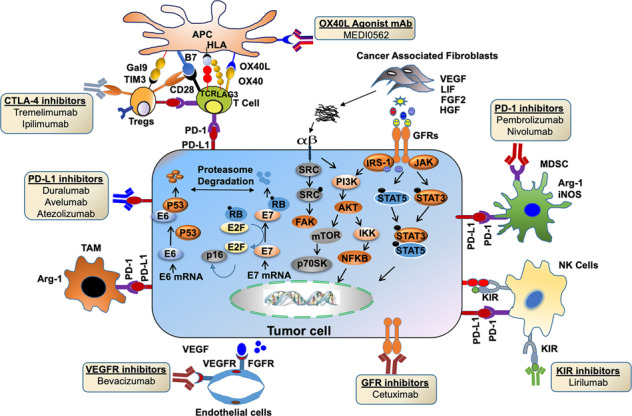
Schematic showing the immunotherapeutic approaches targeting the HNSCC TME. Inactivation of CTLs by secretory immunomodulators or immune checkpoints promotes immune escape of cancer cells. While B7 ligands on APCs interact with CD28 on CTL and provide a secondary signal for their activation and immune response, CTLA-4 on Tregs interfere in this interaction, suppresses CTLs activity and enhances Treg activity. In addition, increased PD-L1 on cancer cells or immune cells like NK, MDSCs, M2 macrophages, and Tregs bind to its receptor PD-1 on activated T cells to promote the state of anergy in CTLs. The OX40 ligand on the APC cells interacts with the OX40 receptor on T cells and increase CTL activation. Cancer cells secrete VEGF, which, upon binding with its receptor VEGFR on endothelial cells, promotes angiogenesis. Many pharmacological inhibitors, including monoclonal antibodies/agonists of immune checkpoints, are being tested in HNSCC and shown to restore CTL antitumor activity and relieve immunosuppression
Besides inducing tumor cell death, the standard-of-care CRT promote antitumoral immune responses by depleting and exhaustion of CTLs, development of CRT-resistant Tregs, increasing MDSCs and enhancing tumor cell MHC I expression.206 However, recent studies have shown that lower doses of CRT also trigger adaptive and innate immune response207 and improve immune recognition of dying cancer cells by mobilizing bone marrow hematopoietic and myeloid cells, increasing infiltration of DCs, CD4+, and CD8+ T cells208 associated with IFN-γ and IL-2 secretion,209 and elimination of MDSCs.210 These studies suggest that the combination of lower CT or RT doses and ICI will improve response in patients with poor immune-infiltrated tumors. Cetuximab administration is associated with decreased tumor cell proliferation, inhibition of angiogenesis, and reduced metastasis. It also binds to the CD16 receptor on NK cells and DCs and improves immune response by promoting T-cell priming, ADCC, and NK cell activation associated with increased HNSCC patient survival.211 However, the limited survival benefits of cetuximab are linked to increased expression of CTLA-4 by Tregs associated with poor prognosis in HNSCC patients.212 In this context, the use of ipilimumab was shown to reverse cetuximab resistance in HNSCC by activating NK cells thereby eliminating Tregs.212 A combination of pembrolizumab with platinum-based CT and 5FU with or without cetuximab is currently being investigated in HNSCCs (NCT02358031). RT is known to increase PD-L1 expression rendering HNSCC TME immunosuppressive.213 Based on the diverse immunomodulatory effects of RT, the combination of ICI with RT is under intense investigation. Many clinical trials combining ICI including nivolumab with stereotactic body radiotherapy (STBR) (NCT02684253) or intensity-modulated radiation therapy (IMRT) along with chemotherapeutic agents like cisplatin (NCT02764593) have recently been started. A combination of ipilimumab with IMRT or with nivolumab or cetuximab is currently being established in HNSCC (NCT01860430, NCT02741570, and NCT01935921). In addition, efficacy of concurrent vs sequential pembrolizumab, cisplatin, and IMRT is being evaluated in HNSCC (NCT02777385). Besides targeting immune cells, clinical trial for use of humanized IgG1 anti-PD-L1 mAb atezolizumab along with bevacizumab (mAb against VEGF) is currently enrolling HPV or EBV-associated HNSCC patients (NCT03074513). Similarly, use of FAP (CAF-specific protein) through PT100 combined with oxaliplatin improved CT, reduced recruitment of MDSCs, Tregs, and inhibited angiogenesis.214 Besides, inhibition of CAF promoted CXCL12/CXCR4 signaling using CXCR4 antagonist (TN14003), or with the anti-CXCR4 antibody AMD3100, or siRNAs inhibited growth and invasion in vitro.111,215–217
Furthermore, combining ulocuplumab (a fully human anti-CXCR4) with nivolumab is currently being investigated for LA/M solid tumors (NCT02472977). Similarly, a combination of cell-cycle regulator CDK4/6 inhibitors (abemaciclib and palbociclib) and PD-L1 inhibitor avelumab has been started in HNSCC (NCT03498378). Keeping in view the enhanced efficacy of CDK4/6 inhibitors in RB-positive tumors, HPV+ve HNSCCs may respond more effectively to this combination owing to their high RB and PD-L1 expression. As mentioned previously, HPV+ve tumors exhibit increased glycolysis and OXPHOS.55 Use of mTOR (regulator of OXPHOS) inhibitor rapamycin along with anti-PD-L1 mAb showed improved survival by increasing IFN-γ production in tumor-infiltrating CD8+ T cells.218 In addition to these studies, several other clinical trials using combinations of ICI and CRT are underway and included in Table 2.
Epidermal growth factor receptor (EGFR), a transmembrane tyrosine kinase receptor (TKR) of the ErbB family, is overexpressed or amplified in ~90% of HNSCC patients219,220 and is associated with aggressive tumor behavior,221 resistance to radiation,222 and poor prognosis.223,224 The constitutive activation of EGFR result in the activation of several downstream signaling pathways, including Ras/Raf/MAPK/ERK, PI3K/Akt, STAT, and the PLC-γ signaling pathways,225,226 those are involved in proliferation, survival, CSCs maintenance227 etc. While the relationship between HPV status, EGFR expression and patient outcome are still ambiguous in HNSCC, many studies have reported increased EGFR amplification in p16−ve (a surrogate biomarker for HPV infection) oropharyngeal cancers.228,229 In this direction, several studies have conclusively established better clinical outcomes in HPV+ve HNSCC patients with low EGFR expression.229–232 Interestingly, the use of cetuximab against EGFR is the first FDA approved targeted therapeutics in HNSCC. However, constitutively active EGFR variant (EGFRvIII) promotes resistance against cetuximab in HNSCC.233 While the therapeutic efficacy of EGFR inhibition remains unclear and controversial in HPV+ve HNSCC,234 many other EGFR inhibitors including mabs (panitumumab or nimotuzumab) and small-molecule inhibitors (lapatinib, erlotinib, gefitinib, and afatinib), either alone or along with CRT are currently being investigated in many clinical trials (reviewed in ref. 235).
Conclusion and future perspective
The primary goal of HNSCC treatment is to inhibit growth and prevent metastasis. However, most of the previous preclinical studies based more on aberrant genetic and epigenetic mutations, altered gene expression in tumor cells have failed in clinical trials. The underlying reasons being the limited understanding of the (1) HNSCC tumor biology and (2) the importance of TME in the progression and development of therapeutic resistance. Recent clinical, genomic, and cellular studies have demonstrated HNSCC TME as highly heterogeneous and immunosuppressive. Although the current clinical trials involving the combination of CT or RT with immune checkpoint blockers are underway, low ORR warrants a better understanding of the role of the immune system in HNSCC. Many recent studies have shown the dynamic nature of the TME during tumor progression or upon the administration of therapeutic interventions. High-throughput analysis should be utilized to comprehensively investigate the spatiotemporal, in-depth characterization of phenotypic, functional features of diverse cell types, and their dynamic cross talk in HNSCC TME during tumor progression and metastasis.
The emerging knowledge of the differential molecular and immune landscape between HPV+ve and HPV−ve HNSCC tumors has created newer opportunities to develop a personalized, targeted CIT approach. Therefore, designing of CIT trials based on genetic, molecular, and immunological landscape and topography of immune cell distribution in the TME will help develop new strategies for effective antitumor immunity and improved clinical outcomes. Future rational combination studies should consider the impact of biological and mechanistic cross talk between cancer cells—-TME—-and therefore include coculture, organoids/tumoroids, and humanized patient-derived xenograft (PDX) models in preclinical studies. Besides, the use of immunocompetent animal models ((KrasG12D;Trp53R172H/+;K14-CreERtam) and E6/E7;KrasG12D;K14-CreERtam)) that histopathologically recapitulate the tobacco/alcohol236 and HPV-mediated237 human HNSCC pathogenesis, respectively, should be considered for developing CITs. Considering the importance of HPV infection on immune HNSCC milieu and, therefore CIT response, future clinical trials should take into account HPV status and immune phenotype to escalate a precision regimen to clinics and ensure better therapeutic outcomes in HNSCC patients. While the development of therapeutic resistance is unavoidable, identification of reliable response markers and understanding resistance is essential.
Acknowledgements
This study was supported by Ramalingaswami Fellowship (Grant number: D.O.NO.BT/HRD/35/02/2006) from the Department of Biotechnology, Government of India, New Delhi to Muzafar A. Macha. Mohammad Haris is funded by a grant (5071012001) from Sidra Medicine Doha, Qatar. Ajaz A. Bhat is supported by Sidra Medicine internal grant (5011041002). We thank Dr. Vineeta Tanwar (Ohio State University, Columbus, Ohio, USA) for her professional assistance in editing the paper.
Author contributions
P.Y., M.A.M., and A.A.B.: prepared the scientific material, wrote the paper, and generated figures. N.A.W., A.R., S.S.C., M.A.S., D.B., W.ER., M.P.F., and S.K.B.: critical revision and editing of the scientific contents. M.A.M. and M.H. conceived of and designed the review contents and contributed to the paper writing and editing. All authors read and approved the final paper.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Ajaz A. Bhat, Parvaiz Yousuf
Contributor Information
Mohammad Haris, Email: mharis@sidra.org.
Muzafar A. Macha, Email: muzafar.aiiims@gmail.com
References
- 1.Fitzmaurice C, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol. 2017;3:524–548. doi: 10.1001/jamaoncol.2017.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghantous Y, Abu Elnaaj I. Global incidence and risk factors of oral cancer. Harefuah. 2017;156:645–649. [PubMed] [Google Scholar]
- 3.Bray F, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 4.Noggle FT, Jr., Clark CR, DeRuiter J. Liquid chromatographic and mass spectral analysis of the anabolic 17-hydroxy steroid esters. J. Chromatographic Sci. 1990;28:263–268. doi: 10.1093/chromsci/28.5.263. [DOI] [PubMed] [Google Scholar]
- 5.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat. Rev. Cancer. 2011;11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 6.Blot WJ, et al. Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res. 1988;48:3282–3287. [PubMed] [Google Scholar]
- 7.Chaturvedi AK, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011;29:4294–4301. doi: 10.1200/JCO.2011.36.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leemans CR, Snijders PJF, Brakenhoff RH. The molecular landscape of head and neck cancer. Nat. Rev. Cancer. 2018;18:269–282. doi: 10.1038/nrc.2018.11. [DOI] [PubMed] [Google Scholar]
- 9.Fakhry C, Gillison ML. Clinical implications of human papillomavirus in head and neck cancers. J. Clin. Oncol. 2006;24:2606–2611. doi: 10.1200/JCO.2006.06.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hashibe M, et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol., Biomark. Prev. 2009;18:541–550. doi: 10.1158/1055-9965.EPI-08-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maier H, Dietz A, Gewelke U, Heller WD, Weidauer H. Tobacco and alcohol and the risk of head and neck cancer. Clin. Investig. 1992;70:320–327. doi: 10.1007/BF00184668. [DOI] [PubMed] [Google Scholar]
- 12.Sturgis EM, Wei Q. Genetic susceptibility-molecular epidemiology of head and neck cancer. Curr. Opin. Oncol. 2002;14:310–317. doi: 10.1097/00001622-200205000-00010. [DOI] [PubMed] [Google Scholar]
- 13.Preston-Martin S, Thomas DC, White SC, Cohen D. Prior exposure to medical and dental x-rays related to tumors of the parotid gland. J. Natl. Cancer Inst. 1988;80:943–949. doi: 10.1093/jnci/80.12.943. [DOI] [PubMed] [Google Scholar]
- 14.Yu MC, Yuan JM. Epidemiology of nasopharyngeal carcinoma. Semin. Cancer Biol. 2002;12:421–429. doi: 10.1016/S1044579X02000858. [DOI] [PubMed] [Google Scholar]
- 15.Boffetta P, et al. Occupation and larynx and hypopharynx cancer: an international case-control study in France, Italy, Spain, and Switzerland. Cancer Causes Control. 2003;14:203–212. doi: 10.1023/A:1023699717598. [DOI] [PubMed] [Google Scholar]
- 16.Guha N, et al. Oral health and risk of squamous cell carcinoma of the head and neck and esophagus: results of two multicentric case-control studies. Am. J. Epidemiol. 2007;166:1159–1173. doi: 10.1093/aje/kwm193. [DOI] [PubMed] [Google Scholar]
- 17.Chien YC, et al. Serologic markers of Epstein-Barr virus infection and nasopharyngeal carcinoma in Taiwanese men. N. Engl. J. Med. 2001;345:1877–1882. doi: 10.1056/NEJMoa011610. [DOI] [PubMed] [Google Scholar]
- 18.Martin L, Zoubir M, Le Tourneau C. Recurrence of upper aerodigestive tract tumors. Bull. du Cancer. 2014;101:511–520. doi: 10.1684/bdc.2014.1970. [DOI] [PubMed] [Google Scholar]
- 19.Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61–68. doi: 10.1016/j.canlet.2016.01.043. [DOI] [PubMed] [Google Scholar]
- 20.Soysal SD, Tzankov A, Muenst SE. Role of the tumor microenvironment in breast cancer. Pathobiology: J. Immunopathol., Mol. Cell. Biol. 2015;82:142–152. doi: 10.1159/000430499. [DOI] [PubMed] [Google Scholar]
- 21.Denton AE, Roberts EW, Fearon DT. Stromal cells in the tumor microenvironment. Adv. Exp. Med. Biol. 2018;1060:99–114. doi: 10.1007/978-3-319-78127-3_6. [DOI] [PubMed] [Google Scholar]
- 22.Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell144, 646–674 (2011). [DOI] [PubMed]
- 23.Giancotti FG. Deregulation of cell signaling in cancer. FEBS Lett. 2014;588:2558–2570. doi: 10.1016/j.febslet.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arneth, B. Tumor microenvironment. Medicina (Kaunas, Lithuania)56, 10.3390/medicina56010015 (2019). [DOI] [PMC free article] [PubMed]
- 25.Cristina, V., Herrera-Gómez, R. G., Szturz, P., Espeli, V. & Siano, M. Immunotherapies and future combination strategies for head and neck squamous cell carcinoma. Int. J. Mol. Sci.20, 10.3390/ijms20215399 (2019). [DOI] [PMC free article] [PubMed]
- 26.Curry JM, et al. Tumor microenvironment in head and neck squamous cell carcinoma. Semin. Oncol. 2014;41:217–234. doi: 10.1053/j.seminoncol.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Peltanova B, Raudenska M, Masarik M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: a systematic review. Mol. Cancer. 2019;18:63. doi: 10.1186/s12943-019-0983-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaudhary S, et al. Immunometabolic alterations by HPV infection: new dimensions to head and neck cancer disparity. J. Natl. Cancer Inst. 2019;111:233–244. doi: 10.1093/jnci/djy207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobrenis K, Gauthier LR, Barroca V, Magnon C. Granulocyte colony-stimulating factor off-target effect on nerve outgrowth promotes prostate cancer development. Int. J. Cancer. 2015;136:982–988. doi: 10.1002/ijc.29046. [DOI] [PubMed] [Google Scholar]
- 30.Hu P, et al. Intratumoral polymorphonuclear granulocyte is associated with poor prognosis in squamous esophageal cancer by promoting epithelial-mesenchymal transition. Future Oncol. 2015;11:771–783. doi: 10.2217/fon.14.306. [DOI] [PubMed] [Google Scholar]
- 31.Oft M, Heider KH, Beug H. TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis. Curr. Biol. 1998;8:1243–1252. doi: 10.1016/S0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- 32.Cheng L, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153:139–152. doi: 10.1016/j.cell.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev. Cell. 2010;18:884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quezada SA, Peggs KS, Simpson TR, Allison JP. Shifting the equilibrium in cancer immunoediting: from tumor tolerance to eradication. Immunol. Rev. 2011;241:104–118. doi: 10.1111/j.1600-065X.2011.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen F, et al. New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med. 2015;13:45. doi: 10.1186/s12916-015-0278-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fang J, et al. Prognostic significance of tumor infiltrating immune cells in oral squamous cell carcinoma. BMC Cancer. 2017;17:375. doi: 10.1186/s12885-017-3317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen N, et al. Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head Neck. 2016;38:1074–1084. doi: 10.1002/hed.24406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonomi M, Patsias A, Posner M, Sikora A. The role of inflammation in head and neck cancer. Adv. Exp. Med. Biol. 2014;816:107–127. doi: 10.1007/978-3-0348-0837-8_5. [DOI] [PubMed] [Google Scholar]
- 39.Johnson SD, De Costa AM, Young MR. Effect of the premalignant and tumor microenvironment on immune cell cytokine production in head and neck cancer. Cancers. 2014;6:756–770. doi: 10.3390/cancers6020756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chaudhary M, et al. Comparison of myofibroblasts expression in oral squamous cell carcinoma, verrucous carcinoma, high risk epithelial dysplasia, low risk epithelial dysplasia and normal oral mucosa. Head Neck Pathol. 2012;6:305–313. doi: 10.1007/s12105-012-0335-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kapse SC, et al. Quantitative assessment of myofibroblast in severe dysplasia, microinvasion and oral squamous cell carcinoma: an immunohistochemical study. J. Contemp. Dent. Pract. 2013;14:34–38. doi: 10.5005/jp-journals-10024-1265. [DOI] [PubMed] [Google Scholar]
- 42.De Costa AM, Schuyler CA, Walker DD, Young MR. Characterization of the evolution of immune phenotype during the development and progression of squamous cell carcinoma of the head and neck. Cancer Immunol., immunothe. 2012;61:927–939. doi: 10.1007/s00262-011-1154-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Juretić M, et al. Salivary levels of TNF-α and IL-6 in patients with oral premalignant and malignant lesions. Folia Biologica. 2013;59:99–102. [PubMed] [Google Scholar]
- 44.Mehanna H, et al. Prevalence of human papillomavirus in oropharyngeal and nonoropharyngeal head and neck cancer-systematic review and meta-analysis of trends by time and region. Head Neck. 2013;35:747–755. doi: 10.1002/hed.22015. [DOI] [PubMed] [Google Scholar]
- 45.Carpén T, et al. Presenting symptoms and clinical findings in HPV-positive and HPV-negative oropharyngeal cancer patients. Acta oto-laryngologica. 2018;138:513–518. doi: 10.1080/00016489.2017.1405279. [DOI] [PubMed] [Google Scholar]
- 46.Näsman A, et al. Incidence of human papillomavirus (HPV) positive tonsillar carcinoma in Stockholm, Sweden: an epidemic of viral-induced carcinoma? Int. J. Cancer. 2009;125:362–366. doi: 10.1002/ijc.24339. [DOI] [PubMed] [Google Scholar]
- 47.Panwar A, Batra R, Lydiatt WM, Ganti AK. Human papilloma virus positive oropharyngeal squamous cell carcinoma: a growing epidemic. Cancer Treat. Rev. 2014;40:215–219. doi: 10.1016/j.ctrv.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 48.Pytynia KB, Dahlstrom KR, Sturgis EM. Epidemiology of HPV-associated oropharyngeal cancer. Oral. Oncol. 2014;50:380–386. doi: 10.1016/j.oraloncology.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature517, 576–582 (2015). [DOI] [PMC free article] [PubMed]
- 50.Seiwert TY, et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res. 2015;21:632–641. doi: 10.1158/1078-0432.CCR-13-3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gunduz M, Gunduz E, Tamagawa S, Enomoto K, Hotomi M. Identification and chemoresistance of cancer stem cells in HPV-negative oropharyngeal cancer. Oncol. Lett. 2020;19:965–971. doi: 10.3892/ol.2019.11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weinberger PM, et al. Molecular classification identifies a subset of human papillomavirus-associated oropharyngeal cancers with favorable prognosis. J. Clin. Oncol. 2006;24:736–747. doi: 10.1200/JCO.2004.00.3335. [DOI] [PubMed] [Google Scholar]
- 53.Mandal, R. et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight1 (2016). [DOI] [PMC free article] [PubMed]
- 54.Koneva LA, et al. HPV integration in HNSCC correlates with survival outcomes, immune response signatures, and candidate drivers. Mol. Cancer Res. 2018;16:90–102. doi: 10.1158/1541-7786.MCR-17-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krupar R, et al. Immunologic and metabolic characteristics of HPV-negative and HPV-positive head and neck squamous cell carcinomas are strikingly different. Virchows Arch. 2014;465:299–312. doi: 10.1007/s00428-014-1630-6. [DOI] [PubMed] [Google Scholar]
- 56.Allen CT, Clavijo PE, Van Waes C, Chen Z. Anti-tumor immunity in head and neck cancer: understanding the evidence, how tumors escape and immunotherapeutic approaches. Cancers. 2015;7:2397–2414. doi: 10.3390/cancers7040900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mandal R, et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight. 2016;1:e89829. doi: 10.1172/jci.insight.89829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Ruiter EJ, Ooft ML, Devriese LA, Willems SM. The prognostic role of tumor infiltrating T-lymphocytes in squamous cell carcinoma of the head and neck: a systematic review and meta-analysis. Oncoimmunology. 2017;6:e1356148. doi: 10.1080/2162402X.2017.1356148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gameiro SF, et al. Treatment-naïve HPV+ head and neck cancers display a T-cell-inflamed phenotype distinct from their HPV- counterparts that has implications for immunotherapy. Oncoimmunology. 2018;7:e1498439. doi: 10.1080/2162402X.2018.1498439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Horton JD, Knochelmann HM, Day TA, Paulos CM, Neskey DM. Immune evasion by head and neck cancer: foundations for combination therapy. Trends Cancer. 2019;5:208–232. doi: 10.1016/j.trecan.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kindt N, et al. High stromal Foxp3-positive T cell number combined to tumor stage improved prognosis in head and neck squamous cell carcinoma. Oral. Oncol. 2017;67:183–191. doi: 10.1016/j.oraloncology.2017.02.023. [DOI] [PubMed] [Google Scholar]
- 62.Punt S, et al. A beneficial tumor microenvironment in oropharyngeal squamous cell carcinoma is characterized by a high T cell and low IL-17(+) cell frequency. Cancer Immunol., immunotherapy. 2016;65:393–403. doi: 10.1007/s00262-016-1805-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seminerio, I. et al. Infiltration of FoxP3+ regulatory T cells is a strong and independent prognostic factor in head and neck squamous cell carcinoma. Cancers11, 227 (2019). [DOI] [PMC free article] [PubMed]
- 64.Oguejiofor K, et al. Stromal infiltration of CD8 T cells is associated with improved clinical outcome in HPV-positive oropharyngeal squamous carcinoma. Br. J. Cancer. 2015;113:886–893. doi: 10.1038/bjc.2015.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Kempen PM, et al. Oropharyngeal squamous cell carcinomas differentially express granzyme inhibitors. Cancer Immunol., Immunother. 2016;65:575–585. doi: 10.1007/s00262-016-1819-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heusinkveld M, et al. Systemic and local human papillomavirus 16-specific T-cell immunity in patients with head and neck cancer. Int. J. Cancer. 2012;131:E74–E85. doi: 10.1002/ijc.26497. [DOI] [PubMed] [Google Scholar]
- 67.Albers A, et al. Antitumor activity of human papillomavirus type 16 E7–specific T cells against virally infected squamous cell carcinoma of the head and neck. Cancer Res. 2005;65:11146–11155. doi: 10.1158/0008-5472.CAN-05-0772. [DOI] [PubMed] [Google Scholar]
- 68.Masterson L, et al. CD8+ T cell response to human papillomavirus 16 E7 is able to predict survival outcome in oropharyngeal cancer. Eur. J. Cancer. 2016;67:141–151. doi: 10.1016/j.ejca.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 69.Yin, W., Duluc, D., Joo, H. & Oh, S. Dendritic cell targeting vaccine for HPV-associated cancer. Cancer Cell Microenviron.3 (2016). [PMC free article] [PubMed]
- 70.Partlová S, et al. Distinct patterns of intratumoral immune cell infiltrates in patients with HPV-associated compared to non-virally induced head and neck squamous cell carcinoma. Oncoimmunology. 2015;4:e965570. doi: 10.4161/21624011.2014.965570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lechien, J. R. et al. HPV Involvement in the tumor microenvironment and immune treatment in head and neck squamous cell carcinomas. Cancers12, 10.3390/cancers12051060 (2020). [DOI] [PMC free article] [PubMed]
- 72.Costa NL, et al. Tumor-associated macrophages and the profile of inflammatory cytokines in oral squamous cell carcinoma. Oral Oncol. 2013;49:216–223. doi: 10.1016/j.oraloncology.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 73.Wolf GT, et al. Tumor infiltrating lymphocytes (TIL) and prognosis in oral cavity squamous carcinoma: a preliminary study. Oral Oncol. 2015;51:90–95. doi: 10.1016/j.oraloncology.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen X, et al. Immunological network analysis in HPV associated head and neck squamous cancer and implications for disease prognosis. Mol. Immunol. 2018;96:28–36. doi: 10.1016/j.molimm.2018.02.005. [DOI] [PubMed] [Google Scholar]
- 75.Seminerio I, et al. High infiltration of CD68+ macrophages is associated with poor prognoses of head and neck squamous cell carcinoma patients and is influenced by human papillomavirus. Oncotarget. 2018;9:11046–11059. doi: 10.18632/oncotarget.24306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wagner S, et al. CD56‐positive lymphocyte infiltration in relation to human papillomavirus association and prognostic significance in oropharyngeal squamous cell carcinoma. Int. J. Cancer. 2016;138:2263–2273. doi: 10.1002/ijc.29962. [DOI] [PubMed] [Google Scholar]
- 77.Kindt N, et al. Langerhans cell number is a strong and independent prognostic factor for head and neck squamous cell carcinomas. Oral Oncol. 2016;62:1–10. doi: 10.1016/j.oraloncology.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 78.Guess JC, McCance DJ. Decreased migration of Langerhans precursor-like cells in response to human keratinocytes expressing human papillomavirus type 16 E6/E7 is related to reduced macrophage inflammatory protein-3alpha production. J. Virol. 2005;79:14852–14862. doi: 10.1128/JVI.79.23.14852-14862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang HF, et al. The double-edged sword-how human papillomaviruses interact with immunity in head and neck cancer. Front. Immunol. 2019;10:653. doi: 10.3389/fimmu.2019.00653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wellenstein MD, de Visser KE. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity. 2018;48:399–416. doi: 10.1016/j.immuni.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 81.Kanodia S, Fahey LM, Kast WM. Mechanisms used by human papillomaviruses to escape the host immune response. Curr. Cancer Drug Targets. 2007;7:79–89. doi: 10.2174/156800907780006869. [DOI] [PubMed] [Google Scholar]
- 82.Patel D, Huang SM, Baglia LA, McCance DJ. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 1999;18:5061–5072. doi: 10.1093/emboj/18.18.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou C, Tuong ZK, Frazer IH. Papillomavirus immune evasion strategies target the infected cell and the local immune system. Front. Oncol. 2019;9:682. doi: 10.3389/fonc.2019.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Matthews K, et al. Depletion of Langerhans cells in human papillomavirus type 16-infected skin is associated with E6-mediated down regulation of E-cadherin. J. Virol. 2003;77:8378–8385. doi: 10.1128/JVI.77.15.8378-8385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miura S, et al. CD1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: a possible mechanism for immune evasion by HPV. J. Virol. 2010;84:11614–11623. doi: 10.1128/JVI.01053-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Outh-Gauer S, et al. The microenvironment of head and neck cancers: papillomavirus involvement and potential impact of immunomodulatory treatments. Head Neck Pathol. 2020;14:330–340. doi: 10.1007/s12105-020-01147-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bernadt CT, Collins BT. Fine-needle aspiration biopsy of HPV-related squamous cell carcinoma of the head and neck: current ancillary testing methods for determining HPV status. Diagnostic Cytopathol. 2017;45:221–229. doi: 10.1002/dc.23668. [DOI] [PubMed] [Google Scholar]
- 88.Syrjänen S. HPV infections and tonsillar carcinoma. J. Clin. Pathol. 2004;57:449–455. doi: 10.1136/jcp.2003.008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sun Y, et al. Serum antibodies to human papillomavirus 16 proteins in women from Brazil with invasive cervical carcinoma. Cancer Epidemiol., Biomark. Prev. 1999;8:935–940. [PubMed] [Google Scholar]
- 90.Olsen AO, Dillner J, Gjøen K, Magnus P. Seropositivity against HPV 16 capsids: a better marker of past sexual behaviour than presence of HPV DNA. Genitourin. Med. 1997;73:131–135. doi: 10.1136/sti.73.2.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ho GY, Studentsov YY, Bierman R, Burk RD. Natural history of human papillomavirus type 16 virus-like particle antibodies in young women. Cancer Epidemiol., Biomark. Prev. 2004;13:110–116. doi: 10.1158/1055-9965.EPI-03-0191. [DOI] [PubMed] [Google Scholar]
- 92.Gillison ML, et al. Tobacco smoking and increased risk of death and progression for patients with p16-positive and p16-negative oropharyngeal cancer. J. Clin. Oncol. 2012;30:2102–2111. doi: 10.1200/JCO.2011.38.4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pagès F, et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J. Clin. Oncol. 2009;27:5944–5951. doi: 10.1200/JCO.2008.19.6147. [DOI] [PubMed] [Google Scholar]
- 94.Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. 2017;377:2500–2501. doi: 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goodman AM, et al. Prevalence of PDL1 amplification and preliminary response to immune checkpoint blockade in solid tumors. JAMA Oncol. 2018;4:1237–1244. doi: 10.1001/jamaoncol.2018.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hanna, G. J. et al. Frameshift events predict anti-PD-1/L1 response in head and neck cancer. JCI Insight3, 10.1172/jci.insight.98811 (2018). [DOI] [PMC free article] [PubMed]
- 97.Seiwert TY, et al. Abstract LB-339: biomarkers predictive of response to pembrolizumab in head and neck cancer (HNSCC) J. Cancer Res. 2018;78:LB-339–LB-339. [Google Scholar]
- 98.Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J. Clin. Oncol. 2006;24:2666–2672. doi: 10.1200/JCO.2005.04.8306. [DOI] [PubMed] [Google Scholar]
- 99.Zimmermann M, Zouhair A, Azria D, Ozsahin M. The epidermal growth factor receptor (EGFR) in head and neck cancer: its role and treatment implications. Radiat. Oncol. 2006;1:11. doi: 10.1186/1748-717X-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Diepenbruck M, Christofori G. Epithelial-mesenchymal transition (EMT) and metastasis: yes, no, maybe? Curr. Opin. Cell Biol. 2016;43:7–13. doi: 10.1016/j.ceb.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 101.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 102.Yokoyama K, et al. Reverse correlation of E-cadherin and snail expression in oral squamous cell carcinoma cells in vitro. Oral Oncol. 2001;37:65–71. doi: 10.1016/S1368-8375(00)00059-2. [DOI] [PubMed] [Google Scholar]
- 103.Gao L, et al. CCL2/EGF positive feedback loop between cancer cells and macrophages promotes cell migration and invasion in head and neck squamous cell carcinoma. Oncotarget. 2016;7:87037–87051. doi: 10.18632/oncotarget.13523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Finke J, et al. MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int. Immunopharmacol. 2011;11:856–861. doi: 10.1016/j.intimp.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Toh B, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011;9:e1001162. doi: 10.1371/journal.pbio.1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dohadwala M, et al. The role of ZEB1 in the inflammation-induced promotion of EMT in HNSCC. Otolaryngol.-Head Neck Surg. 2010;142:753–759. doi: 10.1016/j.otohns.2010.01.034. [DOI] [PubMed] [Google Scholar]
- 107.St John MA, et al. Proinflammatory mediators upregulate snail in head and neck squamous cell carcinoma. Clin. Cancer Res. 2009;15:6018–6027. doi: 10.1158/1078-0432.CCR-09-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Oliveira-Neto HH, et al. Involvement of CXCL12 and CXCR4 in lymph node metastases and development of oral squamous cell carcinomas. Tumour Biol.: J. Int. Soc. Oncodev. Biol. Med. 2008;29:262–271. doi: 10.1159/000152944. [DOI] [PubMed] [Google Scholar]
- 109.Schmitz S, Bindea G, Albu RI, Mlecnik B, Machiels JP. Cetuximab promotes epithelial to mesenchymal transition and cancer associated fibroblasts in patients with head and neck cancer. Oncotarget. 2015;6:34288–34299. doi: 10.18632/oncotarget.5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Onoue T, et al. Epithelial-mesenchymal transition induced by the stromal cell-derived factor-1/CXCR4 system in oral squamous cell carcinoma cells. Int. J. Oncol. 2006;29:1133–1138. [PubMed] [Google Scholar]
- 111.Yoon Y, et al. CXC chemokine receptor-4 antagonist blocks both growth of primary tumor and metastasis of head and neck cancer in xenograft mouse models. Cancer Res. 2007;67:7518–7524. doi: 10.1158/0008-5472.CAN-06-2263. [DOI] [PubMed] [Google Scholar]
- 112.Koontongkaew S, Amornphimoltham P, Monthanpisut P, Saensuk T, Leelakriangsak M. Fibroblasts and extracellular matrix differently modulate MMP activation by primary and metastatic head and neck cancer cells. Med. Oncol. 2012;29:690–703. doi: 10.1007/s12032-011-9871-6. [DOI] [PubMed] [Google Scholar]
- 113.Jung DW, et al. Tumor-stromal crosstalk in invasion of oral squamous cell carcinoma: a pivotal role of CCL7. Int. J. Cancer. 2010;127:332–344. doi: 10.1002/ijc.25060. [DOI] [PubMed] [Google Scholar]
- 114.Yu C, et al. TGF-β1 mediates epithelial to mesenchymal transition via the TGF-β/Smad pathway in squamous cell carcinoma of the head and neck. Oncol. Rep. 2011;25:1581–1587. doi: 10.3892/or.2011.1144. [DOI] [PubMed] [Google Scholar]
- 115.Hinsley EE, Kumar S, Hunter KD, Whawell SA, Lambert DW. Endothelin-1 stimulates oral fibroblasts to promote oral cancer invasion. Life Sci. 2012;91:557–561. doi: 10.1016/j.lfs.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 116.Smith A, Teknos TN, Pan Q. Epithelial to mesenchymal transition in head and neck squamous cell carcinoma. Oral Oncol. 2013;49:287–292. doi: 10.1016/j.oraloncology.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ishida T, Hijioka H, Kume K, Miyawaki A, Nakamura N. Notch signaling induces EMT in OSCC cell lines in a hypoxic environment. Oncol. Lett. 2013;6:1201–1206. doi: 10.3892/ol.2013.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhu G, et al. Hypoxia promotes migration/invasion and glycolysis in head and neck squamous cell carcinoma via an HIF-1α-MTDH loop. Oncol. Rep. 2017;38:2893–2900. doi: 10.3892/or.2017.5949. [DOI] [PubMed] [Google Scholar]
- 119.Yang MH, et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010;12:982–992. doi: 10.1038/ncb2099. [DOI] [PubMed] [Google Scholar]
- 120.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Frisch SM, Screaton RA. Anoikis mechanisms. Curr. Opin. Cell Biol. 2001;13:555–562. doi: 10.1016/S0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- 122.Kupferman ME, et al. Molecular analysis of anoikis resistance in oral cavity squamous cell carcinoma. Oral Oncol. 2007;43:440–454. doi: 10.1016/j.oraloncology.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 123.Liu XW, Bernardo MM, Fridman R, Kim HR. Tissue inhibitor of metalloproteinase-1 protects human breast epithelial cells against intrinsic apoptotic cell death via the focal adhesion kinase/phosphatidylinositol 3-kinase and MAPK signaling pathway. J. Biol. Chem. 2003;278:40364–40372. doi: 10.1074/jbc.M302999200. [DOI] [PubMed] [Google Scholar]
- 124.Cabral A, et al. Structural organization and regulation of the small proline-rich family of cornified envelope precursors suggest a role in adaptive barrier function. J. Biol. Chem. 2001;276:19231–19237. doi: 10.1074/jbc.M100336200. [DOI] [PubMed] [Google Scholar]
- 125.Moriwaki K, et al. TRKB tyrosine kinase receptor is a potential therapeutic target for poorly differentiated oral squamous cell carcinoma. Oncotarget. 2018;9:25225–25243. doi: 10.18632/oncotarget.25396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ganjre, A., Sarode, G. S., Sarode, S. C. & Patil, S. Oral squamous cell carcinoma and anoikis: a brief review on recent advances. J. Int. Oral Health8, 1043 (2016).
- 127.Walsh MF, Thamilselvan V, Grotelueschen R, Farhana L, Basson M. Absence of adhesion triggers differential FAK and SAPKp38 signals in SW620 human colon cancer cells that may inhibit adhesiveness and lead to cell death. Cell Physiol. Biochem. 2003;13:135–146. doi: 10.1159/000071864. [DOI] [PubMed] [Google Scholar]
- 128.Knowles LM, et al. HGF and c-Met participate in paracrine tumorigenic pathways in head and neck squamous cell cancer. Clin. Cancer Res. 2009;15:3740–3750. doi: 10.1158/1078-0432.CCR-08-3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zeng Q, et al. Hepatocyte growth factor inhibits anoikis in head and neck squamous cell carcinoma cells by activation of ERK and Akt signaling independent of NFkappa B. J. Biol. Chem. 2002;277:25203–25208. doi: 10.1074/jbc.M201598200. [DOI] [PubMed] [Google Scholar]
- 130.Neiva KG, et al. Cross talk initiated by endothelial cells enhances migration and inhibits anoikis of squamous cell carcinoma cells through STAT3/Akt/ERK signaling. Neoplasia. 2009;11:583–593. doi: 10.1593/neo.09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Campos MS, Neiva KG, Meyers KA, Krishnamurthy S, Nör JE. Endothelial derived factors inhibit anoikis of head and neck cancer stem cells. Oral Oncol. 2012;48:26–32. doi: 10.1016/j.oraloncology.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liao YH, et al. Epidermal growth factor-induced ANGPTL4 enhances anoikis resistance and tumour metastasis in head and neck squamous cell carcinoma. Oncogene. 2017;36:2228–2242. doi: 10.1038/onc.2016.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lian L, et al. Inhibition of MCF-7 breast cancer cell-induced platelet aggregation using a combination of antiplatelet drugs. Oncol. Lett. 2013;5:675–680. doi: 10.3892/ol.2012.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dey KK, et al. Mechanistic attributes of S100A7 (psoriasin) in resistance of anoikis resulting tumor progression in squamous cell carcinoma of the oral cavity. Cancer Cell Int. 2015;15:74. doi: 10.1186/s12935-015-0226-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 135.Zhang Y, Lu H, Dazin P, Kapila Y. Squamous cell carcinoma cell aggregates escape suspension-induced, p53-mediated anoikis: fibronectin and integrin alphav mediate survival signals through focal adhesion kinase. J. Biol. Chem. 2004;279:48342–48349. doi: 10.1074/jbc.M407953200. [DOI] [PubMed] [Google Scholar]
- 136.Bozzo C, et al. Activation of caspase-8 triggers anoikis in human neuroblastoma cells. Neurosci. Res. 2006;56:145–153. doi: 10.1016/j.neures.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 137.Bresnick AR, Weber DJ, Zimmer DB. S100 proteins in cancer. Nat. Rev. Cancer. 2015;15:96–109. doi: 10.1038/nrc3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Petruzzelli GJ. Tumor angiogenesis. Head neck. 1996;18:283–291. doi: 10.1002/(SICI)1097-0347(199605/06)18:3<283::AID-HED11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 139.Kaneko T, et al. Bcl-2 orchestrates a cross-talk between endothelial and tumor cells that promotes tumor growth. Cancer Res. 2007;67:9685–9693. doi: 10.1158/0008-5472.CAN-07-1497. [DOI] [PubMed] [Google Scholar]
- 140.Karl E, et al. Unidirectional crosstalk between Bcl-xL and Bcl-2 enhances the angiogenic phenotype of endothelial cells. Cell Death Differ. 2007;14:1657–1666. doi: 10.1038/sj.cdd.4402174. [DOI] [PubMed] [Google Scholar]
- 141.Liang Z, et al. CXCR4/CXCL12 axis promotes VEGF-mediated tumor angiogenesis through Akt signaling pathway. Biochem. Biophys. Res. Commun. 2007;359:716–722. doi: 10.1016/j.bbrc.2007.05.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lu SL, et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006;20:1331–1342. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yang JG, et al. Lymphotoxin-α promotes tumor angiogenesis in HNSCC by modulating glycolysis in a PFKFB3-dependent manner. Int. J. Cancer. 2019;145:1358–1370. doi: 10.1002/ijc.32221. [DOI] [PubMed] [Google Scholar]
- 144.Sato, S. et al. EPHB2 carried on small extracellular vesicles induces tumor angiogenesis via activation of ephrin reverse signaling. JCI Insight4, 10.1172/jci.insight.132447 (2019). [DOI] [PMC free article] [PubMed]
- 145.Ludwig N, Yerneni SS, Razzo BM, Whiteside TL. Exosomes from HNSCC promote angiogenesis through reprogramming of endothelial cells. Mol. Cancer Res. 2018;16:1798–1808. doi: 10.1158/1541-7786.MCR-18-0358. [DOI] [PubMed] [Google Scholar]
- 146.Andrade SP, Hart IR, Piper PJ. Inhibitors of nitric oxide synthase selectively reduce flow in tumor-associated neovasculature. Br. J. Pharmacol. 1992;107:1092–1095. doi: 10.1111/j.1476-5381.1992.tb13412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Burnet FM. The concept of immunological surveillance. Prog. Exp. tumor Res. 1970;13:1–27. doi: 10.1159/000386035. [DOI] [PubMed] [Google Scholar]
- 148.Lorusso G, Rüegg C. The tumor microenvironment and its contribution to tumor evolution toward metastasis. Histochem. Cell Biol. 2008;130:1091–1103. doi: 10.1007/s00418-008-0530-8. [DOI] [PubMed] [Google Scholar]
- 149.Abd Hamid M, et al. Enriched HLA-E and CD94/NKG2A interaction limits antitumor CD8(+) tumor-infiltrating T lymphocyte responses. Cancer Immunol. Res. 2019;7:1293–1306. doi: 10.1158/2326-6066.CIR-18-0885. [DOI] [PubMed] [Google Scholar]
- 150.Merlano MC, Denaro N, Garrone O. Immune escape mechanisms in head and neck squamous cell carcinoma and implication for new immunotherapy approach. Curr. Opin. Oncol. 2020;32:203–209. doi: 10.1097/CCO.0000000000000623. [DOI] [PubMed] [Google Scholar]
- 151.Baruah P, et al. Decreased levels of alternative co-stimulatory receptors OX40 and 4-1BB characterise T cells from head and neck cancer patients. Immunobiology. 2012;217:669–675. doi: 10.1016/j.imbio.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 152.Kassouf N, Thornhill MH. Oral cancer cell lines can use multiple ligands, including Fas-L, TRAIL and TNF-alpha, to induce apoptosis in Jurkat T cells: possible mechanisms for immune escape by head and neck cancers. Oral Oncol. 2008;44:672–682. doi: 10.1016/j.oraloncology.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 153.Duray A, Demoulin S, Hubert P, Delvenne P, Saussez S. Immune suppression in head and neck cancers: a review. Clin. Dev. Immunol. 2010;2010:701657. doi: 10.1155/2010/701657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bergmann C, et al. Tumor-derived microvesicles in sera of patients with head and neck cancer and their role in tumor progression. Head Neck. 2009;31:371–380. doi: 10.1002/hed.20968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sukumar M, Kishton RJ, Restifo NP. Metabolic reprograming of anti-tumor immunity. Curr. Opin. Immunol. 2017;46:14–22. doi: 10.1016/j.coi.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chikamatsu K, et al. Immunosuppressive activity of CD14+ HLA-DR- cells in squamous cell carcinoma of the head and neck. Cancer Sci. 2012;103:976–983. doi: 10.1111/j.1349-7006.2012.02248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J. Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Luo X, et al. HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J. Clin. Investig. 2020;130:1635–1652. doi: 10.1172/JCI129497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat. Rev. Cancer. 2009;9:285–293. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sleeman JP. The metastatic niche and stromal progression. Cancer Metastasis Rev. 2012;31:429–440. doi: 10.1007/s10555-012-9373-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sleeman JP, et al. Concepts of metastasis in flux: the stromal progression model. Semin. Cancer Biol. 2012;22:174–186. doi: 10.1016/j.semcancer.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 162.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006;18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 163.Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 2006;8:1369–1375. doi: 10.1038/ncb1507. [DOI] [PubMed] [Google Scholar]
- 164.Erler JT, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gao D, et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res. 2012;72:1384–1394. doi: 10.1158/0008-5472.CAN-11-2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Du R, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chang KP, et al. Overexpression of macrophage inflammatory protein-3α in oral cavity squamous cell carcinoma is associated with nodal metastasis. Oral Oncol. 2011;47:108–113. doi: 10.1016/j.oraloncology.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 168.Wang C, Liu XQ, Hou JS, Wang JN, Huang HZ. Molecular mechanisms of chemoresistance in oral cancer. Chin. J. Dent. Res. 2016;19:25–33. doi: 10.3290/j.cjdr.a35694. [DOI] [PubMed] [Google Scholar]
- 169.Masui T, et al. Snail-induced epithelial-mesenchymal transition promotes cancer stem cell-like phenotype in head and neck cancer cells. Int. J. Oncol. 2014;44:693–699. doi: 10.3892/ijo.2013.2225. [DOI] [PubMed] [Google Scholar]
- 170.Rausch MP, Sertil AR. A stressful microenvironment: opposing effects of the endoplasmic reticulum stress response in the suppression and enhancement of adaptive tumor immunity. Int. Rev. Immunol. 2015;34:104–122. doi: 10.3109/08830185.2015.1018415. [DOI] [PubMed] [Google Scholar]
- 171.Wang HC, Chan LP, Cho SF. Targeting the immune microenvironment in the treatment of head and neck squamous cell carcinoma. Front. Oncol. 2019;9:1084. doi: 10.3389/fonc.2019.01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Olson OC, Kim H, Quail DF, Foley EA, Joyce JA. Tumor-associated macrophages suppress the cytotoxic activity of antimitotic agents. Cell Rep. 2017;19:101–113. doi: 10.1016/j.celrep.2017.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Arce Vargas F, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity. 2017;46:577–586. doi: 10.1016/j.immuni.2017.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lyford-Pike S, et al. Evidence for a role of the PD-1:PD-L1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Res. 2013;73:1733–1741. doi: 10.1158/0008-5472.CAN-12-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017;52:71–81. doi: 10.1016/j.ctrv.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 176.Koyama S, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016;7:10501. doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zaretsky JM, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Palazón A, Aragonés J, Morales-Kastresana A, de Landázuri MO, Melero I. Molecular pathways: hypoxia response in immune cells fighting or promoting cancer. Clin. Cancer Res. 2012;18:1207–1213. doi: 10.1158/1078-0432.CCR-11-1591. [DOI] [PubMed] [Google Scholar]
- 179.Macha MA, et al. MicroRNAs (miRNAs) as biomarker(s) for prognosis and diagnosis of gastrointestinal (GI) cancers. Curr. Pharm. Des. 2014;20:5287–5297. doi: 10.2174/1381612820666140128213117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lajer CB, et al. The role of miRNAs in human papilloma virus (HPV)-associated cancers: bridging between HPV-related head and neck cancer and cervical cancer. Br. J. Cancer. 2012;106:1526–1534. doi: 10.1038/bjc.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wan Y, et al. Salivary miRNA panel to detect HPV-positive and HPV-negative head and neck cancer patients. Oncotarget. 2017;8:99990–100001. doi: 10.18632/oncotarget.21725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lajer CB, et al. Different miRNA signatures of oral and pharyngeal squamous cell carcinomas: a prospective translational study. Br. J. Cancer. 2011;104:830–840. doi: 10.1038/bjc.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wald AI, Hoskins EE, Wells SI, Ferris RL, Khan SA. Alteration of microRNA profiles in squamous cell carcinoma of the head and neck cell lines by human papillomavirus. Head Neck. 2011;33:504–512. doi: 10.1002/hed.21475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Dai Y, et al. MicroRNA expression profiles of head and neck squamous cell carcinoma with docetaxel-induced multidrug resistance. Head Neck. 2011;33:786–791. doi: 10.1002/hed.21540. [DOI] [PubMed] [Google Scholar]
- 185.Yu ZW, et al. MicroRNAs contribute to the chemoresistance of cisplatin in tongue squamous cell carcinoma lines. Oral Oncol. 2010;46:317–322. doi: 10.1016/j.oraloncology.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 186.Liu J, Tang Q, Li S, Yang X. Inhibition of HAX-1 by miR-125a reverses cisplatin resistance in laryngeal cancer stem cells. Oncotarget. 2016;7:86446–86456. doi: 10.18632/oncotarget.13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zhao L, et al. Deregulation of the miR-222-ABCG2 regulatory module in tongue squamous cell carcinoma contributes to chemoresistance and enhanced migratory/invasive potential. Oncotarget. 2015;6:44538–44550. doi: 10.18632/oncotarget.6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sun X, Xiao D, Xu T, Yuan Y. miRNA-24-3p promotes cell proliferation and regulates chemosensitivity in head and neck squamous cell carcinoma by targeting CHD5. Future Oncol. 2016;12:2701–2712. doi: 10.2217/fon-2016-0179. [DOI] [PubMed] [Google Scholar]
- 189.Maia D, et al. Expression of miR-296-5p as predictive marker for radiotherapy resistance in early-stage laryngeal carcinoma. J. Transl. Med. 2015;13:262. doi: 10.1186/s12967-015-0621-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Shiiba M, et al. MicroRNA-125b regulates proliferation and radioresistance of oral squamous cell carcinoma. Br. J. Cancer. 2013;108:1817–1821. doi: 10.1038/bjc.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Li G, et al. MicroRNA-324-3p regulates nasopharyngeal carcinoma radioresistance by directly targeting WNT2B. Eur. J. Cancer. 2013;49:2596–2607. doi: 10.1016/j.ejca.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 192.Li G, et al. miR-185-3p regulates nasopharyngeal carcinoma radioresistance by targeting WNT2B in vitro. Cancer Sci. 2014;105:1560–1568. doi: 10.1111/cas.12555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Suh YE, et al. MicroRNA-196a promotes an oncogenic effect in head and neck cancer cells by suppressing annexin A1 and enhancing radioresistance. Int. J. Cancer. 2015;137:1021–1034. doi: 10.1002/ijc.29397. [DOI] [PubMed] [Google Scholar]
- 194.Zhang T, et al. MiR-451 increases radiosensitivity of nasopharyngeal carcinoma cells by targeting ras-related protein 14 (RAB14) Tumour Biol.: J. Int. Soc. Oncodev. Biol. Med. 2014;35:12593–12599. doi: 10.1007/s13277-014-2581-x. [DOI] [PubMed] [Google Scholar]
- 195.Qu C, et al. MiR-205 determines the radioresistance of human nasopharyngeal carcinoma by directly targeting PTEN. Cell Cycle. 2012;11:785–796. doi: 10.4161/cc.11.4.19228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Vahabi M, et al. miR-96-5p targets PTEN expression affecting radio-chemosensitivity of HNSCC cells. J. Exp. Clin. Cancer Res. 2019;38:141. doi: 10.1186/s13046-019-1119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Qu JQ, et al. MiR-23a sensitizes nasopharyngeal carcinoma to irradiation by targeting IL-8/Stat3 pathway. Oncotarget. 2015;6:28341–28356. doi: 10.18632/oncotarget.5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Qu JQ, et al. MiRNA-203 reduces nasopharyngeal carcinoma radioresistance by targeting IL8/AKT signaling. Mol. Cancer therapeutics. 2015;14:2653–2664. doi: 10.1158/1535-7163.MCT-15-0461. [DOI] [PubMed] [Google Scholar]
- 199.Spanos WC, et al. Immune response during therapy with cisplatin or radiation for human papillomavirus–related head and neck cancer. Arch. Otolaryngol.–Head Neck Surg. 2009;135:1137–1146. doi: 10.1001/archoto.2009.159. [DOI] [PubMed] [Google Scholar]
- 200.O’Sullivan B, et al. Deintensification candidate subgroups in human papillomavirus–related oropharyngeal cancer according to minimal risk of distant metastasis. J. Clin. Oncol. 2013;31:543–550. doi: 10.1200/JCO.2012.44.0164. [DOI] [PubMed] [Google Scholar]
- 201.Meulendijks D, et al. HPV-negative squamous cell carcinoma of the anal canal is unresponsive to standard treatment and frequently carries disruptive mutations in TP53. Br. J. Cancer. 2015;112:1358. doi: 10.1038/bjc.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Baruah P, et al. TLR9 mediated tumor-stroma interactions in human papilloma virus (HPV)-positive head and neck squamous cell carcinoma up-regulate PD-L1 and PD-L2. Front. Immunol. 2019;10:1644. doi: 10.3389/fimmu.2019.01644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zandberg DP, et al. Durvalumab for recurrent or metastatic head and neck squamous cell carcinoma: results from a single-arm, phase II study in patients with >/=25% tumour cell PD-L1 expression who have progressed on platinum-based chemotherapy. Eur. J. Cancer. 2019;107:142–152. doi: 10.1016/j.ejca.2018.11.015. [DOI] [PubMed] [Google Scholar]
- 204.Powell, S. F. et al. Safety and efficacy of pembrolizumab with chemoradiotherapy in locally advanced head and neck squamous cell carcinoma: a phase IB study. J. Clin. Oncol. Jco1903156, 10.1200/jco.19.03156 (2020). [DOI] [PMC free article] [PubMed]
- 205.Ferris RL, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016;375:1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Hanoteau A, et al. Tumor microenvironment modulation enhances immunologic benefit of chemoradiotherapy. J. Immunother. Cancer. 2019;7:10. doi: 10.1186/s40425-018-0485-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Scharovsky OG, Mainetti LE, Rozados VR. Metronomic chemotherapy: changing the paradigm that more is better. Curr. Oncol. 2009;16:7–15. doi: 10.3747/co.v16i2.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tsuchikawa T, et al. The immunological impact of neoadjuvant chemotherapy on the tumor microenvironment of esophageal squamous cell carcinoma. Ann. Surgical Oncol. 2012;19:1713–1719. doi: 10.1245/s10434-011-1906-x. [DOI] [PubMed] [Google Scholar]
- 209.Bracci L, et al. Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation, and specific tumor infiltration. Clin. Cancer Res. 2007;13:644–653. doi: 10.1158/1078-0432.CCR-06-1209. [DOI] [PubMed] [Google Scholar]
- 210.Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014;21:15–25. doi: 10.1038/cdd.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Concu R, Cordeiro M. Cetuximab and the head and neck squamous cell cancer. Curr. Top. Medicinal Chem. 2018;18:192–198. doi: 10.2174/1568026618666180112162412. [DOI] [PubMed] [Google Scholar]
- 212.Jie HB, et al. CTLA-4+ regulatory T cells increased in cetuximab-treated head and neck cancer patients suppress NK cell cytotoxicity and correlate with poor prognosis. Cancer Res. 2015;75:2200–2210. doi: 10.1158/0008-5472.CAN-14-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Dovedi SJ, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014;74:5458–5468. doi: 10.1158/0008-5472.CAN-14-1258. [DOI] [PubMed] [Google Scholar]
- 214.Li M, et al. Targeting of cancer‑associated fibroblasts enhances the efficacy of cancer chemotherapy by regulating the tumor microenvironment. Mol. Med. Rep. 2016;13:2476–2484. doi: 10.3892/mmr.2016.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Albert S, et al. Focus on the role of the CXCL12/CXCR4 chemokine axis in head and neck squamous cell carcinoma. Head Neck. 2013;35:1819–1828. doi: 10.1002/hed.23217. [DOI] [PubMed] [Google Scholar]
- 216.Yu T, et al. RNAi targeting CXCR4 inhibits tumor growth through inducing cell cycle arrest and apoptosis. Mol. Ther.: J. Am. Soc. Gene Ther. 2012;20:398–407. doi: 10.1038/mt.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Schmitz S, Machiels JP. Targeting the tumor environment in squamous cell carcinoma of the head and neck. Curr. Treat. Options Oncol. 2016;17:37. doi: 10.1007/s11864-016-0412-6. [DOI] [PubMed] [Google Scholar]
- 218.Moore EC, et al. Enhanced tumor control with combination mTOR and PD-L1 inhibition in syngeneic oral cavity cancers. Cancer Immunol. Res. 2016;4:611–620. doi: 10.1158/2326-6066.CIR-15-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53:3579–3584. [PubMed] [Google Scholar]
- 220.Ishitoya J, et al. Gene amplification and overexpression of EGF receptor in squamous cell carcinomas of the head and neck. Br. J. Cancer. 1989;59:559–562. doi: 10.1038/bjc.1989.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Liang K, Ang KK, Milas L, Hunter N, Fan Z. The epidermal growth factor receptor mediates radioresistance. Int. J. Radiat. Oncol., Biol., Phys. 2003;57:246–254. doi: 10.1016/S0360-3016(03)00511-X. [DOI] [PubMed] [Google Scholar]
- 223.Şimşek H, Han Ü, Önal B, Şimişek G. The expression of EGFR, cerbB2, p16, and p53 and their relationship with conventional parameters in squamous cell carcinoma of the larynx. Turkish J. Med. Sci. 2014;44:411–416. doi: 10.3906/sag-1305-9. [DOI] [PubMed] [Google Scholar]
- 224.Ang KK, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002;62:7350–7356. [PubMed] [Google Scholar]
- 225.Bussink J, Kaanders JH, van der Kogel AJ. Microenvironmental transformations by VEGF- and EGF-receptor inhibition and potential implications for responsiveness to radiotherapy. Radiother. Oncol. J. Eur. Soc. Therapeutic Radiol. Oncol. 2007;82:10–17. doi: 10.1016/j.radonc.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 226.Rodemann HP, Dittmann K, Toulany M. Radiation-induced EGFR-signaling and control of DNA-damage repair. Int. J. Radiat. Biol. 2007;83:781–791. doi: 10.1080/09553000701769970. [DOI] [PubMed] [Google Scholar]
- 227.Ma L, et al. Cancer stem-like cell properties are regulated by EGFR/AKT/β-catenin signaling and preferentially inhibited by gefitinib in nasopharyngeal carcinoma. FEBS J. 2013;280:2027–2041. doi: 10.1111/febs.12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Young RJ, et al. Relationship between epidermal growth factor receptor status, p16(INK4A), and outcome in head and neck squamous cell carcinoma. Cancer Epidemiol., Biomark. Prev. 2011;20:1230–1237. doi: 10.1158/1055-9965.EPI-10-1262. [DOI] [PubMed] [Google Scholar]
- 229.Reimers N, et al. Combined analysis of HPV-DNA, p16 and EGFR expression to predict prognosis in oropharyngeal cancer. Int. J. Cancer. 2007;120:1731–1738. doi: 10.1002/ijc.22355. [DOI] [PubMed] [Google Scholar]
- 230.Kong CS, et al. The relationship between human papillomavirus status and other molecular prognostic markers in head and neck squamous cell carcinomas. Int. J. Radiat. Oncol., Biol., Phys. 2009;74:553–561. doi: 10.1016/j.ijrobp.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Kumar B, et al. EGFR, p16, HPV Titer, Bcl-xL and p53, sex, and smoking as indicators of response to therapy and survival in oropharyngeal cancer. J. Clin. Oncol. 2008;26:3128–3137. doi: 10.1200/JCO.2007.12.7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kumar B, et al. Response to therapy and outcomes in oropharyngeal cancer are associated with biomarkers including human papillomavirus, epidermal growth factor receptor, gender, and smoking. Int. J. Radiat. Oncol., Biol., Phys. 2007;69:S109–S111. doi: 10.1016/j.ijrobp.2007.05.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Sok JC, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin. Cancer Res. 2006;12:5064–5073. doi: 10.1158/1078-0432.CCR-06-0913. [DOI] [PubMed] [Google Scholar]
- 234.Misiukiewicz K, Posner M. The SPECTRUM of findings in treatment options for recurrent/metastatic head and neck cancer. J. Comp. Effectiveness Res. 2013;2:533–535. doi: 10.2217/cer.13.72. [DOI] [PubMed] [Google Scholar]
- 235.Bernier J, Bentzen SM, Vermorken JB. Molecular therapy in head and neck oncology. Nat. Rev. Clin. Oncol. 2009;6:266–277. doi: 10.1038/nrclinonc.2009.40. [DOI] [PubMed] [Google Scholar]
- 236.Raimondi AR, Molinolo A, Gutkind JS. Rapamycin prevents early onset of tumorigenesis in an oral-specific K-ras and p53 two-hit carcinogenesis model. Cancer Res. 2009;69:4159–4166. doi: 10.1158/0008-5472.CAN-08-4645. [DOI] [PubMed] [Google Scholar]
- 237.Zhong R, Pytynia M, Pelizzari C, Spiotto M. Bioluminescent imaging of HPV-positive oral tumor growth and its response to image-guided radiotherapy. Cancer Res. 2014;74:2073–2081. doi: 10.1158/0008-5472.CAN-13-2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Lechner A, et al. Tumor-associated B cells and humoral immune response in head and neck squamous cell carcinoma. Oncoimmunology. 2019;8:1535293. doi: 10.1080/2162402X.2018.1535293. [DOI] [PMC free article] [PubMed] [Google Scholar]