

Abstract
Introduction
Adults with Down syndrome (DS) are predisposed to Alzheimer's disease (AD) and the relationship between cognition and glucose metabolism in this population has yet to be evaluated.
Methods
Adults with DS (N = 90; mean age [standard deviation] = 38.0 [8.30] years) underwent [C‐11]Pittsburgh compound B (PiB) and [F‐18]fluorodeoxyglucose (FDG) positron emission tomography scans. Associations among amyloid beta (Aβ), FDG, and measures of cognition were explored. Interregional FDG metabolic connectivity was assessed to compare cognitively stable DS and mild cognitive impairment/AD (MCI‐DS/AD).
Results
Negative associations between Aβ and FDG were evident in regions affected in sporadic AD. A positive association was observed in the putamen, which is the brain region showing the earliest increases in Aβ deposition. Both Aβ and FDG were associated with measures of cognition, and metabolic connectivity distinguished cases of MCI‐DS/AD from cognitively stable DS.
Discussion
Associations among Aβ, FDG, and cognition reveal that neurodegeneration in DS resembles sporadic AD with the exception of the putamen, highlighting the usefulness of FDG in monitoring neurodegeneration in DS.
Keywords: Alzheimer's disease, amyloid positron emission tomography, cognitive decline, Down syndrome, fluorodeoxyglucose, individual metabolic brain network
1. INTRODUCTION
Adults with Down syndrome (DS) are predisposed to Alzheimer's disease (AD), with a sharp increase in prevalence of dementia after age 50. 1 The triplication of chromosome 21 results in overexpression of the gene encoding production of the amyloid precursor protein (APP), and an earlier presence of amyloid beta (Aβ) plaques in the brain. 2 , 3 Histopathological studies have revealed that Aβ deposition in DS begins early in life, with severe cortical prominence evident by age 40, 4 , 5 which is decades earlier than reported in cases of sporadic AD. 6 In sporadic AD, the deposition of Aβ precedes symptoms of dementia by roughly two decades, 7 and it is postulated that there is a temporal latency between Aβ presence and the progression of other AD biomarkers such as neurofibrillary tangles, glucose hypometabolism, gray matter atrophy, and cognitive decline. 8
The spatial extent of Aβ can be monitored in vivo with positron emission tomography (PET) throughout the time course of AD using Aβ‐targeting radioligands, such as [C‐11]Pittsburgh compound B (PiB). 9 In non‐demented DS, a pattern of striatum‐first Aβ retention was identified using PiB PET with the youngest case of Aβ‐positivity (Aβ+) in the striatum at age 38. 10 This is reminiscent of the pattern seen in other early‐onset forms of AD including autosomal dominant AD 11 , 12 and APP duplication. 13 With the exception of the striatum, PET studies in DS revealed that the spatial distribution of Aβ in the brain closely resembled the pattern observed in sporadic AD. 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 Longitudinal PET imaging in DS identified Aβ increases of 3% to 4% per year with the striatum showing the earliest and most prominent change compared to other cortical regions, 26 , 27 highlighting the striatum as a target region of interest to monitor early AD progression in DS. 28
Changes in cerebral glucose metabolism are well established in sporadic AD (non‐DS) when measured with [F‐18]fluorodeoxyglucose (FDG). 29 , 30 , 31 , 32 The most prominent regions showing glucose hypometabolism early on during preclinical AD progression are the parietal cortex, precuneus, and posterior cingulate, followed by the medial temporal lobe. 33 These regions also show the greatest FDG hypometabolism during the transition from cognitively stable to mild cognitive impairment (MCI). 34 The transition from MCI to AD is associated with further FDG hypometabolism in these regions as well as in the frontal lobe. 35 The regional patterns of glucose hypometabolism in familial AD are identical to that of sporadic AD; 36 however, frontal lobe hypometabolism is more prominent in these populations. 37 , 38 In DS, glucose hypometabolism has been observed in the parietal cortex, temporal cortex, and posterior cingulate. 25 , 39 , 40 To better characterize the transition from cognitively stable to MCI or AD in DS, it is important to relate imaging findings to neuropsychological measures of cognition. Previous studies in DS have shown that cognitively stable individuals tolerate elevated Aβ deposition without impact on cognition, 16 and that adults with DS that were consistently Aβ+ evidenced worsening cognitive performance. 41 Additionally, a previous study in DS attempted to relate FDG hypometabolism to cognitive function, 42 but found no observable association due to both the limited sample of MCI‐DS/AD participants and the use of a single receptive language measure to represent overall cognition.
RESEARCH IN CONTEXT
Systematic review: Previous [F‐18]fluorodeoxyglucose (FDG) studies in Down syndrome (DS) have revealed glucose hypometabolism with increased amyloid beta (Aβ) in regions implicated in Alzheimer's disease (AD). However, no association between Aβ and FDG has been observed in the striatum, a region subject to early and prominent Aβ retention in DS. Additionally, associations between FDG and cognitive performance have yet to be evaluated in this population.
Interpretation: Our findings in DS reveal the pattern of glucose hypometabolism resembles that of sporadic AD. A positive association between Aβ and FDG in the putamen was observed, suggesting this region is spared from neurodegeneration in DS. FDG hypometabolism was greatly associated with lower cognitive performance, and interregional metabolic connectivity distinguished mild cognitive impairment/AD (MCI‐DS/AD) from cognitively stable DS.
Future directions: Future research should evaluate longitudinal FDG change in DS to characterize early regional metabolism change during the transition between cognitively stable and MCI‐DS.
FDG hypometabolism in regions implicated in sporadic AD has been shown to be localized to areas of elevated Aβ plaques. 29 In sporadic AD imaging studies, there are reported cases of FDG hypermetabolism with elevated Aβ in frontal, temporal, parietal, and thalamic regions in cognitively stable and early‐MCI populations. 43 , 44 , 45 This hypermetabolism may reflect a compensatory response to the presence of Aβ 44 or higher pre‐morbid brain reserve/resistance, 43 and poses as a potential marker of early metabolic change during the preclinical AD phases. 29 In DS, however, only negative associations between Aβ and FDG have been observed in the typical AD regions. 42 Despite the early and prominent retention of Aβ in the striatum in DS, no associations with FDG have been identified, suggesting this region may be spared from AD‐associated metabolic change. 42 The same phenomenon was also observed in cases of autosomal dominant AD (ADAD), which shares similar striatum‐dominant Aβ retention with DS. 46 However, this failure to detect an association between striatal Aβ and FDG in DS may be a consequence of both small sample sizes and evaluating changes in the striatum as a whole rather than within each striatal subunit. A DS study using an attention task during FDG imaging revealed increased glucose metabolic rate in typical AD‐associated regions and the caudate that correlated with gray matter volume reduction and indicators of dementia rating, suggesting a compensatory brain response may be evident during preclinical AD progression. 47 A large DS study evaluating cerebrospinal fluid, plasma, and PET‐based biomarkers revealed decreases in glucose metabolism with symptomatic AD compared to asymptomatic individuals, suggesting there is a lengthy preclinical AD phase during which biomarkers follow predictable trajectories similar to those seen in sporadic AD. 48
The Alzheimer's Biomarker Consortium–Down Syndrome (ABC‐DS) is an ongoing longitudinal study aimed at characterizing the natural history of AD‐related biomarker change in a large DS cohort. 49 The objective of the current study was to examine the associations among Aβ, glucose metabolism, and early AD‐related cognitive decline and to evaluate the use of FDG for monitoring subtle changes in AD‐related progression. Due to the nature of early Aβ accumulation in the striatum, FDG uptake across the caudate and putamen was evaluated to identify any localized metabolic change. Finally, interregional FDG metabolic connectivity was assessed using an individual metabolic brain network and compared to a control group of age‐matched siblings without DS to classify AD‐related neurodegeneration in DS. In regard to cognition, we examined episodic memory, as this domain has been found to be especially sensitive to change in AD biomarkers early on in the transition to AD in DS, 41 , 50 as well as a composite measure of the varied cognitive domains implicated in MCI in DS. 51 , 52 , 53
2. METHODS
2.1. Participants
A total of N = 90 participants with DS (mean age [standard deviation (SD)] = 38.0 [8.30] years) and N = 14 age‐matched siblings without DS (mean age [SD] = 46.6 [13.4] years) were recruited from the University of Wisconsin‐Madison and University of Pittsburgh imaging sites of the ABC‐DS. 49 Age‐matched sibling controls without DS and free of symptoms of dementia were enrolled in the study to act as a biomarker reference group. Institutional review board approval and informed consent were obtained during enrollment into the study by the participant or legally designated caregiver. Inclusion criteria included age ≥ 25 years and having receptive language ≥ 3 years. Genetic testing was performed to confirm cases of DS (trisomy 21, mosaicism, or partial translocation). Exclusion criteria included having a prior diagnosis of dementia or a psychiatric condition that impaired cognitive functioning. In the current study, 10 participants were classified having MCI‐DS or AD, 77 were cognitively stable (CS‐DS), and the remaining 3 showed cognitive decline but possibly due to non‐AD reasons (eg, life stressors or medical conditions). These diagnostic classifications were performed independent of imaging findings and based on case consensus processing informed by directly administered and caregiver‐reported measures as previously described. 49 The three participants with cognitive decline possibly due to non‐AD reasons evidenced low to moderate Aβ, but no FDG hypometabolism and were included in analyses associating Aβ and FDG with cognition. However, these three participants were excluded from analyses requiring a definitive cognitive consensus. DS participant demographics are outlined in Table 1.
TABLE 1.
Down syndrome and sibling control participant demographics by age, sex, cognitive status, and Aβ status. Hypometabolism on FDG defined as having FDG SUVR lower than the mean SUVR from the Aβ– group by 1.5 standard deviations
| Sibling controls | All participants | Aβ– | Striatal Aβ+ | Global Aβ+ | |
|---|---|---|---|---|---|
| N | 14 | 90 | 65 | 25 | 16 |
| Age (mean [SD]) years | 46.6 (13.4) | 38.0 (8.3) | 35.1 (5.81) | 48.3 (5.87) | 50.4 (4.24) |
| Sex (M/F) | 1/13 | 45/45 | 33/32 | 12/13 | 9/7 |
| MCI‐DS/AD consensus | 0 | 10 | 2 | 8 | 7 |
| Hypometabolism on FDG | 0 | 12 | 2 | 10 | 10 |
| Episodic memory Z‐score | NA | 0.00 (1.89) | 0.57 (1.45) | –1.38 (2.11) | –2.15 (2.20) |
| Overall cognition Z‐score | NA | 0.00 (5.32) | 1.57 (4.25) | –4.09 (5.67) | –5.46 (6.01) |
Abbreviations: Aβ, amyloid beta; FDG, fluorodeoxyglucose; MCI‐DS, mild cognitive impairment‐Down syndrome; SD, standard deviation; SUVR, standardized uptake value ratio.
2.2. Sociodemographics
Sex was reported by caregivers and coded as M/F. Chronological age was coded in years. The Peabody Picture Vocabulary Test–Fourth Edition (PPVT) 54 administered at the first time point of the study was used to assess lifetime cognitive ability and has shown to be a valid measure of receptive language in adults with DS that strongly correlates with IQ. 55
Highlights:
Glucose hypometabolism in Down syndrome (DS) resembled that of sporadic Alzheimer's disease (AD).
A positive association was observed between amyloid beta and fluorodeoxyglucose (FDG) in the putamen.
FDG hypometabolism strongly correlates with lower cognitive performance in DS.
Metabolic connectivity distinguishes mild cognitive impairment‐DS/AD from cognitively stable DS.
2.3. Cognitive measures
Episodic memory was measured using the Cued Recall Test which has been shown to be reliable in DS. 56 A composite for overall cognition was generated by summing Z‐scores from a variety of cognitive tests that includes the Cued Recall Test (episodic memory), Down Syndrome Mental Status Examination (dementia symptoms/mental status), 57 Developmental Test of Visual‐Motor Integration–Fifth Edition (visual perception, fine motor skills, and hand–eye coordination), 58 the Cat and Dog Modified Stroop Task (executive functioning), 59 Purdue Pegboard (motor planning and coordination), 60 and Developmental Neuropsychological Assessment Word Generation Semantic Fluency test (verbal fluency). 61 These assessment tools have previously been shown to be promising outcome measures early on in the transition to AD in DS. 41 , 50
2.4. Magnetic resonance imaging
T1‐weighted magnetic resonance imaging (MRI) scans were acquired on a 3T GE Discovery MR750 (Wisconsin) and a Siemens Trio or Prisma (Pittsburgh) scanner. MRI scans were acquired the same day as neuropsychological evaluation. MRI images were processed using FreeSurfer v5.3.0 for the purpose of extracting volumes from the lateral ventricles, and no DS template was used for the FreeSurfer spatial normalization. MRI from all 90 participants were used in the analysis.
2.5. PET imaging
PET scans were performed on a Siemens ECAT HR+ scanner (Wisconsin). For the Pittsburgh site, both a Siemens ECAT HR+ and Siemens 4‐ring Biograph mCT were used for PET imaging. All PET imaging was performed 1 day after the neuropsychological evaluation and MRI scan. A target dose of 15 mCi of [C‐11] PiB was injected intravenously, and PET scans to measure brain Aβ plaques were acquired 50 to 70 minutes post‐injection (four 5‐minute frames). Sixteen months after PiB imaging, participants were imaged with FDG PET to assess brain glucose metabolism. A target dose of 5 mCi of FDG was injected intravenously, and scans were acquired 30 to 60 minutes post‐injection (six 5‐minute frames). PET images from all 90 participants were used in the analysis. PET frames were re‐aligned to correct for motion, averaged, and spatially normalized to the Montreal Neurological Institute 152 space (MNI152) via DS‐specific PET templates for PiB and FDG as previously described. 20 Spatial normalization was required for all PET images for regional analysis using template space regions of interest (ROIs) that account for differences in DS brain morphology compared to conventional atlas‐based ROIs, which have been previously validated for PET quantification in DS. 20 For PiB images specifically, spatial normalization was required to calculate the amyloid load (AβL), a global measure of total Aβ computed by the linear least squares method between the PET image, and images of specific radioligand binding and nonspecific/off‐target binding defined in a template space. 62 , 63 Standardized uptake value ratio (SUVR) images were generated by voxel normalization to cerebellar gray matter (PiB) or a cerebral global mean (FDG). Global Aβ was calculated from the PiB SUVR images using the AβL index following methodology specific to DS brains. 64 Regional values of SUVR were calculated for PiB and FDG in early‐stage AD regions (parietal cortex, precuneus/posterior cingulate), late‐stage regions (frontal cortex, temporal cortex, anterior cingulate), the caudate, and putamen. Aβ+ derived from prior work in the DS population was defined in the striatum for SUVR ≥ 1.43, and globally for AβL ≥ 20.0. 64
2.6. Individual metabolic brain network
A novel method for generating a metabolic brain network from a single FDG image was recently developed 65 and applied to the DS data. First, a correlation coefficient matrix was generated for FDG SUVR values across all ROIs in the sibling control group (MC). The mean (XC) and standard deviation (sC) SUVR for each individual ROI was then calculated across all sibling controls. For a single FDG image, an effect size difference (ESD) matrix between the individual DS participant and the control group was calculated:
| (1) |
where xi and xj are the regional SUVR for regions i and j from an individual DS participant, XC,i and XC,j are the mean regional SUVR from the control group, and sp(i, j) is the pooled standard deviation between the regions in the control group. Using the Fisher transformation, ESD values were converted to correlation coefficient values (R) as follows:
| (2) |
A higher value of ESD corresponds to a stronger difference of SUVR variation between regions, resulting in a weaker regional correlation coefficient. 65 A weighting factor was then applied across the regional correlation coefficients between the DS subject and the control group as W(i, j) = 1–R(i, j), such that 0 < W(i, j) < 1. The final individual connectivity matrix M was calculated as
| (3) |
where ⊙ represents element‐by‐element multiplication.
2.7. Statistical analysis
To assess the association between global AβL and FDG SUVR in striatal, early, and late‐stage AD regions in DS (described above), Pearson's correlation coefficients were calculated. Pearson's correlation coefficients were also used to evaluate the associations between striatal FDG SUVR and ventricular volume. To assess the association between AD biomarker progression and cognition, multiple linear regression models were performed. The models were performed as follows: each cognitive measure (episodic memory, overall cognition composite) was used as an outcome with AβL and regional FDG SUVR as independent variables. Regression models were repeated to adjust for chronological age and lifetime cognitive ability level (ie, PPVT). Associations were considered statistically significant for p‐values ≤ 0.05 (adjusted for Holm‐Bonferroni correction). Spearman's correlations were then performed between regional PiB and FDG SUVR across groups of Aβ– and Aβ+ individuals to assess the influence of localized Aβ on glucose metabolism. Spearman's correlations were repeated to assess the influence of global AβL on regional FDG SUVR. For the individual metabolic brain network analysis, a threshold value for connectivity was selected as the minimum from the CS‐DS group as described previously, 65 which was calculated as M = 0.10. For the subsequent analyses, only connectivity values exceeding this threshold were analyzed. Interregional connectivity from ROI data was compared across the sibling control, DS‐Aβ– and DS‐Aβ+ groups using analysis of covariance (ANCOVA) adjusting for age and sex. Post hoc Student's t‐tests were then performed across the individual groups while adjusting for Bonferroni correction. The analyses were repeated comparing the sibling control, CS‐DS, and MCI‐DS/AD groups. Because different scanners were used for image collection, all models were corrected for imaging site. Statistical analyses were performed using SAS v9.4.
3. RESULTS
3.1. Aβ and FDG correlations
Negative associations between global AβL and regional FDG SUVR were observed in early‐stage (Pearson's r [95% confidence interval (CI)] = –0.70[–0.79, –0.58]; P‐value: .00001) and late‐stage AD regions (Pearson's r = –0.33[–0.50, –0.13]; P‐value: .0015; Figure 1). For the striatum, a negative association with a large magnitude effect size (Cohen's d) 66 , 67 was observed in the caudate (Pearson's r = –0.63[–0.74, –0.49]; P‐value: .00001), while a positive association with a lower magnitude effect size was observed in the putamen (Pearson's r = 0.24[0.04, 0.43]; P‐value: .022). FDG in the caudate was associated with increased ventricle volume (Pearson's r = –0.73[–0.81, –0.62]; P‐value: .00001), with a large magnitude effect association, and a low effect magnitude association was observed between ventricle volume and FDG in the putamen (Pearson's r = 0.19[–0.02, 0.38]; P‐value: .068; Figure 2). This suggests that the positive association observed between Aβ and FDG in the putamen is independent of ventricular enlargement. Additionally, the association observed between FDG SUVR in the caudate and in the putamen was very small (Pearson's r = –0.073[–0.28, 0.14]; P‐value: .49), suggesting that putamen FDG was not associated with the observed caudate hypometabolism. To account for the partial volume effect, the striatal analysis was repeated using the Rousset geometric transfer matrix (GTM) method. 68 After GTM correction, the association observed between AβL and FDG in the caudate (Pearson's r = 0.08[–0.13, 0.28]; P‐value: .45) was also extremely small while the putamen showed a positive association (Pearson's r = 0.74[0.63, 0.82]; P‐value: .00001) with a large effect size (Figure 2). For all associations, imaging site did not influence the model outcomes.
FIGURE 1.
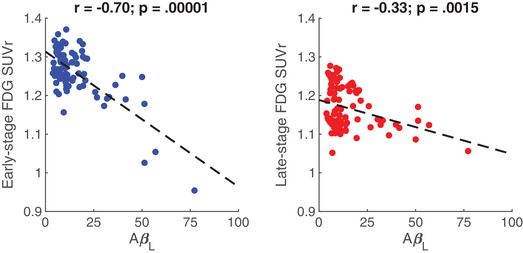
Pearson's correlations (with corresponding P‐values) between global amyloid load (AβL) and fluorodeoxyglucose standardized uptake value ratio (FDG SUVR) in early (parietal cortex, precuneus/posterior cingulate; left) and late‐stage (frontal cortex, temporal cortex, anterior cingulate; right) Alzheimer's disease regions
FIGURE 2.
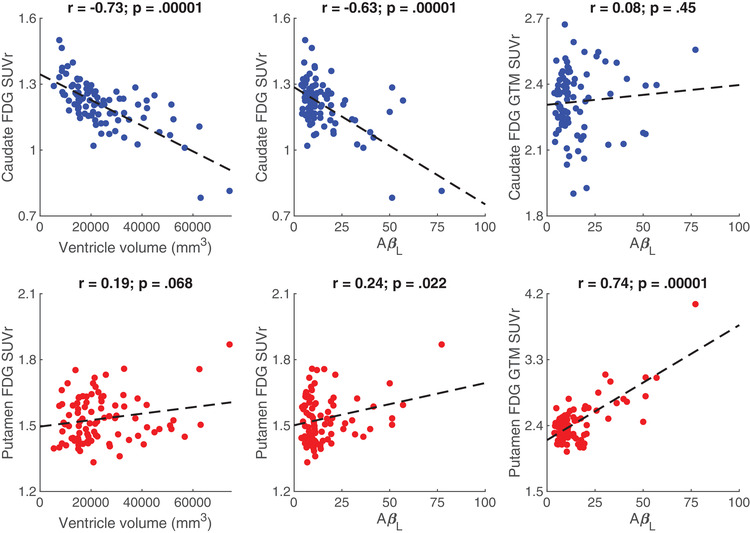
Pearson's correlations (with corresponding p‐values) for fluorodeoxyglucose standardized uptake value ratio (FDG SUVR) and ventricle volume, FDG SUVR and global amyloid load (AβL), and geometric transfer matrix‐corrected (GTM) FDG SUVR and global AβL in the caudate (top row) and putamen (bottom row)
Putamen FDG was then compared across the Aβ–, Aβ+, and sibling control groups using analysis of variance (ANOVA) with post hoc Student's t‐tests while adjusting for Bonferroni correction. The Aβ– and Aβ+ groups showed elevated putaminal FDG compared to the sibling controls (ANOVA; P = .0003). From the post hoc tests, no significant difference was observed between putamen FDG in the Aβ– and Aβ+ groups (P > .05 adjusted for Bonferroni correction).
Due to the large magnitude effect association between FDG in the caudate and ventricle volume, multiple linear regressions were performed between these measures considering linear, quadratic, and cubic polynomials. The association between caudate FDG and ventricle volume was best represented by the model that included both the quadratic and cubic terms (R2 = 0.57) compared to model considering only the linear term (one‐way ANOVA F = 9.04; P = .0003) and the model considering the linear and quadratic terms (one‐way ANOVA F = 18.1; P = .00005).
3.2. Impact of Aβ and FDG on cognition
First‐level analysis with Pearson's correlations revealed significant associations between AβL and between regional FDG SUVR with cognition in both early and late‐stage AD regions (all P < .05). No associations were observed between FDG SUVR and cognition in the caudate or putamen, and these regions were excluded from the regression analysis. From the regression models (Table 2), each variable, AβL and then FDG SUVR in both early‐ and late‐stage regions, showed significant associations (presented as slope estimates [regression coefficients] with 95% CIs) with episodic memory and overall cognition (all P < 0.05 adjusted for Holm‐Bonferroni correction). Associations with episodic memory and overall cognition remained significant for the AβL regressions and early‐stage FDG SUVR regressions after adjusting for chronological age and lifetime ability (ie, PPVT; Table 2). For late‐stage FDG regions, significant associations with overall cognition survived age adjustment. Inclusion of sex in the model parameters did not influence the outcome for all regressions performed. For all associations, imaging site did not influence the model outcomes.
TABLE 2.
Linear regression coefficient estimates (with 95% CIs) for models using cognitive measures as the outcome variable and AβL and FDG SUVR as independent variables. Regressions were repeated for each outcome variable while adjusting for chronological age and lifetime cognitive ability (PPVT). P‐values were adjusted for multiple comparisons using the Holm‐Bonferroni method
| Outcome | AβL | Early‐stage FDG | Late‐stage FDG |
|---|---|---|---|
| Episodic memory | –4.2[–5.3,–3.0]** | 0.016[0.0096,0.022]** | 0.009[0.0033,0.015]* |
| Age adjusted | –2.4[–3.5,–1.3]** | 0.013[0.0055,0.020]** | 0.0081[0.0015,0.015] |
| Age and PPVT adjusted | –2.6[–3.9,–1.2]** | 0.013[0.0042,0.022]* | 0.0048[‐0.0035,0.013] |
| Overall cognition | –1.5[–1.9,–1.0]** | 0.0061[0.0038,0.0083]** | 0.0038[0.0018,0.0058]** |
| Age adjusted | –0.89[–1.3,–0.53]** | 0.0049[0.0025,0.0072]** | 0.0035[0.0013,0.0057]* |
| Age and PPVT adjusted | –1.4[–2.0,–0.82]** | 0.0077[0.0039,0.012]** | 0.0035[–0.00013,0.0070] |
Significance: *P < .05; **P < .01.
Abbreviations: AβL, amyloid beta load; CI, confidence interval; FDG, fluorodeoxyglucose; PPVT, Peabody Picture Vocabulary Test–Fourth Edition; SUVR, standardized uptake value ratio.
3.3. FDG in relation to Aβ status
Regional PiB SUVR and global AβL were compared against regional FDG SUVR across groups of Aβ– and Aβ+ individuals (considering both striatal and global Aβ+ cutoffs) using Spearman correlations (Table 3). For both the striatal Aβ+ and global Aβ+ groups, large effect sizes were observed between regional PiB and FDG SUVR in early‐stage AD regions (striatal Aβ+: Ρ = –.60; global Aβ+: Ρ = –.50), and late‐stage AD regions (striatal Aβ+: Ρ = –.62; global Aβ+: Ρ = –.67). Large effect sizes were also displayed between global AβL with early‐stage AD regions (striatal Aβ+: Ρ = –.73; global Aβ+: Ρ = –.68) and late‐stage AD regions (striatal Aβ+: Ρ = –.68; global Aβ+: Ρ = –.68). A medium effect size was observed between global AβL and putaminal FDG SUVR in only the striatal Aβ+ group (Ρ = .45). Caudate FDG was not associated with caudate PiB or global AβL at the group level. The striatal Aβ– group displayed small effect sizes for regional PiB and FDG SUVR (Ρ = –.26) as well as global AβL and regional FDG SUVr (Ρ = –.24) in the late‐stage AD regions. Compared to the global Aβ–/Aβ+ groups, the striatal Aβ–/Aβ+ groups displayed larger effect sizes when associating Aβ and FDG. Compared to regional PiB SUVr, the global AβL metric displayed larger effect sizes in relation to regional FDG.
TABLE 3.
Spearman correlation coefficients (with 95% CIs) comparing regional PiB and FDG SUVR, as well as global AβL and regional FDG SUVR across different Aβ– and Aβ+ groups
| Striatum Aβ– N = 65 | Striatum Aβ+ N = 25 | Global Aβ– N = 74 | Global Aβ+ N = 16 | |
|---|---|---|---|---|
| Early‐stage PiB vs Early‐stage FDG | –0.082[–0.32,0.17] | –0.60[–0.80,–0.27]** | –0.061[–0.29,0.17] | –0.50[–0.80,–0.0057]* |
| Late‐stage PiB vs Late‐stage FDG | –0.26[–0.47,–0.019]* | –0.62[–0.82,–0.30]*** | –0.20[–0.41,0.031] | –0.67[–0.88,–0.27]** |
| Putamen PiB vs Putamen FDG | –0.12[–0.35,0.13] | 0.38[–0.014,0.68] | –0.18[–0.39,0.052] | 0.012[–0.49,0.50] |
| Caudate PiB vs Caudate FDG | –0.21[–0.44, 0.031] | 0.11[–0.30,0.48] | –0.18[–0.40,0.047] | 0.39[–0.13,0.74] |
| Global AβL vs Early‐stage FDG | –0.23[–0.45,0.014] | –0.73[–0.87,–0.47]*** | –0.16[–0.38,0.070] | –0.68[–0.88,–0.28]** |
| Global AβL vs Late‐stage FDG | –0.24[–0.46,–0.0011]* | –0.68[–0.85,–0.39]*** | –0.15[–0.37,0.080] | –0.68[–0.88,–0.28]** |
| Global AβL vs Putamen FDG | –0.095[–0.33,0.15] | 0.45[0.070,0.72]* | –0.15[–0.37,0.078] | 0.079[–0.43,0.55] |
| Global AβL vs Caudate FDG | –0.17[–0.40,0.073] | 0.058[–0.35,0.44] | –0.13[–0.35,0.10] | –0.056[–0.54,0.45] |
Significance: *P < .05; **P < .01; ***P < .001.
Abbreviations: AβL, amyloid beta load; CI, confidence interval; FDG, fluorodeoxyglucose; PiB, Pittsburgh comound B; SUVR, standardized uptake value ratio.
3.4. Individual metabolic brain network
Relative to the sibling control group, reductions in the number of interregional FDG connections were evident in the DS‐Aβ–, DS‐Aβ+, CS‐DS, and MCI‐DS/AD groups (Figure 3). From ANCOVA, significant differences between all groups were observed (all P < .05) surviving adjustment for age, but not sex. Inclusion of imaging site did not influence the model outcomes. From the post hoc analysis, the frequency of significant connections (presented as mean [SD]) for the sibling controls (0.60 [0.23]), DS‐Aβ– (0.44 [0.23]) and DS‐Aβ+ (0.28 [0.21]) were significantly different across all group pairings (P < .05 adjusted for Bonferroni correction). Significant differences were also observed between the sibling controls and the CS‐DS (0.43 [0.23]) and MCI‐DS/AD (0.29 [0.20]) groups (P < .05 adjusted for Bonferroni correction). Within the Aβ+ and MCI‐DS/AD groups, the weakest interregional connectivity values were evident in the early‐stage AD regions (parietal cortex, precuneus/posterior cingulate).
FIGURE 3.
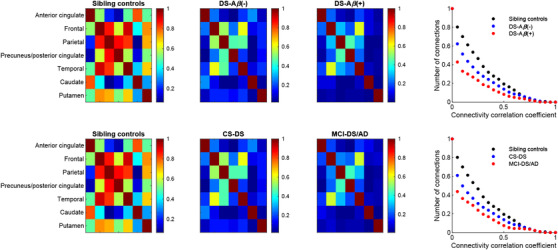
Interregional fluorodeoxyglucose connectivity matrices across the sibling control and Down syndrome groups. The plots display the number of interregional connections across all individuals in each group
4. DISCUSSION
In the DS population, we report FDG hypometabolism with elevated Aβ in typical AD regions (parietal cortex, precuneus/posterior cingulate, frontal cortex, temporal cortex) similar to the patterns observed in sporadic AD. However, a positive association between global AβL and putamen FDG SUVR emerged. Previous imaging studies in DS have attempted to relate Aβ with striatal FDG and found no association; 42 however, this is possibly a consequence of examining the striatum as a whole rather than across individual striatal subunits. Our data revealed a significant relation between lower glucose metabolism in the caudate and higher volume of the lateral ventricles. Thus, with striatal regions grouped, it is conceivable that this positive association between AβL and FDG in the putamen could be offset by caudate hypometabolism related to ventricular enlargement. After partial volume correction, the positive association between global AβL and FDG in the putamen remained significant, while the signal in the caudate showed no association with AβL. This effect may result from correction of signal spill‐in from white matter, which shows a greater effect in the putamen because it is bounded by large white matter tracts and reveals little atrophy. The analysis was then repeated using the cerebellum as the reference region for FDG SUVr calculation to ensure that the finding was not an artifact of region for normalization. The FDG and Aβ associations in the caudate and putamen were similar to those observed with the global normalization, suggesting that these regions are spared from hypometabolism during the progression of AD.
Histopathological studies in DS have revealed the presence of diffuse and cored Aβ plaques in the striatum, which accumulates larger amounts of diffuse plaques compared to the surrounding neocortical areas. 69 The abundance of diffuse plaques compared to the more neurotoxic cored plaques may spare the striatum from AD‐related neurodegeneration, as observed by the lack of FDG hypometabolism. Compared to sibling controls, the putamen showed elevated baseline FDG in DS; however, no significant difference in FDG was observed between Aβ– and Aβ+ groups. This may indicate higher basal metabolism in DS sparing the putamen from neurodegeneration. Because no measurable increase in FDG is observed in DS, the positive association between Aβ and FDG cannot be used as a direct confirmation of hypermetabolism in the putamen. However, the positive association between Aβ and FDG in the putamen may reflect an inflammatory response to Aβ, although a similar response would be expected in the caudate. These possibilities should be examined in future research that uses PET imaging of neuroinflammation (eg, translocator protein ligand) in the DS population. While striatal Aβ is indicative of preclinical AD progression in DS, the lack of FDG hypometabolism suggests this region would not be a useful marker to monitor early AD neurodegeneration.
The regression analysis revealed significant associations between Aβ and FDG and AD‐related domains of cognitive functioning in DS. A negative association was observed between Aβ and episodic memory and the overall composite of cognitive functioning. FDG hypometabolism in both early‐stage (parietal cortex, precuneus/posterior cingulate) and late‐stage (frontal cortex, temporal cortex, anterior cingulate) AD regions was associated with lower cognitive performance, and these associations were independent of chronological age. Due to the modest size of the slope estimates between FDG and our measures of cognition, a correlation analysis was performed across groups based on Aβ+ status and AD clinical status (cognitively stable, MCI‐DS, or AD). The associations between measures were found to be primarily influenced by individuals that were globally Aβ+, which includes those classified as having MCI‐DS or AD, suggesting that FDG is useful to monitor subtle changes in AD progression. Measures of episodic memory in particular have shown to be sensitive indicators of the transition between preclinical and prodromal AD in non‐DS populations, 70 and the relations of these measures in DS with FDG highlight their utility in monitoring AD progression.
For the group analysis, DS participants were classified as Aβ– or Aβ+ using either striatal or global thresholds. Large effect sizes were observed between Aβ and FDG across both striatal and global Aβ+ groups, suggesting that both classifications of Aβ+ are useful in monitoring AD progression. In the putamen, a greater association between Aβ and FDG was evident in the striatal Aβ+ group, indicating that metabolic change occurs in this region early in the course of Aβ deposition. While Aβ deposition in the striatum is detectable with PET prior to that in the neocortex, the occurrence of histologically detectable diffuse and cored plaques in the neocortex precedes that in the striatum based on post mortem studies. 69 Thus, use of a striatal classification for Aβ+ may provide more information on early metabolic change in neocortical regions. In general, FDG hypometabolism was not evident prior to the onset of Aβ+, suggesting Aβ may be a precursor to AD‐related metabolic change in DS and that the trajectories of these biomarkers are in accordance with the disease staging in sporadic AD. 8
Regional PiB SUVR and global AβL were compared against regional FDG across the different Aβ+ groups to evaluate whether any local associations are lost when using a global measure. While regional PiB SUVR was able to distinguish FDG change across Aβ+ and Aβ– groups, the global AβL measure displayed larger effect sizes and may be a more sensitive metric to predict FDG change. Previous studies have shown that the AβL metric improves quantification due to its suppression of nonspecific radioligand binding signal, resulting in greater sensitivity to detect small increases in Aβ. 63 , 64 This improved sensitivity to measure Aβ with AβL may translate to a more sensitive prediction of FDG change during the early stages of AD progression.
For sporadic AD, FDG PET has been used as a proxy for neurodegeneration as described by the AT(N) (Aβ/neurofibrillary tau/neurodegeneration) classification scheme for AD. 71 To assess the potential of FDG PET for classifying neurodegeneration in DS, an individual metabolic brain network was used to compare interregional metabolic connectivity from the FDG scan of a single participant to a group of healthy sibling controls. The metabolic brain network revealed that the number and strength of interregional connections were lower in both cognitively stable DS and MCI‐DS or AD compared to the sibling controls. Also evident was a significant difference in connectivity between the cognitively stable DS and MCI‐DS or AD that was independent of normal aging effects. One limitation to the current study was the limited sample size of participants classified as having MCI‐DS or AD (n = 10). Given the small number of adults with DS with MCI and AD, these classification groups were combined in the analysis to improve statistical power. However, in future studies it will be important to determine whether FDG PET differences are observed in the MCI group prior to conversion to AD. Additionally, n = 2 participants classified as having MCI‐DS/AD were considered Aβ– (Table 1); however, both participants had global AβL values >19 at the baseline visit and likely surpassed the Aβ+ threshold by the time of FDG imaging, which occurred 16 months after baseline. This may also suggest that our global cutoff for Aβ+ in DS may be too conservative, which is evidenced by the FDG hypometabolism observed in participants that are Aβ+ in the striatum but not globally. After consideration of a lower baseline metabolic connectivity, the individual metabolic brain network was capable of distinguishing cases of MCI and AD from cognitively stable DS, suggesting FDG PET is a useful marker for neurodegeneration in DS within the AT(N) framework.
5. CONCLUSION
Evaluating FDG PET in a large DS population revealed that the regional patterns of glucose hypometabolism throughout AD progression are largely similar to the observations in sporadic AD. Compared to Aβ PET, regional hypometabolism was not evident prior to the onset of Aβ+ status. However, a positive association between Aβ and FDG emerged in the putamen, a region subject to early and rapid accumulation of diffuse Aβ plaques in DS. FDG PET showed significant associations with measures of episodic memory and overall cognition, suggesting the utility of FDG for monitoring declines in cognition. Finally, FDG was capable of distinguishing individuals with DS with a prior diagnosis of MCI and AD from those who were cognitively stable, highlighting the utility of FDG as a marker of AD progression.
CONFLICTS OF INTEREST
GE Healthcare holds a license agreement with the University of Pittsburgh based on the technology described in this manuscript. Dr. Klunk is a co‐inventor of PiB and, as such, has a financial interest in this license agreement. GE Healthcare provided no grant support for this study and had no role in the design or interpretation of results or preparation of this manuscript. All other authors have no conflicts of interest with this work and had full access to all of the data in the study, and take responsibility for the integrity of the data and the accuracy of the data analysis.
ACKNOWLEDGMENTS
ABC‐DS: The Alzheimer's Biomarkers Consortium—Down Syndrome (ABC‐DS) project is a longitudinal study of cognition and blood‐based, genetic, and imaging biomarkers of Alzheimer's disease. This study is funded by the National Institute on Aging (NIA) grants (U01AG051406, R01AG031110, U54HD090256) and the National Institute for Child Health and Human Development (NICHD). We thank the ABC‐DS study participants and the ABC‐DS research and support staff for their contributions to this study.
This manuscript has been reviewed by ABC‐DS investigators for scientific content and consistency of data interpretation with previous ABC‐DS study publications. We acknowledge the ABC‐DS study participants and the ABC‐DS research and support staff for their contributions to this study. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Zammit MD, Laymon CM, Tudorascu DL, et al. Patterns of glucose hypometabolism in Down syndrome resemble sporadic Alzheimer's disease except for the putamen. Alzheimer's Dement. 2020;12:e12138 10.1002/dad2.12138
REFERENCES
- 1. Schupf N. Genetic and host factors for dementia in Down's syndrome. Br J Psychiatry. 2002;180:405‐410. [DOI] [PubMed] [Google Scholar]
- 2. Oyama F, Cairns NJ, Shimada H, Oyama R, Titani K, Ihara Y. Down's Syndrome: up‐Regulation of β‐Amyloid protein precursor and τ mRNAs and their defective coordination. J Neurochem. 1994;62:1062‐1066. 4159.1994.62031062.x. [DOI] [PubMed] [Google Scholar]
- 3. Rumble B, Retallack R, Hilbich C, et al. Amyloid A4 protein and its precursor in Down's Syndrome and Alzheimer's disease. N Engl J Med. 1989;320:1446‐1452. [DOI] [PubMed] [Google Scholar]
- 4. Mann DMA. Alzheimer's disease and Down's syndrome. Histopathology. 1988;13:125‐137. [DOI] [PubMed] [Google Scholar]
- 5. Wisniewski KE, Wisniewski HM, Wen GY. Occurrence of neuropathological changes and dementia of Alzheimer's disease in Down's syndrome. Ann Neurol. 1985;17:278‐282. [DOI] [PubMed] [Google Scholar]
- 6. Jack CR, Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132:1355‐1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013;12:357‐367. [DOI] [PubMed] [Google Scholar]
- 8. Jack CR, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound‐B. Ann Neurol. 2004;55:306‐319. 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 10. Handen BL, Cohen AD, Channamalappa U, et al. Imaging brain amyloid in nondemented young adults with Down syndrome using Pittsburgh compound B. Alzheimers Dement. 2012;8:496‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klunk WE, Price JC, Mathis CA, et al. Amyloid deposition begins in the striatum of presenilin‐1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007;27:6174‐6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Villemagne VL, Ataka S, Mizuno T, et al. High striatal amyloid β‐Peptide deposition across different autosomal Alzheimer disease mutation types. Arch Neurol. 2009;66:1537‐1544. [DOI] [PubMed] [Google Scholar]
- 13. Remes AM, Laru L, Tuominen H, et al. Carbon 11–Labeled Pittsburgh compound B positron emission tomographic amyloid imaging in patients with APP locus duplication. Arch Neurol. 2008;65:540‐544. [DOI] [PubMed] [Google Scholar]
- 14. Annus T, Wilson LR, Hong YTet al. The pattern of amyloid accumulation in the brains of adults with Down syndrome. Alzheimers Dement. 2016;12:538‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cole JH, Annus T, Wilson LR, et al. Brain‐predicted age in Down syndrome is associated with beta amyloid deposition and cognitive decline. Neurobiol Aging. 2017;56:41‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hartley SL, Handen BL, Devenny DA, et al. Cognitive functioning in relation to brain amyloid‐β in healthy adults with Down syndrome. Brain. 2014;137:2556‐2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jennings D, Seibyl J, Sabbagh M, et al. Age dependence of brain β‐amyloid deposition in Down syndrome. Neurology. 2015;84:500. [DOI] [PubMed] [Google Scholar]
- 18. Landt J, D'Abrera JC, Holland AJ, et al. Using positron emission tomography and carbon 11–Labeled pittsburgh compound B to image brain fibrillar β‐Amyloid in adults With Down Syndrome: safety, acceptability, and feasibility. Arch Neurol. 2011;68:890‐896. [DOI] [PubMed] [Google Scholar]
- 19. Lao PJ, Betthauser TJ, Hillmer AT, et al. The effects of normal aging on amyloid‐β deposition in nondemented adults with Down syndrome as imaged by carbon 11–labeled Pittsburgh compound B. Alzheimers Dement. 2016;12:380‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lao PJ, Handen BL, Betthauser TJ, et al. Imaging neurodegeneration in Down syndrome: brain templates for amyloid burden and tissue segmentation. Brain Imaging Behav. 2018. 10.1007/s11682-018-9888-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mak E, Bickerton A, Padilla C, et al. Longitudinal trajectories of amyloid deposition, cortical thickness, and tau in Down syndrome: a deep‐phenotyping case report. Alzheimers Dement Diagn Assess Dis Monit. 2019;11:654‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matthews DC, Lukic AS, Andrews RD, et al. Dissociation of Down syndrome and Alzheimer's disease effects with imaging. Alzheimers Dement Transl Res Clin Interv. 2016;2:69‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rafii M, Wishnek H, Brewer J, et al. The down syndrome biomarker initiative (DSBI) pilot: proof of concept for deep phenotyping of Alzheimer's disease biomarkers in down syndrome. Front Behav Neurosci. 2015;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rafii MS, Lukic AS, Andrews RD, et al. PET imaging of tau pathology and relationship to amyloid, longitudinal MRI, and cognitive change in Down Syndrome: results from the Down Syndrome biomarker initiative (DSBI). J Alzheimers Dis. 2017;60:439‐450. [DOI] [PubMed] [Google Scholar]
- 25. Sabbagh MN, Chen K, Rogers J, et al. Florbetapir PET, FDG PET, and MRI in Down syndrome individuals with and without Alzheimer's dementia. Alzheimers Dement. 2015;11:994‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lao PJ, Handen BL, Betthauser TJ, et al. Longitudinal changes in amyloid positron emission tomography and volumetric magnetic resonance imaging in the nondemented Down syndrome population. Alzheimers Dement Diagn Assess Dis Monit. 2017;9:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tudorascu DL, Minhas DS, Lao PJ, et al. The use of Centiloids for applying [11C]PiB classification cutoffs across region‐of‐interest delineation methods. Alzheimers Dement Diagn Assess Dis Monit. 2018;10:332‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cohen AD, McDade E, Christian B, et al. Early striatal amyloid deposition distinguishes Down syndrome and autosomal dominant Alzheimer's disease from late‐onset amyloid deposition. Alzheimers Dement. 2018;14:743‐750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cohen AD, Klunk WE. Early detection of Alzheimer's disease using PiB and FDG PET. Neurobiol Dis. 2014;72:117‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Minoshima S. Imaging Alzheimer's disease: clinical applications. Neuroimaging Clin N Am. 2003;13:769‐780. [DOI] [PubMed] [Google Scholar]
- 31. Mosconi L, De Santi S, Li Y, et al. Visual rating of medial temporal lobe metabolism in mild cognitive impairment and Alzheimer's disease using FDG‐PET. Eur J Nucl Med Mol Imaging. 2006;33:210‐221. [DOI] [PubMed] [Google Scholar]
- 32. Silverman DHS, Alavi A. PET imaging in the assessment of normal and impaired cognitive function. Radiol Clin North Am. 2005;43:67‐77. [DOI] [PubMed] [Google Scholar]
- 33. Mosconi L, Berti V, Glodzik L, Pupi A, De Santi S, de Leon MJ. Pre‐Clinical detection of Alzheimer's disease using FDG‐PET, with or without Amyloid Imaging. J Alzheimers Dis JAD. 2010;20:843‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de Leon MJ, Convit A, Wolf OT, et al. Prediction of cognitive decline in normal elderly subjects with 2‐[18F]fluoro‐2‐deoxy‐d‐glucose/positron‐emission tomography (FDG/PET). Proc Natl Acad Sci U S A. 2001;98:10966‐10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer's disease. Ann Neurol. 1997;42:85‐94. [DOI] [PubMed] [Google Scholar]
- 36. Verclytte S, Lopes R, Lenfant P, et al. Cerebral hypoperfusion and hypometabolism detected by arterial spin labeling MRI and FDG‐PET in early‐onset Alzheimer's disease. J Neuroimaging. 2016;26:207‐212. [DOI] [PubMed] [Google Scholar]
- 37. Kalpouzos G, Eustache F, de la Sayette V, Viader F, Chételat G, Desgranges B. Working memory and FDG–PET dissociate early and late onset Alzheimer disease patients. J Neurol. 2005;252:548‐558. [DOI] [PubMed] [Google Scholar]
- 38. Kennedy AM, Newman SK, Frackowiak RSJ, et al. Chromosome 14 linked familial Alzheimer's diseaseA clinico‐pathological study of a single pedigree. Brain. 1995;118:185‐205. [DOI] [PubMed] [Google Scholar]
- 39. Haier RJ, Alkire MT, White NS, et al. Temporal cortex hypermetabolism in Down syndrome prior to the onset of dementia. Neurology. 2003;61:1673‐1679. [DOI] [PubMed] [Google Scholar]
- 40. Schapiro MB, Haxby JV, Grady CL. Nature of mental retardation and dementia in down syndrome: study with PET, CT, and neuropsychology. Neurobiol Aging. 1992;13:723‐734. [DOI] [PubMed] [Google Scholar]
- 41. Hartley SL, Handen BL, Devenny D, et al. Cognitive decline and brain amyloid‐β accumulation across 3 years in adults with Down syndrome. Neurobiol Aging. 2017;58:68‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lao PJ, Handen BL, Betthauser TJ, et al. Alzheimer‐Like pattern of hypometabolism emerges with Elevated Amyloid‐β burden in Down Syndrome. J Alzheimers Dis JAD. 2018;61:631‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cohen AD, Price JC, Weissfeld LA, et al. Basal cerebral metabolism may modulate the cognitive effects of Aβ in mild cognitive impairment: an example of brain reserve. J Neurosci. 2009;29:14770‐14778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Johnson SC, Christian BT, Okonkwo OC, et al. Amyloid burden and neural function in people at risk for Alzheimer's Disease. Neurobiol Aging. 2014;35:576‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oh H, Habeck C, Madison C, Jagust W. Covarying alterations in Aβ deposition, glucose metabolism, and gray matter volume in cognitively normal elderly. Hum Brain Mapp. 2014;35:297‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Benzinger TLS, Blazey T, Jack CR, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc Natl Acad Sci. 2013;110:E4502‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Haier RJ, Head K, Head E, Lott IT. Neuroimaging of individuals with Down's syndrome at‐risk for dementia: evidence for possible compensatory events. NeuroImage. 2008;39:1324‐1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fortea J, Vilaplana E, Carmona‐Iragui M, et al. Clinical and biomarker changes of Alzheimer's disease in adults with Down syndrome: a cross‐sectional study. The Lancet. 2020;395:1988‐1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Handen BL, Lott IT, Christian BT, et al. The Alzheimer's biomarker consortium‐Down Syndrome: rationale and methodology. Alzheimers Dement Diagn Assess Dis Monit. 2020;12:e12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hartley SL, Handen BL, Devenny D, et al. Cognitive indicators of transition to preclinical and prodromal stages of Alzheimer's disease in Down syndrome. Alzheimers Dement Diagn Assess Dis Monit. 2020;12:e12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Firth NC, Startin CM, Hithersay R, et al. Aging related cognitive changes associated with Alzheimer's disease in Down syndrome. Ann Clin Transl Neurol. 2018;5:741‐751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Startin CM, Lowe B, Hamburg S, et al. Validating the cognitive scale for Down Syndrome (CS‐DS) to detect longitudinal cognitive decline in adults with Down Syndrome. Front Psychiatry. 2019:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Krinsky‐McHale SJ, Zigman WB, Lee JH, et al. Promising outcome measures of early Alzheimer's dementia in adults with Down syndrome. Alzheimers Dement Diagn Assess Dis Monit. 2020;12:e12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dunn LM, Dunn DM. Peabody picture vocabulary test. 4th ed San Antonio, TX: NCD Pearson, Inc.; 2007. [Google Scholar]
- 55. Phillips BA, Loveall SJ, Channell MM, Conners FA. Matching variables for research involving youth with Down syndrome: leiter‐R versus PPVT‐4. Res Dev Disabil. 2014;35:429‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zimmerli E, Devenny DA. Cued recall as a screen for dementia in the MR population. Gatlinburg Conf Res Theory Ment Retard Dev Disabil. 1995. [Google Scholar]
- 57. Haxby JV. Neuropsychological evaluation of adults with Down's syndrome: patterns of selective impairment in non‐demented old adults. J Intellect Disabil Res. 1989;33:193‐210. [DOI] [PubMed] [Google Scholar]
- 58. Beery KE, Buktenica NA, Beery NA. Beery VMI: The Beery‐Buktenica developmental test of visual‐motor integration. 5th ed. Minneapolis, MN: Pearson; 2004. [Google Scholar]
- 59. Nash HM, Snowling MJ. Semantic and phonological fluency in children with Down syndrome: atypical organization of language or less efficient retrieval strategies?. Cogn Neuropsychol. 2008;25:690‐703. [DOI] [PubMed] [Google Scholar]
- 60. Vega A. Use of Purdue Pegboard and finger tapping performance as a rapid screening test for brain damage. J Clin Psychol. 1969;25:255‐258. [DOI] [PubMed] [Google Scholar]
- 61. Korkman M, Kirk U, Kemp S. NEPSY. 2nd ed. San Antonio, TX: Harcourt Assessment, Inc.; 2007. [Google Scholar]
- 62. Whittington A, Sharp DJ, Gunn RN. Spatiotemporal distribution of β‐Amyloid in Alzheimer disease is the result of heterogeneous regional carrying capacities. J Nucl Med. 2018;59:822‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Whittington A, Gunn RN. Amyloid Load – a more sensitive biomarker for amyloid imaging. J Nucl Med. 2018. 10.2967/jnumed.118.210518.jnumed.118.210518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zammit MD, Laymon CM, Betthauser TJ, et al. Amyloid accumulation in Down syndrome measured with amyloid load. Alzheimers Dement Diagn Assess Dis Monit. 2020. 10.1002/dad2.12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Huang S‐Y, Hsu J‐L, Lin K‐J, Hsiao I‐T. A novel individual metabolic brain network for 18F‐FDG PET imaging. Front Neurosci. 2020;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cohen J. A power primer. Psychol Bull. 1992;112:155‐159. [DOI] [PubMed] [Google Scholar]
- 67. Cohen J. Statistical power analysis for the behavioral sciences. 2nd ed. Hillsdale, N.J: L. Erlbaum Associates; 1988. [Google Scholar]
- 68. Rousset OG, Ma Y, Evans AC. Correction for partial volume effects in PET: principle and validation. J Nucl Med. 1998;39:904‐911. [PubMed] [Google Scholar]
- 69. Abrahamson EE, Head E, Lott IT, et al. Neuropathological correlates of amyloid PET imaging in Down syndrome. Dev Neurobiol. 2019;79:750‐766.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gagliardi G, Epelbaum S, Houot M, et al. Which episodic memory performance is associated with Alzheimer's disease biomarkers in elderly cognitive complainers? evidence from a longitudinal observational study with four episodic memory tests (Insight‐PreAD). J Alzheimers Dis. 2019;70:811‐824. [DOI] [PubMed] [Google Scholar]
- 71. Jack CR, Bennett DA, Blennow K, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 2016;87:539‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]