

Abstract
Tumor-induced osteomalacia (TIO), caused by phosphaturic mesenchymal tumors (PMTs), is a rare paraneoplastic syndrome characterized by frequent bone fractures, bone pain, muscle weakness, and affected gait. These tumors typically secrete high levels of Fibroblastic Growth Factor 23 (FGF23), a hormone which acts on the kidney to cause hypophosphatemia, ultimately impairing bone mineralization. In this case report, we present a 41-year-old female with FGF23-mediated hypophosphatemia with a 26-year delay in TIO diagnosis and a concurrent misdiagnosis of X-linked hypophosphatemic rickets (XLH). Given an absence of family history of hypophosphatemia, a 13-gene hypophosphatemia panel including XLH (PHEX gene) was performed and came back negative prompting a diagnostic search for a PMT causing TIO. A 68Ga-DOTATATE PET/CT scan revealed the presence of a 9th right rib lesion, for which she underwent rib resection. The patient's laboratory values (notably serum phosphorus, calcium, and vitamin D) normalized, with FGF23 decreasing immediately after surgery, and symptoms resolving over the next three months. Chromogenic in situ hybridization (CISH) and RNA-sequencing of the tumor were positive for FGF23 (CISH) and the transcriptional marker FN1-FGFR1, a novel fusion gene between fibronectin (FN1) and Fibroblast Growth Factor Receptor 1 (FGFR1), previously determined to be present in the majority of TIO-associated tumors. This case demonstrates the notion that rare and diagnostically challenging disorders like TIO can be undiagnosed and/or misdiagnosed for many years, even by experienced clinicians and routine lab testing. It also underscores the power of novel tools available to clinicians such as gene panels, CISH, and RNA sequencing, and their ability to characterize TIO and its related tumors in the context of several phenotypically similar diseases.
Keywords: Tumor-induced osteomalacia, TIO, Fibroblast growth factor 23, FGF23, Paraneoplastic, Phosphaturic mesenchymal tumor, 68Ga-DOTATATE, Burosumab, Chromogenic in situ hybridization, RNA sequencing, X-linked hypophosphatemic rickets, XLH, PHEX, FN1-FGFR1, fibronectin, FN1, Fibroblast growth factor receptor 1, FGFR1, phosphorous, phosphate wasting disorders
Graphical abstract
(A) CT (white arrows = mass). (B) PET (white arrows = activity). (C) 68 Ga-DOTATE Scan Axial (red arrows = activity). (D) 68 Ga-DOTATE Scan Coronal (A/P) (red arrows = activity). (E) Post-operative image of 9th right rib removed during the second, successful surgery. White stars (*) denote the two areas of abnormality, red arrow denotes the primary lesion of concern for PMT shown histologically in F and G. (F) Histological appearance of the Phosphaturic Mesenchymal Tumor (PMT) specifically showing infiltration of rib bone and proliferation of uniform short spindle to ovoid cells with a rich vascular background and cystic changes. (G) FGF23 Chromogenic in situ hybridization (CISH) of the PMT.

Highlights
-
•
TIO is caused by FGF23 hypersecretion by phosphaturic mesenchymal tumors (PMTs).
-
•
TIO can go undiagnosed and/or be misdiagnosed with similar disorders (e.g. XLH).
-
•
Family history, biochemistry, gene panels, and imaging may help distinguish TIO.
-
•
FN1-FGF1 and FN1-FGFR1 are transcribed and synthesized by the majority of PMTs.
-
•
Burosumab (anti-FGF23) and FGF/R inhibitors show promise; resection is preferred.
1. Introduction
Osteomalacia is the progressive softening of bones due to decreased mineralization of calcium and phosphates at sites of bone remodeling and growth, commonly resulting in bone pain and muscle weakness (Michigami, 2019). Fibroblast Growth Factor 23 (FGF23) is a protein secreted by osteocytes mainly in response to calcitriol and phosphate (Martin et al., 2012). FGF23 regulates plasma phosphate concentration predominately by reducing serum calcitriol which mediates intestinal phosphate absorption and by reducing NPT2-mediated phosphate reabsorption in the kidney. Tumor-Induced Osteomalacia (TIO) is a rare paraneoplastic disease in which a phosphaturic mesenchymal tumor (PMT) secretes significant amounts of FGF23 into the bloodstream, leading to downstream osteomalacia through renal phosphate wasting and defective Vitamin D metabolism (Yin et al., 2018). While less than 500 cases of TIO have been reported worldwide, and formal epidemiological studies of the disease have not been performed, surgical resection offers a potentially curative treatment in contrast to similarly presenting disorders (Folpe, 2019).
X-linked hypophosphatemia (XLH) is a phosphate wasting disorder caused by mutations in the PHEX gene which expresses the enzyme phosphate-regulating neutral endopeptidase (PHEX). Studies suggest that the PHEX enzyme may be involved in the regulation of FGF23 (Beck-Nielsen et al., 2019; Liu et al., 2003). Importantly, diagnosing phosphate wasting disorders based purely on clinical criteria is confounded by the similarity of clinical and radiological presentations of this family of disorders. Knowing the distinct genetic basis for a patient's phosphate wasting disorder often allows clinicians to tailor their treatment/management strategy accordingly, potentially saving years of hardship for affected patients. As such, recent emphasis has been placed on using gene panels to accelerate clinicians' diagnosis of these disorders.
In particular, a potential for misdiagnosis of TIO as XLH exists because FGF23 levels are increased in both, PMT tumors are notoriously difficult to find due to their small size and variable location, and (albeit still rare) the incidence of XLH far exceeds that of TIO (Yin et al., 2018; Carpenter, 1997). Here, we present a 41-year-old female in whom a 26-year delayed TIO diagnosis and concurrent misdiagnosis of XLH was identified via a negative hypophosphatemia gene panel and corrected to TIO through identification of a PMT of her 9th right rib. Surgical resection resulted in biochemical and clinical symptom improvement. Histological analysis was positive for FGF23 (CISH) and the FN1-FGFR1 transcriptional marker. Finally, we highlight recent key advances in diagnosing phosphate wasting disorders, molecular characterization of PMTs, and TIO treatment and management.
2. Case presentation
A 41-year-old woman with a prior clinical diagnosis of hypophosphatemic rickets presented to our institution with a 26-year history of low phosphorus, bone pain, proximal muscle weakness, gait abnormalities, and multiple traumatic and insufficiency fractures of the hip, humerus, and spine (Fig. 1, Fig. 7). She had no history of hearing loss. She had lost all her secondary teeth (edentulous). The patient reported no family history of hypophosphatemia, including her 8-year-old male child and 17-year-old female child, neither of whom showed clinical or laboratory indications of musculoskeletal conditions or phosphate wasting disorders. Her maximum height was 5′2″ but she presented to us at 4′8″ due to progressive spine deformities.
Fig. 1.

Patient musculoskeletal radiology. X-ray AP Pelvis (A) shows evidence of prior bilateral obturator ring fractures with bilateral femoral head/neck angular deformities. X-ray Spine Total AP/Lateral (B-C) shows normal trabecular bone with vertebral body alignments and interspacing intact, but there is mild flattening of the mid vertebral body at multiple levels. There is evidence of previous insufficiency fractures.
Fig. 7.
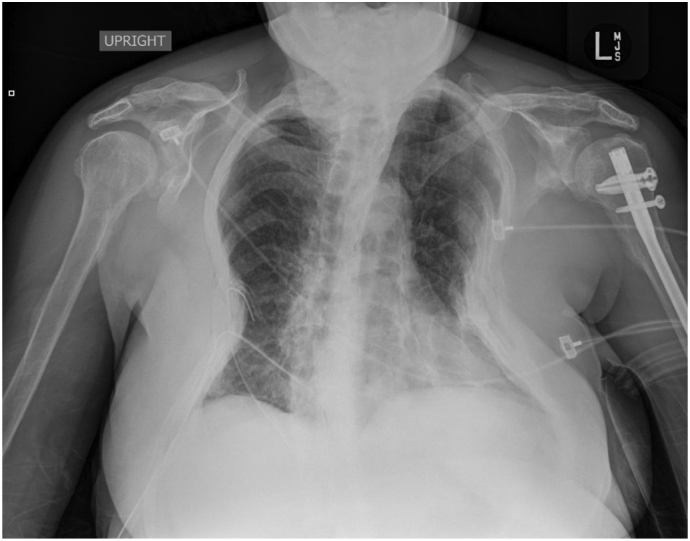
Post-operative portable chest X-ray after rib removal (second successful surgery). A left humeral rod and right chest tube are observed. No pneumothorax is seen. The lungs appear clear except for volume loss related to the overlying thoracic deformity. Extensive bone deformities identified, and multiple performed ribs are seen bilaterally but both appear similar to pre-operative radiographs.
Despite intermittent treatment with conventional therapy with phosphorus supplements and calcitriol during her teens and 20s, she developed progressive gait abnormalities and decreased hip strength and ultimately sustained bilateral hip fractures (Fig. 1A) and small fractures throughout her spine (Fig. 1B-C).
Despite treatment, the patient's symptoms deteriorated further in her 20s, when she had stability rods and screws placed bilaterally in her femurs and hips, respectively (Fig. 1A). Ten years later, the patient presented with a traumatic left humerus fracture, for which she underwent surgery (Figure 7). Post-operatively, the patient was placed on the osteoporosis medication teriparatide.
Teriparatide therapy failed to prevent more spinal fractures (Fig. 1B-C), bone pain, and muscle weakness over the next year, and the patient was eventually confined to a wheelchair. Orthopedic physicians reevaluated her history, officially diagnosed her with X-linked Hypophosphatemia (XLH) (although patient claims having an “unofficial” XLH diagnosis years prior by previous physicians), and started her on Burosumab (60 mg subcutaneous injection every 28 days) - the monoclonal antibody targeted against FGF23. Unfortunately, laboratory values relevant to phosphate wasting (e.g. TmP/GFR, FGF23) were not reported prior to initiation of Burosumab therapy or referral to our institution for further evaluation and treatment.
The patient remained on Burosumab for 3 months. While on Burosumab, the patient reported mild symptomatic improvement with no reported adverse effects, but still had severe limitations in mobility (difficulty from sit to stand, Video 1), fatigue, and pain. On therapy, FGF23 levels were 33,900 RU/mL (normal <180RU/mL) and 36,900 RU/mL two months later as measured by ELISA (Quidel Corporation). Other notable preoperative laboratory values include a normal serum phosphorus, normal calcium, normal total vitamin D, elevated 1,25 dihydroxyvitamin D (333 pg/mL; normal 19.9–79.3 pg/mL), high alkaline phosphatase (168 U/mL; normal 40–150 U/mL), and high PTH (189 pg/mL, normal 16–77 pg/mL) (Table 2). These findings are abnormal for a patient with TIO, but they should be viewed in the context of Burosumab therapy. After discontinuation, her total FGF23 declined to 1532 RU/mL after discontinuing Burosumab but before excision of her PMT which was still considerably elevated above reference range.
Table 2.
Longitudinal patient laboratory values. Op 1 denotes the first unsuccessful 10th rib removal operation. Op 2 denotes the second successful 9th rib removal operation. PTH = parathyroid hormone. Note = Pre-Op 1 labs were taken while the patient was on Burosumab, all other labs were taken while the patient was off Burosumab, and Post-Op 2 labs were taken 3 months after resection.
| Laboratory value | Normal range | Pre-Op 1 (on Burosumab) | Post-Op 1, Pre-Op 2 | Post-Op 2 (3 months after 9th right rib resection) |
|---|---|---|---|---|
| Serum phosphorus | 2.3–4.7 mg/dL | 2.5 | 1.4 | 3.7 |
| Calcium | 8.4–10.5 mg/dL | 9.7 | 9.3 | 10.4 |
| Total vitamin D | 25–80 ng/mL | 40 | 38 | 35 |
| 1,25 dihydroxyvitamin D | 19.9–79.3 pg/mL | 333 | 15 | 327 |
| Alkaline phosphatase | 40–150 U/L | 168 | 111 | 130 |
| Chloride | 96–106 mmol/L | 108 | 109 | 105 |
| PTH | 16–77 pg/mL | 189 | Not collected | 194 |
| FGF23 | <180 RU/mL | 36,900 | 1532 | 245 |
At our clinic, areal bone density of the lumbar spine and left forearm (bone density of the femurs could not be performed due to bilateral hip prostheses) were assessed by dual-energy X-ray absorptiometry (DXA) using a GE Lunar Prodigy Advanced densitometer. Densitometry revealed normal bone density in the spine and significantly lower than expected density in the forearm (Fig. 2A-B). A renal ultrasound showed a 4-mm non-obstructing calculus in the lower pole of the right kidney, indicating nephrolithiasis, a well-documented complication of long-term phosphorus and calcitriol therapy (Jan de Beur, 2005).
Fig. 2.

Pre-operative DXA scans measuring bone mineral density (BMD). (A) Pre-operative bone density of the lumbar spine, (L1-L4): 1.202 g per square cm, T-score (SD of peak BMD): 0.1 Z-score (SD of age-matched BMD): 0.1. (B) Pre-operative bone density of the left forearm (radius 33%) is: 0.694 g per square cm T-score (SD of peak BMD): −2.1 Z-score (SD of age-matched BMD): −2.1. Bone density of the femurs could not be performed due to bilateral hip prostheses.
Additionally, a 13-gene Invitae hypophosphatemia panel (https://www.invitae.com/en/hypophosphatemia) (Table 1) was performed to adjudicate her clinical diagnosis of XLH. No pathogenic variants were detected prompting our team to suspect TIO and re-evaluate her previous XLH diagnosis.
Table 1.
Genes tested on the Invitae hypophosphatemia next-generation sequencing panel (https://www.invitae.com/en/hypophosphatemia), with associated localization, diseases, and pathological mutations (Lloyd et al., 1996; Priante et al., 2017; Root, 2018). Panel is sensitive to deletions, insertions, duplications and copy number variants, and single-nucleotide polymorphisms (SNPs). AD = autosomal dominant, AR = autosomal recessive, VDDR = Vitamin D-dependent rickets.
| Gene | Primary location(s) of expression | Type of mutation precipitating disease | Associated hypophosphatemic/osteomalacic disease |
|---|---|---|---|
| ALPL | Osteoblasts | Loss of function | Hypophosphatasia |
| CLCN5 | Proximal renal tubule | Loss of function | X-linked recessive hypophosphatemic rickets |
| CYP27B1 | Many cell types | Loss of function | AR VDDR type 1A |
| CYP2R1 | Hepatocytes | Loss of function | AR VDDR type 1B |
| DMP1 | Osteoblasts, osteocytes | Loss of function | AR hypophosphatemic rickets |
| ENPP1 | Chondrocytes, osteocytes, plasma cells | Loss of function | AR hypophosphatemic rickets |
| FAH | Hepatocytes | Loss of function | AR tyrosinemia type 1 |
| FAM20C | Osteoblasts, osteocytes | Loss of function | AR osteosclerotic bone dysplasia (Raine syndrome) |
| FGF23 | Osteoblasts, osteocytes | Gain of function | AD hypophosphatemic rickets |
| FGFR1 | Many cell types | Gain of function | AD type 1 Pfeiffer syndrome and osteoglophonic dysplasia; synthesis upregulated in many TIO-associated tumors |
| PHEX | Osteoblasts | Loss of function, X-linked | XLH, increased FGF23 secretion |
| SLC34A3 | Proximal renal tubule | Loss of function | AR hypophosphatemic rickets with hypercalciuria |
| VDR | Loss of function | Calcitriol resistance, VDDR type 2A |
Due to suspected TIO, a combined 68Ga-DOTATATE PET/CT scan was performed. The CT Scan (Fig. 3A) revealed an unusual lesion in her 9th lateral right rib; PET scan (Fig. 3B) showed that this lesion had high activity; and a 68Ga-DOTATATE scan (an indirect measure of somatostatin receptors which are highly expressed in most TIO-causing tumors) demonstrated activity in the same region (Fig. 3C-D).
Fig. 3.

Diagnostic radiologic characterization of the right rib mass. (A) CT (white arrows = mass). (B) PET (white arrows = activity). (C) 68 Ga-DOTATE Scan Axial (red arrows = activity). (D) 68 Ga-DOTATE Scan Coronal (A/P) (red arrows = activity).
Some TIO patients with unresectable or unidentifiable PMTs have benefited greatly from Burosumab therapy (Jan De Beur et al., 2019). However, the accessibility of our patient's tumor prompted us to refer her to Thoracic Surgery for complete resection of the tumor and rib. Further, Burosumab therapy was discontinued prior to measuring baseline laboratory values due to potential confounding, especially with the FGF23 level.
Intra-operatively, surgeons identified a more concerning lesion on the patient's 10th right rib (Fig. 4A) that had no imaging correlate. This lesion was removed, and the surgeons deferred removal of the 9th right rib to ensure the 10th right rib lesion was not the culprit of her TIO symptoms or other pathology warranting alternative intervention. The patient's hypophosphatemia persisted post-operatively. 4-months after surgery, notable lab values included low serum phosphorus (1.4 mg/dL; normal 2.3–4.7 mg/dL), low 1,25 dihydroxyvitamin D (15 pg/mL; 19.9–79.3 pg/mL), normal total vitamin D (38 ng/mL; 25–80 ng/mL), and elevated total FGF23 (1532 RU/mL; <180 RU/mL). Histopathology of the 10th right rib mass showed only non-specific reactive changes, possibly representing an old fracture site.
Fig. 4.

Gross appearance of surgical specimens. (A) Intra-operative image of 10th right rib mass (no imaging correlate) removed during the first, unsuccessful surgery. White star (*) denotes the large abnormality that prompted surgeons to remove the 10th rib mass. (B-C) Post-operative images of 9th right rib removed during the second, successful surgery. (B) White stars (*) denote the two areas of abnormality, red arrow denotes the primary lesion of concern for PMT shown in Fig. 5, Fig. 6. (C) Significant thickening observed in the 9th right rib.
These findings further implicated the active 9th rib lesion in her pathology (Figure 3), prompting Thoracic Surgery to remove the 9th right rib (Fig. 4B-C) 5 months after the first unsuccessful surgery. Gross appearance of the 9th right rib showed two areas of abnormality with one denoting the PMT lesion (Fig. 4B) and significant rib thickening along the lateral surface (Fig. 4C). Histological sections of the 9th right rib showed a morphologically classical phosphaturic mesenchymal tumor, consisting of bland spindled cells in a highly vascular background, with unusually hyalinized to occasionally calcified matrix (Fig. 5). Chromogenic in situ hybridization (CISH) of the tumor and RNA-sequencing for the presence of rearrangements in 138 target genes were pursued using methods previously published (Martinez et al., 2019; Carter et al., 2015). FGF23 CISH was strongly positive (Fig. 6). Sequencing was positive for the transcriptional marker FN1-FGFR1, a novel fusion gene between fibronectin (FN1) and Fibroblast Growth Factor Receptor 1 (FGFR1). Taken together, these results are characteristic of a typical PMT.
Fig. 5.

Histological appearance of 9th right rib phosphaturic mesenchymal tumor (PMT). A) Lower magnification showing infiltration of rib bone (pink, paucicellular) by the PMT. B) Higher magnification (black box of A) of the PMT specifically showing proliferation of uniform short spindle to ovoid cells with a rich vascular background and cystic changes. C) Low magnification of the PMT from a different depth of sectioning.
Fig. 6.
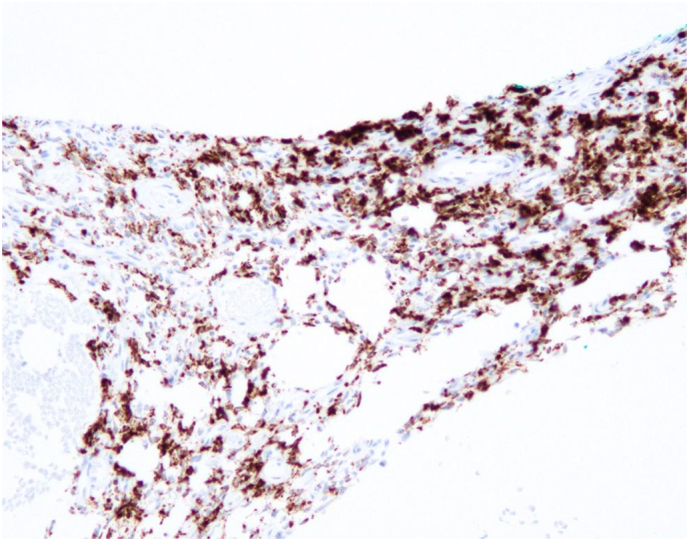
FGF23 chromogenic in situ hybridization (CISH) of 9th right rib phosphaturic mesenchymal tumor (PMT).
3 months post-resection, chest x-rays showed no pneumothorax and no significant changes compared to prior radiographs (Fig. 7). FGF23 levels decreased again but remained slightly elevated (245 RU/mL), serum phosphorus normalized (3.7 mg/dL), alkaline phosphatase normalized (130 U/mL), and total vitamin D was 35 ng/mL. Parathyroid hormone remained elevated (194 pg/mL) (Table 2). Clinical assessments of mobility markedly improved (Video 1, Video 2), and the patient has stated she is in less pain and is more active and feels stronger than ever. She will continue to follow up longitudinally in our Endocrinology clinic.
3. Discussion
TIO is a paraneoplastic disease resulting in elevated FGF23, renal phosphate wasting, defective Vitamin D metabolism, and subsequent osteomalacia. Clinically, patients with TIO present with progressive bone pain, muscle weakness, fatigue, and sporadic insufficiency fractures (Yin et al., 2018). TIO can be suggested by serum hypophosphatemia, hyperphosphaturia, and low or inappropriately normal calcitriol levels, but the identification of a phosphaturic mesenchymal tumor (PMT) in the soft tissue or bone is necessary for definitive diagnosis (Feng et al., 2017). Here, we presented a 41-year-old woman suffering from TIO with a 26-year delay in diagnosis with a concurrent misdiagnosis of XLH.
Diagnosing TIO is often complicated by small tumor size and similar initial clinical presentation to more common phosphate wasting disorders such as XLH. A retrospective study of 144 TIO cases found that TIO was misdiagnosed in over 95% (137/144) of cases, with intervertebral disc herniation, spondyloarthritis, and osteoporosis being the most common misdiagnoses (Feng et al., 2017). In this case, our patient was misdiagnosed for 26 years with little symptomatic improvement with either conventional therapy or Burosumab, and subsequent gene paneling at our institution revealed no genetic evidence of her XLH diagnosis.
Patients who present with bone pain, muscle weakness, and multiple fractures should be evaluated for hypophosphatemia, and TMP/GFR should be performed for hypophosphatemic patients. While diagnostically burdensome, a low TMP/GFR is a powerful indicator of renal phosphate wasting in the absence of secondary hyperthyroidism (Jagtap et al., 2012). Though less sensitive than TMP/GFR, calculating a percent tubular reabsorption of phosphate (TRP) is a more easily collected alternative (Takeda et al., 2015). Additionally, 24-h urine phosphorus and calcium can differentiate between malabsorption and phosphate wasting disorders (Payne, 1998).
Patient FGF23 levels and family/personal histories are also important tools for differentiating between phosphate wasting disorders (Table 3). As discussed in our previous report, TIO distinguishes itself from other FGF23-high phosphate wasting disorders in that it often presents with rapid symptom onset later in life, often with a negative family history. On the other hand, XLH is usually inherited as an X-linked dominant inherited disorder that typically presents in early childhood (MD, 2012). As such, genetic testing and associated costs can often be avoided if a clear X-linked pattern is identified by pedigree. That said, XLH can still occur due to de novo mutations, so absence of a family history should not entirely rule out XLH (Durmaz et al., 2013).
Table 3.
Phosphate wasting disorder differential diagnosis. A differential diagnosis table (not extensive) for phosphate wasting disorders, including TIO, distinguished by serum FGF23 level, reproduced from Colazo et al. (n.d.), Imel and Econs (2012), and Zoller et al. (2017).
| Phosphate wasting disorders with low FGF23 | Phosphate wasting disorders with high or “inappropriately normal” FGF23 |
|---|---|
| Fanconi syndrome | X-linked hypophosphatemia (XLH) |
| Hyperparathyroidism | Tumor-induced osteomalacia (TIO) |
| Diuretics (e.g. acetazolamide) | Autosomal dominant/autosomal recessive rickets |
| Myeloma | McCune Albright syndrome |
| Copper disorders (Menkes) | Cutaneous-skeletal hypophosphatemia syndrome (CSHS) (e.g., epidermal nevus syndromes) |
| Alcohol consumption | Iron use (iron polymaltose infusions) |
| Genetic disorders (e.g. Npt2a mutations) |
Finally, in the absence of genetic data, indication of several bone and calcification disorders can help distinguish XLH from TIO, though these criteria are highly variable. Enthesopathy (calcification of the joint capsule, tendon insertions, and ligaments) is a common feature of XLH but not of TIO and results in increased apparent BMD (Beck-Nielsen et al., 2019). By contrast, typical TIO patients have remarkably low BMD, often being labelled osteoporotic. Dentition is another clinical differentiator, in that patients with XLH can have recurrent abscesses and dental loss, while dentition in TIO is usually normal (Lee et al., 2017). However, the patient presented here was edentulous, demonstrating that TIO should not be ruled out based on these features.
Suspected TIO should prompt an investigation for active PMTs in the hands, feet, ribs, nasal cavities, and brain (Colazo et al., n.d). In recent literature, a full body 68Ga-DOTATATE scan has been shown to be a specific imaging test for identifying culprit tumors in osteomalacia (Zhang et al., 2015). The 68Ga-DOTATATE scan works by utilizing 68Gallium (68Ga) conjugated to somatostatin peptide analogues, which allows for somatostatin receptor imaging by PET scan. DOTATATE scans are most often used for diagnosing somatostatin-receptor positive neuroendocrine tumors, but PMTs preferentially express somatostatin type 2 receptors (Houang et al., 2013). Hence, this technique allows for targeted imaging of TIO-causing lesions. That said, false positivity can still occur in hypersplenism, fractures, sarcoidosis, or vertebral hemangiomas (Hofman et al., 2015). If very small and/or multiple tumors of unknown significance are present, selective FGF23 venous sampling has shown promise in deciphering the FGF23-producing tumor (Colazo et al., n.d.; Schober et al., 2017). In this case, since two suspicious lesions were encountered, FGF23 venous sampling could have been utilized, but the anatomical location and proximity of both lesions to each other would have made the process difficult to perform and most likely futile.
Misdiagnosis can also occur after tumor identification, especially in cases in which non-PMT tumors morphologically mimic PMTs in the context of hypophosphatemia (Lee et al., 2016). Recent studies have identified novel gene fusions FN1-FGF1 and FN1-FGFR1 that are transcribed and synthesized by the majority of PMTs. The fusion of FN1 and FGFR1 leads to constitutive overexpression of the 3′ portion of FGFR1 through constitutive activation of the FN1 promoter, and the fusion protein products have suspected roles in autocrine signaling (Lee et al., 2016). These fusion products can be detected by RNA sequencing or FISH of the tumor, and several emerging FGF/R inhibitors have been shown to be therapeutically beneficial in relapsing TIO (Hartley et al., 2020).
While diagnosis of TIO depends solely on postoperative biochemical resolution of laboratory abnormalities, the classification of the resected tumor can be helpful. Our patient's tumor tested positive for FN1-FGFR1 by RNAseq and for FGF23 by chromogenic in situ hybridization (CISH), further suggesting PMT (Martinez et al., 2019; Carter et al., 2015). These results support the use of FN1-FGF1/R1 gene fusions and FGF23 CISH as an additional diagnostic tool for TIO and can be used for comparison between the original and a recurrent tumor.
To date, surgical PMT resection is the only curative therapy for TIO. To mitigate risk of recurrence or metastasis, resection should be performed with wide margins where possible, and patients should be monitored longitudinally post-operatively (Hautmann et al., 2015). Here, our patient had dramatic improvements 3 months post-resection in symptoms and mobility (Video 1, Video 2), as well as in serum FGF23, phosphate, and Vitamin D levels (Table 2). Patients with unresectable or undetectable tumors have historically been placed on supplemental phosphate and calcitriol; however, Burosumab is a novel human anti-FGF23 monoclonal antibody initially approved by the FDA to treat XLH that has been recently approved for treatment of such complex TIO presentations (Carpenter et al., 2016; Day et al., 2020). In particular, Kyowa Kirin and Ultragenyx's Crysvita (burosumab-twza) has been approved for the treatment of TIO associated with PMTs that cannot be curatively resected or localized in adult and pediatric patients 2 years of age and older. Burosumab has been shown to improve serum phosphate levels and histomorphology of bone biopsies in patients with TIO, but mild side effects have been reported, including reactive overproduction of FGF23 and consequent Vitamin D deficiency (Day et al., 2020). For these reasons, monitoring FGF23 levels is not a reliable way to monitor patients on Burosumab; clinicians should focus on serum phosphorous, Alkaline phosphatase, renal phosphate wasting, 1,25 Dihydroxyvitamin D, and clinical signs/symptoms.
Indeed, in the 3 months prior to tumor identification, our patient was placed on Burosumab therapy and enjoyed mild symptomatic improvement and stable lab values (notably a normal serum phosphorus, see Table 2). While no labs were taken pre-induction, her condition deteriorated while off Burosumab between her two rib resections, indicating a positive impact of Burosumab on her condition. Her marked post-operative improvement supports the use of PMT resection when possible.
In this report, we have demonstrated the utility of two powerful genetic tools – gene paneling and RNA sequencing – at guiding clinicians to a challenging diagnosis of a non-heritable condition. In the 26-year history of this patient's disease, genetic tools have become increasingly cost-effective, sensitive, and precise. A lack of available tests at initial presentation motivated a diagnosis of XLH (a germline genetic condition) based on clinical evidence alone. The similar clinical presentations of many phosphate wasting disorders, the uniqueness of their genetic identifiers like FN1-FGFR1 or PHEX, and the illumination of known, curative therapies through such tests all support the expanded use of gene paneling and tumor sequencing alongside clinical reasoning around phosphate wasting disorders. This fact is further evidenced by the requirement of genetic diagnosis for enrollment in most clinical trials targeting phosphate wasting disorders. Additionally, the advancements in the field over the past 3 decades may warrant the re-evaluation of poorly managed, long-term diagnoses through the lens of a gene panel or specific genetic marker.
4. Conclusion
Tumor-induced osteomalacia (TIO) is a paraneoplastic disease driven by hypersecretion of Fibroblast Growth Factor 23 (FGF23) from a phosphaturic mesenchymal tumor (PMT). In this case report, we present a patient with what we believe to be the longest history of a delayed TIO diagnosis reported in the literature, outlined current diagnostic strategies, and demonstrated the increasing utility of genetic testing in the diagnosis of phosphate wasting disorders.
We highlight the importance of the personal and family histories, as well as potential pitfalls of using exclusively clinical criteria in diagnosing phosphate wasting disorders, as our patient had several clinical findings suggestive of XLH yet had a negative family history of phosphate wasting disorders. Further, we stress the utility of full body imaging, especially a 68Ga-DOTATATE PET/CT scan, in probing cases of high clinical suspicion for TIO, and we applied novel genetic screening methods (i.e. gene paneling and tumor RNA sequencing) to confirm our diagnosis. When possible, performing these tests provides powerful support for a TIO diagnosis. Finally, while our patient has enjoyed a marked recovery 3 months post-surgical resection, we discuss Burosumab, an emerging therapeutic antibody against FGF23 that may be an attractive treatment for undetectable or unresectable tumors in TIO.
The following are the supplementary data related to this article.
Pre-operative functionality of patient
Post-operative functionality of patient
Transparency document
Transparency document.
Acknowledgments
Acknowledgments
Two of the authors (JMC, JAD) are supported by NIGMS of the National Institutes of Health under award number T32GM007347. The content in this report is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The Transparency document associated with this article can be found, in online version.
Contributor Information
Juan M. Colazo, Email: juan.m.colazo@vanderbilt.edu.
Joseph A. DeCorte, Email: joseph.a.decorte@vanderbilt.edu.
Erin A. Gillaspie, Email: erin.a.gillaspie@vumc.org.
Andrew L. Folpe, Email: folpe.andrew@mayo.edu.
Kathryn M. Dahir, Email: kathryn.dahir@vumc.org.
References
- Beck-Nielsen S.S. FGF23 and its role in X-linked hypophosphatemia-related morbidity. Orphanet Journal of Rare Diseases. 2019;14:58. doi: 10.1186/s13023-019-1014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter T.O. New perspectives on the biology and treatment of X-linked hypophosphatemic rickets. Pediatr. Clin. N. Am. 1997;44:443–466. doi: 10.1016/s0031-3955(05)70485-5. [DOI] [PubMed] [Google Scholar]
- M. P. Carpenter T.O., Weber T., Peacock M., Ruppe M., Insogna K., Osei S., Luca D., Skrinar A., San Martin J. (2016) Effects of KRN23, and anti-FGF23 antibody, in patients with tumor induced osteomalacia and epidermal nevus syndrome: results from an ongoing phase 2 study. in Jan De Beur S. Annual Meeting of the American Society for Bone and Mineral Research 1098.
- Carter J.M., Caron B.L., Dogan A., Folpe A.L. A novel chromogenic in situ hybridization assay for FGF23 mRNA in phosphaturic mesenchymal tumors. Am. J. Surg. Pathol. 2015;39:75–83. doi: 10.1097/PAS.0000000000000290. [DOI] [PubMed] [Google Scholar]
- J. M. Colazo, R. C. Thompson, N. V. Covington, K. M. Dahir, An Intracranial Mass Causing Tumor-induced Osteomalacia (TIO): Rapid and Complete Resolution of Severe Osteoporosis After Surgical Resection. doi: 10.1016/j.radcr.2020.01.039. [DOI] [PMC free article] [PubMed]
- Day A.L., Gutiérrez O.M., Guthrie B.L., Saag K.G. Burosumab in tumor-induced osteomalacia: a case report. Joint Bone Spine. 2020;87:81–83. doi: 10.1016/j.jbspin.2019.07.012. [DOI] [PubMed] [Google Scholar]
- Durmaz E. Novel and de novo PHEX mutations in patients with hypophosphatemic rickets. Bone. 2013;52:286–291. doi: 10.1016/j.bone.2012.10.012. [DOI] [PubMed] [Google Scholar]
- Feng J. The diagnostic dilemma of tumor induced osteomalacia: a retrospective analysis of 144 cases. Endocr. J. 2017;64:675–683. doi: 10.1507/endocrj.EJ16-0587. [DOI] [PubMed] [Google Scholar]
- Folpe A.L. Phosphaturic mesenchymal tumors: a review and update. Semin. Diagn. Pathol. 2019;36:260–268. doi: 10.1053/j.semdp.2019.07.002. [DOI] [PubMed] [Google Scholar]
- Hartley I.R. Targeted FGFR blockade for the treatment of tumor-induced osteomalacia. N. Engl. J. Med. 2020;383:1387–1389. doi: 10.1056/NEJMc2020399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hautmann A.H., Hautmann M.G., Kölbl O., Herr W., Fleck M. Tumor-induced osteomalacia: an up-to-date review. Curr. Rheumatol. Rep. 2015;17:37. doi: 10.1007/s11926-015-0512-5. [DOI] [PubMed] [Google Scholar]
- Hofman M.S., Lau W.F., Hicks R.J. Somatostatin receptor imaging with 68Ga DOTATATE PET/CT: clinical utility, normal patterns, pearls, and pitfalls in interpretation. Radiographics. 2015;35:500–516. doi: 10.1148/rg.352140164. [DOI] [PubMed] [Google Scholar]
- Houang M. Phosphaturic mesenchymal tumors show positive staining for somatostatin receptor 2A (SSTR2A) Hum. Pathol. 2013;44:2711–2718. doi: 10.1016/j.humpath.2013.07.016. [DOI] [PubMed] [Google Scholar]
- Imel E.A., Econs M.J. Approach to the hypophosphatemic patient. The Journal of Clinical Endocrinology & Metabolism. 2012;97:696–706. doi: 10.1210/jc.2011-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagtap V.S. Hypophosphatemic rickets. Indian J Endocrinol Metab. 2012;16:177–182. doi: 10.4103/2230-8210.93733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan de Beur S.M. Tumor-induced osteomalacia. JAMA. 2005;294:1260–1267. doi: 10.1001/jama.294.10.1260. [DOI] [PubMed] [Google Scholar]
- S. Jan De Beur et al., OR13-1 Burosumab improves the biochemical, skeletal, and clinical symptoms of tumor-induced osteomalacia syndrome. Journal of the Endocrine Society 3 (2019).
- Lee J.C. Characterization of FN1-FGFR1 and novel FN1-FGF1 fusion genes in a large series of phosphaturic mesenchymal tumors. Mod. Pathol. 2016;29:1335–1346. doi: 10.1038/modpathol.2016.137. [DOI] [PubMed] [Google Scholar]
- Lee B.N., Jung H.Y., Chang H.S., Hwang Y.C., Oh W.M. Dental management of patients with X-linked hypophosphatemia. Restor Dent Endod. 2017;42:146–151. doi: 10.5395/rde.2017.42.2.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J. Biol. Chem. 2003;278:37419–37426. doi: 10.1074/jbc.M304544200. [DOI] [PubMed] [Google Scholar]
- Lloyd S.E. A common molecular basis for three inherited kidney stone diseases. Nature. 1996;379:445–449. doi: 10.1038/379445a0. [DOI] [PubMed] [Google Scholar]
- Martin A., David V., Quarles L.D. Regulation and function of the FGF23/Klotho endocrine pathways. Physiol. Rev. 2012;92:131–155. doi: 10.1152/physrev.00002.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A.P. Histiocyte-rich rhabdomyoblastic tumor: rhabdomyosarcoma, rhabdomyoma, or rhabdomyoblastic tumor of uncertain malignant potential? A histologically distinctive rhabdomyoblastic tumor in search of a place in the classification of skeletal muscle neoplasms. Mod Pathol. 2019;32:446–457. doi: 10.1038/s41379-018-0145-0. [DOI] [PubMed] [Google Scholar]
- R. MD, "X-Linked hypophosphatemia" in GeneReviews [Internet], A. H. Adam MP, Pagon RA, et al., Ed. (University of Washington, Seattle, WA, 2012). [PubMed]
- Michigami T. Skeletal mineralization: mechanisms and diseases. Ann Pediatr Endocrinol Metab. 2019;24:213–219. doi: 10.6065/apem.2019.24.4.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne R.B. Renal tubular reabsorption of phosphate (TmP/GFR): indications and interpretation. Ann. Clin. Biochem. 1998;35(Pt 2):201–206. doi: 10.1177/000456329803500203. [DOI] [PubMed] [Google Scholar]
- C. M. Priante G, Terrin L, Gianesello L, Quaggio F, Del Prete D, Anglani F., "Understanding the pathophysiology of nephrocalcinosis" in Updates and Advances in Nephrolithiasis - Pathophysiology, Genetics, and Treatment Modalities. (InTech Open, 2017).
- Root A.W. Genetic disorders of calcium, phosphorus, and bone homeostasis. Translational Science of Rare Diseases. 2018;3:1–36. [Google Scholar]
- Schober H.-C., Kneitz C., Fieber F., Hesse K., Schroeder H. Selective blood sampling for FGF-23 in tumor-induced osteomalacia. Endocrinol Diabetes Metab Case Rep. 2017;2017 doi: 10.1530/EDM-17-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- M. K. Takeda R, Takagi M, Goto M, Ariyasu D, Izawa M, Igaki J, Suzuki E, Nakamura Y, Hasegawa Y, TmP/GFR is a useful marker in making a clinical diagnosis of X-linked hypophosphataemic rickets caused by the PHEX gene mutation. ESPE Abstracts 84 (2015).
- Yin Z., Du J., Yu F., Xia W. Tumor-induced osteomalacia. Osteoporos Sarcopenia. 2018;4:119–127. doi: 10.1016/j.afos.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. 68Ga DOTATATE PET/CT is an accurate imaging modality in the detection of culprit tumors causing osteomalacia. Clin. Nucl. Med. 2015;40:642–646. doi: 10.1097/RLU.0000000000000854. [DOI] [PubMed] [Google Scholar]
- Zoller H., Schaefer B., Glodny B. Iron-induced hypophosphatemia: an emerging complication. Curr. Opin. Nephrol. Hypertens. 2017;26:266–275. doi: 10.1097/MNH.0000000000000329. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pre-operative functionality of patient
Post-operative functionality of patient
Transparency document.