

Abstract
Objective:
The aim of this study was to uncover the mediators and mechanistic events that facilitate the browning of white adipose tissue (WAT) in response to burns.
Background:
In hypermetabolic patients (eg, burns, cancer), the browning of WAT has presented substantial clinical challenges related to cachexia, atherosclerosis, and poor clinical outcomes. Browning of the adipose tissue has recently been found to induce and sustain hypermetabolism. Although browning appears central in trauma-, burn-, or cancer-induced hypermetabolic catabolism, the mediators are essentially unknown.
Methods:
WAT and blood samples were collected from patients admitted to the Ross Tilley Burn Centre at Sunnybrook Hospital. Wild type, CCR2 KO, and interleukin (IL)-6 KO male mice were purchased from Jax laboratories and subjected to a 30% total body surface area burn injury. WAT and serum collected were analyzed for browning markers, macrophages, and metabolic state via histology, gene expression, and mitochondrial respiration.
Results:
In the present study, we show that burn-induced browning is associated with an increased macrophage infiltration, with a greater type 2 macrophage profile in the fat of burn patients. Similar to our clinical findings in burn patients, both an increase in macrophage recruitment and a type 2 macrophage profile were also observed in post burn mice. Genetic loss of the chemokine CCR2 responsible for macrophage migration to the adipose impairs burn-induced browning. Mechanistically, we show that macrophages recruited to burn-stressed subcutaneous WAT (sWAT) undergo alternative activation to induce tyrosine hydroxylase expression and catecholamine production mediated by IL-6, factors required for browning of sWAT.
Conclusion:
Together, our findings uncover macrophages as the key instigators and missing link in trauma-induced browning.
Keywords: browning, burns, hypermetabolism, macrophages, trauma
Hypermetabolism is a debilitating and complex pathologic response but commonly observed in individuals with advanced cancer, burns, and heart disease.1–3 Significant catabolism, insulin resistance with hyperglycemia, lipolysis, hyperinflammation, and organ dysfunction characterize this hypermetabolic response.4–6 Persistent and exaggerated hypermetabolism contributes to increased morbidity and mortality.7,8 Despite advances in these areas, severe hypermetabolism still persists and the underlying cause of hypermetabolism is poorly understood and essentially unknown.
Recently, the phenomenon of white adipose tissue (WAT) browning, in which subcutaneous white adipose converts to a more brown-like adipose termed beige/brite adipose tissue, has been implicated in facilitating persistent hypermetabolism in burns and cancer.9–11 In response to injury (burns, cancer) or stress (cold temperatures), white adipocyte cells undergo a “browning process” and adopt brown-like features, including multilocular lipid droplets and high uncoupling protein 1 (UCP1) expression.9–11 Activation of browning leads to a change in mitochondrial function to a predominant production of heat by uncoupling the mitochondrial respiratory chain from ATP synthesis, thereby sending these already hypermetabolic patients to metabolic overdrive. This browning process has been suggested to have a paradoxical impact on health.12 For instance, in obesity and diabetes, beige activation has been intensively studied to harness its thermogenic power to facilitate weight loss and improve metabolic health in these patients.13 Conversely, burn-triggered and cancer beige activation is detrimental as it fuels high metabolic rates and leads to accelerated development and progression of cachexia in the latter.10,14,15 We therefore hypothesize that browning is a crucial mediating process in the pathologic hypermetabolic response.
Unfortunately, the exact cellular mechanisms that regulate browning during hypermetabolic states (burns, cancer) are unknown. Previous studies, in cold-induced browning, have suggested that recruited monocytes are reprogramed and differentiated into catecholamine-secreting macrophages that eventually drive WAT browning.16,17 These studies have also reported that this reprogramming of macrophages is mediated by anti-inflammatory cytokines such as interleukin (IL)-4 and IL-13.16,17 In hypermetabolic conditions, IL-4 and IL-13 are not as prevalent,7,18 therefore leading to our hypothesis that an alternative cytokine or mechanism must be involved in hypermetabolic conditions. Although there is strong evidence that IL-6 is involved in the browning response of hypermetabolic patients, the cellular and effector cells involved are unclear. We therefore aimed to investigate the effector cells involved in the IL-6-browning of adipose tissue circuitry that occurs in response to burn injury.
METHODS
Burn Patients
Patients admitted to the Ross Tilley Burn Centre at Sunnybrook Hospital (Toronto, Canada) or nonburn patients undergoing elective surgery were consented preoperatively for tissue collection. Approval for our study was obtained from the Research Ethics Board at Sunnybrook Hospital under study #194–2010. We enrolled 15 severely burned adults with burns encompassing 45% ± 16% of their total body surface area (TBSA). Adipose tissue (subcutaneous depots) obtained from the last OR (≥10 days) was immediately transferred to the laboratory and either frozen (−80°C) or transferred into fixative until time of analysis. Resting energy expenditure was measured with a Sensor Medics 2900 metabolic measurement cart.
Mouse Thermal Injury Model
Male C57BL/6, IL6 KO, and CCR2 KO mice (Jackson) were housed at ambient temperature and cared in accordance with the Guide for the Care and Use of Laboratory Animals. All procedures were approved by the Sunnybrook Research Institute Animal Care Committee under AUP 467 (Toronto, Ontario, Canada). Mice received a 30% TBSA, full thickness burn. Burnt mice were subsequently housed individually in sterile cages and fed ad libitum until sacrifice. Adipose tissue (inguinal adipose depots and interscapular brown adipose) was harvested 6 days post burn injury and used for analysis. Interscapular brown adipose tissue was also harvested from these mice for quality control purposes in regards to testing out specificity of the UCP1 primer and antibody utilized in this study.
Histology and Immunohistochemistry
Adipose tissue collected (inguinal adipose depots in mice and subcutaneous depots in patients) was immediately fixed in 10% formalin and then maintained in 70% ethanol before paraffin embedding. Subsequently, tissues were sectioned and stained with hematoxylin and eosin (H&E) or incubated with UCP1 (Sigma), F480 (AbD Serotec), and CD11c (Abcam) antibody followed by DAB staining. Imaging was performed on a LSM confocal microscope (Zeiss, Germany).
Quantitative Polymerase Chain Reaction
Total RNA isolated from adipose tissue and primary macrophages was analyzed by quantitative real-time polymerase chain reaction (RT-PCR). RNA was isolated from tissue and cells using TRIzol-chloroform (Life Technologies) with subsequent purification using the RNeasy Kit (Qiagen) according to the manufacturer’s instructions. RNA (2 mg) was transcribed to cDNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems). RT-PCR was performed using the Applied Biosystems Step One Plus Real-Time PCR System. Primer sequences used are available upon request.
Primary Macrophage Culture and Conditioned Medium Preparation
Mice were sacrificed by cervical dislocation, and then were rinsed in 70% (vol/vol) ethanol, and bone marrow was isolated from femurs and tibias. Bone marrow cells were plated at a density of 1 × 106 to 2 × 106 cells/mL in RPMI-1640 medium (supplemented with 10% FCS, 1% glutamine, 1% penicillin-streptomycin, and 10 ng/mL of macrophage colony-stimulating factor [PeproTech]) on 6-well plates and were allowed to differentiate for 7 days. Post-differentiations, these bone marrow-derived macrophages (BMDMs) were stimulated with recombinant IL-6 (R&D Systems) for 24 hours. Conditioned medium from BMDMs treated with IL-6 was collected, filtered, and spun down. Also human immortalized THP-1 monocytes were maintained in RPMI 1640 supplemented with 10% heat-inactivated FCS and 10 μg/mL penicillin/streptomycin in a humidified atmosphere at 37°C and 5% CO2. Cells were passaged every 3 to 4 days before differentiation. For macrophage differentiation, THP1 cells were incubated for 72 hours with 100 nM phorbol 12-myristate 13-acetate and cultured in 6 well plates. These differentiated macrophages were stimulated with vehicle, or recombinant Ie I (3ïmg/mL L-6 (R&D Systems) for 24 hours.
Primary Adipocytes Culture
C3H10T1/2 cells (ATCC) were maintained in Dulbecco modified Eagle medium with 10% fetal bovine serum (FBS; Invitrogen) at 37°C in a 5% CO2 environment. Adipocyte differentiation was induced in preadipocyte cultures by treating confluent cells for 48 hours in medium containing 10% FBS, 0.5 mM isobutylmethylxanthine, 125 nM indomethacin, 1 mM dexamethasone, 850 nM insulin, and with 1 mM rosiglitazone (Alexis Biochemicals). After 48 hours, the cells were moved to the medium containing 10% FBS and 850 nM insulin. Adipocytes were allowed to differentiate for 10 days and then washed with phosphate-buffered saline (PBS) before treatment with either vehicle or IL-6 BMDM conditioned medium for 24 hours. Total RNAwas extracted from adipocytes post-treatment using TRIzol-chloroform (Life Technologies) with subsequent purification using the RNeasy Kit (Qiagen) according to the manufacturer’s instructions.
Flow Cytometry
Adipose tissues were minced and digested with collagenas, Worthington) for 45 minutes at 37°C in a shaker (400 rpm). The digested cell suspension was centrifuged at 1800 rpm for 5 minutes to separate stromal-vascular fraction from adipocytes. Pelleted cells were resuspended in FACS buffer (PBS containing 5% FBS and 1% l-glutamine) and passed through a 40-μm strainer (BD Biosciences) to remove large cellular debris. Antibodies directed against mouse CD45 (BD Pharmingen); CD4 (BD Pharmingen); CD11b (BD Pharmingen); CD206 (BD Pharmingen); F4/80 (BD Pharmingen); Tyrosine 3-Hydroxylase (TA303716, OriGene); anti-rabbit (BD Pharmingen) were used for flow cytometric analysis.
Catecholamines and Lipids
Catecholamines (Rocky Mountain Diagnotics) and free fatty acids (Biovision) were quantified in duplicate as per the manufacturers’ protocols. For catecholamine enzyme-linked immunosorbent assays, tissues were homogenized by sonication in homogenization buffer (0.01N HCl, 1 mM EDTA, 4 mM Na2S2O5), and cellular debris was pelleted by centrifugation at 13,000 rpm for 15 minutes at 4 C. The cleared homogenates were collected and stored in −80 C freezer before quantification. All samples were normalized to total tissue protein concentration.
Cytokine Profile
Rodent and human sera, as well as macrophage-conditioned medium, were collected and using a Multiplex platform (Millipore, MA) and cytokine profiles were measured.
Statistics
All data are presented as mean ± standard error mean (SEM) and analyzed using Prism (Graphpad). Statistical differences between 2 groups were evaluated using 2-tailed, unpaired t test for single variables. Statistical differences between ≥3 groups were evaluated using a 2-way analysis of variance followed by Bonferroni post-tests. Significant results were established for P < 0.05 (*).
RESULTS
Browning in Response to Burn Injury Is Associated With Increased Infiltration of Macrophages in the Adipose
Burn Patients
We first examined the activation of browning of the adipose tissue in burn patients and compared it to control patients who did not have a burn injury (Tables 1 and 2, Supplementary Figure 1, http://links.lww.com/SLA/B313). In agreement with previous reports, burn patients expressed increased hypermetabolism with elevated resting energy expenditure and fat oxidation in conjunction with browning of subcutaneous WAT (sWAT) in response to burn (Fig. 1A–D). Having established burn-induced browning in these patients, we next sought to define which effector cells mediate this browning process in response to trauma. Innate immune cells, particularly macrophages, have recently been implicated in mediating cold-induced browning.16,17 This prompted us to ask whether burn-induced browning operates in a similar macrophage-dependent pathway. We therefore next examined sWAT from burn patients and from non-burn controls by immunohistochemical analysis with a mAb to cd11b, a marker specific for myeloid. The antibody detected large multinucleate cells (accumulated macrophages) in sWAT from burn patients but not controls (Fig. 1E). To corroborate our histological findings, we then studied the transcript levels of various myeloid and macrophage markers in sWAT, post-burn. Gene expression studies of myeloid marker CD11b and macrophage marker EMR-1 indicated that there are increased levels of these transcripts in sWAT of burn patients when compared to controls (Fig. 1F and G). Flow cytometry analysis further affirmed that the percentage of CD45+/CD14 (key macrophage markers) cells within isolated stroma vascular fraction (SVF) was elevated >2-fold in sWAT from burn patients relative to controls (Fig. 1H).
TABLE 1.
Demographics for Burn Patients
| No. of Burn Patients | 13 |
|---|---|
| Age, mean ± SD | 42 ± 12 |
| Male, no. (%) | 11 (85%) |
| TBSA, mean ± SD | 45 ± 16 |
| Inhalation injury, no. (%) | 8 (62%) |
| Etiology, no. (%) | |
| Flame | 12 (92%) |
| Scald | 1 (8%) |
TABLE 2.
Demographics for Non-burn Controls
| Non-burn Controls | 8 |
|---|---|
| Age, mean ± SD | 46 ± 7 |
| Male, no. (%) | 5 (63%) |
FIGURE 1.
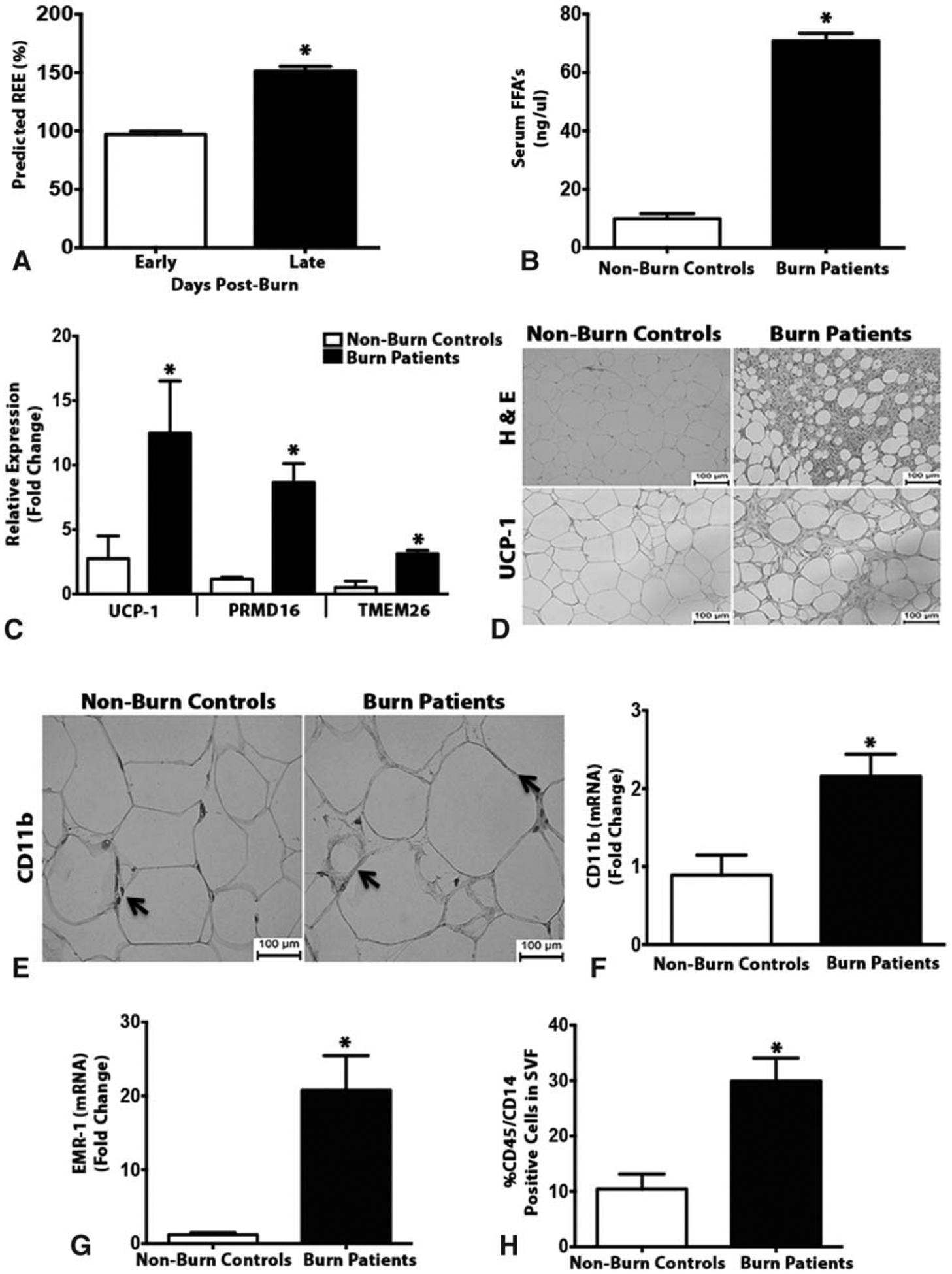
Browning of white adipose tissue in burn patients is associated with increased macrophage infiltration. (A) Average measured predicted resting energy expenditure (REE) as a function of days post-burn injury was measured in burn patients. (B) Serum concentration of free fatty acids in burned patients and non-burn controls. (C) Quantitative RT-PCR analysis of browning genes in subcutaneous WAT isolated from burn patients and non-burn controls. (D) H&E and uncoupling protein 1 (UCP1) staining in subcutaneous WAT of burned patients and non-burn controls. (E) Immunohistochemical staining of macrophage marker cd11b in subcutaneous WAT isolated from burn patients and non-burn controls. (F and G) Quantitative RT-PCR analysis of macrophage markers cd11b and EMR-1 in subcutaneous WAT isolated from burn patients and non-burn controls. (H) Flow cytometry analysis of macrophage markers CD45+/CD14+ cells gated in the subcutaneous WAT SVF of burn patients and non-burn controls. Data represented as mean ± SEM, P < 0.05 *Significant difference non-burn controls vs burn patients (n = 8 non-burn controls; n = 20 burn patients).
Burn Mice
To further uncover the significance of macrophages in burn-induced browning, we utilized a burn injury mouse model. C57BL/6 mice were subjected to a 30% TBSA thermal injury and the inguinal white adipose (iWAT) was collected and assessed for the browning response (Supplementary Figure 2A and B, http://links.lww.com/SLA/B313). A similar browning process observed earlier in burn patients was also observed in post-burn mice. Specifically, elevated expression of browning genes UCP1, CIDEA, PPARγ, and PRDM16 and histological staining for UCP1+ adipocytes in the inguinal white adipose depots (iWAT) of post-burn mice (Fig. 2A and B). Consistent with our observations in burn patients, WT mice exposed to a burn injury showed increased macrophage infiltration in the iWAT post injury (Fig. 2C). This was further confirmed by flow cytometry analysis of SVF obtained from post-burn mice, in which a >3-fold elevation of F480+/CD11b (key macrophage markers) cells was observed (Fig. 2D). Together, these results suggest that the browning of adipose tissue in response to burn injury might be attributable to infiltrated macrophages.
FIGURE 2.
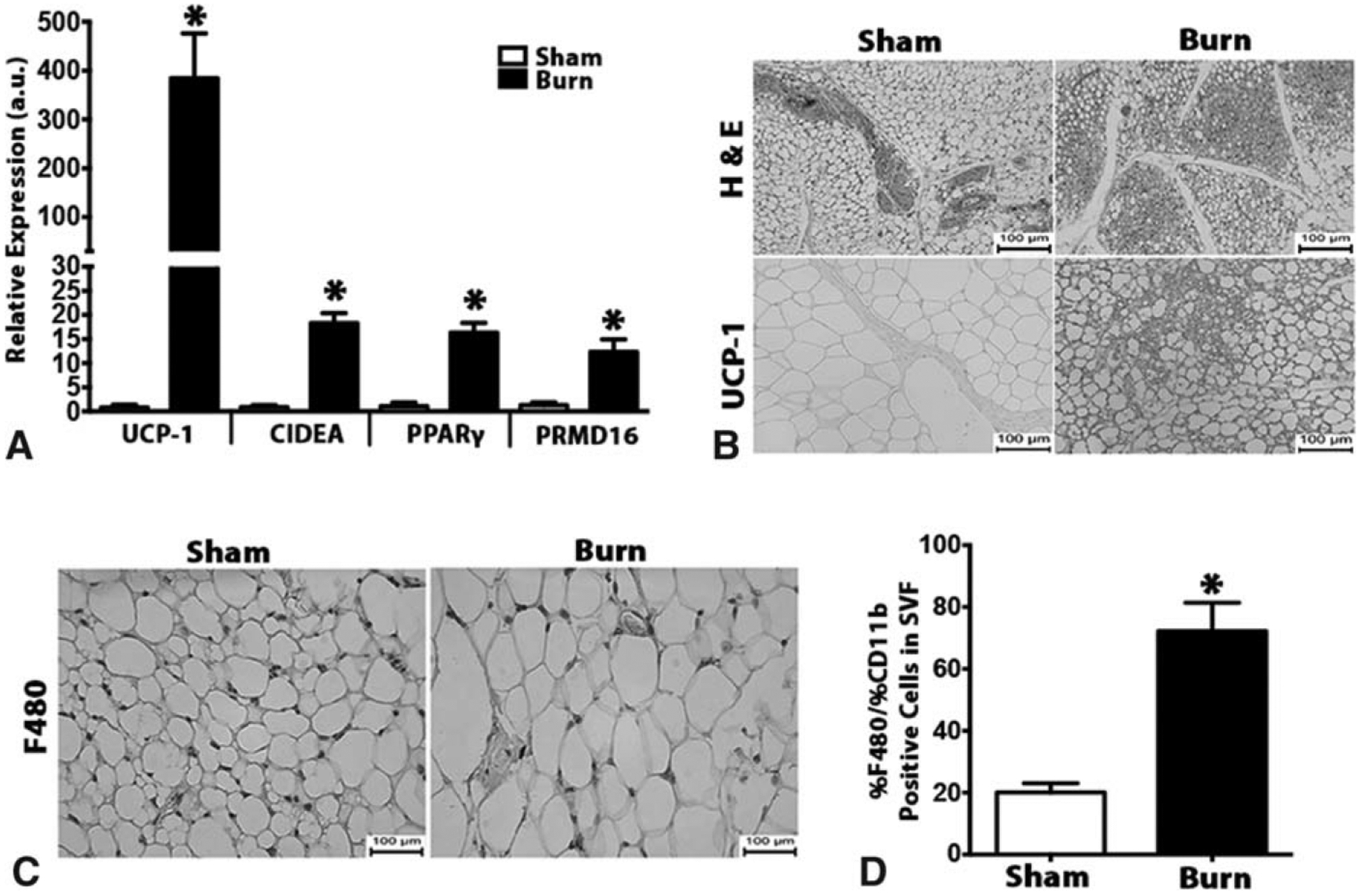
Browning of white adipose tissue in post-burn mice is associated with increased macrophage infiltration. (A) Quantitative RT-PCR analysis of browning genes in inguinal WAT of burned mice and littermate controls. (B) H&E and uncoupling protein 1 (UCP1) staining in inguinal WAT of burned mice and littermate controls. (C) Immunohistochemical staining of macrophage marker F480 in inguinal WAT of burned mice and littermate controls. (D) Flow cytometry analysis of macrophage markers CD11b+F4/80+ cells gated in the inguinal WAT SVF of burned mice and littermate controls. Data are represented as mean ± SEM, P < 0.05. *Significant difference burn vs littermate controls (n = 6).
Burn-induced Browning Requires Adipose Tissue Macrophage Recruitment Via the Chemokine CCR2
In type 2 immune responses, resident macrophages can expand solely through in situ proliferation, without the need to recruit monocytes.19 Given the presence of increased macrophages in the adipose tissue in response to burn injury, we sought to examine whether local expansion or recruitment was the driving factor in adipose macrophage infiltration post-injury. Macrophages are primarily recruited to injury sites via the C-C chemokine receptor type 2 (CCR2). Using CCR2−/− mice that have an inherent disruption in monocyte recruitment post-injury, we assessed the significance of myeloid migration to burn-induced browning (Fig. 3A and B). Burn-induced expression of the core browning gene UCP1 was reduced in the iWAT of CCR2 KO mice (Fig. 3C and D). This impairment in beige fat biogenesis was also evident histologically, as iWAT of CCR2 KO mice had fewer multilocular, UCP1+ adipocytes (Fig. 3C and D). These results suggest that monocyte recruitment via CCR2 to the adipose tissue is associated with the browning of WAT in response to burn injury.
FIGURE 3.
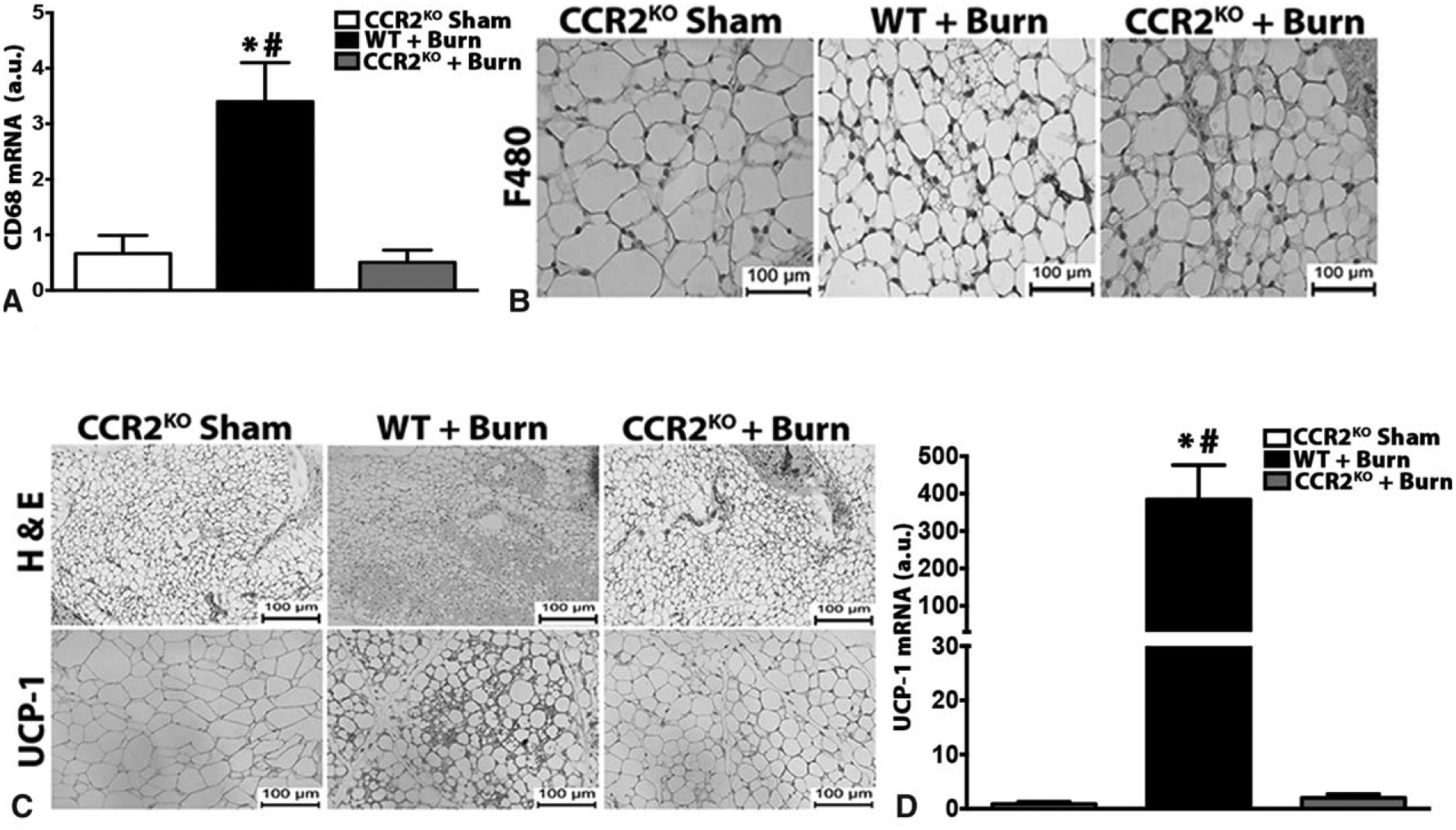
Macrophage recruitment via CCR2 is required for burn-induced browning. (A) Quantitative RT-PCR analysis of macrophage marker CD68 in inguinal WAT of burn or sham treated CCR2-KO and post burn WT mice. (B) Immunohistochemical staining of macrophage marker F480 in the inguinal WAT of burn or sham treated WT or CCR2-KO mice. (C) H&E and uncoupling protein 1 (UCP1) staining in inguinal WAT of burn or sham treated WT or CCR2-KO mice. (D) Quantitative RT-PCR analysis of browning marker UCP-1 in inguinal WAT of burn or sham treated WT or CCR2-KO mice. Data are represented as mean ± SEM, P < 0.05. *Significant difference burn vs littermate controls (n = 4). CCR2-KO: P < 0.05. *Significant difference burn vs CCR2-KO sham, P < 0.05. #WT + burn vs CCR2-KO burn (n = 3–5).
Enrichment of Alternatively Activated Macrophages in the Adipose Tissue Post-burn Injury
Having established that monocyte recruitment through CCR2 is associated with browning of adipose, we next wanted to characterize the type of macrophages that were expressed in response to burn injury. This is important because macrophages display a spectrum of activation states, which are broadly described as classical M1 (proinflammatory), and/or alternative M2 (anti-inflammatory). RT-PCR analysis revealed a progressive increase in type 2 macrophage markers including Arg1, transforming growth factor-β, and IL-10 in sWAT obtained from burn patients relative to controls (Fig. 4A). Consistently, flow cytometry analysis of SVF from burned patients demonstrated a significantly elevated accumulation of CD206, a well-established type 2 macrophage marker (Fig. 4B). Comparable results were present in post-burn mice, which showed a higher expression of “M2-like” macrophages including Arg1, IL-10, and Mrc1in iWAT (Fig. 4C). This was further confirmed by flow cytometry analysis revealing increased CD206 positive cells in SVF of post burn mice (Fig. 4D). Thus, these findings suggest that browning is not only associated with recruitment of macrophages but more specifically the expression of alternatively activated macrophages.
FIGURE 4.
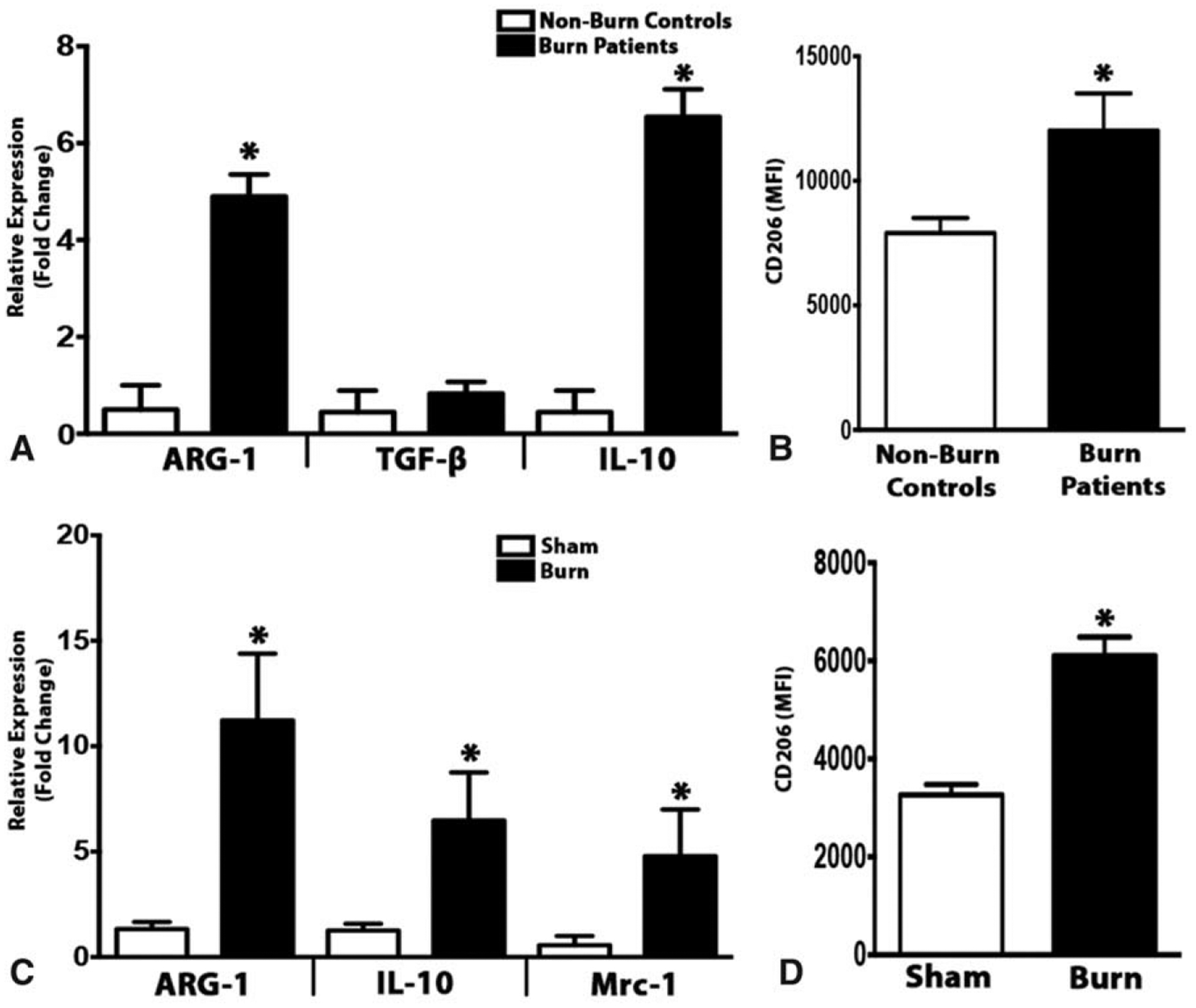
Increased expression of alternatively activated macrophages in the adipose tissue in response to burn injury. (A) Quantitative RT-PCR analysis of M2 macrophage markers in subcutaneous WAT of burn patients and non-burn controls. (B) Flow cytometry analysis of CD206 expression in the subcutaneous WAT SVF of burn patients and non-burn controls. (C) Quantitative RT-PCR analysis of M2 macrophage markers in inguinal WAT of burned mice and littermate controls. (D) Flow cytometry analysis of CD206 expression in the inguinal WAT SVF of burned mice and littermate controls. Human data represented as mean ± SEM, P < 0.05. *Significant difference non-burn controls vs burn patients (n = 5 non-burn controls; n = 10 burn patients). Mice data are represented as mean ± SEM, P < 0.05. *Significant difference burn vs littermate controls (n = 6).
IL-6 Signaling Is Required for the Alternative Activation of Macrophages in Response to Burn Injury
The cytokine microenvironment in which a macrophage is found often provides the signal that shifts the macrophage’s phenotype either toward “classically activated” (M1) or “alternatively activated” (M2). In fact, a recent report from Chawla et al has observed that cold-induced browning was mediated by eosinophils that produced IL-4 and IL-13, which in turn triggered the alternative activation of macrophages. Although we recently reported that the proinflammatory cytokine IL-6 is critical to burn-induced browning,20 it is currently unknown how IL-6 accomplishes this phenotypic switch in adipose tissue. We therefore sough to examine whether IL-6 regulated burn-induced browning via effects on recruitment or polarization of macrophages in the adipose tissue. Consistent with our previous findings, IL-6−/− mice show impairments in burn-induced browning, as confirmed by genomic and histological analysis that revealed diminished multilocular, UCP1+ adipocytes in the adipose of these mice (Fig. 5A and B). These IL-6−/− mice were also protected against burn-induced weight and fat loss (Supplementary Fig. 3A and B, http://links.lww.com/SLA/B313). Interestingly, in IL-6−/− mice post-burn injury, we found no significant impairments in the magnitude of macrophage infiltration in the iWAT (Fig. 5C and D). However, in contrast to the dramatic increase in M2 macrophage markers IL-10, Arg1, and Mrc1 observed earlier in WT mice, these changes were abrogated in IL-6−/− mice (Fig. 5E). This was further confirmed by flow cytometry analysis revealing IL-6−/− mice had no significant increase in CD206-positive cells in their iWAT post-burn injury (Fig. 5F). These findings suggest that IL-6 likely works in conjunction with recruited macrophages by shifting them to the M2 phenotype to induce burn-induced browning.
FIGURE 5.
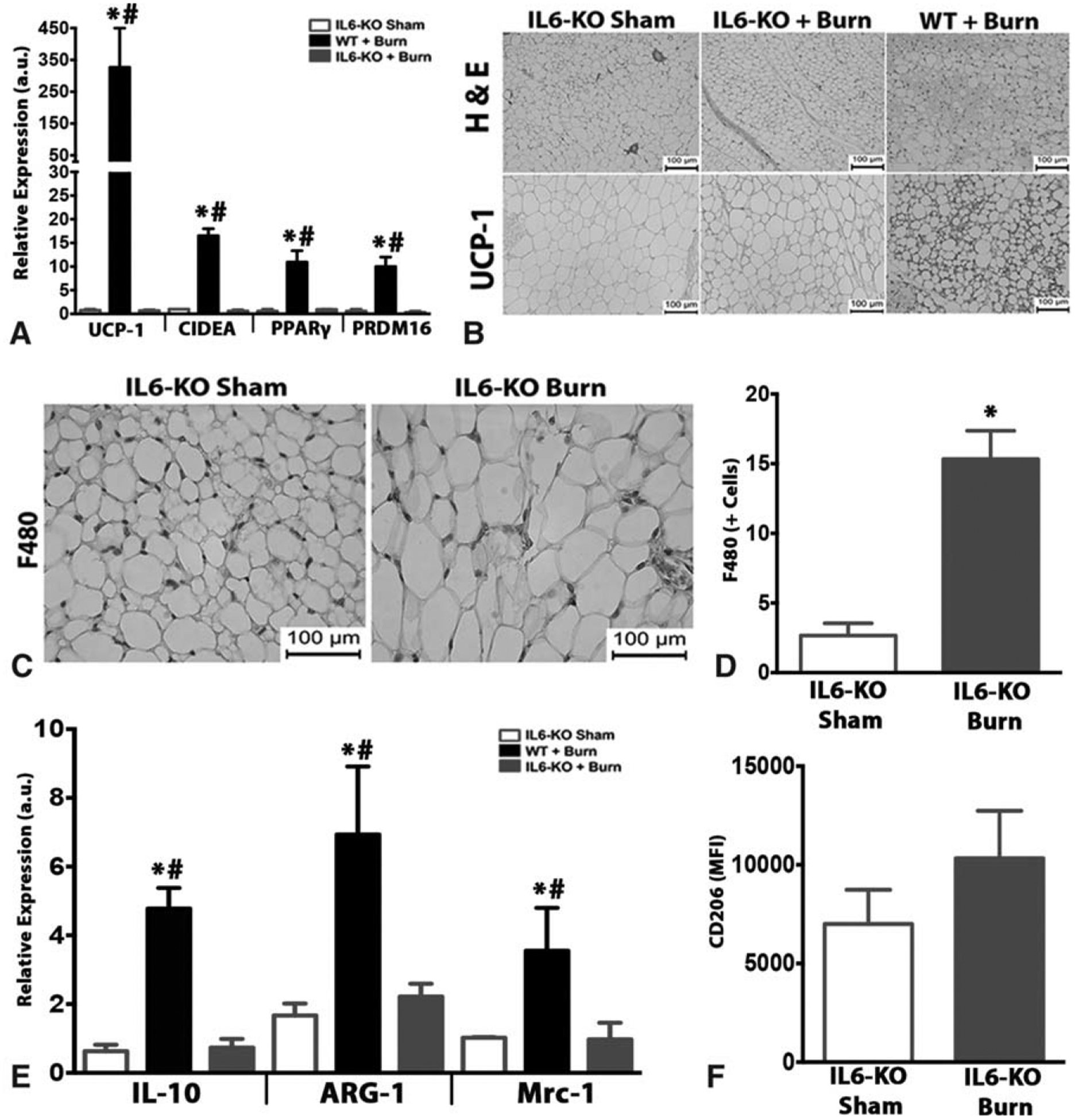
The adipose tissue of IL-6−/− mice show impaired browning and reductions in alternatively activated macrophages post burn injury. (A) Quantitative RT-PCR analysis of browning genes in inguinal WAT of WT and IL-6−/− burned mice and littermate controls. (B) H&E and uncoupling protein 1 (UCP1) staining in inguinal WAT of IL-6−/− burned mice and IL-6−/− littermate controls. (C and D) Immunohistochemical staining of macrophage marker F480 and quantification of the inguinal WAT in burn or sham treated IL-6−/− mice. (E) Quantitative RT-PCR analysis of M2 macrophage markers in inguinal WAT of burn treated WT and IL-6−/− mice, as well as littermate controls. (F) Flow cytometry analysis of CD206 expression in the inguinal WAT SVF of burn treated IL-6−/− mice and littermate controls. Data are represented as mean ± SEM, P < 0.05. *Significant difference burn vs littermate controls, P < 0.05. #WT + burn vs IL-6−/− burn (n = 6).
IL-6 Induces the Polarization of Macrophages in Vitro
To further implicate the requirement of IL-6 in the polarization of recruited macrophages to the “M2-like” phenotype post-burn injury, an in-vitro human and mouse myeloid model was utilized. Bone marrow-derived macrophages were isolated from WT mice and stimulated with IL-6 an assessed for macrophage polarization. IL-6-stimulated macrophages showed a robust expression of well-established type 2 macrophage markers IL-10, Arg-1, and CD206 (Fig. 6A and B). Additionally, IL-6-stimulated macrophages showed no secretion of the proinflammatory cytokine IL-1β (Fig. 6C). Consistent with our primary mice macrophage data, stimulating human THP-1 differentiated macrophages with IL-6 also elicited a robust type 2 macrophage profile and expression (Fig. 6D–F). Thus, these experiments indicate that IL-6 acts in cells of the myeloid lineage to promote “M2-like” polarization.
FIGURE 6.
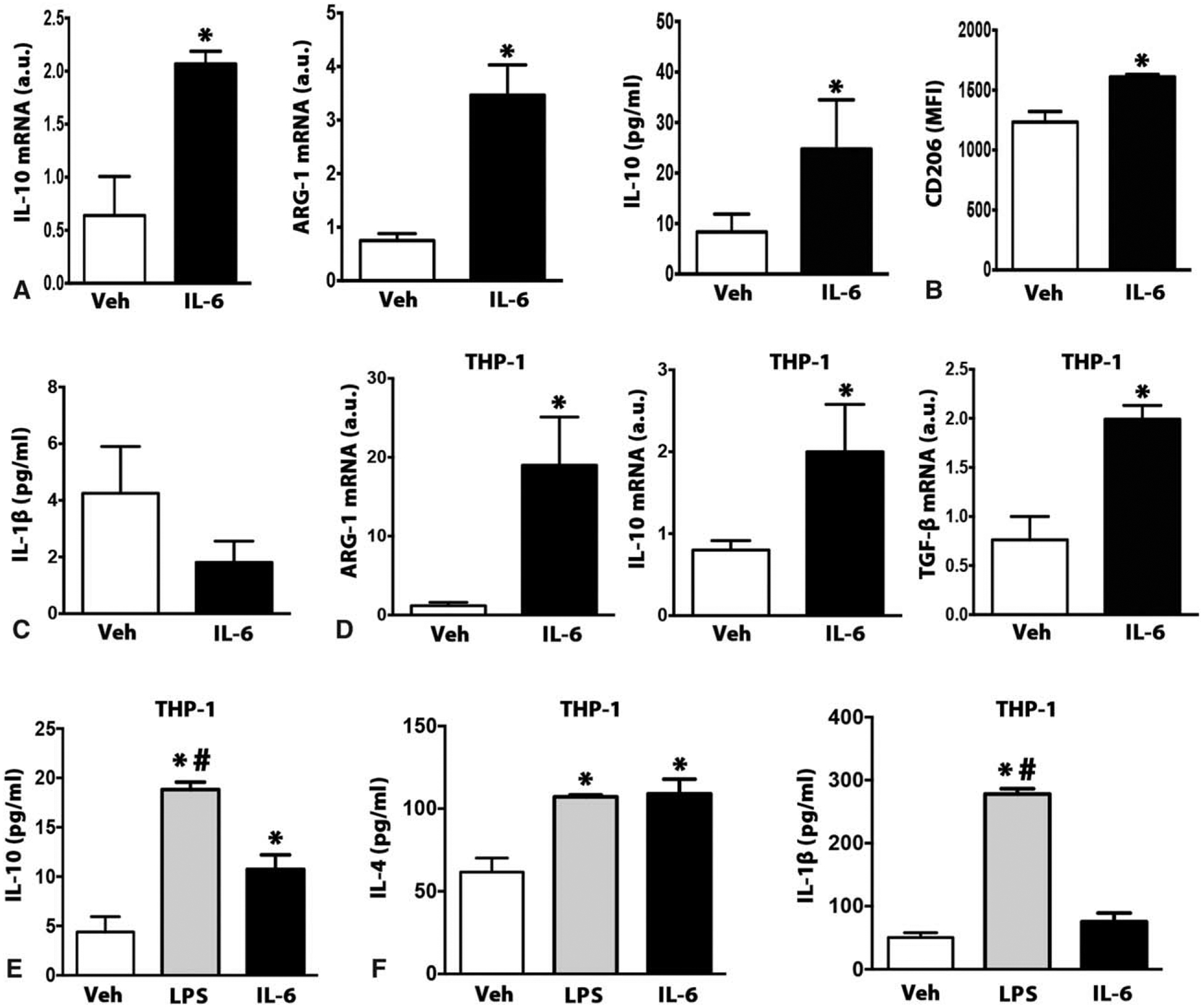
IL-6 induces alternative activation in myeloid cells. (A) Quantitative RT-PCR analysis of M2 macrophage markers and IL-10 cytokine secretion in WT isolated BMDMs stimulated with either Veh or IL-6 cytokine for 24 hours. (B) Flow cytometry analysis of CD206 expression in WT (BMDMs) stimulated with either Veh or IL-6 cytokine for 24 hours. (C) Type 1 cytokine marker IL-1β level in the conditioned medium of WT (BMDMs) stimulated with either Veh or IL-6 cytokine for 24 hours. (D) Quantitative RT-PCR analysis of M2 macrophage markers in differentiated THP-1 human macrophages stimulated with either Veh or IL-6 cytokine for 24 hours. (E) Type 2 cytokine (IL-10, IL-4) markers in the conditioned medium of differentiated THP-1 human macrophages stimulated with either Veh or IL-6 cytokine for 24 hours. (F) Type 1 cytokine marker IL-1β level in the conditioned medium of differentiated THP-1 human macrophages stimulated with either Veh or IL-6 cytokine for 24hrs. Data represented as mean ± SEM, P < 0.05 *=significant difference IL-6 vs Veh (N = 6).
Alternatively Activated Macrophages Induce Browning in Adipocytes Via Release of Catecholamines
To directly link polarized macrophages as the effector cells driving browning and UCP1 activation, we used differentiated multi-potent C3H10T1/2 adipocytes as an in vitro model of browning (Fig. 7A). Media conditioned by polarized macrophages via IL-6 (IL-6 CM) was able to induce browning in differentiated adipocytes, as evident by greater UCP1 expression and co-localization with mitochondria (Fig. 7B and C). In contrast, media conditioned by vehicle-treated macrophages (CTL-CM) did not alter the expression of UCP1 nor mitochondrial colocalization (Fig. 7B and C). These findings suggest that the browning-mediated effects of type 2 macrophages are not dependent on direct cell effects, but rather arise from a circulating/soluble factor able to influence the browning of adipocytes.
FIGURE 7.
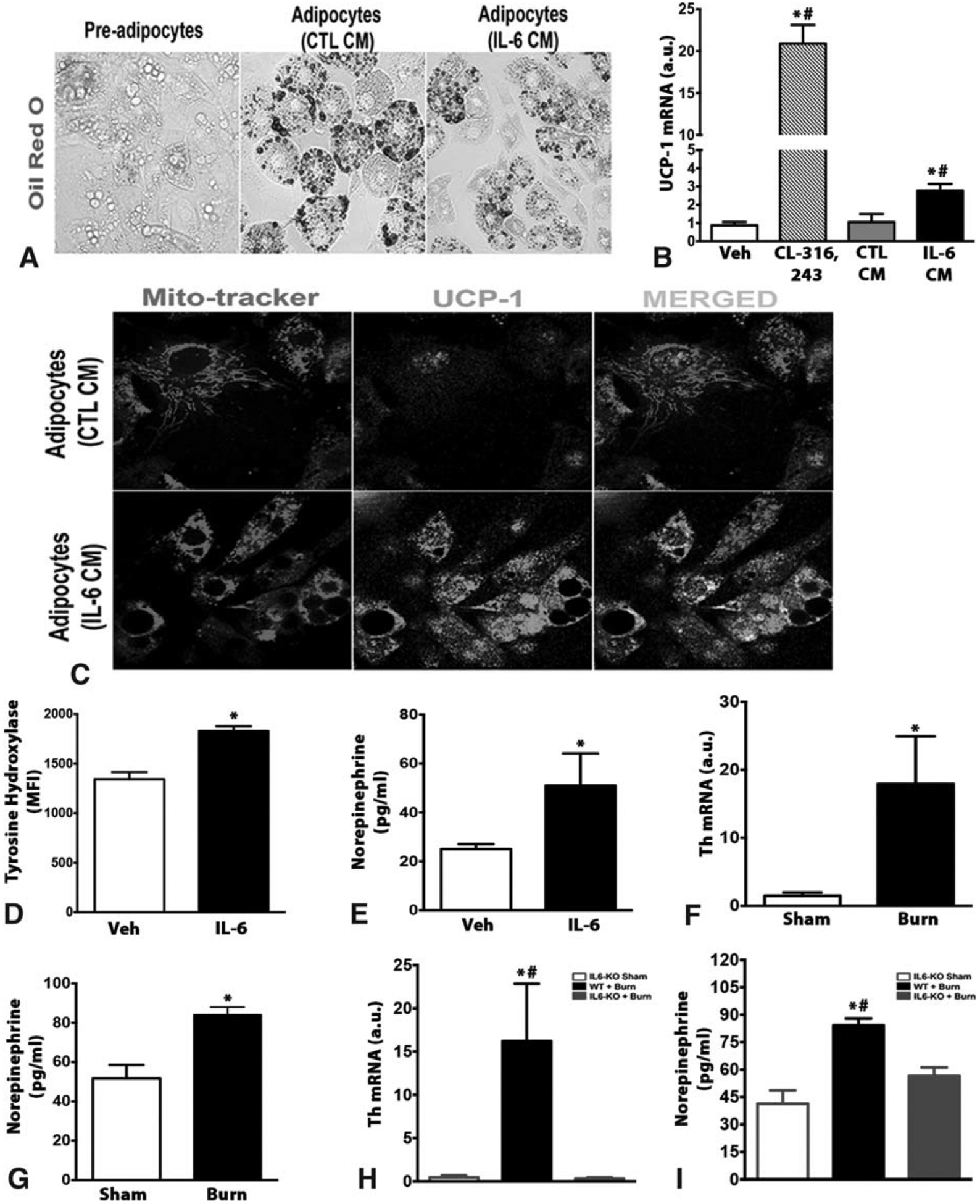
Alternatively activated macrophages secrete catecholamines to induce white adipose tissue browning post burn injury. (A) C3H10T1/2 cells were fixed after treatment, then subjected to oil red O staining (original magnification 40×). (B) Quantitative RT-PCR analysis of browning marker UCP1 in differentiated adipocytes treated with conditioned medium from BMDMs stimulated with either Veh or IL-6 cytokine for 24 hours. CL-316,243 β3-agonist was used a positive control. (C) Differentiated adipocytes were fixed after treatment with conditioned medium from BMDMs stimulated with either Veh or IL-6 cytokine for 24 hours, and then stained for mito-tracker (Red), and UCP1 (Green), original magnification 60×. (D) Flow cytometry analysis of TH in BMDMs stimulated with either Veh or IL-6 cytokine for 24 hours. (E) Norepinephrine content in the conditioned medium of BMDMs stimulated with either Veh or IL-6 cytokine for 24 hours. (F and G) Quantitative RT-PCR analysis of TH (F), and norepinephrine content (G) in inguinal WAT of burned mice and littermate controls. (H and I) Quantitative RT-PCR analysis of TH (H), and norepinephrine content (I) in inguinal WAT of IL-6−/− burned mice and littermate controls. Data are represented as mean ± SEM, P < 0.05. *Significant difference IL-6 CM vs CTL CM (or IL-6 vs Veh), P < 0.05. #Significant difference IL-6 CM vs CL316243. WT: P < 0.05. *Significant difference burn vs littermate controls (n = 4–5).
There are multiple potential sources for such a circulating factor, including hormones, small molecules, and factors produced by macrophages. However, recently, it was shown that alternatively activated macrophages promote browning in response to cold exposure by expressing the enzyme tyrosine hydroxylase (TH), the rate-limiting enzyme in catecholamine production. Thus, we hypothesized that a similar mechanism might underlie the browning effects of type 2 macrophages in burn-induced browning. To assess this, we looked at the expression of TH in macrophages polarized by IL-6. Our analysis revealed increased expression of TH in macrophages treated with IL-6 compared to vehicle-treated macrophages (Fig. 7D). Importantly, the increase in TH expression in IL-6-treated macrophages translated into increased catecholamine (norepinephrine) appearance in the conditioned media of these cells (Fig. 7E). Consistently, iWAT from WT mice subjected to a burn injury showed increased expression of TH, in addition to increased norepinephrine content (Fig. 7F and G). In contrast, IL-6−/− mice that are protected from burn-induced browning showed decreased TH gene expression and norepinephrine content in iWAT (Fig. 7H and I). Taken together, our in-vitro studies and animal work support that macrophage-derived catecholamines are one source of the browning “factor” that stimulates burn-induced remodeling of sWAT into thermogenic beige fat.
DISCUSSION
Chronic hypermetabolism characterized by severe weight loss, chronic inflammation, and muscle/adipose atrophy is a signature response of burns and cancer.2,6 Numerous studies have detailed the devastating metabolic changes (systemic and local insulin resistance) and outcome (complications, multiorgan dysfunction, sepsis) driven by this debilitating response.7,8 However, the mechanisms, and in particular the signaling circuitry that facilitates hypermetabolism in these patients, are essentially unknown. Our study reveals a critical role of both IL-6 and recruited macrophages in regulating the browning of adipose tissue in response to burn injury. Specifically, our findings show that IL-6 and recruited macrophages work in conjunction to facilitate burn-induced browning, the latter (IL-6) reprogramming recruited macrophages (via CCR2) into the alternative M2 phenotype.
Currently hailed as the savior of metabolic syndromes, WAT browning has been described as a beneficial event that promotes weight loss and improves insulin sensitivity in obese and diabetic patients.21 However, it was recently shown that this browning phenomena can be a double-edged sword, in that browning is deleterious in the context of burns and cancer, as it fuels the hypermetabolic response and metabolic dysfunction.12 Exactly how browning is regulated and activated in these hypermetabolic conditions (burns, cancer) is not yet clear. As previously reported, cold-induced browning is mediated by increases in circulating eosinophils and type 2 cytokines (IL-4 and IL-13).16 We have previously reported that IL-6 and not IL-4 is the distinct cytokine that regulates burn-induced WAT browning in a mouse burn injury model.20 However, we highlight in this study that a commonality between IL-6 and other cytokines that have previously been implicated in browning is the reliance of alternatively activated macrophages. Our results here uncover that macrophages are the missing link in the efferent circuit by which IL-6 drives the browning of adipose in burns. This finding may appear unexpected, given the general assumption that only anti-inflammatory cytokines (IL-4, IL-13) are the major drivers of browning. In both in vitro and ex vivo models, we showed that IL-6 potently induced M2 polarization of macrophages that was necessary for beige fat development post-burn injury.
Similar to the recent realization that WAT browning is a double-edged sword, the role of IL-6 in metabolism continues to remain controversial. This controversy stems from different studies in obesity that have shown IL-6 has contradictory metabolic functions.22,23 The metabolic role of IL-6 becomes significantly less clear, however, when the browning of WAT is considered, with paradoxical results seen. For instance, our previous findings in burns and others in cancer have shown that the IL-6 cytokine is critical to adipose browning.9,20 In contrast, recent findings in brown adipose tissue suggest that IL-6 functions as a brown adipokine, or “batokine,” in which increases in BAT-derived IL-6 improves systemic glucose metabolism in obese mice.13 Although it is difficult to reconcile the putative paracrine or endocrine metabolic effects of IL-6, it does appear that these metabolic benefits depend on the location of IL-6 secretion.
In conclusion, the control mechanisms regulating the hypermetabolic response to injury are extensive and will ultimately require the integration of immune, neuroendocrine, and metabolic systems to advance our understanding. Here, we provide one component of this complex immune-metabolism interplay that gravely affects patient outcome. Inhibition of WAT browning might have therapeutic potential for treating patients with burns by attenuating burn-induced hypermetabolism. In this regard, future studies should determine whether selectively blocking IL-6 attenuates hypermetabolism via its effects on macrophage polarization and subsequent browning in burn patients. Indeed, given that the IL-6 cytokine has undeniably been linked to being the master regulator of both burn and cancer induced browning, tocilizumab (IL-6 blocker) use and the subsequent inhibition of the IL-6 signaling maybe therapeutically beneficial in these patients. As with any intervention, the key to the therapeutic utilization of blocking IL-6 clinically will be specificity and efficacy. Exploiting this avenue has great potential for success in these patients.
Supplementary Material
Acknowledgments
This study was supported by National Institutes of Health R01-GM087285-01. CFI Leader’s Opportunity Fund: Project #25407 and Canadian Institutes of Health Research (CIHR) grant #123336.
A.A. is a recipient of the Vanier Canada Graduate Scholarship. M.G.J. holds grants from Canadian Institutes of Health Research, Canada Fund for Innovation (CFI) Leader’s Opportunity Fund Project, and the National Institutes of Health.
Footnotes
The authors report no conflicts of interest.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.annalsofsurgery.com).
REFERENCES
- 1.Ishida J, Konishi M, Saito M, et al. Hypermetabolism: should cancer types, pathological stages and races be considered in assessing metabolism and could elevated resting energy expenditure be the therapeutic target in patients with advanced cancer? J Cachexia Sarcopenia Muscle. 2015;6:391–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jeschke MG. Postburn hypermetabolism: past, present, and future. J Burn Care Res. 2016;37:86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lees MH, Bristow JD, Griswold HE, et al. Relative hypermetabolism in infants with congenital heart disease and undernutrition. Pediatrics. 1965;36:183–191. [PubMed] [Google Scholar]
- 4.Jeschke MG, Gauglitz GG, Kulp GA, et al. Long-term persistance of the pathophysiologic response to severe burn injury. PloS One. 2011;6:e21245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeschke MG. The hepatic response to thermal injury: is the liver important for postburn outcomes? Mol Med. 2009;15:337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dodson S, Baracos VE, Jatoi A, et al. Muscle wasting in cancer cachexia: clinical implications, diagnosis, and emerging treatment strategies. Annu Rev Med. 2011;62:265–279. [DOI] [PubMed] [Google Scholar]
- 7.Jeschke MG, Gauglitz GG, Jeschke MG, et al. Survivors versus nonsurvivors postburn: differences in inflammatory and hypermetabolic trajectories. Ann Surg. 2014;259:814–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deans C, Wigmore SJ. Systemic inflammation, cachexia and prognosis in patients with cancer. Curr Opin Clin Nutr Metab Care. 2005;8:265–269. [DOI] [PubMed] [Google Scholar]
- 9.Petruzzelli M, Schweiger M, Schreiber R, et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014;20:433–447. [DOI] [PubMed] [Google Scholar]
- 10.Patsouris D, Qi P, Abdullahi A, et al. Burn induces browning of the subcutaneous white adipose tissue in mice and humans. Cell Rep. 2015;13:1538–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sidossis LS, Porter C, Saraf MK, et al. Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell Metab. 2015;22:219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdullahi A, Jeschke MG. White adipose tissue browning: a double-edged sword. Trends Endocrinol Metab. 2016;27:542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stanford KI, Middelbeak RJ, Townsend KL, et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest. 2013;123:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong M, Yang X, Lim S, et al. Cold exposure promotes atherosclerotic plaque growth and instability via UCP1-dependent lipolysis. Cell Metab. 2013;18:118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kir S, White JP, Kleiner S, et al. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature. 2014;513:100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiu Y, Nguyen KD, Odegaard JI, et al. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell. 2014;157:1292–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fabbiano S, Suarez-Zamorano N, Rigo D, et al. Caloric restriction leads to browning of white adipose tissue through type 2 immune signaling. Cell Metab. 2016;24:434–446. [DOI] [PubMed] [Google Scholar]
- 18.Li N, Grivennikov SI, Karin M. The unholy trinity: inflammation, cytokines, and STAT3 shape the cancer microenvironment. Cancer Cell. 2011;19:429–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jenkins SJ, Ruckerl D, Cook PC, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science (New York N Y). 2011;332:1284–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abdullahi A, Chen P, Stanojcic M, et al. IL-6 signal from the bone marrow is required for the browning of white adipose tissue post burn injury. Shock. 2011;332:1284–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chondronikola M, Volpi E, Borsheim E, et al. Brown adipose tissue activation is linked to distinct systemic effects on lipid metabolism in humans. Cell Metab. 2016;23:1200–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skurk T, Alberti-Huber C, Herder C, et al. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab. 2007;92:1023–1033. [DOI] [PubMed] [Google Scholar]
- 23.Wallenius V, Wallenius K, Ahren B, et al. Interleukin-6-deficient mice develop mature-onset obesity. Nature Med. 2002;8:75–79. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.