

Key Points
We observed excellent survival in LAD-I and -III after allo-HSCT from 10/10 HLA-matched sibling, nonsibling family, or unrelated donor.
Acute GVHD remains a relevant complication, affecting patient’s outcome.
Abstract
Type I and III leukocyte adhesion deficiencies (LADs) are primary immunodeficiency disorders resulting in early death due to infections and additional bleeding tendency in LAD-III. The curative treatment of LAD-I and LAD-III is allogeneic hematopoietic stem cell transplantation (allo-HSCT). In this retrospective multicenter study, data were collected using the European Society for Blood and Marrow Transplantation registry; we analyzed data from 84 LAD patients from 33 centers, all receiving an allo-HSCT from 2007 to 2017. The 3-year overall survival estimate (95% confidence interval [CI]) was 83% (74-92) for the entire cohort: 84% (75-94) and 75% (50-100) for LAD-I and LAD-III, respectively. We observed cumulative incidences (95% CI) of graft failure (GF) at 3 years of 17% (9%-26%) and grade II to IV acute graft-versus-host disease (aGVHD) at 100 days of 24% (15%-34%). The estimate (95% CI) at 3 years for GF- and GVHD-II to IV–free survival as event-free survival (EFS) was 56% (46-69) for the entire cohort; 58% (46-72) and 56% (23-88) for LAD-I and LAD-III, respectively. Grade II to IV acute GVHD was a relevant risk factor for death (hazard ratio 3.6; 95% CI 1.4-9.1; P = .006). Patients’ age at transplant ≥13 months, transplantation from a nonsibling donor, and any serological cytomegalovirus mismatch in donor-recipient pairs were significantly associated with severe acute GVHD and inferior EFS. The choice of busulfan- or treosulfan-based conditioning, type of GVHD prophylaxis, and serotherapy did not impact overall survival, EFS, or aGVHD. An intrinsic inflammatory component of LAD may contribute to inflammatory complications during allo-HSCT, thus providing the rationale for considering anti-inflammatory therapy pretreatment.
Visual Abstract

Introduction
Leukocyte endothelial adherence and transendothelial migration to the site of infection require a well-organized interaction of multiple cellular and humoral factors. Leukocyte adhesion deficiency (LAD) syndromes are a group of rare primary immunodeficiency disorders.1 The spectrum includes LAD-I (MIM 116920), with mutations in ITGB2, causing dysfunctional β2 integrins (CD11/CD18) with a clinical phenotype of ulcerative deep tissue infections, leukocytosis with impaired pus formation, delayed detachment of the umbilical cord, and omphalitis. The severity of the infectious complications correlates with the level of CD11/CD18 expression on leukocytes (<10%).2,3 Patients with a less severe infectious disease course develop severe periodontitis and bone loss. A distinct hyperinflammatory component of this disease involving the interleukin-12 (IL-12)/IL-23 pathway was recently suggested, while emphasizing the role of macrophages in regulating inflammatory responses beyond microbial defense.4 LAD-III (LAD-I variant) is caused by mutations in FERMT3 resulting in an activation defect of all β-integrins on immune cells and thrombocytes. LAD-III is characterized by an additional Glanzmann thrombasthenia-like bleeding tendency and may resemble osteopetrosis.5-7 LAD-II is caused by mutations in SLC35C1, a gene that encodes a GDP-fucose transporter of the Golgi system, resulting in syndromic features, mental retardation, and typical Bombay blood type.8 A further type of LAD was linked to cystic fibrosis with a monocyte-specific adhesion deficiency suggesting a CFTR-dependent LAD-type (LAD-IV).9
Most affected individuals with LAD-I and III suffer a substantially shortened life expectancy and a mortality >75% by 2 years of age.10 Therapeutic options include antibiotics, local wound management, enzyme replacement for LAD-II, and cystic fibrosis-specific treatment for LAD-IV. Recently, ustekinumab (a monoclonal antibody binding the common P40 unit of IL-12 and IL-23) ameliorated inflammatory symptoms in an adult LAD-I patient, resulting in better wound management and teeth remineralization.11 Recombinant factor VIIa might be beneficial for preventing bleeding in LAD-III.12
A successful allogeneic hematopoietic stem cell transplantation (allo-HSCT) can restore leukocyte function without the need for further treatment in LAD-I and LAD-III patients.13-16 Gene therapy may provide a possible alternative to allo-HSCT and is currently being explored in a phase 1/2 study with a retroviral-based gene therapy (RP-L201) for LAD-I patients (Rocket Pharma). In this retrospective multinational study, we report the clinical outcome and details of transplant modalities in a cohort of 84 LAD patients.
Patients and methods
In this multicenter retrospective study (European Society for Blood and Marrow Transplantation [EBMT]-7427004), data of all registered LAD patients undergoing allo-HSCT between 2007 and 2017 were collected using EBMT extended questionnaires. LAD type was diagnosed by the responsible physician based on CD11/CD18 expression and/or genetic confirmation. In LAD-III, bleeding tendency was taken as a leading symptom in addition to genetic determinants. Event-free survival (EFS) was analyzed as survival without graft failure (GF) and without acute graft-versus-host disease (aGVHD) II to IV.
Conditioning regimens were reported as myeloablative (MA) or nonmyeloablative (non-MA) by the centers. Data on drug dosages were available in 81/84 patients. Busulfan (Bu) cumulative exposure was reported only in 1 case. In line with the literature,17,18 EBMT, and Stem Cell Transplantation for Immunodeficiencies in Europe (SCETIDE) reporting, conditioning regimens, including total Bu dose >8 mg/kg as well as treosulfan total dose ≥30g/m2, were considered MA (which was the case in all patients who received treosulfan). Conditioning with total Bu dose ≤8 mg/kg (n = 2) as well as regimens containing only fludarabin and melphalan (n = 15) was considered non-MA.
Fisher's exact, χ2, and Wilcoxon-Mann-Whitney tests were used for comparisons. Median follow-up time was obtained via the reverse Kaplan-Meier method. Probabilities of overall survival (OS) and EFS were estimated by Kaplan-Meier method. Nonparametric cumulative incidence function of Fine and Gray was used to estimate incidences of neutrophil engraftment, GF, of grade II to IV aGVHD and of chronic aGVHD in a competing-risks setting. Univariate analyses were performed via the log-rank test and the cumulative incidence functions via the Gray test. Cox proportional hazard models were used to examine associations between patient or transplant-related factors and outcomes. Multivariate Cox regression model for EFS included covariates age, stem cell source, and donor. All P values are 2 sided, and P < .05 was considered significant. Statistical software R version 3.6.1 was used.
Results
Patient characteristics
This cohort included 84 LAD patients receiving allo-HSCT from 2007 to 2017; 69 patients (82%) were identified as LAD-I, and 11 (13%) were identified as LAD-III. For 4 (5%) patients, no LAD-type classification was available. Females accounted for 48% of the whole cohort. Median age at diagnosis was 3 months (range, 0-131 months), whereas that at allo-HSCT was 13 months (range, 1-354 months) (Table 1). Cohort stratification by age is given in supplemental Tables 5 and 6.
Table 1.
Patient’s characteristics before allo-HSCT
| No. (%) of cases with available information | Overall | LAD type I | LAD type III | P | |
|---|---|---|---|---|---|
| N (%) | 84 (100) | 69 (82) | 11 (13) | ||
| Sex, n (%) | 83 (99) | .858 | |||
| Female | 40 (48) | 32 (46) | 6 (55) | ||
| Male | 43 (52) | 37 (54) | 5 (45) | ||
| Age at diagnosis, mo | 84 (100) | .214 | |||
| Median | 3 | 3 | 7 | ||
| IQR | 1-12 | 1-10 | 3-17 | ||
| Range | 0-131 | 0-131 | 0-50 | ||
| Time between diagnosis and allo-HSCT, mo | 84 (100) | ||||
| Median | 7 | 8 | 7 | .442 | |
| IQR | 3-17 | 3-20 | 2-13 | ||
| Range | 1-353 | 1-353 | 1-46 | ||
| Infections before allo-HSCT, n (%) | 51 (61) | .224 | |||
| Yes | 45 (88) | 37 (93) | 7 (78) | ||
| No | 6 (12) | 3 (7) | 2 (22) | ||
| Recurrence of infections before allo-HSCT, n (%) | 32 (71)* | .120 | |||
| Once with 1 infection | 1 (3) | 0 | 1 (33) | ||
| Once with 2 or more simultaneous infections | 21 (66) | 19 (68) | 2 (67) | ||
| Twice with 2 or more simultaneous infections | 8 (25) | 7 (25) | 0 | ||
| More than twice with more simultaneous infections | 2 (6) | 2 (7) | 0 |
IQR, interquartile range.
This percentage corresponds to n = 45 patients with infections.
Transplantation characteristics
A matched donor was available for most subjects (N = 65; 79%), including siblings (matched sibling donor [MSD]; N = 32; 39%), unrelated donor (matched unrelated donor [MUD]; N = 24, 29%), or a nonsibling family donor (matched family donor [MFD]; N = 9; 11%). Within the MUD group, there were 19 (79%) 10/10 and 5 (21%) 9/10 HLA-matched donors. Mismatched donors (MMD) included 13 (16%) cord blood grafts with >1 HLA-locus mismatch (mismatched unrelated donor [MMUD]) and 4 patients (5%) with HLA-haploidentical grafts from a mismatched family donor (MMFD). The stem cell source was unmanipulated bone marrow (BM) in 44 (55%) patients, peripheral blood in 20 (25%) patients, and cord blood in 16 (20%) patients.
Conditioning was based on Bu in 42 (51%) patients, treosulfan in 26 (31%) patients, and other chemotherapies in 15 (18%) patients. Bu was mainly combined with fludarabine (N = 21; 25%) or cyclophosphamide (N = 17; 21%). Treosulfan-based conditioning included additional fludarabine (N = 14; 17%) or fludarabine and thiotepa (N = 10; 12%). The 15 remaining regimens consisted of fludarabine combined with melphalan (N = 13; 16%) as a non-MA regimen or combined with melphalan and thiotepa (N = 2; 2%). In most cases (81%; n = 66), the combination and the dosage of the drugs were MA. Serotherapy (75% of the cohort [n = 62]) included antithymocyte globulin (ATG) in 51 (61%) and alemtuzumab in 11 (13%) patients. For GVHD prophylaxis, cyclosporine A (CSA) as monotherapy was given to 20 (25%) patients, CSA + steroids to 8 (10%) patients, CSA + mycophenolate mofetil (MMF) to 15 (19%) patients, and CSA + methotrexate (MTX) to 27 (34%) patients. Two (2%) patients receiving a haploidentical graft did not receive any drugs as GVHD prevention. Eight patients (10%) received “other” GVHD prophylaxis without reported drug specification (Table 2).
Table 2.
Characteristics of first allo-HSCT
| Characteristics | No. (%) of cases with available information | Overall | LAD type I | LAD type III | P |
|---|---|---|---|---|---|
| N (%) | 84 (100) | 69 (82) | 11 (13) | ||
| Allo-HSCT y, n (%) | 84 (100) | 1 | |||
| 2007-2012 | 41 (49) | 34 (49) | 5 (45) | ||
| 2013-2017 | 43 (51) | 35 (51) | 6 (55) | ||
| Age at allo-HSCT, mo | 84 (100) | .872 | |||
| Median | 13 | 12 | 15 | ||
| IQR | 0-29 | 6-32 | 8-29 | ||
| Range | 1-354 | 1-354 | 2-95 | ||
| Lansky/Karnofsky performance, n (%) | 63 (75) | .560 | |||
| Score 100 | 22 (35) | 18 (35) | 3 (43) | ||
| Score 90 | 22 (35) | 19 (37) | 1 (14) | ||
| Score 80 | 16 (25) | 12 (23) | 3 (43) | ||
| Score 70 | 1 (2) | 1 (2) | 0 | ||
| Score 60 | 2 (3) | 2 (4) | 0 | ||
| Allo-HSCT source, n (%) | 80 (95) | .267 | |||
| BM | 44 (55) | 38 (58) | 4 (40) | ||
| CB | 16 (20) | 10 (15) | 4 (40) | ||
| PB | 20 (25) | 18 (27) | 2 (20) | ||
| Donor, n (%) | 82 (98) | .419 | |||
| MSD | 32 (39) | 29 (43) | 3 (27) | ||
| 10/10 MUD + MFD | 28 (34) | 23 (34) | 3 (27) | ||
| MUD (9/10) | 5 (6) | 4 (6) | 1 (9) | ||
| MMD (CB + MMFD) | 17 (21) | 11 (16) | 4 (36) | ||
| CMV recipient-donor, n (%) | 70 (83) | .378 | |||
| −/− | 19 (27) | 16 (27) | 1 (14) | ||
| −/+ | 9 (13) | 8 (14) | 0 | ||
| +/− | 10 (14) | 7 (12) | 2 (29) | ||
| +/+ | 32 (46) | 28 (47) | 4 (57) | ||
| Conditioning regimen, n (%) | 81 (96) | .247 | |||
| Non-MA | 15 (19) | 14 (21) | 0 | ||
| MA | 66 (81) | 53 (79) | 10 (100) | ||
| Conditioning drugs, n (%) | 83 (99) | .205 | |||
| Bu based | 42 (51) | 36 (53) | 4 (36) | ||
| Bu + Cy | 17 (21) | 16 (24) | 1 (9) | ||
| Bu + Flu | 21 (25) | 18 (26) | 2 (18) | ||
| Bu + Flu + Cy | 1 (1) | 1 (1) | 0 | ||
| Bu + Flu + TT | 3 (4) | 1 (1) | 1 (9) | ||
| Treosulfan based | 26 (31) | 19 (28) | 6 (55) | ||
| Treo + Cy | 1 (1) | 1 (1) | 0 | ||
| Treo + Flu | 14 (17) | 13 (19) | 1 (9) | ||
| Treo + Flu + TT | 10 (12) | 4 (6) | 5 (45) | ||
| Treo + Mel + Flu | 1 (1) | 1 (1) | 0 | ||
| Others | 15 (18) | 13 (19) | 1 (9) | ||
| Mel + Flu | 13 (16) | 12 (18) | 0 | ||
| Mel + Flu + TT | 2 (2) | 1 (1) | 1 (9) | ||
| Serotherapy, n (%) | 83 (99) | .898 | |||
| No | 21 (25) | 18 (26) | 3 (27) | ||
| Yes | 62 (75) | 50 (74) | 8 (73) | ||
| ATG | 51 (61) | 41 (60) | 6 (55) | ||
| Alemtuzumab | 11 (13) | 9 (13) | 2 (18) | ||
| GVHD prophylaxis, n (%) | 80 (95) | .442 | |||
| No GvHD prevention | 2 (2) | 1 (2) | 1 (10) | ||
| Yes | 78 (98) | 65 (98) | 9 (90) | ||
| CSA alone | 20 (25) | 19 (29) | 1 (10) | ||
| CSA + corticosteroids | 8 (10) | 5 (8) | 1 (10) | ||
| CSA + MMF | 15 (19) | 10 (15) | 4 (40) | ||
| CSA + MTX | 27 (34) | 24 (36) | 2 (20) | ||
| Other | 8 (10) | 7 (11) | 1 (10) |
P values determined using Fisher's exact test or Wilcoxon-Mann-Whitney test as appropriate; missing values were excluded.
Cy, cyclophosphamide; Flu, fludarabine; Mel, melphalan; PB, peripheral blood; Treo, treosulfan, TT, thiotepa.
OS
The median follow-up after allo-HSCT was 32 months (range, 3-120). The 3-year OS probability was 83% (95% confidence interval [CI], 74% to 92%) without any difference in LAD type (LAD-I 84% vs LAD-III 75%; P = .770) (Figure 1A; Table 3). The OS was significantly different according to the type of donor chosen (P = .001), being similar in MSD and 10/10 MUD (P = .918), both superior to any ≤9/10 mismatched unrelated and family donors (Figure 1B; supplemental Table 2).
Figure 1.

Overall survival. (A) OS of the entire cohort without a significant difference in OS after allo-HSCT in LAD-I vs LAD-III (P = .770). (B) Patients receiving a graft from MSD or 10/10 MD were similar in their OS and were significanlty superior to other donor types (P = .001).
Table 3.
Outcomes
| Overall | LAD type I | LAD type III | P | |
|---|---|---|---|---|
| Neutrophil engraftment | .346 | |||
| 28-d cumulative incidence, % (95% CI) | 86 (78-93) | 88 (81-96) | 73 (46-99) | |
| Median time to engraftment, d | 17 | 17 | 17 | |
| Range time to engraftment, d | 8-61 | 8-61 | 9-39 | |
| PGF | .117 | |||
| 28-d cumulative incidence, % (95% CI) | 3 (0-8) | 3 (0-7) | 9 (0-26) | |
| SGF | .651 | |||
| 3-y estimate, % (95% CI) | 15 (7-23) | 13 (5-22) | 20 (13-45) | |
| Range time to SGF, mo | 1-72 | 1-72 | 1-2 | |
| Overall survival | .770 | |||
| 3-y OS estimate, % (95% CI) | 83 (74-92) | 84 (75-94) | 75 (50-100) | |
| EFS | .934 | |||
| 3-y EFS estimate, % (95% CI) | 56 (46-69) | 58 (46-70) | 56 (23-88) | |
| Acute GVHVD | ||||
| 100-d cumulative incidence grade II to IV, % (95% CI) | 24 (15-34) | 24 (14-35) | 11 (0-32) | .318 |
| 100-d cumulative incidence grade III to IV, % (95% CI) | 13 (6-20) | 14 (5-22) | 0 | .320 |
| Chronic GVHD | .815 | |||
| 3-y cumulative incidence, % (95% CI) | 12 (5-20) | 13 (5-21) | 11 (0-32) | |
| Yes, n | 9 | 7 | 2 | |
| Limited/extensive/not specified | 4/3/2 | 4/2/1 | 0/1/1 | |
| No, n | 64 | 54 | 8 | |
| Not applicable (OS < 100 d), N | 5 | 5 | ||
| No data, n | 6 | 3 | 1 |
P values determined using log-rank test or Gray test as appropriate.
Any cytomegalovirus (CMV) mismatches (positive or negative CMV immunoglobulin G serology in recipient-donor pairs) showed a significant negative correlation with survival (P < .001), although CMV infection/reactivation was not reported as a cause of death in any patient included in this analysis. The age at allo-HSCT, sex, source, conditioning regimens, serotherapy, and GVHD prophylaxis did not impact the OS. GF was analyzed as a time-dependent covariate and was not significantly associated with OS because almost all patients experiencing GF were rescued by a second allo-HSCT (hazard ratio [HR], 2.3; 95% CI, 0.63-8.56; P = .201) (supplemental Table 1).
EFS and disease clearance
Based on available data of 79 patients, we observed an EFS probability of 58% (95% CI, 46-70) for LAD-I and 56% (23-88) for LAD-III patients. EFS was significantly different according to the type of donor employed (P = .003). The MSD group had superior EFS compared with 10/10 matched donors, including MUD or MFD (P = .011) (Figure 2A). Furthermore, age at transplant <13 months was significantly associated with a better EFS (P = .004) (Figure 2B).
Figure 2.
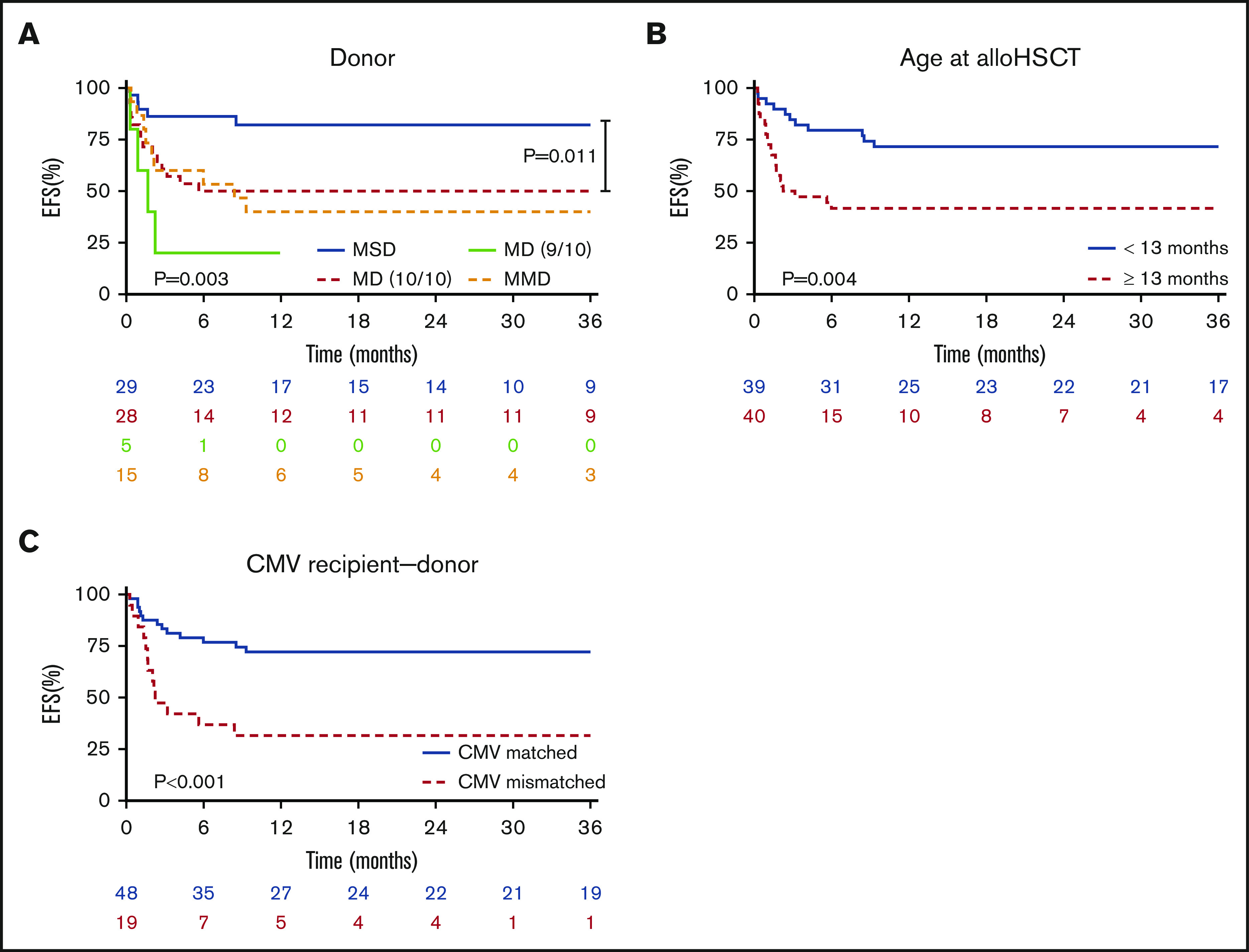
EFS as GF-GVHD II-IV–free survival. (A) Patients receiving a graft from MSD were significantly superior in EFS (P = .003). In direct comparison of MSD and 10/10 MD we also observed a significant superiority in EFS for MSD (P = .011). (B) Patients receiving an allo-HSCT during infancy (<13 months) showed a significantly better EFS compared to older patients (P = .004). (C) EFS was also significantly higher in patients having a donor with a matching CMV serology status (+/+ or −/−) (P < .001).
Seropositivity for CMV in both donor and recipient pairs was associated with better EFS (supplemental Table 2), and any CMV mismatches (donor vs recipient −/+ or +/−) correlated with an inferior EFS (P < .001) (Figure 2C). However, in the multivariate analysis of factors possibly impacting EFS (age, stem cell source, and donor type), we observed a better outcome when patients were transplanted at a young age (<13 months), received an MSD graft, and received BM as the stem cell source (Table 4). At last follow-up, 69 patients were alive and well, and 2 patients were alive despite secondary graft failure (SGF) without a further transplant. In total, 13 patients died.
Table 4.
Multivariate analysis of EFS
| n | HR (95% CI) | P | |
|---|---|---|---|
| Age at allo-HSCT | |||
| Agelog10* | 79 | 2.47 (1.14-5.32) | .021 |
| Allo-HSCT source | |||
| BM | 44 | 1 | |
| CB | 15 | 3.03 (0.90-10.36) | .077 |
| PB | 19 | 1.92 (0.71-5.14) | .195 |
| Donor | |||
| MSD | 29 | 1 | |
| MUD 10/10 + MFD | 28 | 6.78 (1.93-23.81) | .003 |
| MUD 9/10 | 5 | 8.37 (1.85-37.82) | .006 |
| MMD (CB + MMFD) | 15 | 4.99 (1.03-24.13) | .045 |
Log transformation was used in setting of a Cox regression model, because there was no normal (Gaussian) distribution of the age (age skewness with a range of 0.20-29.46 y with a median of 1.075).
Causes of death
Thirteen patients died at a median of 4 months (range, 0.3-26) after allo-HSCT. HSCT-related death was reported in 12 patients; 1 patient died in an accident. aGVHD was reported as the cause of death in 6 children; of those, 5/6 suffered from grade III to IV aGVHD. Other deaths were due to infections (n = 6). Three patients developed infections during primary or secondary GF; 3 patients suffered from lethal infections despite sustained engraftment (Table 5).
Table 5.
Deceased patients
| Sex | LAD | Age, mo | Source, HLA-matching | Donor | CMV patient/donor | Conditioning | Serotherapy | GVHD prophylaxis | Engraftment, d | GF, mo | aGVHD | 2nd allo-HSCT | Cause of death, mo |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M | I | 11 | BM, 10/10 | MSD | −/+ | Treo + Flu | ATG | CSA | Yes, +27 | No | Grade 0 | No | Infection, +1 |
| M | I | 23 | BM, 10/10 | MUD | +/− | Bu + Cy | ATG | CSA + corticosteroids | Yes, +49 | SGF, +2 | Grade I | SGF, +4 | Infection, +26 |
| F | I | 3 | BM, 10/10 | MFD | n.a. | Treo + Flu + TT | ATG | CSA + MMF | No | PGF, +0.3 | No | No | Infection, +0.3 |
| M | III | 25 | CB, <5/6 | MMUD | +/+ | Bu + Flu | ATG | CSA + MMF | No | PGF, +0.7 | No | PGF | Infection, +4 |
| F | 12 | CB, <5/6 | MMUD | +/− | Bu + Flu | ATG | CSA + corticosteroids | Yes, +15 | No | Grade II | No | Infection, +8 | |
| F | I | 30 | BM | MSD | −/+ | Bu + Cy | None | CSA + MTX | Yes, +12 | No | Grade III | No | septic shock, +10 |
| F | I | 77 | PB, 9/10 | MUD | +/− | Bu + Flu | ATG | CSA + MTX | Yes, +14 | No | Grade IV | No | GVHD, +3 |
| M | I | 137 | PB, 10/10 | MUD | n.a. | Bu + Flu + Cy | ATG | Other | Yes, +15 | No | Grade IV | No | GVHD, +3 |
| M | I | 20 | PB, 9/10 | MUD | n.a. | Mel + Flu | Alemtuzumab | CSA+MMF | Yes, +61 | No | Grade IV | No | GVHD, +3 |
| M | III | 15 | CB, 5/6 | MMUD | +/− | Bu + Flu | ATG | CSA+corticosteroids | Yes, +35 | No | Grade II | No | cGVHD, +13 |
| F | I | 5 | BM | n.a. | −/+ | Mel + Flu | ATG | CSA | Yes, +26 | No | Grade III | No | GVHD, +9 |
| F | I | 47 | PB | n.a. | n.a. | Mel + Flu | ATG | CSA + MTX | Yes, +19 | No | Grade IV | No | GVHD, +2 |
| M | I | 6 | CB, 5/6 | MMUD | −/− | Bu + Cy | ATG | n.a. | Yes, +19 | No | Grade I | No | Accident, +9 |
cGVHD, chronic GVHD; F, female; M, male; n.a., data not available.
GVHD
Twenty patients developed grade II to IV aGVHD with a median onset at day +30 (range, 6-126). This resulted in a cumulative incidence of 24% at day +100 (95% CI, 15%-34%). Severe aGVHD (grade III-IV) occurred in 11/79, resulting in a 100-day CI of 13% (range, 6-20). Chronic GVHD affected 9 patients and was extensive in 3 patients (3-year cumulative incidence 12% [5-20]) (Table 3).
With regard to donors, we observed the lowest 100-day cumulative incidence of grade II to IV aGVHD in MSD (10%, 0-21). Comparing MSD with 10/10 MUD + MFD, there was a trend toward lower incidence of aGVHD in MSD (P = .087) (Figure 3A; supplemental Table 3). The patient’s age at transplantation correlated significantly with incidence of aGVHD (100-day grade II-IV incidence aGVHD for patients ≥13 months 38% vs 10% for patients <13 months; P = .008) (Figure 3B; supplemental Table 3).
Figure 3.
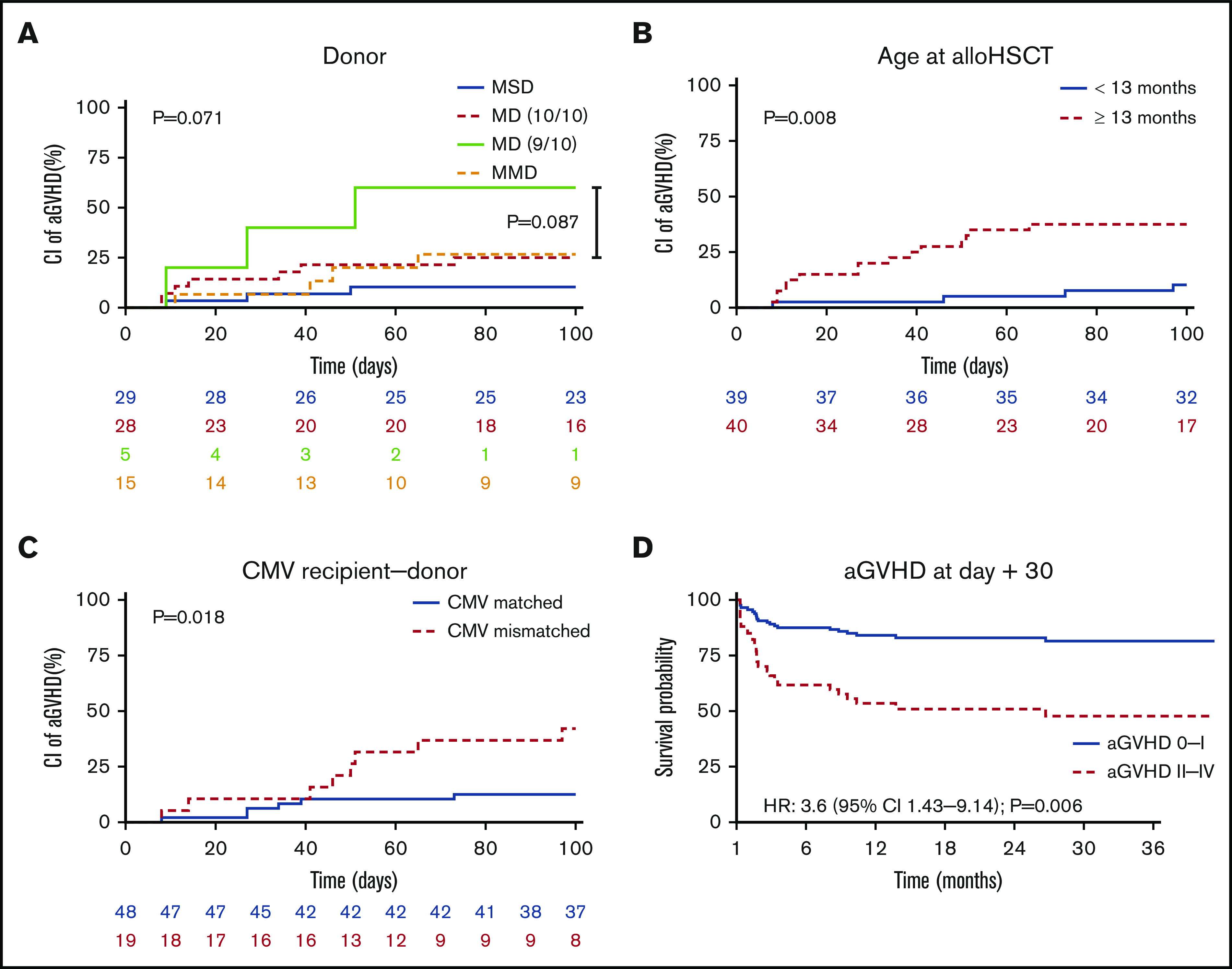
aGVHD II-IV. (A) We observed a tendency towards less aGVHD in patients having MSD (P = .071). (B) Patients receiving allo-HSCT during infancy (<13 months) suffered significanlty less aGVHD than older patients (P = .008). (C) A matched CMV serostatus correlated to less aGVHD (P = .018). (D) Predicted OS in patients according to patients’ aGVHD at day 30. The survival probability of OS was significatly reduced in patients developing aGVHD II-IV by day 30 (HR = 3.6 [95% CI 1.43-9.14]; P = .006).
As mentioned above for EFS, any mismatches in CMV serology (donor/recipient pairs) correlated with a higher incidence of grade II to IV aGVHD at day 100 (CMV+/+ or CMV −/−, 13%; CMV+/− or CMV −/+ 42%; P = .018) (Figure 3C; supplemental Table 3). Further factors, including conditioning regimens, and the use of serotherapy and/or type of GVHD prophylaxis did not impact the incidence of aGVHD II to IV (supplemental Table 3).
Among patients experiencing grade II to IV and III to IV aGVHD, 40% and 55%, respectively, died. The time-dependent Cox regression indicated an increased risk of death for patients suffering from aGVHD II to IV of 3.6-fold (HR, 3.6; 95% CI, 1.43-9.14; P = .006) vs patients with aGVHD 0-I (Figure 3D). In severe aGVHD (III+IV), the risk for death was increased with HR 7.15 (95% CI, 2.56-20; P < .001). There was a tendency toward more aGVHD in non-MA–treated patients (P = .066)
Engraftment and GF
Overall, 81 (96%) patients reached neutrophil engraftment (>0.5 × 103/µL). Median time to neutrophil and thrombocyte engraftment (>20 × 103/µL) was 17 (range, 8-61) and 26 (range, 6-118) days, respectively. Primary GF occurred in 2 LAD-I and 1 LAD-III patients. One patient died early, and the other two received a second transplant. SGF occurred in 14 patients reported as complete graft loss with autologous reconstitution at a median of 3 months after allo-HSCT (range, 1-72), resulting in an estimated probability of SGF at 36 months of 15% (95% CI, 7-23) (Table 3; supplemental Figures 1 and 2). The cumulative incidence of GF (CI-GF) considering both primary graft failure (PGF) and SGF was 17% (95% CI, 9-26) at 36 months after allo-HSCT.
Regarding donor choice, patients transplanted from MSD or 10/10 MUD suffered less GF than patients with a ≤9/10 donor (HR = 2.94; 95% CI, 1.16-7.46; P = .023). Patients <13 months, the median age in this cohort, showed a tendency toward lower CI-GF than older patients (<13 months, 13%; ≥13 months, 26%; P = .074).
With regard to conditioning regimen, 15 patients in MA and 1 patient in the non-MA group suffered a GF. Among MA-treated patients, eight had received a treosulfan-based and seven a Bu-based regimen. The intensity of conditioning (non-MA vs MA; P = .225), type of conditioning regimen (Bu-based, treosulfan-based, or other), and GVHD prophylaxis did not affect CI-GF significantly (supplemental Table 4).
Second allo-HSCT
Fifteen of 17 patients with GF (PGF; N = 2 or SGF; N = 13) underwent a second allo-HSCT at a median of 9 months after the first allo-HSCT (range, 2-95). Two patients did not receive a second allo-HSCT: one because of early death after GF, and the other after SGF with disease activity at last follow-up.
Among the second transplant recipients, 1 patient with PGF did not reach neutrophil engraftment after the second transplant and died. Three patients with SGF experienced another GF after the second transplant. One died, and the other two were alive at last follow-up without a third transplant. The other 11 patients reached stable engraftment after a second transplant and were all alive at last follow-up. In summary, 13 of 15 (87%) patients were alive at last follow-up after the second allo-HSCT (supplemental Figure 2; Table 6).
Table 6.
Second allo-HSCT
| First allo-HSCT | Second allo-HSCT | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | LAD type | Age at HSCT, mo | Source, HLA-matching | Donor | Conditioning | Serotherapy | GVHD prophylaxis | Engraftment, d | aGVHD grade | GF, mo | 2nd HSCT, mo | Source, HLA-matching | Donor | Conditioning | Serotherapy | GVHD prophylaxis | Engraftment, d | GF, mo | aGVHD grade | LFU since 1st allo-HSCT, mo |
| M | I | 19 | PB, <9/10 | MMFD (haplo) | Treo + Flu + TT | Alemtuzumab | No GVHD prevention | No | — | PGF | +1 | PB, <9/10 | MMFD | Flu + Cy + TBI | ATG | No GVHD prevention | Yes, +32 | No | I | Alive, +6 |
| M | III | 25 | CB, <5/6 | MMUD | Bu + Flu | ATG | CSA + MMF | No | — | PGF | +1 | CB, <5/6 | MMUD | n.a. | CSA + MMF | No | PGF | 0 | Dead, +4 | |
| F | I | 4 | n.a. | MSD | Bu + Flu | ATG | n.a. | Yes, +14 | SGF, +9 | +11 | n.a. | MSD | Treo + Flu | No | n.a. | n.a. | n.a. | n.a. | Alive, +25 | |
| F | I | 36 | n.a. | MSD | Mel + Flu | ATG | CSA + corticosteroids | Yes, +14 | II | SGF, +71 | +86 | BM, 10/10 | MSD | Treo + Flu + TT | No | CSA + MTX | Yes, +19 | No | I | Alive, +121 |
| M | I | 14 | BM, 10/10 | MSD | Treo + Flu | ATG | CSA | Yes, +17 | III | SGF, +12 | +15 | BM, 10/10 | MSD | n.a. | n.a. | Yes, +14 | No | 0 | Alive, +31 | |
| M | I | 16 | CB, 6/6 | MUD | Bu + Flu | ATG | CSA | Yes, +25 | 0 | SGF, +1 | +5 | PB, 10/10 | MUD | Treo + Flu | Alemtuzumab | CSA + MMF | Yes, +9 | No | 0 | Alive, +54 |
| F | Other | 12 | BM, 10/10 | MUD | Bu + Flu + TT | ATG | CSA + MMF | Yes, +43 | 0 | SGF, +3 | +5 | PB, 10/10 | MUD | n.a. | No | Other | Yes, +16 | No | II | Alive, +30 |
| M | I | 23 | BM, 10/10 | MUD | Bu + Cy | ATG | CSA + corticosteroids | Yes, +49 | I | SGF, +2 | +14 | PB, <9/10 | MMUD | Mel + Flu | Alemtuzumab | Other | Yes, +17 | SGF, +4 | 0 | Dead, +26 |
| M | I | 354 | BM, 10/10 | MUD | Treo + Flu | ATG | CSA + MTX | Yes, +22 | 0 | SGF, +3 | +13 | PB, 10/10 | MUD | Treo + Flu + TT | ATG | CSA + MTX | Yes, +23 | No | 0 | Alive, +16 |
| M | I | 24 | PB, 10/10 | MUD | Treo + Flu | ATG | CSA + MTX | Yes, +27 | 0 | SGF, +2 | +5 | BM, 10/10 | MUD | Bu + Flu + TT | ATG | CSA + MTX | Yes, +24 | No | 0 | Alive, +29 |
| M | I | 14 | PB, <9/10 | MMUD | Treo + Flu | ATG | Other | Yes, +18 | 0 | SGF, +6 | +6 | BM, <9/10 | MMUD | Flu + Cy | ATG | Other | Yes, +8 | No | 0 | Alive, +19 |
| M | I | 5 | CB, 5/6 | MMUD | Bu + Cy | ATG | CSA + corticosteroids | Yes, +34 | 0 | SGF, +8 | +8 | CB n.a. | n.a. | Cy + TBI | ATG | CSA + corticosteroids | Yes, +35 | No | 0 | Alive, +55 |
| F | I | 8 | CB, <5/6 | MMUD | Treo + Flu | ATG | CSA | Yes, +27 | 0 | SGF, +43 | +95 | PB, <9/10 | MMUD | Mel + Flu | Alemtuzumab | CSA + MMF | Yes, +20 | SGF, +32 | I | Alive, +128 |
| F | III | 22 | CB, 5/6 | MMUD | Bu + Cy | ATG | CSA + MMF | Yes, +16 | 0 | SGF, +1 | +27 | BM, <9/10 | MMUD | Treo + Flu + TT | ATG | CSA + MMF | Yes, +14 | No | IV | Alive, +78 |
| F | III | 44 | PB, <9/10 | MMFD | Mel + Flu + TT | ATG | No GVHD prevention | Yes, +9 | 0 | SGF, +2 | +2 | PB, <9/10 | MMFD | Bu + Cy + TBI | No | No GVHD prevention | n.a. | SGF, +3 | 0 | Alive, +5 |
LFU, last follow-up; TBI, total body irradiation.
Chimerism
Chimeric analyses at 6 months after allo-HSCT were reported in 46/84 patients. The cell sources for the hematopoietic chimerism analysis were heterogeneous. These analyses were performed in whole blood in 27 patients; thereof, 18 showed complete chimerism (CC), and 9 patients had a mixed chimerism (MC). In 7 patients, chimerism was investigated in peripheral blood mononuclear cells (5 patients had CC and 2 patients had MC). In 10 patients, chimerism was assessed in myeloid cells showing CC in 8 patients vs MC 2 patients. In 2 patients, chimerism was assessed in BM, both being MC. There were no significant associations between chimerism and the type of conditioning regimen, donor type, and the source of stem cells.
Discussion
Here, we present the largest cohort of LAD patients transplanted over a 10-year period. We observed a 3-year OS estimate of 83% (95% CI, 74-92) for the entire cohort. Survival was excellent (>90%) for patients with a sibling, matched family, or 10/10 MUD and when transplanted in infancy.
Patient characteristics of our cohort were similar to those of the previous EBMT study of LAD transplantations (1997 to 2007) in which 36 patients were reported,16 from whom 75% had received BM from a matched related or unrelated donor (78%). The OS of the 1997 to 2007 cohort was 75% and was slightly better than the historic cohort of 1982 to 1993. The authors reported a high rate of GF especially in the setting of haploidentical transplantation, as well as 9 (25%) deaths, including infections (N = 5), hepatic venoocclusive disease (N = 3), and 1 case of EBV-PTLD. Although GF, without being impacted by the type of conditioning, remained a relevant complication of transplant in our cohort (CI-GF 17%), toxicities, especially veno-occlusive disease, were not reported. This was possibly due to the use of less toxic conditioning regimens during our study period.
Almost all patients with GF could be rescued by a second transplant; aGVHD remained the most relevant factor correlating to inferior survival. The previous study reported aGVHD (grade II-IV) in 25% of the cohort,16 which is similar to our cohort; however, no deaths were reported because of aGVHD in the previous cohort, whereas aGVHD was one of the major causes of death in our patients. Factors related to GVHD II to IV (and to severe aGVHD [III-IV]) were age at transplant (≥13 months) and a nonsibling donor. In the cohort of patients suffering from aGVHD, we observed a higher proportion of donor-recipient pairs having mismatched CMV serology.
Detailed chimerism analyses were not possible because of a large proportion of missing chimerism but also because of a very heterogeneous data set (BM vs peripheral blood mononuclear cells and missing myeloid chimerism), which is a limitation of this study.
Death related to allo-HSCT occurred in 12 patients. Causes of death were infections (3 with GF and 3 despite stable engraftment), and 6 patients with aGVHD. The estimated 3-year EFS was 58% (46 to 72) and 56% (23-88) in LAD-I and LAD-III, respectively. In multivariate analysis, we observed a better EFS in patients transplanted from an MSD, using BM as a stem cell source, and with an age <13 months at time of allo-HSCT.
While in children transplanted for leukemia, a 9/10 or 10/10 HLA match was shown to offer an OS/EFS comparable to that of an MSD,19 in this cohort, we observed that patients receiving a graft from an MSD or from a 10/10 MUD and MFD had a similar OS, whereas patients who received a transplant from a donor who was matching in ≤9/10 HLA loci had a lower probability to survive. However, these findings need to be interpreted with caution because of the low number (N = 5) of 9/10 MUD. Even though the group of older (≥13 months) patients did have 5 (9/10) MUD vs no 9/10 MUD within the younger (<13 months) individuals, there was no statistical significance (supplemental Table 5). Furthermore, the multivariate analysis did confirm the significance of age as an independent parameter impacting EFS. Comparing MSD to 10/10 MUD and MFD, there was a trend toward less grade II to IV aGVHD in MSD without reaching the level of significance (P = .087). Because of low numbers of 9/10 MUD, no direct comparison of 9/10 vs 10/10 MUD was possible. Still, EFS is significantly better in MSD compared with all other donor types. This is in line with observations in other nonmalignant diseases, for example, chronic granulomatous disease (CGD). Nonetheless, the average reported incidence of severe aGVHD in CGD seems to be lower. Two large studies are available on allo-HSCT in CGD (both using RIC regimens) reporting an incidence of severe aGVHD (III-IV) of ∼4% to 8% (in our LAD study 13%).20,21 A recently published CGD study reported a 2-year cumulative incidence of grade II to IV aGVHD and grade III to IV aGVHD of 20.1% (95% CI, 17.1-23.2) and 9% (95% CI, 6.9-11.2).22 For DOCK 8 (dedicator of cytokinesis 8) deficiency, 1 recent study reported 11% rate of grade III to IV aGVHD.23 Taken together, we observed that allo-HSCT in LAD-I and -III can be complicated by a relevant treatment-related mortality because of inflammatory issues in terms of GF and aGVHD despite well-matched donors, mainly MA treatment and the use of serotherapy.
The intrinsic role of inflammation in LAD has been discussed in human and animal models of this disease. Especially, the origin of hyperleukocytosis in this condition has been postulated not only as a consequence of an impaired neutrophil migration but also because of a severely impaired IL-12/IL-23 pathway resulting in an IL-17–mediated inflammatory disease. A hyperinflammatory microenvironment has also been shown in gingiva of LAD patients’ periodontitis.4,22 Nevertheless, in this study, we could not assess the level of pre-/peritransplant inflammatory status of the cohort; therefore, a link between disease duration and increased inflammation, as well as treatment-related mortality can only be speculated, but not formally proven. aGVHD remains a challenge that will require specific approaches, including controlling the inflammatory component pretransplant. Whether ustekinumab may be an interesting tool needs to be evaluated further. Furthermore, gene therapy might be an option in the future for patients at risk for severe GVHD.
The age at transplant was the main factor associated with EFS, being defined as survival without GF, and aGVHD II to IV. With regard to stem cell source, we believe that cord blood (CB) correlated with lower EFS because of the higher rate of mismatches. A direct analysis of matched CB and mismatched CB was not possible because of low case numbers in each group. Likewise, a further analysis of stem cell sources within the mismatched donor group was also not possible for any of our endpoints, including GF. However, 9/10 MUD group did not include any CB.
In summary, our data show that patients with LAD-I and -III who received an allo-HSCT from either an MSD or a 10/10 matched donor had an excellent chance to survive and cure the disease, even though GF and aGVHD remain relevant issues. It seems that age >13 months at allo-HSCT and CMV mismatch remain independent risk factors for treatment failure. Gene therapy for LAD-I is currently being tested in a phase 2 study. Depending on the final safety and efficacy data obtained in this trial, gene therapy may become an interesting alternative for HSCT, particularly in patients without a suitable donor and/or with significant comorbidity at risk for high transplant-related mortality and morbidity.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the data managers from the different centers for their support in data collection. The authors are grateful to all medical and nurse personnel of the participating clinical and transplant centers for patients’ care, as well as to biotherapy departments for processing the stem cell grafts.
This work was supported by Goethe University Frankfurt and the EBMT Office Leiden.
B.S.’s position was funded by the Australian Government Research Training Program, The Garvan Institute of Medical Research, and Hadassah Australia.
Footnotes
Data from this paper may be acquired by contacting the corresponding author, Shahrzad Bakhtiar, at shahrzad.bakhtiar@kgu.de.
Authorship
Contribution: S.B. and P.B. designed the study; S.B., P.B., A.L., and E.S.-M. wrote the manuscript; P.B., A.L., and M.H.A. revised the manuscript; and all authors of the EBMT/SCETIDE registry (H.-J.B., D.-J.E., S.H., M.A., A. Toren, G.G., D.M., F.L., P.M., G.M., G.O., A.S., C.H., M.I., R.W., O.A., Y.B., A. Tbakhi, P.V., M.K., A.K., A.G., R.H., E.U., A.P.-M., M.G., F.P., T.A., G.K., I.B., P.L., E.S., A.Y., T.Z., R.R.G.B., P.S., B.S., M.S., A.R.G., and M.H.A.) provided clinical data and revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
All authors are members of the Pediatric Diseases Working Party and/or the Inborn Errors Working Party of EBMT.
Correspondence: Shahrzad Bakhtiar, Division for Stem Cell Transplantation, Immunology, and Intensive Care Medicine, Department for Children and Adolescents Medicine, University Hospital Frankfurt, Thoedor-Stern-Kai 7, 60590 Frankfurt am Main, Germany; e-mail: shahrzad.bakhtiar@kgu.de.
References
- 1.Etzioni A. Genetic etiologies of leukocyte adhesion defects. Curr Opin Immunol. 2009;21(5):481-486. [DOI] [PubMed] [Google Scholar]
- 2.Hayward AR, Harvey BA, Leonard J, Greenwood MC, Wood CB, Soothill JF. Delayed separation of the umbilical cord, widespread infections, and defective neutrophil mobility. Lancet. 1979;1(8126):1099-1101. [DOI] [PubMed] [Google Scholar]
- 3.Almarza Novoa E, Kasbekar S, Thrasher AJ, et al. . Leukocyte adhesion deficiency-I: A comprehensive review of all published cases. J Allergy Clin Immunol Pract. 2018;6(4):1418-1420.e10. [DOI] [PubMed] [Google Scholar]
- 4.Moutsopoulos NM, Konkel J, Sarmadi M, et al. . Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med. 2014;6(229):229ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robert P, Canault M, Farnarier C, et al. . A novel leukocyte adhesion deficiency III variant: kindlin-3 deficiency results in integrin- and nonintegrin-related defects in different steps of leukocyte adhesion. J Immunol. 2011;186(9):5273-5283. [DOI] [PubMed] [Google Scholar]
- 6.Kuijpers TW, Van Lier RA, Hamann D, et al. . Leukocyte adhesion deficiency type 1 (LAD-1)/variant. A novel immunodeficiency syndrome characterized by dysfunctional beta2 integrins. J Clin Invest. 1997;100(7):1725-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Essa MF, Elbashir E, Alroqi F, Mohammed R, Alsultan A. Successful hematopoietic stem cell transplant in leukocyte adhesion deficiency type III presenting primarily as malignant infantile osteopetrosis. Clin Immunol. 2020;213:108365. [DOI] [PubMed] [Google Scholar]
- 8.Etzioni A, Frydman M, Pollack S, et al. . Brief report: recurrent severe infections caused by a novel leukocyte adhesion deficiency. N Engl J Med. 1992;327(25):1789-1792. [DOI] [PubMed] [Google Scholar]
- 9.Fan Z, Ley K. Leukocyte adhesion deficiency IV. Monocyte integrin activation deficiency in cystic fibrosis. Am J Respir Crit Care Med. 2016;193(10):1075-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van de Vijver E, van den Berg TK, Kuijpers TW. Leukocyte adhesion deficiencies. Hematol Oncol Clin North Am. 2013;27(1):101-116, viii. [DOI] [PubMed] [Google Scholar]
- 11.Moutsopoulos NM, Zerbe CS, Wild T, et al. . Interleukin-12 and interleukin-23 blockade in leukocyte adhesion deficiency type 1. N Engl J Med. 2017;376(12):1141-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saultier P, Szepetowski S, Canault M, et al. . Long-term management of leukocyte adhesion deficiency type III without hematopoietic stem cell transplantation. Haematologica. 2018;103(6):e264-e267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamidieh AA, Pourpak Z, Hosseinzadeh M, et al. . Reduced-intensity conditioning hematopoietic SCT for pediatric patients with LAD-1: clinical efficacy and importance of chimerism. Bone Marrow Transplant. 2012;47(5):646-650. [DOI] [PubMed] [Google Scholar]
- 14.Horikoshi Y, Umeda K, Imai K, et al. . Allogeneic hematopoietic stem cell transplantation for leukocyte adhesion deficiency. J Pediatr Hematol Oncol. 2018;40(2):137-140. [DOI] [PubMed] [Google Scholar]
- 15.Stepensky PY, Wolach B, Gavrieli R, et al. . Leukocyte adhesion deficiency type III: clinical features and treatment with stem cell transplantation. J Pediatr Hematol Oncol. 2015;37(4):264-268. [DOI] [PubMed] [Google Scholar]
- 16.Qasim W, Cavazzana-Calvo M, Davies EG, et al. . Allogeneic hematopoietic stem-cell transplantation for leukocyte adhesion deficiency [published correction appears in Pediatrics. 2009;123(5):143]. Pediatrics. 2009;123(3):836-840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giralt S, Ballen K, Rizzo D, et al. . Reduced-intensity conditioning regimen workshop: defining the dose spectrum. Report of a workshop convened by the center for international blood and marrow transplant research. Biol Blood Marrow Transplant. 2009;15(3):367-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bacigalupo A, Ballen K, Rizzo D, et al. . Defining the intensity of conditioning regimens: working definitions. Biol Blood Marrow Transplant. 2009;15(12):1628-1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peters C, Schrappe M, von Stackelberg A, et al. . Stem-cell transplantation in children with acute lymphoblastic leukemia: a prospective international multicenter trial comparing sibling donors with matched unrelated donors-The ALL-SCT-BFM-2003 trial. J Clin Oncol. 2015;33(11):1265-1274. [DOI] [PubMed] [Google Scholar]
- 20.Morillo-Gutierrez B, Beier R, Rao K, et al. . Treosulfan-based conditioning for allogeneic HSCT in children with chronic granulomatous disease: a multicenter experience [published correction appears in Blood. 2016;128(3):440-448]. Blood. 2016;128(3):440-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Güngör T, Teira P, Slatter M, et al. ; Inborn Errors Working Party of the European Society for Blood and Marrow Transplantation . Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. 2014;383(9915):436-448. [DOI] [PubMed] [Google Scholar]
- 22.Chiesa R, Wang J, Blok H-J, et al. Allogeneic hematopoietic stem cell transplantation in children and adults with chronic granulomatous disease (CGD): a study of the Inborn Errors Working Party (IEWP) of the EBMT [abstract]. Blood. 2018;132(suppl 1). Abstract 970.
- 23.Aydin SE, Freeman AF, Al-Herz W, et al. ; Inborn Errors Working Party of the European Group for Blood and Marrow Transplantation and the European Society for Primary Immunodeficiencies . Hematopoietic stem cell transplantation as treatment for patients with DOCK8 deficiency. J Allergy Clin Immunol Pract. 2019;7(3):848-855. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.