

Abstract
As one of the most abundant immune cell populations in the tumor microenvironment (TME), tumor‐associated macrophages (TAMs) play important roles in multiple solid malignancies, including breast cancer, prostate cancer, liver cancer, lung cancer, ovarian cancer, gastric cancer, pancreatic cancer, and colorectal cancer. TAMs could contribute to carcinogenesis, neoangiogenesis, immune‐suppressive TME remodeling, cancer chemoresistance, recurrence, and metastasis. Therefore, reprogramming of the immune‐suppressive TAMs by pharmacological approaches has attracted considerable research attention in recent years. In this review, the promising pharmaceutical targets, as well as the existing modulatory strategies of TAMs were summarized. The chemokine–chemokine receptor signaling, tyrosine kinase receptor signaling, metabolic signaling, and exosomal signaling have been highlighted in determining the biological functions of TAMs. Besides, both preclinical research and clinical trials have suggested the chemokine–chemokine receptor blockers, tyrosine kinase inhibitors, bisphosphonates, as well as the exosomal or nanoparticle‐based targeting delivery systems as the promising pharmacological approaches for TAMs deletion or reprogramming. Lastly, the combined therapies of TAMs‐targeting strategies with traditional treatments or immunotherapies as well as the exosome‐like nanovesicles for cancer therapy are prospected.
Keywords: chemokine–chemokine receptor, exosomeimmune suppression, metabolism, tumor‐associated macrophages, tyrosine kinase receptor
The biofunctions and clinical implications of TAMs in cancer progression as well as the current pharmaceutical targets in TAMs regulation were summarized. The latest advancements of agents that were effective in TAMs depletion or reprogramming were summarized and discussed. The promising future of exosomal signaling for TAMs‐targeted therapy was discussed and expected.
Abbreviations
- TAMs
tumor‐associated macrophages
- TME
tumor microenvironment
- PAMP
pathogen‐associated patterns
- LPS
lipopolysaccharides
- TNF‐α
tumor necrosis factor α
- GM‐CSF
granulocyte‐macrophage colony‐stimulating factor
- IL
interleukin
- DAMP
damage‐associated patterns
- ARG1
arginase
- DMBA
12‐dimethylbenz‐(a)anthracene
- sCD163
soluble CD163
- irAEs
immune‐related adverse events
- VEGFA
vascular endothelial growth factor A
- ADM
adrenomedullin
- bFGF
basic fibroblast growth factor
- PDGF
platelet‐derived growth factor
- MIF
macrophage‐inhibitory factor
- PAF
platelet‐activating factor
- monocyte chemoattractant protein‐1 MCP‐1
monocyte chemoattractant protein‐1
- MMPs
matrix metalloproteinases
- PD‐L1
program cell death ligand 1
- CTLA‐4
cytotoxic T‐lymphocyte‐associated protein 4
- OS
overall survival
- DFS
disease free survival
- PSGL‐1
P‐selectin glycoprotein ligand‐1
- ICAM‐1
intracellular cell adhesion molecule‐1
- ERK5
extracellular‐regulated protein kinase 5
- EMT
epithelial–mesenchymal transition
- ECM
extracellular matrix
- PMN
premetastatic niche
- GPCRs
G‐protein‐coupled receptors
- MDSCs
myeloid‐derived suppressor cells
- CTCL
cutaneous T‐cell lymphoma
- IFNs
interferons
- RTKs
Tyrosine kinase receptors
- CSF‐1R
colony stimulation factor‐1 receptor
- MSP
macrophage‐stimulating protein
- MerTK
Mer tyrosine kinase
- HIF
Hypoxia‐inducible factors
- BMDMs
bone marrow–derived macrophages
- PPAR
peroxisome proliferator‐activated receptor
- PDK
pyruvate dehydrogenase kinase
- IDO
indoleamine‐2, 3‐dioxygenase
- RAGE
receptor for advanced glycation end‐product
- DAG
diacylglycerol
- MG
methylglyoxal
- AGEs
advanced glycation end products
- ELISA
enzyme‐linked immunosorbent assays
- dt‐GCTs
diffuse‐type tenosynovial giant‐cell tumors
- exoASO
exosome carrying antisense oligonucleotide
- TRL7/8
toll‐like receptor 7 and 8
- SIRPα
signal regulatory protein‐α
- TREM2
triggering receptor expressed on myeloid cells 2
- ERα
estrogen receptor α
- ERK
extracellular signal‐regulated kinase
- PKC
protein kinase C
- PI3K
phosphoinositide‐3 kinase
- AKT
the serine/threonine kinase
- MAPK
mitogen‐activated protein kinase
- mTOR
mammalian/mechanistic target of rapamycin
- NF‐κB
nuclear factor κB
- PDGF‐BB
platelet‐derived growth factor subunit B homodimer
- Ezh2
enhancer of zeste homolog 2
- CSCs
cancer stem cells
- ECs
endothelial cells
- STAT3
signal transducer and activator of transcription 3
- CSN5
COP9 signalosome subunit 5
- FOXP3
forkhead box P3
- KLF5
kruppel‐like factor 5
- TGF‐β
transforming growth factor β
- GSK‐3β
Glycogen synthase kinase‐3β
- ANXA2
annexin A2
- PITPNM3
phosphatidylinositol transfer protein 3
- FOXO1
Forkhead box O1
- CEBPB
CCAAT/enhancer binding protein beta
- TRAIL
tumor necrosis factor‐related apoptosis‐inducing ligand
- ABCB1
ATP binding cassette subfamily B member 1
- HuR
human antigen R
- JAK2
Janus protein tyrosine kinases
- Pyk2
proline‐rich tyrosine kinase 2
- ELMO1
engulfment and cell motility 1
- SAPK
stress‐activated protein kinase
- JNK
Jun N‐terminal kinase
- HDAC1
histone deacetylase 1
- TACE
tumor necrosis factor alpha‐converting enzyme
- SOX4
SRY‐box transcription factor 4
- S100A8/9
S100 calcium binding protein A8/9
- TAK1
transforming growth factor‐beta‐activated kinase 1
- EGFR
epidermal growth factor receptor
- FAK
focal adhesion kinase
- RhoA
ras homolog family member A
- PGE2
Prostaglandin E2
- ARHGAP10
Rho GTPase Activating Protein 10
- MALAT1
metastasis‐associated lung adenocarcinoma transcript 1
- TOPK
T‐LAK Cell‐originated Protein Kinase
- JNK
c‐Jun N‐terminal kinase
- MST1R
macrophage‐stimulating 1 receptor
- HK2
hexokinase 2
- GPR132
G protein‐coupled receptor 132
- REDD1
DNA damage response‐1
- HMGB1
high‐mobility group box 1
- RIPK3
receptor‐interacting protein kinase‐3
- FABP
fatty acid binding protein
- MCAD
medium‐chain acyl‐coA dehydrogenase
- GS
glutamine synthetase
- ARG
arginase
- FPN
ferroportin
- LCN
lipocalin
- BRG1
brahma‐related gene 1
- LIPA
lipase A
- PTEN
phosphatase and tensin homolog
- TGFBR3
transforming growth factor beta receptor 3
1. BACKGROUND
Cancer is a heterogeneous disease that is composed of numerous cell types, including both cancer and noncancer cells. Although macrophage content is highly variable in different tumor entities, macrophages are among the most prominent tumor‐associated noncancer cell type in the tumor microenvironment (TME), 1 , 2 known as tumor‐associated macrophages (TAMs). Current evidence suggests that TAMs engage in complex network interactions with cancer stem cells, cancer cells, endothelial cells, fibroblasts, T cells, B cells, and natural killer cells. 3 Their cross talks accelerate the formation of the immune‐suppressive TME that not only stimulates cancer cell proliferation, neoangiogenesis, lymphangiogenesis, drug resistance, and distant metastasis, but also further recruits macrophages to establish a vicious feedback loop that continually reinforces the immune‐suppressive TME. Recently, multiple signaling pathways have been identified as critical nodes mediating TAMs polarization and interactions with malignant cells. Small molecule inhibitors or monoclonal antibodies targeting TAMs signaling have been demonstrated to effectively inhibit cancer development and metastasis. Therefore, TAMs have emerged as a central drug target for cancer therapy. Here, our review focuses on discussing the biological functions of TAMs, critical signaling and targets regulating TAMs activities, and current therapeutic strategies for treating malignancies that affect TAMs.
HIGHLIGHT
The biofunctions and clinical implications of TAMs in cancer progression were summarized.
The current pharmaceutical targets in TAMs regulation were introduced and discussed.
The latest advancements of agents that were effective in TAMs depletion or reprogramming were summarized and discussed.
The promising future of exosomal signaling for TAMs‐targeted therapy was discussed and expected.
2. MACROPHAGE POLARIZATION IN CANCER
Since the first description by llya llyich Mechnikov in 1882, phagocytes have been reported to be present throughout the body and to perform specific biological functions. For example, liver macrophages (Kupffer cells) eliminate pathogenic and waste products from circulation. 4 Brain‐resident macrophages (microglia) contribute to the maintenance of engrams (i.e., the neural bases of memories) 5 by facilitating synaptic pruning. Langerhans cells in the skin are responsible for activating local inflammatory reactions and clearing pathogenic substances. 6 Phagocytes can also be infiltrated into the TME to perform pro‐ and/or antitumor functions. Usually, macrophages are considered as a plastic cell type capable of activating or polarizing into different statuses. Macrophages change their activation or polarization statuses in response to any potential entity, which is capable of being recognized by macrophages. The common stimuli including growth factors, cytokines/chemokines, hypoxia, exosomes, microbes, microbial products, nucleotide derivatives, antibody‐Fc receptor stimulation, glucocorticoids, infection, and phagocytosis. 7 Therefore, it is hard to establish the common nomenclatures or standards for describing the activation or polarization properties of macrophages induced by various stimuli. 7 There are mainly four definitions of macrophage activation or polarization statuses, including terms such as M1‐like and M2‐like, alternative and classical activation, “regulatory” macrophages, and subdivisions originating from the parent terms. 7 In 2012, Mills proposed the M1‐like/M2‐like dichotomy to describe the two major and opposing activities of macrophages. 8 At present, this definition method of macrophage activation or polarization statuses is widely used by the majority of researchers. M1‐like polarization is reported to be induced by pathogen‐associated patterns, including lipopolysaccharides (LPS), interferon‐γ, granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), and tumor necrosis factor α (TNF‐α). M1‐like activation causes the release of various cytokines, such as interleukin 6 (IL‐6), IL‐1β, and TNF‐α, that facilitate a proinflammatory response to defense against pathogenic insults. 9 In contrast, the M2‐like phenotype is induced by damage‐associated patterns (DAMPs) from cytokines such as IL‐4 and IL‐13, which subsequently activate the JAK‐STAT pathway and turn on the expression of anti‐inflammatory cytokines, such as resistin‐like molecule α, IL‐10, and arginase 1 (ARG1). 10 These signals further accelerate the remodeling of the TME to promote cancer angiogenesis, growth, and immune suppression. Usually, M1‐like is considered to be an inhibitory phenotype due to its function in promoting Th1 responses that exert tumoricidal and microbicidal functions, whereas M2‐like represents a restorative phenotype due to its effects in activating Th2 responses that contribute to tissue remodeling/repair, angiogenesis, immunosuppression, and tumor progression. 11 Generally, both M1‐like and M2‐like phenotypes can exist within the same microenvironment; therefore, the molecular targets controlling the polarization balance are considered as an important approach for cancer therapy. The polarization biomarkers of M1‐like macrophages include CD86 and CD80, while the polarization biomarkers of M2‐like macrophages include CD163, CD204, CD206, CD115, and CD301. 12 Particularly, CD163 can simultaneously be present both in a membrane‐bound form on M2‐like phenotype TAMs and a soluble form in plasma. Because of its macrophage‐specific expression characteristic and highly elevated concentrations during various pathological conditions, soluble CD163 (sCD163) level is also regarded as a reliable biomarker for monitoring macrophage activity. 13 For example, CD163+ TAMs were reported to express high levels of immune‐checkpoint molecules PD1 and PD‐L1. Blocking PD1/PD‐L1 signaling in CD163+ TAMs with antibodies could induce M1‐like polarization and increase macrophage phagocytosis, reduce tumor burden and prolong survival, 14 , 15 and result in an increased release of sCD163 in the lesional skin of melanoma. 13 Meanwhile, Fujimura et al. identified serum sCD163 as a reliable predictive biomarker for immune‐related adverse events (irAEs) and efficacy of nivolumab in patients with advanced melanoma. 16 , 17 These findings suggest that macrophage polarization modulation may also be the potential pharmacological mechanism of immune‐checkpoint inhibitors. The combination treatment of macrophage‐targeted strategy and PD‐1/PD‐L1 blockade may provide a synergetic antitumor efficacy, which deserves further investigations.
3. CLINICAL IMPLICATIONS OF TAMs IN CANCER DEVELOPMENT
Over the past decade, TAMs have attracted rising research attention for their modulatory potential on angiogenesis, cytokines, and immune regulation that either inhibit or facilitate tumor progression. Clinically, elevated infiltration levels of M1‐like macrophages within tumor tissues predict a favorable prognosis, while elevations in the M2‐like macrophages usually predict poor outcomes. 18 A tissue microarray analysis of 553 primary non‐small cell lung cancer patients revealed that M1‐like macrophages in metastatic lymph nodes were a predictor of improved survival. 19 In esophageal cancer patients, cases with high CD163 and CD204 expression levels showed a significantly shorter overall survival than those with comparatively lower CD163 and CD204 levels. 20 In addition, in triple‐negative breast cancer patients with either high CD163 expression or low CD163 expression, the 5‐year overall survival ratio was 72.3% or 82.5%, respectively. 21 These clinical observations have been well supported by experimental studies using macrophage depletion or overexpression strategies. For example, genetic ablation of GM‐CSF in different murine tumor models—such as the MMTV‐PyVT+/‐ breast cancer model, the spontaneous colon cancer model, and the osteosarcoma xenotransplant model—significantly reduces M2‐like macrophage density in solid tumors, which in turn inhibits cancer growth and progression. 22 , 23 , 24 In addition, macrophage depletion with clodronate‐encapsulated liposomes also yields a significant reduction of tumor volume in several malignancies. 25 , 26 In contrast, overexpression of CSF‐1 remarkably increases TAMs recruitment and accelerates cancer development and metastasis. 27 It should be noted that the effect of TAMs on tumor tumorigenesis and progression may fluctuate due to their differentiation heterogeneity. 28 Based on these clinical observations and experimental findings, numerous studies have investigated the underlying mechanisms of TAMs in controlling cancer development. At present, M2‐like macrophages have been found to participate in all steps of malignant development including carcinogenesis, neoangiogenesis, overall immune suppression, drug resistance, and later recurrence and/or metastasis (Figure 1).
FIGURE 1.
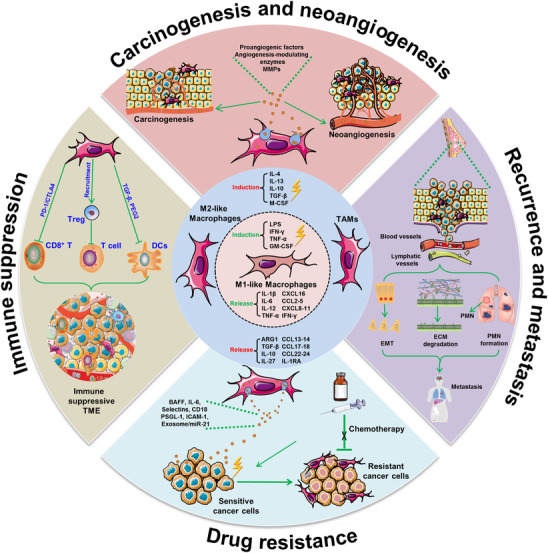
Pro‐oncogenic role of TAMs in cancer development. Macrophages could be differentiated into M1‐like and M2‐like phenotypes under different stimuli. M1‐like macrophages were usually considered as a “killer” phenotype while M2‐like macrophages were known as the “healing” phenotype. M2‐like macrophages have been reported to participate in the process of carcinogenesis and neoangiogenesis via releasing proangiogenic factors, enzymes, and MMPs. Meanwhile, M2‐like macrophages promoted cancer recurrence and metastasis via modulating epithelial–mesenchymal transition, extracellular matrix degradation, and facilitating premetastatic niche formation. On the other hand, M2‐like macrophages could induce chemoresistance by exosomal signaling or cell–cell contact. Most importantly, M2‐like macrophages contributed to establishing the immune suppression microenvironment by elevating the PD‐1/CTLA4 signaling or inhibiting the bio‐functions of cytotoxic T cells or dendritic cells
3.1. TAMs contribute to carcinogenesis and neoangiogenesis
TAMs have been found to be involved in the first step of carcinogenic lesion formation during neoplasia. Macrophage infiltration has been found to be upregulated in a murine chemically induced skin carcinogenesis model. 29 Similarly, a massive accumulation of CD206+ or ARG1+ macrophages has also been found in an inflammation‐mediated skin tumorigenesis mice model, while macrophage ablation has been shown to significantly reduce tumor incidence. 30 In an EGFR‐driven lung carcinogenesis model, sustained macrophage recruitment has been observed and macrophage depletion causes a significant reduction in tumor burden. 31 Neoangiogenesis is also a critical step during carcinogenesis, in which macrophage infiltration is also involved. Various studies have suggested that TAMs are predominantly located near the blood vessels of malignant solid tumors, and TAMs numbers are usually positively correlated with blood vessel density. 32 , 33 , 34 , 35 Functional studies have also demonstrated that TAMs elimination causes the reduction of neoangiogenesis, 36 while TAMs enhancement aggravates this process. 36 Mechanistic studies imply that TAMs can release multiple proangiogenic factors, such as vascular endothelial growth factor A (VEGF‐A), macrophage‐inhibitory factor (MIF), adrenomedullin (ADM), platelet‐activating factor (PAF), platelet‐derived growth factor (PDGF), basic fibroblast growth factor (bFGF), and TGF‐β, as well as numerous cytokines such as TNF‐α, IL‐1, IL‐8, and monocyte chemoattractant protein‐1 (MCP‐1). 37 , 38 , 39 , 40 Additionally, TAMs also release numerous angiogenesis‐modulating enzymes including iNOS, 41 COX‐2, and matrix metalloproteinases (MMPs), 42 , 43 , 44 all of which have been associated in matrix degradation and endothelial cell invasion.
3.2. TAMs facilitate the formation of the immune‐suppressive microenvironment
TAMs recruitment not only supports cancer growth via neoangiogenesis induction but also facilitates the establishment of the immune‐suppressive microenvironment. Recent studies have suggested that TAMs express PD‐L1, PD‐L2, CD86, and CD80, all of which induce CD8+ T cell dysfunction upon binding to immune‐checkpoint receptors such as PD1 or cytotoxic T‐lymphocyte‐associated protein 4 (CTLA4). 44 , 45 In addition, TAMs release multiple cytokines, enzymes, and chemokines that inhibit T‐cell activity through natural regulatory T cell recruitment or L‐arginine depletion in the TME. For example, IL‐10 produced by TAMs could suppress IL‐12 secretion from myeloid cells and promote Th2‐type immune response. 46 The secretion of TGF‐β and PGE2 can impair the maturation process of dendritic cells, which subsequently compromise the balance between innate and adaptive immunity. 47 , 48 Immune‐checkpoint inhibitors have revealed successful therapeutic responses in multiple malignant tumors such as melanoma and lung cancers. 49 Unfortunately, only approximately 20% of cancer patients respond to immunotherapy, and mixed responses can limit therapeutic efficacies and lead to local recurrences and/or distant metastases. 50 Given the abundance and immune‐suppressive properties of TAMs, targeting TAMs has been suggested as a promising approach to promote the efficacy of checkpoint antagonists. For example, anti‐PD1/anti‐CTLA4 treatment can decrease pancreatic tumor growth by approximately 50%, while their combination with PLX3397 (CSF1R inhibitor) can dramatically attenuate tumor expansion and even results in tumor regression by 15%. 51 FcγR is a receptor typically expressed by TAMs. Similarly, a PD1 antibody also results in tumor growth inhibition in colon cancer xenografts, although this efficacy typically varies among mice. Strikingly, when a PD1 antibody and FcγR antagonist were simultaneously administrated, tumor growth was completely inhibited in all mice. 52 Additionally, suppressing IL‐10 signaling in TAMs can promote CD8+ T cell‐mediated antitumor immune responses in breast cancer receiving chemotherapy. 46 Given these encouraging preclinical results, utilization of TAMs‐targeting strategies is promising for future clinical application as a combination therapy with checkpoint inhibitors.
3.3. TAMs aggravate cancer drug resistance
TAMs density is also closely correlated with therapeutic responses and is highlighted in cancer drug resistance. Multiple studies have found that TAMs populations are enriched following toxic cancer‐killing treatments. In aromatase‐inhibitor–resistant breast cancer, TAMs density is much higher in primary tumor tissues and significantly correlates with decreased disease‐free survival (DFS) and overall survival (OS). 53 TAMs infiltration was also found to be elevated following chemotherapy in a pancreatic ductal adenocarcinoma model, and TAMs pretreatment could render pancreatic cancer cells resistant to gemcitabine whereas this efficacy was strikingly enhanced by TAMs depletion via clodronate liposomes. 54 In colon cancer, TAMs infiltration has frequently detected been in chemoresistant patients and is significantly associated with poor survival. 55 The macrophage marker, CD68, is positively correlated with the expression of MDR1. Notably, blocking IL‐6 secreted from TAMs is capable of resensitizing colon cancer cells to chemotherapeutic drugs. Meanwhile, B‐cell activating factor released by TAMs is also effective in mediating bortezomib resistance in myeloma. In addition to cytokine secretion, ligand–receptor interactions between cells are also involved in mediating chemoresistance. 56 In a macrophage–myeloma contact model, plasma‐membrane protein/gene profiling assays have demonstrated that CD18 and selectins in macrophages remarkably contribute to chemoresistance via combining with intracellular cell adhesion molecule‐1 (ICAM‐1) and P‐selectin glycoprotein ligand‐1 (PSGL‐1), respectively, in myeloma cells. Pharmacological inhibition or genetic knockdown of PSGL‐1 or ICAM‐1 signaling in these myeloma cells can diminish the protective effects of TAMs from cytotoxic cancer‐killing agents including melphalan, doxorubicin, and bortezomib. Recent findings also suggest that exosomes are also implicated in TAMs‐induced chemoresistance. It is found that macrophages‐derived exosomal miR‐21 signal could be directly delivered into gastric cancer cells, and thus results in apoptosis inhibition and cisplatin resistance of gastric cancer cells. 57 Therefore, it may be worthwhile to apply TAMs‐targeting therapies with traditional cancer‐killing strategies in future clinical applications.
3.4. TAMs accelerate cancer recurrence and metastasis
Local recurrence and distant metastasis are not solely determined by the malignant behavior of cancer cells because recent studies have suggested that stromal cells, particularly TAMs, act as an important driving force in these processes. For example, Giurisato et al. proved that extracellular‐regulated protein kinase 5 (ERK5)‐mediated macrophage proliferation could support melanoma invasiveness and metastasis in vivo, suggesting TAMs renewal as an integral component of tumor metastasis. 58 Mechanistically, numerous researches have demonstrated the crucial role of TAMs in regulating the epithelial–mesenchymal transition (EMT) process. 59 , 60 , 61 Cocultured hepatocellular cancer cells with TAMs could enhance the expression levels of mesenchymal markers including N‐cadherin and vimentin, whereas attenuating the epithelial marker E‐cadherin. 60 A similar phenomenon was also observed in breast, gastric, colon, and pancreatic malignancies. 62 Biologically, multiple TAMs‐secreted cytokines including IL‐1β, IL‐8, EGF, TNF‐α, and TGF‐β have been validated to promote the EMT process. 60 , 63 , 64 Meanwhile, TAMs can secrete various kinds of proteolytic enzymes including MMPs, cathepsins, and serine proteases to degrade the extracellular matrix (ECM). 65 , 66 For instance, cathepsin B and S secreted by TAMs were found to be critical in promoting pancreatic cancer invasion. 67 An earlier study also demonstrated that TAMs synthesize SPARC/osteonectin to deposit collagen IV, resulting in enhanced tumor invasion and adhesion to other ECM components. 68 TAMs have also been shown to be necessary for facilitating cancer cell intravasation and extravasation. Multiphoton intravital imaging techniques have shown that intravasating cancer cells are invariably accompanied by a macrophage within one cell diameter. 69 Interestingly, macrophages loss can significantly impair the extravasation rates and metastasis of cancer cells. 70 Additionally, TAMs have also been found to be critical in supporting the survival of cancer cells in the circulation. TAMs depletion via genetic methods can dramatically suppress cancer cell survival in pulmonary capillaries as well as the subsequent lung metastasis formation. 71 , 72 The underlying molecular mechanisms of their synergistic interaction are as follows. First, TAMs may trigger PI3K/Akt survival signaling in cancer cells to circumvent proapoptotic cytokines such as TRAIL. 73 Second, TAMs may alleviate survival stress from NK or cytotoxic T cells in the circulation. 72 Following extravasation, it has been found that TAMs are one of the key determinants in establishing the premetastatic niche (PMN). 74 Circulatory or residential TAMs can release chemokines that guide the localization of cancer cells into the PMN with increased expression levels of MMPs, fibronectins, S100A8, and S100A9. 75 , 76 , 77 Our recent report demonstrated that TAMs‐derived CXCL1 exerted an important role in recruiting breast cancer cells into the PMN. 78 Thus, TAMs act as an indispensable factor in fostering cancer cells traveling from the primary site to metastatic lesions. Molecular elucidation of TAMs regulation in cancer development is thus warranted and critical for further developing cancer‐targeting strategies.
4. PHARMACEUTICAL TARGETS OF TAMS
To date, several key pathways including chemokine–chemokine receptor signaling, tyrosine kinase receptor (RTK) signaling, metabolic signaling, and exosomal signaling have been highlighted in determining the biofunctions of TAMs. The promising pharmacological targets of each signaling were described as following and summarized in Table 1.
TABLE 1.
The pharmaceutical targets of TAMs
| Signaling | Targets | Tumors | Mechanisms | References |
|---|---|---|---|---|
| Chemokine–chemokine receptor signaling | CCL2/ CCR2 | Breast cancer | Facilitating cancer progression via activation of ERα and PI3K/AKT/NF‐κB signaling; inducing tamoxifen resistance by activating PI3K/Akt/mTOR signaling; enhancing growth and cell‐cycle progression through SRC and PKC activation | 177 , 178 , 179 |
| CCL2/ CCR2 | Prostate cancer | Recruiting and regulating macrophages via the MAPK/ ERK signaling pathway; protecting cancer cells from autophagic death via the PI3K/Akt/survivin pathway; promoting cancer progression through the induction of MMP‐2 activity | 180 , 181 , 182 | |
| CCL2/ CCR2 | Liver cancer | Inducing invasion and EMT through activation of the Hedgehog pathway; inducing migration and invasion depending on MMP‐2 and MMP‐9; | 183 , 184 | |
| CCL2/ CCR2 | Lung cancer | Increasing invasion via ERα; promoting TAMs infiltration via NF‐κB/CCL2 signaling; promoting invasion mediated by autocrine loop of PDGF‐BB | 185 , 186 , 187 | |
| CCL2/ CCR2 | Ovarian cancer | Promoting peritoneal metastasis through P38‐MAPK pathway | 188 | |
| CCL2/ CCR2 | Gastric cancer | Inhibiting proapoptotic autophagy by activating PI3K‐Akt‐mTOR signaling | 189 | |
| CCL5/CCR1 | Colorectal cancer | Promoting cancer progression through CCL5/β‐catenin/Slug pathway | 190 | |
| CCL5/CCR1 | Prostate cancer | Promoting invasion by increasing MMP‐2/9 and activating ERK/Rac signaling | 191 | |
| CCL5/CCR5 | Lung cancer | Increasing migration via PI3K/AKT/NF‐κB signaling; Promoting metastasis and macrophages infiltration via EZH2 | 192 , 193 | |
| CCL5/CCR5 | Breast cancer | Promoting immune cell infiltration driven by TNFα/NF‐κB signaling; enhancing Trastuzumab resistance by ERK pathway activation; promoting cancer proliferation through the mTOR pathway | 194 , 195 , 196 | |
| CCL5/CCR5 | Ovarian cancer | Promoting invasion and migration via NF‐κB–mediated MMP‐9 upregulation; mediating differentiation of CSCs into ECs via the NF‐κB and STAT3 signaling | 197 , 198 | |
| CCL5/CCR5 | Liver cancer | Inducing EMT through activation of the Akt pathway | 199 | |
| CCL5/CCR5 | Pancreatic cancer | Recruiting Treg cells via cancer‐FOXP3; inducing proliferation of cancer cells through F‐actin polymerization | 200 , 201 | |
| CCL5/CCR5 | Colorectal cancer | Enhancing TGF‐β‐mediated killing of CD8+ T cells; Facilitating immune escape via the p65/STAT3‐CSN5‐PD‐L1 pathway | 91 , 202 | |
| CCL5/CCR5 | Gastric cancer | Enhancing aberrant DNA methylation via STAT3 signaling; inducing invasion and proliferation via KLF5 | 203 , 204 | |
| CCL5/CCR5 | Melanoma | Activating apoptotic pathway involving release of cytochrome c and activation of caspase‐9 and caspase‐3 | 205 | |
| CCL18 | Breast cancer | Inducing EMT via PI3K/Akt/GSK3β/Snail signaling through AnxA2; recruiting Treg cells and promoting metastasis and via PITPNM3; enhancing EMT via N‐Ras/ERK/PI3K/NF‐κB/Lin28b signaling | 206 , 207 , 208 | |
| CCL18 | Oral squamous cell carcinoma | Promoting growth and metastasis by activating the JAK2/STAT3 signaling; promoting migration via mTOR signaling through Slug | 209 , 210 | |
| CCL18 | Ovarian cancer | Promoting migration through Pyk2 signaling or mTORC2 signaling | 211 , 212 | |
| CCL18 | Lung cancer | Promoting migration and invasion via Nir1 through Nir1‐ELMO1/DOC180 signaling | 213 | |
| CCL18 | Bladder cancer | Promoting lymphangiogenesis by increasing the production of VEGF‐C and MMP‐2 | 214 | |
| CCL18 | Pancreatic cancer | Promoting progression and the Warburg effect via NF‐κB/VCAM‐1 pathway | 215 | |
| CCL18 | Gastric cancer | Promoting invasion and migration via ERK1/2/NF‐κB signaling | 216 | |
| CCL20/CCR6 | Lung cancer | Promoting migration via ERK1/2‐MAPK and PI3K pathways; promoting progression mediated by lncRNA‑u50535 through CCL20/ERK signaling | 217 , 218 | |
| CCL20/CCR6 | Renal cell carcinoma | Inducing EMT through Akt activation | 219 | |
| CCL20/CCR6 | Colorectal cancer | Promoting chemoresistance via FOXO1/CEBPB/NF‐κB signaling; promoting growth and metastasis mediated by lncRNA‑u50535; promoting proliferation and migration through ERK‐1/2, SAPK/JNK, and Akt signaling | 220 , 221 , 222 | |
| CCL20/CCR6 | Pancreatic cancer | Increasing TRAIL resistance via RelA‐CCL20 pathway; promoting migration, EMT, and invasion through PI3K/AKT‐ERK1/2 signaling | 223 , 224 | |
| CCL20/CCR6 | Breast cancer | Increasing chemoresistance via ABCB1/ NF‐κB signaling; promoting bone metastasis mediated by HuR; inducing EMT via PKC‐α/Src/Akt/NF‐kB/Snail signaling; promoting invasion by PKC‐α through EGFR and ERK1/2/MAPK pathway | 225 , 226 , 227 , 228 | |
| CCL20/CCR6 | Gastric cancer | Inducing EMT mediated by CRKL via Akt pathway | 229 | |
| CCL22/CCR4 | Prostate cancer | Promoting migration and invasion via phosphorylation of Akt | 230 | |
| CCL22/CCR4 | Colorectal cancer | Enhancing chemoresistance via PI3K/AKT pathway and caspase‐mediated apoptosis | 231 | |
| CCL22/CCR4 | Liver cancer | Recruiting regulatory T cells through p65/miR‐23a/CCL22 axis | 232 | |
| CXCL1/CXCR2 | Lung cancer | Recruiting Treg cells via miR141‐CXCL1‐CXCR2 signaling | 92 | |
| CXCL1/CXCR2 | Breast cancer | Promoting metastasis via activating NF‐κB/SOX4 signaling; promoting cancer cell survival mediated by TNF‐αvia NF‐κB signaling; promoting metastasis and chemoresistance through myeloid cell‐derived S100A8/9; stimulating migration and invasion via activation of the ERK/MMP2/9 signaling | 105 , 233 , 234 , 235 | |
| CXCL1/CXCR2 | Pancreatic cancer | Promoting migration and invasion by CXCL1‐mediated Akt phosphorylation | 236 | |
| CXCL1/CXCR2 | Ovarian cancer | Recruiting MDSC mediated by Snail; Enhancing metastatic potential via the TAK1/NF‐κB signaling; driving cancer progression by NF‐κB activation via EGFR‐transactivated Akt signaling | 237 , 238 , 239 | |
| CXCL1/CXCR2 | Prostate cancer | Promoting migration via the Src activation; increasing migration and invasion by fibulin‐1 downregulation through NF‐κB/HDAC1 | 240 , 241 | |
| CXCL1/CXCR2 | Colorectal cancer | Forming a premetastatic niche stimulated by VEGFA | 93 | |
| CXCL1/CXCR2 | Gastric cancer | Promoting migration and metastasis through activation of CXCR2/STAT3 signaling; promoting lymph node metastasis through integrin β1/FAK/AKT signaling; promoting tumor growth through VEGF pathway activation | 242 , 243 , 244 | |
| CXCL12/CXCR4 | Colorectal cancer | Promoting proliferation, invasion, angiogenesis via MAPK/PI3K/AP‐1 signaling; enhancing metastatic potential via PI3K/Akt/mTOR pathway; promoting progression by lncRNA XIST/ miR‐133a‐3p/ RhoA signaling; promoting growth and metastasis through activation of the Wnt/β‐catenin pathway; promoting chemoresistance via surviving | 245 , 246 , 247 , 248 , 249 | |
| CXCL12/CXCR4 | Breast cancer | Promoting metastasis by Pit‐1‐CXCL12‐CXCR4 axis; driving the metastatic phenotype through activation of MEK/PI3K pathway | 250 , 251 | |
| CXCL12/CXCR4 | Ovarian cancer | Recruiting MDSCs mediated by PGE2; promoting invasion through suppressing ARHGAP10 expression; promoting growth and migration through Notch pathway | 94 , 252 , 253 | |
| CXCL12/CXCR4 | Lung cancer | Suppressing cisplatin‐induced apoptosis through JAK2/STAT3 signaling | 254 | |
| CXCL12/CXCR4 | Prostate cancer | Inducing myofibroblast phenoconversion through EGFR/MEK/ERK signaling; promoting migration and invasion through SLUG | 255 , 256 | |
| CXCL12/CXCR4 | Gastric cancer | Inducing migration via SRC‐mediated CXCR4‐EGFR; increasing invasiveness via integrin β1 clustering; promoting migration by F‐actin reorganization and RhoA activation through mTOR signaling | 257 , 258 , 259 | |
| CXCL12/CXCR4 | Thyroid papillary cancer | Enhancing proliferation and invasion through Akt and snail signaling | 260 | |
| CXCL8/ CXCR1/2 | Gastric cancer | Inhibiting CD8+ T cells function by inducing the expression of PD‐L1 | 261 | |
| CXCL8/ CXCR2 | Lung cancer | Stimulating cell proliferation via transactivation of the EGFR involving the MAPK pathways | 262 | |
| CXCL8/ CXCR1/2 | Breast cancer | Increases the activity of cancer stem‐like cells by transactivation of HER2 | 263 | |
| CXCL8/ CXCR1/2 | Melanoma | Increasing tumor growth and metastasis by activating MMP‐2 | 264 | |
| CXCL8/ CXCR1/2 | Prostate cancer | Promoted tumorigenesis via STAT3/MALAT1 pathway | 265 | |
| CXCL8/ CXCR1/2 | Colorectal cancer | Enhancing resistance of cancer cells to anoikis through AKT/TOPK/ERK signaling; enhancing migration by increasing αvβ6 integrin expression; promoting proliferation and migration through heparin binding EGF | 266 , 267 , 268 | |
| CX3CL1/CX3CR1 | Breast cancer | Promoting the migration and invasion via Src/FAK signaling; triggering proliferation through transactivation of EGF pathway | 269 , 270 | |
| CX3CL1/CX3CR1 | Lung cancer | Promoting the migration and invasion via Src/FAK signaling; promoting lymph node metastasis via JNK and MMP2/MMP9 pathway | 271 , 272 | |
| CX3CL1/CX3CR1 | Ovarian cancer | Promoting EMT mediated by HIF‐1α | 273 | |
| CX3CL1/CX3CR1 | Prostate cancer | Promoting EMT via TACE/TGF‐α/EGFR pathway and upregulation of Slug; promoting metastasis via EGFR through activation of the Src/FAK pathway | 274 , 275 | |
| CX3CL1/CX3CR1 | Pancreatic cancer | Enhancing growth and migration through JAK/STAT signaling; promoting motility, invasion, and growth via AKT activation; promoting tumor cell survival and TRAIL resistance via RelA/NF‐κB signaling; stimulating HIF‐1α expression through the PI3K/Akt and MAPK pathways | 276 , 277 , 278 , 279 | |
| Tyrosine kinase receptor signaling | CSF‐1/CSF‐1R | Gliomas | Promoting tumor cell survival through IGF‐1R/ PI3K signaling | 280 |
| CSF‐1/CSF‐1R | Breast cancer | Driving tumor progression through Kindlin‐2/TGFβ/CSF‐1 signaling | 281 | |
| CSF‐1/CSF‐1R | Ovarian cancer | Promoting metastasis through the induction of MMP‐9 activity | 282 | |
| CSF‐1/CSF‐1R | Pancreatic cancer | Remodeling tumor immune microenvironment via PI3K‐γ and CSF‐1/CSF‐1R pathways | 283 | |
| Ron | Prostate cancer | Promoting prostate cancer cell growth via MST1R | 284 | |
| Ron | Pancreatic cancer | Promoting macrophage polarization through MST1‐MST1R signaling | 285 | |
| Metabolic signaling | Glycolysis | Pancreatic cancer | Conferring a prometastatic phenotype by HK2; | 286 |
| Glycolysis | Breast cancer | Promoting M2‐TAMs polarization through activation of GPR132 by lactate; promoting tumor angiogenesis by hypoxia‐induced REDD1/ mTOR signaling | 287 , 288 | |
| Glycolysis | Melanoma | Promoting M2‐TAMs accumulation by HMGB1; inducing TME acidification involving GPCR | 141 , 289 | |
| Glycolysis | Thyroid carcinoma | Inducing reprogramming of TAMs and inflammation through AKT1/mTOR pathway | 140 | |
| Glycolysis | Lung cancer | Increasing glucose uptake and glycolysis flux by activation of AMPK; promoting M2‐TAMs polarization mediated by HIF1α | 290 , 291 | |
| Fatty acid metabolism | Bladder carcinoma | Promoting cancer growth and metastasis through COX‐2/PGE2 pathway | 292 | |
| Fatty acid metabolism | Liver cancer | Promoting cell migration by enhancing IL‐1β secretion; enhancing the accumulation and polarization of M2‐like TAMs by RIPK3 | 293 , 294 | |
| Fatty acid metabolism | Breast cancer | Promoting cancer progression by FABP by favoring IL6 / STAT3 signaling; promoting TAMs differentiation through caspase‐1/PPARγ/MCAD pathway | 295 , 296 | |
| Fatty acid metabolism | Ovarian cancer | Promoting the M2‐like polarization through the PPARγ/NF‐κB pathway | 297 | |
| Glutamine synthesis | Lung cancer | Promoting M2‐like polarization through GS | 298 | |
| Tryptophan metabolism | Colon, gastrointestinal cancer | Inhibiting T‐cell functions through IDO upregulation | 299 , 300 | |
| Arginine metabolism | Melanoma | Promoting tumor cell proliferation through ARG1 | 301 | |
| Arginine metabolism | Breast cancer | Promoting cell growth by ARG induction and suppressing NO‐mediated tumor cytotoxicity | 302 | |
| Iron metabolism | Breast cancer | Releasing iron mediated by FPN in the TME; supporting proliferation by LCN | 303 , 304 | |
| RAGE | Breast cancer | Enhancing tumor growth and metastasis through S100A7 signaling | 305 | |
| RAGE | Glioma | Promoting angiogenesis through MMP9 signaling | 306 | |
| RAGE | Melanoma | Promoting invasiveness through S100A4 signaling | 307 | |
| Exosomal signaling | miR‐21‐5‐p, miR‐155‐5p | Colorectal cancer | Promoting migration and invasion by downregulating expression of BRG1 | 164 |
| miR‐25‐3p, miR‐130b‐3p, miR‐425‐5p | Colorectal cancer | Inducing M2‐like macrophage polarization by regulating PTEN/PI3K/Akt signaling | 308 | |
| Wnt | Colorectal cancer | Promoting chemoresistance through Wnt signaling | 309 | |
| miR‐125b‐5p | Melanoma | Educating TAMs by targeting LIPA | 310 | |
| miR‐21 | Gastric cancer | Suppressing apoptosis through PI3K/AKT signaling by down‐regulation of PTEN | 57 | |
| miR‐301a | Pancreatic cancer | Inducing M2‐like macrophage polarization via PTEN/PI3Kγ signaling | 311 | |
| miRNA‐501‐3p | Pancreatic cancer | Promoting progression through the TGFBR3‐mediated TGF‐β signaling | 312 | |
| miR‐223 | Ovarian cancer | Promoting drug resistance via the PTEN‐PI3K/AKT pathway | 313 | |
| miR‐95 | Prostate cancer | Promote cell proliferation, invasion, and EMT via miR‐95/JunB axis | 314 |
4.1. Chemokine–chemokine receptor signaling
Chemokines, which are small and soluble (8–14 kDa) signaling proteins, are a family of chemotactic cytokines responsible for cellular trafficking. More than 50 kinds of chemokines and 20 kinds of chemokine receptors have been identified until now. 79 According to the positions of conserved cysteine residues, chemokines can be classified into four groups including CXC, CC, CX3C, and C. 80 Correspondingly, chemokine receptor nomenclature essentially follows that of chemokines. Eleven kinds of CC chemokine receptors (CCR1–CCR11) recognize the CC subfamily chemokines, while seven kinds of CXC receptors (CXCR1–CXCR7) recognize the CXC subfamily chemokines, respectively. 80 Similarly, the CX3C subfamily chemokine (CX3CL1) only binds to CX3CR1 receptor while the C subfamily chemokines (XCL1/2) only bind to XCR1 receptor, respectively. Chemokine receptors are typical G‐protein‐coupled receptors (GPCRs) with seven transmembrane domains. Chemokine receptors can relay their signals through heterotrimeric G proteins that result in the directional migration of cells along a concentration gradient of ligands. 79 The interactions between chemokine and chemokine receptors are summarized in Figure 2 and Table 1. In the TME, TAMs are considered as the main stromal cells that secrete chemokines. As indicated by chemokine arrays, highly expressed chemokines of TAMs have been reported to consist of CCL2, CCL3, CCL5, CCL18, CXCL1, and CXCL12. Currently, the ligand receptor for CCL2 has been identified as CCR2. CCR1 and CCR5 have been found to be the receptors for both CCL3 and CCL5. Meanwhile, CCR3 has also been identified as a receptor for CCL5. By immunoprecipitation methods, the cognate receptor for CCL18 has been identified as PITPNM3, which has been suggested to be a putative six‐transmembrane protein that is sufficient to exert GPCR‐related functions. Additionally, the receptors for CXCL1 and CXCL12 have been identified to be CXCR2 and CXCR4, respectively.
FIGURE 2.
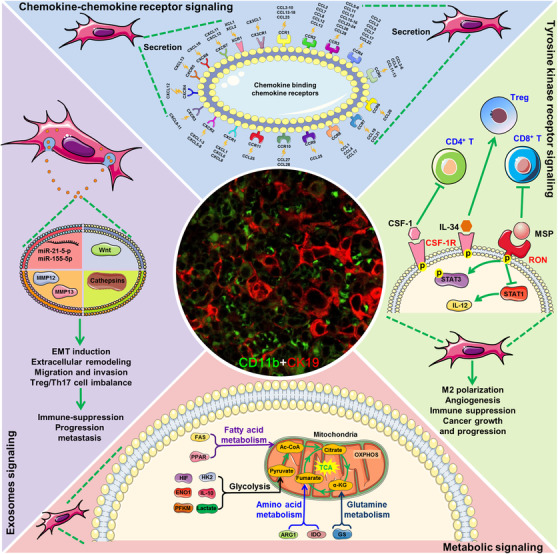
Pharmaceutical targets involved in TAMs regulation. Chemokine–chemokine receptor signaling was the most important target for TAMs‐targeting therapy, particularly for CCL2‐CCR2, CCL5‐CCR5, CXCL1/CXCR2, and CXCL12/CXCR4 signaling. Tyrosine kinase receptors including CSF‐1R and RON were also highly expressed on the cell membrane of TAMs and perfect for drug intervention. Molecular targets involved in glycolysis, fatty acid oxidation, and amino acid metabolism in TAMs were also being focused on drug development. Notably, exosomes act as the ideal drug‐loading and delivery vehicle for modulating biofunctions of TAMs
Chemokines and chemokine receptors are important for attracting infiltrating leukocytes to the TME. CCL2 and CCL5 are the primary chemokines responsible for monocytic precursors recruitment in tumors. 81 , 82 , 83 Their expression levels are closely associated with TAMs infiltration, lymph‐node metastasis, as well as unfavorable prognosis. CCL2 production from macrophages is critical for recruiting myeloid‐derived suppressor cells (MDSCs) to build a local immunosuppressive microenvironment in gliomas. 84 Moreover, in mixed‐bone marrow chimeric assays, CCR2‐deficient MDSCs failed to accumulate in gliomas. 84 In addition, the CCL2‐CCR2 axis was also found to be critical in mobilizing dendritic cell‐like antigen‐presenting cells into fibrosarcomas, as well as tumor‐infiltrating lymphocytes. 85 Multiple studies have demonstrated that CCL5 can inhibit the antitumor immune response and is correlated with poor outcomes in multiple malignancies. 86 , 87 , 88 , 89 CCL5/CCR3 signaling has been found to promote Th2 immune polarization and results in luminal breast cancer. 90 The CCL5/CCR5 chemokine axis is also capable of enhancing TGF‐β‐mediated killing of cytotoxic CD8+ T cells in colon cancer through regulatory T cells. 91 With regard to CXC chemokines, CXCL1 chemokines have been demonstrated to be effective in recruiting CXCR2+ Treg cells and lead to metastasis in nonsmall cell lung cancer. 92 CXCL1 is also critical in facilitating PMN formation in colorectal cancer by recruiting CXCR2+ MDSCs. 93 Meanwhile, CXCL12/CXCR4 signaling has been reported to induce the recruitment of MDSCs in the TME of human ovarian cancer. 94 Besides shaping the tumor immune milieu, chemokine axis is also critical in regulating the neoangiogenic process. CXC chemokines have been reported to be directly chemotactic for endothelial cells and to stimulate angiogenesis in vivo. 95 CXCR4 receptor is expressed on endothelial cells and its activation after CXCL12 binding can induce the proliferation and migration of endothelial cells. Moreover, CXCL12 recruits CXCR4+ circulating or bone‐marrow endothelial precursors and therefore promotes tumor angiogenesis. 96 Similarly, CXCL1 was also reported to promote neoangiogenesis in breast, liver, and colorectal cancers. 97 , 98 , 99 Finally, chemokine signaling can directly act on chemokine receptors expressed on the plasma membrane of cancer cells, thus controlling their malignancy‐related functions. For example, a variety of chemokine receptors—including CXCR2, CXCR4, CCR1, CCR2, and CCR5—have been found in breast tumor cells. 100 , 101 , 102 , 103 , 104 It is found that CXCL1 can induce chemoresistance and metastasis of breast cancer via CXCR2 activation. 105 Furthermore, CCL2 induces hepatocellular carcinoma invasion and EMT via CCR2 activation. 106 CXCL12 is also capable of regulating glioblastoma‐stem‐like cells by modulating CXCR4. 107 Moreover, CXCL12‐CXCR4 signaling has also been highlighted in guiding the homing of cancer cells to their specific metastatic organs. 108 Altogether, these diverse chemokine functions establish crosstalk between cancer cells and the TME, which represents a promising target for the successful development of novel therapeutic strategies.
It should be pointed out that multiple chemokines play multifaceted roles within the complicated TME, usually dividing into two directions: paracrine signaling for immune activation and autocrine signaling for proliferation and metastasis of cancer cells. 109 , 110 , 111 For example, the CXCL9‐10 axis could induce the migration, differentiation, and activation of the CXCR3+ activated T or NK cells, leading to increased antitumor immune response. 112 Additionally, CCL5/CCR5 axis is also a double‐edged sword in cancer. On one hand, CCL5 could promote CSCs self‐renewal and recruit the immunosuppressive cells (such as CCR5+ TAMs and Tregs) to form a tumor‐protecting TME. On the other hand, CCL5 could also promote antitumor immunity by recruiting CCR5+ activated T cells and DCs to the TME and therefore enhances the immunotherapy response in different tumor types. 110 , 111
TAMs‐related chemokine signals have been widely investigated in skin area because of their reliable prognosis and therapeutic values for skin tumors. For example, Wu et al. reported that TAMs‐related chemokine signals played key roles in the development and progression of cutaneous T‐cell lymphoma (CTCL). Compared with normal controls, mycosis fungoides (the most common variant of CTCL) skin displayed abundant TAMs infiltration as well as increased expression of a subset of macrophage‐related chemokines and associated receptors. 113 Selective depletion of M2‐phenotype TAMs using clodronate‐containing liposomes remarkably delayed mycosis fungoides development in vivo. Interferons (IFNs) have been widely used for mycosis fungoides and malignant melanoma treatment in the clinic. Growing evidence has suggested that modulating TAMs‐related chemokine signals may be the possible mechanism of the therapeutic effects of IFNs. For example, Furudate et al. reported that IFN‐α2a and IFN‐γ stimulation could decrease the release of CCL17/18, whereas elevating the production of CXCL10/11 in monocyte‐derived TAMs in vitro. More importantly, the subcutaneous administration of IFN‐α2a could increase the CXCL11‐secreting cell population in the lesional skin of advanced mycosis fungoides patients. 114 Kakizaki et al. reported that peritumoral injection of IFN‐β remarkably suppressed the recruitment of PD‐L1+ TAMs in B16F10 melanoma xenograft in vivo. 115 Meanwhile, IFN‐β administration dramatically attenuated the production of CCL17/22, whereas elevated the secretion of CXCL9/11 from TAMs. More importantly, these immunomodulatory effects of IFN‐β on TAMs were also observed in the lesional skin of patients with in‐transit melanoma. It should be noted that the clinical application of IFNs for the treatment of cancer is still controversial. For example, Numerous clinical trials have examined the antitumor potential of recombinant IFN‐γ as an adjuvant to surgery or conventional chemotherapy. 116 However, these clinical trials often reported conflicting outcomes in patients with the same tumor type. 116 Extensive efforts are still needed to better understand the complex roles of IFNs in cancer treatment.
4.2. RTK signaling
RTKs play a vital role in signal transduction that supports cellular communication and survival. Dysregulation of RTKs, such as receptor mutation or overexpression, contributes to the initiation, tumorigenesis, and progression of multiple malignancies. To date, it has been reported that 58 kinds of RTKs are edited by the human genome. 117 , 118 Notably, colony stimulation factor‐1 receptor (CSF‐1R), one of the most important members of the RTKs family, is highly expressed by TAMs. 119 Upon binding to its ligands including CSF‐1 or IL‐34, CSF‐1R undergoes dimerization and therefore causes a signaling cascade that facilitates the proliferation, functioning, and survival of macrophages. High expression of CSF‐1 has been found in breast, prostate, pancreatic, gastric, and many other types of cancers, and its level has been closely associated with TAMs density and poor prognosis. 120 , 121 , 122 , 123 It is found that CSF‐1‐positive cancer cells colocalize with abundant CSF‐1R‐positive TAMs in invasive breast cancer, and the CSF‐1/CSF‐1R axis is significantly correlated with a higher grade, basal‐like phenotype, and lymph node metastasis. 124 In CSF‐1‐knockout MMTV‐PyMT+/‐ spontaneous breast cancer mice, the perivascular macrophage density was declined with a sixfold reduction, accompanied by a 16‐fold decrease of circulating tumor cells in the blood. 125 Moreover, CSF‐1/CSF‐1R signaling of macrophages could suppress CD4+ T‐cell responses and induce the generation of Treg cells, which facilitated the immune‐suppressive TME. 126 Additionally, CSF‐1 is also involved in the pro‐angiogenic process since it promotes M2‐like polarization of macrophages and secretes a variety of angiogenic factors. 127 Therefore, blocking CSF‐1/CSF‐1R activation is becoming an important therapeutic strategy to inhibit cancer growth and metastasis.
RON is another RTK expressed on macrophages surface that specifically recognizes macrophage‐stimulating protein (MSP). Overexpression of RON has been implicated in the progression and metastasis of diverse malignancies. In breast cancer, MSP/RON overactivation is sufficient to promote cancer growth as well as metastasis to the lungs, liver, brain, and bone. Conversely, RON inactivation is capable of inhibiting the growth, angiogenesis, and metastasis of breast cancer xenografts. 128 The cancer‐promoting effects of MSP/RON signaling are mainly attributed to the M2‐like phenotype polarization of macrophages. RON activation can cause the phosphorylation of STAT3, which is essential for the tumor‐promoting efficacies and immunosuppressive functions of TAMs. 129 By contrast, the MSP/RON pathway can downregulate STAT1 activity, which actively participates in antitumor immune responses via up‐regulating IL‐12. 130 Eyob et al. has demonstrated that RON signaling in macrophages is capable of impairing antitumor functions of CD8+ T cells. 131 Altogether, both STAT3 activation and STAT1 inactivation induced by MSP/RON signaling may contribute to tumor immune tolerance and lead to cancer progression. Considering the important role of RON receptor signaling in TAMs modulation, pharmacological inhibition of RON kinases has received more attention from research groups around the world.
Mer tyrosine kinase (MerTK) is also a macrophage‐specific RTK and its overexpression usually correlates with poor prognosis in cancer. 132 MerTK signaling activation can promote tumor immune tolerance by initiating macrophages efferocytosis and promoting the clearance of immunogenic antigens. Meanwhile, MerTK signaling activation can suppress inflammatory cytokines production and polarize macrophages toward the anti‐inflammatory M2‐like phenotype. 133 In contrast, macrophages MerTK signaling blockage can induce the repolarization of macrophages toward the M1‐like phenotype and enhance antitumor immunogenic cell death, a crucial step in T‐cell priming and activation. 134 Thus, MerTK blockade may represent a promising antitumor strategy that could significantly increase tumor immunogenicity and potentiate antitumor immunity. It has been reported that MerTK knockdown or MerTK neutralizing antibody treatment could remodel the cellular immune profile, bringing a more inflamed TME with increased T cell infiltration and T cell‐mediated cytotoxicity. 132 Additionally, anti‐MerTK antibody treatment could stimulate T cell activation and synergize with anti‐PD‐1 or anti‐PD‐L1 therapies in tumor‐bearing mice. 135 Altogether, macrophages MerTK blockade could promote tumor immunogenicity and potentiate antitumor immunity, which represents a promising strategy for tumor immunotherapy.
4.3. Metabolic signaling
A typical feature of cancer cells is their abnormal metabolism caused by oxygen deprivation and nutrient limitation. 136 Hypoxic TME not only induces the metabolism pattern shifting of cancer cells but also reprograms the polarization balance and metabolic mode of macrophages, which facilitate the formation of immune‐suppressive TME. In hypoxic regions of tumors, a more dominant M2‐like phenotype is observed, which might indicate the potential effects of hypoxia on macrophage polarization. 137 Hypoxia‐inducible factors (HIFs) are major transcriptional factors that are responsive to hypoxia and control aerobic glycolysis. 138 Enhanced glycolytic activity allows macrophages to synthesize sufficient ATP and various biosynthetic intermediates to maintain their tumor‐prone functions. A recent study demonstrated the enhanced glycolytic activity of TAMs when compared with that of bone marrow–derived macrophages (BMDMs). Glycolytic proteins, such as hexokinase‐2, phosphofructokinase, and enolase 1, were all increased in TAMs. 139 Similar findings were also observed in a transwell co‐culture model of macrophages with cancer cells or tumor‐extract solutions. 140 In addition to intrinsic HIF activation, cytokines and lactate released from cancer cells under hypoxic conditions also contribute to the glycolytic phenotype of TAMs. In hypoxic melanoma cells, HIF‐1α accumulation induces HMGB1 translocation that causes IL‐10 production, which consequently promotes M2‐like macrophage activation. 141 Cancer‐cell–derived lactate has also been reported to reprogram macrophages to a tumor‐prone phenotype by inhibiting the NF‐κB pathway that in turn may impair M1‐like activation and suppress the immune surveillance functions of infiltrating T cells and NK cells. 142 , 143 , 144 Interestingly, the OXPHOS chain has also been found to be elevated in a coculture model of thyroid‐carcinoma cells and macrophages, characterized by enhanced oxygen consumption rate. 140 The concentrations of AcCoA and succinate are significantly increased in the mitochondria of M2‐like macrophages, suggesting that TAMs have an intact OXPHOS despite an impaired TCA cycle. 145 The enhanced OXPHOS chain may not only yield ROS‐induced DNA damage but may also provide citrates for fatty acid synthesis. However, more studies are still required to reveal the precise switching mechanisms between glycolysis and OXPHOS in TAMs depending on cancer types and stages.
Fatty acid metabolism is important in cancer development. It has been suggested that increased fatty acid metabolism provides cancer cells with sufficient membranes and energy to support cancer growth. 146 It is found that fatty acid oxidation is necessary for IL‐4‐induced macrophage activation, as CPT1 suppression could attenuate the expression of the M2‐like marker proteins, CD301, and CD206. 147 Meanwhile, fatty acid oxidation can activate the downstream COX2/PGE2 pathway to promote cancer growth and metastasis. TAMs express high levels of fatty acid synthase and increased PPAR signaling, which promote fatty acid oxidation and tumor growth. 148 PPARs are composed of three isoforms including PPAR‐α, PPAR‐β/δ, and PPAR‐γ. PPAR‐α manages lipid oxidation and clearance. PPAR‐γ facilitates lipogenesis while PPAR‐β/δ controls lipid uptake and storage. 149 PPAR‐β/δ plays a crucial role in mediating M2‐like polarization. 150 Ovarian cancer patient‐derived TAMs exhibited increased expression levels of multiple PPAR‐β/δ target genes when compared with that of monocyte‐derived macrophages. 151 Lipidomic analysis revealed that the deregulation of PPAR‐β/δ target genes did not result from PPAR‐β/δ overexpression or its promotion effect on target genes, but was attributed to high levels of polyunsaturated fatty acids, especially arachidonic acid and linoleic acid in tumor ascites. 151 The internalization of polyunsaturated fatty acids and the subsequent lipid droplet formation may provide a reservoir of PPAR‐β/δ ligands to TAMs, leading to a consistent activation of PPAR‐β/δ target genes, such as pyruvate dehydrogenase kinase 4 (PDK4). Interestingly, PDK4 can induce glucose catabolism shifting toward aerobic glycolysis, further rendering TAMs to adapt to hypoxic TME. 151 PPAR‐γ is also known to regulate metabolic pathways and M2‐like polarization. Macrophage deficiencies in PPAR‐γ and δ and their coactivator, PGC1β, are ineffective for IL‐4‐induced fatty acid oxidation and M2‐like polarization. 152 Hence, targeting of PPAR signaling is a promising approach to control macrophage activation and cancer progression.
In addition to enhanced glycolysis and fatty acid oxidation, M2‐like macrophages have also been found to increase glutamine synthesis through glutamate, which has not been demonstrated in M1‐like macrophages. 153 Inhibition of glutamine syntheses results in a 50% reduction of M2‐like polarization, which might be ascribed to the role of glutamine in protein glycosylation. 154 Meanwhile, glutamine is extensively fluxed into the TCA cycle to provide enough carbons to satisfy energetic needs. 155 Besides glutamate, another amino acid, arginine, is also required in the ARG1 pathway of M2‐like macrophages. 156 Interestingly, ARG1 is also the downstream gene of HIF‐1α. ARG1 overexpression not only enables cancer cells to rapidly adapt to the hypoxic microenvironment but also results in the production of ornithine and urea. 156 Ornithine can be further utilized in the synthesis of proline and polyamine, which are required for cellular proliferation, tissue remodeling, and collagen biogenesis. 157 Moreover, the arginine clearance by ARG1 suppresses T‐cell immune responses, favoring the establishment of the immunosuppression microenvironment. 157 Besides, tryptophan metabolism has been shown to be deregulated in cancers. It was found that the enzyme accounting for tryptophan catabolism, indoleamine‐2, 3‐dioxygenase (IDO), was frequently overexpressed in M2‐like macrophages. 158 IDO upregulation was reported to inhibit T‐cell functions, whereas tryptophan treatment or administration of IDO inhibitors could restore T‐cell proliferation. 158
RAGE (receptor for advanced glycation end‐product) is a member of the immunoglobulin superfamily and has been reported to be highly expressed on the surface of macrophages. 159 It has been reported that RAGE overexpression could induce the accumulation of TAMs and thus accelerate the growth of lung cancer xenografts in vivo. 160 Mechanistically, RAGE expression strictly correlates to the metabolic switch in cancer and immune cells. For example, cancer cells usually exhibit a metabolism switch characterized by decreased mitochondrial oxidative phosphorylation as well as elevated glycolysis, leading to the overproduction of toxic glucose metabolites. 161 The toxic glucose metabolites such as lactate, sorbitol, diacylglycerol (DAG), and methylglyoxal (MG) could further contribute to the development of advanced glycation end products (AGEs). On one hand, the formation of AGEs could impair the phagocytosis function of M1‐like macrophages within the TME. 162 On the other hand, AGEs could induce RAGE expression in macrophages and their interaction could further promote cancer progression and metastasis by recruiting TAMs. 160 , 163 Therefore, AGEs/RAGE signaling may also represent a metabolism‐related target for the pharmacological modulation of TAMs, which still needs further investigations. Collectively, the above findings suggest that the metabolic pathways and targets can also be applied to modulate the biological functions of TAMs, which may provide valuable opportunities for treating multiple malignancies (Figure 2 and Table 1).
4.4. Exosomal signaling
In recent years, exosomal signaling has evoked increased interest in cancer research. Exosomes are defined as small extracellular vesicles that are 30–100 nm in diameter. 164 Similar to cells, exosomes are composed of a lipid bilayer containing all known molecular components including DNA, RNA, and proteins. 165 Exosomes can be released by all cell types and widely exist in human bodily fluids such as blood, urine, amniotic fluid, bronchoalveolar lavage fluid, and malignant fluid. In particular, normal human blood is expected to contain approximately 2,000 trillion exosomes, while this value is elevated to 4,000 trillion in the blood of cancer patients. 166 , 167 , 168 Therefore, exosomal signaling is highly implicated in intercellular communication and carcinogenesis. Recent studies have also demonstrated that exosomal signaling plays a vital role in tumor angiogenesis, 169 metastasis, 170 drug resistance, 171 and immune regulation, 172 particularly for exosomes derived from macrophages. It has been reported that TAMs‐derived exosomes exhibit high expression levels of miR‐21‐5‐p and miR‐155‐5p, which significantly promote cellular migration and invasion of colorectal cancer cells. 164 Besides, exosomes from macrophages also contain a high level of Wnt, which is considered to provide critical signaling in EMT induction and mesenchymal‐phenotype maintenance. 173 , 174 In addition to metastasis, TAMs‐derived exosomes also contribute to extracellular‐matrix remodeling. It has been shown that MMP‐12 and MMP‐13, as well as cathepsin B, D, K, L, S, and Z, are all upregulated in TAMs‐derived exosomes, suggesting their positive roles in extracellular remodeling. 175 Notably, exosomes released from TAMs also contain miRNAs that lead to Treg/Th17 cell imbalance in ovarian cancer, resulting in the generation of an immune‐suppressive TME and thus promoting cancer progression and metastasis. 176 Altogether, targeting TAMs‐derived exosomal signaling represents a novel and promising approach for cancer treatment (Table 1).
5. PHARMACOLOGICAL MODULATION OF TAMS
5.1. Agents targeting chemokine–chemokine receptors
Chemokine–chemokine receptor signaling constitutes an important network that regulates cancer growth, angiogenesis, immune suppression, and metastasis. Targeting of chemokine–chemokine receptor networks has been evaluated in multiple preclinical models and clinical trials, particularly in terms of the CCL2‐CCR2, CCL5‐CCR5, and CXCL12‐CXCR4 axes (Table 2). Blockade of CCL2 with a neutralizing antibody inhibited inflammatory monocyte recruitment, reduced lung metastasis, and prolonged OS in breast cancer–bearing mice. 315 In a metastatic prostate cancer model, combined treatment with an anti‐CCL2 antibody and docetaxel caused reduced tumor burden and bone resorption, as well as an improved survival period. 316 The development of CCR2 inhibitors has also yielded encouraging results in cancer treatments. Oral administration of the CCR2 inhibitor, PF‐04136309, in a preclinical pancreatic cancer model reduced the number of tumor‐infiltrating macrophages and inflammatory monocytes. 317 Significantly, PF‐04136309 acted synergistically with the chemotherapeutic drug, gemcitabine, to inhibit metastasis, increase anti‐tumor T‐cell responses, and ultimately lead to an enhanced chemotherapeutic efficacy. 318 Similarly, the synergistic effects between PF‐04136309 and paclitaxel or the standard chemotherapy, FOLFIRINOX, have also been reported in pancreatic cancer patients. 319 Another CCR2 inhibitor, CCX872, was demonstrated to improve anti‐PD‐1 treatment in preclinical settings and positive results were also obtained when used in combination with the FOLFIRINOX strategy in pancreatic cancer patients. 320 In murine models of hepatocellular carcinoma, combined treatment with the CCR2 antagonists, RDC018 or 747, with sorafenib was validated to inhibit tumor growth and metastasis, accompanied by a significant reduction of macrophage infiltration. 321 , 322 It should be pointed out that there are also some disappointing results in the clinical trials of CCL2/CCR2 blockers. For example, carlumab, a human IgG1κ anti‐CCL2 mAb, did not show antitumor activity as a single agent in metastatic castration‐resistant prostate cancer patients in a phase II study. 323 Overall, the above results suggest that the CCL2‐CCR2 axis may be a promising target for cancer treatment, especially when used in combination with traditional cancer‐killing strategies or immunotherapies. However, the successful pharmaceutical development of CCL2/CCR2 blockers still has a long way to go.
TABLE 2.
Chemokine–chemokine receptor targeting agents in clinical trials
| Drug/description | Sponsors | Phase/status | Tumor type | Treatment | ClinicalTrials.govIdentifier | First received |
|---|---|---|---|---|---|---|
| CCL2‐CCR2 inhibitor | ||||||
| Carlumab/CNTO 888 (anti‐CCL2 antibody) | Centocor Research & Development | 2 a | Prostate cancer | Monotherapy | NCT00992186 | 2009‐10‐09 |
| MLN1202/S0916 (anti‐CCR2 antibody) | Southwest Oncology Group | 2 a | Metastatic cancer | Monotherapy | NCT01015560 | 2009‐11‐08 |
| Navarixin/MK‐7123/ SCH 527123(CXCR1 and CCR2 antagonist) | Merck Sharp & Dohme | 2 b | Advanced/Metastatic solid tumors | With pembrolizumab | NCT03473925 | 2018‐03‐22 |
| PF‐04136309/PF6309 (CCR2 antagonist) | Washington University School of Medicine and National Cancer Institute | 1b a | Locally advanced or borderline resectable pancreatic cancer | With FOLFIRINOX chemotherapy | NCT01413022 319 | 2011‐08‐09 |
| CCX872‐B (CCR2 antagonist) | ChemoCentryx | 1b b | Pancreatic cancer | With FOLFIRINOX chemotherapy | NCT02345408 | 2015‐01‐26 |
| AZD5069 (CCR2 antagonist) | Institute of Cancer Research, UK | 1/2 b | Metastatic castration‐resistant prostate cancer | With enzalutamide | NCT03177187 | 2017‐06‐06 |
| Reparixin (CXCR1 and CCR2 antagonist) | Dompé Farmaceutici S.p.A | 1 a | Metastatic breast cancer | With paclitaxel | NCT02001974 354 | 2013‐12‐05 |
| SX‐682(CXCR1 and CCR2 antagonist) | Syntrix Biosystems | 1 b | Metastatic melanoma | With pembrolizumab | NCT03161431 | 2017‐05‐19 |
| CCL5‐CCR5 inhibitor | ||||||
| Maraviroc/Celsentri (CCR5 antagonist) | Abramson Cancer Center of the University of Pennsylvania | 2 a | Hematologic malignancy | Monotherapy | NCT01785810 | 2013‐02‐07 |
| University Hospital Heidelberg | 1 a | Metastatic colorectal cancer | With Pembrolizumab | NCT03274804 | 2017‐09‐07 | |
| National Center for Tumor Diseases, Heidelberg | 1 a | Colorectal cancer patients with liver metastases | Monotherapy | NCT01736813 | 2012‐11‐29 | |
| Leronlimab/PRO‐140 (anti‐CCR5 antibody) | CytoDyn | 1b/2 b | Triple negative breast neoplasms | With carboplatin | NCT03838367 | 2019‐02‐12 |
| CXCL12‐CXCR4 inhibitor | ||||||
| Mogamulizumab/ KW‐0761 (anti‐CCR4 antibody) | Kyowa Kirin | 3 b | Cutaneous T‐cell lymphoma | Monotherapy | NCT01728805 355 | 2012‐11‐20 |
| European Organisation for Research and Treatment of Cancer | 2 b | Stage IB‐IIB cutaneous T‐cell lymphoma | With radiation (total skin electron beam therapy) | NCT04128072 | 2019‐10‐16 | |
| Kyowa Kirin | 2 a | Adult T‐cell leukemia‐lymphoma | Monotherapy | NCT00920790 356 | 2009‐06‐15 | |
| Kyowa Kirin | 2 a | Peripheral T/NK‐cell lymphoma | Monotherapy | NCT01192984 | 2010‐09‐01 | |
| Kyowa Kirin | 1/2 a | Advanced solid tumors | With nivolumab | NCT02705105 | 2016‐03‐10 | |
| National Cancer Institute | 1/2 b | Diffuse large B cell lymphoma | With pembrolizumab | NCT03309878 | 2017‐10‐16 | |
| Kyowa Kirin | 1 a | Non‐small cell lung cancer | With docetaxel | NCT02358473 | 2015‐02‐09 | |
| Kyowa Kirin | 1 a | Adult T‐cell leukemia‐lymphoma and peripheral T‐cell lymphoma | Monotherapy | NCT00355472 357 | 2006‐07‐24 | |
| Ulocuplumab/BMS‐936564/MDX‐1338 (Anti‐CCR4 antibody) | Bristol‐Myers Squibb | 1 a | Acute myelogenous leukemia and selected B‐cell cancers | Monotherapy | NCT01120457 | 2010‐05‐11 |
| Bristol‐Myers Squibb | 1 a | Multiple myeloma | With Revlimid/ Velcade and dexamethasone | NCT01359657 | 2011‐05‐25 | |
| AMD3100/Plerixafor (CXCR4 antagonist) | Massachusetts General Hospital | 2 b | Head and neck cancer | With pembrolizumab | NCT04058145 | 2019‐08‐15 |
| Washington University School of Medicine | 2 a | Hematologic neoplasms | With leukopheresis | NCT00914849 358 | 2009‐06‐05 | |
| Washington University School of Medicine | 1/2 a | Acute myeloid leukemia | With mitoxantrone, etoposide, and cytarabine | NCT00512252 359 | 2007‐08‐07 | |
| LY2510924 (CXCR4 antagonist) | M.D. Anderson Cancer Center | 1b a | Relapsed or refractory acute myeloid leukemia | With idarubicin and cytarabine | NCT02652871 | 2016‐01‐12 |
| Eli Lilly and Company | 2 a | Extensive stage small cell lung carcinoma | With carboplatin and etoposide | NCT01439568 360 | 2011‐09‐23 | |
| Motixafortide/BKT140/BL‐8040 (CXCR4 antagonist) | Biokine Therapeutics | 1/2a a | Multiple myeloma | Monotherapy | NCT01010880 | 2009‐11‐10 |
| M.D. Anderson Cancer Center | 2b b | Metastatic and recurrent pancreatic adenocarcinoma | With pembrolizumab | NCT02907099 | 2016‐09‐20 | |
| PTX‐9908 (CXCR4 antagonist) | TCM Biotech International Corp. | 1/2 b | Hepatocellular carcinoma | Monotherapy | NCT03812874 | 2019‐01‐23 |
| USL‐311 (CXCR4 antagonist) | Proximagen | 1/2 b | Solid tumors and relapsed/recurrent GBM | Monotherapy or with lomustine | NCT02765165 | 2016‐05‐06 |
| Balixafortide/POL6326 (CXCR4 antagonist) | Polyphor | 3 b | HER2‐ locally recurrent or metastatic breast cancer | With Eribulin | NCT03786094 | 2018‐12‐24 |
| Polyphor | 2 a | Multiple myeloma | Monotherapy | NCT01105403 | 2010‐04‐16 | |
| Polyphor | 1 a | Metastatic breast cancer | With Eribulin | NCT01837095 361 | 2013‐04‐22 | |
| GMI‐1359 (CXCR4 antagonist) | GlycoMimetics | 1b b | HR+ metastatic breast cancer | Monotherapy | NCT04197999 | 2019‐12‐13 |
| TG‐0054 (CXCR4 antagonist) | TaiGen Biotechnology | 2 a | Multiple myeloma and non‐Hodgkin lymphoma and Hodgkin disease | With G‐CSF | NCT02104427 | 2014‐04‐04 |
Source: clinicaltrials.gov.
Completed stage.
Ongoing stage, including both recruiting and nonrecruiting stages.
With regard to CCL5‐CCR5 signaling, systemic treatment of mice with CCL5‐directed antibodies inhibit colon cancer growth, lung metastasis, and peritoneal dissemination. 324 CD45‐immunoreactive cells in tumor tissues and adjacent healthy tissues have been found to be upregulated following CCL5 blockade. Meanwhile, CCL5 neutralization renders colon tumors more sensitive to PDGFRβ‐targeted therapy. 324 CCL5‐targeted blockade has also been demonstrated to be effective in several malignancies including breast, 325 gastric, 326 and prostate cancers. 327 Concurrently, much attention has been paid to the discovery of CCR5 antagonists. Maraviroc, an FDA‐approved CCR5 antagonist, has been applied as an antiretroviral therapy strategy for HIV infection. 328 Recent studies have demonstrated its efficacy to suppress cancer cell invasiveness in a variety of cancers. Maraviroc administration resulted in cell‐cycle arrest and apoptosis in both colorectal and breast cancer cell lines in vitro. 325 , 329 In gastric cancer, treatment with maraviroc effectively reduced tumor burden, inhibited peritoneal dissemination, and prolonged the survival period. 326 It was also observed that maraviroc significantly inhibited pancreatic cancer liver metastasis, accompanied by marked cell‐cycle arrest at the G1/S checkpoint. 330 TAK779 is another synthetic CCR5 antagonist that was initially developed for HIV treatments. 331 Menu et al. demonstrated that TAK779 has direct inhibitory effects on melanoma growth in vitro and in vivo. 332 Another study suggested that TAK779 could efficiently block CCR‐5‐dependent T‐cell migration. 333 In a pancreatic cancer mouse model, TAK779 administration resulted in significant suppression of Treg recruitment and cancer growth. 334 Compared to synthetic CCR5 inhibitors, aibamine is the first natural CCR5 antagonist with an IC50 of 1 μM. 335 Alibamine was found to suppress the invasion and metastasis of prostate cancer cells in mice. 327 By analyzing the chemical structure differences between anibamine and other CCR5 inhibitors, the main difference has been shown to consist of the side chains of anibamine being simple, undecorated, and aliphatic chains. 336 Therefore, further drug development based on the structure of anibamine may bring a novel candidate drug for cancer treatment.
CXCL12‐CXCR4 is the most commonly overexpressed signaling pathway in a variety of cancers and a number of small‐molecule drugs and peptide inhibitors that target CXCR4 have been developed. AMD3100 is the first CXCR4 antagonist that is used for hematopoietic stem cell mobilization during transplantation in patients with non‐Hodgkin's lymphoma or multiple myeloma. 337 Furthermore, AMD3100 and its derivate, AMD3465, are also capable of mobilizing cancer cells in the bone marrow to ultimately enhance the efficacies of conventional therapies. 338 AMD3100 treatment in leukemic mice induced a ninefold increase in circulating leukemic cells. 339 The combined treatment of chemotherapy and AMD3100 led to a reduced tumor burden and improved OS compared with those in mice received chemotherapy alone. A similar phenomenon was also observed in a phase 1/2 study of refractory acute myeloid leukemia. 339 In addition to its chemosensitizing effects, AMD3100 synergistically interacts with antibodies targeting PD‐L1 and CTLA‐4 results in enhanced T‐cell infiltration into tumor tissues, yielding a greater anticancer response. 340 Another CXCR4 antagonist, LY2510924, is also able of suppressing cancer growth and metastasis in multiple preclinical models. 341 , 342 Notably, LY2510924 was demonstrated to be clinically safe and well‐tolerated in advanced solid cancers including colorectal, lung, breast, and prostate cancers. 343 Meanwhile, several CXCR4 small‐molecule antagonists—such as BKT140, PRX177561, and POL5551—are also capable of inhibiting cancer growth and metastasis. 344 , 345 , 346 Recent studies have also suggested that peptide inhibitors targeting the amino‐terminal region of CXCR4 are also effective in inhibiting cancer growth, such as T22, TN14003, and CTCE‐9908. 347 , 348 , 349 In preclinical models, TN14003 significantly inhibited pulmonary metastasis of melanomas, as well as breast cancers. 350 , 351 Similarly, CTCE‐9908 showed considerable inhibitory effects on cancer growth and metastasis in malignancies including osteosarcomas and melanomas, as well as breast and prostate cancers. 347 , 352 , 353 These results suggest that disrupting the CXCL12‐CXCR4 axis represents a viable strategy for further anticancer drug developments. However, most of the above‐mentioned studies were carried out in the early course of cancer progression, and preclinical models may not fully simulate the clinical pathological process. Therefore, further clinical trials investigating the efficacies and safeties of CXCR4 antagonists are urgently needed in the context of advanced cancers.
5.2. Agents targeting tyrosine kinases
Given the activated CSF‐1/CSF‐1R signaling in multiple malignancies and their significant implications in predicting poor prognoses, developing agents to block the CSF‐1/CSF‐1R axis has attracted increasing research attention. Currently, both CSF‐1/CSF‐1R antibodies and CSF‐1R kinase inhibitors have been successfully developed for therapeutic regulation of macrophages. The key advantage of antibodies over kinase inhibitors is their higher selectivity and safety. Currently, several CSF‐1/CSF‐1R targeted antibodies have been developed. H27K15 is a novel competitive monoclonal antibody for CSF‐1 blockage that has been generated for cancer immunotherapy. Enzyme‐linked immunosorbent assays (ELISAs) have shown that H27K15 produces no cross‐reaction with other tyrosine kinases and can inhibit CSF‐1R phosphorylation by blocking CSF‐1. 362 In preclinical models, H27K15 can decrease the secretion of both MCP‐1 and IL‐6, which play important roles in recruiting M2‐like macrophages. H27K15 is also capable of preventing monocyte differentiation into CD163+CD64+ M2‐like macrophages and induces their differentiation into CD14‐CD1a+ dendritic cells. 363 RG7155 is designed as a humanized monoclonal antibody against CSF‐1R, with an IC50 of 0.3 nM. 364 RG7155 can suppress CSF‐1R dimerization and therefore interrupt the formation of the CSF1/CSF1‐R complex. Consequently, RG7155 results in a significant reduction of CD68/CD163 macrophages. 363 It has been reported that objective responses were observed in 83% of patients with diffuse‐type tenosynovial giant‐cell tumors (dt‐GCTs) following RG7155 treatment, even during a short‐course treatment in patients who are unable to undergo surgery. 365 Additionally, RG7155 is also effective in increasing the CD8/CD4 ratio in multiple solid malignancies. 363 , 364 FPA008 is also a monoclonal antibody that disrupts the binding of CSF‐1R to its ligands and thus suppressing downstream receptor activation. The modulatory effects of FPA008 on macrophages and other immune cells in a variety of cancers are currently under evaluation by phase‐Ia/Ib clinical trials. 363 With regard to adverse effects, increases in short‐lived enzymes (e.g., AST, LDH) are detected, which might be due to spontaneous inhibitory effects of antibodies on CSF‐1R+ Kupffer cells. 366 Furthermore, common adverse effects also include fatigue, asthenia, facial edema, rash, and pruritus, but most of the events are only of grade 1 or 2 severities.
In addition to antibody therapies, multiple CSF‐1/CSF‐1R inhibitors are under development at clinical stages (Table 3). JNJ‐40346527 is a selective inhibitor of CSF‐1R with an IC50 of 3.2 nM. In a phase‐I/II clinical study of relapsed or refractory Hodgkin lymphoma, one patient out of 21 showed complete remission after JNJ‐40346527 treatment (150 mg/day) for approximately 1 year. Furthermore, 11 patients remained with disease stabilization. 367 Notably, in vivo pharmacological activity showed that CSF‐1R phosphorylation was inhibited by 95% following JNJ‐40346527 treatment (150 mg/day). Meanwhile, higher doses (150–600 mg/day) also demonstrated acceptable tolerability and safety in healthy volunteers. 363 GW2580 is an orally bioavailable CSF‐1R inhibitor with an IC50 of 60 nM. 368 However, GW2580 shows limited inhibitory effects on macrophage recruitment and tumor growth. Priceman et al. 369 treated mice to test the effects of GW2580 in blocking TAMs infiltration into growing tumors. Despite the concentration reaching as high as 160 mg/kg, only a slight decrease in TAMs was observed, while no notable changes in monocytes infiltration levels were observed in peripheral tissues including bone marrow, blood, and the spleen. Nevertheless, the combination treatment of GW2580 and VEGFR2 antibody efficiently abrogated the recruitment of TAMs and monocytic MDSCs and synergistically enhanced the antiangiogenesis response. 369 Meanwhile, GW2580 was also found to increase gemcitabine therapeutic efficacy in pancreatic cancer compared to that of GW2580 alone. 370 GW2580 was found to inhibit gemcitabine‐induced CSF‐1 overproduction in cancer cells and thus inhibited the recruitment of immunosuppressive myeloid cells into tumors. 370
TABLE 3.
Tyrosine kinases targeting agents in clinical trials
| Drug/description | Sponsors | Phase/status | Tumor type | Treatment | Clinical trials.govIdentifier | First received |
|---|---|---|---|---|---|---|
| CSF‐1R/CSF‐1 inhibitor | ||||||
| Emactuzumab/RG7155/RO5509554 (anti‐ CSF1R antibody) | M.D. Anderson Cancer Center | 2 b | Ovarian, fallopian tube, or primary peritoneal cancer | With paclitaxel and bevacizumab | NCT02923739 | 2016‐10‐05 |
| Hoffmann‐La Roche | 1 b | Advanced or metastatic solid cancers | With atezolizumab | NCT02323191 | 2014‐12‐23 | |
| Hoffmann‐La Roche | 1 a | Advanced solid tumors | With RO7009789 | NCT02760797 | 2016‐05‐04 | |
| Hoffmann‐La Roche | 1 a | Advanced solid tumors | Monotherapy | NCT01494688 378 | 2011‐12‐19 | |
| FPA008/Cabiralizumab (anti‐CSF1R antibody) | Bristol‐Myers Squibb | 2 b | Advanced pancreatic cancer | With nivolumab | NCT03336216 | 2017‐12‐08 |
| Five Prime Therapeutics | 1/2 b | Tenosynovial giant cell tumor | Monotherapy | NCT02471716 | 2015‐06‐15 | |
| Five Prime Therapeutics | 1a/b b | Advanced lung cancer, head and neck cancer, pancreatic cancer, ovarian cancer, renal cell carcinoma, and malignant glioma | With nivolumab | NCT02526017 | 2015‐08‐18 | |
| Bristol‐Myers Squibb | 1 a | Advanced malignancies | With nivolumab | NCT03158272 | 2017‐05‐18 | |
| LY3022855/IMC‐CS4 (anti‐CSF1R antibody) | Eli Lilly and Company | 1a/b a | Solid tumor | With durvalumab or tremelimumab | NCT02718911 | 2016‐03‐24 |
| Dana‐Farber Cancer Institute | 1/2 b | Melanoma | With vemurafenib and cobimetinib | NCT03101254 | 2017‐04‐05 | |
| Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins | 1 b | Pancreatic cancer | With GVAX, cyclophosphamide, and pembrolizumab | NCT03153410 | 2017‐05‐15 | |
| Eli Lilly and Company | 1 a | Advanced breast or prostate cancer | Monotherapy | NCT02265536 | 2014‐10‐16 | |
| Eli Lilly and Company | 1 a | Advanced solid tumors | Monotherapy | NCT01346358 | 2011‐05‐03 | |
| AMG820 (anti‐CSF1R antibody) | Amgen | 1b/2 a | Pancreatic cancer, colorectal cancer and non‐small cell lung cancer | With pembrolizumab | NCT02713529 | 2016‐03‐18 |
| Amgen | 1 a | Advanced solid tumors | Monotherapy | NCT01444404 | 2011‐09‐30 | |
| MCS110 (anti‐ CSF1 antibody) | Yonsei University | 2 b | Squamous cell carcinoma | With PDR001 | NCT03785496 | 2018‐12‐24 |
| Seoul National University Hospital | 2 b | Gastric cancer | With PDR001 | NCT03694977 | 2018‐10‐03 | |
| Novartis Pharmaceuticals | 2 b | Advanced triple‐negative breast cancer | With carboplatin and gemcitabine | NCT02435680 | 2015‐05‐06 | |
| Dana‐Farber Cancer Institute | 1/2 b | Melanoma | With dabrafenib and trametinib | NCT03455764 | 2018‐03‐07 | |
| Novartis Pharmaceuticals | 1b/2 b | Triple‐negative breast cancer, pancreatic carcinoma, melanoma, and endometrial carcinoma | With PDR001 | NCT02807844 | 2016‐06‐21 | |
| JNJ‐40346527 (CSF‐1R kinase inhibitor) | OHSU Knight Cancer Institute | 2 b | Relapsed/Refractory acute myeloid leukemia | Monotherapy | NCT03557970 | 2018‐06‐15 |
| Janssen Research & Development | 1/2 a | Relapsed or refractory Hodgkin lymphoma | Monotherapy | NCT01572519 | 2012‐04‐06 | |
| M.D. Anderson Cancer Center | 1 b | Prostate cancer | Monotherapy | NCT03177460 | 2017‐06‐06 | |
| Pexidartinib/PLX3397 (CSF‐1R kinase inhibitor) | Daiichi Sankyo | 3 b | Pigmented villonodular synovitis or giant cell tumor of the tendon sheath | Monotherapy | NCT02371369 379 | 2015‐02‐25 |
| Daiichi Sankyo | 2 a | Hodgkin lymphoma | Monotherapy | NCT01217229 | 2010‐10‐08 | |
| Daiichi Sankyo | 1/2 b | Melanoma | Monotherapy | NCT02975700 | 2016‐12‐29 | |
| National Cancer Institute | 1/2 b | Plexiform neurofibroma, precursor cell lymphoblastic leukemia lymphoma and acute prolymphocytic leukemia | Monotherapy | NCT02390752 | 2015‐03‐18 | |
| Hope Rugo, MD | 1b/2 a | Metastatic breast cancer | With Eribulin | NCT01596751 | 2012‐05‐11 | |
| Daiichi Sankyo | 1b/2 b | Glioblastoma | With radiation therapy and temozolomide | NCT01790503 | 2013‐02‐13 | |
| Daiichi Sankyo | 1/2 a | Acute myeloid leukemia | Monotherapy | NCT01349049 380 | 2011‐05‐06 | |
| Gulam Manji | 1 b | Sarcoma and malignant peripheral nerve sheath tumors | With sirolimus | NCT02584647 | 2015‐10‐22 | |
| Plexxikon | 1 a | Advanced solid tumors | With Paclitaxel | NCT01525602 | 2012‐02‐03 | |
| Daiichi Sankyo | 1 b | Solid tumor | Monotherapy | NCT01004861 373 | 2009‐10‐30 | |
| Centre Leon Berard | 1 a | Advanced and metastatic colorectal cancer and pancreatic cancer | With durvalumab | NCT02777710 | 2016‐05‐19 | |
| ARRY‐382 (CSF‐1R kinase inhibitor) | Array Biopharma | 1 a | Metastatic cancer | Monotherapy | NCT01316822 | 2011‐03‐16 |
| Array BioPharma | 1b/2 a | Advanced solid tumors | With pembrolizumab | NCT02880371 | 2016‐08‐26 | |
| BLZ945 (CSF‐1R kinase inhibitor) | Novartis Pharmaceuticals | 1/2 b | Advanced solid tumors | With PDR001 | NCT02829723 | 2016‐07‐12 |
| DCC‐3014 (CSF‐1R kinase inhibitor) | Deciphera Pharmaceuticals | 1/2 b | Malignant solid tumors and tenosynovial giant cell tumor | Monotherapy | NCT03069469 | 2017‐03‐03 |
| ENMD‐2076 (CSF‐1R kinase inhibitor) | University Health Network, Toronto | 2#a | Ovarian clear cell carcinoma | Monotherapy | NCT01914510 | 2013‐08‐02 |
| University Health Network, Toronto | 2 a | Advanced/metastatic soft tissue sarcoma | Monotherapy | NCT01719744 | 2012‐11‐01 | |
| CASI Pharmaceuticals | 2 a | Advanced adult hepatocellular carcinoma and advanced fibrolamellar carcinoma | Monotherapy | NCT02234986 | 2014‐09‐09 | |
| CASI Pharmaceuticals | 2 a | Triple‐negative breast cancer | Monotherapy | NCT01639248 381 | 2012‐07‐12 | |
| RON inhibitor | ||||||
| Narnatumab/ IMC‐RON8 (anti‐RON antibody) | Eli Lilly and Company | 1 a | Solid tumors | Monotherapy | NCT01119456 | 2010‐05‐07 |
| ASLAN002/BMS 777607 (RON kinase inhibitor) | Bristol‐Myers Squibb | 1/2 a | Advanced or metastatic solid tumor | Monotherapy | NCT00605618 | 2008‐01‐31 |
| Aslan Pharmaceuticals | 1 a | Advanced or metastatic solid tumor | Monotherapy | NCT01721148 382 | 2012‐12‐04 | |
| Crizotinib/ PF‐02341066 (RON kinase inhibitor) | UNICANCER | 2 b | Hematologic cancers and solid tumors and metastatic cancer | Monotherapy | NCT02034981 383 | 2014‐01‐14 |
| Dana‐Farber Cancer Institute | 1 b | Castration‐resistant prostate cancer | With enzalutamide | NCT02207504 | 2014‐08‐04 | |
| Foretinib/GSK1363089 (RON kinase inhibitor) | NCIC Clinical Trials Group | 2 a | Recurrent breast cancer | Monotherapy | NCT01147484 384 | 2010‐06‐22 |
| GlaxoSmithKline | 2 a | Head and neck cancer | Monotherapy | NCT00725764 | 2008‐07‐30 | |
| GlaxoSmithKline | 2 a | Papillary Renal‐Cell Carcinoma | Monotherapy | NCT00726323 385 | 2008‐07‐31 | |
| GlaxoSmithKline | 2 a | Metastatic gastric carcinoma | Monotherapy | NCT00725712 386 | 2008‐07‐30 | |
| GlaxoSmithKline | 1 a | Solid Tumors | Monotherapy | NCT00742131 | 2008‐08‐27 | |
| GlaxoSmithKline | 1 a | Hepatocellular carcinoma | Monotherapy | NCT00920192 387 | 2009‐06‐15 | |
Source: clinicaltrials.gov.
Completed stage.
Ongoing stage, including both recruiting and nonrecruiting stages.
Apart from the above highly selective CSF‐1R inhibitors, several CSF‐1R inhibitors with lower specificity have been identified, which also exhibit broader inhibitory activity on other kinases (Table 3). PLX3397 represents one such inhibitor and has an IC50 of 13 nM on CSF‐1R. Additionally, PLX3397 also exhibits activities against KIT, FLT3, and PDGFRβ. 364 , 371 , 372 PLX3397 displays anticancer effects in multiple malignancies. In patients with tenosynovial giant‐cell tumors, 52% of cases significantly responded to PLX3397 treatment with the disease control period lasting more than 8 months. 373 In glioblastoma, PLX3397 not only prolonged the survival time but also slowed the disease progression rate. 374 PLX3397 can easily cross the blood‐brain barrier and reach glioblastoma tissue, resulting in a significant reduction of macrophage numbers and invasiveness of cancer cells. In a phase‐II study of recurrent glioblastoma, PLX3397 treatment at a daily dose of 1000 mg showed acceptable tolerability and led to both CSF‐1R activity inhibition and TAMs infiltration reduction in tumor tissues. 372 At present, multiple clinical trials are being designed and conducted to test the clinical efficacies of CSF‐1R inhibitors on cancer inhibition. However, CSF‐1R blockade alone usually achieves marginal therapeutic benefit, leading to the delay of tumor growth at most. Therefore, more clinical trials are of an urgent need to evaluate the combined effects of CSF‐1/CSF‐1R blockage and other treatment approaches including immunotherapies and standard treatment modalities. Particularly, results for combinations of CSF‐1/CSF‐1R blockage with immune‐checkpoint inhibitors or other immunotherapeutic strategies are eagerly expected.
Several investigators have focused on the development of inhibitors targeting RON receptors (Table 3). The small‐molecule ASLAN002 is a selective inhibitor of RON receptor with an IC50 < 500 nM. 375 ASLAN002 can block MSP‐induced phosphorylation of RON. In a PyMT‐MSP breast cancer mouse model, ASLAN002 administration (50 mg/kg) remarkably inhibited lung metastasis by two‐ to threefold. 376 Even when ASLAN002 was administrated after the formation of metastatic colonization, a four‐fold inhibitory response was observed, indicating that RON inhibition might be a novel therapeutic option to inhibit metastatic growth as an adjuvant treatment. A mechanistic study found that ASLAN002 resulted in a proinflammatory milieu in the TME, particularly in terms of an increase in TNF‐α‐positive macrophages. 376 Additionally, CD8+ T cells act as critical factors influencing the efficacy of ASLAN002. The anti‐tumor colonization ability of ASLAN002 was greatly suppressed in the absence of cytotoxic CD8+ T cells, indicating the immune‐regulating cells may be primarily responsible for the anti‐cancer effects of ASLAN002. However, future research is necessary to explore the synergistic effects between ASLAN002 and immune‐checkpoint inhibitors. Another study revealed that in breast tumors harboring PIK3CA mutations, the anticancer effects of ASLAN002 were greatly limited, which were overcome by treatment with the PI3K inhibitor, NVP‐BKM120. 377 Meanwhile, concurrent inhibition of RON and PI3K provided a more durable response in PIK3CA‐wild‐type breast cancer. These results suggest that further investigation focusing on the combined applications of small‐molecule inhibitors is worthwhile for improving clinical prognoses of cancer patients.
5.3. Bisphosphonates
Bisphosphonates are a family of inhibitors of osteoclast‐mediated bone resorption and are widely applied in the treatment of bone diseases including postmenopausal osteoporosis, Paget disease, and bone metastasis. The first generation of nonamino bisphosphonates, such as etidronate and clodronate, are metabolized intracellularly into cytotoxic adenosine‐triphosphate analogs that induce osteoclast cell death. 388 Conversely, the second and third generations of bisphosphonates—such as ibandronate, risedronate, pamidronate, and zoledronic acid—are much more potent than nonamino compounds by inhibiting farnesyl‐pyrophosphate synthases, which also results in osteoclast apoptosis. 389 Moreover, preclinical studies demonstrate that bisphosphonates may also exhibit extraskeletal therapeutic effects in mouse breast cancer xenografts. 390 For example, zoledronic acid was found to be phagocyted by TAMs and thus induced TAMs apoptosis and repolarization toward M1‐like phenotype. 9 In breast cancer patients, radiolabeled bisphosphonate was also found in the resected tumor infiltrated with TAMs and microcalcifications. Several studies have demonstrated the proinflammatory activity of amino‐bisphosphonates in vitro. Ibandronate was found to enhance LPS‐stimulated IL‐6 and IL‐1β secretion from mouse Raw264.7 macrophages. 391 A remarkable elevation in TNFα expression was also observed following pamidronate and zoledronic‐acid administrations. 392 In mouse xenografts, ibandronate treatment led to increased infiltration of extravasated leukocytes and rolling leukocytes, accompanied by the stimulated production of TNFα and IFN‐γ in peripheral blood, indicating its proinflammatory effects in vivo. 393 In cancer patients treated with zoledronic acid, it was also found that the circulating concentrations of IFN‐γ, TNFα, and IL‐6 were all significantly elevated following drug administration, further confirming that amino‐bisphosphonates might promote M2‐like to M1‐like repolarization to trigger inflammatory responses. 394 By contrast, nonamino bisphosphonates have been demonstrated to inhibit cancer progression via suppressing the growth of macrophages. Clodronate inhibits iNOS expression in Raw264.7 macrophages, accompanied by a significant reduction of IL‐6, IL‐1β, TNFα, and NO secretions at noncytotoxic concentrations. 395 Therefore, nonamino bisphosphonates might suppress macrophages in a cytostatic but not cytotoxic manner. Based on the above findings, bisphosphonates have been commonly applied to deplete TAMs in preclinical models. Although bisphosphonates have yielded improved survival in various kinds of preclinical animal models, bisphosphonates treatment alone is not able to induce complete regression of tumors. In fact, the application of bisphosphonates may induce side effects on residential macrophages in normal tissues, thereby influencing antitumor innate immunity. Considering that the complete depletion of macrophages in tumors could lead to worse therapeutic outcomes in some cases, TAMs‐targeted reprogramming strategies that spare the potentially beneficial macrophages may be more preferable in the future.
5.4. Nanoparticle or exosomal‐targeted delivery
Although pharmaceutical inhibitors and monoclonal antibodies have been developed for TAMs‐targeting treatments, their therapeutic efficacies have been greatly limited by difficulties in penetrating biological barriers. Progress in nanotechnology has provided a valuable opportunity for improving drug‐loading/drug‐releasing parameters, biocompatibilities, and drug‐circulation time. The engineering of new nanoparticles with the ability to target and kill or reeducate TAMs has stood up as a promising strategy for cancer treatment in recent years. As an important member of the mononuclear phagocytic system, macrophages play a major role in nanomaterials recognition, uptake, processing, and clearance. 396 Therefore, the loading of macrophages with nanomedicines may be used to profit from the high infiltration and the innate tumor homing abilities of macrophages, allowing the drug release in the bulk of the tumor. For example, Rao et al. reported that the magnetic nanoparticles can be used to promote the repolarization of the M2‐like TAMs as well as the systemic circulation and tumor accumulation of the loaded drugs. 397 Notably, there are still some challenges for the effective targeting of TAMs with nanoparticles and their application in the clinic. The major one can be attributed to the undesirable clearance of nanoparticles by the mononuclear phagocyte system (macrophages) in clearance organs (liver, lung, or spleen) upon their intravenous injection. In vivo studies have demonstrated high nanomaterials sequestration by macrophages in clearance organs such as liver, spleen, and kidney. 396 This is because that the immune system may recognize the components of nanoparticles (e.g., shell, core, surface‐decorating moieties, and cargoes) as foreign, and initiates an immune response through a complex process. 398 Therefore, because of the synthetic origin, nanoparticles are usually recognized by the host immune system and are preferentially sequestered by macrophages in filtering organs.
Recently, exosomes have emerged as natural nano‐sized vesicles for drug delivery. Given their nanoscale size, excellent biocompatibility, potential capacity to express targeting ligands, and natural capacity to carry macromolecules, the use of exosomes for cancer treatment has raised considerable interest. 399 , 400 Due to the lipid bilayered membrane, exosomes exhibit good permeability and can cross most biological membranes, including the blood‐brain barrier. 401 After reaching recipient cells, exosomes can activate certain signaling pathways by fusion to the plasma membrane and subsequent secretion of the contents into the cytoplasm of recipient cells. Alternatively, exosomes can enter cells through endocytic processes including receptor‐mediated endocytosis, micropinocytosis, and phagocytosis. Moreover, exosomes have significant advantages in drug delivery areas without inducing proinflammatory responses or adverse immune reactions. More importantly, the composition of exosomal surfaces permits modifications for enhancing interactions between recipient cells and exosomes. Surface modification of naturally purified exosomes with GE11, an epidermal growth factor, was demonstrated to increase the targeting potential of exosomes to EGFR+ breast cancer cells both in vitro and in vivo. 402 Given these priorities, exosomes represent a promising candidate tool for the delivery of chemotherapeutics, as well as nucleic acids and phytochemicals. For example, doxorubicin treatment through the exosome delivery system induced a 10‐fold increase in cancer cell death when compared with free doxorubicin. 403 Similarly, exosome‐loaded anti‐miR‐214 led to a remarkable inhibition of gastric tumor growth. 404 Interestingly, curcumin has also been shown to be encapsulated into exosomes and to interfere with colon carcinogenesis (NCT01294072).
Given that exosomes can be preferentially sequestered by macrophages, 405 , 406 it is becoming an attractive vehicle for delivering genetic drugs or cytotoxic agents to inhibit TAMs. Exosomes containing miRNA‐155 and 125b2 derived from pancreatic cancer cells induced macrophage differentiation to the M1‐like phenotype. 407 Similarly, exosomes transfected with wt‐p53 and miRNA‐125b secreted from colon cancer cells also mediated macrophage repolarization toward the M1‐like phenotype. 408 Interestingly, epigallocatechin‐gallate‐treated breast cancer cells‐derived exosomes were also found to impair tumor growth by suppressing TAMs infiltration and M2‐like phenotype polarization. 137 Recently, a commercialized engineered exosome carrying antisense oligonucleotide (exoASO) was developed by Codiak Bioscience. It was designed to deliver ASO to TAMs and to specifically inhibit the expression of immune‐suppressive transcription factors, including STAT6 and C/EBPT. 137 Preclinical studies demonstrate that exoASO effectively reprograms M2‐like macrophages to a pro‐inflammatory M1‐like phenotype, promoting targeted anti‐tumor activity in vivo. In vitro, exoASO is preferentially taken up by M2‐like macrophages to a significantly greater extent than that of free ASO, resulting in a greater decrease of STAT6 and C/EBPβ mRNA levels. Subsequent gene expression analysis and cytokine assays showed a 40‐fold increase in TNFα and a 29‐fold decrease in IL‐10 associated with exoASO treatment, consistent with repolarization from immunosuppressive M2‐like macrophages to immune‐stimulatory M1‐like macrophages. All of these findings suggest that exosomes may represent promising drug‐delivery systems for modulating the biological functions of TAMs. Notably, there are still many difficulties to overcome before the successful application of the exosomal delivery system for TAMs modulation in the future. The most prominent one may be that different exosome isolation techniques have reported poor yield and loss of functional properties. 409 To solve this limitation, reengineering TAMs‐targeted exosomes with the synthetic liposomes as a refined biomimetic nanostructure is suggested. 409
5.5. Other therapeutic strategies
In recent years, multiple novel TAMs reprogramming strategies have been reported with promising future. For example, Christopher B et al. reported that R848, an agonist of toll‐like receptor 7 and 8 (TRL7/8), could significantly induce the M1‐like phenotype polarization of macrophages in vitro, while the R848‐loaded β‐cyclodextrin nanoparticles could promote the repolarization of TAMs toward an M1‐like phenotype in multiple tumor models in vivo. 410 Feng et al. synthesized the novel TLR agonist of acGM using glucomannan polysaccharide with acetyl modification, which could specifically activate the TLR2 signaling and thus induce TAMs repolarization into an antitumor phenotype. 411 Macrophages can also be targeted via CD40 agonists to boost the antitumor response. Gregory et al. reported that the CD40 agonist FGK45 could reactivate TAMs, which became tumoricidal and thus rapidly facilitated the depletion of tumor stroma and the tumor regression of pancreatic ductal adenocarcinoma. 412 Blocking checkpoints on T cells have recently shown unprecedented, durable cures in the clinic. A similar strategy targeting macrophages is also developing by blocking CD47 from binding its receptor signal regulatory protein‐α (SIRPα) on macrophage surface. 413 It has been reported that interfering of CD47‐SIRPα interaction by blocking antibodies could promote the phagocytosis of TAMs and consequently suppresses cholangiocarcinoma growth and metastasis. 414 Nowadays, a number of CD47‐SIRPα axis blockers, such as Hu5F9‐G4, CC‐90002, SRF231, ALX148, and IBI188, are under clinical trials. 413
6. CONCLUSIONS, CHALLENGES, AND PERSPECTIVES
With a deeper understanding of cancer immunology, targeting TAMs provides a novel approach to improve anticancer therapies. However, since there exists complex intercellular crosstalk involving TAMs in the TME, TAMs elimination may elicit multifaceted stromal reactions in the host that are unable to predict and may vary among patients. Additionally, some patients may be refractory to TAMs‐targeting therapies due to gene mutations. For example, it has been reported that certain single‐nucleotide polymorphisms in CSF‐1R can decrease the therapeutic efficacy of emactuzumab. 137 Therefore, it is important and significant to explore the synergistic effects of TAMs‐targeting approaches with other immunomodulatory strategies, especially for checkpoint‐blockade–based therapies. Checkpoint inhibition unleashes the brakes, such as CTLA4 and PD‐1, to enhance the effective recognition and killing of tumor cells by immune cells. However, only approximately 20% of cancer patients respond to checkpoint‐inhibition therapies, where mixed responses can limit treatment effectiveness and cause drug resistance and/or distant metastasis. TAMs may act as important cellular components influencing the responsiveness of checkpoint‐blockade treatments. On one hand, TAMs express PD‐L1 and PD‐L2, which could aggravate the immunosuppressive TME and impair cytotoxic T lymphocytes against cancer cells. 14 , 15 , 415 On the other hand, TAMs also expressed TREM2 (triggering receptor expressed on myeloid cells 2) that remodel the tumor myeloid landscape and weaken the responsiveness of anti‐PD‐1 immunotherapy. 416 Currently, a variety of clinical trials are ongoing to examine the synergistic effects of TAMs‐targeting approaches with PD‐L1 or CTLA‐4 blockades. Preclinical studies have also demonstrated that CSF1R antagonists can boost the efficacy of checkpoint inhibition in mouse models of pancreatic, breast, cervical, and ovarian cancers. Moreover, in immunotherapy‐resistant pancreatic cancer patients, CSF‐1R antagonists combined with PD‐1‐targeting approaches result in partial responses in some patients. All of these findings indicate the promising future and significance of TAMs‐targeting strategies in cancer immunotherapies.
With an increasing number of available drugs for modulating TAMs to induce anti‐tumor responses by targeting different molecular targets, the next challenge will be how to deliver these immunoregulatory drugs into TAMs effectively and selectively while minimizing the harmful off‐target side effects. Although nanovesicles, such as exosomes, provide an ideal platform for drug loading and delivery and can be primarily internalized by macrophages, their biodistributions in vivo are largely affected by residential macrophages outside of tumors, such as Kupffer cells in the liver. To improve TAMs selectivity over residential macrophages, several studies have utilized the unique physical and chemical properties of the TME to optimize their targeting systems. For example, the acid‐sensitive PEG 2000 was applied to modify nanoparticles to selectively eliminate TAMs as PEG 2000 can be cleaved in the acidic TME to release cytotoxic drugs. 417 This therapeutic strategy can minimize the off‐target effects of PEG 2000‐modified nanoparticles on resident macrophages. Additionally, targeting ligands have also been applied to improve the preferential delivery to TAMs via utilizing the specific ligand–receptor interactions. CD206 represents one of the most commonly targeted receptors for macrophage delivery because of its TAMs‐specific overexpression characteristic. As the native ligand of CD206, mannose can be conjugated to nanovesicles easily and result in an enhanced uptake by TAMs. 417 TAMs also overexpress folate receptor β, the ligand of which is folic acid. Therefore, folic‐acid‐modified nanovesicles have been developed as an important cancer‐targeting strategy. 418 Another factor influencing the anti‐tumor efficacy of nanovesicles is the ability of drug‐loaded macrophages to infiltrate into tumors. A recent study suggested that the size of nanovesicles can significantly alter the motilities of macrophages. 419 Although smaller nanovesicles (30–50 nm) usually have higher uptake into macrophages compared to that of larger nanocarriers (100–500 nm), smaller nanovesicles were found to significantly retard the motilities of macrophages. This study suggested that 100‐nm nanovesicles provide a good balance for effective drug loading and macrophage migration. Hence, exosomes are an ideal and promising type of nanovesicle for developing TAMs‐targeting strategies.
Taken together, the pharmaceutical targets of TAMs and their corresponding drugs are rapidly being explored and developed. With the development of single‐cell sequencing, various “omic” analyses, and nanotechnology, our understanding of the heterogeneity of TAMs in the tumor milieu will be considerably enhanced. It is expected that future studies will further elucidate the clinical benefits resulting from combined therapies of TAMs‐targeting strategies with traditional treatments or immunotherapies. Meanwhile, a more advanced drug delivery system via exosome‐like nanovesicles is anticipated to help reduce the off‐target/side effects of TAMs‐targeting approaches.
AUTHOR CONTRIBUTIONS
NW and ZYW conceived, wrote, and revised the article. SQW drew the figures and revised the article. XW generated the tables and added references. YFZ, BWY, JPZ, BP, and JLG participated in the work of reference selection and article check.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
ACKNOWLEDGMENTS
We thank Letpub for its linguistic assistance during the preparation of this article.
Wang N, Wang S, Wang X, et al. Research trends in pharmacological modulation of tumor‐associated macrophages. Clin Transl Med. 2021;11:e288 10.1002/ctm2.288
Neng Wang and Shengqi Wang contributed equally to this work.
REFERENCES
- 1. Ahmed F, Steele JC, Herbert JMJ, Steven NM, R B. Tumor stroma as a target in cancer. Curr Cancer Drug Targets. 2008;8(6):447‐453. [DOI] [PubMed] [Google Scholar]
- 2. Wagner J, Rapsomaniki MA, Chevrier S, et al. A single‐cell atlas of the tumor and immune ecosystem of human breast cancer. Cell. 2019;177(5):1330‐1345 e1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Komohara Y, Fujiwara Y, Ohnishi K, Takeya M. Tumor‐associated macrophages: potential therapeutic targets for anti‐cancer therapy. Adv Drug Deliv Rev. 2016;99(Pt B):180‐185. [DOI] [PubMed] [Google Scholar]
- 4. Kolios G, Valatas V, Kouroumalis E. Role of Kupffer cells in the pathogenesis of liver disease. World J Gastroenterol. 2006;12(46):7413‐7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang X, Zhao L, Zhang J, et al. Requirement for microglia for the maintenance of synaptic function and integrity in the mature retina. J Neurosci. 2016;36(9):2827‐2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kimber I, Cumberbatch M, Dearman RJ. Langerhans cells and skin irritation In: Ai‐LeanChew HM, ed. Irritant Dermatitis. Berlin, New York, Heidelberg: Springer‐Verlag; 2006:383–391. [Google Scholar]
- 7. Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol. 2012;32(6):463‐488. [DOI] [PubMed] [Google Scholar]
- 9. Coscia M, Quaglino E, Iezzi M, et al. Zoledronic acid repolarizes tumour‐associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J Cell Mol Med. 2010;14(12):2803‐2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23(3):277‐286. [DOI] [PubMed] [Google Scholar]
- 11. Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett. 2008;267(2):204‐215. [DOI] [PubMed] [Google Scholar]
- 12. Trombetta AC, Soldano S, Contini P, et al. A circulating cell population showing both M1 and M2 monocyte/macrophage surface markers characterizes systemic sclerosis patients with lung involvement. Respir Res. 2018;19(1):186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Van Gorp H, Delputte PL, Nauwynck HJ. Scavenger receptor CD163, a Jack‐of‐all‐trades and potential target for cell‐directed therapy. Mol Immunol. 2010;47(7‐8):1650‐1660. [DOI] [PubMed] [Google Scholar]
- 14. Gordon SR, Maute RL, Dulken BW, et al. PD‐1 expression by tumour‐associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cai J, Qi Q, Qian X, et al. The role of PD‐1/PD‐L1 axis and macrophage in the progression and treatment of cancer. J Cancer Res Clin Oncol. 2019;145(6):1377‐1385. [DOI] [PubMed] [Google Scholar]
- 16. Fujimura T, Sato Y, Tanita K, et al. Serum levels of soluble CD163 and CXCL5 may be predictive markers for immune‐related adverse events in patients with advanced melanoma treated with nivolumab: a pilot study. Oncotarget. 2018;9(21):15542‐15551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fujimura T, Sato Y, Tanita K, et al. Serum level of soluble CD163 may be a predictive marker of the effectiveness of nivolumab in patients with advanced cutaneous melanoma. Front Oncol. 2018;8:530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fransisca L, Curtis LT, James WM, et al. Macrophage polarization contributes to the anti‐tumoral efficacy of mesoporous nanovectors loaded with albumin‐bound paclitaxel. J Frontiers in Immunology. 2017;8:683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rakaee M, Busund LR, Jamaly S, et al. Prognostic value of macrophage phenotypes in resectable non‐small cell lung cancer assessed by multiplex immunohistochemistry. Neoplasia. 2019;21(3):282‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yagi T, Baba Y, Okadome K, et al. Tumour‐associated macrophages are associated with poor prognosis and programmed death ligand 1 expression in oesophageal cancer. Eur J Cancer. 2019;111:38‐49. [DOI] [PubMed] [Google Scholar]
- 21. Zhou J, Wang XH, Zhao YX, et al. Cancer‐associated fibroblasts correlate with tumor‐associated macrophages infiltration and lymphatic metastasis in triple negative breast cancer patients. J Cancer. 2018;9(24):4635‐4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thacker JD, Dedhar S, Hogge DE. The effect of GM‐CSF and G‐CSF on the growth of human osteosarcoma cells in vitro and in vivo. Int J Cancer. 1994;56(2):236‐243. [DOI] [PubMed] [Google Scholar]
- 23. Wang Y, Han G, Wang K, et al. Tumor‐derived GM‐CSF promotes inflammatory colon carcinogenesis via stimulating epithelial release of VEGF. Cancer Res. 2014;74(3):716‐726. [DOI] [PubMed] [Google Scholar]
- 24. Garcia‐Mendoza MG, Inman DR, Ponik SM, et al. Neutrophils drive accelerated tumor progression in the collagen‐dense mammary tumor microenvironment. Breast Cancer Res. 2016;18(1):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fritz JM, Tennis MA, Orlicky DJ, et al. Depletion of tumor‐associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front Immunol. 2014;5:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cottone L, Valtorta S, Capobianco A, et al. Evaluation of the role of tumor‐associated macrophages in an experimental model of peritoneal carcinomatosis using(18)F‐FDG PET. J Nucl Med. 2011;52(11):1770‐1777. [DOI] [PubMed] [Google Scholar]
- 27. Sullivan AR, Pixley FJ. CSF‐1R signaling in health and disease: a focus on the mammary gland. J Mammary Gland Biol Neoplasia. 2014;19(2):149‐159. [DOI] [PubMed] [Google Scholar]
- 28. Franklin RA, Liao W, Sarkar A, et al. The cellular and molecular origin of tumor‐associated macrophages. Science. 2014;344(6186):921‐925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li X, Eckard J, Shah R, Malluck C, Frenkel K. Interleukin‐1α up‐regulation in vivo by a potent carcinogen 7,12‐dimethylbenz(a)anthracene(DMBA) and control of DMBA‐induced inflammatory responses. Cancer Res. 2002;62(2):417‐423. [PubMed] [Google Scholar]
- 30. Weber C, Telerman SB, Reimer AS, et al. Macrophage infiltration and alternative activation during wound healing promote MEK1‐induced skin carcinogenesis. Cancer Res. 2016;76(4):805‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saxon JA, Sherrill TP, Polosukhin VV, Sai J, Blackwell TS. Epithelial NF‐κB signaling promotes EGFR‐driven lung carcinogenesis via macrophage recruitment. Oncoimmunology. 2016;5(6):e1168549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996;56(20):4625‐4629. [PubMed] [Google Scholar]
- 33. Negus RPM, Stamp GWH, Hadley J, Balkwill FR. Quantitative assessment of the leukocyte infiltrate in ovarian cancer and its relationship to the expression of C‐C chemokines. Am J Pathol. 1997;150(5):1723‐1734. [PMC free article] [PubMed] [Google Scholar]
- 34. Leek RD, Harris AL. Tumor‐associated macrophages in breast cancer. J Mammary Gland Biol Neoplasia. 2002;7(2):177‐189. [DOI] [PubMed] [Google Scholar]
- 35. Onita T, Ping GJ, Xuan JW, et al. Hypoxia‐induced, perinecrotic expression of endothelial Per‐ARNT‐Sim domain protein‐1/hypoxia‐inducible factor‐2α correlates with tumor progression, vascularization, and focal macrophage infiltration in bladder cancer. Clin Cancer Res. 2002;8(2):471‐480. [PubMed] [Google Scholar]
- 36. Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony‐stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193(6):727‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dirkx AE, Oude Egbrink MG, Wagstaff J, Griffioen AW. Monocyte/macrophage infiltration in tumors: modulators of angiogenesis. J Leukoc Biol. 2006;80(6):1183‐1196. [DOI] [PubMed] [Google Scholar]
- 38. Sunderkötter C, Goebeler M, Schulze‐Osthoff K, Bhardwaj R, Sorg C. Macrophage‐derived angiogenesis factors. Pharmacol Ther. 1991;51(2):195‐216. [DOI] [PubMed] [Google Scholar]
- 39. Lin EY, Li JF, Bricard G, et al. Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Mol Oncol. 2007;1(3):288‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pilar Cejudo‐Martín MM‐R, Ros J, Navasa M, et al. Hypoxia is an inducer of vasodilator agents in peritoneal macrophages of cirrhotic patients. Hepatology. 2002;36(5):1172‐1179. [DOI] [PubMed] [Google Scholar]
- 41. Klimp AH, Hollema H, Kempinga C, van der Zee AG, de Vries EG, Daemen T. Expression of cyclooxygenase‐2 and inducible nitric oxide synthase in human ovarian tumors and tumor‐associated macrophages. Cancer Res. 2001;61(19):7305‐7309. [PubMed] [Google Scholar]
- 42. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66(2):605‐612. [DOI] [PubMed] [Google Scholar]
- 43. Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103(33):12493‐12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Houghton AM, Grisolano JL, Baumann ML, et al. Macrophage elastase(matrix metalloproteinase‐12) suppresses growth of lung metastases. Cancer Res. 2006;66(12):6149‐6155. [DOI] [PubMed] [Google Scholar]
- 45. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour‐associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14(7):399‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ruffell B, Chang‐Strachan D, Chan V, et al. Macrophage IL‐10 blocks CD8+ T cell‐dependent responses to chemotherapy by suppressing IL‐12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26(5):623‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shalapour S, Karin M. Pas de Deux: control of anti‐tumor immunity by cancer‐associated inflammation. Immunity. 2019;51(1):15‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid‐derived suppressor cells. Blood. 2011;118(20):5498‐5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kawano Y, Roccaro AM, Ghobrial IM, Azzi J. Multiple myeloma and the immune microenvironment. Curr Cancer Drug Targets. 2017;17(9):806‐818. [DOI] [PubMed] [Google Scholar]
- 50. El‐Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma(CheckMate 040): an open‐label, non‐comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492‐2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R blockade reprograms tumor‐infiltrating macrophages and improves response to T‐cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057‐5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. HS S, H B, C B, R V, et al. FcγR interaction is not required for effective anti‐PD‐L1 immunotherapy but can add additional benefit depending on the tumor model. Int J Cancer. 2019;144(2):345. [DOI] [PubMed] [Google Scholar]
- 53. Liu H, Wang J, Zhang M, et al. Jagged1 promotes aromatase inhibitor resistance by modulating tumor‐associated macrophage differentiation in breast cancer patients. Breast Cancer Res Treat. 2017;166(1):95‐107. [DOI] [PubMed] [Google Scholar]
- 54. D'Errico G, Alonso‐Nocelo M, Vallespinos M, et al. Tumor‐associated macrophage‐secreted 14‐3‐3ζ signals via AXL to promote pancreatic cancer chemoresistance. Oncogene. 2019;38(27):5469‐5485. [DOI] [PubMed] [Google Scholar]
- 55. Forssell J, Oberg A, Henriksson ML, Stenling R, Jung A, Palmqvist R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin Cancer Res. 2007;13(5):1472‐1479. [DOI] [PubMed] [Google Scholar]
- 56. Zheng Y, Yang J, Qian J, et al. PSGL‐1/selectin and ICAM‐1/CD18 interactions are involved in macrophage‐induced drug resistance in myeloma. Leukemia. 2013;27(3):702‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zheng P, Chen L, Yuan X, et al. Exosomal transfer of tumor‐associated macrophage‐derived miR‐21 confers cisplatin resistance in gastric cancer cells. J Exp Clin Cancer Res. 2017;36(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Giurisato E, Lonardi S, Telfer B, et al. Extracellular‐regulated protein kinase 5‐mediated control of p21 expression promotes macrophage proliferation associated with tumor growth and metastasis. Cancer Res. 2020;80(16):3319‐3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Su S, Liu Q, Chen J, et al. A positive feedback loop between mesenchymal‐like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell. 2014;25(5):605‐620. [DOI] [PubMed] [Google Scholar]
- 60. Fu XT, Dai Z, Song K, et al. Macrophage‐secreted IL‐8 induces epithelial‐mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int J Oncol. 2015;46(2):587‐596. [DOI] [PubMed] [Google Scholar]
- 61. Ravi J, Elbaz M, Wani NA, Nasser MW, Ganju RK. Cannabinoid receptor‐2 agonist inhibits macrophage induced EMT in non‐small cell lung cancer by downregulation of EGFR pathway. Mol Carcinog. 2016;55(12):2063‐2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Helm O, Held‐Feindt J, Grage‐Griebenow E, et al. Tumor‐associated macrophages exhibit pro‐ and anti‐inflammatory properties by which they impact on pancreatic tumorigenesis. Int J Cancer. 2014;135(4):843‐861. [DOI] [PubMed] [Google Scholar]
- 63. Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF‐kappaB is required for inflammation‐induced cell migration and invasion. Cancer Cell. 2009;15(5):416‐428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kawata M, Koinuma D, Ogami T, et al. TGF‐beta‐induced epithelial‐mesenchymal transition of A549 lung adenocarcinoma cells is enhanced by pro‐inflammatory cytokines derived from RAW 264.7 macrophage cells. J Biochem. 2012;151(2):205‐216. [DOI] [PubMed] [Google Scholar]
- 65. Vasiljeva O, Papazoglou A, Kruger A, et al. Tumor cell‐derived and macrophage‐derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res. 2006;66(10):5242‐5250. [DOI] [PubMed] [Google Scholar]
- 66. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gocheva V, Wang HW, Gadea BB, et al. IL‐4 induces cathepsin protease activity in tumor‐associated macrophages to promote cancer growth and invasion. Genes Dev. 2010;24(3):241‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sangaletti S, Di Carlo E, Gariboldi S, et al. Macrophage‐derived SPARC bridges tumor cell‐extracellular matrix interactions toward metastasis. Cancer Res. 2008;68(21):9050‐9059. [DOI] [PubMed] [Google Scholar]
- 69. Sidani M, Wyckoff J, Xue C, Segall JE, Condeelis J. Probing the microenvironment of mammary tumors using multiphoton microscopy. J Mammary Gland Biol Neoplasia. 2006;11(2):151‐163. [DOI] [PubMed] [Google Scholar]
- 70. Qian B, Deng Y, Im JH, et al. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One. 2009;4(8):e6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nierodzik ML, Karpatkin S. Thrombin induces tumor growth, metastasis, and angiogenesis: evidence for a thrombin‐regulated dormant tumor phenotype. Cancer Cell. 2006;10(5):355‐362. [DOI] [PubMed] [Google Scholar]
- 72. Gil‐Bernabe AM, Ferjancic S, Tlalka M, et al. Recruitment of monocytes/macrophages by tissue factor‐mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood. 2012;119(13):3164‐3175. [DOI] [PubMed] [Google Scholar]
- 73. Chen Q, Zhang XH, Massague J. Macrophage binding to receptor VCAM‐1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell. 2011;20(4):538‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kaplan RN, Riba RD, Zacharoulis S, et al. VEGFR1‐positive haematopoietic bone marrow progenitors initiate the pre‐metastatic niche. Nature. 2005;438(7069):820‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sceneay J, Smyth MJ, Moller A. The pre‐metastatic niche: finding common ground. Cancer Metastasis Rev. 2013;32(3‐4):449‐464. [DOI] [PubMed] [Google Scholar]
- 76. Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the “pre‐metastatic niche”: within bone and beyond. Cancer Metastasis Rev. 2006;25(4):521‐529. [DOI] [PubMed] [Google Scholar]
- 77. Eisenblaetter M, Flores‐Borja F, Lee JJ, et al. Visualization of tumor‐immune interaction—target‐specific imaging of S100A8/A9 reveals pre‐metastatic niche establishment. Theranostics. 2017;7(9):2392‐2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zheng Y, Wang N, Wang S, et al. XIAOPI formula inhibits the pre‐metastatic niche formation in breast cancer via suppressing TAMs/CXCL1 signaling. Cell Commun Signal. 2020;18(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Balkwill FR. The chemokine system and cancer. J Pathol. 2012;226(2):148‐157. [DOI] [PubMed] [Google Scholar]
- 80. Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12(2):121‐127. [DOI] [PubMed] [Google Scholar]
- 81. Mollaoglu G, Jones A, Wait SJ, et al. The lineage‐defining transcription factors SOX2 and NKX2‐1 determine lung cancer cell fate and shape the tumor immune microenvironment. Immunity. 2018;49(4):764‐779 e769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Conti I, Rollins BJ. CCL2(monocyte chemoattractant protein‐1) and cancer. Semin Cancer Biol. 2004;14(3):149‐154. [DOI] [PubMed] [Google Scholar]
- 83. Sawanobori Y, Ueha S, Kurachi M, et al. Chemokine‐mediated rapid turnover of myeloid‐derived suppressor cells in tumor‐bearing mice. Blood. 2008;111(12):5457‐5466. [DOI] [PubMed] [Google Scholar]
- 84. Fujita M, Kohanash G, Fellows W, et al. COX‐2 blockade suppresses gliomagenesis by inhibiting CCL2‐mediated accumulation of myeloid‐derived suppressor cells. Cancer Res. 2011;71:1788‐1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ma Y, Mattarollo SR, Adjemian S, et al. CCL2/CCR2‐dependent recruitment of functional antigen‐presenting cells into tumors upon chemotherapy. Cancer Res. 2014;74(2):436‐445. [DOI] [PubMed] [Google Scholar]
- 86. Luboshits G, Shina S, Kaplan O, et al. Elevated expression of the CC chemokine regulated on activation, normal T cell expressed and secreted(RANTES) in advanced breast carcinoma. Cancer Res. 1999;59(18):4681‐4687. [PubMed] [Google Scholar]
- 87. Duell EJ, Casella DP, Burk RD, Kelsey KT, Holly EA. Inflammation, genetic polymorphisms in proinflammatory genes TNF‐A, RANTES, and CCR5, and risk of pancreatic adenocarcinoma. Cancer Epidemiol Biomarkers Prev. 2006;15(4):726‐731. [DOI] [PubMed] [Google Scholar]
- 88. Tsukishiro S, Suzumori N, Nishikawa H, Arakawa A, Suzumori K. Elevated serum RANTES levels in patients with ovarian cancer correlate with the extent of the disorder. Gynecol Oncol. 2006;102(3):542‐545. [DOI] [PubMed] [Google Scholar]
- 89. Vaday GG, Peehl DM, Kadam PA, Lawrence DM. Expression of CCL5(RANTES) and CCR5 in prostate cancer. Prostate. 2006;66(2):124‐134. [DOI] [PubMed] [Google Scholar]
- 90. Zhang Q, Qin J, Zhong L, et al. CCL5‐Mediated Th2 immune polarization promotes metastasis in luminal breast cancer. Cancer Res. 2015;75(20):4312‐4321. [DOI] [PubMed] [Google Scholar]
- 91. Chang LY, Lin YC, Mahalingam J, et al. Tumor‐derived chemokine CCL5 enhances TGF‐beta‐mediated killing of CD8(+) T cells in colon cancer by T‐regulatory cells. Cancer Res. 2012;72(5):1092‐1102. [DOI] [PubMed] [Google Scholar]
- 92. Lv M, Xu Y, Tang R, et al. miR141‐CXCL1‐CXCR2 signaling‐induced Treg recruitment regulates metastases and survival of non‐small cell lung cancer. Mol Cancer Ther. 2014;13(12):3152‐3162. [DOI] [PubMed] [Google Scholar]
- 93. Wang D, Sun H, Wei J, Cen B, DuBois RN. CXCL1 is critical for premetastatic niche formation and metastasis in colorectal cancer. Cancer Res. 2017;77(13):3655‐3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE(2)‐induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. 2011;71(24):7463‐7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Martins‐Green M, Feugate JE. The 9E3/CEF4 gene product is a chemotactic and angiogenic factor that can initiate the wound‐healing cascade in vivo. Cytokine. 1998;10(7):522‐535. [DOI] [PubMed] [Google Scholar]
- 96. Teicher BA, Fricker SP. CXCL12(SDF‐1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16(11):2927‐2931. [DOI] [PubMed] [Google Scholar]
- 97. Wang Y, Liu J, Jiang Q, et al. Human adipose‐derived mesenchymal stem cell‐secreted CXCL1 and CXCL8 facilitate breast tumor growth by promoting angiogenesis. Stem Cells. 2017;35(9):2060‐2070. [DOI] [PubMed] [Google Scholar]
- 98. Tang KH, Ma S, Lee TK, et al. CD133(+) liver tumor‐initiating cells promote tumor angiogenesis, growth, and self‐renewal through neurotensin/interleukin‐8/CXCL1 signaling. Hepatology. 2012;55(3):807‐820. [DOI] [PubMed] [Google Scholar]
- 99. Wang D, Wang H, Brown J, et al. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J Exp Med. 2006;203(4):941‐951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Azenshtein E, Luboshits G, Shina S, et al. The CC chemokine RANTES in breast carcinoma progression: regulation of expression and potential mechanisms of promalignant activity. Cancer Res. 2002;62(4):1093‐1102. [PubMed] [Google Scholar]
- 101. Youngs SJ, Ali SA, Taub DD, Rees RC. Chemokines induce migrational responses in human breast carcinoma cell lines. Int J Cancer. 1997;71(2):257‐266. [DOI] [PubMed] [Google Scholar]
- 102. Prest SJ, Rees RC, Murdoch C, et al. Chemokines induce the cellular migration of MCF‐7 human breast carcinoma cells: subpopulations of tumour cells display positive and negative chemotaxis and differential in vivo growth potentials. Clin Exp Metastasis. 1999;17(5):389‐396. [DOI] [PubMed] [Google Scholar]
- 103. Miller LJ, Kurtzman SH, Wang Y, Anderson KH, Lindquist RR, Kreutzer DL. Expression of interleukin‐8 receptors on tumor cells and vascular endothelial cells in human breast cancer tissue. Anticancer Res. 1998;18(1A):77‐81. [PubMed] [Google Scholar]
- 104. Kato M, Kitayama J, Kazama S, Nagawa H. Expression pattern of CXC chemokine receptor‐4 is correlated with lymph node metastasis in human invasive ductal carcinoma. Breast Cancer Res. 2003;5(5):R144‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Acharyya S, Oskarsson T, Vanharanta S, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150(1):165‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhuang H, Cao G, Kou C, Liu T. CCL2/CCR2 axis induces hepatocellular carcinoma invasion and epithelial‐mesenchymal transition in vitro through activation of the Hedgehog pathway. Oncol Rep. 2018;39(1):21‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lee CC, Lai JH, Hueng DY, et al. Disrupting the CXCL12/CXCR4 axis disturbs the characteristics of glioblastoma stem‐like cells of rat RG2 glioblastoma. Cancer Cell Int. 2013;13(1):85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ben‐Baruch A. The multifaceted roles of chemokines in malignancy. Cancer Metastasis Rev. 2006;25(3):357‐371. [DOI] [PubMed] [Google Scholar]
- 109. Tokunaga R, Zhang W, Naseem M, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—a target for novel cancer therapy. Cancer Treat Rev. 2018;63:40‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Aldinucci D, Borghese C, Casagrande N. The CCL5/CCR5 axis in cancer progression. Cancers(Basel). 2020;12(7):1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Huang R, Wang S, Wang N, et al. CCL5 derived from tumor‐associated macrophages promotes prostate cancer stem cells and metastasis via activating beta‐catenin/STAT3 signaling. Cell Death Dis. 2020;11(4):234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Neo SY, Lundqvist A. The multifaceted roles of CXCL9 within the tumor microenvironment. Adv Exp Med Biol. 2020;1231:45‐51. [DOI] [PubMed] [Google Scholar]
- 113. Wu X, Schulte BC, Zhou Y, et al. Depletion of M2‐like tumor‐associated macrophages delays cutaneous T‐cell lymphoma development in vivo. J Invest Dermatol. 2014;134(11):2814‐2822. [DOI] [PubMed] [Google Scholar]
- 114. Furudate S, Fujimura T, Kakizaki A, Hidaka T, Asano M, Aiba S. Tumor‐associated M2 macrophages in mycosis fungoides acquire immunomodulatory function by interferon alpha and interferon gamma. J Dermatol Sci. 2016;83(3):182‐189. [DOI] [PubMed] [Google Scholar]
- 115. Kakizaki A, Fujimura T, Furudate S, et al. Immunomodulatory effect of peritumorally administered interferon‐beta on melanoma through tumor‐associated macrophages. Oncoimmunology. 2015;4(11):e1047584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Miller CH, Maher SG, Young HA. Clinical use of interferon‐gamma. Ann N Y Acad Sci. 2009;1182:69‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Schmidt T, Ben‐Batalla I, Schultze A, Loges S. Macrophage‐tumor crosstalk: role of TAMR tyrosine kinase receptors and of their ligands. Cell Mol Life Sci. 2012;69(9):1391‐1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lemieux SM, Hadden MK. Targeting the fibroblast growth factor receptors for the treatment of cancer. Anticancer Agents Med Chem. 2013;13(5):748‐761. [DOI] [PubMed] [Google Scholar]
- 119. Im D, Jung K, Yang S, Aman W, Hah JM. Discovery of 4‐arylamido 3‐methyl isoxazole derivatives as novel FMS kinase inhibitors. Eur J Med Chem. 2015;102:600‐610. [DOI] [PubMed] [Google Scholar]
- 120. Okugawa Y, Toiyama Y, Ichikawa T, et al. Colony‐stimulating factor‐1 and colony‐stimulating factor‐1 receptor co‐expression is associated with disease progression in gastric cancer. Int J Oncol. 2018;53(2):737‐749. [DOI] [PubMed] [Google Scholar]
- 121. Steins A, van Mackelenbergh MG, van der Zalm AP, et al. High‐grade mesenchymal pancreatic ductal adenocarcinoma drives stromal deactivation through CSF‐1. Embo Rep. 2020;21(5):e48780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Jang JY, Lee JK, Jeon YK, Kim CW. Exosome derived from epigallocatechin gallate treated breast cancer cells suppresses tumor growth by inhibiting tumor‐associated macrophage infiltration and M2 polarization. BMC Cancer. 2013;13:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Achkova D, Maher J. Role of the colony‐stimulating factor(CSF)/CSF‐1 receptor axis in cancer. Biochem Soc Trans. 2016;44(2):333‐341. [DOI] [PubMed] [Google Scholar]
- 124. Scholl SM, Pallud C, Beuvon F, et al. Anti‐colony‐stimulating factor‐1 antibody staining in primary breast adenocarcinomas correlates with marked inflammatory cell infiltrates and prognosis. J Natl Cancer Inst. 1994;86(2):120‐126. [DOI] [PubMed] [Google Scholar]
- 125. Wyckoff JB, Wang Y, Lin EY, et al. Direct visualization of macrophage‐assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67(6):2649‐2656. [DOI] [PubMed] [Google Scholar]
- 126. Wiehagen KR, Girgis NM, Yamada DH, et al. Combination of CD40 agonism and CSF‐1R blockade reconditions tumor‐associated macrophages and drives potent antitumor immunity. Cancer Immunol Res. 2017;5(12):1109‐1121. [DOI] [PubMed] [Google Scholar]
- 127. Zhong WQ, Chen G, Zhang W, et al. M2‐polarized macrophages in keratocystic odontogenic tumor: relation to tumor angiogenesis. Sci Rep. 2015;5:15586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Welm AL, Sneddon JB, Taylor C, et al. The macrophage‐stimulating protein pathway promotes metastasis in a mouse model for breast cancer and predicts poor prognosis in humans. Proc Natl Acad Sci U S A. 2007;104(18):7570‐7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Yuan ZL, Guan YJ, Wang L, Wei W, Kane AB, Chin YE. Central role of the threonine residue within the p+1 loop of receptor tyrosine kinase in STAT3 constitutive phosphorylation in metastatic cancer cells. Mol Cell Biol. 2004;24(21):9390‐9400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Liu QP, Fruit K, Ward J, Correll PH. Negative regulation of macrophage activation in response to IFN‐gamma and lipopolysaccharide by the STK/RON receptor tyrosine kinase. J Immunol. 1999;163(12):6606‐6613. [PubMed] [Google Scholar]
- 131. Eyob H, Ekiz HA, Welm AL. RON promotes the metastatic spread of breast carcinomas by subverting antitumor immune responses. Oncoimmunology. 2013;2(9):e25670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Davra V, Kumar S, Geng K, et al. Axl and Mertk receptors cooperate to promote breast cancer progression by combined oncogenic signaling and evasion of host anti‐tumor immunity. Cancer Res. 2020. 10.1158/0008-5472.can-20-2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Zizzo G, Hilliard BA, Monestier M, Cohen PL. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol. 2012;189(7):3508‐3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Caetano MS, Younes AI, Barsoumian HB, et al. Triple therapy with MerTK and pd1 inhibition plus radiotherapy promotes abscopal antitumor immune responses. Clin Cancer Res. 2019;25(24):7576‐7584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Zhou Y, Fei M, Zhang G, et al. Blockade of the phagocytic receptor MerTK on tumor‐associated macrophages enhances P2×7R‐dependent STING activation by tumor‐derived cGAMP. Immunity. 2020;52(2):357‐373 e359. [DOI] [PubMed] [Google Scholar]
- 136. Dehne N, Mora J, Namgaladze D, Weigert A, Brune B. Cancer cell and macrophage cross‐talk in the tumor microenvironment. Curr Opin Pharmacol. 2017;35:12‐19. [DOI] [PubMed] [Google Scholar]
- 137. Jang JY, Lee JK, Jeon YK, Kim CW. Exosome derived from epigallocatechin gallate treated breast cancer cells suppresses tumor growth by inhibiting tumor‐associated macrophage infiltration and M2 polarization. BMC Cancer. 2013;13(1):421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wang T, Liu H, Lian G, Zhang SY, Wang X, Jiang C. HIF1alpha‐induced glycolysis metabolism is essential to the activation of inflammatory macrophages. Mediators Inflamm. 2017;2017:9029327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Liu D, Chang C, Lu N, et al. Comprehensive proteomics analysis reveals metabolic reprogramming of tumor‐associated macrophages stimulated by the tumor microenvironment. J Proteome Res. 2017;16(1):288‐297. [DOI] [PubMed] [Google Scholar]
- 140. Arts RJ, Plantinga TS, Tuit S, et al. Transcriptional and metabolic reprogramming induce an inflammatory phenotype in non‐medullary thyroid carcinoma‐induced macrophages. Oncoimmunology. 2016;5(12):e1229725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Huber R, Meier B, Otsuka A, et al. Tumour hypoxia promotes melanoma growth and metastasis via high mobility group Box‐1 and M2‐like macrophages. Sci Rep. 2016;6:29914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Brand A, Singer K, Koehl GE, et al. LDHA‐associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016;24(5):657‐671. [DOI] [PubMed] [Google Scholar]
- 143. Lim SO, Li CW, Xia W, et al. EGFR Signaling enhances aerobic glycolysis in triple‐negative breast cancer cells to promote tumor growth and immune escape. Cancer Res. 2016;76(5):1284‐1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Kellum JA, Song M, Li J. Lactic and hydrochloric acids induce different patterns of inflammatory response in LPS‐stimulated RAW 264.7 cells. Am J Physiol Regul Integr Comp Physiol. 2004;286(4):R686‐692. [DOI] [PubMed] [Google Scholar]
- 145. Artyomov MN, Sergushichev A, Schilling JD. Integrating immunometabolism and macrophage diversity. Semin Immunol. 2016;28(5):417‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Currie E, Schulze A, Zechner R, Walther TC, Farese RV, Jr . Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18(2):153‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Huang SC, Everts B, Ivanova Y, et al. Cell‐intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15(9):846‐855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Castegna A, Gissi R, Menga A, et al. Pharmacological targets of metabolism in disease: opportunities from macrophages. Pharmacol Ther. 2020;210:107521. [DOI] [PubMed] [Google Scholar]
- 149. Li AC, Binder CJ, Gutierrez A, et al. Differential inhibition of macrophage foam‐cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J Clin Invest. 2004;114(11):1564‐1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Kang K, Reilly SM, Karabacak V, et al. Adipocyte‐derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008;7(6):485‐495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Schumann T, Adhikary T, Wortmann A, et al. Deregulation of PPARbeta/delta target genes in tumor‐associated macrophages by fatty acid ligands in the ovarian cancer microenvironment. Oncotarget. 2015;6(15):13416‐13433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Yakeu G, Butcher L, Isa S, et al. Low‐intensity exercise enhances expression of markers of alternative activation in circulating leukocytes: roles of PPARgamma and Th2 cytokines. Atherosclerosis. 2010;212(2):668‐673. [DOI] [PubMed] [Google Scholar]
- 153. Palmieri EM, Menga A, Martin‐Perez R, et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1‐like phenotype and inhibits tumor metastasis. Cell Rep. 2017;20(7):1654‐1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Kornyeyev DY. Inhibition of glutamine synthetase activity by phosphinothricin results in disappearance of the peak M2 of the chlorophyll fluorescence induction curve. Photosynthetica. 2000;36(4):601‐604. [Google Scholar]
- 155. Jha AK, Huang SC, Sergushichev A, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42(3):419‐430. [DOI] [PubMed] [Google Scholar]
- 156. Rath M, Muller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. 2014;5:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Curran JN, Winter DC, Bouchier‐Hayes D. Biological fate and clinical implications of arginine metabolism in tissue healing. Wound Repair Regen. 2006;14(4):376‐386. [DOI] [PubMed] [Google Scholar]
- 158. Zhao Q, Kuang DM, Wu Y, et al. Activated CD69+ T cells foster immune privilege by regulating IDO expression in tumor‐associated macrophages. J Immunol. 2012;188(3):1117‐1124. [DOI] [PubMed] [Google Scholar]
- 159. Song F, Hurtado del Pozo C, Rosario R, et al. RAGE regulates the metabolic and inflammatory response to high‐fat feeding in mice. Diabetes. 2014;63(6):1948‐1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Chen MC, Chen KC, Chang GC, et al. RAGE acts as an oncogenic role and promotes the metastasis of human lung cancer. Cell Death Dis. 2020;11(4):265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Wang S, Wang N, Zheng Y, et al. Caveolin‐1 inhibits breast cancer stem cells via c‐Myc‐mediated metabolic reprogramming. Cell Death Dis. 2020;11(6):450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Wang Q, Zhu G, Cao X, Dong J, Song F, Niu Y. Blocking AGE‐RAGE signaling improved functional disorders of macrophages in diabetic wound. J Diabetes Res. 2017;2017:1428537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Jin X, Yao T, Zhou Z, et al. Advanced glycation end products enhance macrophages polarization into M1 phenotype through activating RAGE/NF‐kappaB pathway. Biomed Res Int. 2015;2015:732450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Lan J, Sun L, Xu F, et al. M2 macrophage‐derived exosomes promote cell migration and invasion in colon cancer. Cancer Res. 2019;79(1):146‐158. [DOI] [PubMed] [Google Scholar]
- 165. Li X, Wang X. The emerging roles and therapeutic potential of exosomes in epithelial ovarian cancer. Mol Cancer. 2017;16(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Melo SA, Luecke LB, Kahlert C, et al. Glypican‐1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523(7559):177‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Melo SA, Sugimoto H, O'Connell JT, et al. Cancer exosomes perform cell‐independent microRNA biogenesis and promote tumorigenesis. Cancer Cell. 2014;26(5):707‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Caradec J, Kharmate G, Hosseini‐Beheshti E, Adomat H, Gleave M, Guns E. Reproducibility and efficiency of serum‐derived exosome extraction methods. Clin Biochem. 2014;47(13‐14):1286‐1292. [DOI] [PubMed] [Google Scholar]
- 169. Aslan C, Maralbashi S, Salari F, et al. Tumor‐derived exosomes: Implication in angiogenesis and antiangiogenesis cancer therapy. J Cell Physiol. 2019;234(10):16885‐16903. [DOI] [PubMed] [Google Scholar]
- 170. Kosaka N, Iguchi H, Hagiwara K, Yoshioka Y, Takeshita F, Ochiya T. Neutral sphingomyelinase 2(nSMase2)‐dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J Biol Chem. 2013;288(15):10849‐10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Challagundla KB, Wise PM, Paolo N, et al. Exosome‐mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J Natl Cancer Inst. 2015;107(7):djv135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Fabbr M. MicroRNAs bind to toll‐like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A. 2012;109(31):E2110‐E2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Saha S, Aranda E, Hayakawa Y, et al. Macrophage‐derived extracellular vesicle‐packaged WNTs rescue intestinal stem cells and enhance survival after radiation injury. Nat Commun. 2016;7:13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Monica F, Laura C, Gennaro C. The epithelial‐to‐mesenchymal transition in breast cancer: focus on basal‐like carcinomas. Cancers. 2017;9(10):134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Jinjie W, Haiyan L, Hongyu X, Xianrui W, Ping L. The malignant role of exosomes in the communication among colorectal cancer cell, macrophage and microbiome. Carcinogenesis. 2019;40(5):601‐610. [DOI] [PubMed] [Google Scholar]
- 176. Jieru, Zhou , Xiaoduan, Li , Xiaoli, Wu , et al. Exosomes released from tumor‐associated macrophages transfer miRNAs that induce a Treg/Th17 cell imbalance in epithelial ovarian cancer. Cancer Immunol Res. 2018;6(12):1578‐1592. [DOI] [PubMed] [Google Scholar]
- 177. Han R, Gu S, Zhang Y, et al. Estrogen promotes progression of hormone‐dependent breast cancer through CCL2‐CCR2 axis by upregulation of Twist via PI3K/AKT/NF‐κB signaling. Sci Rep. 2018;8(1):9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Li D, Ji H, Niu X, et al. Tumor‐associated macrophages secrete CC‐chemokine ligand 2 and induce tamoxifen resistance by activating PI3K/Akt/mTOR in breast cancer. Cancer Sci. 2020;111(1):47‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Yao M, Fang W, Smart C, et al. CCR2 chemokine receptors enhance growth and cell‐cycle progression of breast cancer cells through SRC and PKC activation. Mol Cancer Res. 2019;17(2):604‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Lee GT, Kwon SJ, Kim J, et al. WNT5A induces castration‐resistant prostate cancer via CCL2 and tumour‐infiltrating macrophages. Br J Cancer. 2018;118(5):670‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Roca H, Varsos Z, Pienta KJ. CCL2 protects prostate cancer PC3 cells from autophagic death via phosphatidylinositol 3‐kinase/AKT‐dependent survivin up‐regulation. J Biol Chem. 2008;283(36):25057‐25073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Ito Y, Ishiguro H, Kobayashi N, et al. Adipocyte‐derived monocyte chemotactic protein‐1(MCP‐1) promotes prostate cancer progression through the induction of MMP‐2 activity. Prostate. 2015;75(10):1009‐1019. [DOI] [PubMed] [Google Scholar]
- 183. Zhuang H, Cao G, Kou C, Liu T. CCL2/CCR2 axis induces hepatocellular carcinoma invasion and epithelial‐mesenchymal transition in vitro through activation of the Hedgehog pathway. Oncol Rep. 2018;39(1):21‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Dagouassat M, Suffee N, Hlawaty H, et al. Monocyte chemoattractant protein‐1(MCP‐1)/CCL2 secreted by hepatic myofibroblasts promotes migration and invasion of human hepatoma cells. Int J Cancer. 2010;126(5):1095‐1108. [DOI] [PubMed] [Google Scholar]
- 185. He M, Yu W, Chang C, et al. Estrogen receptor α promotes lung cancer cell invasion via increase of and cross‐talk with infiltrated macrophages through the CCL2/CCR2/MMP9 and CXCL12/CXCR4 signaling pathways. Mol Oncol. 2020;14(8):1779‐1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Zhou L, Jiang Y, Liu X, et al. Promotion of tumor‐associated macrophages infiltration by elevated neddylation pathway via NF‐κB‐CCL2 signaling in lung cancer. Oncogene. 2019;38(29):5792‐5804. [DOI] [PubMed] [Google Scholar]
- 187. Ding M, He SJ, Yang J. MCP‐1/CCL2 mediated by autocrine loop of PDGF‐BB promotes invasion of lung cancer cell by recruitment of macrophages via CCL2‐CCR2 axis. J Interferon Cytokine Res. 2019;39(4):224‐232. [DOI] [PubMed] [Google Scholar]
- 188. Mizutani K, Sud S, McGregor NA, et al. The chemokine CCL2 increases prostate tumor growth and bone metastasis through macrophage and osteoclast recruitment. Neoplasia. 2009;11(11):1235‐1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Xu W, Wei Q, Han M, et al. CCL2‐SQSTM1 positive feedback loop suppresses autophagy to promote chemoresistance in gastric cancer. Int J Biol Sci. 2018;14(9):1054‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Chen K, Liu Q, Tsang LL, et al. Human MSCs promotes colorectal cancer epithelial‐mesenchymal transition and progression via CCL5/β‐catenin/Slug pathway. Cell Death Dis. 2017;8(5):e2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Kato T, Fujita Y, Nakane K, et al. CCR1/CCL5 interaction promotes invasion of taxane‐resistant PC3 prostate cancer cells by increasing secretion of MMPs 2/9 and by activating ERK and Rac signaling. Cytokine. 2013;64(1):251‐257. [DOI] [PubMed] [Google Scholar]
- 192. Huang CY, Fong YC, Lee CY, et al. CCL5 increases lung cancer migration via PI3K, Akt and NF‐kappaB pathways. Biochem Pharmacol. 2009;77(5):794‐803. [DOI] [PubMed] [Google Scholar]
- 193. Xia L, Zhu X, Zhang L, Xu Y, Chen G, Luo J. EZH2 enhances expression of CCL5 to promote recruitment of macrophages and invasion in lung cancer. Biotechnol Appl Biochem. 2020; 10.1002/bab.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Walens A, DiMarco AV, Lupo R, Kroger BR, Damrauer JS, Alvarez JV. CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. Elife. 2019;8:e43653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Zazo S, González‐Alonso P, Martín‐Aparicio E, et al. Autocrine CCL5 effect mediates trastuzumab resistance by ERK pathway activation in HER2‐positive breast cancer. Mol Cancer Ther. 2020;19(8):1696‐1707. [DOI] [PubMed] [Google Scholar]
- 196. Murooka TT, Rahbar R, Fish EN. CCL5 promotes proliferation of MCF‐7 cells through mTOR‐dependent mRNA translation. Biochem Biophys Res Commun. 2009;387(2):381‐386. [DOI] [PubMed] [Google Scholar]
- 197. Long H, Xie R, Xiang T, et al. Autocrine CCL5 signaling promotes invasion and migration of CD133+ ovarian cancer stem‐like cells via NF‐κB‐mediated MMP‐9 upregulation. Stem Cells. 2012;30(10):2309‐2319. [DOI] [PubMed] [Google Scholar]
- 198. Tang S, Xiang T, Huang S, et al. Ovarian cancer stem‐like cells differentiate into endothelial cells and participate in tumor angiogenesis through autocrine CCL5 signaling. Cancer Lett. 2016;376(1):137‐147. [DOI] [PubMed] [Google Scholar]
- 199. Singh SK, Mishra MK, Rivers BM, Gordetsky JB, Bae S, Singh R. Biological and clinical significance of the CCR5/CCL5 axis in hepatocellular carcinoma. Cancers(Basel). 2020;12(4):883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Wang X, Lang M, Zhao T, et al. Cancer‐FOXP3 directly activated CCL5 to recruit FOXP3(+)Treg cells in pancreatic ductal adenocarcinoma. Oncogene. 2017;36(21):3048‐3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Singh SK, Mishra MK, Eltoum IA, Bae S, Lillard JW, Jr. , Singh R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci Rep. 2018;8(1):1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Liu C, Yao Z, Wang J, et al. Macrophage‐derived CCL5 facilitates immune escape of colorectal cancer cells via the p65/STAT3‐CSN5‐PD‐L1 pathway. Cell Death Differ. 2020;27(6):1765‐1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Wang HC, Chen CW, Yang CL, et al. Tumor‐associated macrophages promote epigenetic silencing of gelsolin through DNA methyltransferase 1 in gastric cancer cells. Cancer Immunol Res. 2017;5(10):885‐897. [DOI] [PubMed] [Google Scholar]
- 204. Yang T, Chen M, Yang X, et al. Down‐regulation of KLF5 in cancer‐associated fibroblasts inhibit gastric cancer cells progression by CCL5/CCR5 axis. Cancer Biol Ther. 2017;18(10):806‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Mellado M, de Ana AM, Moreno MC, Martínez C, Rodríguez‐Frade JM. A potential immune escape mechanism by melanoma cells through the activation of chemokine‐induced T cell death. Curr Biol. 2001;11(9):691‐696. [DOI] [PubMed] [Google Scholar]
- 206. Zhao C, Zheng S, Yan Z, Deng Z, Wang R, Zhang B. CCL18 promotes the invasion and metastasis of breast cancer through Annexin A2. Oncol Rep. 2020;43(2):571‐580. [DOI] [PubMed] [Google Scholar]
- 207. Chen J, Yao Y, Gong C, et al. CCL18 from tumor‐associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell. 2011;19(4):541‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Lin X, Chen L, Yao Y, et al. CCL18‐mediated down‐regulation of miR98 and miR27b promotes breast cancer metastasis. Oncotarget. 2015;6(24):20485‐20499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Jiang X, Huang Z, Sun X, et al. CCL18‐NIR1 promotes oral cancer cell growth and metastasis by activating the JAK2/STAT3 signaling pathway. BMC Cancer. 2020;20(1):632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Wang H, Liang X, Li M, et al. Chemokine(CC motif) ligand 18 upregulates Slug expression to promote stem‐cell like features by activating the mammalian target of rapamycin pathway in oral squamous cell carcinoma. Cancer Sci. 2017;108(8):1584‐1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Wang Q, Tang Y, Yu H, et al. CCL18 from tumor‐cells promotes epithelial ovarian cancer metastasis via mTOR signaling pathway. Mol Carcinog. 2016;55(11):1688‐1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Lane D, Matte I, Laplante C, et al. CCL18 from ascites promotes ovarian cancer cell migration through proline‐rich tyrosine kinase 2 signaling. Mol Cancer. 2016;15(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Shi L, Zhang B, Sun X, et al. CC chemokine ligand 18(CCL18) promotes migration and invasion of lung cancer cells by binding to Nir1 through Nir1‐ELMO1/DOC180 signaling pathway. Mol Carcinog. 2016;55(12):2051‐2062. [DOI] [PubMed] [Google Scholar]
- 214. Liu X, Xu X, Deng W, et al. CCL18 enhances migration, invasion and EMT by binding CCR8 in bladder cancer cells. Mol Med Rep. 2019;19(3):1678‐1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Ye H, Zhou Q, Zheng S, et al. Tumor‐associated macrophages promote progression and the Warburg effect via CCL18/NF‐kB/VCAM‐1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018;9(5):453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Hou X, Zhang Y, Qiao H. CCL18 promotes the invasion and migration of gastric cancer cells via ERK1/2/NF‐κB signaling pathway. Tumour Biol. 2016;37(1):641‐651. [DOI] [PubMed] [Google Scholar]
- 217. Wang B, Shi L, Sun X, Wang L, Wang X, Chen C. Production of CCL20 from lung cancer cells induces the cell migration and proliferation through PI3K pathway. J Cell Mol Med. 2016;20(5):920‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Wei W, Zhao X, Zhu J, et al. lncRNAu50535 promotes the progression of lung cancer by activating CCL20/ERK signaling. Oncol Rep. 2019;42(5):1946‐1956. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 219. Kadomoto S, Izumi K, Hiratsuka K, et al. Tumor‐associated macrophages induce migration of renal cell carcinoma cells via activation of the CCL20‐CCR6 axis. Cancers(Basel). 2019;12(1):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Yu X, Yuan Z, Yang Z, et al. The novel long noncoding RNA u50535 promotes colorectal cancer growth and metastasis by regulating CCL20. Cell Death Dis. 2018;9(7):751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Brand S, Olszak T, Beigel F, et al. Cell differentiation dependent expressed CCR6 mediates ERK‐1/2, SAPK/JNK, and Akt signaling resulting in proliferation and migration of colorectal cancer cells. J Cell Biochem. 2006;97(4):709‐723. [DOI] [PubMed] [Google Scholar]
- 222. Wang D, Yang L, Yu W, et al. Colorectal cancer cell‐derived CCL20 recruits regulatory T cells to promote chemoresistance via FOXO1/CEBPB/NF‐κB signaling. J Immunother Cancer. 2019;7(1):215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Geismann C, Grohmann F, Dreher A, et al. Role of CCL20 mediated immune cell recruitment in NF‐κB mediated TRAIL resistance of pancreatic cancer. Biochim Biophys Acta Mol Cell Res. 2017;1864(5):782‐796. [DOI] [PubMed] [Google Scholar]
- 224. Liu B, Jia Y, Ma J, et al. Tumor‐associated macrophage‐derived CCL20 enhances the growth and metastasis of pancreatic cancer. Acta Biochim Biophys Sin(Shanghai). 2016;48(12):1067‐1074. [DOI] [PubMed] [Google Scholar]
- 225. Chen W, Qin Y, Wang D, et al. CCL20 triggered by chemotherapy hinders the therapeutic efficacy of breast cancer. PLoS Biol. 2018;16(7):e2005869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Lee SK, Park KK, Kim HJ, et al. Human antigen R‐regulated CCL20 contributes to osteolytic breast cancer bone metastasis. Sci Rep. 2017;7(1):9610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Marsigliante S, Vetrugno C, Muscella A. Paracrine CCL20 loop induces epithelial‐mesenchymal transition in breast epithelial cells. Mol Carcinog. 2016;55(7):1175‐1186. [DOI] [PubMed] [Google Scholar]
- 228. Marsigliante S, Vetrugno C, Muscella A. CCL20 induces migration and proliferation on breast epithelial cells. J Cell Physiol. 2013;228(9):1873‐1883. [DOI] [PubMed] [Google Scholar]
- 229. Han G, Wu D, Yang Y, Li Z, Zhang J, Li C. CrkL meditates CCL20/CCR6‐induced EMT in gastric cancer. Cytokine. 2015;76(2):163‐169. [DOI] [PubMed] [Google Scholar]
- 230. Maolake A, Izumi K, Shigehara K, et al. Tumor‐associated macrophages promote prostate cancer migration through activation of the CCL22‐CCR4 axis. Oncotarget. 2017;8(6):9739‐9751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Wei C, Yang C, Wang S, et al. M2 macrophages confer resistance to 5‐fluorouracil in colorectal cancer through the activation of CCL22/PI3K/AKT signaling. Onco Targets Ther. 2019;12:3051‐3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Li ZQ, Wang HY, Zeng QL, et al. p65/miR‐23a/CCL22 axis regulated regulatory T cells recruitment in hepatitis B virus positive hepatocellular carcinoma. Cancer Med. 2020;9(2):711‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Wang N, Liu W, Zheng Y, et al. CXCL1 derived from tumor‐associated macrophages promotes breast cancer metastasis via activating NF‐κB/SOX4 signaling. Cell Death Dis. 2018;9(9):880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Cabrero‐de Las Heras S, Martínez‐Balibrea E. CXC family of chemokines as prognostic or predictive biomarkers and possible drug targets in colorectal cancer. World J Gastroenterol. 2018;24(42):4738‐4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Yang C, Yu H, Chen R, et al. CXCL1 stimulates migration and invasion in ERnegative breast cancer cells via activation of the ERK/MMP2/9 signaling axis. Int J Oncol. 2019;55(3):684‐696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Wen Z, Liu Q, Wu J, et al. Fibroblast activation protein α‐positive pancreatic stellate cells promote the migration and invasion of pancreatic cancer by CXCL1‐mediated Akt phosphorylation. Ann Transl Med. 2019;7(20):532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Taki M, Abiko K, Baba T, et al. Snail promotes ovarian cancer progression by recruiting myeloid‐derived suppressor cells via CXCR2 ligand upregulation. Nat Commun. 2018;9(1):1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Yung MM, Tang HW, Cai PC, et al. GRO‐α and IL‐8 enhance ovarian cancer metastatic potential via the CXCR2‐mediated TAK1/NFκB signaling cascade. Theranostics. 2018;8(5):1270‐1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Dong YL, Kabir SM, Lee ES, Son DS. CXCR2‐driven ovarian cancer progression involves upregulation of proinflammatory chemokines by potentiating NF‐κB activation via EGFR‐transactivated Akt signaling. PLoS One. 2013;8(12):e83789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Lu Y, Dong B, Xu F, et al. CXCL1‐LCN2 paracrine axis promotes progression of prostate cancer via the Src activation and epithelial‐mesenchymal transition. Cell Commun Signal. 2019;17(1):118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Kuo PL, Shen KH, Hung SH, Hsu YL. CXCL1/GROα increases cell migration and invasion of prostate cancer by decreasing fibulin‐1 expression through NF‐κB/HDAC1 epigenetic regulation. Carcinogenesis. 2012;33(12):2477‐2487. [DOI] [PubMed] [Google Scholar]
- 242. Zhou Z, Xia G, Xiang Z, et al. A C‐X‐C Chemokine receptor type 2‐dominated cross‐talk between tumor cells and macrophages drives gastric cancer metastasis. Clin Cancer Res. 2019;25(11):3317‐3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Wang Z, Wang Z, Li G, et al. CXCL1 from tumor‐associated lymphatic endothelial cells drives gastric cancer cell into lymphatic system via activating integrin β1/FAK/AKT signaling. Cancer Lett. 2017;385:28‐38. [DOI] [PubMed] [Google Scholar]
- 244. Wei ZW, Xia GK, Wu Y, et al. CXCL1 promotes tumor growth through VEGF pathway activation and is associated with inferior survival in gastric cancer. Cancer Lett. 2015;359(2):335‐343. [DOI] [PubMed] [Google Scholar]
- 245. Ma J, Su H, Yu B, et al. CXCL12 gene silencing down‐regulates metastatic potential via blockage of MAPK/PI3K/AP‐1 signaling pathway in colon cancer. Clin Transl Oncol. 2018;20(8):1035‐1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Ma JC, Sun XW, Su H, et al. Fibroblast‐derived CXCL12/SDF‐1α promotes CXCL6 secretion and co‐operatively enhances metastatic potential through the PI3K/Akt/mTOR pathway in colon cancer. World J Gastroenterol. 2017;23(28):5167‐5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Yu X, Wang D, Wang X, et al. CXCL12/CXCR4 promotes inflammation‐driven colorectal cancer progression through activation of RhoA signaling by sponging miR‐133a‐3p. J Exp Clin Cancer Res. 2019;38(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Song ZY, Gao ZH, Chu JH, Han XZ, Qu XJ. Downregulation of the CXCR4/CXCL12 axis blocks the activation of the Wnt/β‐catenin pathway in human colon cancer cells. Biomed Pharmacother. 2015;71:46‐52. [DOI] [PubMed] [Google Scholar]
- 249. Wang D, Jiao C, Zhu Y, et al. Activation of CXCL12/CXCR4 renders colorectal cancer cells less sensitive to radiotherapy via up‐regulating the expression of survivin. Exp Biol Med(Maywood). 2017;242(4):429‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Martinez‐Ordoñez A, Seoane S, Cabezas P, et al. Breast cancer metastasis to liver and lung is facilitated by Pit‐1‐CXCL12‐CXCR4 axis. Oncogene. 2018;37(11):1430‐1444. [DOI] [PubMed] [Google Scholar]
- 251. Sobolik T, Su YJ, Wells S, Ayers GD, Cook RS, Richmond A. CXCR4 drives the metastatic phenotype in breast cancer through induction of CXCR2 and activation of MEK and PI3K pathways. Mol Biol Cell. 2014;25(5):566‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Luo N, Chen DD, Liu L, Li L, Cheng ZP. CXCL12 promotes human ovarian cancer cell invasion through suppressing ARHGAP10 expression. Biochem Biophys Res Commun. 2019;518(3):416‐422. [DOI] [PubMed] [Google Scholar]
- 253. Chiaramonte R, Colombo M, Bulfamante G, et al. Notch pathway promotes ovarian cancer growth and migration via CXCR4/SDF1α chemokine system. Int J Biochem Cell Biol. 2015;66:134‐140. [DOI] [PubMed] [Google Scholar]
- 254. Wang M, Lin T, Wang Y, et al. CXCL12 suppresses cisplatin‐induced apoptosis through activation of JAK2/STAT3 signaling in human non‐small‐cell lung cancer cells. Onco Targets Ther. 2017;10:3215‐3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Rodríguez‐Nieves JA, Patalano SC, Almanza D, Gharaee‐Kermani M, Macoska JA. CXCL12/CXCR4 axis activation mediates prostate myofibroblast phenoconversion through non‐canonical EGFR/MEK/ERK signaling. PLoS One. 2016;11(7):e0159490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Uygur B, Wu WS. SLUG promotes prostate cancer cell migration and invasion via CXCR4/CXCL12 axis. Mol Cancer. 2011;10:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Cheng Y, Qu J, Che X, et al. CXCL12/SDF‐1α induces migration via SRC‐mediated CXCR4‐EGFR cross‐talk in gastric cancer cells. Oncol Lett. 2017;14(2):2103‐2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Izumi D, Ishimoto T, Miyake K, et al. CXCL12/CXCR4 activation by cancer‐associated fibroblasts promotes integrin β1 clustering and invasiveness in gastric cancer. Int J Cancer. 2016;138(5):1207‐1219. [DOI] [PubMed] [Google Scholar]
- 259. Chen G, Chen SM, Wang X, Ding XF, Ding J, Meng LH. Inhibition of chemokine(CXC motif) ligand 12/chemokine(CXC motif) receptor 4 axis(CXCL12/CXCR4)‐mediated cell migration by targeting mammalian target of rapamycin(mTOR) pathway in human gastric carcinoma cells. J Biol Chem. 2012;287(15):12132‐12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Chen F, Yang D, Ru Y, Cao S, Gao A. MicroRNA‐101 Targets CXCL12‐mediated akt and snail signaling pathways to inhibit cellular proliferation and invasion in papillary thyroid carcinoma. Oncol Res. 2019;27(6):691‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Lin C, He H, Liu H, et al. Tumour‐associated macrophages‐derived CXCL8 determines immune evasion through autonomous PD‐L1 expression in gastric cancer. Gut. 2019;68(10):1764‐1773. [DOI] [PubMed] [Google Scholar]
- 262. Luppi F, Longo AM, de Boer WI, Rabe KF, Hiemstra PS. Interleukin‐8 stimulates cell proliferation in non‐small cell lung cancer through epidermal growth factor receptor transactivation. Lung Cancer. 2007;56(1):25‐33. [DOI] [PubMed] [Google Scholar]
- 263. Singh JK, Simões BM, Clarke RB, Bundred NJ. Targeting IL‐8 signalling to inhibit breast cancer stem cell activity. Expert Opin Ther Targets. 2013;17(11):1235‐1241. [DOI] [PubMed] [Google Scholar]
- 264. Luca M, Huang S, Gershenwald JE, Singh RK, Reich R, Bar‐Eli M. Expression of interleukin‐8 by human melanoma cells up‐regulates MMP‐2 activity and increases tumor growth and metastasis. Am J Pathol. 1997;151(4):1105‐1113. [PMC free article] [PubMed] [Google Scholar]
- 265. Zheng T, Ma G, Tang M, Li Z, Xu R. IL‐8 Secreted from M2 macrophages promoted prostate tumorigenesis via STAT3/MALAT1 pathway. Int J Mol Sci. 2018;20(1):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Xiao YC, Yang ZB, Cheng XS, et al. CXCL8, overexpressed in colorectal cancer, enhances the resistance of colorectal cancer cells to anoikis. Cancer Lett. 2015;361(1):22‐32. [DOI] [PubMed] [Google Scholar]
- 267. Sun Q, Sun F, Wang B, et al. Interleukin‐8 promotes cell migration through integrin αvβ6 upregulation in colorectal cancer. Cancer Lett. 2014;354(2):245‐253. [DOI] [PubMed] [Google Scholar]
- 268. Itoh Y, Joh T, Tanida S, et al. IL‐8 promotes cell proliferation and migration through metalloproteinase‐cleavage proHB‐EGF in human colon carcinoma cells. Cytokine. 2005;29(6):275‐282. [DOI] [PubMed] [Google Scholar]
- 269. Tardáguila M, Mira E, García‐Cabezas MA, et al. CX3CL1 promotes breast cancer via transactivation of the EGF pathway. Cancer Res. 2013;73(14):4461‐4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Liang Y, Yi L, Liu P, et al. CX3CL1 involves in breast cancer metastasizing to the spine via the Src/FAK signaling pathway. J Cancer. 2018;9(19):3603‐3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Liu W, Liang Y, Chan Q, Jiang L, Dong J. CX3CL1 promotes lung cancer cell migration and invasion via the Src/focal adhesion kinase signaling pathway. Oncol Rep. 2019;41(3):1911‐1917. [DOI] [PubMed] [Google Scholar]
- 272. Su YC, Chang H, Sun SJ, et al. Differential impact of CX3CL1 on lung cancer prognosis in smokers and non‐smokers. Mol Carcinog. 2018;57(5):629‐639. [DOI] [PubMed] [Google Scholar]
- 273. Singh SK, Mishra MK, Singh R. Hypoxia‐inducible factor‐1α induces CX3CR1 expression and promotes the epithelial to mesenchymal transition(EMT) in ovarian cancer cells. J Ovarian Res. 2019;12(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Tang J, Xiao L, Cui R, et al. CX3CL1 increases invasiveness and metastasis by promoting epithelial‐to‐mesenchymal transition through the TACE/TGF‐α/EGFR pathway in hypoxic androgen‐independent prostate cancer cells. Oncol Rep. 2016;35(2):1153‐1162. [DOI] [PubMed] [Google Scholar]
- 275. Liu P, Liang Y, Jiang L, Wang H, Wang S, Dong J. CX3CL1/fractalkine enhances prostate cancer spinal metastasis by activating the Src/FAK pathway. Int J Oncol. 2018;53(4):1544‐1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Ren H, Zhao T, Sun J, et al. The CX3CL1/CX3CR1 reprograms glucose metabolism through HIF‐1 pathway in pancreatic adenocarcinoma. J Cell Biochem. 2013;114(11):2603‐2611. [DOI] [PubMed] [Google Scholar]
- 277. Geismann C, Erhart W, Grohmann F, et al. TRAIL/NF‐κB/CX3CL1 mediated onco‐immuno crosstalk leading to TRAIL resistance of pancreatic cancer cell lines. Int J Mol Sci. 2018;19(6):1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Stout MC, Narayan S, Pillet ES, Salvino JM, Campbell PM. Inhibition of CX(3)CR1 reduces cell motility and viability in pancreatic adenocarcinoma epithelial cells. Biochem Biophys Res Commun. 2018;495(3):2264‐2269. [DOI] [PubMed] [Google Scholar]
- 279. Huang LY, Chen P, Xu LX, Zhou YF, Zhang YP, Yuan YZ. Fractalkine upregulates inflammation through CX3CR1 and the Jak‐Stat pathway in severe acute pancreatitis rat model. Inflammation. 2012;35(3):1023‐1030. [DOI] [PubMed] [Google Scholar]
- 280. Quail DF, Bowman RL, Akkari L, et al. The tumor microenvironment underlies acquired resistance to CSF‐1R inhibition in gliomas. Science. 2016;352(6288):aad3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Sossey‐Alaoui K, Pluskota E, Bialkowska K, et al. Kindlin‐2 regulates the growth of breast cancer tumors by activating CSF‐1‐mediated macrophage infiltration. Cancer Res. 2017;77(18):5129‐5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Lu X, Meng T. Depletion of tumor‐associated macrophages enhances the anti‐tumor effect of docetaxel in a murine epithelial ovarian cancer. Immunobiology. 2019;224(3):355‐361. [DOI] [PubMed] [Google Scholar]
- 283. Li M, Li M, Yang Y, et al. Remodeling tumor immune microenvironment via targeted blockade of PI3K‐γ and CSF‐1/CSF‐1R pathways in tumor associated macrophages for pancreatic cancer therapy. J Control Release. 2020;321:23‐35. [DOI] [PubMed] [Google Scholar]
- 284. Gurusamy D, Gray JK, Pathrose P, Kulkarni RM, Finkleman FD, Waltz SE. Myeloid‐specific expression of Ron receptor kinase promotes prostate tumor growth. Cancer Res. 2013;73(6):1752‐1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Babicky ML, Harper MM, Chakedis J, et al. MST1R kinase accelerates pancreatic cancer progression via effects on both epithelial cells and macrophages. Oncogene. 2019;38(28):5599‐5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Penny HL, Sieow JL, Adriani G, et al. Warburg metabolism in tumor‐conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology. 2016;5(8):e1191731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Chen P, Zuo H, Xiong H, et al. Gpr132 sensing of lactate mediates tumor‐macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci U S A. 2017;114(3):580‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Mantovani A, Locati M. Macrophage metabolism shapes angiogenesis in tumors. Cell Metab. 2016;24(6):887‐888. [DOI] [PubMed] [Google Scholar]
- 289. Bohn T, Rapp S, Luther N, et al. Tumor immunoevasion via acidosis‐dependent induction of regulatory tumor‐associated macrophages. Nat Immunol. 2018;19(12):1319‐1329. [DOI] [PubMed] [Google Scholar]
- 290. Zheng X, Mansouri S, Krager A, et al. Metabolism in tumour‐associated macrophages: a quid pro quo with the tumour microenvironment. Eur Respir Rev. 2020;29(157):200134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Colegio OR, Chu NQ, Szabo AL, et al. Functional polarization of tumour‐associated macrophages by tumour‐derived lactic acid. Nature. 2014;513(7519):559‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD‐L1 expression in tumor‐associated macrophages and myeloid‐derived suppressor cells. Proc Natl Acad Sci U S A. 2017;114(5):1117‐1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Zhang Q, Wang H, Mao C, et al. Fatty acid oxidation contributes to IL‐1β secretion in M2 macrophages and promotes macrophage‐mediated tumor cell migration. Mol Immunol. 2018;94:27‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Wu L, Zhang X, Zheng L, et al. RIPK3 Orchestrates fatty acid metabolism in tumor‐associated macrophages and hepatocarcinogenesis. Cancer Immunol Res. 2020;8(5):710‐721. [DOI] [PubMed] [Google Scholar]
- 295. Hao J, Yan F, Zhang Y, et al. Expression of adipocyte/macrophage fatty acid‐binding protein in tumor‐associated macrophages promotes breast cancer progression. Cancer Res. 2018;78(9):2343‐2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Niu Z, Shi Q, Zhang W, et al. Caspase‐1 cleaves PPARγ for potentiating the pro‐tumor action of TAMs. Nat Commun. 2017;8(1):766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Deng X, Zhang P, Liang T, Deng S, Chen X, Zhu L. Ovarian cancer stem cells induce the M2 polarization of macrophages through the PPARγ and NF‐κB pathways. Int J Mol Med. 2015;36(2):449‐454. [DOI] [PubMed] [Google Scholar]
- 298. Palmieri EM, Menga A, Martín‐Pérez R, et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1‐like phenotype and inhibits tumor metastasis. Cell Rep. 2017;20(7):1654‐1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Santhanam S, Alvarado DM, Ciorba MA. Therapeutic targeting of inflammation and tryptophan metabolism in colon and gastrointestinal cancer. Transl Res. 2016;167(1):67‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Mazzone M, Menga A, Castegna A. Metabolism and TAM functions‐it takes two to tango. FEBS J. 2018;285(4):700‐716. [DOI] [PubMed] [Google Scholar]
- 301. Massi D, Marconi C, Franchi A, et al. Arginine metabolism in tumor‐associated macrophages in cutaneous malignant melanoma: evidence from human and experimental tumors. Hum Pathol. 2007;38(10):1516‐1525. [DOI] [PubMed] [Google Scholar]
- 302. Chang CI, Liao JC, Kuo L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide‐mediated tumor cytotoxicity. Cancer Res. 2001;61(3):1100‐1106. [PubMed] [Google Scholar]
- 303. Dong D, Zhang G, Yang J, et al. The role of iron metabolism in cancer therapy focusing on tumor‐associated macrophages. J Cell Physiol. 2019;234(6):8028‐8039. [DOI] [PubMed] [Google Scholar]
- 304. Jung M, Mertens C, Brüne B. Macrophage iron homeostasis and polarization in the context of cancer. Immunobiology. 2015;220(2):295‐304. [DOI] [PubMed] [Google Scholar]
- 305. Nasser MW, Wani NA, Ahirwar DK, et al. RAGE mediates S100A7‐induced breast cancer growth and metastasis by modulating the tumor microenvironment. Cancer Res. 2015;75(6):974‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Chen X, Zhang L, Zhang IY, et al. RAGE expression in tumor‐associated macrophages promotes angiogenesis in glioma. Cancer Res. 2014;74(24):7285‐7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Haase‐Kohn C, Wolf S, Herwig N, Mosch B, Pietzsch J. Metastatic potential of B16‐F10 melanoma cells is enhanced by extracellular S100A4 derived from RAW264.7 macrophages. Biochem Biophys Res Commun. 2014;446(1):143‐148. [DOI] [PubMed] [Google Scholar]
- 308. Wang D, Wang X, Si M, et al. Exosome‐encapsulated miRNAs contribute to CXCL12/CXCR4‐induced liver metastasis of colorectal cancer by enhancing M2 polarization of macrophages. Cancer Lett. 2020;474:36‐52. [DOI] [PubMed] [Google Scholar]
- 309. Hu YB, Yan C, Mu L, et al. Exosomal Wnt‐induced dedifferentiation of colorectal cancer cells contributes to chemotherapy resistance. Oncogene. 2019;38(11):1951‐1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Gerloff D, Lützkendorf J, Moritz RKC, et al. Melanoma‐derived exosomal miR‐125b‐5p educates tumor associated macrophages (TAMs) by targeting lysosomal acid Lipase A(LIPA). Cancers(Basel). 2020;12(2):464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Wang X, Luo G, Zhang K, et al. Hypoxic tumor‐derived exosomal miR‐301a mediates M2 macrophage polarization via PTEN/PI3Kγ to promote pancreatic cancer metastasis. Cancer Res. 2018;78(16):4586‐4598. [DOI] [PubMed] [Google Scholar]
- 312. Yin Z, Ma T, Huang B, et al. Macrophage‐derived exosomal microRNA‐501‐3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3‐mediated TGF‐β signaling pathway. J Exp Clin Cancer Res. 2019;38(1):310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Zhu X, Shen H, Yin X, et al. Macrophages derived exosomes deliver miR‐223 to epithelial ovarian cancer cells to elicit a chemoresistant phenotype. J Exp Clin Cancer Res. 2019;38(1): 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Guan H, Peng R, Fang F, et al. Tumor‐associated macrophages promote prostate cancer progression via exosome‐mediated miR‐95 transfer. J Cell Physiol. 2020;235(12):9729‐9742. [DOI] [PubMed] [Google Scholar]
- 315. Lu X, Kang Y. Chemokine(C‐C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J Biol Chem. 2009;284(42):29087‐29096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Loberg RD, Ying C, Craig M, et al. CCL2, Prostate cancer, docetaxel, CNTO888. Cancer Res. 2007;67(19):9417‐9424. [DOI] [PubMed] [Google Scholar]
- 317. Sanford DE, Belt BA, Panni RZ, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013;19(13):3404‐3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Farina S, Yang H, Tu G, et al. Targeting tumor associated myeloid cells with CCR2 inhibitor PF‐04136309 enhances gemcitabine/paclitaxel and doxorubicin anti‐tumor activity. Cancer Res. 2017;77:LB‐194. [Google Scholar]
- 319. Nywening TM, Wang‐Gillam A, Sanford DE, et al. Targeting tumour‐associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single‐centre, open‐label, dose‐finding, non‐randomised, phase 1b trial. Lancet Oncol. 2016;17(5):651‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Duliege A‐M, Sleijfer S, Bischof A, et al. CCX872: Pharmacodynamic study of a potent and selective CCR2 antagonist in human volunteers and plans for phase Ib trial in patients with pancreatic cancer. Cancer Res. 2015;75:CT223‐CT223. [Google Scholar]
- 321. Yao W, Ba Q, Li X, et al. A Natural CCR2 Antagonist relieves tumor‐associated macrophage‐mediated immunosuppression to produce a therapeutic effect for liver cancer. EBioMedicine. 2017;22:58‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Li X, Yao W, Yuan Y, et al. Targeting of tumour‐infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66(1):157‐167. [DOI] [PubMed] [Google Scholar]
- 323. Pienta KJ, Machiels JP, Schrijvers D, et al. Phase 2 study of carlumab(CNTO 888), a human monoclonal antibody against CC‐chemokine ligand 2(CCL2), in metastatic castration‐resistant prostate cancer. Invest New Drugs. 2013;31(3):760‐768. [DOI] [PubMed] [Google Scholar]
- 324. Cambien B, Richard‐Fiardo P, Karimdjee BF, et al. CCL5 neutralization restricts cancer growth and potentiates the targeting of PDGFRbeta in colorectal carcinoma. PLoS One. 2011;6(12):e28842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Pervaiz A, Zepp M, Mahmood S, Ali DM, Berger MR, Adwan H. CCR5 blockage by maraviroc: a potential therapeutic option for metastatic breast cancer. Cell Oncol(Dordr). 2019;42(1):93‐106. [DOI] [PubMed] [Google Scholar]
- 326. Mencarelli A, Graziosi L, Renga B, et al. CCR5 antagonism by maraviroc reduces the potential for gastric cancer cell dissemination. Transl Oncol. 2013;6(6):784‐793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Zhang X, Haney KM, Richardson AC, et al. Anibamine, a natural product CCR5 antagonist, as a novel lead for the development of anti‐prostate cancer agents. Bioorg Med Chem Lett. 2010;20(15):4627‐4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Yost R, Pasquale TR, Sahloff EG. Maraviroc: a coreceptor CCR5 antagonist for management of HIV infection. Am J Health Syst Pharm. 2009;66(8):715‐726. [DOI] [PubMed] [Google Scholar]
- 329. Pervaiz A, Ansari S, Berger MR, Adwan H. CCR5 blockage by maraviroc induces cytotoxic and apoptotic effects in colorectal cancer cells. Med Oncol. 2015;32(5):158. [DOI] [PubMed] [Google Scholar]
- 330. Huang H, Zepp M, Georges RB, et al. The CCR5 antagonist maraviroc causes remission of pancreatic cancer liver metastasis in nude rats based on cell cycle inhibition and apoptosis induction. Cancer Lett. 2020;474:82‐93. [DOI] [PubMed] [Google Scholar]
- 331. Aquaro S, Hatse S, Princen K, et al. Potent anti‐HIV‐1 activity of TAK‐779 in human primary macrophages. Antivir Res. 2002;53:A45‐A45. [Google Scholar]
- 332. Menu E, De Leenheer E, De Raeve H, et al. Role of CCR1 and CCR5 in homing and growth of multiple myeloma and in the development of osteolytic lesions: a study in the 5TMM model. Clin Exp Metastasis. 2006;23(5‐6):291‐300. [DOI] [PubMed] [Google Scholar]
- 333. Ni J, Zhu YN, Zhong XG, et al. The chemokine receptor antagonist, TAK‐779, decreased experimental autoimmune encephalomyelitis by reducing inflammatory cell migration into the central nervous system, without affecting T cell function. Br J Pharmacol. 2009;158(8):2046‐2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Tan MC, Goedegebuure PS, Belt BA, et al. Disruption of CCR5‐dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182(3):1746‐1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Zhang F, Arnatt CK, Haney KM, et al. Structure activity relationship studies of natural product chemokine receptor CCR5 antagonist anibamine toward the development of novel anti prostate cancer agents. Eur J Med Chem. 2012;55:395‐408. [DOI] [PubMed] [Google Scholar]
- 336. Li G, Haney KM, Kellogg GE, Zhang Y. Comparative docking study of anibamine as the first natural product CCR5 antagonist in CCR5 homology models. J Chem Inf Model. 2009;49(1):120‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Moreb JS. Plerixafor in non‐Hodgkin's lymphoma and multiple myeloma patients undergoing autologous stem cell transplantation. Oncology Rev. 2011;5(1):67‐73. [Google Scholar]
- 338. Szczucinski A, Losy J. Chemokines and chemokine receptors in multiple sclerosis. Potential targets for new therapies. Acta Neurol Scand. 2007;115(3):137‐146. [DOI] [PubMed] [Google Scholar]
- 339. Nervi B, Ramirez P, Rettig MP, et al. Chemosensitization of acute myeloid leukemia(AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113(24):6206‐6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP‐expressing carcinoma‐associated fibroblasts synergizes with anti‐PD‐L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212‐20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Ling X, Spaeth E, Chen Y, et al. The CXCR4 antagonist AMD3465 regulates oncogenic signaling and invasiveness in vitro and prevents breast cancer growth and metastasis in vivo. PLoS One. 2013;8(3):e58426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Peng SB, Zhang X, Paul D, et al. Identification of LY2510924, a novel cyclic peptide CXCR4 antagonist that exhibits antitumor activities in solid tumor and breast cancer metastatic models. Mol Cancer Ther. 2015;14(2):480‐490. [DOI] [PubMed] [Google Scholar]
- 343. Galsky MD, Vogelzang NJ, Conkling P, et al. A phase I trial of LY2510924, a CXCR4 peptide antagonist, in patients with advanced cancer. Clin Cancer Res. 2014;20(13):3581‐3588. [DOI] [PubMed] [Google Scholar]
- 344. Beider K, Ribakovsky E, Abraham M, et al. Targeting the CD20 and CXCR4 pathways in non‐hodgkin lymphoma with rituximab and high‐affinity CXCR4 antagonist BKT140. Clin Cancer Res. 2013;19(13):3495‐3507. [DOI] [PubMed] [Google Scholar]
- 345. Gravina GL, Mancini A, Marampon F, et al. The brain‐penetrating CXCR4 antagonist, PRX177561, increases the antitumor effects of bevacizumab and sunitinib in preclinical models of human glioblastoma. J Hematol Oncol. 2017;10(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Gagner JP, Sarfraz Y, Ortenzi V, et al. Multifaceted C‐X‐C chemokine receptor 4(CXCR4) inhibition interferes with anti‐vascular endothelial growth factor therapy‐induced glioma dissemination. Am J Pathol. 2017;187(9):2080‐2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Kim SY, Lee CH, Midura BV, et al. Inhibition of the CXCR4/CXCL12 chemokine pathway reduces the development of murine pulmonary metastases. Clin Exp Metastasis. 2008;25(3):201‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Porvasnik S, Sakamoto N, Kusmartsev S, et al. Effects of CXCR4 antagonist CTCE‐9908 on prostate tumor growth. Prostate. 2009;69(13):1460‐1469. [DOI] [PubMed] [Google Scholar]
- 349. Murakami T, Maki W, Cardones AR, et al. Expression of CXC chemokine receptor‐4 enhances the pulmonary metastatic potential of murine B16 melanoma cells. Cancer Res. 2002;62(24):7328‐7334. [PubMed] [Google Scholar]
- 350. Di Cesare S, Marshall JC, Fernandes BF, et al. In vitro characterization and inhibition of the CXCR4/CXCL12 chemokine axis in human uveal melanoma cell lines. Cancer Cell Int. 2007;7:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Liang Z, Wu T, Lou H, et al. Inhibition of breast cancer metastasis by selective synthetic polypeptide against CXCR4. Cancer Res. 2004;64(12):4302‐4308. [DOI] [PubMed] [Google Scholar]
- 352. Richert MM, Vaidya KS, Mills CN, et al. Inhibition of CXCR4 by CTCE‐9908 inhibits breast cancer metastasis to lung and bone. Oncol Rep. 2009;21(3):761‐767. [PubMed] [Google Scholar]
- 353. Wong D, Kandagatla P, Korz W, Chinni SR. Targeting CXCR4 with CTCE‐9908 inhibits prostate tumor metastasis. BMC Urol. 2014;14:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Schott AF, Goldstein LJ, Cristofanilli M, et al. Phase Ib pilot study to evaluate Reparixin in combination with weekly Paclitaxel in patients with HER‐2‐negative metastatic breast cancer. Clin Cancer Res. 2017;23(18):5358‐5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Kim YH, Bagot M, Pinter‐Brown L, et al. Mogamulizumab versus vorinostat in previously treated cutaneous T‐cell lymphoma(MAVORIC): an international, open‐label, randomised, controlled phase 3 trial. Lancet Oncol. 2018;19(9):1192‐1204. [DOI] [PubMed] [Google Scholar]
- 356. Ishida T, Utsunomiya A, Jo T, et al. Mogamulizumab for relapsed adult T‐cell leukemia‐lymphoma: updated follow‐up analysis of phase I and II studies. Cancer Sci. 2017;108(10):2022‐2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Yano H, Ishida T, Inagaki A, et al. Defucosylated anti CC chemokine receptor 4 monoclonal antibody combined with immunomodulatory cytokines: a novel immunotherapy for aggressive/refractory Mycosis fungoides and Sezary syndrome. Clin Cancer Res. 2007;13(21):6494‐6500. [DOI] [PubMed] [Google Scholar]
- 358. Schroeder MA, Rettig MP, Lopez S, et al. Mobilization of allogeneic peripheral blood stem cell donors with intravenous plerixafor mobilizes a unique graft. Blood. 2017;129(19):2680‐2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Uy GL, Rettig MP, Motabi IH, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119(17):3917‐3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Salgia R, Stille JR, Weaver RW, et al. A randomized phase II study of LY2510924 and carboplatin/etoposide versus carboplatin/etoposide in extensive‐disease small cell lung cancer. Lung Cancer. 2017;105:7‐13. [DOI] [PubMed] [Google Scholar]
- 361. Pernas S, Martin M, Kaufman PA, et al. Balixafortide plus eribulin in HER2‐negative metastatic breast cancer: a phase 1, single‐arm, dose‐escalation trial. Lancet Oncol. 2018;19(6):812‐824. [DOI] [PubMed] [Google Scholar]
- 362. Hélène Haegel CT, Hallet R, Geist M, et al. A unique anti‐CD115 monoclonal antibody which inhibits osteolysis and skews human monocyte differentiation from M2‐polarized macrophages toward dendritic cells. MAbs. 2013;5(5):736‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Mohammed E‐G, K A‐AS, M A‐KD, G HM. Recent advances of colony‐stimulating factor‐1 receptor(CSF‐1R) kinase and its inhibitors. J Med Chem. 2018;61(13):5450‐5466. [DOI] [PubMed] [Google Scholar]
- 364. Ries CH, Cannarile MA, Hoves S, et al. Targeting tumor‐associated macrophages with anti‐CSF‐1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25(6):846‐859. [DOI] [PubMed] [Google Scholar]
- 365. Cassier PA, Italiano A, Gomez‐Roca CA, et al. CSF1R inhibition with emactuzumab in locally advanced diffuse‐type tenosynovial giant cell tumours of the soft tissue: a dose‐escalation and dose‐expansion phase 1 study. Lancet Oncol. 2015;16(8):949‐956. [DOI] [PubMed] [Google Scholar]
- 366. Genovese MC, Hsia E, Belkowski SM, et al. Results from a phase IIA parallel group study of JNJ‐40346527, an oral CSF‐1R inhibitor, in patients with active rheumatoid arthritis despite disease‐modifying antirheumatic drug therapy. J Rheumatol. 2015;42(10):1752‐1760. [DOI] [PubMed] [Google Scholar]
- 367. von Tresckow B, Morschhauser F, Ribrag V, et al. An open‐label, multicenter, phase I/II study of JNJ‐40346527, a CSF‐1R inhibitor, in patients with relapsed or refractory Hodgkin lymphoma. Clin Cancer Res. 2015;21(8):1843‐1850. [DOI] [PubMed] [Google Scholar]
- 368. Conway JG, McDonald B, Parham J, et al. Inhibition of colony‐stimulating‐factor‐1 signaling in vivo with the orally bioavailable cFMS kinase inhibitor GW2580. Proc Natl Acad Sci U S A. 2005;102(44):16078‐16083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Priceman SJ, Sung JL, Shaposhnik Z, et al. Targeting distinct tumor‐infiltrating myeloid cells by inhibiting CSF‐1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. 2010;115(7):1461‐1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Mitchem JB, Brennan DJ, Knolhoff BL, et al. Targeting tumor‐infiltrating macrophages decreases tumor‐initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128‐1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Hatzimichael E, Georgiou G, Benetatos L, Briasoulis E. Gene mutations and molecularly targeted therapies in acute myeloid leukemia. Am J Blood Res. 2013;3(1):29‐51. [PMC free article] [PubMed] [Google Scholar]
- 372. Butowski N, Colman H, De Groot JF, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 2016;18(4):557‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Tap WD, Wainberg ZA, Anthony SP, et al. Structure‐guided blockade of CSF1R kinase in tenosynovial giant‐cell tumor. N Engl J Med. 2015;373(5):428‐437. [DOI] [PubMed] [Google Scholar]
- 374. Butowski N, Colman H, Groot J, et al. A phase 2 study of orally administered PLX3397 in patients with recurrent glioblastoma. ASCO Meeting Abstracts; 2014. [Google Scholar]
- 375. Schroeder GM, An Y, Cai ZW, et al. Discovery of N‐(4‐(2‐amino‐3‐chloropyridin‐4‐yloxy)‐3‐fluorophenyl)‐4‐ethoxy‐1‐(4‐fluorophenyl)‐2‐oxo‐1,2‐dihydropyridine‐3‐carboxamide(BMS‐777607), a selective and orally efficacious inhibitor of the Met kinase superfamily. J Med Chem. 2009;52(5):1251‐1254. [DOI] [PubMed] [Google Scholar]
- 376. Eyob H, Ekiz HA, Derose YS, Waltz SE, Williams MA, Welm AL. Inhibition of ron kinase blocks conversion of micrometastases to overt metastases by boosting antitumor immunity. Cancer Discov. 2013;3(7):751‐760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Bieniasz M, Radhakrishnan P, Faham N, De La O J‐P, Welm AL. Pre‐clinical efficacy of Ron kinase inhibitors alone and in combination with PI3K inhibitors for treatment of sfRon‐expressing breast cancer patient‐derived xenografts. Clin Cancer Res. 2015;21(24):5588‐5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Gomez‐Roca CA, Italiano A, Le Tourneau C, et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2‐like macrophages. Ann Oncol. 2019;30(8):1381‐1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Tap WD, Gelderblom H, Palmerini E, et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour(ENLIVEN): a randomised phase 3 trial. Lancet. 2019;394(10197):478‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Smith CC, Levis MJ, Frankfurt O, et al. A phase 1/2 study of the oral FLT3 inhibitor pexidartinib in relapsed/refractory FLT3‐ITD‐mutant acute myeloid leukemia. Blood Adv. 2020;4(8):1711‐1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Diamond JR, Eckhardt SG, Pitts TM, et al. A phase II clinical trial of the Aurora and angiogenic kinase inhibitor ENMD‐2076 for previously treated, advanced, or metastatic triple‐negative breast cancer. Breast Cancer Res. 2018;20(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Roohullah A, Cooper A, Lomax AJ, et al. A phase I trial to determine safety and pharmacokinetics of ASLAN002, an oral MET superfamily kinase inhibitor, in patients with advanced or metastatic solid cancers. Invest New Drugs. 2018;36(5):886‐894. [DOI] [PubMed] [Google Scholar]
- 383. Moro‐Sibilot D, Cozic N, Perol M, et al. Crizotinib in c‐MET‐ or ROS1‐positive NSCLC: results of the AcSe phase II trial. Ann Oncol. 2019;30(12):1985‐1991. [DOI] [PubMed] [Google Scholar]
- 384. Rayson D, Lupichuk S, Potvin K, et al. Canadian Cancer Trials Group IND197: a phase II study of foretinib in patients with estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2‐negative recurrent or metastatic breast cancer. Breast Cancer Res Treat. 2016;157(1):109‐116. [DOI] [PubMed] [Google Scholar]
- 385. Choueiri TK, Vaishampayan U, Rosenberg JE, et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013;31(2):181‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Shah MA, Wainberg ZA, Catenacci DV, et al. Phase II study evaluating 2 dosing schedules of oral foretinib(GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PLoS One. 2013;8(3):e54014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Singh RP, Patel B, Kallender H, Ottesen LH, Adams LM, Cox DS. Population pharmacokinetics modeling and analysis of foretinib in adult patients with advanced solid tumors. J Clin Pharmacol. 2015;55(10):1184‐1192. [DOI] [PubMed] [Google Scholar]
- 388. Roelofs AJ, Thompson K, Gordon S, Rogers MJ. Molecular mechanisms of action of bisphosphonates: current status. Clin Cancer Res. 2006;12(20 Pt 2):6222s‐6230s. [DOI] [PubMed] [Google Scholar]
- 389. Stresing V, Fournier PG, Bellahcène A, et al. Nitrogen‐containing bisphosphonates can inhibit angiogenesis in vivo without the involvement of farnesyl pyrophosphate synthase. Bone. 2011;48(2):259‐266. [DOI] [PubMed] [Google Scholar]
- 390. Junankar S, Shay G, Jurczyluk J, et al. Real‐time intravital imaging establishes tumor‐associated macrophages as the extraskeletal target of bisphosphonate action in cancer. Cancer Discov. 2015;5(1):35‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Pietschmann P, Stohlawetz P, Brosch S, Steiner G, Smolen JS, Peterlik M. The effect of alendronate on cytokine production, adhesion molecule expression, and transendothelial migration of human peripheral blood mononuclear cells. Calcif Tissue Int. 1998;63(4):325‐330. [DOI] [PubMed] [Google Scholar]
- 392. Thiébaud D, Sauty A, Burckhardt P, et al. AnIn vitroandin vivostudy of cytokines in the acute‐phase response associated with bisphosphonates. Calcified Tissue Int. 1997;61(5):386‐392. [DOI] [PubMed] [Google Scholar]
- 393. Zysk SP, Durr HR, Gebhard HH, et al. Effects of ibandronate on inflammation in mouse antigen‐induced arthritis. Inflamm Res. 2003;52(5):221‐226. [DOI] [PubMed] [Google Scholar]
- 394. Dicuonzo G, Vincenzi B, Santini D, Avvisati G, Tonini G. Fever after zoledronic acid administration is due to increase in TNF‐α and IL‐6. J Interferon Cytokine Res. 2003;23(11):649‐654. [DOI] [PubMed] [Google Scholar]
- 395. Pennanen N, Lapinjoki S, Urtti A, Mönkkönen J. Effect of liposomal and free bisphosphonates on the IL‐1β, IL‐6 and TNFα secretion from RAW 264 cells in vitro. Pharm Res. 1995;12(6):916‐922. [DOI] [PubMed] [Google Scholar]
- 396. Pallardy MJ, Turbica I, Biola‐Vidamment A. Why the immune system should be concerned by nanomaterials? Front Immunol. 2017;8:544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Rao L, Zhao SK, Wen C, et al. Activating macrophage‐mediated cancer immunotherapy by genetically edited nanoparticles. Adv Mater. 2020;32(47):e2004853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Elsabahy M, Wooley KL. Cytokines as biomarkers of nanoparticle immunotoxicity. Chem Soc Rev. 2013;42(12):5552‐5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Sander AA Kooijmans P, Vader SM, van Dommelen WW, van Solinge, Schiffelers RM. Exosome mimetics: a novel class of drug delivery systems. Int J Nanomedicine. 2012;7:1525‐1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400. Hood JL, Wickline SA. A systematic approach to exosome‐based translational nanomedicine. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2012;4(4):458‐467. [DOI] [PubMed] [Google Scholar]
- 401. Kooijmans SAA, Fliervoet LAL, van der Meel R. et al. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J Control Release. 2016;224:77‐85. [DOI] [PubMed] [Google Scholar]
- 402. Shin‐ichiro O, Masakatsu T, Katsuko S, et al. Systemically injected exosomes targeted to EGFR deliver antitumor MicroRNA to breast cancer cells. Mol Ther. 2013;21(1):185‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Gomari H, Forouzandeh Moghadam M, Soleimani M. Targeted cancer therapy using engineered exosome as a natural drug delivery vehicle. Onco Targets Ther. 2018;11:5753‐5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404. Wang X, Zhang H, Bai M, et al. Exosomes serve as nanoparticles to deliver anti‐miR‐214 to reverse chemoresistance to cisplatin in gastric cancer. Mol Ther. 2018;26(3):774‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. McKelvey KJ, Powell KL, Ashton AW, Morris JM, McCracken SA. Exosomes: mechanisms of uptake. J Circ Biomark. 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Zhuang X, Xiang X, Grizzle W, et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti‐inflammatory drugs from the nasal region to the brain. Mol Ther. 2011;19(10):1769‐1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Su MJ, Aldawsari H, Amiji M. Pancreatic cancer cell exosome‐mediated macrophage reprogramming and the role of microRNAs 155 and 125b2 transfection using nanoparticle delivery systems. Sci Rep. 2016;22(6):30110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Trivedi M, Talekar M, Shah P, Ouyang Q, Amiji M. Modification of tumor cell exosome content by transfection with wt‐p53 and microRNA‐125b expressing plasmid DNA and its effect on macrophage polarization. Oncogenesis. 2016;5(8):e250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Rayamajhi S, Nguyen TDT, Marasini R, Aryal S. Macrophage‐derived exosome‐mimetic hybrid vesicles for tumor targeted drug delivery. Acta Biomater. 2019;94:482‐494. [DOI] [PubMed] [Google Scholar]
- 410. Rodell CB, Arlauckas SP, Cuccarese MF, et al. TLR7/8‐agonist‐loaded nanoparticles promote the polarization of tumour‐associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. 2018;2:578‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Feng Y, Mu R, Wang Z, et al. A toll‐like receptor agonist mimicking microbial signal to generate tumor‐suppressive macrophages. Nat Commun. 2019;10(1):2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612‐1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Andrechak JC, Dooling LJ, Discher DE. The macrophage checkpoint CD47 : SIRPalpha for recognition of “self” cells: from clinical trials of blocking antibodies to mechanobiological fundamentals. Philos Trans R Soc Lond B Biol Sci. 2019;374(1779):20180217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Vaeteewoottacharn K, Kariya R, Pothipan P, et al. Attenuation of CD47‐SIRPalpha signal in cholangiocarcinoma potentiates tumor‐associated macrophage‐mediated phagocytosis and suppresses intrahepatic metastasis. Transl Oncol. 2019;12(2):217‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Hartley GP, Chow L, Ammons DT, Wheat WH, Dow SW. Programmed cell death ligand 1(PD‐L1) signaling regulates macrophage proliferation and activation. Cancer Immunol Res. 2018;6(10):1260‐1273. [DOI] [PubMed] [Google Scholar]
- 416. Molgora M, Esaulova E, Vermi W, et al. TREM2 modulation remodels the tumor myeloid landscape enhancing anti‐PD‐1 immunotherapy. Cell. 2020;182(4):886‐900 e817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417. Zhu S, Niu M, O'Mary H, Cui Z. Targeting of tumor‐associated macrophages made possible by PEG‐sheddable, mannose‐modified nanoparticles. Mol Pharm. 2013;10(9):3525–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Xia Y, Xu T, Zhao M, et al. Delivery of doxorubicin for human cervical carcinoma targeting therapy by folic acid‐modified selenium nanoparticles. Int J Mol Sci. 2018;19(11):3582‐3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Van DB, G J , Schlee M, Coch C, Hartmann G. SiRNA delivery with exosome nanoparticles. Nat Biotechnol. 2011;29(4):325‐326. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.