

ABSTRACT
Preeclampsia remains a challenge without an effective therapy. Evidence supports targetability of soluble fms‐like tyrosine kinase‐1 (sFlt‐1) and soluble endoglin (sEng), which are released excessively from the placenta under ischemic and hypoxic stresses. We compared four trophoblast cell lines, BeWo, Jar, Jeg‐3, and HTR‐8/SVneo, in order to identify a suitable model for drug screening. Cultured trophoblasts were exposed to 1% oxygen vs. normoxia for 24‐48 hr; human umbilical vein and aortic endothelial cells were included for comparison. Supernatant sFlt‐1 and sEng concentrations were measured by ELISA, and sFlt‐1 mRNA expression determined by RT‐PCR. Cellular responses to experimental therapeutics were explored. All four trophoblast lines secreted sEng, which did not increase by hypoxia. BeWo, Jar, and Jeg‐3 exhibited significantly enhanced expression of sFlt‐1 i13 and e15a mRNA in response to hypoxia; however, only BeWo released a detectable level of sFlt‐1 protein, which was doubled by hypoxia. In contrast, hypoxia decreased sFlt‐1 mRNA expression and protein release in HTR‐8/SVneo, similarly to endothelial cells. The cellular mechanism involved HIFα. BeWo responded to representative agents similarly to human primary placental tissues in the literature. These data support that the BeWo‐hypoxia model mimics a key pathogenic mechanism of preeclampsia and has potential value for translational drug discovery.
Keywords: disease model, hypoxia, preeclampsia, soluble fms‐like tyrosine kinase‐1, trophoblast
Abbreviations
- DMOG
dimethyloxalylglycine
- ELISA
enzyme‐linked immunosorbent assay
- HAoECs
human aortic endothelial cells
- HIFα
hypoxia inducible factor alpha
- HUVECs
human umbilical vein endothelial cells
- LPS
lipopolysaccharides
- PBS
phosphate‐buffered saline
- sEng
soluble endoglin
- sFlt‐1
soluble fms‐like tyrosine kinase‐1
- TGFβ1
transforming growth factor β1
- VEGF
vascular endothelial growth factor
1. INTRODUCTION
Preeclampsia remains a major complication of pregnancy that causes significant mortality and morbidity. 1 , 2 It affects 3%‐5% of pregnant women in general, but the risk is much higher in those with predisposing conditions such as diabetes. 3 It is characterized by de novo hypertension after 20 weeks of gestation in a previously normotensive woman, accompanied by proteinuria or end‐organ damage including thrombocytopenia, renal insufficiency, liver dysfunction, pulmonary edema, or cerebral or visual symptoms. 4 The etiology of preeclampsia is still unknown and there is no truly effective therapy. The current standard of care is resource‐intensive, relying heavily on risk assessment and monitoring. Available treatments, including antihypertensives and anticonvulsants, are “reactive” and do not address mechanisms. Delivery is the only known cure. Although low‐dose aspirin has some prophylactic value in high‐risk women, its overall efficacy is modest. 5 Therefore, there is an urgent need for new, targeted therapies that address pathogenesis.
Development of effective therapies relies on careful selection of “druggable” targets and adequate screening models that are of translational value. In the past, drug discovery for preeclampsia (and for maternity indications in general) was largely hindered in part by poor understanding of disease biology, in addition to safety concerns; however, recent scientific advances provide new opportunities. Preeclampsia is generally considered a two‐stage disease: (1) in early pregnancy, deficient remodeling of uterine spiral arteries results in narrowed vessels; and (2) later, as demands increase, the placenta suffers ischemia, hypoxia, and oxidative stress, and releases “toxic” circulating factors into the maternal circulation, causing vascular and glomerular endothelial injury (endotheliosis) leading to clinical preeclampsia. 6 Seminal work in the last 15 years has led to the identification of at least two such circulating factors, including soluble fms‐like tyrosine kinase‐1 (sFlt‐1) and soluble endoglin (sEng): these are the extracellular fragments of vascular endothelial growth factor (VEGF) receptor 1 and endoglin (co‐receptor for transforming growth factor β1; TGFβ1), respectively. 7 , 8 , 9 Specifically, sFlt‐1 is produced by alternative splicing and sEng is thought to be an enzymatic cleavage product. Elevated in blood circulation, they scavenge VEGF, placental growth factor, and TGFβ1, causing a deficiency of angiogenic factors that in turn leads to generalized endothelial dysfunction and vasospasm characteristic of preeclampsia, and eventually, eclampsia.
Numerous clinical studies have established sFlt‐1 and sEng as preeclampsia biomarkers; they are elevated weeks before disease onset. 3 , 7 , 8 , 9 Ample evidence also supports an important mechanistic role especially for sFlt‐1 in preeclampsia development. Karumanchi et al 10 showed that higher plasma sFlt‐1 and sEng levels were associated with more severe hypertension, kidney injury, and adverse outcomes in preeclampsia patients. Experimentally induced placental ischemia in rats and primates produced symptoms analogous to human preeclampsia, accompanied by elevated sFlt‐1 and sEng. 11 , 12 , 13 In rodents, administration of sFlt‐1 or VEGF‐neutralizing antibody mimicked preeclampsia. 14 , 15 The findings of preeclampsia‐like symptoms in non‐pregnant women, and also men, receiving anti‐VEGF therapies provided unique insights into the role of the angiogenic/antiangiogenic axis in disease development. 16 The biological evidence was also corroborated by human genomic findings. 17 , 18 Encouragingly, some interventional strategies to mitigate sFlt‐1 effects have been explored with favorable outcomes. In a mouse preeclampsia model induced by sFlt‐1, VEGF supplementation lowered plasma sFlt‐1 to a subthreshold level and prevented vascular and renal damage. 19 In women with severe preeclampsia, removal of circulating sFlt‐1 by apheresis improved symptoms and prolonged pregnancy, without apparent adverse effects. 20 , 21 Thus, these studies provide important, albeit preliminary, proof‐of‐concept evidence to support sFlt‐1 as a potential drug target. However, apheresis is an invasive procedure, and its effect in pregnant women was transient, likely because it only passively filters out circulating sFlt‐1 without addressing placental overproduction or release. Newer therapies to intervene at the placental level could have more sustained efficacy.
To exploit these promising targets, it is important to identify a suitable model that can be employed for drug screening. While a simple cell model mimicking human preeclampsia is highly desirable, such an ideal model hardly exists. Human primary cytotrophoblasts are typically considered the gold standard, but they undergo rapid phenotypic changes once in culture, are individually variable, and do not proliferate; thus, their utility in drug screening is limited. Immortalized cell lines (either of tumor origin or virally transfected) are generally considered less representative of human placenta, but they are readily available and can be expanded to accommodate large‐scale applications with good reproducibility. In the present study, we examined several commonly used immortalized trophoblast cell lines in order to identify one (or more) surrogate model that best simulates the human disease pathogenesis, specifically, the placental expression and secretion of the soluble anti‐antigenic factors in response to hypoxia. We also compared such a model with the available data from human primary cytotrophoblasts and ex vivo placental tissues in the literature, and evaluated its translational potential using experimental therapeutic agents.
2. MATERIALS AND METHODS
2.1. Cell culture
The choriocarcinoma trophoblast cell lines, BeWo, Jar, and Jeg‐3, were from American Type Culture Collection (ATCC). The non‐tumor trophoblast HTR‐8/SVneo was a gift from Prof. Charles Graham (Queen's University at Kingston, Canada). Human umbilical vein endothelial cells (HUVECs; originally from ATCC) and aortic endothelial cells (HAoECs; originally from PromoCell GmbH) were provided by Dr. Andriana Margariti and Prof. David Grieve, respectively (Queen's University Belfast, UK). BeWo cells were maintained in DMEM/F12 Gibco (Thermo Fisher) supplemented with 2 mmol/L L‐glutamate. Jar and HTR‐8/SVneo were cultured in RPMI 1640 (Sigma‐Aldrich), and Jeg‐3 was cultured in EMEM (ATCC). Both HUVECs and HAoECs were cultured in EGM‐2 (Lonza) with all vendor‐provided supplements in flasks or plates coated with 0.1% gelatin (Sigma‐Aldrich). All growth media contained 10% fetal calf serum. There was no mycoplasma contamination with the cells.
2.2. Hypoxia and drug treatments
For investigations of hypoxia effects, cells were harvested at 90% confluency after trypsinization, and seeded into 6‐well culture plates overnight prior to treatment with ambient oxygen or 1% oxygen in a humidified, temperature‐controlled hypoxia chamber (Coy Laboratories) for 24‐48 hr. Initial cell densities in 6‐well plates with 1.5 ml growth media were optimized to ensure subconfluency at 48 hr, that is, 30 × 104 for BeWo, Jeg‐3, and HTR‐8/SVneo, 20 × 104 for Jar, and 5 × 104 for HUVECs and HAoECs. To determine time‐related cell viability under hypoxia, 6 × 104 BeWo cells were seeded into a 48‐well plate. At 6, 12, 24, and 48 hr, cells were treated with cold phosphate‐buffered saline (PBS) for 10 min before being harvested, followed by immediate cell number quantification with a hemocytometer and trypan blue.
For studies of the effects of dimethyloxalylglycine (DMOG; Sigma‐Aldrich), cells were seeded into 6‐well plates overnight before DMOG treatment for 24 hr. Other agents including chetomin (Santa Cruz Biotechnology), Trolox, heparin, lipopolysaccharides (LPS) (all from Sigma‐Aldrich), pravastatin, and resveratrol (both from Tocris) were added to cultured cells 1 hr before treatment with hypoxia or ambient oxygen.
2.3. Enzyme‐linked immunosorbent assay (ELISA)
Culture supernatants were collected and clarified by centrifugation for 10 min at 4°C. sFlt‐1 concentrations were initially quantified using the Quantikine ELISA kit DVR100B, and later by the newer version Quantikine ELISA kit DVR100C (both from R&D Systems) when the manufacturer discontinued the former. All samples were assayed in duplicate per manufacturer's instructions. For the DVR100B kit, a detection range of 7.8‐2000 pg/ml was established to accommodate the lower protein concentrations in some samples. Per the vendor, the DVR100C kit uses a polyclonal antibody that improves the detectability of sFlt‐1 e15a isoform, and thus has approximately 7‐fold higher sensitivity than DVR100B, using a conversion formula y = 7.2175 × + 32.06 (y, new kit and x, old kit). We have compared the measurements from both kits using the same set of samples, which were generally comparable after conversion (Figure S1). Therefore, relative fold changes were reported for the purpose of data presentation. sEng protein levels were quantified using the Quantikine ELISA kit DNDG00 (R&D Systems).
2.4. Real‐time quantitative PCR (RT‐PCR)
Total RNA was isolated using an RNeasy mini kit (Qiagen) per manufacturer's protocol, and concentrations were assessed by a NanoDrop 1000 spectrophotometer (NanoDrop Technologies). One microgram RNA was reverse‐transcribed into cDNA using the First Strand cDNA synthesis kit (Thermo Fisher). mRNA expression was quantified by RT‐PCR using the LightCycler 480 system with SYBR Green I Master (Roche Diagnostics). The primers included sFlt‐1 i13 forward: ACAATCAGAGGTGAGCACTGCAA, sFlt‐1 i13 reverse: TCCGAGCCTGAAAGTTAGCAA, sFlt‐1 e15a forward: ACACAGTGGCCATCAGCAGTT, sFlt‐1 e15a reverse: CCCGGCCATTTGTTATTGTTA, 18S forward: TAACGAACGAGACTCTGGCAT, and 18S reverse: CGGACATCTAAGGGCATCACAG. 18S was used as a reference house‐keeping gene. Gene expression fold changes were calculated by dividing normalized values of treated samples by the normalized values of control samples.
2.5. Western blotting
Cells were washed with cold PBS, lysed with the RIPA Lysis and Extraction Buffer in the presence of the Halt Protease and Phosphatase Inhibitor (1:1000) (Thermo Fisher). Protein concentrations were determined by the Pierce BCA assay (Thermo Fisher). Protein (10 μg) was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane, and then probed with the primary antibodies against HIF‐1α (1:1000) and tubulin (1:10000) (Cell Signalling Technology). Tubulin was used as a loading control. The membrane was developed by Pierce ECL Western Blotting Substrate (Thermo Fisher), and images were captured with the UVP BioSpectrum Imaging System (UVP).
2.6. Data analysis
Data were presented as means ± SEM. Unpaired, two‐tailed Student's t‐test was used to compare experimental and control groups. For measurements over time, two‐way ANOVA analysis was conducted. P values <0.05 were considered statistically significant.
3. RESULTS
3.1. Hypoxia differentially modulates sFlt‐1 protein release from human trophoblast cell lines
To determine the effect of hypoxia on sFlt‐1 release from cultured trophoblasts, we first measured supernatant sFlt‐1 levels by ELISA following incubation under hypoxia (1% oxygen) vs. normoxia (21% oxygen) for 24‐48 hr. We found that sFlt‐1 was undetectable in the supernatant from either Jar or Jeg‐3, after culture with various cell densities ranging from 10 × 104 to 40 × 104 in 6‐well plates. For BeWo, sFlt‐1 concentration was approximately 10 pg/ml (average of n = 6, based on Quantikine ELISA kit DVR100B; cell density 30 × 104) at 24 hr under normoxia. Following 24 hr hypoxia treatment, sFlt‐1 concentration was doubled; however, the difference between hypoxia and normoxia disappeared at 48 hr (Figure 1A). In HTR‐8/SVneo, sFlt‐1 concentration was approximately 38 pg/ml (average of n = 6; cell density 30 × 104) at 24 hr under normoxia. Surprisingly, hypoxia reduced supernatant sFlt‐1 levels in HTR‐8/SVneo by approximately 25% and 40% at 24 hr and 48 hr, respectively, as compared to normoxia (Figure 1B). All four cell lines had detectable sEng protein in the supernatant as measured by ELISA, which, however, did not increase in response to hypoxia; therefore, we did not explore it further.
FIGURE 1.
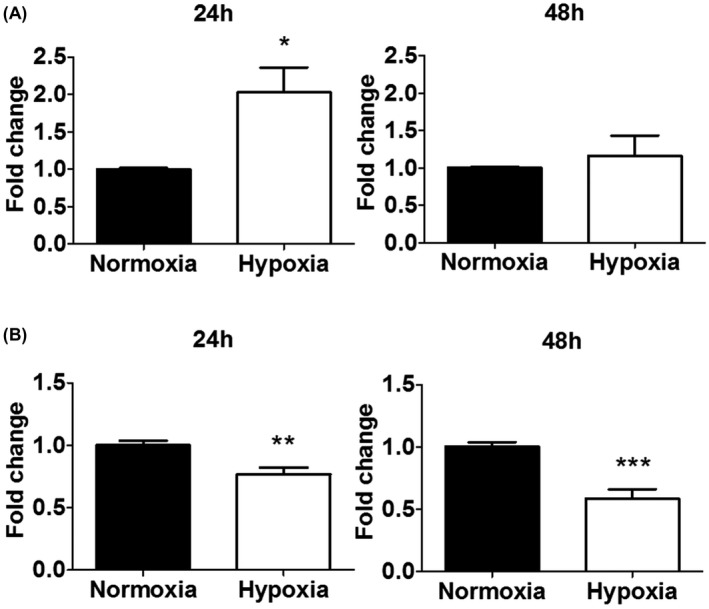
Effects of hypoxia on sFlt‐1 protein secretion from BeWo and HTR‐8/SVneo cells. A, Hypoxia increased sFlt‐1 release from BeWo at 24 hr, but not at 48 hr, relative to normoxia at the same time points. B, Hypoxia reduced sFlt‐1 release from HTR‐8/SVneo at both 24 and 48 hr, relative to normoxia at the same time points. Data are presented as means ± SEM, n = 6. *p < 0.05, **p < 0.01 and ***p < 0.001.
3.2. Hypoxia differentially modulates sFlt‐1 gene expression in human trophoblast cell lines
Since sFlt‐1 i13 and e15a are the two predominantly expressed isoforms in trophoblasts, and both are implicated in preeclampsia, we investigated the effect of hypoxia on their expression. Overall, hypoxia treatment significantly enhanced sFlt‐1 gene expression in BeWo, Jar, and Jeg‐3 (Figure 2A‐C): both i13 and e15a transcripts were upregulated relative to normoxia at both time points (except for i13 in Jar at 48 hr).
FIGURE 2.
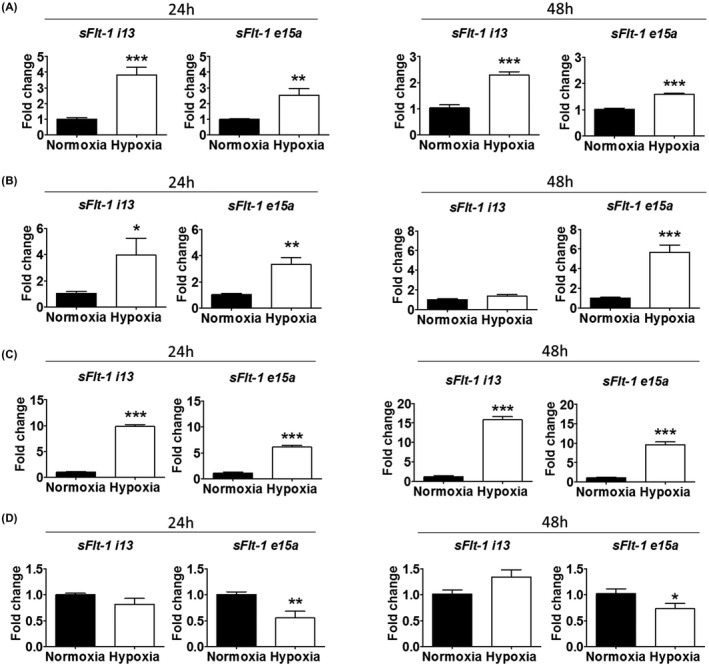
Effects of hypoxia, relative to normoxia, on sFlt‐1 i13 and e15a mRNA expression in four trophoblast cell lines. A, In BeWo, hypoxia increased sFlt‐1 i13 and e15a at both 24 and 48 hr. B, In Jar, hypoxia increased both sFlt‐1 i13 and e15a at 24 hr, and sFlt‐1 e15a at 48 hr. C, In Jeg‐3, hypoxia increased sFlt‐1 i13 and e15a at both 24 and 48 hr. D, In HTR‐8/SVneo, hypoxia did not significantly affect sFlt‐1 i13 expression, but decreased sFlt‐1 e15a expression at 24 and 48 hr. Data are presented as means ± SEM, n = 6. *p < 0.05, **p < 0.01 and ***p < 0.001.
In HTR‐8/SVneo, hypoxia did not significantly affect sFlt‐1 i13 gene expression, but reduced sFlt‐1 e15a gene expression at both 24 and 48 hr, relative to normoxia (Figure 2D). This is consistent with the above observation of a reduction of sFlt‐1 protein in this cell line under hypoxia (Figure 1B).
3.3. Hypoxia down‐regulates sFlt‐1 protein and mRNA expression in vascular endothelial cells
Because HTR‐8/SVneo, as an extravillous cytotrophoblast, shares some endothelial characteristics during placental development, we included two endothelial cell lines for comparison. In HUVECs, supernatant sFlt‐1 was approximately 2290 pg/ml (mean of n = 6; cell density 5 × 104) at 24 hr under normoxia. Hypoxia did not affect sFlt‐1 protein or gene expression at 24 hr, but reduced protein levels by 55% at 48 hr relative to normoxia (Figure 3A). In line with this, hypoxia also decreased sFlt‐1 i13 (but not e15a) gene expression vs. normoxia at 48 hr.
FIGURE 3.
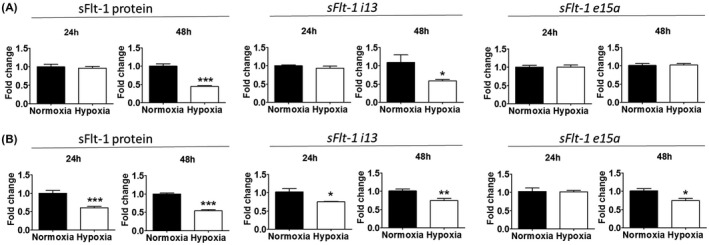
Effects of hypoxia, relative to normoxia, on sFlt‐1 secretion and gene expression in endothelial cells. A, Hypoxia reduced sFlt‐1 protein release and sFlt‐1 i13 expression from HUVECs at 48 hr, but did not affect sFlt‐1 e15a expression. B, Hypoxia reduced sFlt‐1 release from HAoECs at both 24 and 48 hr. It also down‐regulated sFlt‐1 i13 expression at both 24 and 48 hr, and sFlt‐1 e15a expression at 48 hr. Data are presented as means ± SEM, n = 6. *p < 0.05, **p < 0.01 and ***p < 0.001.
In HAoECs, supernatant sFlt‐1 protein level was approximately 4381 pg/ml (mean of n = 6; cell density 5 × 104) at 24 hr under normoxia. Hypoxia treatment caused 40%‐45% reduction of supernatant sFlt‐1 at 24 and 48 hr, relative to normoxia (Figure 3B). Consistently, hypoxia also down‐regulated sFlt‐1 gene expression, that is, i13 at both 24 and 48 hr, and e15a at 48 hr. Thus, hypoxia did not cause an upregulation or release of sFlt‐1 in these two endothelial cells.
3.4. Hypoxia induces sFlt‐1 protein release from BeWo in a time‐related fashion
Apart from HTR‐8/SVneo, all other three trophoblast cell lines we investigated (BeWo, Jar, and Jeg‐3) showed an enhanced sFlt‐1 gene expression in response to hypoxia, but only BeWo secreted a quantity of sFlt‐1 protein that was detectable by ELISA. To further explore its potentiality to be used as a drug‐testing model, we profiled the time‐course effect of hypoxia. As shown in Figure 4A, sFlt‐1 protein concentrations in BeWo supernatant increased over time, approximating 10‐fold at 48 hr. At 6 hr, the concentrations were comparable between hypoxia and normoxia, but hypoxia significantly enhanced sFlt‐1 release vs. normoxia at 12 and 24 hr. However, the gap between hypoxia and normoxia disappeared at 48 hr, likely because the cells were less healthy following longer‐term hypoxia treatment. During this period, there was a modest degree of cell proliferation (Figure 4B), but the rate of change was significantly less than that of sFlt‐1. Hypoxia did not significantly alter cell proliferation. Therefore, 12‐24 hr appeared to be an optimal window to simulate hypoxia effects for drug evaluation.
FIGURE 4.
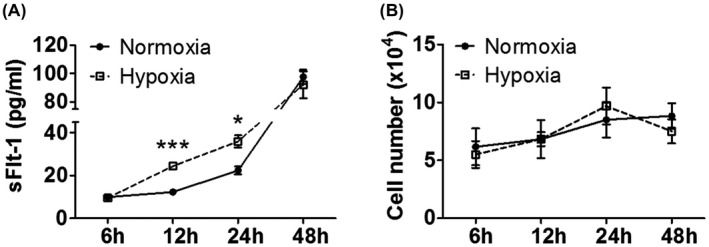
Time‐course effects of hypoxia on sFlt‐1 secretion from BeWo. A, Hypoxia significantly increased sFlt‐1 protein release from BeWo relative to normoxia at 12 and 24 hr. B, Hypoxia did not significantly alter BeWo cell proliferation relative to normoxia over 48 hr. Data are presented as means ± SEM, n = 3. *p < 0.05 and ***p < 0.001.
3.5. HIFα is involved in the regulation of sFlt‐1 in BeWo
Since HIFα mediates hypoxic responses in numerous tissues including sFlt‐1 regulation in human placenta, 22 we investigated if this property was also shared by BeWo. We found that HIF1α protein was significantly upregulated under hypoxia for 24 hr (Figure 5). DMOG, a HIFα prolyl hydroxylase inhibitor, induced sFlt‐1 protein release under normoxia (Figure 6A) to a degree that was comparable to true hypoxia treatment. It also upregulated both sFlt‐1 i13 and e15a gene expression. We further tested the effect of a HIFα inhibitor, chetomin. Under normoxia, chetomin did not significantly alter sFlt‐1 protein secretion from BeWo, although it reduced sFlt‐1 i13 and e15a mRNA expression at 10 nmol/L (Figure 6B). Under hypoxia, chetomin suppressed sFlt‐1 protein release in a dose‐related manner, together with a reduction of sFlt‐1 i13 and e15a mRNA expression (Figure 6C). Thus, these data confirm an involvement of HIFα in the BeWo‐hypoxia model.
FIGURE 5.
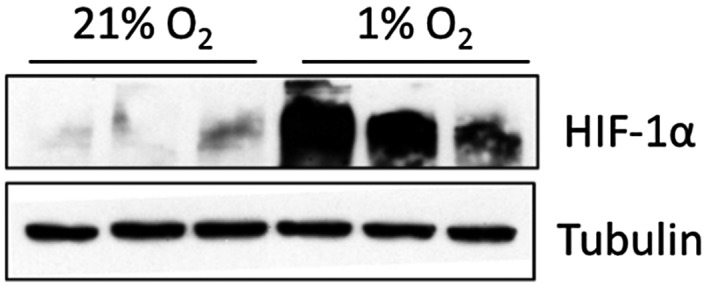
Upregulation of HIF‐1α in BeWo cells under hypoxia vs. normoxia for 24 hr. Western blotting, n = 3.
FIGURE 6.
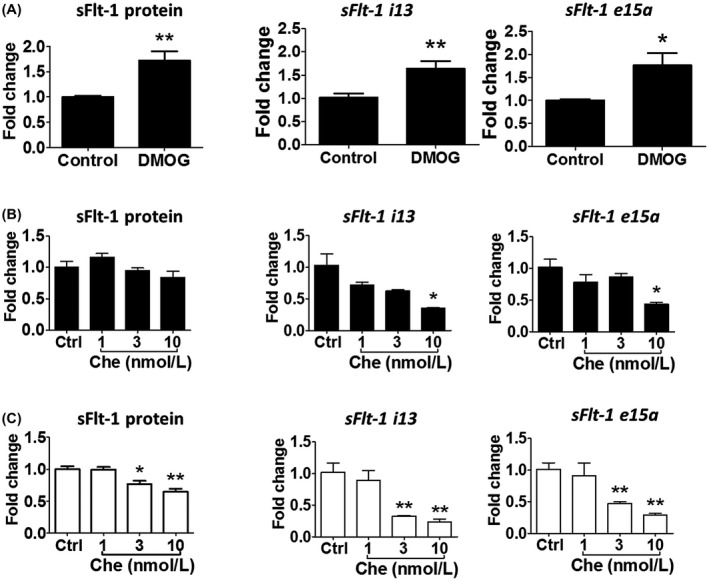
Regulation of sFlt‐1 expression by the HIF pathway. A, The HIF stabilizer DMOG (10 μmol/L, 24 hr) increased sFlt‐1 protein release and mRNA expression for both sFlt‐1 i13 and e15a in BeWo (n = 3). B, Under normoxia, the HIF inhibitor chetomin (1‐10 ‐nmol/L, 24 hr) did not significantly affect sFlt‐1 release from BeWo; however, it reduced sFlt‐1 i13 and e15a expression at the high concentration of 10 nmol/L (n = 6).C, Under hypoxia, chetomin (1‐10 nmol/L, 24 hr) significantly reduced sFlt‐1 protein release and gene expression (both i13 and e15a) in BeWo in a concentration‐dependent manner (n = 6). Data are presented as means ± SEM. *p < 0.05 and **p < 0.01. Che, chetomin.
3.6. Effects of representative agents on hypoxia‐induced sFlt‐1 response in BeWo
To determine whether BeWo responds to some of the previously reported agents, we evaluated their effects in this model. We found that supernatant sFlt‐1 was suppressed by resveratrol at 50 μmol/L under hypoxia (Figure 7A). Similarly, the synthetic vitamin E, Trolox, inhibited sFlt‐1 protein release and gene expression in a concentration‐related fashion (Figure 7B). However, pravastatin was unable to reduce sFlt‐1 up to 1000 μmol/L tested, but modestly increased it at the higher concentration range (Figure 7C). Pravastatin also did not inhibit sFlt‐1 i13 or e15a mRNA expression. In addition, BeWo responded to heparin treatment with a dramatically enhanced sFlt‐1 release, but was not responsive to LPS (Figure S2).
FIGURE 7.
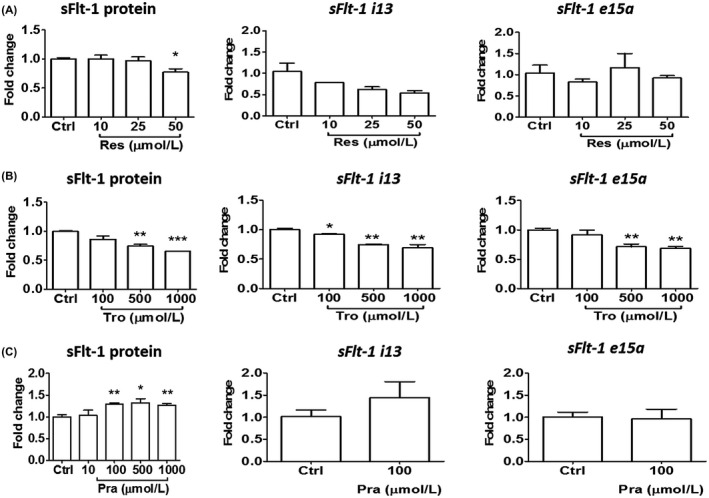
Responses of BeWo to representative chemical agents in terms of sFlt‐1 protein release and gene expression under hypoxia. A, Resveratrol reduced sFlt‐1 protein release at 50 μmol/L, and also showed a trend to reduction of sFlt‐1 i13 gene expression. B, Trolox‐elicited concentration‐dependent reductions of sFlt‐1 protein release and gene expression (both i13 and e15a). C, Pravastatin did not reduce, but instead elicited a modest increase of sFlt‐1 protein release at higher concentrations. All drug treatments lasted 24 hr. Data are presented as means ± SEM, n = 3. *p < 0.05, **p < 0.01, and ***p < 0.001. Res, resveratrol; Tro, Trolox; Pra, pravastatin.
4. DISCUSSION
Developing new therapies for preeclampsia and the underlying placental insufficiency continues to be challenging in light of the lack of success in clinical trials. 23 , 24 , 25 Effective translational research requires reliable preclinical models that simulate human disease pathogenesis as closely as possible. In this study, we explored the feasibility of establishing a cell‐based phenotypic model to simulate a key pathogenic feature of preeclampsia, that is, placental release of sFlt‐1 in exposure to hypoxic stress. Considering the need for high assay throughput, reproducibility and scalability, we chose commercially available immortalized cell lines that included three choriocarcinoma trophoblasts (BeWo, Jar, and Jeg‐3) 26 , 27 , 28 and a non‐tumor cell line HTR‐8/SVneo. 29 We also evaluated the potential of the cell‐based model for translation to the clinical setting using representative agents. We found that while all three choriocarcinoma trophoblasts exhibited significantly enhanced sFlt‐1 mRNA expression in response to hypoxia, only BeWo secreted a detectable quantity of sFlt‐1 protein. HTR‐8/SVneo produced measurable protein, but had a modest yet statistically significant reduction of both sFlt‐1 mRNA expression and protein release. Therefore, BeWo showed the greatest promise. BeWo also responded to several probe agents in a manner that is generally comparable to human primary placental tissues in the literature. Thus, our data support that the BeWo‐hypoxia model mimics a key pathogenic mechanism of human preeclampsia with potential translational value.
Previous comparative studies have revealed a multitude of genetic, epigenetic, transcriptomic, and phenotypic similarities and dissimilarities across trophoblast cell types. 30 , 31 , 32 , 33 , 34 , 35 , 36 Specifically, controversies abound with regard to sFlt‐1 biology: while some studies showed that hypoxiaelicited sFlt‐1 release or gene expression in primary trophoblasts 37 , 38 , 39 , 40 and trophoblast cell lines, such as BeWo, Jar, and Jeg‐3, 38 , 41 , 42 , 43 others found no effect of hypoxia on sFlt‐1 release or gene expression in HTR‐8/SVneo, 44 Jar, or BeWo. 45 One study reported no measurable sFlt‐1 protein in culture supernatants of BeWo, Jar, and Jeg‐3. 39 An interesting observation from our study is that all three tumor cell lines exhibited enhanced sFlt‐1 mRNA expression following hypoxia exposure, consistent with the current understanding of preeclampsia pathogenesis. Two of the predominant sFlt‐1 transcripts, i13 and e15a, were both upregulated, generally at 24 and 48 hr. However, among these tumor cell lines, only BeWo secreted sFlt‐1 protein that was measurable by ELISA in the supernatant, with an optimal window identified at 12‐24 hr for the hypoxia effect. This difference is most likely attributable to the lower basal level of sFlt‐1 mRNA expression in Jar and Jeg‐3 (Ct values for i13 were 31.5 and 33.3, respectively, vs. 28.0 in BeWo, and for e15a were 32.1 and 33.3, respectively, vs. 28.6 in BeWo). This coincides with that, of the cell lines examined, only BeWo forms syncytium, 46 and that sFlt‐1 is released primarily from syncytiotrophoblasts. 22 It is noted that both BeWo and Jeg‐3 originated from the same choriocarcinoma specimen of a patient more than a half century ago, and thus, genetic or epigenetic changes at certain stage might have caused differential degrees of sFlt‐1 gene expression. Additionally, we found that hypoxia also enhanced membrane Flt‐1 mRNA expression in BeWo (data not shown), suggesting a regulatory mechanism primarily at the transcriptional level.
The mechanism for the hypoxia‐elicited sFlt‐1 overexpression and release in BeWo involved the HIFα pathway, as shown by the effect of the HIFα inhibitor, chetomin. The hypoxia‐induced sFlt‐1 response was also reproduced by the HIFα stabilizer DMOG. This profile is in line with the concept that placental hypoxia is a major pathogenic factor of preeclampsia. 22
It was surprising that hypoxia inhibited sFlt‐1 expression and release in HTR‐8/SVneo, a non‐tumor cell line that has been thought to mimic human trophoblasts more closely than tumor cell lines. HTR‐8/SVneo is a first trimester human trophoblast cell line that was immortalized by transfection of Simian virus 40 large T antigen. 29 It is unclear why HTR‐8/SVneo possesses a distinct sFlt‐1 biology. We speculate that because HTR‐8/SVneo represents extravillous cytotrophoblasts that invade the decidua and remodel uterine spiral arteries by replacing the endothelium during placental development, it might assume some aspects of vascular endothelial phenotypes. Indeed, we found that both HUVECs (fetal endothelial cells) and HAoECs (adult endothelial cells) behaved similarly to HTR‐8/SVneo in their response to hypoxia. These findings are generally in line with some 40 , 44 but not all studies. 47 One study reported reduced sFlt‐1 expression in dermal microvascular endothelial cells under hypoxia 48 ; in other preliminary work, we found that hypoxia also did not enhance sFlt‐1 expression in human retinal endothelial cells (data not shown). Thus, HTR‐8/SVneo differs from villous trophoblasts in this respect. HTR‐8/SVneo cells are able to incorporate with endothelial cells to form capillary‐like tubes 49 ; a reduction (but not increase) of sFlt‐1 may facilitate vascularization. In addition, a recent study found that HTR‐8/SVneo contains stromal cells and thus might not represent a uniform cell type 50 ; whether the present observation was complicated by stromal cells requires further investigation.
In order to assess the general responsiveness of the BeWo‐hypoxia model to pharmacologic interventions, we tested several agents that have previously been evaluated in human primary placental tissues or clinical studies. We found that sFlt‐1 protein release and mRNA expression were attenuated by both resveratrol (at 50 μmol/L) and Trolox (a synthetic vitamin E, at 500‐1000 μmol/L), consistent with their antioxidant properties. Oxidative stress in preeclamptic placentas contributes to hypoxia‐elicited sFlt‐1 secretion. 51 Resveratrol has previously been shown to reduce the release of sFlt‐1 (at 25‐100 μmol/L) and sEng (at 100 μmol/L) from human primary placental trophoblasts and villous explants. 52 , 53 Trolox at 1000 μmol/L has been reported to suppress hypoxia‐induced reactive oxygen species formation and sEng release in Jar cells, 54 and tocopherol supplementation in pregnant ewes suppressed sFlt‐1 mRNA expression and improved angiogenesis in the placenta. 55 While effective in preclinical scenarios, these antioxidants have yet to establish definitive clinical efficacy in preventing or treating preeclampsia, considering their low potencies relative to the clinical dose exposures. 56 , 57 , 58 , 59
The discovery of the role of soluble antiangiogenic factors, particularly sFlt‐1, in preeclampsia development has greatly stimulated therapeutic discovery efforts in recent years. One of the most investigated clinical candidates is pravastatin, which has been evaluated as a potential prevention or treatment for preeclampsia. 60 , 61 , 62 In a pilot randomized controlled pharmacokinetic study of high‐risk pregnancies, Costantine et al 63 showed promising, although non‐significant, sFlt‐1 and sEng lowering in 10 women who received 10 mg pravastatin daily (vs. 10 placebo subjects) starting from early second trimester. A few other small trials and case studies also reported encouraging data. 64 , 65 , 66 However, the recent randomized controlled trial of “Statins to Ameliorate Pre‐Eclampsia (StAmP)” found no evidence of sFlt‐1 reduction in 30 women with early‐onset preeclampsia who received 40 mg pravastatin daily in the third trimester (vs. 32 placebo subjects). 25 Intriguingly, in most studies with cultured human primary trophoblasts or villous explants, pravastatin appeared to reduce sFlt‐1 and sEng only at sub‐millimolar to millimolar concentrations, 67 , 68 , 69 , 70 substantially higher than the sub‐micromolar plasma concentrations achievable in humans taking the drug. 63 , 71 In the present BeWo‐hypoxia model, we were also unable to confirm sFlt‐1 inhibition by pravastatin. However, it is important to note that the BeWo model is not designed to detect non‐trophoblast mechanisms of action (e.g., placental vascular effects), which may occur in vivo and underlie pravastatin's potentially prophylactic effect for preeclampsia.
Additionally, we found that BeWo responded to heparin treatment with dramatically enhanced sFlt‐1 release, a known phenomenon that was also observed in human primary placental tissues. 72 , 73 It was suggested that hypoxia might promote sFlt‐1 release from BeWo via heparanase activation rather than transcriptional upregulation, 42 although our present data are explicable by the latter effect. Also, we found that BeWo did not respond to LPS stimulation in regard to sFlt‐1 regulation; this is consistent with the earlier findings that Toll‐like receptor 4 is not responsive in this cell line. 74 , 75 Thus, the BeWo‐hypoxia model exhibits characteristics that are generally comparable to the published human primary placental trophoblasts and tissues. Although our present focus was on the placenta‐derived antiangiogenic factors, others have documented similarities between BeWo and human primary trophoblasts in regard to placental protein‐13 and β‐hCG secretion and the effect of vitamin C. 76
The novelty of this work includes the identification and characterization of BeWo to be a suitable surrogate model for the hypoxia‐mediated sFlt‐1 release from the placenta, a promising therapeutic target already with early proof‐of‐concept evidence, and the validation of its translational potential using clinically relevant agents. Using a tumor cell line, rather than primary cells or tissues, is expected to greatly facilitate drug development for preeclampsia. There are some un‐answered questions and limitations in our study. First, previous studies showed that human primary cytotrophoblasts could release up to ng/mL of sFlt‐1, 37 , 40 but trophoblast cell lines appeared to secret much lower concentrations. This likely had led to the earlier conclusion that most trophoblast cell lines do not secrete sFlt‐1. 39 Second, there are controversies with regard to the oxygen level to be used in cell culture studies: some adopted 8% oxygen as the normoxia control, which represents the intervillous blood oxygen level during the second and third trimesters when preeclampsia occurs. 77 However, the work from Chen et al. 78 shows that the steady‐state pericellular oxygen tension of cultured human trophoblasts is much lower than ambient air, reflecting an inefficient gas diffusion in the medium, where no hemoglobin is present to facilitate oxygen delivery as in vivo. They estimated that even 21% O2 in cell culture might not be sufficient to reproduce the intervillous O2 level of humans. Therefore, we used ambient air as the normoxia baseline. This has the additional benefits to allow for comparison of our results vs. the primary trophoblast cell data in the literature, as well as a convenient drug screening setup without the unnecessary phenotypic or epigenetic alterations potentially caused by an artificial adjustment of the baseline oxygen level. It is also possible that acute hypoxia treatment in cell culture may not fully mimic chronic placental ischemia in preeclampsia patients. Nevertheless, the hypoxia‐elicited sFlt‐1 response (~2‐fold) was in good agreement with the literature data from human primary trophoblasts and other experimental conditions, 37 , 39 , 40 , 44 as well as that in humans, 3 , 7 lending further support to the utility of BeWo as a surrogate model. Third, while BeWo exhibits sFlt‐1 biology consistent with human preeclampsia and fulfils the throughput requirement for drug screening, it remains a tumor cell line and thus caution is always needed in preclinical‐to‐clinical translation. This limitation may be mitigated by secondary verifications using human primary trophoblasts or additional cells/tissues, as well as multiple species integrative pharmacokinetic/pharmacodynamic analyses.
In summary, by comparing four common human placental trophoblast cell lines, we found that BeWo responded to hypoxia with an enhanced expression and release of sFlt‐1, a major pathogenic factor in preeclampsia development. BeWo responses were generally consistent with known properties of human primary placental trophoblasts and tissues. The BeWo‐hypoxia model has potential as a convenient cell‐based platform for medium‐to‐high throughput drug testing (e.g., in 48‐96 well plates) for the development of new, targeted therapies for preeclampsia.
CONFLICT OF INTEREST
The authors declare that there is no duality of interest associated with this manuscript.
AUTHOR CONTRIBUTIONS
JYY, TJL, and JZ contributed to study design, data interpretation, and manuscript writing. JZ, RPC, RHM, and MBH performed experiments, collected data, and analyzed data. RPC collected data using the newer sFlt‐1 ELISA kit. All authors reviewed and approved the final manuscript. JYY is the guarantor of this work.
Supporting information
Fig S1‐S2
ACKNOWLEDGMENTS
This work was supported by Saving Lives at Birth (a partnership between the United States Agency for International Development, the Norwegian Ministry of Foreign Affairs, the Bill & Melinda Gates Foundation, Grand Challenges Canada, and the Department for International Development of United Kingdom) under award #0703‐03, and by the Eunice Kennedy Shriver National Institute Of Child Health & Human Development (NICHD) of the National Institutes of Health under award #R01HD096501. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. The authors are also grateful to Professor Charles Graham (Queen’s University at Kingston, Canada), Dr Andriana Margariti, and Professor David Grieve (Queen's University Belfast, UK) for generously providing the cell lines in this study.
REFERENCES
- 1. Kassebaum NJ, Barber RM, Bhutta ZA, et al. Global, regional, and national levels of maternal mortality, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1775‐1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shih T, Peneva D, Xu X, et al. The rising burden of preeclampsia in the United States impacts both maternal and child health. Am J Perinatol. 2016;33:329‐338. [DOI] [PubMed] [Google Scholar]
- 3. Yu Y, Jenkins AJ, Nankervis AJ, et al. Anti‐angiogenic factors and pre‐eclampsia in type 1 diabetic women. Diabetologia. 2009;52:160‐168. [DOI] [PubMed] [Google Scholar]
- 4. American College of, O., Gynecologists, and Task Force on Hypertension in, P . Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet Gynecol. 2013;122:1122‐1131. [DOI] [PubMed] [Google Scholar]
- 5. Henderson JT, Whitlock EP, O'Connor E, Senger CA, Thompson JH, Rowland MG. Low‐dose aspirin for prevention of morbidity and mortality from preeclampsia: a systematic evidence review for the U.S. Preventive services task force. Ann Intern Med. 2014;160:695‐703. [DOI] [PubMed] [Google Scholar]
- 6. Roberts JM, Hubel CA. The two stage model of preeclampsia: variations on the theme. Placenta. 2009;30:32‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Levine RJ, Maynard SE, Qian C, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350:672‐683. [DOI] [PubMed] [Google Scholar]
- 8. Levine RJ, Lam C, Qian C, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006;355:992‐1005. [DOI] [PubMed] [Google Scholar]
- 9. Naljayan MV, Karumanchi SA. New developments in the pathogenesis of preeclampsia. Adv Chronic Kidney Dis. 2013;20:265‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rana S, Schnettler WT, Powe C, et al. Clinical characterization and outcomes of preeclampsia with normal angiogenic profile. Hypertens Pregnancy. 2013;32:189‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Makris A, Thornton C, Thompson J, et al. Uteroplacental ischemia results in proteinuric hypertension and elevated sFLT‐1. Kidney Int. 2007;71:977‐984. [DOI] [PubMed] [Google Scholar]
- 12. Gilbert JS, Babcock SA, Granger JP. Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble fms‐like tyrosine kinase‐1 expression. Hypertension. 2007;50:1142‐1147. [DOI] [PubMed] [Google Scholar]
- 13. Gilbert JS, Gilbert SA, Arany M, Granger JP. Hypertension produced by placental ischemia in pregnant rats is associated with increased soluble endoglin expression. Hypertension. 2009;53:399‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sugimoto H, Hamano Y, Charytan D, et al. Neutralization of circulating vascular endothelial growth factor (VEGF) by anti‐VEGF antibodies and soluble VEGF receptor 1 (sFlt‐1) induces proteinuria. J Biol Chem. 2003;278:12605‐12608. [DOI] [PubMed] [Google Scholar]
- 15. Maynard SE, Min JY, Merchan J, et al. Excess placental soluble fms‐like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vigneau C, Lorcy N, Dolley‐Hitze T, et al. All anti‐vascular endothelial growth factor drugs can induce ‘pre‐eclampsia‐like syndrome’: a RARe study. Nephrol Dial Transplant. 2014;29:325‐332. [DOI] [PubMed] [Google Scholar]
- 17. McGinnis R, Steinthorsdottir V, Williams NO, et al. Variants in the fetal genome near FLT1 are associated with risk of preeclampsia. Nat Genet. 2017;49:1255‐1260. [DOI] [PubMed] [Google Scholar]
- 18. Lokki AI, Daly E, Triebwasser M, et al. Protective low‐frequency variants for preeclampsia in the Fms related tyrosine kinase 1 gene in the Finnish population. Hypertension. 2017;70:365‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bergmann A, Ahmad S, Cudmore M, et al. Reduction of circulating soluble Flt‐1 alleviates preeclampsia‐like symptoms in a mouse model. J Cell Mol Med. 2010;14(6b):1857‐1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thadhani R, Kisner T, Hagmann H, et al. Pilot study of extracorporeal removal of soluble fms‐like tyrosine kinase 1 in preeclampsia. Circulation. 2011;124:940‐950. [DOI] [PubMed] [Google Scholar]
- 21. Thadhani R, Hagmann H, Schaarschmidt W, et al. Removal of soluble Fms‐like tyrosine kinase‐1 by dextran sulfate apheresis in preeclampsia. J Am Soc Nephrol. 2016;27:903‐913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nevo O, Soleymanlou N, Wu Y, et al. Increased expression of sFlt‐1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF‐1. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1085‐1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sharp A, Cornforth C, Jackson R, et al. Maternal sildenafil for severe fetal growth restriction (STRIDER): a multicentre, randomised, placebo‐controlled, double‐blind trial. Lancet Child Adolesc Health. 2018;2:93‐102. [DOI] [PubMed] [Google Scholar]
- 24. Cluver CA, Hannan NJ, van Papendorp E, et al. Esomeprazole to treat women with preterm preeclampsia: a randomized placebo controlled trial. Am J Obstet Gynecol. 2018;219:388.e381‐388.e317. [DOI] [PubMed] [Google Scholar]
- 25. Ahmed A, Williams DJ, Cheed V, et al. Pravastatin for early‐onset pre‐eclampsia: a randomised, blinded, placebo‐controlled trial. BJOG. 2020;127:478‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pattillo RA, Gey GO, Delfs E, Mattingly RF. Human hormone production in vitro. Science. 1968;159:1467‐1469. [DOI] [PubMed] [Google Scholar]
- 27. Pattillo RA, Ruckert A, Hussa R, Bernstein R, Delfs E. The Jar cell line – continuous human multihormone production and controls. In Vitro. 1971;6:398‐399. [Google Scholar]
- 28. Kohler PO, Bridson WE. Isolation of hormone‐producing clonal lines of human choriocarcinoma. J Clin Endocrinol Metab. 1971;32:683‐687. [DOI] [PubMed] [Google Scholar]
- 29. Graham CH, Hawley TS, Hawley RG, et al. Establishment and characterization of first trimester human trophoblast cells with extended lifespan. Exp Cell Res. 1993;206:204‐211. [DOI] [PubMed] [Google Scholar]
- 30. Vegh GL, Fulop V, Liu Y, et al. Differential gene expression pattern between normal human trophoblast and choriocarcinoma cell lines: downregulation of heat shock protein‐27 in choriocarcinoma in vitro and in vivo. Gynecol Oncol. 1999;75:391‐396. [DOI] [PubMed] [Google Scholar]
- 31. Burleigh DW, Kendziorski CM, Choi YJ, et al. Microarray analysis of BeWo and JEG3 trophoblast cell lines: identification of differentially expressed transcripts. Placenta. 2007;28:383‐389. [DOI] [PubMed] [Google Scholar]
- 32. Bilban M, Tauber S, Haslinger P, et al. Trophoblast invasion: assessment of cellular models using gene expression signatures. Placenta. 2010;31:989‐996. [DOI] [PubMed] [Google Scholar]
- 33. Novakovic B, Gordon L, Wong NC, et al. Wide‐ranging DNA methylation differences of primary trophoblast cell populations and derived cell lines: implications and opportunities for understanding trophoblast function. Mol Hum Reprod. 2011;17:344‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Szklanna PB, Wynne K, Nolan M, Egan K, Ainle FN, Maguire PB. Comparative proteomic analysis of trophoblast cell models reveals their differential phenotypes, potential uses, and limitations. Proteomics. 2017;17:e1700037. [DOI] [PubMed] [Google Scholar]
- 35. Kallol S, Moser‐Haessig R, Ontsouka CE, Albrecht C. Comparative expression patterns of selected membrane transporters in differentiated BeWo and human primary trophoblast cells. Placenta. 2018;72–73:48‐52. [DOI] [PubMed] [Google Scholar]
- 36. Msheik H, El Hayek S, Bari MF, et al. Transcriptomic profiling of trophoblast fusion using BeWo and JEG‐3 cell lines. Mol Hum Reprod. 2019;25:811‐824. [DOI] [PubMed] [Google Scholar]
- 37. Li H, Gu B, Zhang Y, Lewis DF, Wang Y. Hypoxia‐induced increase in soluble Flt‐1 production correlates with enhanced oxidative stress in trophoblast cells from the human placenta. Placenta. 2005;26:210‐217. [DOI] [PubMed] [Google Scholar]
- 38. Thomas CP, Andrews JI, Raikwar NS, et al. A recently evolved novel trophoblast‐enriched secreted form of fms‐like tyrosine kinase‐1 variant is up‐regulated in hypoxia and preeclampsia. J Clin Endocrinol Metab. 2009;94:2524‐2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaitu'u‐Lino TJ, Tong S, Beard S, et al. Characterization of protocols for primary trophoblast purification, optimized for functional investigation of sFlt‐1 and soluble endoglin. Pregnancy Hypertens. 2014;4:287‐295. [DOI] [PubMed] [Google Scholar]
- 40. Nagamatsu T, Fujii T, Kusumi M, et al. Cytotrophoblasts up‐regulate soluble fms‐like tyrosine kinase‐1 expression under reduced oxygen: an implication for the placental vascular development and the pathophysiology of preeclampsia. Endocrinology. 2004;145:4838‐4845. [DOI] [PubMed] [Google Scholar]
- 41. Jung JJ, Tiwari A, Inamdar SM, Thomas CP, Goel A, Choudhury A. Secretion of soluble vascular endothelial growth factor receptor 1 (sVEGFR1/sFlt1) requires Arf1, Arf6, and Rab11 GTPases. PLoS One. 2012;7:e44572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eddy AC, Chapman H, George EM. Heparanase regulation of sFLT‐1 release in trophoblasts in vitro. Placenta. 2019;85:63‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sasagawa T, Nagamatsu T, Morita K, et al. HIF‐2alpha, but not HIF‐1alpha, mediates hypoxia‐induced up‐regulation of Flt‐1 gene expression in placental trophoblasts. Sci Rep. 2018;8:17375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Munaut C, Lorquet S, Pequeux C, et al. Hypoxia is responsible for soluble vascular endothelial growth factor receptor‐1 (VEGFR‐1) but not for soluble endoglin induction in villous trophoblast. Hum Reprod. 2008;23:1407‐1415. [DOI] [PubMed] [Google Scholar]
- 45. Gillham HE, Banek CT, Needham K, Gilbert J. Differential effects of pravastatin on secretion of pro‐ and anti‐angiogenic factors from trophoblast and endothelial cells. Faseb J. 2013;27(907):905. [Google Scholar]
- 46. Wice B, Menton D, Geuze H, Schwartz AL. Modulators of cyclic AMP metabolism induce syncytiotrophoblast formation in vitro. Exp Cell Res. 1990;186:306‐316. [DOI] [PubMed] [Google Scholar]
- 47. Xiong Y, Liebermann DA, Tront JS, et al. Gadd45a stress signaling regulates sFlt‐1 expression in preeclampsia. J Cell Physiol. 2009;220:632‐639. [DOI] [PubMed] [Google Scholar]
- 48. Ikeda T, Sun L, Tsuruoka N, et al. Hypoxia down‐regulates sFlt‐1 (sVEGFR‐1) expression in human microvascular endothelial cells by a mechanism involving mRNA alternative processing. Biochem J. 2011;436:399‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kalkunte S, Lai Z, Tewari N, et al. In vitro and in vivo evidence for lack of endovascular remodeling by third trimester trophoblasts. Placenta. 2008;29:871‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Abou‐Kheir W, Barrak J, Hadadeh O, Daoud G. HTR‐8/SVneo cell line contains a mixed population of cells. Placenta. 2017;50:1‐7. [DOI] [PubMed] [Google Scholar]
- 51. George EM, Colson D, Dixon J, Palei AC, Granger JP. Heme oxygenase‐1 attenuates hypoxia‐induced sFlt‐1 and oxidative stress in placental villi through its metabolic products CO and bilirubin. Int J Hypertens. 2012;2012:486053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cudmore MJ, Ramma W, Cai M, et al. Resveratrol inhibits the release of soluble fms‐like tyrosine kinase (sFlt‐1) from human placenta. Am J Obstet Gynecol. 2012;206(253):e210‐255. [DOI] [PubMed] [Google Scholar]
- 53. Hannan NJ, Brownfoot FC, Cannon P, et al. Resveratrol inhibits release of soluble fms‐like tyrosine kinase (sFlt‐1) and soluble endoglin and improves vascular dysfunction ‐ implications as a preeclampsia treatment. Sci Rep. 2017;7:1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Valbuena‐Diez AC, Blanco FJ, Oujo B, et al. Oxysterol‐induced soluble endoglin release and its involvement in hypertension. Circulation. 2012;126:2612‐2624. [DOI] [PubMed] [Google Scholar]
- 55. Kasimanickam RK, Kasimanickam VR, Haldorson GJ, Tibary A. Effect of tocopherol supplementation during last trimester of pregnancy on mRNA abundances of interleukins and angiogenesis in ovine placenta and uterus. Reprod Biol Endocrinol. 2012;10:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tenorio MB, Ferreira RC, Moura FA, Bueno NB, Goulart MOF, Oliveira ACM. Oral antioxidant therapy for prevention and treatment of preeclampsia: Meta‐analysis of randomized controlled trials. Nutr Metab Cardiovasc Dis. 2018;28:865‐876. [DOI] [PubMed] [Google Scholar]
- 57. Walle T, Hsieh F, DeLegge MH, Oatis JE Jr, Walle UK. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab Dispos. 2004;32:1377‐1382. [DOI] [PubMed] [Google Scholar]
- 58. Roberts JM, Myatt L, Spong CY, et al. Vitamins C and E to prevent complications of pregnancy‐associated hypertension. N Engl J Med. 2010;362:1282‐1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McCance David R, Holmes Valerie A, Maresh Michael JA, et al. Vitamins C and E for prevention of pre‐eclampsia in women with type 1 diabetes (DAPIT): a randomised placebo‐controlled trial. Lancet. 2010;376:259‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Costantine MM, Cleary K, Eunice Kennedy Shriver National Institute of Child, H., and Human Development Obstetric‐Fetal Pharmacology Research Units, N . Pravastatin for the prevention of preeclampsia in high‐risk pregnant women. Obstet Gynecol. 2013;121:349‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ramma W, Ahmed A. Therapeutic potential of statins and the induction of heme oxygenase‐1 in preeclampsia. J Reprod Immunol. 2014;101–102:153‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ma'ayeh M, Rood KM, Kniss D, Costantine MM. Novel interventions for the prevention of preeclampsia. Curr Hypertens Rep. 2020;22:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Costantine MM, Cleary K, Hebert MF, et al.Eunice Kennedy Shriver National Institute of Child, H., and Human Development Obstetric‐Fetal Pharmacology Research Units, N . Safety and pharmacokinetics of pravastatin used for the prevention of preeclampsia in high‐risk pregnant women: a pilot randomized controlled trial. Am J Obstet Gynecol. 2016;214(6):720.e1‐720.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lefkou E, Mamopoulos A, Fragakis N, et al. Clinical improvement and successful pregnancy in a preeclamptic patient with antiphospholipid syndrome treated with pravastatin. Hypertension. 2014;63:e118‐e119. [DOI] [PubMed] [Google Scholar]
- 65. Chaiworapongsa T, Romero R, Korzeniewski SJ, et al. Pravastatin to prevent recurrent fetal death in massive perivillous fibrin deposition of the placenta (MPFD). J Matern Fetal Neonatal Med. 2016;29:855‐862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Otten LA, van der Ven K, Kuhr M, Gembruch U, Merz WM. Pravastatin for prevention of HELLP syndrome: a case report. Medicine. 2017;96:e8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Brownfoot FC, Tong S, Hannan NJ, et al. Effects of pravastatin on human placenta, endothelium, and women with severe preeclampsia. Hypertension. 2015;66:687‐697. discussion 445. [DOI] [PubMed] [Google Scholar]
- 68. Brownfoot FC, Tong S, Hannan NJ, Hastie R, Cannon P, Kaitu'u‐Lino TJ. Effects of simvastatin, rosuvastatin and pravastatin on soluble fms‐like tyrosine kinase 1 (sFlt‐1) and soluble endoglin (sENG) secretion from human umbilical vein endothelial cells, primary trophoblast cells and placenta. BMC Pregnancy Childbirth. 2016;16:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gangooly S, Muttukrishna S, Jauniaux E. In‐vitro study of the effect of anti‐hypertensive drugs on placental hormones and angiogenic proteins synthesis in pre‐eclampsia. PLoS One. 2014;9:e107644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Balan A, Szaingurten‐Solodkin I, Swissa SS, et al. The effects of pravastatin on the normal human placenta: lessons from ex‐vivo models. PLoS One. 2017;12:e0172174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bellosta S, Paoletti R, Corsini A. Safety of statins: focus on clinical pharmacokinetics and drug interactions. Circulation. 2004;109:III‐50‐III‐57. [DOI] [PubMed] [Google Scholar]
- 72. Drewlo S, Levytska K, Sobel M, Baczyk D, Lye SJ, Kingdom JC. Heparin promotes soluble VEGF receptor expression in human placental villi to impair endothelial VEGF signaling. J Thromb Haemost. 2011;9:2486‐2497. [DOI] [PubMed] [Google Scholar]
- 73. Rosenberg VA, Buhimschi IA, Lockwood CJ, et al. Heparin elevates circulating soluble fms‐like tyrosine kinase‐1 immunoreactivity in pregnant women receiving anticoagulation therapy. Circulation. 2011;124:2543‐2553. [DOI] [PubMed] [Google Scholar]
- 74. Koh YQ, Chan HW, Nitert MD, Vaswani K, Mitchell MD, Rice GE. Differential response to lipopolysaccharide by JEG‐3 and BeWo human choriocarcinoma cell lines. Eur J Obstet Gynecol Reprod Biol. 2014;175:129‐133. [DOI] [PubMed] [Google Scholar]
- 75. Gierman LM, Stodle GS, Tangeras LH, et al. Toll‐like receptor profiling of seven trophoblast cell lines warrants caution for translation to primary trophoblasts. Placenta. 2015;36:1246‐1253. [DOI] [PubMed] [Google Scholar]
- 76. Orendi K, Gauster M, Moser G, Meiri H, Huppertz B. Effects of vitamins C and E, acetylsalicylic acid and heparin on fusion, beta‐hCG and PP13 expression in BeWo cells. Placenta. 2010;31:431‐438. [DOI] [PubMed] [Google Scholar]
- 77. Schneider H. Oxygenation of the placental‐fetal unit in humans. Respir Physiol Neurobiol. 2011;178:51‐58. [DOI] [PubMed] [Google Scholar]
- 78. Chen B, Longtine MS, Nelson DM. Pericellular oxygen concentration of cultured primary human trophoblasts. Placenta. 2013;34:106‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S2