

Abstract
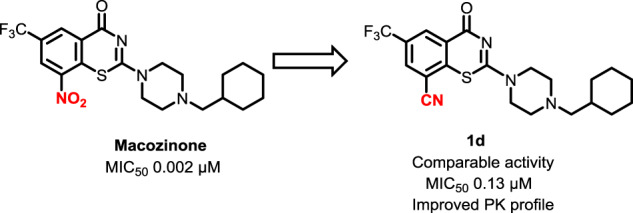
8-Nitrobenzothiazinones (BTZs) exemplified by macozinone are a new class of antitubercular agents with exceptionally potent activity. The aryl nitro group has been considered indispensable for activity since this is bioactivated within mycobacteria by the flavoenzyme DprE1 to a reactive nitroso metabolite that covalently labels Cys387. However, the aryl nitro group is a potential liability with regards to safety, stability, and resistance. In this paper, we introduced a nitrile as a bioisosteric replacement of the nitro group, which we hypothesize can maintain a similar covalent mechanism of inhibition, but mitigate against the aforementioned concerns. 8-cyanobenzothiazinone 1d displayed potent antitubercular activity with an MIC of 130 nM and had an improved volume of distribution in mice that increased the intrinsic half-life by twofold compared to macozinone. Analysis of the C-2 substituent of 1d revealed similar structure–activity relationships as observed for macozinone. Overall, the results confirm the 8-nitro group of benzothiazinones can be successfully replaced with a nitrile to retain useful activity and favorable pharmacokinetic properties.
Introduction
Tuberculosis (TB) is one of the top 10 causes of death worldwide and the leading cause from a single infectious agent [1]. Moreover, nearly one quarter of the world’s population are latently infected with Mycobacterium tuberculosis (Mtb). Reactivation of latent TB occurs when the immune system is weakened through aging, HIV co-infection, malnutrition, or diabetes. The Covid-19 pandemic and continued rise of drug-resistant TB threatens the modest gains made in recent years to bring TB back under control [1]. The development of new therapeutic agents, improved diagnostics, and effective vaccines will be required to achieve the WHO End TB strategy to decrease the TB incidence rate and the number of TB deaths by 80–90% within the next decade [1].
The benzothiazinones initially reported in 2009 have attracted consider attention for their potent antimycobacterial activity against drug-susceptible and resistant strains, synergy with other TB drugs, and outstanding in vivo activity in murine TB models [2–5]. Optimization of the initial lead candidate BTZ043 led to Macozinone (PBTZ169) with improved drug disposition properties and enhanced in vitro and in vivo activity [5, 6]. Macozinone successfully completed phase I clinical studies (NCT03036163 and NCT03423030); however phase 2a clinical trials to evaluate the early bactericidal activity were halted due to low enrollment (NCT03334734).
The benzothiazinones inhibit the biosynthesis of the arabinans, which form an integral part of the mycobacterial cell envelope [3, 4, 7–9]. The flavoenzyme DprE1 catalyzes the penultimate biosynthetic step of the decaprenyl-phospho-D-arabinose building block for construction of the highly branched arabinoglycan polymer, which links the inner peptidoglycan layer to the outer mycomembrane. Following binding to DprE1, the benzothiazinone 8-nitro group is reduced by the flavin cofactor of DprE1 to a nitroso metabolite, which covalently reacts with Cys387 of DprE1 to form a stable semimercaptal adduct (Fig. 1a) [10, 11]. The initially reported structure–activity relationships (SAR) demonstrated a requirement for a nitro group at C-8, a strongly electron-withdrawing group at C-6, no substituents at C-5 or C-7, a carbonyl at C-4 and sulfur at the first position [12–14]. The amino group at the C-2 position is the most tolerant to modification, consequently this position has been the most extensively explored to modulate potency and drug disposition properties [3, 6, 15–33]. However, the 8-nitro group remains a potential liability for idiosyncratic toxicity, metabolism by abundant commensal bacterial nitroreductases, reductive activation by glutathione and other biological thiolates and observed drug resistance by mutation of Cys387 to Ser387 [34–37].
Fig. 1.

Mechanism of benzothiazinone inactivation of DprE1. a Mechanism of 8-nitrobenzothiazinones. b Proposed covalent mechanism of 8-cyano benzothiazinones
Replacement of the C-8 nitro group of the benzothiazinones with pyrrole and triazole groups, which cannot covalently react with Cys387, afforded compounds with modest micromolar activity suggesting ‘the invariant C-8 nitro group’ may be more tolerant to modification than originally suggested [12, 17]. We hypothesize that bioisosteric replacement of the 8-nitro group with a nitrile will maintain activity by enabling reaction with Cys387 or the Cys387Ser mutant to form a covalent (thio)imidate adduct, but mitigate some of the undesirable features of the nitro group (Fig. 1b). The use of a nitrile to react with a nucleophilic serine or cysteine residue in an enzyme active site to form a reversible, but covalent, adduct has been demonstrated for Saxagliptin, a proline dipeptidase inhibitor used for type-2 diabetes and for Odanacatib, an investigator drug for osteoporosis that inhibits cathepsin K [38]. Qiao and co-workers recently disclosed the first successful bioisosteric replacement of the 8-nitro group with a nitrile [39]. Herein, we report our independent studies confirming this initial finding as well as the first SAR studies of 8-cyano derivatives.
Benzothiazinones containing an 8-cyano substituent were synthesized using an adaptation of the initial Makarov route starting from 2-chloro-5-(trifluoromethyl)benzoic acid, which was nitrated using concentrated sulfuric and nitric acid to provide known intermediate 2 (Scheme 1) [40]. Subsequent nitro reduction with iron powder in aqueous ethanol provided aniline 3 that was smoothly converted to aryl nitrile 4 through diazotization followed by Sandmeyer reaction with CuCN. Formation of the resultant benzamide derivative 5 was accomplished under mild conditions employing di-tert-butyl dicarbonate and ammonium bicarbonate [41]. Condensation of benzamide 5 with a variety of sodium carbodithioates 6a–i in DMF through nucleophilic aromatic substitution followed by intramolecular cyclization and dehydrosulfidation yielded target compounds 1a–i in good yields without competitive reaction with the nitrile [13, 42].
Scheme 1.
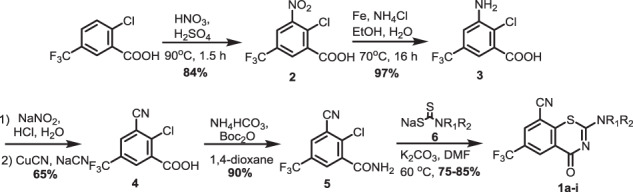
Synthesis of 8-cyano-benzothiazinones 1a–i

The 8-cyanobenzothiazinone derivatives and macozinone as a positive control were screened for antimycobacterial activity against Mtb H37Rv to determine the minimum inhibitory concentration (MIC) resulting in complete inhibition of observable mycobacterial growth (Table 1). Compound 1d containing the optimal cyclohexylmethylpiperazine substituent found in macozinone displayed an MIC of 130 nM providing a rare example of a des-nitro benzothiazinone analog with potent nanomolar activity serving to validate our design principle. We next investigated the SAR of the cyclohexyl group with ring homologues 1a–c, but only cyclopentyl methyl 1c retained activity with an MIC of 200 nM while 1a–b were inactive. While oxetanylmethyl 1f was inactive consistent with the data obtained from cyclobutylmethyl 1b, we were pleasantly surprised to observe tetrahydrofuran-3-ylmethyl 1e incorporating a polar oxygen atom retained moderate activity showing only a 10-fold loss of potency relative to cyclopentyl methyl 1c. Modification of the piperazine by methylation in 1g–h or replacement with the bicyclic isostere in 1i abolished activity. Taken together, the limited SAR of this nitrile series demonstrates the cyclohexylmethylpiperazine substituent is optimal, only very conservative modifications are tolerated in the cycloalkyl substituent, and the piperazine must be strictly maintained. The active compound 1c, 1d, and 1e were further evaluated for potential cytotoxicity against Vero cells, but none demonstrated any inhibition of cellular growth at the highest concentration evaluated (CC50 > 150 µM) furnishing a therapeutic index (MIC/CC50) of greater than 1000 for 1d.
Table 1.
MIC and CC50 of compound 1a–i
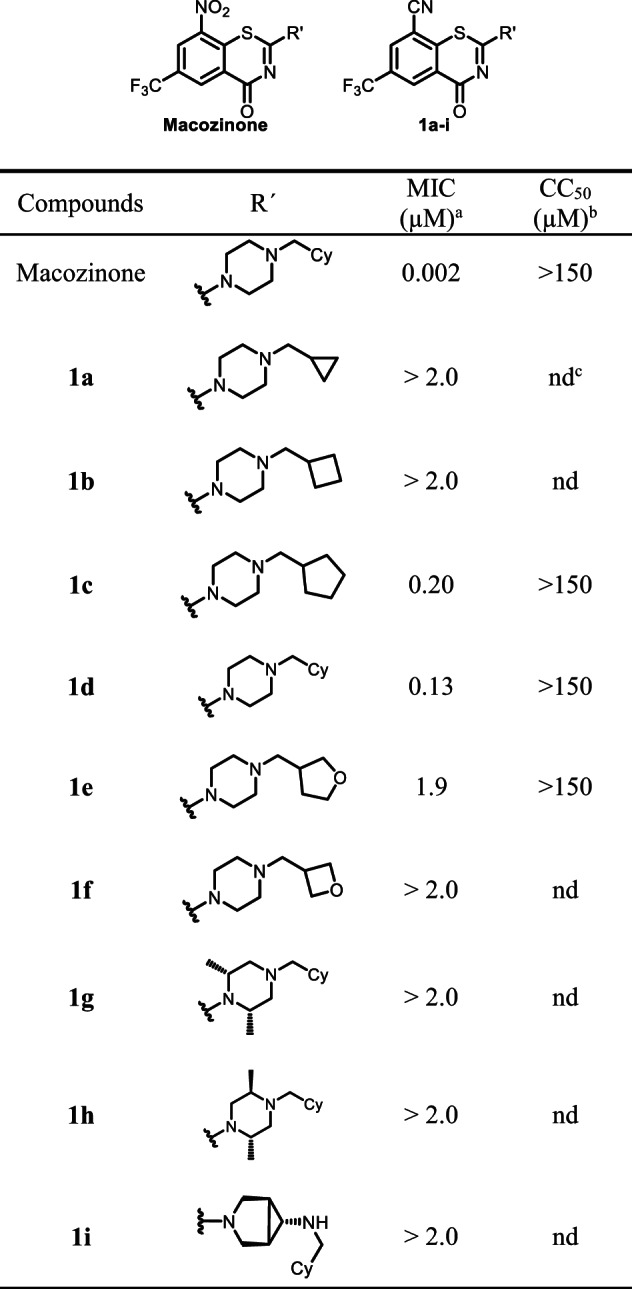
aMinimum inhibitory concentration against M. tuberculosis H37Rv
bThe concentration required to decrease cell viability of Vero cells by 50%; cnd = not determined
The calculated and experimentally determined physicochemical properties of the analogs and macozinone are shown in Tables 2 for comparison. We calculated the overall lipophilicity as measured by the partition coefficient clogP, the lipophilic ligand efficiency (LLE, logMIC -logP), the total polar surface area (tPSA) and experimentally measured the melting points as a proxy for compound solubility [43]. Replacement of the C-8 nitro group of macozinone with a nitrile in 1d results in a modest decrease in clogP of only 0.3 units. However, the LLE for 1d decreases by 1.5 units relative to macozinone, due to the loss of antimycobacterial activity. The nitrile analog does significantly reduce the tPSA, which is expected to enhance membrane permeability. Among the other active nitrile analogs, compound 1e containing a tetrahydrofuran-3-ylmethyl substituent had an improved LLE attributed to the dramatically lower clogP that offset the loss of potency.
Table 2.
The ClogP, LLE, and melting point of 1a–i
| Compounds | ClogPa | LLEb | Mp (oC) | tPSA |
|---|---|---|---|---|
| Macozinone | 5.1 | 3.6 | 185–187 | 87.7 |
| 1a | 3.1 | – | 151–153 | 59.7 |
| 1b | 3.7 | – | 162–165 | 59.7 |
| 1c | 4.2 | 2.5 | 196–198 | 59.7 |
| 1d | 4.8 | 2.1 | 218–221 | 59.7 |
| 1e | 2.0 | 3.8 | 173–175 | 68.9 |
| 1f | 1.3 | – | 172–174 | 68.9 |
| 1g | 5.8 | – | 182–185 | 59.7 |
| 1h | 5.8 | – | 180–182 | 59.7 |
| 1i | 4.1 | – | 193–195 | 68.5 |
aClogP and tPSA were determined by Chemdraw Professional Version 16.0
bLipophilic Ligand Efficiency (LLE) was calculated from the equation: LLE = log10MIC − Clog10P
The metabolic stability of 1d and macozinone were evaluated using mouse and human liver microsomes with and without NADPH. Macozinone was included as a positive control while samples without NADPH served as negative controls. In the absence of NADPH neither macozinone nor 1d were metabolized. The metabolic stability of macozinone was consistent with previous reports showing a half-life of 34–43 min [30]. Compound 1d was metabolized approximately twice as quickly in human liver microsomes, but nearly to the same extent in mouse liver microsomes compared to macozinone (Table 3).
Table 3.
Microsomal stability of 1d
| Compounds | Mouse | Human | ||
|---|---|---|---|---|
| (% remaining at 30 min) | ||||
| With NADPH | Without NADPH | With NADPH | Without NADPH | |
| Macozinone | 50.7 | 102 | 46.8 | 101 |
| 1d | 38.6 | 96.4 | 26.3 | 102 |
Compound 1d and macozinone were evaluated in male ICR mice to determine their pharmacokinetic parameters. Compound 1d reached a maximum plasma concentration of 110 ng/mL or 240 nM, following oral dosing at 10 mg/mL and showed a 2.1-fold increase in the intrinsic half-life relative to macozinone (determined from intravenously (i.v.) dosing). The increased half-life of 1d was driven by a corresponding 2.0-fold increase in the volume of distribution (V) since the clearance (Cl) remained nearly unchanged. The enhanced volume of distribution may in turn reflect the reduced tPSA that can enhance membrane permeability. However, the oral bioavailability of 1d was decreased twofold relative to macozinone that may in turn potentially reflect lower solubility of 1d, which is predicted based on the substantially higher melting point of 1d compared to macozinone. While the increased volume of distribution is favorable, compounds 1d was deemed to possess inadequate PK parameters due to its low oral bioavailability. To the best of our knowledge, this report also provides the first data for the oral bioavailability of macozinone (Table 4).
Table 4.
Pharmacokinetic parameters of macozinone and 1d in male ICR mice following oral or intravenous administration
| Parameters | Units | Macozinone | 1d | ||
|---|---|---|---|---|---|
| PO | iv | PO | iv | ||
| t1/2β | h | 1.68 | 0.86 | 1.74 | 1.84 |
| Tmax | h | 0.25 | 0.033 | 0.25 | 0.033 |
| Cmax | ng/ml | 251 | 1230 | 110 | 1335 |
| AUC(0-t) | hang/ml | 354 | 423 | 174 | 445 |
| Vd | L/kg | – | 5.64 | – | 11.3 |
| Cl | ml/min/kg | – | 75.8 | – | 71.2 |
| F | % | 16.7 | – | 7.8 | – |
aThe doses of oral and intravenous administration were 10 and 2 mg/kg, respectively
In conclusion, we successfully replaced the 8-nitro group of macozinone with an isosteric nitrile moiety to address potential concerns of safety, stability, and resistance. Compound 1d, containing an identical C-2 substituent as found in macozinone, possessed respectable antimycobacterial activity with an MIC of 130 nM, displayed no cytotoxicity, and an improved intrinsic half-life owing to its greater volume of distribution. However, 1d has approximately twofold lower oral bioavailability than macozinone, which we demonstrated was also low. We explored the SAR of the C-2 substituent of 1d and showed only conservative modifications were tolerated. Further optimization at C-2 will be required to enhance oral bioavailability and exposure as well as potency before the 8-cyano benzothiazinones can be advanced.
Methods
General
Unless otherwise noted, reagents and materials were obtained from commercial suppliers and were used without further purification. Solvents were dried by the appropriate drying agents prior to use. Anhydrous tetrahydrofuran and dichloromethane were obtained from commercial sources. All solvents used for routine isolation of products and for chromatography were reagent grade. Moisture- and air-sensitive reactions were carried out under an atmosphere of Argon. All reactions were monitored by thin layer chromatography (TLC) and column chromatography purification was performed using 230-400 mesh silica gel. NMR spectra were measured on Bruker AV400 spectrometer at 400 or 300 MHz for 1H spectra and at 100 or 75 MHz for 13C spectra using CDCl3, CD3OD, and D2O, and calibrated from the residual solvent signal. All final products were characterized by 1H NMR, 13C NMR, and MS analyses. Except for the known compounds, all new compounds were also characterized and confirmed by HRMS.
2-chloro-3-nitro-5-(trifluoromethyl)benzoic acid (2)
To a mixture of conc. H2SO4 (3.0 mL) and conc. HNO3 (3.0 mL) was added 2-chloro-5-(trifluoromethyl)benzoic acid (0.52 g, 2.32 mmol). After stirred for 1.5 h at 90 °C, the reaction was cooled to room temperature and poured into ice water (10 mL), filtered and washed with cold water (5.0 mL), dried in vacuo to afford the title compound (0.52 g, 84%) as a white solid: 1H NMR (400 MHz, DMSO-d6) δ 8.72 (s, 1H), 8.41 (s, 1H); 13C NMR (DMSO-d6) δ 164.8, 150.1, 136.1, 130.2 (d, J = 3.8 Hz), 129.4, 129.0, 127.7, and 124.4.
3-amino-2-chloro-5-(trifluoromethyl)benzoic acid (3)
A mixture of intermediate 2 (0.5 g, 1.9 mmol, 1.0 equiv.), reductive iron powder (0.5 g, 8.7 mmol, 4.6 equiv.), NH4Cl (0.7 g, 14 mmol, 7.4 equiv.) in 60 mL of ethanol aqueous solution (ethanol: water, 5:1) was stirred at 70 °C and monitored by TLC. After 16 h, the solvent was removed, and the residue was suspended in 10 mL of ethyl acetate. The suspension was filtered, and the filtrate was washed with 1 N HCl (10 mL) and brine (15 mL), dried over Na2SO4, filtered and concentrated in vacuo to afford the title compound (0.40 g, 91%) as a brown solid: Rf = 0.36 (DCM:MeOH, 8:1 with a trace amount of acetic acid); 1H NMR (400 MHz, DMSO-d6) δ 13.53 (s, 1H), 7.21 (s, 1H), 7.09 (s, 1H), 6.09 (s, 2H); 13C NMR (DMSO-d6) δ 166.9, 147.0, 134.2, 128.3 (d, J = 32.4 Hz), 125.5, 122.8, 118.4, and 112.8 (d, J = 20.2 Hz).
2-chloro-3-cyano-5-(trifluoromethyl)benzoic acid (4)
To a mixture of intermediate 3 (0.63 g, 2.63 mmol, 1.0 equiv.) in H2O (5.0 mL) and conc. HCl (0.4 mL), was added dropwise a solution of NaNO2 (0.24 g, 3.42 mmol, 1.3 equiv.) in H2O (0.5 mL) at 0 °C. After stirred at 0–10 °C for 2 h, the solution was added to a mixture of CuCN (0.23 g, 2.63 mmol, 1.0 equiv.) and NaCN (0.19 g, 3.95 mmol, 1.5 equiv.) in H2O (3.0 mL) at 0 °C. The mixture was stirred at 70 °C for 6 h and cooled to room temperature. The pH value was adjusted to 1 with conc. HCl. The crude product was extracted with ethyl acetate (3 × 10 mL), washed with 1 N HCl (1 mL), brine (10 mL), dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by silica gel column chromatography (eluting with 100: 1 DCM:MeOH) to afford the title compound (0.43 g, 65%) as a colorless solid: Rf = 0.47 (DCM:MeOH, 8:1 with a trace amount of acetic acid). 1H NMR (400 MHz, CD3OD) δ 8.37 (s, 1H), 8.35 (s, 1H); 13C NMR (151 MHz, CD3OD) δ 164.3, 139.1, 133.8, 133.2 (d, J = 3.9 Hz), 131.5 (d, J = 3.7 Hz), 129.8, 129.5, 116.4, and 114.0.
2-chloro-3-cyano-5-(trifluoromethyl)benzamide (5)
To a stirred solution of 2-chloro-3-cyano-5-(trifluoromethyl)-benzoic acid (0.25 g, 1.0 mmol, 1.0 equiv.), pyridine (0.05 mL, 0.62 mmol, 0.62 equiv.), and Boc2O (0.28 g, 1.3 mmol, 1.3 equiv.) in 1,4-dioxane (2.0 mL) at room temperature was added ammonium bicarbonate (0.10 g, 1.26 mmol, 1.26 equiv.). The reaction was stirred overnight at room temperature and then partitioned between EtOAc (10.0 mL) and H2O (10.0 mL). The organic layer was separated, washed consecutively with water (10.0 mL) and 0.6 N aqueous HCl (10.0 mL), dried over anhydrous Mg2SO4, and filtered. The filtrate was concentrated under reduced pressure to afford the title compound (0.22 g, 90%) as an off-white solid: 1H NMR (600 MHz, CD3OD) δ 8.30 (d, J = 1.0 Hz, 1H), 8.09 (d, J = 1.0 Hz, 1H); 13C NMR (151 MHz, CD3OD) δ 168.8, 138.3, 133.2 (q, J = 3.6 Hz), 131.3 (q, J = 34.4 Hz), 130.7 (q, J = 4.0 Hz), 124.0 (q, 1JC-F = 272.5 Hz), 116.8, and 115.4.
General procedure for the synthesis of compounds 6a–i
To a pre-cooled mixture of amine (2.04 mmol, 1 equiv) in EtOAc was added 30% aqueous NaOH (0.68 mL, 5.09 mmol, 2.5 equiv) followed by CS2 (0.15 mL, 2.45 mmol, 1.2 equiv). The reaction mixture was stirred for 3 h at 0 °C, then at room temperature for another 3 h. The reaction mixture was filtered and the filter cake was washed by ethyl acetate and dried under vacuum to afford intermediates 6a-i as published in our previous papers [30].
General procedure for the synthesis of compounds 1a–i
A mixture of intermediate 5 (0.16 g, 0.61 mmol, 1.2 equiv), 6a–i (0.51 mmol, 1.0 equiv) and anhydrous K2CO3 (0.08 g, 0.56 mmol, 1.1 equiv) was stirred in anhydrous DMF (5 mL) at 60 °C. After 2 h, the reaction mixture was cooled down to room temperature and poured into ice water. The product was extracted with CH2Cl2 (5 × 20 mL). The organic layer was combined, washed by saturated brine (3 × 5 mL) and dried by anhydrous sodium sulfate which was removed by filtration after 20 min. The solvent CH2Cl2 was removed under reduced pressure. The product was purified by silica flash column chromatography with the indicated solvent system (petroleum ether/ethyl acetate 10:1–>5:1–>2:1–>1:1–>1:2–>1:10–>100% ethyl acetate).
2-(4-(cyclopropylmethyl)piperazin-1-yl)-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazine-8-carbonitrile (1a)
The title compound was prepared from sodium 4-(cyclopropylmethyl)piperazine-1-carbodithioate 6a (0.08 g, 0.34 mmol, 1.0 equiv) and 2-chloro-3-cyano-5-(trifluoromethyl)benzamide 5 (0.10 g, 0.41 mmol, 1.2 equiv) using the general procedure for cyclization to afford compound 1a (0.12 g, 86%) as a light yellow solid: mp 151–153 °C; Rf = 0.20 (1:2 Hexane-EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.91 (d, J = 2.0 Hz, 1H), 8.08 (d, J = 2.0 Hz, 1H), 4.19 (br s, 2H), 3.83 (br s, 2H), 2.69 (br s, 4H), 2.34 (d, J = 6.6 Hz, 2H), 0.91–0.86 (m, 1H), 0.60–0.54 (m, 2H), 0.16-0.11 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 166.4, 159.5, 140.6, 132.9 (q, J = 2.6 Hz), 131.6 (q, J = 2.6 Hz), 130.9 (q, J = 25.9 Hz), 124.8, 122.5 (q, 1JC-F = 203.7 Hz), 114.0, 111.1, 63.3, 52.5, 47.0–46.3 (m), 29.8, 8.2, and 4.1; HRMS (ESI+) m/z [M + H] + calcd for C18H18F3N4OS: 395.1148; found 395.1144 (error 1.0 ppm).
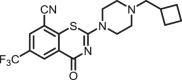
2-(4-(cyclobutylmethyl)piperazin-1-yl)-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazine-8-carbonitrile (1b)
The title compound was prepared from sodium 4-(cyclobutylmethyl)piperazine-1-carbodithioate 6b (0.09 g, 0.34 mmol, 1.0 equiv) and 2-chloro-3-cyano-5-(trifluoromethyl)benzamide 5 (0.10 g, 0.41 mmol, 1.2 equiv) using the general procedure for cyclization to afford compound 1b (0.12 g, 85%) as light yellow solid: mp 162–165 °C; Rf = 0.20 (1:2 Hexane-EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.91 (d, J = 2.0 Hz, 1H), 8.08 (d, J = 2.0 Hz, 1H), 4.13 (m, 2H), 3.77 (m, 2H), 2.55–2.46 (m, 7H), 2.10–2.04 (m, 2H), 1.98–1.82 (m, 2H), 1.75–1.67 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 166.2, 159.2, 140.5, 132.7 (q, J = 3.6 Hz), 131.4 (q, J = 3.6 Hz), 130.5 (q, J = 34.7 Hz), 124.6, 122.4 (q, 1JC-F = 271.4 Hz), 113.9, 110.9, 64.5, 52.6, 47.0–46.4 (m), 33.4, 27.6, and 18.8; HRMS (ESI+) m/z [M + H] + calcd for C19H20F3N4OS: 409.1304; found 409.1302 (error 0.6 ppm).
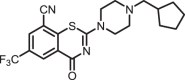
2-(4-(cyclopentylmethyl)piperazin-1-yl)-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazine-8-carbonitrile (1c)
The title compound was prepared from sodium 4-(cyclopentylmethyl)piperazine-1-carbodithioate 6c (0.09 g, 0.34 mmol, 1.0 equiv) and 2-chloro-3-cyano-5-(trifluoromethyl)benzamide 5 (0.10 g, 0.41 mmol, 1.2 equiv) using the general procedure for cyclization to afford compound 1c (0.12 g, 86%) as light yellow solid: mp 196–198 °C; Rf = 0.20 (1:2 Hexane-EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.91 (d, J = 2.0 Hz, 1H), 8.08 (d, J = 2.0 Hz, 1H), 4.15 (m, 2H), 3.78 (m, 2H), 2.58 (m, 4H), 2.33–2.30 (m, 2H), 2.13–2.03 (m, 1H), 1.77–1.75 (m, 2H), 1.61–1.55 (m, 4H), and 1.24–1.18 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 166.3, 159.3, 140.6, 132.7 (q, J = 3.6 Hz), 131.4 (q, J = 3.6 Hz), 130.6 (q, J = 34.7 Hz), 124.5, 122.4 (q, 1JC-F = 271.4 Hz), 113.9, 110.9, 63.9, 52.7, 47.1–46.5 (m), 36.9, and 31.1, 25.1; HRMS (ESI+) m/z [M + H] + calcd for C20H22F3N4OS: 423.1461; found 423.1457 (error 0.9 ppm).
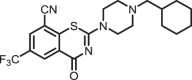
2-(4-(cyclohexylmethyl)piperazin-1-yl)-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazine-8-carbonitrile (1d)
The title compound was prepared from sodium 4-(cyclohexylmethyl)piperazine-1-carbodithioate 6d (0.10 g, 0.34 mmol, 1.0 equiv) and 2-chloro-3-cyano-5-(trifluoromethyl)benzamide 5 (0.10 g, 0.41 mmol, 1.2 equiv) using the general procedure for cyclization to afford compound 1d (0.13 g, 89%) as light yellow solid: mp 218–221 °C; Rf = 0.20 (1:2 Hexane-EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.91 (d, J = 2.0 Hz, 1H), 8.07 (d, J = 2.0 Hz, 1H), 4.14 (s, 2H), 3.77 (s, 2H), 2.53 (s, 4H), 2.18 (d, J = 7.1 Hz, 2H), 1.80–1.66 (m, 5H), 1.52–1.46 (m, 1H), 1.29–1.15 (m, 3H), 0.93–0.84 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 166.5, 159.4, 140.7, 132.9 (q, J = 3.7 Hz), 131.6 (q, J = 3.6 Hz), 130.8 (q, J = 34.9 Hz), 124.8, 122.5 (q, 1JC-F = 274.2 Hz), 114.1, 111.1, 65.2, 53.1, 47.2–46.7 (m), 35.1, 31.8, and 26.8, 26.1; HRMS (ESI+) m/z [M + H] + calcd for C21H24F3N4OS: 437.1617; found 437.1613 (error 1.0 ppm).
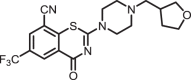
4-Oxo-2-(4-((tetrahydrofuran-3-yl)methyl)piperazin-1-yl)-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazine-8-carbonitrile (1e)
The title compound was prepared from sodium 4-((tetrahydrofuran-3-yl)methyl)piperazine-1-carbodithioate 6e (0.09 g, 0.34 mmol, 1.0 equiv) and 2-chloro-3-cyano-5-(trifluoromethyl)benzamide 5 (0.10 g, 0.41 mmol, 1.2 equiv) using the general procedure for cyclization to afford compound 1e (0.11 g, 76%) as light yellow solid: mp 173–175 °C; Rf = 0.20 (1:2 Hexane-EtOAc); 1H NMR (300 MHz, CDCl3): δ 8.92 (d, J = 2.0 Hz, 1H), 8.08 (d, J = 2.0 Hz, 1H), 4.16–4.11 (m, 2H), 3.91–3.72 (m, 5H), 3.59–3.54 (m, 1H), 2.62 (m, 4H), 2.50–2.43 (m, 2H), 2.10–1.99 (m, 1H), 1.68–1.58 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 166.4, 159.5, 140.6, 132.9 (q, J = 2.7 Hz), 131.6 (q, J = 2.7 Hz), 130.9 (q, J = 26.0 Hz), 124.8, 122.5 (q, 1JC-F = 203.5 Hz), 114.0, 111.1, 72.1, 67.8, 61.5, 52.8, 47.0–46.6 (m), 36.7, 30.6, and 29.8; HRMS (ESI+) m/z [M + H] + calcd for C19H20F3N4O2S: 425.1254; found 425.1249 (error 1.1 ppm).
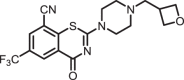
2-(4-(oxetan-3-ylmethyl)piperazin-1-yl)-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazine-8-carbonitrile (1f)
The title compound was prepared from sodium 4-(oxetan-3-ylmethyl)piperazine-1-carbodithioate 6f (0.09 g, 0.34 mmol, 1.0 equiv) and 2-chloro-3-cyano-5-(trifluoromethyl)benzamide 5 (0.10 g, 0.41 mmol, 1.2 equiv) using the general procedure for cyclization to afford compound 1f (0.10 g, 72%) as light yellow solid: mp 172–174 °C; Rf = 0.20 (1:2 Hexane-EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.92 (d, J = 2.0 Hz, 1H), 8.08 (d, J = 2.0 Hz, 1H), 4.85–4.81 (m, 2H), 4.43–4.39 (m, 2H), 4.13 (m, 2H), 3.78 (m, 2H), 3.24 (m, 1H), 2.80–2.77 (m, 2H), 2.56 (m, 3H), and 1.57 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 166.4, 159.5, 140.5, 132.9 (q, J = 2.6 Hz), 131.7 (q, J = 2.7 Hz), 130.9 (q, J = 26.2 Hz), 124.7, 122.5 (q, 1JC-F = 203.7 Hz), 114.0, 111.1, 61.5, 52.7, 46.5, 32.9, and 29.8; HRMS (ESI+) m/z [M + H] + calcd for C18H18F3N4O2S: 411.1097; found 411.1094 (error 0.8 ppm).
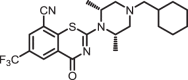
2-((2S,6R)-4-(cyclohexylmethyl)-2,6-dimethylpiperazin-1-yl)-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazine-8-carbonitrile (1g)
The title compound was prepared from sodium (2S,6R)-4-(cyclohexylmethyl)-2,6-dimethylpiperazine-1-carbodithioate 6i (0.10 g, 0.34 mmol, 1.0 equiv) and 2-chloro-3-cyano-5-(trifluoromethyl)benzamide 5 (0.10 g, 0.41 mmol, 1.2 equiv) using the general procedure for cyclization to afford compound 1g (0.13 g, 83%) as light yellow solid: mp 182–185 °C; Rf = 0.20 (1:2 Hexane-EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.93 (d, J = 2.0 Hz, 1H), 8.07 (d, J = 2.0 Hz, 1H), 5.21 (m, 1H), 4.21 (m, 1H), 2.80–2.74 (m, 2H), 2.31–2.15 (m, 4H), 1.83–1.67 (m, 5H), 1.59–1.44 (m, 7H), 1.32–1.11 (m, 3H), 0.98–0.85 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 166.1, 159.1, 141.1, 132.7 (q, J = 3.6 Hz), 131.5 (q, J = 3.6 Hz), 130.5 (q, J = 34.6 Hz), 124.8, 122.5 (q, J = 271.4 Hz), 114.1, 110.8, 64.9, 58.3, 57.8, 51.4, 50.8, 35.4, 31.6, 26.8, 26.1, 20.5, and 20.1; HRMS (ESI+) m/z [M + H] + calcd for C23H28F3N4OS: 465.1930; found 465.1928 (error 0.5 ppm).
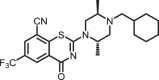
2-((2S,5R)-4-(cyclohexylmethyl)-2,5-dimethylpiperazin-1-yl)-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazine-8-carbonitrile (1h)
The title compound was prepared from sodium (2S,5R)-4-(cyclohexylmethyl)-2,5-dimethylpiperazine-1-carbodithioate 6m (0.10 g, 0.34 mmol, 1.0 equiv) and 2-chloro-3-cyano-5-(trifluoromethyl)benzamide 5 (0.10 g, 0.41 mmol, 1.2 equiv) using the general procedure for cyclization to afford compound 1h (0.13 g, 80%) as light yellow solid: mp 180–182 °C; Rf = 0.20 (1:2 Hexane-EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.92 (d, J = 2.0 Hz, 1H), 8.07 (d, J = 2.0 Hz, 1H), 5.35–3.11 (m, 3H), 3.11 (m, 1H), 2.84–2.80 (m, 1H), 2.44–2.40 (m, 1H), 2.21–2.16 (m, 2H), 1.87–1.83 (m, 1H), 1.75–1.71 (m, 4H), 1.57–1.36 (m, 4H), 1.21–1.14 (m, 3H), 0.99–0.82 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 166.4, 160.0, 140.7, 132.8 (q, J = 3.6 Hz), 131.5 (q, J = 3.6 Hz), 130.6 (q, J = 34.6 Hz), 124.8, 122.4 (q, J = 271.4 Hz), 114.0, 110.9, 61.1, 53.2, 50.8, 49.3, 47.5, 35.3, 31.6, 31.6, 26.8, 26.1, 26.0, 16.4, and 7.7; HRMS (ESI+) m/z [M + H]+ calcd for C23H28F3N4OS: 465.1930; found 465.1930 (error 0.1 ppm).
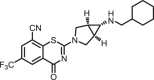
2-((1R,5S,6s)-6-((cyclohexylmethyl)amino)-3-azabicyclo[3.1.0]hexan-3-yl)-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazine-8-carbonitrile (1i)
The title compound was prepared from sodium (1R,5S,6S)-6-((cyclohexylmethyl)amino)-3-azabicyclo[3.1.0]hexane-3-carbodithioate 6p (0.10 g, 0.34 mmol, 1.0 equiv) and 2-chloro-3-cyano-5-(trifluoromethyl)benzamide 5 (0.10 g, 0.41 mmol, 1.2 equiv) using the general procedure for cyclization to afford compound 1i (0.11 g, 71%) as light yellow solid: mp 193–195 °C; Rf = 0.20 (1:2 Hexane-EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.77 (d, J = 2.1 Hz, 1H), 8.63 (d, J = 2.1 Hz, 1H), 3.95–3.91 (m, 1H), 3.84–3.72 (m, 3H), 3.34 (m, 2H), 2.04 (m, 1H), 1.85–1.81 (m, 2H), 1.73–1.63 (m, 5H), 1.42 (m, 1H), 1.23–1.09 (m, 4H), 0.91–0.79 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 174.0, 166.5, 150.6, 143.4, 139.5, 138.1 (q, J = 35.0 Hz), 133.5, 124.2 (q, J = 277.7 Hz), 123.6, 120.0, 74.5, 64.3, 61.3, 58.7, 50.8, 45.5, 40.2, 38.5, 35.6, 34.9, 32.4, 31.5, and 28.2; HRMS (ESI+) m/z [M + H]+ calcd for C22H24F3N4OS: 449.1617; found 449.1613 (error 1.0 ppm).
MIC determination
The test compound MICs against Mtb H37Rv were assessed by the microplate alamar blue assay protocol using macozinone as positive control. Compound stock solutions were prepared in DMSO at a concentration of 32 µg/mL, and the final test concentrations ranged from 3.2 to 0.002 µg/mL. Twofold dilutions of compounds were prepared in 7H9-ADC-TG in a volume of 100 µL in 96-well microplates (BD OptiluxTM, 96-well Microplates, black/clear flat bottom). The TB cultures (100 µL inoculua of 2 × 105 cfu/mL) were added to the media, yielding a final testing volume of 200 µL. The plates were incubated at 37 °C. On the seventh day of incubation, 12.5 µL of 20% Tween 80, and 20 µL of alamar blue (Invitrogen BioSource™) were added to the wells of test plate. After incubation at 37 °C for 16–24 h, fluorescence of the wells was measured at 530 nm (excitation) and 590 nm (emission). The MICs are defined as the lowest concentration effecting a reduction in fluorescence of ≥90% relative to the mean of replicate bacteria-only controls.
Microsomal stability
The selected compounds were dissolved in DMSO to make 10 mM of stock solution. Midazolam, Dextromethorphan, Diclofenac, Omeprazole, and Phenacetin are used as controls. The 5 µL of 10 mM stock solution of the target compounds was diluted to 100 μM of compound solution by adding 495 µL of DMSO. The 2.5 µL control/test compound was mixed with 197.5 µL liver microsome (mouse and human) gently and preincubate at 37 °C for 5 min. The reaction was initiated by adding 50 µL buffer/NADPH working solution (5 mM) and mixed well. At each time point of 0 min and 30 min, 30 µL of reaction solution was taken out and quenched by adding 300 µL of internal standard (10 ng/mL). The mixture was centrifuged at 4000 rpm at 4 °C for 15 min. 100 µL of the supernatant was mixed with 100 µL distilled water and then analyzed by LC-MS/MS.
LC-MS/MS analysis was performed on a Kinetex C18 100 A column (3.0 mm × 30 mm, 2.6 μm) with a gradient mobile phase of acetonitrile/water containing 0.1% formic acid at 0.8 mL/min flow rate. LC condition: solvent A = H2O (0.1% formic acid), solvent B = acetonitrile (0.1% formic acid). Gradient elution: 0.00 min, 95.0% A; 0.50 min, 95% A; 0.80 min, 5% A; 1.50 min, 5% A; 1.51 min, 95% A; 2.00 min, 95% A. The injection volume was 5 μL.
In vivo pharmacokinetic profiles
Animal Care and Welfare Committee of Institute of Materia Medica, Chinese Academy of Medical Sciences approved all animal protocols (1 Xian nong tan Street, Xicheng District, Beijing, China; protocol #SYXK 2014-0023). All animal programs were in compliance with the Guide for the Care and Use of Laboratory Animals issued by Beijing Association on Laboratory Animal Care (BALAC). SPF male ICR mice weighing 22–23 g were divided into two groups with three mice each: one for oral administration and intravenous injection, separately. The tested compound was formulated at a concentration of 1.0 mg/mL for a dose of 10 mg/kg given orally (p.o.) and at 0.4 mg/mL for a dose of 2 mg/kg given i.v. The tested compound was formulated by 0.5% carboxymethyl cellulose and 0.5% Tween 80 for p.o. administration and with 20% HP-β-CD with 4 mol/L HCl for i.v. administration, respectively. Plasma samples were extracted with acetonitrile containing Terfenadine as an internal standard using a 20:1 extractant-to-plasma ratio. Analyte quantitation was performed by a LC/TSQ Quantum Access mass spectrometer (AB Sciex 5500). Chromatographic separation was performed on a Kinetex C18 100 A column (30 mm × 3 mm, 2.6 µm) with an isocratic mobile phase of acetonitrile/water (80:20, v/v) containing 0.1% formic acid at 0.8 mL/min flow rate. The pharmacokinetic parameters were calculated using WinNonlin software version 6.3 based on non-compartmental analysis (Pharsight Corporation, Mountain View, USA). The oral bioavailability was calculated as the ratio between the area under the curve (AUC) following intravenous administration corrected for dose (F = (AUCp.o.×dosei.v.)/(AUCi.v.×dosep.o.)).
Supplementary information
Acknowledgements
This work was financially supported by a grant from the CAMS Innovation Fund for Medical Sciences (CAMS-2017-I2M-1-011).
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1007/s00044-020-02676-4) contains supplementary material, which is available to authorized users.
References
- 1.Global Tuberculosis Report 2020. Geneva: World Health Organization; 2020.
- 2.Stehr M, Elamin AA, Singh M. Filling the pipeline-new drugs for an old disease. Current Topics Med Chem. 2014;14:110–29.. doi: 10.2174/1568026613666131113152908. [DOI] [PubMed] [Google Scholar]
- 3.Makarov V, Manina G, Mikusova K, Möllmann U, Ryabova O, Saint-Joanis B, et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science. 2009;324:801–4. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manina G, Pasca MR, Buroni S, De Rossi E, Riccardi G. Decaprenylphosphoryl-β-D-ribose 2’-epimerase from Mycobacterium tuberculosis is a magic drug target. Current Med Chem. 2010;17:3099–108. doi: 10.2174/092986710791959693. [DOI] [PubMed] [Google Scholar]
- 5.Mikusová K, Makarov V, Neres J. DprE1-from the discovery to the promising tuberculosis drug target. Current Pharm Design. 2014;20:4379–403. doi: 10.2174/138161282027140630122724. [DOI] [PubMed] [Google Scholar]
- 6.Makarov V, Lechartier B, Zhang M, Neres J, van der Sar AM, Raadsen SA, et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol Med. 2014;6:372–83.. doi: 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolucka BA. Biosynthesis of D-arabinose in mycobacteria – a novel bacterial pathway with implications for antimycobacterial therapy. The FEBS J. 2008;275:2691–711. doi: 10.1111/j.1742-4658.2008.06395.x. [DOI] [PubMed] [Google Scholar]
- 8.Mikusová K, Huang H, Yagi T, Holsters M, Vereecke D, D’Haeze W, et al. Decaprenylphosphoryl arabinofuranose, the donor of the D-arabinofuranosyl residues of mycobacterial arabinan, is formed via a two-step epimerization of decaprenylphosphoryl ribose. J Bacteriol. 2005;187:8020–5. doi: 10.1128/jb.187.23.8020-8025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lechartier B, Hartkoorn RC, Cole ST. In vitro combination studies of benzothiazinone lead compound BTZ043 against Mycobacterium tuberculosis. Antimicrobial Agents Chemother. 2012;56:5790–3. doi: 10.1128/aac.01476-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trefzer C, Rengifo-Gonzalez M, Hinner MJ, Schneider P, Makarov V, Cole ST, et al. Benzothiazinones: prodrugs that covalently modify the decaprenylphosphoryl-β-D-ribose 2′-epimerase DprE1 of Mycobacterium tuberculosis. J Am Chem Soc. 2010;132:13663–5.. doi: 10.1021/ja106357w. [DOI] [PubMed] [Google Scholar]
- 11.Sommer R, Neres J, Piton J, Dhar N, van der Sar A, Mukherjee R, et al. Fluorescent benzothiazinone analogues efficiently and selectively label Dpre1 in mycobacteria and actinobacteria. ACS Chem Biol. 2018;13:3184–92. doi: 10.1021/acschembio.8b00790. [DOI] [PubMed] [Google Scholar]
- 12.Makarov V, Neres J, Hartkoorn RC, Ryabova OB, Kazakova E, Šarkan M, et al. The 8-pyrrole-benzothiazinones are noncovalent inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrobial Agents Chemother. 2015;59:4446–52.. doi: 10.1128/AAC.00778-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang G, Aldrich CC. Macozinone: revised synthesis and crystal structure of a promising new drug for treating drug-sensitive and drug-resistant tuberculosis. Acta Crystallographica Section C. 2019;75:1031–5.. doi: 10.1107/S2053229619009185. [DOI] [PubMed] [Google Scholar]
- 14.Richter A, Rudolph I, Möllmann U, Voigt K, Chung CW, Singh OMP, et al. Novel insight into the reaction of nitro, nitroso and hydroxylamino benzothiazinones and of benzoxacinones with Mycobacterium tuberculosis DprE1. Scientific Rep. 2018;8:13473. doi: 10.1038/s41598-018-31316-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao C, Peng C, Shi Y, You X, Ran K, Xiong L, et al. Benzothiazinethione is a potent preclinical candidate for the treatment of drug-resistant tuberculosis. Scientific Rep. 2016;6:1–9.. doi: 10.1038/s41598-016-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao C, Ye T-H, Wang N-Y, Zeng X-X, Zhang L-D, Xiong Y, et al. Synthesis and structure–activity relationships evaluation of benzothiazinone derivatives as potential anti-tubercular agents. Bioorganic Med Chem Lett. 2013;23:4919–22.. doi: 10.1016/j.bmcl.2013.06.069. [DOI] [PubMed] [Google Scholar]
- 17.Tiwari R, Miller PA, Chiarelli LR, Mori G, Šarkan M, Centárová I, et al. Design, syntheses, and anti-TB activity of 1, 3-benzothiazinone azide and click chemistry products inspired by BTZ043. ACS Med Chem Lett. 2016;7:266–70.. doi: 10.1021/acsmedchemlett.5b00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li P, Wang B, Zhang X, Batt SM, Besra GS, Zhang T, et al. Identification of novel benzothiopyranone compounds against Mycobacterium tuberculosis through scaffold morphing from benzothiazinones. Eur J Med Chem. 2018;160:157–70.. doi: 10.1016/j.ejmech.2018.09.042. [DOI] [PubMed] [Google Scholar]
- 19.Karoli T, Becker B, Zuegg J, Möllmann U, Ramu S, Huang JX, et al. Identification of antitubercular benzothiazinone compounds by ligand-based design. J Med Chem. 2012;55:7940–4.. doi: 10.1021/jm3008882. [DOI] [PubMed] [Google Scholar]
- 20.Lv K, Tao Z, Liu Q, Yang L, Wang B, Wu S, et al. Design, synthesis and antitubercular evaluation of benzothiazinones containing a piperidine moiety. Eur J Med Chem. 2018;151:1–8.. doi: 10.1016/j.ejmech.2018.03.060. [DOI] [PubMed] [Google Scholar]
- 21.Lv K, Wang A, Tao Z, Fu L, Liu H, Wang B, et al. hERG optimizations of IMB1603, discovery of alternative benzothiazinones as new antitubercular agents. Eur J Med Chem. 2019;179:208–17.. doi: 10.1016/j.ejmech.2019.06.053. [DOI] [PubMed] [Google Scholar]
- 22.Lv K, You X, Wang B, Wei Z, Chai Y, Wang B, et al. Identification of better pharmacokinetic benzothiazinone derivatives as new antitubercular agents. ACS Med Chem Lett. 2017;8:636–41.. doi: 10.1021/acsmedchemlett.7b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng C-T, Gao C, Wang N-Y, You X-Y, Zhang L-D, Zhu Y-X, et al. Synthesis and antitubercular evaluation of 4-carbonyl piperazine substituted 1, 3-benzothiazin-4-one derivatives. Bioorganic Med Chem Lett. 2015;25:1373–6.. doi: 10.1016/j.bmcl.2015.02.061. [DOI] [PubMed] [Google Scholar]
- 24.Piton J, Vocat A, Lupien A, Foo CS, Riabova O, Makarov V et al. Structure-based drug design and characterization of sulfonyl-piperazine benzothiazinone inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrobial Agents Chemother. 2018;62. [DOI] [PMC free article] [PubMed]
- 25.Tiwari R, Miller PA, Cho S, Franzblau SG, Miller MJ. Syntheses and antituberculosis activity of 1, 3-benzothiazinone sulfoxide and sulfone derived from BTZ043. ACS Med Chem Letters. 2015;6:128–33.. doi: 10.1021/ml5003458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang A, Lv K, Tao Z, Gu J, Fu L, Liu M, et al. Identification of benzothiazinones containing an oxime functional moiety as new anti-tuberculosis agents. Eur J Med Chem. 2019;181:111595.. doi: 10.1016/j.ejmech.2019.111595. [DOI] [PubMed] [Google Scholar]
- 27.Xiong L, Gao C, Shi Y-J, Tao X, Peng C-T, Rong J et al. Metabolism of SKLB-TB1001, a potent antituberculosis agent, in animals. Antimicrobial Agents Chemother. 2018;62. [DOI] [PMC free article] [PubMed]
- 28.Xiong L, Gao C, Shi Y-J, Tao X, Rong J, Liu K-L, et al. Identification of a new series of benzothiazinone derivatives with excellent antitubercular activity and improved pharmacokinetic profiles. RSC Adv. 2018;8:11163–76.. doi: 10.1039/C8RA00720A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang R, Lv K, Wang B, Li L, Wang B, Liu M, et al. Design, synthesis and antitubercular evaluation of benzothiazinones containing an oximido or amino nitrogen heterocycle moiety. RSC Adv. 2017;7:1480–3.. doi: 10.1039/C6RA25712G. [DOI] [Google Scholar]
- 30.Zhang G, Howe M, Aldrich CC. Spirocyclic and bicyclic 8-nitrobenzothiazinones for tuberculosis with improved physicochemical and pharmacokinetic properties. ACS Med Chem Lett. 2019;10:348–51.. doi: 10.1021/acsmedchemlett.8b00634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaikh MH, Subhedar DD, Arkile M, Khedkar VM, Jadhav N, Sarkar D, et al. Synthesis and bioactivity of novel triazole incorporated benzothiazinone derivatives as antitubercular and antioxidant agent. Bioorg Med Chem Lett. 2016;26:561–9. doi: 10.1016/j.bmcl.2015.11.071. [DOI] [PubMed] [Google Scholar]
- 32.Majewski MW, Tiwari R, Miller PA, Cho S, Franzblau SG, Miller MJDesign. syntheses, and anti-tuberculosis activities of conjugates of piperazino-1,3-benzothiazin-4-ones (pBTZs) with 2,7-dimethylimidazo [1,2-a]pyridine-3-carboxylic acids and 7-phenylacetyl cephalosporins. Bioorg Med Chem Lett. 2016;26:2068–71. doi: 10.1016/j.bmcl.2016.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Armenise D, Muraglia M, Florio MA, De Laurentis N, Rosato A, Carrieri A, et al. 4H-1,4-benzothiazine, dihydro-1,4-benzothiazinones and 2-amino-5-fluorobenzenethiol derivatives: design, synthesis and in vitro antimicrobial screening. Archiv der Pharmazie. 2012;345:407–16. doi: 10.1002/ardp.201100309. [DOI] [PubMed] [Google Scholar]
- 34.Tiwari R, Moraski GC, Krchňák V, Miller PA, Colon-Martinez M, Herrero E, et al. Thiolates chemically induce redox activation of BTZ043 and related potent nitroaromatic anti-tuberculosis agents. J Am Chem Soc. 2013;135:3539–49.. doi: 10.1021/ja311058q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foo CS, Lechartier B, Kolly GS, Boy-Röttger S, Neres J, Rybniker J, et al. Characterization of DprE1-mediated Benzothiazinone resistance in mycobacterium tuberculosis. Antimicrobial Agents Chemother. 2016;60:6451–9. doi: 10.1128/aac.01523-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trefzer C, Škovierová H, Buroni S, Bobovská A, Nenci S, Molteni E, et al. Benzothiazinones are suicide inhibitors of mycobacterial decaprenylphosphoryl-β-D-ribofuranose 2’-oxidase DprE1. J Am Chem Soc. 2012;134:912–5. doi: 10.1021/ja211042r. [DOI] [PubMed] [Google Scholar]
- 37.Neres J, Pojer F, Molteni E, Chiarelli LR, Dhar N, Boy-Röttger S, et al. Structural basis for benzothiazinone-mediated killing of Mycobacterium tuberculosis. Sci Transl Med. 2012;4:150ra21. doi: 10.1126/scitranslmed.3004395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fleming FF, Yao L, Ravikumar PC, Funk L, Shook BC. Nitrile-containing Pharmaceuticals: efficacious roles of the nitrile pharmacophore. J Med Chem. 2010;53:7902–17. doi: 10.1021/jm100762r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu L, Kong C, Fumagalli M, Savková K, Xu Y, Huszár S, et al. Design, synthesis and evaluation of covalent inhibitors of DprE1 as antitubercular agents. Eur J Med Chem. 2020;208:112773. doi: 10.1016/j.ejmech.2020.112773. [DOI] [PubMed] [Google Scholar]
- 40.Richter A. Synthese von benzothiazinonen und derivaten als DprE1 hemmstoffe mit antimykobakterieller Aktivität. Martin-Luther-Universität Halle-Wittenberg: Germany, 2017.
- 41.Zhang G, Aldrich CC. Macozinone: revised synthesis and crystal structure of a promising new drug for treating drug-sensitive and drug-resistant tuberculosis. Acta crystallographica Section C. Struct Chem. 2019;75:1031–5. doi: 10.1107/s2053229619009185. [DOI] [PubMed] [Google Scholar]
- 42.Makarov AV, Cole ST, Möllmann U. New benzothiazinone derivatives and their use as antibacterial agents. Google Patents, 2011.
- 43.Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov. 2007;6:881–90.. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.