

Abstract
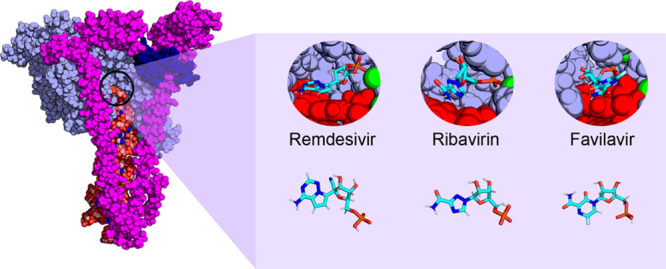
Recent efforts to repurpose drugs to combat COVID-19 have identified Remdesivir as a candidate. It acts on the RNA-dependent, RNA polymerase (RdRp) of the SARS-CoV-2 virus, a protein complex responsible for mediating replication of the virus’s genome. However, its exact action mechanism, and that of other nucleotide analogue inhibitors, is not known. In this study, we examine at the molecular level the interaction of this drug and that of similar nucleotide analogue inhibitors, ribavirin and favilavir, by relying on atomistic molecular simulations and advanced sampling. By analyzing the binding free energies of these different drugs, it is found that all of them bind strongly at the active site. Surprisingly, however, ribavirin and favilavir do not bind the nucleotide on the complementary strand as effectively and seem to act by a different mechanism than remdesivir. Remdesivir exhibits similar binding interactions to the natural base adenine. Moreover, by analyzing remdesivir at downstream positions of the RNA, we also find that, consistent with a “delayed” termination mechanism, additional nucleotides can be incorporated after remdesivir is added, and its highly polar 1′-cyano group induces a set of conformational changes that can affect the normal RdRp complex function. By analyzing the fluctuations of residues that are altered by remdesivir binding, and comparing them to those induced by lethal point mutations, we find a possible secondary mechanism in which remdesivir destabilizes the protein complex and its interactions with the RNA strands.
Short abstract
Through molecular simulation, possible mechanisms of action of the nucleotide-analogue inhibitors remdesivir, favilavir, and ribavirin on the RNA-dependent RNA polymerase of SARS-CoV-2 are elucidated.
Introduction
A new coronavirus of zoonotic origin, SARS-CoV-2, is the etiological agent responsible for the 2019–2020 viral pneumonia COVID-19 outbreak.1−4 At the time of writing, no drug has been established to be completely efficacious against SARS-CoV-2. In some studies, however, remdesivir, from Gilead Sciences Inc., has shown inhibition in vitro and encouraging results in ongoing Phase III trials.5−7 Preliminary data indicate that patients who took remdesivir recovered 4 days faster than patients taking a placebo (median recovery time reduced from 15 days to 11 days with remdesivir).8 In addition, Japan has approved treatment with remdesivir, and the US Food and Drug Administration has authorized remdesivir for emergency use.7,9
Remdesivir is a broad-spectrum antiviral prodrug of an adenosine analogue that has shown inhibitory effects against SARS-CoV and MERS.10−13 As a nucleotide analogue inhibitor, remdesivir acts by inhibiting the coronaviruses’ RNA-dependent RNA polymerase (RdRp), a complex that mediates the replication of their genome and is, therefore, a prime target. RdRp is composed of three different nonstructural proteins: the catalytic subunit nsp12 and its two accessory subunits nsp7 and nsp8.14−16 Recently, several structures of nsp12 in complex with nsp7 and nsp8 have been solved with some differences.14,17 One of these (PDB code: 7BV2) shows nsp12 in a complex with one molecule of nsp8 and nsp7, as well as an 11-base RNA primer-template with remdesivir covalently incorporated at the first base pair of the primer strand; in this report, the authors suggest that remdesivir acts by “immediate” termination of the RNA chain.17 The other structure (PDB code: 6YYT) has nsp12 in a complex with two molecules of nsp8 and one nsp7, where the nsp8 also includes the “sliding poles” of nsp8 and a second turn of RNA. That structure does not incorporate remdesivir. The authors of that study proposed that a “delayed” termination mechanism is more likely than an “immediate” mechanism, as several nucleotides can be added after the addition of remdesivir.14
In order to shed light on these contrasting structures and hypotheses about the mechanism of nucleotide analogue inhibitors, in this work we examine the action of remdesivir, ribavirin, and favilavir. By comparing these three drugs, all of which are nucleotide inhibitors, we seek to (1) work within a comparative framework that might highlight the advantages of one inhibitor over another, (2) identify the advantages of remdesivir, if any, over other drugs, and (3) arrive at general principles that might help the development of other drugs. By relying on atomistic-level molecular simulations, coupled with precise calculations of the free energy, we quantify the binding of these drugs. We revisit the proposed hypotheses for remdesivir action with a detailed model and present additional evidence in support of a “delayed” termination mechanism. Importantly, we have also discovered a previously unknown, secondary mechanism of action for remdesivir. In the first part of our Article, we focus on the drugs in their monophosphate forms, which bind noncovalently to the RdRp complex. We begin by providing insights into the structural differences between the drugs and the natural adenine nucleotide, and we then compare the drugs in terms of their binding free energies obtained by thermodynamic integration (TI) and multistate Bennett’s acceptance ratio (MBAR). Our results are rationalized in terms of the hydrogen bonds that the nucleotide analogues make with their local environment. Whereas ribavirin and favilavir show different binding conformations, we find that remdesivir exhibits the strongest binding and makes similar interactions to adenine in the active site, which points to its potential as a repurposed drug. In the second part of this study, we focus on understanding the mechanism of action of remdesivir at the molecular level. We do so by covalent incorporation of this drug into the RNA nascent strand at different downstream positions. We further explore the stability of the complex in terms of atomic fluctuations and active-site conformational dynamics by comparing the effects of several lethal mutations and remdesivir with the RdRp complex.
Results and Discussion
Molecular dynamics simulations were performed using the Amber18 suite and the ff14SB force field for the proteins,18 ff.RNA.OL3 for the RNA,19 and GAFF for remdesivir, favilavir, and ribavirin missing parameters.20 For the first part of this study, the RdRp complex structure from Yin et al. was used (PDB code: 7BV2). The drugs studied and the natural base adenine that these drugs replace are shown in Figure 1B. In particular, the main difference between remdesivir and adenine is a nitrile group at the ribose part of the compound, whereas favilavir and ribavirin show larger differences.
Figure 1.

(A) Global structure of the RNA at the nsp12 catalytic site: template and nascent strands are shown in cyan, as well as the Mg2+ ions in orange. (B) Chemical structure of the monophosphate form of adenine and the other nucleotide analogues studied here. (C) Zooming in at the nsp12 catalytic site: Mg2+ ions are shown as orange spheres, and the RNA nucleotides are shown as sticks. The last nucleotide in the nascent strand, 3′-end of the RNA chain, is located next to Mg2+ B, and the nucleotide that is being incorporated to the 3′-end of the nascent RNA strand is located next to Mg2+ A. The atoms that are subject to further covalent bond formation, phosphorus (P) and oxygen (O3′), are both highlighted as yellow and red spheres, respectively. (D) Probability distribution for the distances between the phosphate group belonging to the monophosphate nucleotide analogues and Mg2+ A located at the catalytic site. (E) Probability distribution for the distances between phosphorus atom (P) belonging to the phosphate group of the monophosphate analogues and O3′ atom of the 3′-terminal nucleotide. In both cases, green, pink, blue, and black are used for remdesivir, adenine, favilavir, and ribavirin, respectively.
Adenine-MP, Remdesivir-MP, Favilavir-MP, and Ribavirin-MP in Complex with RdRp (PDB code: 7BV2): Structural Analysis and Absolute Free Energy Calculations
It is now known that there is at least one Mg2+ ion in the active site that plays a large role in the stability of the RNA.14,17 To study the stability of these nucleotide analogues at the RdRp catalytic site, we calculated the distance from the monophosphate to the closest Mg2+ (Mg2+ A, see Figure 1A,C for clarification). From Figure 1D, one can appreciate the highly stable interaction between the monophosphate group and the Mg2+ ion, which is essential for RNA stability. Note that all these nucleotide analogue compounds show a narrow distribution centered at 3.6 Å. In addition, we have examined the stability of the active site by assessing the distance between the phosphate group of the monophosphate analogues and the O3′ atom of the 3′-end of the nascent RNA chain (Figure 1A,C shows the nsp12 catalytic site architecture). Typically, such a distance has to be under 4 Å for efficient catalysis when forming the phosphodiester bond (e.g., 3.5–4.0 Å suggested by previous computational studies21,22). Figure 1E shows the distribution of the O3′–P distance for remdesivir-MP (green), favilavir-MP (blue), ribavirin-MP (black), and adenine-MP (pink). The natural nucleotide adenine-MP exhibits the lowest values, centered at 3.6 Å; followed by favilavir (3.8 Å), remdesivir (4.2 Å), and ribavirin (4.4 Å). These results suggest that the incorporation of favilavir-MP should be possible, but with reduced efficiency compared to adenine-MP. Ribavirin-MP and remdesivir-MP exhibit a mean value above 4.0 Å, but the addition should still be possible, albeit with a lower efficiency due to the wide distribution observed in Figure 1E.
We have also relied on thermodynamic integration (TI) and multistate Bennett acceptance ratio (MBAR) to determine the free energy of binding for this series of nucleotide analogues in the monophosphate form.23 This was done in order to verify the validity of our simulations, since both methods should give us similar results at equilibrium. More specifically, we used particle mesh Ewald molecular dynamics (PMEMD) as implemented in Amber 1824 with 11 windows per integration and 150 ns per window. In addition, multiple runs starting from different configurations were considered, all selected from previous long MD trajectories. Three independent replicas were used for each molecule. The results are shown in Table 1.
Table 1. Absolute Binding Free Energies of the Drugs in Their Monophosphate Form Expressed as the Averaged Value from Three Independent Replicasa.
| remdesivir |
favilavir |
ribavirin |
||||
|---|---|---|---|---|---|---|
| method | TI | MBAR | TI | MBAR | TI | MBAR |
| receptor | –130.7 ± 3.9 | –116.4 ± 6.0 | –79.9 ± 1.7 | –69.2 ± 1.9 | –135.6 ± 6.3 | –129.6 ± 4.2 |
| water | –95.7 ± 0.3 | –83.7 ± 0.3 | –62.4 ± 1.2 | –53.9 ± 1.2 | –107.2 ± 1.0 | –100.0 ± 1.3 |
| ΔG | –35.0 ± 3.9 | –32.7 ± 6.0 | –17.4 ± 2.1 | –15.2 ± 2.2 | –28.4 ± 6.4 | –29.6 ± 4.4 |
All values are expressed in kcal/mol. The last row shows the total ΔG, which is the free energy of the drug with the receptor less the free energy of the drug in water.
The data in Table 1 serve to highlight the thermodynamic stability of these drugs at the RdRp complex catalytic site. All the nucleotide analogues considered here exhibit significant, negative absolute binding free energies: remdesivir-MP shows the strongest binding, with ribavirin-MP closely behind. Favilavir-MP exhibits weaker binding energies than both of the other drugs. In all cases, the results of TI and MBAR are in agreement with each other within statistical uncertainty, lending support to the reproducibility of the calculations. Further analysis of the TI and MBAR, including the convergence of each window and smoothness of the energy derivative with respect to λ, can be found in the Supporting Information. While the magnitudes of these free energies are large, especially for remdesivir-MP and ribavirin-MP, they are typical for these types of systems, as shown in other recent computational studies that calculated their free energies using an approximate MM-PBSA scheme.25−27 Note that while our calculations only represent the binding of the monophosphate form of the drugs at the active site, experiments would likely also include some combination of the binding of the triphosphate form, hydrolysis to the active monophosphate form, and transfer of the monophosphate form to RNA strand,28 rendering comparisons of our free energies with experimental binding constants less useful. Instead, these binding free energies should be interpreted relative to each other, as they strictly represent the stability of each drug in their optimal configuration at the active site, and viewed as a measure of their relative success as a nucleotide analogue in translocation.
The free energies shown in Table 1 can be rationalized by closer inspection of the interactions between the drugs and their local environment. The results of our hydrogen bond analyses for all drugs in their monophosphate form bound to their complementary base, and their interactions with surrounding residues, are shown in Figure 2; a more detailed analysis of interactions can be found in the Supporting Information.
Figure 2.

Hydrogen bonds that the different drugs in their monophosphate form and the natural nucleotide adenine do with their environment. (A) Distribution of the base-pair hydrogen bond distances between the drug (remdesivir as an example is shown in the inset) and the base-pair uracil on the complementary strand. The upper and lower panels show the distributions of the different hydrogen bonds as shown in the inset. (B) All of the interactions that the drugs (shown as chemical structure) make with their environment, including hydrogen bonds to their closest nucleotides. The colors of the interactions correspond to the color shown in the legend. The interaction maps were prepared with BIOVIA Discovery Studio Visualizer.29
Figure 2A shows the distribution of distances between the heavy atoms that are hydrogen-bonded to the drug/adenine and its complementary base on the template strand. In all cases, probability distribution curves were obtained from 3 independent replicas of 100 ns each. While adenine and remdesivir display narrow distributions, which reflect the fact that once hydrogen bonds are formed they persist over the vast majority of the simulation time, ribavirin and favilavir rarely form any hydrogen bonds with their base pair nucleotide. Specifically, favilavir shows a wide distribution of distances that are too large to be hydrogen bonds, and ribavirin shows two peaks, one of which corresponds to brief hydrogen bond formation (Figure 2A, upper panel). These wide and doubly peaked distributions for favilavir and ribavirin, respectively, reflect the flexibility of these drugs and complexes but do not explain why the binding free energy of ribavirin is so similar to that of remdesivir. On the lower panel of Figure 2A, we see that remdesivir shows a distribution that peaks at a slightly shorter distance than adenine, forming slightly stronger hydrogen bonds with uracil, which underscores the potential of remdesivir.
To investigate why ribavirin shows a substantial binding free energy without binding strongly to the base on the complementary strand, we further analyzed the interactions that the drugs make with their local environment, as shown in Figure 2B. Ribavirin makes on average six hydrogen bonds, whereas remdesivir and favilavir only make four. Ribavirin adopts a different conformation, which is stabilized by the residues Asp544, Asn612, Thr601, and Arg476. However, both favilavir and ribavirin show some unfavorable interactions with Asn612 and Asp681, respectively, while remdesivir and adenine do not. To summarize, ribavirin and favilavir adopt a nonoptimal configuration, as compared to the natural nucleotide adenine; ribavirin achieves a binding free energy comparable to that of remdesivir, but it does so without forming stable hydrogen bonds with its base on the complementary strand and interacts instead with the surrounding residues. Our results for these three molecules support the view that different nucleotide analogue drugs act by different mechanisms. Favilavir and ribavirin show strong binding free energies in their monophosphate form, with the latter drug being the strongest of the two, and they seem to facilitate a nondelayed chain termination mechanism because they are not aligned with their complementary nucleotide and are therefore unlikely to be translocated. They bind instead to surrounding residues at the active site to stop elongation immediately. However, we also speculate that, because of these poor interactions with the complementary base, conversion from the triphosphate form to the monophosphate form might not be favorable, and ribavirin and favilavir might instead act as competitive inhibitors to ATP in their triphosphate form—an idea that is beyond the scope of this study and would warrant a new investigation.
In short, then, our structural analysis and free energy calculations suggest that remdesivir-MP can be added to the 3′-end of the nascent RNA chain (for clarity, the nsp12 catalytic site architecture is included in Figure 1A,C), while ribavirin and favilavir show unfavorable interactions for this purpose. Remdesivir, therefore, satisfies the prerequisite condition to act as a chain termination inhibitor. We discuss this possibility further and investigate the mechanism of action for remdesivir in what follows.
Remdesivir Mechanism at the Molecular Level: “Delayed” RNA Chain Termination Mechanism
It has been proposed that remdesivir may inhibit the enzyme’s function either through a “delayed” termination mechanism or through an “immediate” mechanism.14,17 In this section, we explore the impact of remdesivir when it is covalently bonded to the nascent strand at different positions; more specifically, we consider bonding at the upstream i+1, i+2, and i+3 locations (see Figure 3D for clarification).
Figure 3.

(A) Schematic representation of base-pair hydrogen bonding interactions between remdesivir and the uracil complementary base. The cyano group in remdesivir is highlighted as a sphere with its van der Waals radius (blue). Probability distribution for the distances between heavy atoms forming hydrogen bonds between the nucleotides under study: (B) hydrogen bond 1, between the carbonyl group of uracil base and the amino group of remdesivir; and (C) hydrogen bond 2, among uracil amino group and remdesivir. In both figures, green, blue, and red show results for remdesivir located at i+1, i+2, and i+3, respectively. (D) Top panel: schematic representation of the RNA template in cyan and the nascent RNA strand in yellow; the position of remdesivir is highlighted in red in all cases (i+1, i+2, and i+3 locations). Lower panel: LIE (linear interaction energy, expressed in kcal/mol) between adenine located at the catalytic site (position i and surroundings: electrostatic and VdW interactions were estimated for all atom pairs). In both cases, a cutoff of 12.0 Å was applied.
Figure 3B,C shows the probability distribution of distances between the heavy atoms that make the hydrogen bonds between remdesivir at different locations on the nascent strand, and its complementary base-pair uracil on the template strand. Figure 3C shows the impact on base-pair hydrogen interactions when remdesivir is located at the i+1 and i+3 positions, where wider distributions and weaker hydrogen bond interactions are observed.
We have also applied an LIE (linear interaction energy) analysis to quantify the effect of remdesivir at the different positions of the nascent strand when a subsequent nucleotide is added. Figure 3D summarizes the four different situations considered here and provides the LIE energies expressed in kcal/mol for each system. For this calculation, electrostatic and VdW interactions were estimated for all atom pairs between the adenine nucleotide located at the catalytic site (position i and the surroundings). In both cases, a cutoff of 12.0 Å was applied. Our results indicate that, after remdesivir is added to the RNA chain and translocated to the adjacent upstream site (i+1), it exerts a pronounced destabilization of the nucleotide located at the catalytic site (much higher LIE energy than the wild type system); this effect might reduce the efficiency of the subsequent addition. In contrast, remdesivir at the i+2 and i+3 sites does not show such an effect.
A recent simulation study of the RdRp–remdesivir complex reported that the highly polar 1′-cyano group on remdesivir, when located at the i+3 site, exerts a strong electrostatic attraction with the salt bridge formed by Lys593 and Asp865.30 It is important to highlight that the main difference between our simulations and that previous report30 resides in the RdRp complex structure: here, we used a recently reported cryo-EM structure (PDB code: 7BV2), as opposed to the homology model built in the aforementioned work. The authors of that study rationalized the destabilization of base-pair interactions in terms of these electrostatic attractions, which they proposed could pull remdesivir away from its canonical conformation. Indeed, in our simulations, we observe a similar deterioration of the hydrogen bonds between remdesivir and its complementary base-pair uracil (Figure 3B). On account of those previous observations,30 we have also explored the occurrence of such electrostatic interactions in our systems. Figure 4A shows a representative configuration for the highly polar 1′-cyano group of remdesivir when it is in close proximity to the salt bridge connecting Lys593 and Asp865.
Figure 4.

(A) Protein–remdesivir interactions when this non-natural nucleotide is located at position i+3: aspartic acid, lysine, and remdesivir are shown with sticks. Atoms belonging to the carboxylate functional group, cyano, and amino are highlighted with spheres (van der Waals radius), where red represents oxygen atoms, blue for nitrogen, and white for hydrogen. (B) Probability distribution distances for salt bridge and hydrogen bond interactions between lysine and aspartic acid when remdesivir is located at the i+3 position (blue), lysine and aspartic acid for the wild type system, without remdesivir (green), and hydrogen bond among the remdesivir cyano group and lysine (red). C) Bar chart: in red the total summary of pairwise interactions for the lysine and remdesivir cyano functional group, in blue the lysine and aspartic acid salt bridge when remdesivir is located at position i+3, in gray the lysine and aspartic salt bridge for the wild type (WT) RNA, and in yellow the repulsive contribution for interactions among the aspartic acid and cyano functional group belonging to the non-natural nucleotide, remdesivir.
Figure 4B shows the probability distribution of distances between the heavy atoms that make the salt bridge (Lys593–Asp865) and the hydrogen bond between the cyano group and Lys593 when remdesivir is located at the i+3 site. In addition, for comparison, the probability distribution of distances for the salt bridge was also estimated in the wild type system, without remdesivir. From the bimodal distribution observed in Figure 4B for the wild type system, we can see that this particular salt bridge exhibits two different conformations, with distances of approximately 3 and 5 Å (green line), respectively. This bimodal pattern is also observed when remdesivir is located at site i+3; however, the constraints induced by this non-natural nucleotide are accompanied by a population increase for the set of conformations where the electrostatic interactions are reinforced (blue line). As expected, the probability distribution for the hydrogen bonding interactions displayed among the remdesivir cyano group and Lys593 follows the same pattern. Figure 4C shows a bar chart for the pairwise, nonbonded interactions among the atoms belonging to the respective functional groups involved in electrostatic interactions: remdesivir’s cyano group and Lys593 hydrogen bond (red), repulsive charge pairwise interactions between remdesivir’s cyano group and Asp865 (orange), Lys593–Asp865 salt bridge in the wild type system (gray), and Lys593–Asp865 salt bridge when remdesivir is located at the i+3 site (blue). The pairwise energy difference in the salt bridge interactions observed for the wild type system (gray) compared to remdesivir at the i+3 location (blue) emphasizes the conformational constraint introduced by this non-natural nucleotide.
Taken together, the data presented in this section are consistent with and supplement previous observations,30 where the presence of remdesivir at the i+1 and i+3 positions was found to lead to destabilization of base-pair interactions. We further propose that, when the non-natural nucleotide is at the i+3 site, there are strong attractive electrostatic interactions between the highly polar 1′-cyano group and the salt bridge formed by Lys593 and Asp865. These results, therefore, support a “delayed” termination mechanism where this set of conformational changes affects the normal RdRp complex function.
RdRp Complex Destabilization by Remdesivir and Three SARS–RdRp Lethal Point Mutations
At the time of preparation of this manuscript, a new structure for the RdRp complex was released. As mentioned in the Introduction, this new structure (PDB code: 6YYT) has nsp12 in complex with two molecules of nsp8 and one nsp7, where the nsp8 also includes the “sliding poles” and a second turn of RNA. The previously used structure (PDB code: 7BV2) showed nsp12 in complex with one molecule of nsp8 and nsp7. Unfortunately, the more recent structure (PDB code: 6YYT) does not incorporate remdesivir. For completeness, in what follows we explore the stability of the RdRp complex in terms of atomic fluctuations and active site conformational dynamics. We do so by relying on what appears to be the most comprehensive structure available at this time, and we incorporate remdesivir by covalently bonding it at the catalytic site. Note that we have also included Mg2+ ions, since these were not solved with the structure. In the two cases, we have aligned both RdRp complex structures, namely, PDB code 7BV2 and PDB code 6YYT, and included remdesivir and Mg2+ ions in the second structure (6YYT).
As before, we compare the role of remdesivir to that of the natural nucleotide, adenine. Figure 5A,B shows the probability distribution of distances between the heavy atoms that make the hydrogen bonds between remdesivir or adenine and its complementary base-pair uracil on the template strand. We can see that the distributions are similar for the natural and non-natural nucleotides. The probability distributions of distances between Mg2+ A and the phosphate group are shown in Figure 5C and illustrate the highly stable interaction between them, essential for RNA stability. It is important to also mention that the RMSD was estimated for all remdesivir heavy atoms, where the average displacement was lower than 0.5 Å. In summary, this structural analysis underscores, again, that remdesivir can be added to the 3′-end of the nascent RNA chain, and it satisfies the prerequisite condition to act as a chain termination inhibitor.
Figure 5.

(A, B) Distribution of distances between the heavy atoms that make the hydrogen bonds between remdesivir or adenine and its complementary base-pair uracil on the template strand. (C) Probability distribution for the distances between the phosphate group belonging to the monophosphate nucleotide analogues and Mg2+ A located at the catalytic site.
It is understood that nsp7 and nsp8 activate and confer processivity to the RNA-synthesizing activity of nsp12. Previous studies have identified three nsp8 residues (K58, P183, and R190) as critical for SARS-CoV genome replication.16 Nsp8 residues P183 and R190 are involved in nsp8/nsp12 interactions, whereas residue K58 modulates the interaction of the polymerase complex with RNA. Point mutations of these residues K58A, P183A, and R190A were all found to be lethal to the previous virus, in the sense that they showed greatly reduced polymerase activity (1–7% of wild type (WT) activity).16 Taking into account that the amino acid sequence alignment for NSP8 of SARS CoV and SARS CoV-2 shows a sequence identity of 97.5% and a sequence similarity of 100.0%,31 we expect some level of RdRp complex destabilization due to these point mutations. We have explored the stability of the nsp7–nsp8–nsp12 complex in terms of backbone heavy atom fluctuations to further elucidate any possible long-range effects on account of the non-natural nucleotide, remdesivir, as well as nsp8 point mutations. Figure 6A,B shows β-factor values (estimated from the root mean squared fluctuation, RMSF), which measure thermal fluctuations for each residue of the nsp8 cofactor.
Figure 6.
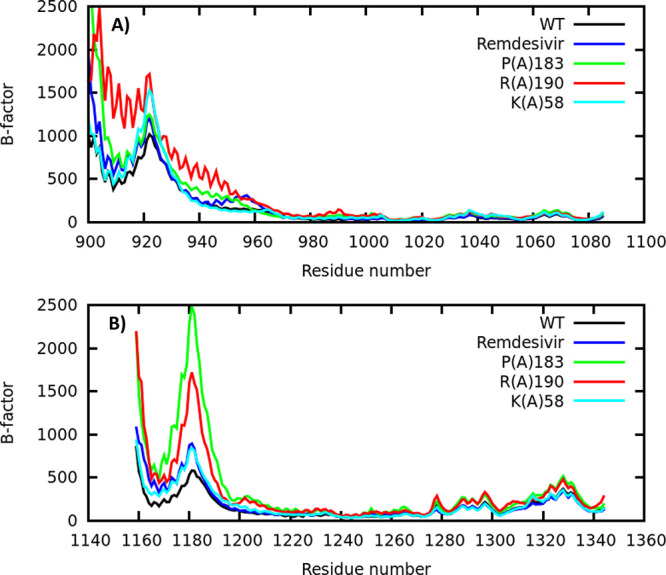
β-factor, estimated from the root mean squared fluctuation, RMSF: (A) nsp8 unit 1 and (B) nsp8 unit 2.
It is evident that the dynamics of nsp8’s flexible regions are significantly altered for all the point mutations considered here, namely, P183A, R190A, and K58A. This finding was previously associated with a critical decrease in the interaction between the polymerase complex and RNA.31
Note, however, that it is still unclear how these distant point mutations affect the function of the RdRp and mediate the replication of the genome at the active site. Importantly, we are also interested in understanding how remdesivir affects the conformation of the active site compared to adenine, and we can do so by comparing the effect of remdesivir to that of these three mutations. With those goals in mind, we performed principal component analysis (PCA) on the residues comprising the active site in the nsp12 subunit to investigate the effect, if any, of the mutations on its conformational dynamics. Briefly, PCA is performed on the Cα distances of the residues comprising the active site and that interact directly with the substrate:17,32 residues 618–626, 679–692, 544–559, 812–814, 758–761, 798, and 836. Then, to project all trajectories onto the same collective coordinates, PCA is performed on the combined trajectories of the RdRp without substrate in the active site: WT, P183A, R190A, and K58A trajectories. The results of the PCA with the distances projected onto the first two principal components are shown in Figure 7.
Figure 7.

Active site conformational dynamics of the RdRp without substrate in the active site for WT, K58A, R190A, and P183A. Cα distances between active site residues described in the main text have been projected onto the first two principal components (PC1 and PC2). The sample density describes the fraction of simulation time spent in a particular state in PC-space.
Figure 7 shows that, even though the point mutations are located far from the active site, they change its conformational dynamics. As expected, the WT RdRp shows a single stable conformation that is presumably optimized for the incorporation of a substrate. However, the mutants show a larger variance in their conformations, displaying a broader profile of the WT conformation at (PC1, PC2) = (0, 0) and visiting several different states during the simulation. This broadened conformation and the departure to different conformational states help explain, at least partially, why these mutations greatly reduced the polymerase activity in vitro, and in vivo for the K58A mutant, on the SARS-CoV RdRp.16 The mutations destabilize the complex, as seen in Figure 6, which leads to the active site conformation becoming broader and less efficient, and allowing for other conformations that are most likely not catalytically efficient or viable (Figure 7). Indeed, upon analysis of the structural differences among the different PCs, we find that PC1 is a measure of catalytic stability, as negative PC1 values show a nonoptimal configuration of the C–G base pair at the i+1 position in the RNA, and at more positive PC1 values, the distance between the hydrogen bonding pairs at the C–G pair becomes shorter. The residues 622–625 also move further away from the active site along PC1, potentially to accommodate the substrate at the active site. However, too large of a distance between these residues and the active site might cause the substrate to become overly flexible at the active site, as these residues interact with the incoming nucleotide/analogue in our simulations with remdesivir and adenine. To investigate the conformational dynamics in the presence of a substrate at the active site, we also performed the same PCA analysis on the trajectories with adenine and remdesivir. The results are shown in Figure 8.
Figure 8.
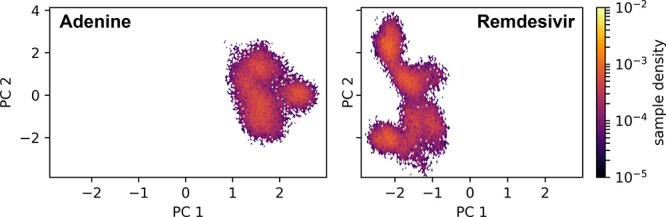
Active site conformational dynamics of the RdRp complex with adenine or remdesivir present. Cα distances between active site residues described in the main text have been projected onto the first two principal components (PC1 and PC2). The sample density describes the fraction of simulation time spent in a particular state in PC-space.
We observe a similar pattern in this case, where the active site exhibits different conformations when adenine or remdesivir is present, respectively. These conformations do not even overlap, which points to the dramatic effect that remdesivir has on the conformation of the active site. Similar to the case with the mutants, PC1 represents the catalytic stability of the complex, where negative PC1 values show larger C–G base pair distances at the i+1 position of the RNA, and positive PC1 values show smaller distances, indicative of more stable interactions. The fact that remdesivir occupies conformations at only negative PC1 values points to a secondary, previously unknown effect of remdesivir where it destabilizes the protein and forces the active site into a catalytically inefficient configuration, with the RNA base pair at the next upstream position being destabilized.
Conclusion
We have investigated the interactions between remdesivir and two other nucleotide analogue inhibitors, ribavirin and favilavir, and the RdRp complex of SARS-CoV-2. By relying on free energy calculations, we have shown that all the nucleotide analogues considered here bind strongly to the active site. Out of the three nucleotide inhibitors, remdesivir binds the strongest, serving to highlight its potential as a repurposed drug, but is closely followed by ribavirin. However, a detailed analysis of the drugs’ interactions with their local environment reveals that remdesivir exhibits strong interactions with its complementary base pair, whereas ribavirin and favilavir display a more dynamic and compliant hydrogen bond network. Even though ribavirin exhibits a binding free energy that is comparable to that of remdesivir, it is stabilized mainly by hydrogen bonds to surrounding residues and shows poor hydrogen bonding with its complementary base, rendering it less likely to succeed as a nucleotide analogue. These results lead us to speculate that ribavirin and favilavir act by different mechanisms from remdesivir, as the former drugs are unable to be fully coupled to the complementary RNA strand. In contrast, remdesivir more closely resembles the interactions that adenine makes with the complementary strand.
To develop a better understanding of the mechanism of action of remdesivir we also incorporated this drug covalently into the RNA nascent strand at different downstream positions. Consistent with the results of a recent study that relied on an approximate, homologous structure derived from a previous virus,30 and in contrast to the findings of a recent, purely structural study,17 we found that remdesivir acts by a delayed RNA chain termination mechanism. An LIE (linear interaction energy) analysis indicates that after remdesivir is added to the RNA chain and translocated to the adjacent upstream site (i+1), it reduces the efficiency of the subsequent addition. This view was inferred from a pronounced destabilization of the nucleotide located at the catalytic site. In addition, the presence of remdesivir at the i+1 and i+3 positions leads to the destabilization of base-pair interactions. This latter effect at the i+3 site was found to be caused by strong attractive electrostatic interactions between the polar 1′-cyano group of remdesivir and the salt bridge formed by Lys593 and Asp865.25 Taken together, all these data support a “delayed” termination mechanism where the aforementioned set of conformational changes might affect the normal RdRp complex function.
It is known that, similar to the RdRp from SARS-CoV, nsp12 does not bind to RNA without nsp7 and nsp8,14,16 which confer processivity to the complex. The RdRp complex is crucial for SARS-CoV-2 replication, and destabilizing it could have detrimental effects on its functions. We found that remdesivir also acts by destabilizing the entire RdRp complex, where the incorporation of this non-natural nucleotide analogue induces significantly higher fluctuations at the protein–protein interfaces and between RdRp and RNA. We also explored and compared the destabilizing effects due to remdesivir and three punctual mutations that are lethal for the previous SARS–RdRp complex:16 K58A, P183A, and R190A. Given the high sequence similarity to SARS-CoV-2, with a sequence identity of 97.5% and a sequence similarity of 100.0%,31 it is likely that they will also be lethal for this new coronavirus. Remdesivir, as well as these mutations, caused nsp8 to exhibit higher β-factors and larger fluctuations, which would lead to destabilization of the nsp8 molecules. Since these mutations are located at positions far from the active site, we used PCA on the active site residues to investigate how they affect the conformational dynamics at the active site. Consistent with an overall destabilization, all of the investigated mutations force the active site to adopt several different conformations. This behavior is to be contrasted with the WT, where only one conformation is observed. These additional conformations show reduced catalytic efficiency in the form of base-pair destabilization. The same analysis, when performed with remdesivir and adenine at the active site, reveals that remdesivir forces the active site into a vastly different conformation compared to the natural nucleotide adenine, consistent with the effect of lethal mutations. All of the remdesivir-induced conformations show a reduced catalytic efficiency as the upstream base-pair interactions are weaker, similar to the action of the point mutations. However, it should be noted that this reduced efficiency due to destabilization is still expected to render elongation to the i+3 site possible, and sufficient to support a delayed termination mechanism, since experimental studies also indicate this.14 We therefore hypothesize that this destabilization acts together with the delayed termination mechanism to render the complex inefficient enough to later terminate translocation, an idea that we will address in future studies.
In summary, these observations suggest that remdesivir acts not only by a delayed RNA chain termination but also by destabilizing the complex and thus reducing the complex’s ability to bind to RNA, similar to how the lethal K58A, R190A, and P183A mutations act. These results suggest that an additional, previously unreported secondary effect of remdesivir is the destabilization of the protein complex and active site, leading to reduced replication efficiency. A complete picture of the mechanism for the destabilization of the RdRp complex by remdesivir is gradually being developed and will be the focus of future studies.
Materials and Methods
A cumulative time of almost 34 μs (4 μs of equilibration and production runs and almost 30 μs of binding free energy calculations) of atomistic MD simulations was run using the AMBER1833 simulation package. In the first part of this Article when different monophosphate nucleotides were explored, the RdRp complex initial configuration was taken from a recently reported structure, PDB code 7BV2, which includes nsp12 in complex with one molecule of nsp8 and nsp7, as well as an 11-base RNA primer-template with remdesivir covalently incorporated at the first base pair of the primer strand. Remdesivir coordinates were taken from this structure, and a slight chemical modification was include in order to consider these non-natural nucleotides in their monophosphate form (noncovalently bound). Force field parameters for all monophosphate nucleotides were described by the General Amber Force Field (GAFF)20 and RNA.OL3 force field.19 The partial atomic charges were determined by the restrained electrostatic potential (RESP) fitting technique. Those electrostatic potential calculations were performed at the HF/6-31G level with Gaussian 09. Approximately 80 000 TIP3P water model molecules and Na+ ions were added. In all simulations, the protein was parametrized using the ff14SB force field14 and RNA using ff.RNA.OL3.19 The simulation protocol included a first minimization of 7000 steps, involving 3500 steepest descent steps followed by 3500 steps of conjugate gradient energy minimization, where constraints were applied on the protein heavy atoms (force constant 500 kcal/(mol Å–2)) and a second minimization (7000 steps) with no constraints of conjugate gradient energy minimization. Next, during the first equilibration, the temperature was gradually increased from 0 to 300 K over 50 ps using a Langevin thermostat with a temperature coupling constant of 1.0 ps in the canonical ensemble. Density equilibration and production runs were carried out using a constant pressure ensemble (NPT). All simulations were performed using periodic boundary conditions and a 2 fs time step. Long-range electrostatic interactions were calculated using the particle mesh Ewald method with a nonbonded cutoff of 10 Å, and the SHAKE algorithm was used to implement rigid constraints. In all cases, three independent replicas of 100 ns were taken into account.
For the second part of our study, where the incorporation of remdesivir at the catalytic site (covalently bound) and three punctual mutations were taken into account, we choose the most recently solved structure for the RdRp complex, PDB code 6YYT, which has nsp12 in complex with two molecules of nsp8 and one nsp7, where the nsp8 also includes the “sliding poles” of nsp8 and a second turn of RNA. Simulations were carried out exactly as previously described.
Free Energy
The absolute binding free energy is defined as ΔGbinding = ΔGL – ΔGRL, where ΔGRL is the free energy change of the natural and non-natural nucleotides annihilation in the RdRp complex, and ΔGL is the free energy change of nucleotide annihilation in water. We run a total of 14.85 μs for drugs in pure water and another 14.85 μs for drugs in the complex with the protein. To calculate these free energy changes, we use Thermodynamic Integration (TI) implemented in PMEMD for AMBER18. We use the one-step annihilation protocol with soft-core potentials.33 The parameters used in the soft-core potentials were α = 0.25 for Lennard-Jones interactions and β = 12 for electrostatic interactions following the recommendations in ref (34) for locally charged environments. In addition, we adopted a simple approach with multiple runs starting from previous MD equilibration simulations. In this way, three independent replicas for each nucleotide were taken into account, as well as three replicas for the nucleotide solvated in pure water. Eleven equally spaced windows were used (Δλ = 0.1) with 150 ns of simulation time per window. To keep the ligand from wandering in TI calculations, we used a soft restraint of 5 kcal/(mol Å2).34 The TI free energy calculations were complemented by a series of multistate Bennett’s acceptance ratio (MBAR) calculations implemented also in AMBER1824 with the aid of the libraries PYMBAR23,35 and Alchemical Analysis.36 For MBAR, the same number of windows and simulation time as in TI was used.
Hydrogen Bond Analysis
Hydrogen bonds were analyzed with the HBonds plugin of VMD 1.9.3,37 where the cutoffs for the distance between heavy atoms and the associated angle were set to 3.5 Å and 30°, respectively. The hydrogen bond distances between the heavy atoms were analyzed with the MDAnalysis package for Python 3.38,39
Principal Component Analysis
The PCA was performed on the Cα pairwise distances between active site residues 618–626, 679–692, 544–559, 812–814, 758–761, 798, and 836, taken from every frame of the trajectories in question. First, the RdRp without any substrate in the active site in the form of WT and the mutants K58A, R190A, and P183A were analyzed to compare the effect of the mutants with the WT. In order to present the results on the same principal components, the PCA was performed on the combined trajectories, and then, the individual trajectories were projected onto these principal components. The same methodology was applied to the case when adenine/remdesivir was present at the active site. The PCA analysis was done using pyEMMA.40
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c01242.
Detailed interactions of remdesivir, favilavir, and ribavirin with nsp12 motifs; and convergence of TI/MBAR (PDF)
Author Contributions
§ F.B., C.A.M., G.R.P-L., and W.A. contributed equally to this work
The study of protein–DNA complexes in epigenetics is supported by the NSF, under grant EFRI CEE 1830969, and the development of new DNA models is supported by BIO/MCB 1818328. The study of allostery and its role in triggerable materials is supported by MURI: W911NF-15-1-0568. The simulations reported here were carried out on the GPU cluster supported by the NSF through grant DMR-1828629. The authors are grateful to the Research Computing Center of the University of Chicago for additional computational resources.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhu N.; Zhang D.; Wang W.; Li X.; Yang B.; Song J.; Zhao X.; Huang B.; Shi W.; Lu R.; Niu P.; Zhan F.; Ma X.; Wang D.; Xu W.; Wu G.; Gao G. F.; Tan W. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Guan X.; Wu P.; Wang X.; Zhou L.; Tong Y.; Ren R.; Leung K. S. M.; Lau E. H. Y.; Wong J. Y.; Xing X.; Xiang N.; Wu Y.; Li C.; Chen Q.; Li D.; Liu T.; Zhao J.; Liu M.; Tu W.; Chen C.; Jin L.; Yang R.; Wang Q.; Zhou S.; Wang R.; Liu H.; Luo Y.; Liu Y.; Shao G.; Li H.; Tao Z.; Yang Y.; Deng Z.; Liu B.; Ma Z.; Zhang Y.; Shi G.; Lam T. T. Y.; Wu J. T.; Gao G. F.; Cowling B. J.; Yang B.; Leung G. M.; Feng Z. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P.; Yang X.-L.; Wang X.-G.; Hu B.; Zhang L.; Zhang W.; Si H.-R.; Zhu Y.; Li B.; Huang C.-L.; Chen H.-D.; Chen J.; Luo Y.; Guo H.; Jiang R.-D.; Liu M.-Q.; Chen Y.; Shen X.-R.; Wang X.; Zheng X.-S.; Zhao K.; Chen Q.-J.; Deng F.; Liu L.-L.; Yan B.; Zhan F.-X.; Wang Y.-Y.; Xiao G.-F.; Shi Z.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F.; Zhao S.; Yu B.; Chen Y.-M.; Wang W.; Song Z.-G.; Hu Y.; Tao Z.-W.; Tian J.-H.; Pei Y.-Y.; Yuan M.-L.; Zhang Y.-L.; Dai F.-H.; Liu Y.; Wang Q.-M.; Zheng J.-J.; Xu L.; Holmes E. C.; Zhang Y.-Z. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remdesivir Clinical Trials. https://www.gilead.com/purpose/advancing-global-health/covid-19/remdesivir-clinical-trials.
- Wang M.; Cao R.; Zhang L.; Yang X.; Liu J.; Xu M.; Shi Z.; Hu Z.; Zhong W.; Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilead Announces Results From Phase 3 Trial of Remdesivir in Patients With Moderate COVID-19. https://www.gilead.com/news-and-press/press-room/press-releases/2020/6/gilead-announces-results-from-phase-3-trial-of-remdesivir-in-patients-with-moderate-covid-19.
- Beigel J. H.; Tomashek K. M.; Dodd L. E.; Mehta A. K.; Zingman B. S.; Kalil A. C.; Hohmann E.; Chu H. Y.; Luetkemeyer A.; Kline S.; Lopez de Castilla D.; Finberg R. W.; Dierberg K.; Tapson V.; Hsieh L.; Patterson T. F.; Paredes R.; Sweeney D. A.; Short W. R.; Touloumi G.; Lye D. C.; Ohmagari N.; Oh M.; Ruiz-Palacios G. M.; Benfield T.; Fätkenheuer G.; Kortepeter M. G.; Atmar R. L.; Creech C. B.; Lundgren J.; Babiker A. G.; Pett S.; Neaton J. D.; Burgess T. H.; Bonnett T.; Green M.; Makowski M.; Osinusi A.; Nayak S.; Lane H. C. N. Engl. J. Med. 2020, 383, 1813–1826. 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolin R.; Hirsch M. S. Remdesivir — An Important First Step. N. Engl. J. Med. 2020, 383, 1886. 10.1056/NEJMe2018715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zhang D.; Du G.; Du R.; Zhao J.; Jin Y.; Fu S.; Gao L.; Cheng Z.; Lu Q.; Hu Y.; Luo G.; Wang K.; Lu Y.; Li H.; Wang S.; Ruan S.; Yang C.; Mei C.; Wang Y.; Ding D.; Wu F.; Tang X.; Ye X.; Ye Y.; Liu B.; Yang J.; Yin W.; Wang A.; Fan G.; Zhou F.; Liu Z.; Gu X.; Xu J.; Shang L.; Zhang Y.; Cao L.; Guo T.; Wan Y.; Qin H.; Jiang Y.; Jaki T.; Hayden F. G.; Horby P. W.; Cao B.; Wang C. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. 10.1016/S0140-6736(20)31022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agostini M. L.; Andres E. L.; Sims A. C.; Graham R. L.; Sheahan T. P.; Lu X.; Smith E. C.; Case J. B.; Feng J. Y.; Jordan R.; Ray A. S.; Cihlar T.; Siegel D.; Mackman R. L.; Clarke M. O.; Baric R. S.; Denison M. R. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. mBio 2018, 9, e00221-18. 10.1128/mBio.00221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. J.; Won J. J.; Graham R. L.; Dinnon K. H.; Sims A. C.; Feng J. Y.; Cihlar T.; Denison M. R.; Baric R. S.; Sheahan T. P. Broad spectrum antiviral remdesivir inhibits human endemic and zoonotic deltacoronaviruses with a highly divergent RNA dependent RNA polymerase. Antiviral Res. 2019, 169, 104541. 10.1016/j.antiviral.2019.104541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan T. P.; Sims A. C.; Leist S. R.; Schäfer A.; Won J.; Brown A. J.; Montgomery S. A.; Hogg A.; Babusis D.; Clarke M. O.; Spahn J. E.; Bauer L.; Sellers S.; Porter D.; Feng J. Y.; Cihlar T.; Jordan R.; Denison M. R.; Baric R. S. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020, 11, 222. 10.1038/s41467-019-13940-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillen H. S.; Kokic G.; Farnung L.; Dienemann C.; Tegunov D.; Cramer P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. 10.1038/s41586-020-2368-8. [DOI] [PubMed] [Google Scholar]
- Ahn D.-G.; Choi J.-K.; Taylor D. R.; Oh J.-W. Biochemical characterization of a recombinant SARS coronavirus nsp12 RNA-dependent RNA polymerase capable of copying viral RNA templates. Arch. Virol. 2012, 157, 2095–2104. 10.1007/s00705-012-1404-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subissi L.; Posthuma C. C.; Collet A.; Zevenhoven-Dobbe J. C.; Gorbalenya A. E.; Decroly E.; Snijder E. J.; Canard B.; Imbert I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E3900–E3909. 10.1073/pnas.1323705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W.; Mao C.; Luan X.; Shen D.-D.; Shen Q.; Su H.; Wang X.; Zhou F.; Zhao W.; Gao M.; Chang S.; Xie Y.-C.; Tian G.; Jiang H.-W.; Tao S.-C.; Shen J.; Jiang Y.; Jiang H.; Xu Y.; Zhang S.; Zhang Y.; Xu H. E. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499. 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgarbová M.; Otyepka M.; Šponer J.; Mládek A.; Banáš P.; Cheatham T. E.; Jurečka P. Refinement of the Cornell et al. Nucleic Acids Force Field Based on Reference Quantum Chemical Calculations of Glycosidic Torsion Profiles. J. Chem. Theory Comput. 2011, 7, 2886–2902. 10.1021/ct200162x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Sgrignani J.; Magistrato A. The Structural Role of Mg2+ Ions in a Class I RNA Polymerase Ribozyme: A Molecular Simulation Study. J. Phys. Chem. B 2012, 116, 2259–2268. 10.1021/jp206475d. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan R. Coupling of Fast and Slow Modes in the Reaction Pathway of the Minimal Hammerhead Ribozyme Cleavage. Biophys. J. 2007, 93, 2391–2399. 10.1529/biophysj.107.104661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirts M. R.; Chodera J. D. Statistically optimal analysis of samples from multiple equilibrium states. J. Chem. Phys. 2008, 129, 124105. 10.1063/1.2978177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaus J. W.; Pierce L. T.; Walker R. C.; McCammon J. A. Improving the Efficiency of Free Energy Calculations in the Amber Molecular Dynamics Package. J. Chem. Theory Comput. 2013, 9, 4131–4139. 10.1021/ct400340s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakchaure P. D.; Ghosh S.; Ganguly B. Revealing the Inhibition Mechanism of RNA-Dependent RNA Polymerase (RdRp) of SARS-CoV-2 by Remdesivir and Nucleotide Analogues: A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2020, 124, 10641–10652. 10.1021/acs.jpcb.0c06747. [DOI] [PubMed] [Google Scholar]
- Koulgi S.; Jani V.; Uppuladinne M. V. N.; Sonavane U.; Joshi R. Remdesivir-bound and ligand-free simulations reveal the probable mechanism of inhibiting the RNA dependent RNA polymerase of severe acute respiratory syndrome coronavirus 2. RSC Adv. 2020, 10, 26792–26803. 10.1039/D0RA04743K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arba M.; Wahyudi S. T.; Brunt D. J.; Paradis N.; Wu C. Mechanistic insight on the remdesivir binding to RNA-Dependent RNA polymerase (RdRp) of SARS-cov-2. Comput. Biol. Med. 2021, 129, 104156. 10.1016/j.compbiomed.2020.104156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougie I.; Bisaillon M. Initial Binding of the Broad Spectrum Antiviral Nucleoside Ribavirin to the Hepatitis C Virus RNA Polymerase. J. Biol. Chem. 2003, 278, 52471–52478. 10.1074/jbc.M308917200. [DOI] [PubMed] [Google Scholar]
- Dassault Systemes BIOVIA . Discovery Studio Visualizer, v20.1.0.19295; Dassault Systemes: San Diego, 2020.
- Zhang L.; Zhang D.; Yuan C.; Wang X.; Li Y.; Jia X.; Gao X.; Yen H.-L.; Cheung P. P.-H.; Huang X.. bioRxiv, 2020, 2020.04.27.063859. 10.1101/2020.04.27.063859. [DOI]
- Yoshimoto F. K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39 (3), 198–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Zhou R.. Binding Mechanism of Remdesivir to SARS-CoV-2 RNA Dependent RNA Polymerase. Preprints, 2020, preprints202003.0267.v1. 10.20944/preprints202003.0267.v1. [DOI] [PubMed]
- Case D. A.; Ben-Shalom I. Y., Brozell S. R.; Cerutti D. S.; Cheatham T. E. III; Cruzeiro V. W. D.; Darden T. A.; Duke R. E.; Ghoreishi D.; Gilson M. K.; Gohlke H.; Goetz A. W.; Greene D.; Harris R.; Homeyer N.; Huang Y.; Izadi S.; Kovalenko A.; Kurtzman T.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Liu J.; Luchko T.; Luo R.; Mermelstein D. J.; Merz K. M.; Miao Y.; Monard G.; Nguyen C.; Nguyen H.; Omelyan I.; Onufriev A.; Pan F.; Qi R.; Roe D. R.; Roitberg A.; Sagui C.; Schott-Verdugo S.; Shen J.; Simmerling C. L.; Smith J.; Salomon-Ferrer R., Swails J.; Walker R. C.; Wang J.; Wei H.; Wolf R. M.; Wu X.; Xiao L.; York D. M.; Kollman P. A.. AMBER 2018; University of California: San Francisco, 2018.
- Steinbrecher T.; Joung I.; Case D. A. Soft-core potentials in thermodynamic integration: Comparing one- and two-step transformations. J. Comput. Chem. 2011, 32, 3253–3263. 10.1002/jcc.21909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliwal H.; Shirts M. R. A Benchmark Test Set for Alchemical Free Energy Transformations and Its Use to Quantify Error in Common Free Energy Methods. J. Chem. Theory Comput. 2011, 7, 4115–4134. 10.1021/ct2003995. [DOI] [PubMed] [Google Scholar]
- Klimovich P. V.; Shirts M. R.; Mobley D. L. Guidelines for the analysis of free energy calculations. J. Comput.-Aided Mol. Des. 2015, 29, 397–411. 10.1007/s10822-015-9840-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Gowers R. J.; Linke M.; Barnoud J.; Reddy T. J. E.; Melo M. N.; Seyler S. L.; Domański J.; Dotson D. L.; Buchoux S.; Kenney I. M.; Beckstein O. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. Proceedings of the 15th Python in Science Conference 2016, 98–105. 10.25080/Majora-629e541a-00e. [DOI] [Google Scholar]
- Michaud-Agrawal N.; Denning E. J.; Woolf T. B.; Beckstein O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer M. K.; Trendelkamp-Schroer B.; Paul F.; Pérez-Hernández G.; Hoffmann M.; Plattner N.; Wehmeyer C.; Prinz J.-H.; Noé F. PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models. J. Chem. Theory Comput. 2015, 11, 5525–5542. 10.1021/acs.jctc.5b00743. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.