

Abstract
Herein we report a transformation that generates an array of enantiomerically enriched, alkyl allyl ethers. Cyclic, acyclic, and heteroatom-bearing alkenyl triflates undergo an enantioselective, palladium-catalyzed C–C bond formation with diverse acyclic O-alkyl enol ethers in good yields and excellent enantioselectivities.
Keywords: Asymmetric Catalysis, C–C Coupling, Heck Reaction
1. Introduction
Enantioenriched alkyl allyl ethers are present in a variety of biologically relevant and active molecules.[1] Therefore, efforts have been reported in the development of new methods to synthesize this class of compounds. Traditionally, chiral allylic ethers have been prepared via the reaction of symmetric π-allyl complexes of Ir,[2] Rh,[3] and Pd[4] with exogenous alcohol nucleophiles, or by enantioselective catalytic SN2’ reactions with allylic trichloroacetimidates.[5] Alternatively, chiral allylic ethers have been synthesized via phosphine-catalyzed γ-oxidation of alkynoates.[6] Despite noteworthy progress, the scope of these transformations has remained limited due to the nature of suitable electrophiles.
Recently, we have reported an alternative approach to allylic ethers through the use of enantioselective, redox-relay Heck reaction of O-aryl enol ethers (B) and alkenyl triflates (Scheme 1A).[7]
Scheme 1.

A. Recent synthesis of enantiomerically enriched aryl allyl ethers using a redox-relay Heck strategy. B. Limitation of previous work. C. Proposed synthesis of optically active allylic and benzylic ethers remote from an aldehyde.
The redox-relay Heck reaction differs from the traditional Heck reaction in that the unsaturation of the olefin is relayed along the carbon chain toward a redox acceptor, such as an alcohol, resulting in oxidation and yielding a carbonyl product.[8] By using an enol ether, it was hypothesized that the oxygen of the alkene would significantly polarize it such that the migratory insertion would occur regioselectively, forming a bond between a cross coupling partner and the proximal alkenyl carbon with respect to the ether (See intermediate C, Scheme 1A). During the course of this work, we noted that O-aryl enol ethers effectively participated in the reaction. However, when employing O-alkyl enol ethers of type D; E1cB products (E) were formed when submitted to our standard reaction conditions (Scheme 1B). We hypothesized that the increased basicity of the alkyl ether compared to aryl ethers may stabilize a six-membered-ring enol intermediate (F), which would ultimately lead to undesired product E. To overcome this challenge, we envisioned adding an additional methylene unit between the alcohol moiety and the ether, thus circumventing the E1cB pathway (Scheme 1C). Herein we disclose the expansion of our alkenylation protocol to the use of O-alkyl enol ethers bearing homoallylic and longer chain alcohols to provide enantiomerically enriched alkyl allylic ether products.
2. Results and Discussion
To begin our study, we subjected O-methyl enol ether 2a to our previously reported reaction conditions developed for the alkenylation of O-aryl enol ethers. However, under these conditions, the redox-relay Heck product was not observed (Scheme 2A) but instead the conjugated diene H was observed in 10% yield by 1H NMR with no further reaction of the starting materials observed. This suggests, after migratory insertion, PdII–alkyl intermediate G undergoes β-oxy elimination to give H (Path A) instead of β-hydride elimination occurring away from the newly generated stereocenter toward the formation of product 3 (Path B). One possible explanation for this is that due to the small size of the methyl substituent, β-oxy elimination preferentially transpires generating H, which may inhibit further catalysis (Scheme 2B).[8b] As a result, we reasoned that increasing the size of the ether substituent could curtail β-oxy elimination and promote β-hydride elimination toward the remote alcohol. To test this, benzyl enol ether 2b was selected and excitingly, provided 80% conversion to the desired product 3b by 1H NMR analysis, while suppressing the amount of the β-oxy elimination product observed. To further support the hypothesis that the substituent size was important in guiding the catalyst in producing the desired product, 2c was prepared containing an O-cyclohexylmethyl enol ether and subjected to the reaction. Good conversion of this substrate to the Heck chain walking product was observed.
Scheme 2.

A. Early observations, percent conversions by 1H NMR. B. Differing reaction pathways favored based on the substituent on the oxygen.
On the basis of these preliminary studies, we examined the efficiency and selectivity of the transformation for a variety of electron-deficient alkenyl triflates with benzyl enol ether 2b (Table 1). Vinylogous lactone triflate 1a provided 3b in 78% yield and >99:1 enantiomeric ratio. An acyclic alkenyl triflate delivered allylic ether 3d in 62% yield and 98:2 er, whereas the benzyl analog gave product 3e in diminished yield (49%) and enantiomeric ratio (87:13). An alkenyl triflate containing a remote protected amine produced product 3f in moderate yield and 90:10 er. An alkenyl triflate containing an alkenyl fluoride was tolerated under the reaction conditions and delivered 3g in 38% yield and 98:2 er. A 5-membered cyclic alkenyl triflate gave product 3h in 56% yield and >99:1 er. Lastly, this method can also be used with enol ether 2d, a substrate that contains a secondary alcohol, to form products bearing a methyl ketone (3i, 3j, and 3k).
Table 1.
Alkenyl Triflate Scope
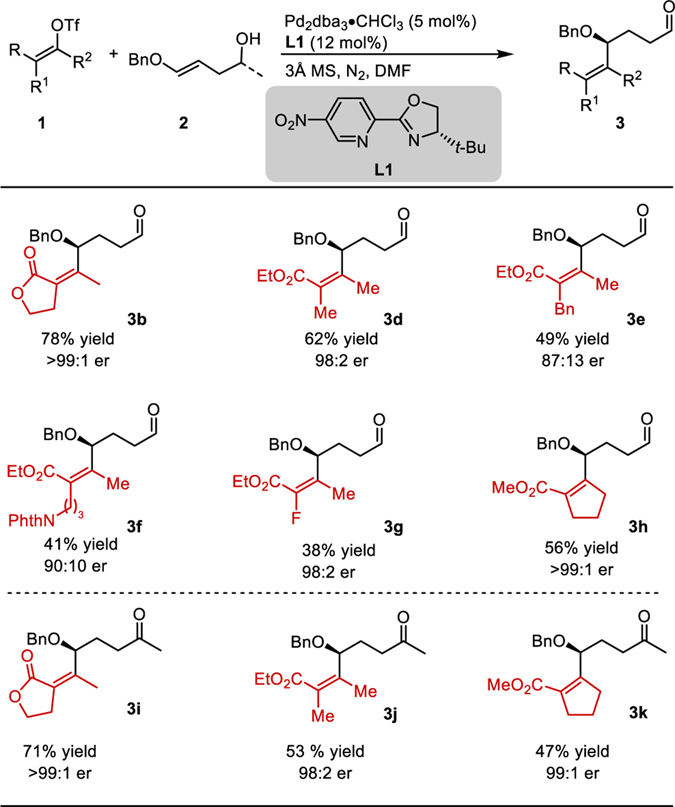 |
Yields are reported as an average of two parallel experiments. Enantioselectivity determined by SFC equipped with a chiral column. Reactions performed on 0.4 mmol scale. Absolute configuration determined to be (S) for 7a (see Sl) arising from the (E)- alkene. All other products were assigned by analogy.
The scope of the O-alkyl substituent was then examined using enol triflate 1a as the standard coupling partner (Table 2). Both electron-rich and electron-poor benzyl ethers were well-tolerated under the reaction conditions (3l and 3m). This suggests that perturbing the electronics of the enol ether has a limited effect on the outcome of the reaction. Trifluoroethyl enol ether provided 3n in 76% yield. Homobenzyl ether reacted in moderate yield to generate product 3o. A Boc-protected amino alcohol derivative delivered product 3p in 49% yield, displaying a method of derivatizing amino alcohols. An enol ether bearing a styrenyl moiety also reacted in 42% yield, which indicates that enol ethers react significantly faster than styrenes with PdII-alkenyl complexes. All of these reactions proceed in high enantioselectivity. These results further support the hypothesis that the size of the ether substituent is an important attribute.
Table 2.
Scope of O-Alkyl Enol Ethers
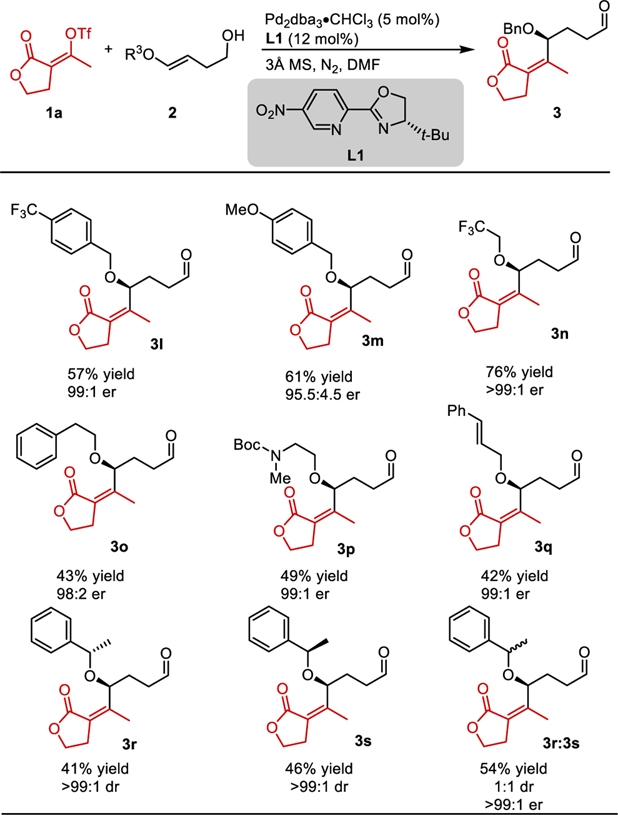 |
Yields are reported as an average of two parallel experiments. Enantioselectivity determined by SFC equipped with a chiral column. Reactions performed on 0.4 mmol scale. Absolute configuration determined to be (S) for 7a (see Sl) arising from the (E)- alkene. All other products were assigned by analogy.
To test if enantiomerically enriched substrates impacted the reaction outcome, both enol ethers 2h and 2i were subjected to the reaction conditions. These enantiomeric substrates performed similarly giving diastereomeric products in high dr. This demonstrates a catalyst controlled, enantioselective bond-forming event, which is confirmed by the use of a racemic mixture resulting in a 1:1 mixture of diastereomers, 3r and 3s. This indicates that the migratory insertion in this case is not sensitive to the adjacent chiral center.
The redox-relay Heck reaction can be used to set remote stereocenters if the Pd-catalyst can effectively migrate towards the alcohol functional group as a result of sequential β-hydride elimination/migratory insertion events. In order to examine the efficiency of this relay process, a bis-homoallylic alcohol substrate was subjected to the reaction conditions and gave δ-alkoxy carbonyl product 4a in 51% yield and 95:5 er (Table 3). Additionally, an O-alkyl enol ether with two additional methylene units (2l) also reacted to give ζ-alkoxy carbonyl product 4b in 54% yield and >99:1 er.
Table 3.
Evaluation of Redox-Relay Process
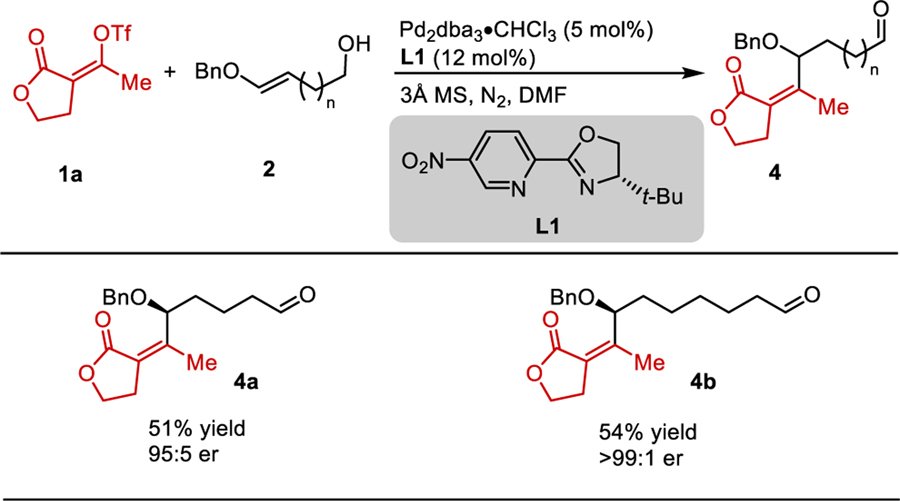 |
Yields are reported as an average of two parallel experiments. Enantioselectivity determined by SFC equipped with a chiral column. Reactions performed on 0.4 mmol scale. Absolute configuration determined to be (S) for 7a (see Sl) arising from the (E)- alkene. All other products were assigned by analogy.
To further explore the scope of this disconnection strategy, we explored the use of aryl coupling partners. It was found that aryl diazonium nucleophiles with enol ether 2b generated the corresponding arylated products in modest yield (6, Table 4). Both electron rich and electron poor aryl diazonium salts were tolerated. However, these reactions gave low levels of enantioselectivity under various conditions with the best enantioselectivity being that of 6c. Control reactions demonstrated that a non-catalytic, background reaction was occurring between the aryl diazonium and enol ether (see supplementary information).
Table 4.
Arylation of O-Alkyl Enol Ethers
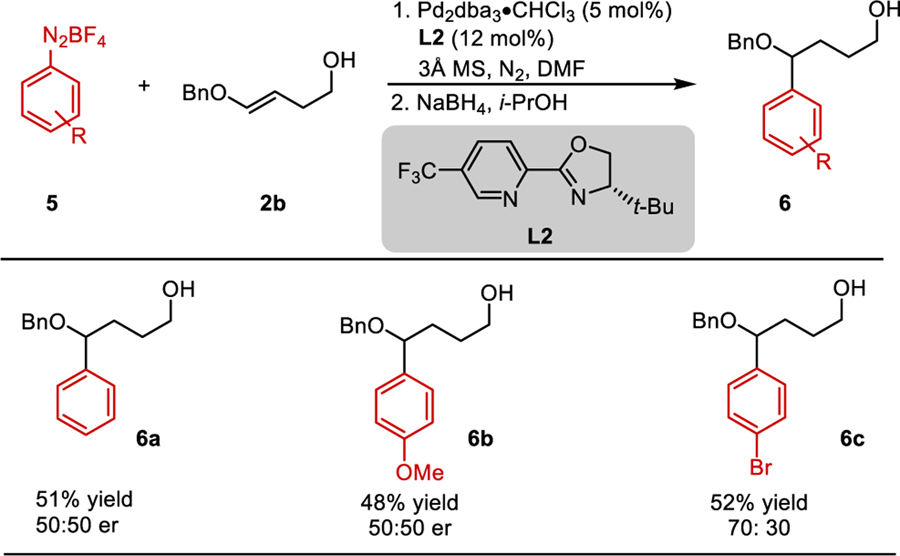 |
Yields are reported as an average of two parallel experiments. Enantioselectivity determined by SFC equipped with a chiral column. Reactions performed on 0.4 mmol scale.
3. Conclusion
In summary, we have expanded the redox-relay Heck reaction to the use of O-alkyl enol ethers as a coupling partner. The majority of the products have been generated in good yields and excellent enantioselectivities. The use of enantiomerically enriched substrates allowed us to determine that the enantioselectivity is under complete catalyst control. Unfortunately, arylation of O-alkyl enol ethers occurred in moderate yields and poor enantioselectivities.
4. Experimental
4.1. General
All reactions were performed using oven-dried or flame-dried glassware equipped with a magnetic stir bar under an atmosphere of nitrogen unless otherwise noted. Tris(dibenzylideneacetone)dipalladium(0)-chloroform adduct (Pd2dba3·CHCl3) was prepared according to the reported literature procedure.[9] All reagents were purchased from commercial suppliers and used without further purification. Dichloromethane, tetrahydrofuran (THF), dimethylformamide (DMF), and acetonitrile were passed through a column of activated alumina immediately prior to use. 1H NMR spectra were obtained in CDCl3 or C6D6 at 500 MHz. Chemical shifts are reported in ppm and referenced to the CHCl3 singlet at 7.26 ppm or C6H6 singlet at 7.15 ppm. 13C NMR spectra were obtained in CDCl3 or C6D6 125 MHz and referenced to the center peak of the CDCl3 triplet at 77.0 ppm or C6D6 triplet at 128.6 ppm. The abbreviations s, d, t, quint, sext, sept, dd, ddd, dddd, dqd dt, and m stand for the resonance multiplicities singlet, doublet, triplet, quintet, sextet, septet, doublet of doublets, doublet of doublet of doublets, doublet of doublet of doublet of doublets, doublet of quartet of doublets, doublet of triplets, and multiplet, respectively. Thin-layer chromatography was performed with EMD silica gel 60 F254 plates eluting with solvents indicated, visualized by a 254 nm UV lamp and stained with phosphomolybdic acid (PMA). Flash chromatography was performed using EM reagent silica 60 (230–400 mesh). IR spectra were recorded using a Thermo Nicolet FT-IR. HRMS data were obtained on a Waters LCP Premier XE instrument by ESI/TOF. SFC (supercritical fluid chromatography) analysis was performed at 40 °C, using a waters UPC2 instrument fitted with AD-H, OZ-H, CEL1, CEL2, AMY1, AY-H, AS-H, OJ-H and OD columns.
4.2. General Procedure A
To a dry 6-dram vial, equipped with a stir bar, was added Pd2dba3·CHCl3 (21 mg, 0.020 mmol, 5.0 mol %), ligand (15 mg, 0.06 mmol, 12 mol %), 3Å MS (200 mg, 50 mg/mmol 1) and sealed using a rubber septum. The reaction vial was evacuated and refilled with N2 three times. To this, DMF (4 mL) was added under nitrogen and the resulting mixture was stirred for 10 min at room temperature. Next, the substrate alkenol (2) (0.4 mmol) and alkenyl triflate 1 (0.4 mmol) were added sequentially, via micro-syringe. The resulting mixture was stirred at room temperature for 12 h. The mixture was then diluted with EtOAc (150 mL) and washed with H2O (2 × 30 ml) and brine (30 mL). The combined aqueous layers were back extracted using EtOAc (30 mL) and the organic layers were combined, dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified using silica gel chromatography.
4.3. Product Synthesis
(Z)-4-(benzyloxy)-5-(2-oxodihydrofuran-3(2H)-ylidene)hexanal (3b):
The general procedure A was followed using (E)-4-(benzyloxy)but-3-en-1-ol (2b, 71 mg, 0.40 mmol) and (Z)-1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl trifluoromethanesulfonate (1a, 110 mg, 0.40 mmol). Purification of this material by chromatography on silica gel (gradient elution: 5→10% EtOAc/hexanes) afforded product 3a as a colorless oil (92 mg, 78% yield): Rf = 0.14 (30% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) δ 9.75 (t, J = 1.9 Hz, 1H), 7.36 – 7.27 (m, 5H), 5.62 (dd, J = 9.0, 4.3 Hz, 1H), 4.41 – 4.31 (m, 4H), 2.90 – 2.84 (m, 2H), 2.65 (m, 1H), 2.48 (m, 1H), 2.02 (m, 1H), 1.87 (s, 3H), 1.80 – 1.73 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 202.09, 169.58, 153.60, 137.98, 128.33, 127.88, 127.72, 121.99, 74.41, 71.70, 64.89, 40.55, 27.79, 26.82, 15.65; IR (neat) 2922, 1723, 1672, 1653, 1602, 1532, 1454, 1154, 1024 cm−1; HRMS (ESI-TOF) m/z calcd for C17H20O4Na (M+Na)+: 311.1259, found 311.1260; [α]D20 = –2.4 (c = 0.4, CHCl3).
(Z)-4-(cyclohexylmethoxy)-5-(2-oxodihydrofuran-3(2H)-ylidene)hexanal (3c):
The general procedure A was followed using (E)-4-(cyclohexylmethoxy)but-3-en-1-ol (2c, 74 mg, 0.40 mmol) and (Z)-1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl trifluoromethanesulfonate (1a, 110 mg, 0.40 mmol). Purification of this material by chromatography on silica gel (gradient elution: 5→10% EtOAc/hexanes) afforded product 3c as a colorless oil (79 mg, 67% yield): Rf = 0.18 (30% EtOAc/hexanes); 1H NMR (500 MHz,CDCl3) δ 9.76 (t, J = 1.8 Hz, 1H), 5.38 (dd, J = 9.2, 4.6 Hz, 1H), 4.36 (ddd, J = 8.4, 6.6, 2.2 Hz, 2H), 3.05 (qd, J = 9.0, 6.3 Hz, 2H), 2.97 – 2.83 (m, 2H), 2.63 (dddd, J = 17.1, 8.4, 6.5, 1.9 Hz, 1H), 2.47 (dddd, J = 17.6, 8.0, 6.0, 1.5 Hz, 1H), 1.95 (dddd, J = 20.4, 14.6, 8.8, 6.6 Hz, 1H), 1.82 (t, J = 1.8 Hz, 3H), 1.71 (dddd, J = 16.7, 14.5, 5.5, 2.8 Hz, 7H), 1.21 (t, J = 7.1 Hz, 2H), 1.14 (tt, J = 11.9, 2.7 Hz, 1H), 0.90 (ddt, J = 13.4, 9.5, 7.0 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 202.31, 169.61, 154.61, 121.51, 75.29, 74.58, 64.88, 40.64, 38.19, 30.21, 30.03, 27.78, 27.02, 26.96, 26.56, 25.84, 15.46. IR (neat) 2974, 2924, 1740, 1654, 1456, 1374, 1265, 1189, 1087, 1035 cm-1. HRMS (ESI-TOF) m/z calcd for C17H26O4Na (M+Na)+: 317.1723, found 317.1726; [α]D20 = –8 (c = 0.2, CHCl3).
ethyl (Z)-4-(benzyloxy)-2,3-dimethyl-7-oxohept-2-enoate (3d):
The general procedure A was followed using (E)-4-(benzyloxy)but-3-en-1-ol (2b, 74 mg, 0.40 mmol) and ethyl (Z)-2-methyl-3-(((trifluoromethyl)sulfonyl)oxy)but-2-enoate (1b, 110 mg, 0.40 mmol). Purification of this material by chromatography on silica gel (gradient elution: 0→10% EtOAc/hexanes) afforded product 3d as a colorless oil (75 mg, 62% yield): Rf = 0.21 (30% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) δ 9.76 (d, J = 1.6 Hz, 1H), 7.37 – 7.28 (m, 5H), 4.57 (dd, J = 9.0, 4.8 Hz, 1H), 4.40 (d, J = 11.7 Hz, 1H), 4.21 (d, J = 11.7 Hz, 1H), 4.19 – 4.12 (m, 2H), 2.65 – 2.56 (m, 1H), 2.50 – 2.42 (m, 1H), 2.09 – 2.00 (m, 1H), 1.93 (s, 3H), 1.89 – 1.80 (m, 1H), 1.79 (s, 3H), 1.27 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 202.34, 169.12, 143.75, 138.12, 128.26, 127.79, 127.54, 127.25, 77.68, 70.58, 60.49, 40.54, 26.49, 16.04, 14.17, 12.89; IR (neat) 2924, 2853, 1716, 1706, 1684, 1457, 1274, 1097 cm−1; HRMS (ESI-TOF) m/z calcd for C18H24O4Na (M+Na)+: 327.1572, found 327.1576; [α]D20 = –16.2 (c = 0.4, CHCl3).
ethyl (Z)-2-benzyl-4-(benzyloxy)-3-methyl-7-oxohept-2-enoate (3e):
The general procedure A was followed using (E)-4-(benzyloxy)but-3-en-1-ol (2b, 74 mg, 0.40 mmol) and ethyl (Z)-2-benzyl-3-(((trifluoromethyl)sulfonyl)oxy)but-2-enoate (1c, 140 mg, 0.40 mmol). Purification of this material by chromatography on silica gel (gradient elution: 0→10% EtOAc/hexanes) afforded product 3e as a colorless oil (75 mg, 49% yield): Rf = 0.24 (30% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) δ 9.75 (t, J = 1.5 Hz, 1H), 7.38 – 7.26 (m, 7H), 7.23 – 7.14 (m, 3H), 4.59 (dd, J = 9.0, 4.7 Hz, 1H), 4.43 (d, J = 11.7 Hz, 1H), 4.24 (d, J = 11.7 Hz, 1H), 4.05 (qd, J = 7.1, 1.7 Hz, 2H), 3.75 (s, 2H), 2.68 – 2.55 (m, 1H), 2.52 – 2.39 (m, 1H), 2.12 – 2.02 (m, 1H), 1.96 – 1.87 (m, 1H), 1.85 (s, 2H), 1.12 (t, J = 7.1 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 202.21, 168.44, 145.24, 138.76, 138.05, 131.05, 128.41, 128.31, 128.12, 127.86, 127.62, 126.17, 77.97, 70.77, 60.52, 40.59, 35.91, 26.65, 14.03, 13.03; IR (neat) 2924, 2851, 1708, 1652, 1495, 1454, 1265, 1206, 1076 cm−1; HRMS (ESI-TOF) m/z calcd for C24H28O4Na (M+Na)+: 403.1885, found 403.1892. [α]D20 = –13.8 (c = 0.5, CHCl3).
ethyl (Z)-4-(benzyloxy)-2-(3-(1,3-dioxoisoindolin-2-yl)propyl)-3-methyl-7-oxohept-2-enoate (3f):
The general procedure A was followed using (E)-4-(benzyloxy)but-3-en-1-ol (2b, 74 mg, 0.40 mmol) and ethyl (Z)-5-(1,3-dioxoisoindolin-2-yl)-2-(1-(((trifluoromethyl)sulfonyl)oxy)ethylidene)pentanoate (1d, 180 mg, 0.40 mmol). Purification of this material by chromatography on silica gel (gradient elution: 0→10% EtOAc/hexanes) afforded product 3f as a colorless oil (78 mg, 41% yield): Rf = 0.16 (30% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) δ 9.72 (t, J = 1.7 Hz, 1H), 7.84 (dd, J = 3.1, 2.4 Hz, 2H), 7.71 (dd, J = 3.1, 2.4 Hz, 2H), 7.34 – 7.28 (m, 2H), 7.27 – 7.23 (m, 3H), 4.42 (dd, J = 7.6, 3.8 Hz, 1H), 4.37 (d, J = 11.7 Hz, 1H), 4.18 (d, J = 11.9 Hz, 1H), 4.15 – 4.07 (m, 2H), 3.72 (t, J = 7.3 Hz, 2H), 2.54 (dddd, J = 16.7, 8.3, 6.9, 1.9 Hz, 1H), 2.46 – 2.33 (m, 3H), 2.01 (ddt, J = 14.2, 8.7, 7.3 Hz, 1H), 1.86 – 1.78 (m, 2H), 1.76 (s, 3H), 1.20 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, cdcl3) δ 202.12, 168.72, 168.28, 143.40, 138.14, 133.93, 132.10, 131.37, 128.28, 127.85, 127.56, 123.19, 78.01, 70.63, 60.58, 40.46, 37.74, 27.71, 27.25, 26.49, 14.11, 12.38. IR 3031, 2931, 2361, 2338, 1772, 1706, 1652, 1437, 1395, 1206, 1037 cm-1. HRMS (ESI-TOF) m/z calcd for C28H31NO6Na (M+Na)+: 500.2049, found: 500.2049. [α]D20 = –14.2 (c = 0.4, CHCl3).
ethyl (E)-4-(benzyloxy)-2-fluoro-3-methyl-7-oxohept-2-enoate (3g):
The general procedure A was followed using (E)-4-(benzyloxy)but-3-en-1-ol (2b, 72 mg, 0.40 mmol) and ethyl (E)-2-fluoro-3-(((trifluoromethyl)sulfonyl)oxy)but-2-enoate (1e, 110 mg, 0.40 mmol). Purification of this material by chromatography on silica gel (gradient elution: 0→8% Et2O/hexanes) afforded product 3g as a colorless oil (47 mg, 38% yield): Rf = 0.42 (30% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) δ 9.75 (t, J = 1.6 Hz, 1H), 7.37 – 7.25 (m, 5H), 5.10 (ddd, J = 9.1, 4.6, 2.0 Hz, 1H), 4.41 (d, J = 11.5 Hz, 1H), 4.31 – 4.21 (m, 3H), 2.60 (dtd, J = 17.2, 8.1, 6.5, 1.6 Hz, 1H), 2.47 (dddd, J = 17.8, 7.8, 6.3, 1.1 Hz, 1H), 2.14 – 2.00 (m, 1H), 1.87 (d, J = 4.6 Hz, 3H), 1.84 – 1.74 (m, 1H), 1.32 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 201.77, 160.70 (d, J = 35.0 Hz), 137.75, 132.73 (d, J = 9.8 Hz), 128.43, 127.88, 127.85, 74.22 (d, J = 4.2 Hz), 71.14, 61.54, 40.40, 29.71, 26.68, 26.65, 9.66 (d, J = 8.5 Hz), 74.22 (d, J = 4.2 Hz), 9.66 (d, J = 8.5 Hz). 19F NMR (471 MHz, CDCl3) δ –120.4 (m). IR (neat) 2924, 2854, 1709, 1696, 1645, 1453, 1375, 1266, 1147, 1046 cm-1. HRMS (ESI-TOF) m/z calcd for C17H21O4NaF (M+Na+) 331.1322, found: 331.1321. [α]D20 = –8.0 (c = 0.1, CHCl3).
methyl 2-(1-(benzyloxy)-4-oxobutyl)cyclopent-1-ene-1-carboxylate (3h):
The general procedure A was followed using (E)-4-(benzyloxy)but-3-en-1-ol (2b, 72 mg, 0.40 mmol) and methyl 2-(((trifluoromethyl)sulfonyl)oxy)cyclopent-1-ene-1-carboxylate (1f, 110 mg, 0.40 mmol). Purification of this material by chromatography on silica gel (gradient elution: 0→10% EtOAc/hexanes) afforded product 3h as a colorless oil (47 mg, 38% yield): Rf = 0.26 (30% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) δ 9.75 (t, J = 1.6 Hz, 1H), 7.39 – 7.26 (m, 5H), 5.04 (dd, J = 8.9, 4.9 Hz, 1H), 4.42 (d, J = 11.6 Hz, 1H), 4.32 (d, J = 11.6 Hz, 1H), 3.72 (s, 3H), 2.72 (dtt, J = 15.3, 7.6, 2.4 Hz, 1H), 2.61 (dtt, J = 24.0, 8.6, 7.4, 2.6 Hz, 3H), 2.47 (dddd, J = 17.6, 7.9, 6.3, 1.5 Hz, 1H), 2.06 (dddd, J = 14.3, 8.9, 7.9, 6.7 Hz, 1H), 1.85 (dq, J = 15.6, 7.8 Hz, 4H). 13C NMR (125 MHz, CDCl3) δ 202.05, 165.89, 158.76, 138.11, 130.95, 128.30, 127.79, 127.63, 126.93, 74.65, 71.36, 51.20, 40.48, 33.83, 32.86, 26.69, 21.48. IR (neat) 2951, 1706, 1638, 1454, 1435, 1353, 1264, 1097 cm-1. HRMS (ESI-TOF) m/z calcd for C18H22O4Na (M+Na)+: 325.1416, found: 325.1421. [α]D20 = –20.6 (c = 0.2, CHCl3).
(Z)-3-(3-(benzyloxy)-6-oxoheptan-2-ylidene)dihydrofuran-2(3H)-one (3i):
The general procedure A was followed using (E)-4-(benzyloxy)pent-4-en-2-ol (2d, 77 mg, 0.40 mmol) and (Z)-1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl trifluoromethanesulfonate (1a, 110 mg, 0.40 mmol). Purification of this material by chromatography on silica gel (gradient elution: 0→10% EtOAc/hexanes) afforded product 3i as a colorless oil (86 mg, 71% yield): Rf = 0.15 (30% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) δ 7.34 – 7.24 (m, 5H), 5.57 (dd, J = 8.9, 5.0 Hz, 1H), 4.38 – 4.31 (m, 4H), 2.85 (t, J = 7.3 Hz, 2H), 2.65 (ddd, J = 17.6, 9.7, 5.6 Hz, 1H), 2.44 (ddd, J = 17.6, 9.6, 5.6 Hz, 1H), 2.09 (s, 3H), 2.00 – 1.91 (m, 1H), 1.85 (t, J = 1.8 Hz, 3H), 1.74 – 1.65 (m, 1H). 13C NMR (125 MHz, CDCl3) δ 208.33, 169.57, 153.88, 138.18, 128.24, 127.75, 127.58, 121.72, 74.36, 71.54, 64.81, 40.03, 29.86, 28.10, 27.74, 15.56. IR (neat) 2919, 2361, 2338, 1737, 1711, 1658, 1372, 1188, 1026 cm-1. HRMS (ESI-TOF) m/z calcd for C18H22O4Na (M+Na+) 325.1416, found: 325.1421. [α]D20 = –14.0 (c = 0.8, CHCl3).
ethyl (Z)-4-(benzyloxy)-2,3-dimethyl-7-oxooct-2-enoate (3j):
The general procedure A was followed using (E)-4-(benzyloxy)pent-4-en-2-ol (2d, 77 mg, 0.40 mmol) and ethyl (Z)-2-methyl-3-(((trifluoromethyl)sulfonyl)oxy)but-2-enoate (1b, 110 mg, 0.40 mmol). Purification of this product by silica gel chromatography gave a clear, colorless oil (67 mg, 53% yield). Rf = 0.34 (20% EtOAc/Hexanes) 1H NMR (500 MHz, CDCl3) δ 7.35 – 7.26 (m, 5H), 4.53 (dd, J = 8.7, 5.2 Hz, 1H), 4.38 (d, J = 11.7 Hz, 1H), 4.20 (d, J = 11.7 Hz, 1H), 4.19 – 4.08 (m, 1H), 2.59 (ddd, J = 17.3, 8.9, 6.1 Hz, 1H), 2.41 (ddd, J = 17.3, 8.7, 5.9 Hz, 1H), 2.10 (s, 3H), 1.99 – 1.89 (m, 1H), 1.90 (s, 3H), 1.80 – 1.74 (m, 1H), 1.75 (s, 3H) 1.25 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 208.47, 169.29, 143.64, 138.42, 128.24, 127.71, 127.45, 127.12, 77.79, 70.51, 60.44, 40.01, 29.86, 27.74, 16.04, 14.19, 12.83. IR (neat) 2928, 1710, 1453, 1365, 1276, 1212, 1164, 1093, 1067 cm-1. HRMS (ESI-TOF) m/z calcd for C19H26O4Na (M+Na+) 341.1729, found: 341.1736. [α]D20 = –8.1 (c = 1.0, CHCl3).
methyl 2-(1-(benzyloxy)-4-oxopentyl)cyclopent-1-ene-1-carboxylate (3k):
The general procedure A was followed using (E)-4-(benzyloxy)pent-4-en-2-ol (2d, 77 mg, 0.40 mmol) and methyl 2-(((trifluoromethyl)sulfonyl)oxy)cyclopent-1-ene-1-carboxylate (1f, 110 mg, 0.40 mmol). Purification of this product by silica gel chromatography gave a clear, colorless oil (60 mg, 47% yield). Rf = 0.36 (20% EtOAc/Hexanes) 1H NMR (500 MHz, CDCl3) δ 7.38 – 7.30 (m, 5H), 5.05 (d, J = 2.3 Hz, 1H), 4.74 (d, J = 11.9 Hz, 1H), 4.48 (d, J = 12.1 Hz, 1H), 3.72 (d, J = 2.2 Hz, 3H), 2.66 – 2.56 (m, 2H), 2.58 – 2.45 (m, 1H), 2.45 – 2.32 (m, 1H), 2.14 – 2.03 (m, 1H), 2.00 – 1.91 (m, 1H), 1.88 – 1.74 (m, 4H), 1.32 (d, J = 6.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 157.00, 138.30, 128.80, 128.27, 127.78, 127.62, 106.25, 74.25, 69.11, 51.13, 46.77, 38.72, 37.24, 35.18, 34.51, 33.66, 20.10. IR (neat) 2959, 2934, 2360, 2336, 1739, 1717, 1653, 1558, 1270, 1114 cm−1 HRMS (ESI-TOF) m/z calcd for C19H24O4Na (M+Na+) 339.1572, found: 339.1574. [α]D20 = +64 (c = 0.2, CHCl3).
(Z)-5-(2-oxodihydrofuran-3(2H)-ylidene)-4-((4-(trifluoromethyl)benzyl)oxy)hexanal (3l):
The general procedure A was followed using (E)-4-((4-(trifluoromethyl)benzyl)oxy)but-3-en-1-ol (2e, 98 mg, 0.40 mmol) and (Z)-1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl trifluoromethanesulfonate (1a, 110 mg, 0.40 mmol). Purification of this product by silica gel chromatography gel (gradient elution: 0→18% EtOAc/hexanes) gave a clear, colorless oil (81 mg, 57% yield); Rf = 0.33 (50% EtOAc/Hexanes) 1H NMR (500 MHz, CDCl3) δ 9.76 (t, J = 1.6 Hz, 1H), 7.59 (d, J = 8.1 Hz, 2H), 7.41 (d, J = 7.9 Hz, 2H), 5.65 (dd, J = 8.9, 4.9 Hz, 1H), 4.42 (q, J = 11.2, 10.2 Hz, 2H), 4.36 (q, J = 7.2 Hz, 2H), 2.88 (td, J = 7.5, 1.9 Hz, 2H), 2.64 (dddd, J = 16.9, 8.4, 6.5, 1.8 Hz, 1H), 2.48 (dddd, J = 17.7, 7.8, 6.1, 1.4 Hz, 1H), 2.06 (ddd, J = 20.7, 8.6, 6.6 Hz, 1H), 1.87 (t, J = 1.8 Hz, 3H), 1.79 (ddt, J = 11.1, 8.4, 6.2, 5.0 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 201.74, 169.55, 152.80, 142.11, 129.4 (q, J = 64 Hz), 127.70, 125.27 (q, J = 3.8 Hz), 74.62, 70.72, 64.87, 40.48, 29.69, 27.79, 26.68, 15.54. HRMS (ESI-TOF) m/z calcd for C18H19O4NaF3 (M+Na+) 379.1133, found: 379.1136. [α]D20 = –9.2 (c = 0.5, CHCl3).
(Z)-4-((4-methoxybenzyl)oxy)-5-(2-oxodihydrofuran-3(2H)-ylidene)hexanal (3m):
The general procedure A was followed using (E)-4-((4-methoxybenzyl)oxy)but-3-en-1-ol (2f, 83 mg, 0.40 mmol) and (Z)-1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl trifluoromethanesulfonate (1a, 110 mg, 0.40 mmol). Purification of this product by silica gel chromatography gel (gradient elution: 0→18% EtOAc/hexanes) gave a clear, colorless oil (78 mg, 61% yield); Rf = 0.31 (50% EtOAc/Hexanes) 1H NMR (500 MHz, CDCl3) δ 9.73 (t, J = 1.7 Hz, 1H), 7.21 (d, J = 8.2 Hz, 2H), 6.86 (d, J = 8.3 Hz, 2H), 5.58 (dd, J = 9.2, 4.7 Hz, 1H), 4.40 – 4.31 (m, 2H), 4.28 (s, 2H), 3.80 (s, 3H), 2.86 (t, J = 7.9 Hz, 2H), 2.61 (ddt, J = 17.2, 6.4, 4.1 Hz, 1H), 2.45 (ddd, J = 17.3, 8.0, 6.3 Hz, 1H), 2.00 (tt, J = 15.6, 7.0 Hz, 2H), 1.85 (t, J = 1.8 Hz, 3H), 1.74 (ddt, J = 19.6, 8.9, 4.7 Hz, 1H). 13C NMR (125 MHz, cdcl3) δ 202.22, 169.70, 153.93, 130.29, 129.72, 121.93, 113.84, 74.28, 71.54, 65.00, 55.39, 40.70, 29.82, 27.93, 27.02, 15.80. IR (neat) 2916, 2849, 1737, 1653, 1612, 1513, 1246, 1180, 1026 cm−1 HRMS (ESI-TOF) m/z calcd for C19H24O4Na (M+Na+) 341.1378, found: 341.1372. [α]D20 = +11 (c = 0.2, CHCl3).
(Z)-5-(2-oxodihydrofuran-3(2H)-ylidene)-4-(2,2,2-trifluoroethoxy)hexanal (3n):
The general procedure A was followed using (E)-4-(2,2,2-trifluoroethoxy)but-3-en-1-ol (2g, 77 mg, 0.40 mmol) and (Z)-1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl trifluoromethanesulfonate (1a, 110 mg, 0.40 mmol). Purification of this product by silica gel chromatography gave a clear, colorless oil (85 mg, 76% yield). 1H NMR (500 MHz, CDCl3) δ 9.73 (t, J = 1.4 Hz, 1H), 5.61 (dd, J = 9.1, 4.7 Hz, 1H), 4.34 (t, J = 7.5 Hz, 2H), 3.75 – 3.57 (m, 2H), 2.94 – 2.81 (m, 3H), 2.63 (dddd, J = 18.0, 8.4, 6.3, 1.5 Hz, 1H), 2.48 (dddd, J = 18.0, 8.5, 6.0, 1.2 Hz, 1H), 2.00 (dtd, J = 14.9, 8.9, 6.2 Hz, 1H), 1.82 (t, J = 1.8 Hz, 3H), 1.72 (ddt, J = 14.1, 8.7, 5.8, 4.6 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 201.36, 169.47, 150.94, 127.80 (q, J = 201.2 Hz), 123.19, 76.12, 66.82 (q, J = 32.2 Hz), 64.94, 40.24, 27.79, 26.34, 15.28. IR (neat) 2931, 1739, 1713, 1660, 1437, 1378, 1270, 1157, 1111, 1035 cm-1. HRMS (ESI-TOF) m/z calcd for C12H15O4NaF3 (M+Na+) 303.0820, found: 303.0825. [α]D20 = +8 (c = 0.1, CHCl3).
(Z)-5-(2-oxodihydrofuran-3(2H)-ylidene)-4-phenethoxyhexanal (3o):
The general procedure A was followed using (E)-4-phenethoxybut-3-en-1-ol (2g, 83 mg, 0.40 mmol) and (Z)-1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl trifluoromethanesulfonate (1a, 110 mg, 0.40 mmol). Purification of this product by silica gel chromatography gel (gradient elution: 0→18% EtOAc/hexanes) gave a clear, colorless oil (52 mg, 43% yield); Rf = 0.34 (50% EtOAc/Hexanes) 1H NMR (500 MHz, CDCl3) δ 9.71 (t, J = 1.5 Hz, 1H), 7.31 – 7.26 (m, 2H), 7.20 (t, J = 6.8 Hz, 3H), 5.46 (dd, J = 9.1, 4.7 Hz, 1H), 4.35 (dd, J = 5.8, 2.7 Hz, 1H), 4.33 (dd, J = 5.8, 2.3 Hz, 1H), 3.56 – 3.43 (m, 2H), 2.84 (m, 4H), 2.55 (dddd, J = 16.8, 8.5, 6.7, 1.8 Hz, 1H), 2.42 (dddd, J = 17.7, 7.8, 6.1, 1.3 Hz, 1H), 1.94 (dtd, J = 17.3, 9.0, 8.3, 6.7 Hz, 1H) 1.71 (t, J = 1.6 Hz, 4H). 13C NMR (125 MHz, CDCl3) δ 202.14, 169.60, 153.99, 138.90, 128.97, 128.26, 126.19, 121.72, 74.48, 70.24, 64.84, 40.54, 36.33, 27.75, 26.82, 15.36. IR (neat) 2923, 2854, 1739, 1717, 1653, 1455, 1376, 1267, 1194, 1091 cm−1 HRMS (ESI-TOF) m/z calcd for C18H22O4Na (M+Na)+: 325.1416, found: 325.1422. [α]D20 = –6 (c = 0.1, CHCl3).
tert-butyl (Z)-methyl(2-((6-oxo-2-(2-oxodihydrofuran-3(2H)-ylidene)hexan-3-yl)oxy)ethyl)carbamate (3p):
The general procedure A was followed using tert-butyl (E)-(2-((4-hydroxybut-1-en-1-yl)oxy)ethyl)(methyl)carbamate (2h, 98 mg, 0.40 mmol) and (Z)-1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl trifluoromethanesulfonate (1a, 110 mg, 0.40 mmol). Purification of this product by silica gel chromatography gel (gradient elution: 0→18% EtOAc/hexanes) gave a clear, colorless oil (70 mg, 49% yield); Rf = 0.21 (50% EtOAc/Hexanes) 1H NMR (500 MHz, CDCl3) δ 9.75 (t, J = 1.6 Hz, 1H), 5.44 (dd, J = 8.7, 4.9 Hz, 1H), 4.35 (td, J = 6.3, 1.7 Hz, 2H), 3.49 – 3.26 (m, 5H), 2.91 – 2.86 (m, 1H, overlapping peaks) 2.89 (s, 3H), 2.61 (dtd, J = 17.5, 10.2, 6.7, 1.7 Hz, 1H), 2.47 (dddd, J = 16.3, 9.3, 6.2, 1.1 Hz, 1H), 1.96 (dtd, J = 17.0, 8.4, 6.6 Hz, 1H), 1.82 (t, J = 1.7 Hz, 3H), 1.74 (dtd, J = 14.0, 8.3, 6.1, 5.0 Hz, 1H), 1.45 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 201.82, 169.54, 155.76, 153.56, 148.16, 122.12, 79.36, 74.76, 67.64, 64.87, 49.48, 40.52, 28.44, 27.80, 26.83, 15.29. IR (neat) 2997, 2924, 1762, 1740, 1692, 1423, 1389, 1202, 1159, 1140, 1098 cm−1 HRMS (ESI-TOF) m/z calcd for C18H29NO6Na (M+Na+) 378.1893, found: 378.1897. [α]D20 = –4.5 (c = 0.5, CHCl3).
(Z)-4-(cinnamyloxy)-5-(2-oxodihydrofuran-3(2H)-ylidene)hexanal (3q):
The general procedure A was followed using tert-butyl (E)-4-(cinnamyloxy)but-3-en-1-ol (2i, 82 mg, 0.40 mmol) and (Z)-1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl trifluoromethanesulfonate (1a, 110 mg, 0.40 mmol). Purification of this product by silica gel chromatography gel (gradient elution: 0→18% EtOAc/hexanes) gave a clear, colorless oil (53 mg, 42% yield); Rf = 0.21 (50% EtOAc/Hexanes) 1H NMR (500 MHz, CDCl3) δ 9.77 (t, J = 1.5 Hz, 1H), 7.42 – 7.34 (m, 2H), 7.34 – 7.26 (m, 2H), 7.23 (tt, J = 7.2, 1.3 Hz, 1H), 6.56 (d, J = 15.9 Hz, 1H), 6.23 (dt, J = 15.9, 6.1 Hz, 1H), 5.57 (dd, J = 8.9, 4.9 Hz, 1H), 4.38 (t, J = 7.3 Hz, 2H), 4.00 (dddd, J = 18.8, 12.4, 6.0, 1.3 Hz, 2H), 2.66 (dddd, J = 16.8, 8.3, 6.5, 1.7 Hz, 1H), 2.50 (dddd, J = 17.6, 9.7, 6.3, 1.7 Hz, 1H), 2.07 – 1.96 (m, 2H), 1.88 (t, J = 1.6 Hz, 3H), 1.82 – 1.71 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 202.02, 169.58, 153.70, 149.60, 132.56, 128.51, 127.70, 126.47, 125.53, 74.26, 70.14, 64.86, 64.33, 40.54, 27.80, 26.70, 15.66. IR (neat) 3056, 3922, 1739, 1713, 1656, 1375, 1265, 1187, 1035, 968 cm-1. [α]D20 = +32.5 (c = 0.4, CHCl3). HRMS (ESI-TOF) m/z calcd for C19H22O4Na (M+Na+) 337.1416, found: 337.1427.
(Z)-5-(2-oxodihydrofuran-3(2H)-ylidene)-4-((S)-1-phenylethoxy)hexanal (3r):
General procedure A was followed using(S,E)-4-(1-phenylethoxy)but-3-en-1-ol ( 2j, 77 mg, 0.40 mmol) and (Z)-1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl trifluoromethanesulfonate (1a, 110 mg, 0.40 mmol). Purification of this material by silica gel chromatography gave a colorless oil (56 mg, 46% yield) Rf = 0.21 (40% EtOAc in Hexanes) 1H NMR (500 MHz, CDCl3) δ 9.65 (t, J = 1.4 Hz, 1H), 7.36 – 7.31 (m, 2H), 7.29 – 7.24 (m, 3H), 5.34 (dd, J = 9.1, 4.8 Hz, 1H), 4.36 (td, J = 7.5, 0.7 Hz, 2H), 4.19 (q, J = 6.5 Hz, 1H), 2.95 (ddd, J = 15.9, 7.6, 1.5 Hz, 1H), 2.88 (ddd, J = 15.9, 7.6, 1.5 Hz, 1H), 2.48 (dddd, J = 17.8, 8.8, 6.1, 1.7 Hz, 1H), 2.24 (dddd, J = 17.8, 8.7, 6.0, 1.3 Hz, 1H), 1.94 (t, J = 1.7 Hz, 3H), 1.89 (ddt, J = 14.8, 9.0, 6.2 Hz, 1H), 1.65 (dddd, J = 14.0, 9.0, 6.1, 5.1 Hz, 1H), 1.41 (d, J = 6.6 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 202.2, 169.1, 154.1, 142.9, 128.4, 127.8, 126.6, 121.6, 76.3, 71.7, 64.8, 40.4, 27.9, 26.8, 23.7, 15.9. IR (neat) 2972, 2927, 1738, 1719, 1658, 1493, 1452, 1374, 1265, 1187 cm−1 HRMS (ESI-TOF) m/z calcd for C18H22O4Na (M+Na+) 325.1416, found: 325.1423. [α]D20 = –59o (c = 0.4, CHCl3).
(Z)-5-(2-oxodihydrofuran-3(2H)-ylidene)-4-((R)-1-phenylethoxy)hexanal (3s):
General procedure A was followed using(R,E)-4-(1-phenylethoxy)but-3-en-1-ol (2k, 77 mg, 0.40 mmol) and (Z)-1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl trifluoromethanesulfonate (1a, 110 mg, 0.40 mmol). Purification of this material by silica gel chromatography gave a colorless oil (56 mg, 46% yield) Rf = 0.21 (40% EtOAc in Hexanes) 1H NMR (500 MHz, CDCl3) δ 9.80 (t, J = 1.7 Hz, 1H), 7.27 – 7.18 (m, 5H), 5.67 (dd, J = 9.2, 4.7 Hz, 1H), 4.36 (q, J = 6.4 Hz, 1H), 4.23 (td, J = 9.2, 4.9 Hz, 1H), 4.10 (td, J = 8.9, 6.6 Hz, 1H), 2.64 (ttd, J = 15.3, 8.9, 6.9, 2.2 Hz, 2H), 2.49 (dddd, J = 17.4, 7.6, 6.1, 1.3 Hz, 2H), 2.01 (dddd, J = 14.8, 9.1, 8.0, 6.9 Hz, 1H), 1.69 (ddt, J = 14.1, 8.1, 6.1, 4.8 Hz, 1H), 1.61 (t, J = 1.6 Hz, 3H), 1.44 (d, J = 6.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 202.18, 169.54, 153.53, 145.03, 143.82, 127.92, 126.53, 78.16, 73.21, 64.67, 40.67, 27.56, 26.95, 25.92, 22.68, 15.96. IR (neat) 3387, 2973, 2925, 2853, 1736, 1719, 1656, 1451, 1374, 1451, 1375, 1027 cm-1. HRMS (ESI-TOF) m/z calcd for C18H22O4Na (M+Na+) 325.1416, found: 325.1423. [α]D20 = +100.3 (c = 0.5, CHCl3).
(Z)-5-(2-oxodihydrofuran-3(2H)-ylidene)-4-(1-phenylethoxy)hexanal (3r:3s):
General procedure A was followed using (E)-4-(1-phenylethoxy)but-3-en-1-ol (2l, 77 mg, 0.40 mmol) and (Z)-1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl trifluoromethanesulfonate (1a, 110 mg, 0.40 mmol). Crude NMR displayed a 1:1 ratio of the two diastereomers (3r and 3s, comparing the peaks at 5.34 and 5.67 ppm respectively). The two diastereomers were separable via slow gradient utilizing silica gel chromatography. (0 → 30% Et2O/Hexanes). Data matched the respective diastereomers (3r and 3s). Combined isolated yield of 65 mg (54% yield).
(Z)-5-(benzyloxy)-6-(2-oxodihydrofuran-3(2H)-ylidene)heptanal (6a):
General procedure A was followed using (E)-5-(benzyloxy)pent-4-en-1-ol (2m, 77 mg, 0.40 mmol) and (Z)-1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl trifluoromethanesulfonate (1a, 110 mg, 0.40 mmol). Purification of this material by silica gel chromatography gave a colorless oil (56 mg, 46% yield) Rf = 0.14 (30% EtOAc in Hexanes) 1H NMR (500 MHz, CDCl3) δ 9.71 (t, J = 1.6 Hz, 1H), 7.34 – 7.24 (m, 5H), 5.64 (dd, J = 8.0, 5.0 Hz, 1H), 4.37 – 4.31 (m, 4H), 2.86 (td, J = 7.6, 7.2, 1.8 Hz, 2H), 2.50 – 2.36 (m, 2H), 1.85 (t, J = 1.7 Hz, 3H), 1.81 – 1.62 (m, 3H). 13C NMR (125 MHz, CDCl3) δ 202.41, 169.56, 154.06, 138.27, 128.25, 127.85, 127.59, 121.64, 74.22, 71.59, 64.77, 43.33, 33.38, 27.75, 18.10, 15.55. IR (neat) 2921, 2853, 1736, 1717, 1658, 1453, 1261, 1189, 1026 cm−1 HRMS (ESI-TOF) m/z calcd for C18H22O4Na (M+Na+) 325.1416, found: 325.1420. [α]D20 = +13.2 (c = 1.0, CHCl3).
(Z)-7-(benzyloxy)-8-(2-oxodihydrofuran-3(2H)-ylidene)nonanal (6b):
General procedure A was followed using (E)-7-(benzyloxy)hept-6-en-1-ol (2n, 88 mg, 0.40 mmol) and (Z)-1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl trifluoromethanesulfonate (1a, 110 mg, 0.40 mmol). Purification of this material by silica gel chromatography gave a colorless oil (71mg, 54% yield) Rf = 0.15 (30% EtOAc in Hexanes) 1H NMR (500 MHz, CDCl3) δ 9.73 (t, J = 1.8 Hz, 1H), 7.35 – 7.22 (m, 5H), 5.63 (dd, J = 8.3, 4.6 Hz, 1H), 4.38 – 4.30 (m, 4H), 2.86 (td, J = 7.6, 1.9 Hz, 2H), 2.39 (td, J = 7.4, 1.8 Hz, 2H), 1.84 (t, J = 1.7 Hz, 3H), 1.73 – 1.65 (m, 1H), 1.64 – 1.56 (m, 2H), 1.49 – 1.38 (m, 2H), 1.36 – 1.28 (m, 3H). 13C NMR (125 MHz, CDCl3) δ 202.76, 169.55, 154.44, 138.49, 128.23, 127.84, 127.52, 121.36, 74.75, 71.61, 64.70, 43.77, 33.83, 28.83, 27.80, 25.23, 21.91, 15.59. IR (neat) 2929, 2858, 2361, 1739, 1723, 1658, 1454, 1188, 1038 cm−1 HRMS (ESI-TOF) m/z calcd for C20H26O4Na (M+Na+) 353.1729, found: 353.1738. [α]D20 = +12.7 (c = 1.0, CHCl3).
4-(benzyloxy)-4-phenylbutan-1-ol (6a):
General procedure A was followed using (E)-4-(benzyloxy)but-3-en-1-ol (2b, 72 mg, 0.40 mmol, 1.0 equiv), phenyldiazonium tetrafluoroborate (5a, 77 mg, 0.40 mmol, 1.0 equiv), and L2 (13.2 mg, 0.048 mmol, 0.12 equiv). General procedure B was used on the crude material to reduce the resulting aldehyde for purification purposes. Purification of this material by silica gel chromatography (0→26% EtOAc/Hexanes) yielded a clear oil (52 mg, 51% yield) Rf = 0.32 (35% EtOAc/Hexanes) 1H NMR (500 MHz, CDCl3) δ 7.41 – 7.26 (m, 10H), 4.47 (d, J = 11.7 Hz, 1H), 4.36 (dd, J = 8.1, 4.7 Hz, 1H), 4.26 (d, J = 11.7 Hz, 1H), 3.63 (t, J = 6.2 Hz, 2H), 1.91 (dtd, J = 13.5, 8.3, 5.7 Hz, 2H), 1.84 – 1.74 (m, 2H, overlapping peaks), 1.77 – 1.68 (m, 1H, overlapping peaks), 1.61 (dq, J = 7.5, 5.3 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 142.25, 138.22, 128.49, 128.38, 127.88, 127.64, 127.62, 126.71, 81.37, 70.54, 62.84, 35.03, 29.33. IR (neat) 3386, 3029, 2925, 2864, 1494, 1452, 1350, 1265, 1056, 1026 cm-1. HRMS (ESI-TOF) m/z calcd for C17H20O2Na (M+Na+) 279.1361, found: 279.1370.
4-(benzyloxy)-4-(4-methoxyphenyl)butan-1-ol (6b):
General procedure A was followed using (E)-4-(benzyloxy)but-3-en-1-ol (2b, 72 mg, 0.40 mmol, 1.0 equiv), 4-methoxyphenyldiazonium tetrafluoroborate (5b, 89 mg, 0.40 mmol, 1.0 equiv), and L2 (13.2 mg, 0.048 mmol, 0.12 equiv). General procedure B was used on the crude material to reduce the resulting aldehyde for purification purposes. Purification of this material by silica gel chromatography (0→28% EtOAc/Hexanes) yielded a clear oil (52 mg, 51% yield) Rf = 0.28 (35% EtOAc/Hexanes) 1H NMR (500 MHz,CDCl3) δ 7.32 (d, J = 7.4 Hz, 2H), 7.31 – 7.24 (m, 5H), 6.91 (d, J = 8.5 Hz, 2H), 4.44 (d, J = 11.7 Hz, 1H), 4.30 (dd, J = 8.0, 4.9 Hz, 1H), 4.24 (d, J = 11.7 Hz, 1H), 3.82 (s, 3H), 3.62 (t, J = 6.2 Hz, 2H), 1.91 (tt, J = 14.0, 8.4, 5.9 Hz, 1H), 1.81 – 1.66 (m, 3H), 1.59 (dtd, J = 13.7, 6.7, 4.0 Hz, 2H). 13C NMR (125 MHz, cdcl3) δ 158.92, 140.29, 135.10, 128.43, 127.99, 127.96, 127.65, 113.91, 80.94, 70.32, 62.94, 55.33, 35.06, 29.45. IR (neat) 3382, 3031, 2926, 2858, 1610, 1510, 1453, 1302, 1243, 1127, 1028 cm-1. HRMS (ESI-TOF) m/z calcd for C18H22O3Na (M+Na+) 309.1467, found: 309.1477.
4-(benzyloxy)-4-(4-bromophenyl)butan-1-ol (6c):
General procedure A was followed using (E)-4-(benzyloxy)but-3-en-1-ol (2b, 72 mg, 0.40 mmol, 1.0 equiv), 4-bromophenyldiazonium tetrafluoroborate (5b, 89 mg, 0.40 mmol, 1.0 equiv), and L2 (13.2 mg, 0.048 mmol, 0.12 equiv). General procedure B was used on the crude material to reduce the resulting aldehyde for purification purposes. Purification of this material by silica gel chromatography (0→28% EtOAc/Hexanes) yielded a clear oil (52 mg, 51% yield) Rf = 0.28 (35% EtOAc/Hexanes) 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 8.3 Hz, 2H), 7.37 – 7.27 (m, 5H), 7.22 (d, J = 8.4 Hz, 2H), 4.45 (d, J = 11.6 Hz, 1H), 4.32 (dd, J = 8.0, 4.6 Hz, 1H), 4.25 (d, J = 11.5 Hz, 1H), 3.63 (t, J = 6.5 Hz, 2H), 1.95 – 1.81 (m, 1H), 1.80 – 1.66 (m, 3H, overlapping peaks), 1.64 – 1.53 (m, 2H, overlapping peaks). 13C NMR (126 MHz, CDCl3) δ 141.30, 137.93, 131.66, 130.45, 128.44, 127.87, 127.75, 121.36, 80.65, 70.64, 62.73, 34.87, 29.14. IR (neat) 3367, 3062, 2926, 2871, 1594, 1489, 1453, 1208, 1072 cm-1. HRMS (ESI-TOF) m/z calcd for C17H19O2NaBr (M+Na+) 357.0466, found: 357.0470.
Supplementary Material
Acknowledgements
The work was supported by National Institute of Health (NIGMS R01GM063540).
REFERENCES
- [1].a) Meng Z., Sun S., Yuan H., Lou H., Liu L., Angew. Chem. Int. Ed. 2013, 126, 553 [Google Scholar]; b) Arai N., Namba T., Kawaguchi K., Matsumoto Y., Ohkuma T., Angew. Chem., Int. Ed. 2018, 57, 1386. [DOI] [PubMed] [Google Scholar]
- [2].a) Stanley LM., Bai C., Ueda M., Hartwig JF., J. Am. Chem. Soc. 2010, 132, 8918. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shekhar S., Trantow B., Leitner A., Hartwig JF., J. Am. Chem. Soc. 2006, 128, 11770. [DOI] [PubMed] [Google Scholar]; c) Leitner A., Shu C., Hartwig JF., Org. Lett. 2005, 7, 1093. [DOI] [PubMed] [Google Scholar]; d) Kiener CA., Incarvito C., Hartwig JF., J. Am. Chem. Soc. 2003, 125, 14272. [DOI] [PubMed] [Google Scholar]; e) Lopez F., Ohmura T., Hartwig JF., J. Am. Chem. Soc. 2003, 125, 3426. [DOI] [PubMed] [Google Scholar]; f) Kim D., Reddy S., Singh OV., Lee JS., Kong SB., Han H., Org. Lett. 2013, 15, 512. [DOI] [PubMed] [Google Scholar]
- [3].a) Li C., Breit B., Chem.: Eur. J. 2016, 22, 14655. [DOI] [PubMed] [Google Scholar]; b) Lysek R., Borsuk K., Furman B., Kaluza Z., Kazimierski A., Chmielewski M., Curr. Med. Chem. 2004, 11, 1813. [DOI] [PubMed] [Google Scholar]; c) Evans PA., Leahy DK., J. Am. Chem. Soc. 2000, 122, 5012. [Google Scholar]
- [4].a) Trost BM., Tsuji HC., Toste FD., J. Am. Chem. Soc. 2000, 122, 3534. [Google Scholar]; b) Trost BM., Toste FD., J. Am. Chem. Soc. 1999, 121, 4545. [Google Scholar]; c) Trost BM., Toste FD., J. Am. Chem. Soc. 1998, 120, 815. [Google Scholar]; d) Uozumi Y., Kimura M., Tetrahedron: Asymmetry 2006, 17, 161. [Google Scholar]; e) Trost BM., Machacek MR., Tsuji HC., J. Am. Chem. Soc. 2005, 127, 7014. [DOI] [PubMed] [Google Scholar]; f) Trost BM., Crawley ML., Chem.-Eur. J. 2004, 10, 2237. [DOI] [PubMed] [Google Scholar]; g) Trost BM., Guzner JL., Dirat O., Rhee YH., J. Am. Chem. Soc. 2002, 124, 10396. [DOI] [PubMed] [Google Scholar]
- [5].a) Kirsch SF., Overman LE., J. Am. Chem. Soc. 2005, 127, 2866. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kirsch SF., Overman LE., White NS., Org. Lett. 2007, 9, 911. [DOI] [PubMed] [Google Scholar]; c) Cannon JS., Overman LE., Acc. Chem. Res. 2016, 49, 2220. [DOI] [PubMed] [Google Scholar]; d) Cannon JS., Kirsch SF., Overman LE., Sneddon HF., J. Am. Chem. Soc. 2010, 132, 15192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ziegler DT., Fu GC., J. Am. Chem. Sosc. 2016, 138, 12069–12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Patel HH., Prater MB., Squire SO., Sigman MS., J. Am. Chem. Soc. 2018, 140, 5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Werner EW., Mei TS., Burckle AJ., Sigman MS., Science 2012, 338, 1455. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Patel HH., Sigman MS., J. Am. Chem. Soc. 2015, 137, 3462. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Patel HH., Sigman MS., J. Am. Chem. Soc. 2016, 138, 14226. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Chen Z-M., Nervig CS., Deluca RJ., Sigman MS., Angew. Chem. Int. Ed. 2017, 56, 6651. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhang C., Santiago CB., Crawford JM., Sigman MS., J. Am. Chem. Soc. 2015, 137, 15668. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Xu L., Hilton MJ., Zhang X., Norrby PO., Wu YD., Sigman MS., Wiest OJ., J. Am. Chem. Soc. 2014, 136, 1960. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Hilton MJ., Xu L-P., Norrby PO., Wu Y-D., Wiest OJ., Sigman MS., J. Org. Chem. 2014, 79, 11841. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Mei TS., Werner EW., Burckle AJ., Sigman MS., J. Am. Chem. Soc. 2013, 135, 6830. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Allen JR., Bahamonde A., Furukawa Y., Sigman MS., J. Am. Chem. Soc. 2019, 141, 8670. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Bahamonde A., Al Rifaie B., Martin-Heras V., Allen JR., Sigman MS., J. Am. Chem. Soc. 2019, 141, 8708. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Race N., Yuan Q., Sigman MS., Chem. Eur. J. 2019, 25, 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zalesskiy SS., Ananikov VP., Organometallics 2012, 31, 2302. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.