

SUMMARY
Eastern equine encephalitis virus (EEEV) is one of the most virulent viruses endemic to North America. No licensed vaccines or antiviral therapeutics are available to combat this infection, which has recently shown an increase in human cases. Here, we characterize human monoclonal antibodies (mAbs) isolated from a survivor of natural EEEV infection with potent (<20 pM) inhibitory activity of EEEV. Cryo-electron microscopy reconstructions of two highly neutralizing mAbs, EEEV-33 and EEEV-143, were solved in complex with chimeric Sindbis/EEEV virions to 7.2 Å and 8.3 Å, respectively. The mAbs recognize two distinct and antigenic sites that are critical for inhibiting viral entry into cells. EEEV-33 and EEEV-143 protect against disease following stringent lethal aerosol challenge of mice with highly pathogenic EEEV. These studies provide insight into the molecular basis for the neutralizing human antibody response against EEEV and can facilitate development of vaccines and candidate antibody therapeutics.
Keywords: Encephalitis, Eastern Equine; Human Antibodies, Monoclonal; Neutralizing; Prophylaxis; Therapy; Aerosol
Graphical Abstract
eTOC Blurb
Eastern equine encephalitis virus (EEEV) causes rare but serious and often fatal infection associated with inflammation of the brain. Monoclonal antibodies identified from a human survivor recognize distinct epitopes to prevent viral entry recognize and demonstrate protection as well as potent neutralization against EEEV, and may inform future vaccine design.
INTRODUCTION
Eastern equine encephalitis virus (EEEV), a mosquito-transmitted encephalitic alphavirus in the Togaviridae family, is one of the most virulent human viruses in North America, with a case fatality rate of 30 to 75% (Armstrong and Andreadis, 2013; Ayres and Feemster, 1949; Lindsey et al., 2020; Lindsey et al., 2018). Of humans surviving infection, up to 90% develop neurological sequelae (Ronca et al., 2016). EEEV is endemic within the Eastern United States and is transmitted between Culiseta melanura mosquitos and avian hosts (Morens et al., 2019; Sherwood et al., 2020). Occasionally, EEEV is transmitted to humans and horses, resulting in, on average, seven human cases per year in the United States (CDC 2020 [July 5th, 2020]). A recent increase in detection of EEEV in mosquitoes that feed on humans (Mitchell et al., 1992; Sherwood et al., 2020) raises concern for more zoonotic spillover events. In 2019 and as of September 9th, 2020, 36 and 5 human EEEV cases, respectively, were reported with a widespread geographical incidence, leading to concern for EEEV as another emergent arbovirus in the United States (CDC 2020 [September 20th, 2020], (Lindsey et al., 2020; Morens et al., 2019). Neither licensed antiviral drugs nor vaccines for EEEV are available for the general population (Morens et al., 2019; Reichert et al., 2009; Trobaugh et al., 2019). EEEV is considered a USDA/CDC Select Agent due to its potential threat as a bioterrorism agent via aerosol spread and absence of treatment (Sidwell and Smee, 2003). Here, we describe human monoclonal antibodies (mAbs) isolated from a survivor of natural EEEV infection that confer protective and therapeutic efficacy against EEEV in a small animal model of aerosol challenge.
Alphaviruses have T = 4 quasi-icosahedral symmetry, in which there are 60 quasi-threefold (“q3”) and 20 icosahedral-threefold (“i3”) trimeric spikes, each consisting of E1 and E2 glycoprotein heterodimers (Fox et al., 2015; Long et al., 2015; Porta et al., 2014; Voss et al., 2010). During infection, alphaviruses attach to cells via attachment factors, including heparan sulfate (Gardner et al., 2011), DC-SIGN (Klimstra et al., 2003), and LC-SIGN (Klimstra et al., 2003), and entry receptors, such as Mxra8 (Zhang et al., 2018), LDLRAD3 (Hongming et al., 2020), and NRAMP2 (Rose et al., 2011). An entry receptor has yet to be identified for EEEV, which suggests that multiple factors may be involved for attachment and entry of EEEV into host cells. Heparan sulfate (HS) has been implicated as an attachment factor that may contribute to the neurovirulence of EEEV (Chen et al., 2020; Gardner et al., 2011). Upon binding to entry receptors, alphaviruses enters cells by receptor-mediated endocytosis, during which conformational changes may occur (Bernard et al., 2010; DeTulleo and Kirchhausen, 1998; Holmes et al., 2020; Flynn et al., 1990; Meyer and Johnston, 1993). The entry dynamics for EEEV are not well understood. For many alphaviruses, the two surface glycoproteins E1 and E2 are the major targets of antibodies.
Antibody responses to alphaviruses contribute to prevention of new infection or clearance of an established infection (Baxter and Griffin, 2016; Griffin, 1995; Griffin et al., 1997; Levine et al., 1991; Metcalf et al., 2013; Metcalf and Griffin, 2011; Nilaratanakul et al., 2018). Previous studies of antibodies to chikungunya virus (CHIKV) (Broeckel et al., 2017; Fong et al., 2014; Fox et al., 2019; Jin et al., 2015; Kose et al., 2019; Long et al., 2015; Pal et al., 2013; Quiroz et al., 2019; Selvarajah et al., 2013; Smith et al., 2015; Sun et al., 2013b), Mayaro virus (MAYV) (Earnest et al., 2019), Ross River virus (RRV) (Powell et al., 2020), Venezuelan equine encephalitis virus (VEEV) (Agapov et al., 1994; Burke et al., 2019; Hunt et al., 2011; Hunt et al., 2006; Hunt et al., 2010; Hunt et al., 1990; Hunt and Roehrig, 1995; Hunt et al., 1991; Porta et al., 2014; Roehrig et al., 1982; Roehrig et al., 1988; Roehrig and Mathews, 1985), and Western equine encephalitis virus (WEEV) (Burke et al., 2018; Hulseweh et al., 2014; Hunt and Roehrig, 1985) helped elucidate the mechanisms of protection mediated by antibodies to alphaviruses. Potently neutralizing antibodies primarily target the E2 glycoprotein and interfere with different steps in the virus replication cycle from receptor attachment to viral egress (Fox et al., 2015; Jin et al., 2015; Kim et al., 2019; Powell et al., 2020). Previous studies of EEEV focused on identification of linear epitopes recognized by murine or avian antibodies induced by immunization with recombinant E2 glycoprotein (EnCheng et al., 2013; Pereboev et al., 1996; Sun et al., 2013a; Zhao et al., 2012). More recently, studies of EEEV E2 glycoprotein-specific murine mAbs elicited through inoculation with the chimeric virus SINV/EEEV, defined conformational epitopes within domains A and B of the E2 glycoprotein and elucidated the mechanisms of neutralization, primarily through fusion inhibition (Hasan et al., 2018; Kim et al., 2019). The conformational epitopes recognized by human antibodies in the context of natural infection of EEEV and mechanisms of neutralization used are unknown. Here, we studied the molecular basis for human antibody recognition and neutralization of EEEV through characterization of human mAbs from the memory B cells of a survivor of natural EEEV infection.
RESULTS
Human anti-EEEV mAbs isolated from a naturally infected EEEV survivor potently neutralize EEEV.
We isolated a panel of forty-eight human anti-EEEV mAbs from the B cells of a peripheral blood sample of a donor who had a prior documented, naturally-acquired EEEV infection. To down-select mAbs from this panel, we focused on those that could efficiently neutralize Sindbis virus (SINV)/EEEV, a chimeric virus for use in BSL2 conditions containing the nonstructural proteins of SINV and the structural proteins of EEEV strain FL93–939 (Kim et al., 2019). Twelve mAbs exhibited neutralizing activity against SINV/EEEV (Figure 1A and D). All mAbs isolated were of the IgG1 subclass except for EEEV-143, which was an IgA1 (Figure 1D). Of these, seven exhibited potent neutralization activity (here defined as half-maximal inhibitory concentration (IC50) <20 pM; <6 ng/mL) and five exhibited moderate neutralization activity (here defined as mAbs with 20 pM – 4 nM [6 ng/mL – 1.2 μg/mL] IC50 values) against SINV/EEEV.
Figure 1. Human anti-EEEV mAbs isolated from an EEEV survivor potently neutralize Sindbis (SINV)/EEEV and WT EEEV.
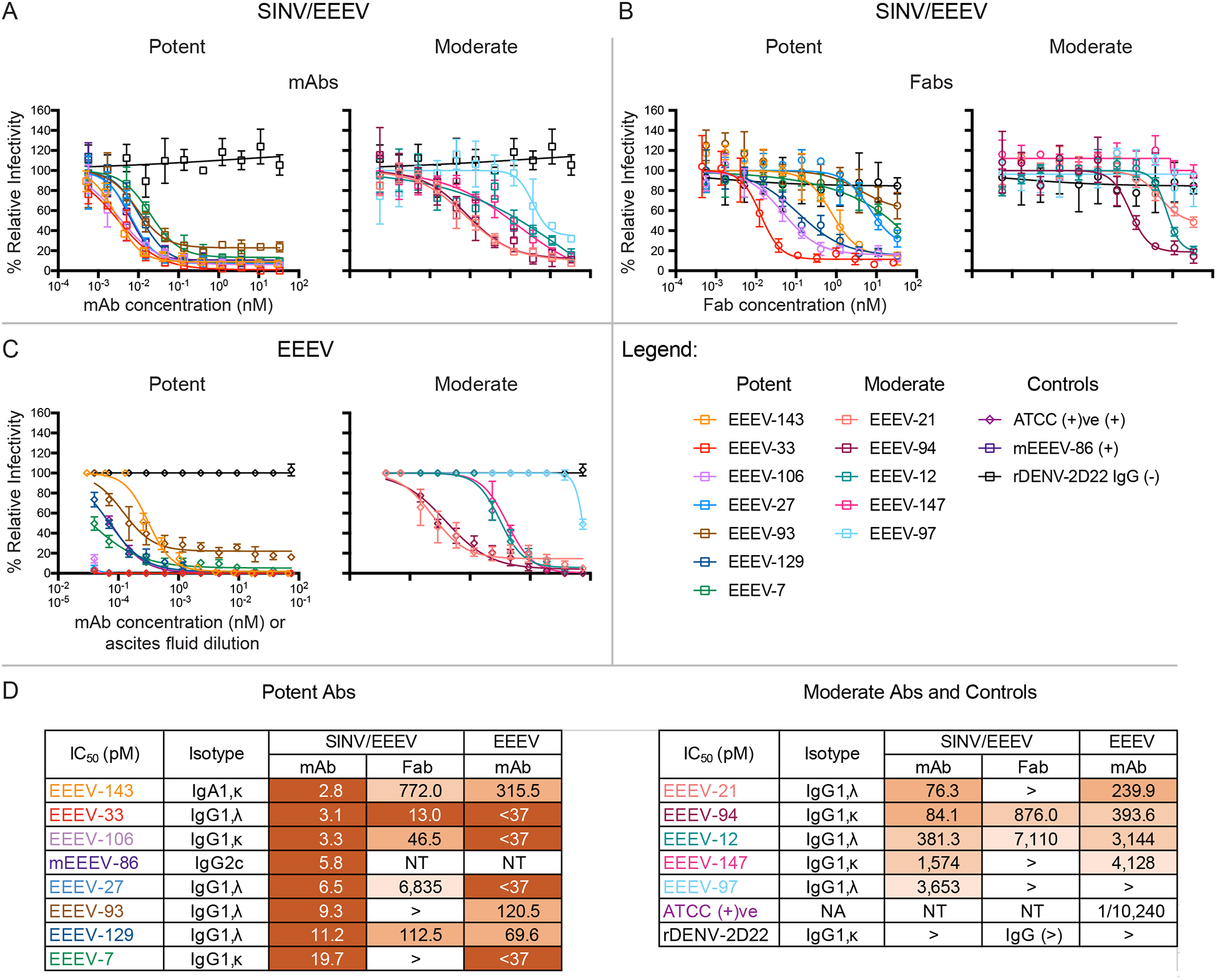
(A–B) Representative neutralization curves of neutralizing human anti-EEEV mAbs (A) or Fabs (B) against SINV/EEEV. Neutralization curves of potent (left) or moderate (right) neutralizing human anti-EEEV mAbs (open squares; A) or Fab molecules (open circles; B) against SINV/EEEV with mAb (A) or Fab (B) concentration (nM) on the x-axis and % relative infectivity on the y-axis. A positive control mouse mAb, EEEV-86 (dark purple) (Kim et al., 2019), and a negative control mAb, rDENV-2D22 (black), were included.
(C) Neutralization curves of human anti-EEEV mAbs against EEEV. Neutralization curves of potent (left) or moderate (right) neutralizing human anti-EEEV mAbs (open diamonds) against EEEV with mAb concentration (nM) on the x-axis and % relative infectivity on the y-axis. A positive control mouse anti-EEEV ascites fluid, ATCC (+)ve, and a negative control mAb (black), were included.
(D) Half-maximal inhibitory concentration (IC50) values (pM) for human anti-EEEV mAbs or Fabs against SINV/EEEV and mAbs against EEEV strain FL93–939. IC50 values (pM) for neutralizing human anti-EEEV mAbs or Fabs against SINV/EEEV or mAbs against EEEV are indicated in the table. Neutralizing human anti-EEEV mAbs are listed in order of increasing IC50 value against SINV/EEEV. IC50 value in pM is indicated by the orange heat map (<33 [dark orange], 33.01 to 333 [medium orange], 333.01 to 3,333 [light orange], <10,000 [lightest orange]). Isotype is indicated as heavy chain (IgG1 or IgA1), light chain (κ or λ) as determined by antibody gene sequencing.
Data in A–B represent mean ± SD of technical triplicates and are representative of three independent focus reduction neutralization test (FRNT) experiments. Data in C represent mean ± SD of technical triplicates of a plaque reduction neutralization test (PRNT) experiment.
The activity of human anti-EEEV mAbs was tested against the pathogenic EEEV strain FL93–939 under BSL-3 conditions. Overall, the neutralization potency with EEEV was consistent with results obtained with SINV/EEEV, supporting the use of chimeric virus for functional analysis (Figure 1C). Nine mAbs neutralized EEEV completely at 75 nM (22.5 μg/mL), such that a residual fraction of infectious virus was not observed (Figure 1C). Several mAbs exhibited extremely potent neutralization activity against EEEV; three mAbs (EEEV-27, −33, and −106) achieved nearly 100% neutralization of EEEV even at the lowest concentration tested, 37 pM (11 ng/mL) (Figure 1C and D). Nine mAbs exhibited neutralization activity of <400 pM (120 ng/mL) IC50 values against EEEV. Additionally, two mAbs exhibited weak neutralization activity with IC50 values in a range of 400 pM – 5 nM (120 ng/mL – 1.5 μg/mL) against EEEV (Figure 1D). EEEV-97 did not neutralize EEEV at any of the concentrations tested, which corresponded with its weak activity against SINV/EEEV.
Neutralizing human anti-EEEV mAbs bind to SINV/EEEV particles and/or recombinant EEEV E2 glycoprotein.
To identify the antigen specificity of the neutralizing human anti-EEEV mAbs, we assessed binding to SINV/EEEV particles and EEEV E2 or E1 glycoproteins (Figure 2 and S1). The ratio of virus/protein half-maximal effective binding concentration (EC50) values for binding was determined (Figure 2B) to identify differences in reactivity of mAbs recognizing virion particle-specific epitopes versus those more accessible in recombinant glycoprotein. Preferential binding to virion-specific epitopes suggests that some mAbs recognize quaternary epitopes on the virion or require bivalent binding with specific geometric orientation. A majority of the mAbs bound strongly to either E2 antigen (isolated protein or SINV/EEEV particles) by ELISA (<100 pM [<30 ng/mL] EC50 values), with a virus/protein EC50 ratio of ~1. EEEV-33 bound weakly to recombinant E2 glycoprotein compared to SINV/EEEV particles (<4 nM vs 11 pM [<1.2 μg/mL vs 3.3 ng/mL] EC50 values), indicated by a virus/protein EC50 ratio of 0.003 (Figure 2B). A few mAbs bound E2 protein better than SINV/EEEV particles, such as EEEV-147 (virus/protein EC50 ratio = 4.4), indicating that these mAbs may recognize an epitope that is less accessible on the virion surface under the conditions tested. Binding to EEEV or CHIKV E1 glycoproteins was not detected.
Figure 2. Neutralizing human anti-EEEV mAbs bind to SINV/EEEV particles and/or recombinant EEEV E2 glycoprotein.
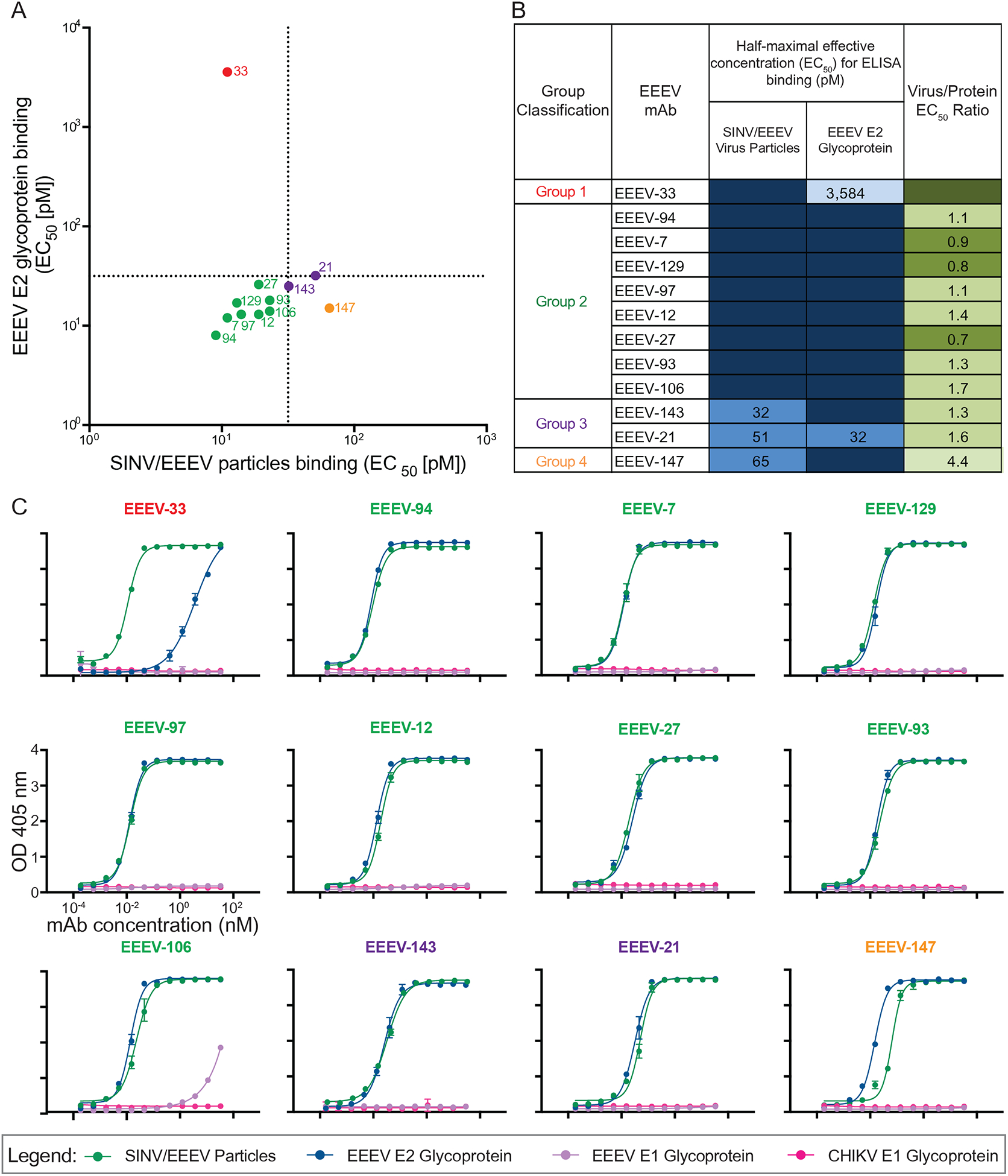
(A) Binding ratio of neutralizing human anti-EEEV mAbs to SINV/EEEV particles versus recombinant monomeric EEEV E2 glycoprotein. A dotted line indicates 32 pM EC50 values for binding, revealing distinct binding patterns of human anti-EEEV mAbs to SINV/EEEV particles and EEEV E2 glycoprotein. Neutralizing human anti-EEEV mAbs are labeled with the anti-EEEV mAb name and are colored according to binding group (Group 1 [red] = virus>protein binding; Group 2 [green] = strong (SINV/EEEV EC50 = <32 pM) virus » protein binding; Group 3 [purple] = weak (SINV/EEEV EC50 = >32 pM) virus » protein binding; Group 4 [orange] = protein>virus binding).
(B) EC50 values (pM) for binding of neutralizing human anti-EEEV mAbs to SINV/EEEV particles or EEEV E2 glycoprotein. Neutralizing human anti-EEEV mAbs are listed in order of binding group and increasing EC50 value for binding to SINV/EEEV particles. EC50 value in pM is indicated by the blue heat map (<32 [dark blue], 32.01 to 100 [medium blue], 100.01 to 320 [light blue], <10,000 [lightest blue]). Ratio of binding to SINV/EEEV particles versus EEEV E2 glycoprotein is indicated as the ratio of EC50 values, corresponding to Figure 2A. Increasing depth of green color indicates lower ratios (<0.1 [dark green], 0.1 to 1.0 [medium green], 1.01 to 2.0 [light green], >2.0 [lightest green]), suggesting recognition of a quaternary epitope on virion particles.
(C) Representative binding curves of neutralizing human anti-EEEV mAbs to four different antigens. Binding curves of neutralizing human anti-EEEV mAbs to SINV/EEEV particles (green) and EEEV E2 glycoprotein (blue), with mAb concentration (nM) on the x-axis and optical density at 405 nm on the y-axis. Binding to EEEV E1 (purple) or CHIKV E1 (pink) glycoproteins was not detected.
Data in A–C represent mean ± SD of technical triplicates and are representative of three independent experiments. See also Figure S1 for recombinant IgG1, IgA1, or Fab binding reactivity to SINV/EEEV particles or EEEV E2 glycoprotein.
Optimal neutralization of SINV/EEEV requires bivalent interactions.
The neutralization potency of Fab and IgG molecules has been compared previously for several alphaviruses (Hasan et al., 2018; Long et al., 2015). In some cases, the neutralizing activity of the Fab form of the mAb is substantially lower than the intact IgG (Hasan et al., 2018). However, we found that the Fab forms for some neutralizing human anti-EEEV mAbs (EEEV-33, −94, −106, −129, −143) still neutralized SINV/EEEV efficiently with <1 nM (<300 ng/mL) IC50 values (Figure 1B and D), indicating that the monovalent Fab molecules may achieve sufficient occupancy of binding for neutralization of EEEV. This finding of similar neutralization potency was consistent with the similar binding strength to SINV/EEEV particles (Figure S1). The neutralization potency of EEEV-94, −106, −129, and −143 as Fab molecules was still reduced (~10 [EEEV-94] to 276 [EEEV-143]-fold change in IC50 values), suggesting bivalent or tetravalent interactions as an IgG (EEEV-94, −106, and −129) or IgA (EEEV-143), respectively, may contribute to optimal neutralization of SINV/EEEV. EEEV-33 had comparable neutralization potency as a Fab or IgG molecule (~4-fold change).
In contrast, EEEV-27 and EEEV-93 exhibited greatly reduced neutralization potency when expressed as Fab molecules, compared to IgG, and showed a corresponding reduction in binding to SINV/EEEV particles (>1,000-fold reduction in IC50 and EC50 values) (Figure S1). These findings suggest that for EEEV-27 and EEEV-93, the monovalent interaction of Fab with SINV/EEEV particles likely is of low affinity and the avidity benefits of interactions achieved though bivalent binding facilitate optimal binding and neutralization of SINV/EEEV. The IC50 values of neutralization and EC50 values for binding to SINV/EEEV did not correspond for all of the inhibitory human anti-EEEV mAbs or Fabs. EEEV-7, −12, −21, −97, and −147 bound SINV/EEEV particles with similar strength as Fab molecules, compared to IgG. However, a reduction in neutralization potency was observed for Fab molecules.
Neutralizing human anti-EEEV mAbs recognize three antigenic determinants on the EEEV E2 glycoprotein.
The E2 glycoprotein consists of three structural domains: 1) a flexible domain B at the apical surface that shields the fusion loop at the distal tip of the E1 glycoprotein, 2) domain A, which is suspected to contain the putative receptor binding site, and 3) domain C that lies proximal to the viral membrane and contains the transmembrane domain (Voss et al., 2010; Chen et al., 2020; Gardner et al., 2013; Gardner et al., 2011; Hasan et al., 2018; Zhang et al., 2011; Fox et al., 2015). To determine the number of major antigenic sites recognized by neutralizing human anti-EEEV mAbs, we performed competition-binding studies utilizing biolayer interferometry and previously described domain B-specific murine anti-EEEV mAbs, including mEEEV-69 and mEEEV-86 (Kim et al., 2019). From this analysis, we identified at least three competition-binding groups on the EEEV E2 glycoprotein (Figure 3A). We observed partial overlap for some of these competition groups, suggesting proximity of the epitopes to each other. A majority of the mAbs competed with murine anti-EEEV mAbs that recognize domain B on the E2 glycoprotein (mEEEV-69 or mEEEV-86), showing that this antigenic determinant was most immunogenic in this individual.
Figure 3. Human anti-EEEV mAbs recognize three neutralizing antigenic determinants on the EEEV E2 glycoprotein.
(A) Competition-binding groups of neutralizing human anti-EEEV mAbs to recombinant EEEV E2 monomeric glycoprotein as determined through biolayer interferometry. Mouse domain B (magenta) and human mAbs were incubated with EEEV E2 glycoprotein to identify the number of antigenic determinants recognized by these mAbs. The first mAb incubated with E2 is shown in the left-hand column and the second mAb is shown in the top column. Black boxes indicate competition, or reduction in maximum signal for binding of the second mAb to <33%. Grey boxes indicate intermediate competition, or reduction in maximum signal for binding of the second mAb to between 33 to 67%. White boxes indicate no competition, or little to no reduction in maximum signal for binding of the second mAb to >67%. Each mAb is colored based on binding group as defined in Figure 2. IC50 (pM) values for neutralization activity against SINV/EEEV are indicated in parentheses (Figure 1D).
(B) Heat map of critical residues for neutralizing human anti-EEEV mAbs as determined through alanine-scanning mutagenesis library analysis. The average percent binding of each neutralizing human anti-EEEV mAbs is indicated for the critical residues identified (<25% binding of mAb in which at least two mAbs exhibited >70% binding to control for expression; D1-L267) and for the previously characterized murine anti-EEEV mAbs (Kim et al., 2019) and the VEEV-specific human mAb, F5 (Hunt et al., 2010; Porta et al., 2014). The heat map displays average % binding relative to WT EEEV E2 glycoprotein with dark blue (>70%), light blue (25–70%), and light green (<25%). Residues are colored based on E2 domain (N-link - purple, Domain A - red, Arch 1 - magenta, Domain B - cyan, and Arch 2 - orange). Each mAb is colored based on binding group as defined in Figure 2 and ordered to correspond with the competition-binding groups as defined in Figure 3A. Data represents mean of at least two independent experiments.
(C) Epitope mapping of critical alanine and arginine residues previously identified for neutralizing murine anti-EEEV mAbs binding to the E2 glycoprotein. Critical residues for binding of murine anti-EEEV mAbs as previously determined through alanine and arginine mutagenesis analyses were mapped onto the 4.2 Å cryo-EM reconstruction of EEEV VLP (EMD-22276; PBD ID: 6XO4) for comparison to the critical alanine residues identified for human anti-EEEV mAbs (see Figure 3D). A trimeric top view of the E2 (green) and E1 (red) glycoproteins is shown with critical residues (spheres) for murine anti-EEEV mAbs that recognize the E2 domains A, B, and A/B. Residues are colored based on E2 domain (Domain A - red and Domain B - cyan).
(D) Epitope mapping of critical alanine residues identified for neutralizing human anti-EEEV mAbs binding to the E2 glycoprotein. Critical residues for binding of human anti-EEEV mAbs as identified through alanine-scanning mutagenesis library analyses (Figure 3B) were mapped as described in Figure 3C. Residues are colored based on E2 domain (N-link - purple, Domain A - red, Arch 1 - magenta, Domain B - cyan, and Arch 2 - orange). Yellow spheres indicate the previously identified SINV/EEEV neutralization escape mutants (M68T, G192R, and L227R) (Kim et al., 2019). Each mAb is presented with its respective E2 domain and is colored based on binding group as defined in Figure 2. See Figure S2 for a bar graph representation of the percent binding of each mAb to the alanine residues described in Figure 3B. See Figure S3 for neutralization activity of mAbs against the SINV/EEEV escape mutants (M68T, G192R, and L227R). See Table S1 for critical alanine residues identified for each mAb.
To identify critical interaction residues for neutralizing human anti-EEEV mAbs, we generated an alanine-scanning mutagenesis library for the EEEV E2 glycoprotein and used it to map residues for which a loss-of-binding phenotype of the antibody occurred. Similar to the competition-binding analysis, three groups emerged. Critical alanine residues identified in this analysis were mapped to the 4.2 Å cryo-EM 3D model of EEEV virus-like particle (VLP) (EMD-22276; PDB ID: 6XO4; Figure S5A–C). The antigenic sites correspond to epitopes on domains A (EEEV-33), B (EEEV-7, −21, −97, −106, −129, −143), or A/B (EEEV-27, −93, −94) of the E2 glycoprotein (Figure 3D and Table S1). Variable gene sequence analysis revealed EEEV-7 and −106 belong to a common lineage, which is supported by their recognition of similar residues for binding. Several mAbs (EEEV-27, −33, −93, −94, and −147) also recognized critical residues in the arch 1 or 2 regions, an acid-sensitive β-connecter region that connects domain A to domain B. Some E2 residues mutated to alanine were identified as critical that are not surface exposed, which may reflect allosteric effects on the epitope that impact antibody binding (Table S1). Critical residues in the EEEV E2 domains were identified previously for murine anti-EEEV mAbs (Kim et al., 2019) (Figure 3C and S2). However, some of the residues identified here are distinct (Table S1), indicating that there are differences in recognition by human and mouse antibodies.
SINV/EEEV neutralization escape mutant viruses with the mutations M68T, G192R, or L227R of the E2 glycoprotein were identified previously using domain A/B murine anti-EEEV mAbs. The mutated residues affected the neutralization potency of domain A and A/B specific murine mAbs to SINV/EEEV. Domain B-specific murine mAbs inhibited the escaped viruses as efficiently as WT SINV/EEEV (Kim et al., 2019). To assess whether viral escape also occurred for the neutralizing human anti-EEEV mAbs, we tested activity of the mAbs against these viruses (Figure S3). Similar to the murine mAbs, mAbs that recognized domain B of the EEEV E2 glycoprotein still neutralized the G192R and L227R escaped viruses with comparable potency to WT SINV/EEEV. The domain B-specific mAb EEEV-97, however, displayed reduced neutralization potency against all three escape mutant viruses. This finding may be due to the weak neutralization potency of EEEV-97 against SINV/EEEV. A number of domain B-specific mAbs (EEEV-7, −106, −129) displayed a >10-fold reduction in neutralization potency to SINV/EEEV (M68T). M68T may lead to an allosteric effect on the epitope for these mAbs, which all depend on the critical alanine residues R205, G207, and H213 for binding. Two other domain B-specific mAbs, EEEV-21 and EEEV-143, also displayed a >5-fold reduction in neutralization potency to SINV/EEEV (M68T), possibly due to an allosteric effect. However, M68T does not affect neutralization potency for EEEV-21 and EEEV-143 to the same extent, which may be due to different critical alanine residues for binding. The domain A-specific EEEV-33 efficiently neutralized the G192R and L227R mutant viruses. However, a >5-fold reduction in neutralization potency was observed for SINV/EEEV (M68T). A loss-of-binding phenotype for clones in the EEEV E2 alanine mutant library was not observed for EEEV-12. However, EEEV-12 displayed a >10-fold or >5-fold reduction in neutralization potency to SINV/EEEV (M68T) or SINV/EEEV (L227R), respectively. Thus, EEEV-12 likely recognizes domain A on the E2 glycoprotein due to reduction of neutralization potency at these mutated residues and observed competition with EEEV-33 for binding to the E2 glycoprotein. Two mAbs, EEEV-94 (domain A/B) and EEEV-147 (N-link/arch 1), displayed comparable neutralization potency of the escaped viruses and WT SINV/EEEV, which correspond with the critical alanine residues identified for these mAbs. EEEV-27 and −93 (domain A/B) displayed reduced neutralization potency for M68T and L227R or comparable potency for the G192R escaped mutant viruses. The observed mutated residues correspond with the epitope identified through alanine-scanning mutagenesis library analysis for EEEV-27 and −93. Thus, through complementary epitope mapping techniques, we defined three major neutralizing E2 antigenic determinants for human mAbs (domain A, B, and A/B).
EEEV-33 and −143 inhibit SINV/EEEV entry into cells.
To elucidate the mechanism of neutralization for human anti-EEEV mAbs, we focused on two potently inhibitory mAbs, EEEV-33 (IgG1; domain A) and EEEV-143 (IgA1; domain B). Entry blockade was assessed by incubating mAbs with SINV/EEEV, allowing the virus to bind and internalize into cells at 37°C, and followed by extensive washing to remove unbound virus and mAb (Figure 4A and 5A). This approach limits exposure of virus to mAb at the attachment, entry and fusion steps of the infection cycle (Fox et al., 2015; Jin et al., 2015). A similar neutralization potency was observed compared to experiments in which the mAb was present at all stages, including egress, suggesting these mAbs act at one of the early entry stages of virus infection.
Figure 4. EEEV-33 recognizes a critical domain A epitope on SINV/EEEV particles for inhibition of viral entry or fusion.
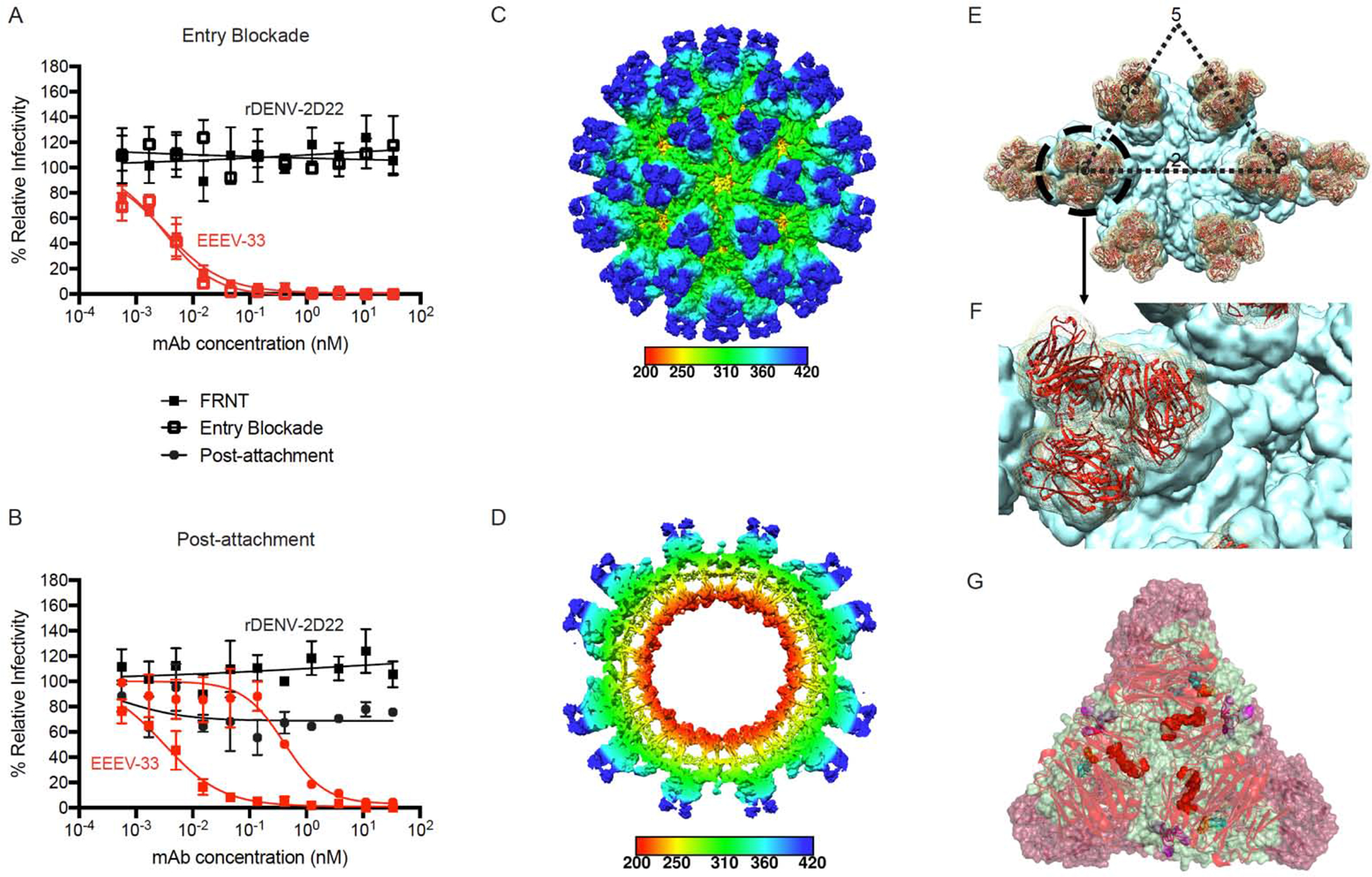
(A) Entry blockade of SINV/EEEV by EEEV-33. An entry blockade assay (open squares) was performed by extensive washing of EEEV-33 (red) or the control mAb (black), from the medium following internalization of SINV/EEEV into Vero cells. Representative neutralization curves are shown for EEEV-33 and rDENV-2D22 as determined through FRNT (closed squares; see Figure 1A) or the entry blockade assay with mAb concentration (nM) on the x-axis and percent relative infectivity on the y-axis.
(B) Post-attachment neutralization of SINV/EEEV. A post-attachment neutralization assay (starred circles) was performed by incubation of Vero cells with SINV/EEEV at 4°C for 1 hour followed by addition of EEEV-33 (red) or rDENV-2D22 (black) at 4°C for 1 hour. Cells were incubated at 37°C for 15 min prior to addition of an overlay and incubation at 37°C for 18 h. Representative neutralization curves are shown for EEEV-33 and rDENV-2D22 as determined through FRNT (closed squares; see Figure 1A) or the post-attachment neutralization assay with mAb concentration (nM) on the x-axis and percent relative infectivity on the y-axis.
(C–D) Cryo-EM reconstruction of SINV/EEEV in complex with EEEV-33 Fab. Cryo-EM structure of EEEV-33 Fab complex (~7.2 Å) showing radially colored surface representation of full (C) and cross section (D) of the map.
(E) EEEV-33 Fab binding footprint to E2 trimeric spikes on SINV/EEEV particles. View of map surface to illustrate binding of EEEV-33 Fab (red) to the q3 and i3 spikes along the icosahedral 2-fold axis.
(F) EEEV-33 Fab constant domain contact interactions. Close-up view of EEEV-33 Fab binding to the i3 spike (black circle in E), in which overlapping Fab constant domain density is observed.
(G) Cryo-EM E2 trimeric view of EEEV-33 Fab binding with critical alanine residues.
Critical alanine residues identified for EEEV-33 are indicated with spheres to illustrate the epitope of EEEV-33 corresponds with the SINV/EEEV (PDB ID: 6MX4) and EEEV-143 Fab (mutated sequence of PDB: 6MWX) docked and rigid body refined cryo-EM model of rEEEV-33 Fab in complex with SINV/EEEV. Sphere color corresponds to E2 domain (N-link - purple, Domain A - red, Arch 1 - magenta, Domain B - cyan, and Arch 2 - orange) as described in Figure 3D.
Data in A–B represent mean ± SD of technical triplicates and are representative of two independent experiments. See Figure S4 for additional views of EEEV-33 Fab binding to the E2 trimer on SINV/EEEV particles.
Figure 5. EEEV-143 recognizes a critical domain B epitope on SINV/EEEV particles for inhibition of viral entry or fusion into cells.
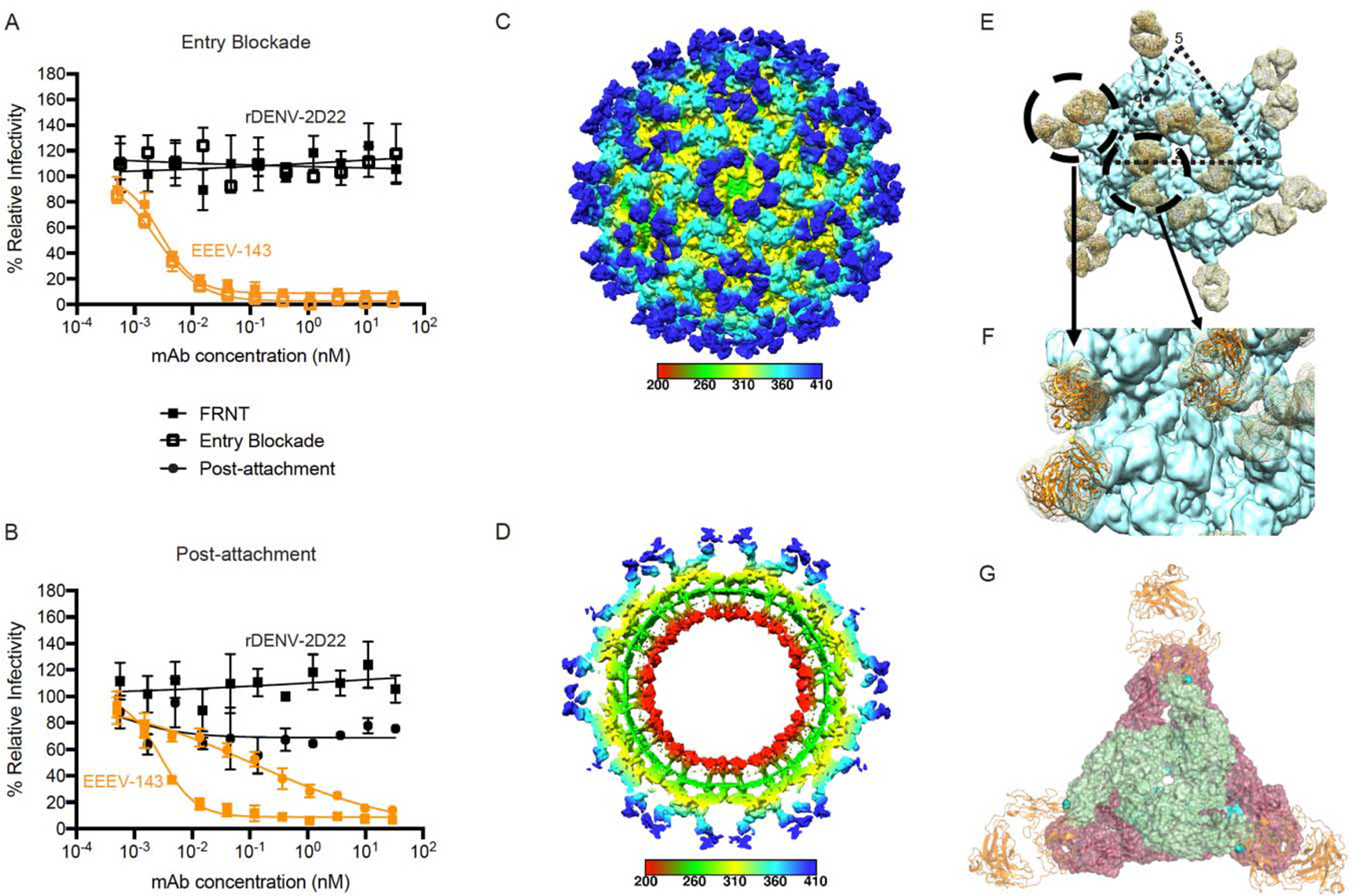
(A) Entry blockade of SINV/EEEV by EEEV-143. An entry blockade assay (open squares) was performed by extensive washing of EEEV-143 (orange) or the control mAb rDENV-2D22 (black), from the medium following internalization of SINV/EEEV. Representative neutralization curves are shown for EEEV-143 and rDENV-2D22 as determined through FRNT (closed squares; see Figure 1A) or the entry blockade assay with mAb concentration (nM) on the x-axis and percent relative infectivity on the y-axis.
(B) Post-attachment neutralization of SINV/EEEV by EEEV-143. A post-attachment neutralization assay (starred circles) was performed by incubation of Vero cells with SINV/EEEV at 4°C for 1 h followed by addition of EEEV-143 (orange) or rDENV-2D22 (black) at 4°C for 1 h. Cells then were incubated at 37°C for 15 min prior to addition of an overlay and incubation at 37°C for 18 h. Representative neutralization curves are shown for EEEV-143 and rDENV-2D22 as determined through FRNT (closed squares; see Figure 1A) or the post-attachment neutralization assay with mAb concentration (nM) on the x-axis and percent relative infectivity on the y-axis.
(C–D) Cryo-EM reconstruction of SINV/EEEV in complex with EEEV-143 Fab. Cryo-EM structure of EEEV-143 Fab complex (~8.3 Å) showing radially colored surface representation of full (C) and cross section (D) of the map.
(E) EEEV-143 Fab binding footprint to E2 trimeric spikes on EEEV virus-like particles (VLPs). View of map surface to illustrate binding of EEEV-143 Fab (in orange) to the q3 and i3 spikes along the icosahedral 2-fold axis.
(F) EEEV-143 Fab constant domain contact interactions. Close-up view of EEEV-143 Fab binding to the q3 and i3 spikes (black circles in E), in which overlapping Fab constant domain density is observed around the 2-fold axis. Fabs bound to the q3 and i3 spikes across the 3-fold axis are ~11 Å apart, in which the flexibility of the Fab may allow for contacts to occur.
(G) Cryo-EM E2 trimeric view of EEEV-143 Fab binding with critical alanine residues.
Critical alanine residues identified for EEEV-143 are indicated with spheres to illustrate the epitope of EEEV-143 corresponds with the EEEV VLP (EMD-22276; PDB ID: 6XO4) and EEEV-143 Fab (mutated sequence of PDB: 6MWX) docked and rigid body refined cryo-EM model. Sphere color corresponds to E2 domain (N-link - purple, Domain A - red, Arch 1 - magenta, Domain B - cyan, and Arch 2 - orange) as described in Figure 3D.
Data in A–B represent mean ± SD of technical triplicates and are representative of two independent experiments. See Figure S4 for additional views of EEEV-143 Fab binding to the E2 trimer on SINV/EEEV particles.
Next, to assess whether EEEV-33 or −143 block virus attachment to cells, a post-attachment neutralization assay was performed (Figure 4B and 5B). In this assay, virus was incubated with cells at 4°C followed by addition of the mAb at 4°C after attachment and removal of free virus. A reduction in neutralization potency of EEEV-33 and −143 was observed, which could indicate that EEEV-33 and −143 block virus attachment to cells to some degree or cannot reach full occupancy of virion binding sites for optimal neutralization due to epitopes blocked by the initial virus attachment to host cells. However, substantial inhibition still occurred post-attachment, indicating that EEEV-33 and −143 can inhibit virus entry into cells after adsorption to the cell surface.
EEEV-33 binds to a critical epitope within domain A of the E2 trimer of SINV/EEEV particles.
We characterized the structural basis for EEEV-33 recognition of SINV/EEEV using single particle cryo-electron microscopy (cryo-EM) and determined a 3D structure of the complex at ~7.2 Å resolution (Figure 4C–G and S7). Analyses above showed that EEEV-33 recognizes domain A of the EEEV E2 glycoprotein (Figure 3). Additionally, the observation that EEEV-33 preferentially recognizes virion particles over E2 glycoprotein suggests that a quaternary interaction between the variable domain of the Fab and E2 protomers in the trimer facilitates binding (Figure 2 and S1). Thus, EEEV-33 may recognize a critical epitope within domain A of the E2 trimer for neutralization of SINV/EEEV. The structural analysis showed that three Fab molecules bound per E2 trimer in a radial orientation. Each E2 protomer was bound to one Fab molecule. The constant domains of each Fab within the trimer appear to clash sterically with one another, such that occupancy might be reduced for the bulkier IgG form of EEEV-33 (Figure 4E and 4F). Additionally, the protomers within the E2 trimer do not move upon EEEV-33 Fab binding, when compared to the apo form of E2 (Figure S4).
EEEV-143 binds to a critical epitope within domain B of the E2 trimer of SINV/EEEV particles and EEEV virus-like particles (VLPs).
We also characterized the structural basis of neutralization of EEEV-143 by studying the Fab in complex with SINV/EEEV particles using single particle cryo-EM to determine a 3D structure at ~8.3 Å resolution (Figure 5C–G and S7). EEEV-143 in complex with EEEV VLPs (Ko et al., 2019) was also determined to ~8.5 Å resolution. Similar results to the SINV/EEEV complex reconstruction above were obtained, indicating the structural conformation of VLPs in development as a candidate vaccine is similar to that of SINV/EEEV (Figure S5). Previous analyses indicated that EEEV-143 recognizes an epitope in domain B of EEEV E2 glycoprotein. The structure shows that three EEEV-143 Fab molecules associate with each E2 trimer with each E2 protomer bound to one Fab molecule in a tangential orientation. The constant domains of Fabs bound between neighboring trimeric spikes across the 2-fold axis appear to make contacts. In addition, Fabs bound to one q3 spike were ~11 Å apart from the constant domain of another Fab bound to the i3 spike across the 3-fold axis (Figure 5E and 5F). Occupancy might be reduced for the IgG form of EEEV-143, due to steric clashes of the Fc regions, and even more so with a polymeric IgA molecule, a model which is supported by the greater binding strength of EEEV-143 IgG1 and Fab molecules to SINV/EEEV (Figure S1). As observed for EEEV-33, alignment of the structural protein asymmetric unit of the solved EEEV VLP (Figure S5A–C; ~4.2 Å) with the EEEV-Fab complex did not show movement of the E2 glycoprotein (Figure S4 and S5I).
EEEV-33 protects against EEEV aerosol challenge in mice.
We next assessed the efficacy for EEEV-33 in vivo using an aerosol-challenge mouse model and a nanoLuciferase-expressing strain of EEEV FL93–939 (Sun et al., 2014). In this model, prophylaxis with a 100-μg dose of EEEV-33 resulted in 91% survival, when administered by the intraperitoneal (i.p.) route 24 hours prior to virus exposure (Figure 6A). Representative in vivo imaging (days 4 or 5 after virus inoculation) is shown in which EEEV replication was not observed in the brain, which differed from animals treated with the control antibody rDENV-2D22 (Figure 6C). Survival was consistent with the body weight patterns of the mice over the course of 14 days (Figure S6A). Additionally, clinical signs of disease (defined as ruffled fur, hunched back/behavioral, seizures/ataxia, moribund, or death) were not observed for the mice that survived. One mouse appeared moribund on day 4 and subsequently died by day 5 (Figure S6C). Survival was reduced (27%) when the same dose of antibody was given 24 hours after exposure compared to rDENV-2D22 (Figure 7A). Representative in vivo imaging (days 4 or 5) is shown for mice that survived infection and reveal EEEV replication was not observed in the brain (Figure 7C). Survival was consistent with the body weight patterns of the mice over the course of 14 days. A reduction in body weight was observed for the mice that died (Figure S6E). Clinical signs of disease were observed for the mice that died (Figure S6G).
Figure 6. Prophylactic administration of mice with EEEV-33 EEEV-143 protects in an aerosol challenge model.
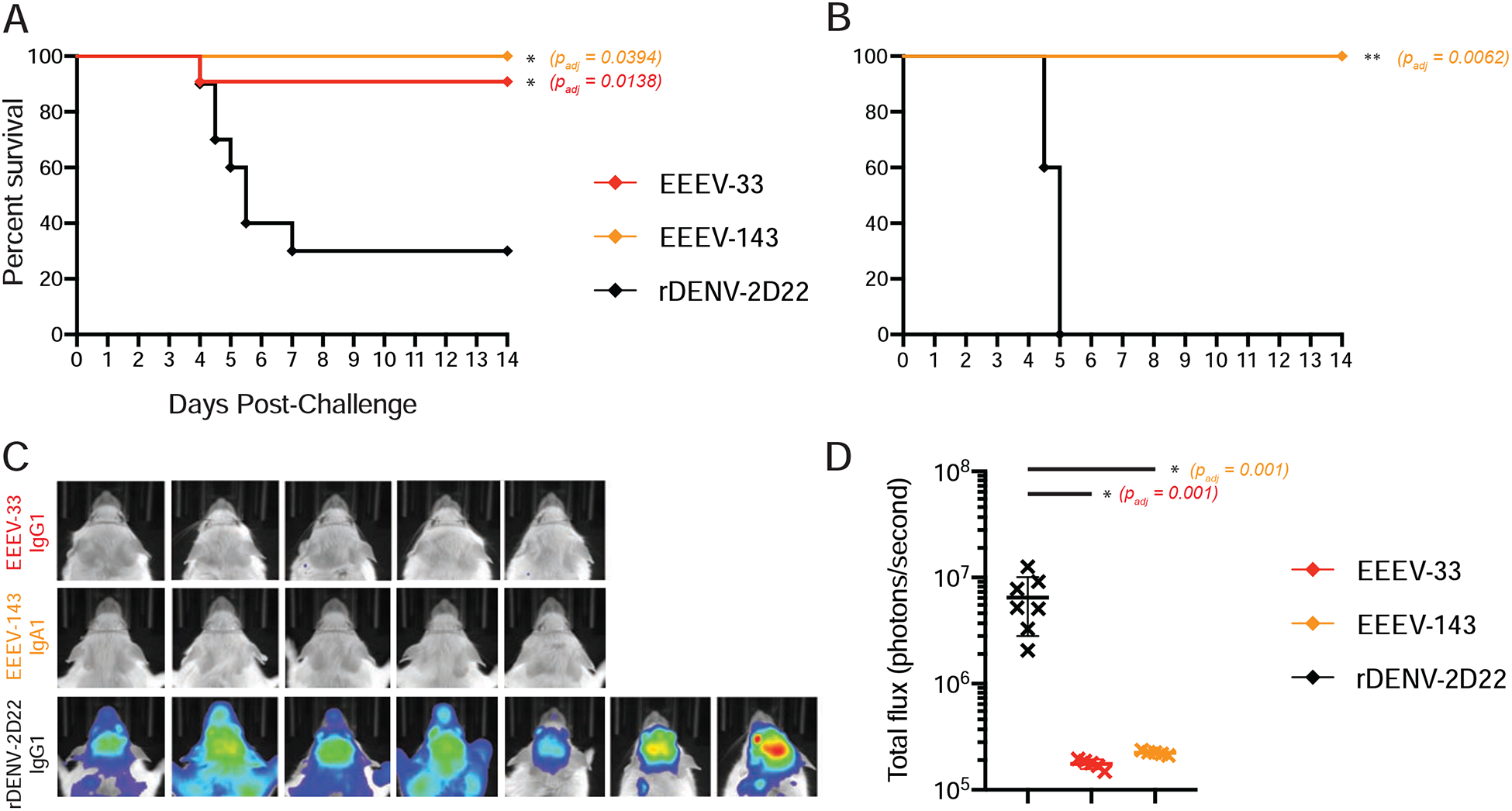
(A) Anti-EEEV mAbs protect against EEEV lethality. EEEV-33 (red; n=11) and EEEV-143 (orange; n=5) were administered prophylactically (24 h prior to virus challenge) at 100 μg via the IP route to CD-1 female mice (4–6-weeks old). EEEV-33 or EEEV-143 protected mice with 91 or 100% survival, respectively, against EEEV (FL93–939) aerosol challenge (1,631 to 1,825 PFU/mouse) compared to the control mAb rDENV-2D22 (black; n=10) (Fibriansah et al., 2015).
(B) EEEV-143 protects against a higher inoculation dose of EEEV. EEEV-143 (orange; n=5) was administered as described in Figure 6A. EEEV-143 exhibited 100% prophylactic survival against EEEV (FL93–939) aerosol challenge (2,739 PFU/mouse). rDENV-2D22 (black; n=5) served as a negative control.
(C) In vivo imaging system (IVIS) images of CD-1 mice for EEEV-33, EEEV-143, and rDENV-2D22 prophylactically treated groups at days 4–5 after EEEV aerosol challenge. IVIS images for EEEV-33 (red), EEEV-143 (orange), and rDENV-2D22 (black). One of the mice in the EEEV-33 group and three of the mice in the rDENV-2D22 negative control group died prior to IVIS imaging on day 5 after virus challenge.
(D) Luminescence intensity of IVIS images. Total flux (photons/second) for the corresponding IVIS images in Figure 6C of the EEEV-33, EEEV-143, and rDENV-2D22 groups is indicated. ~1 × 105 total flux is the background for uninfected mice. One-way ANOVA with Dunnett’s multiple comparisons correction was used to compare luminescence of the images to the rDENV-2D22 control group. *p < 0.01.
Data A, C, D represent combined in vivo data for EEEV-33 (n=6, n=5) or rDENV-2D22 (n=5, n=5) in two independent experiments (1,631 to 1,825 PFU/mouse). Data in A represent in vivo data for EEEV-143 (n=5) in one independent experiment (1,631 to 1,825 PFU/mouse). Data in B–D represent in vivo data for EEEV-143 (n=5) in one independent experiment (2,739 PFU/mouse). Data in A–B, the survival curves were compared using the log-rank test with Bonferroni multiple comparison correction. *p < 0.05, **p < 0.01, ns = not significant.
Figure 7. Post-exposure therapy with EEEV-33 and EEEV-143 partially protects mice in an aerosol challenge model.
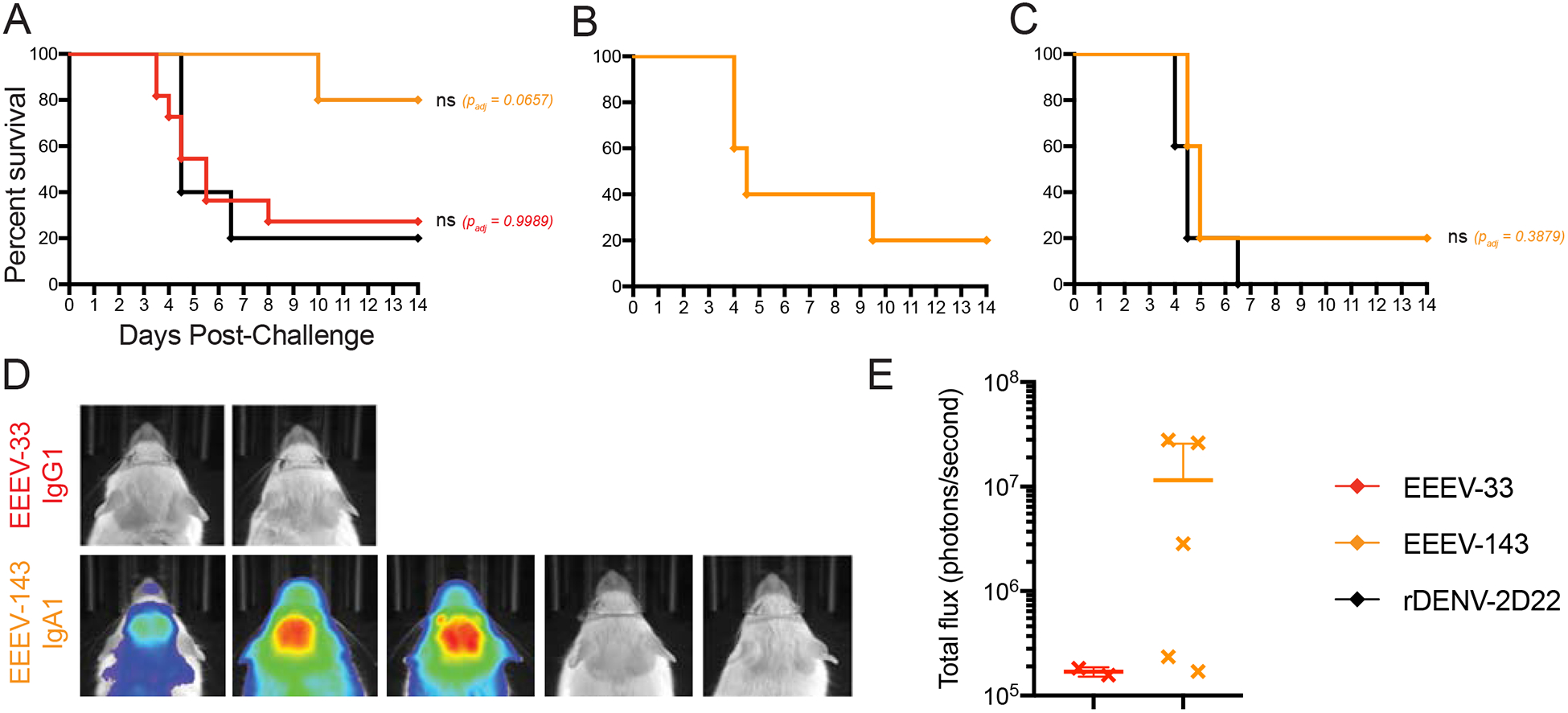
(A) Anti-EEEV mAbs protect against EEEV lethality as post-exposure therapy. EEEV-33 (red; n=11) and EEEV-143 (orange; n=5) were administered therapeutically (24 h post virus challenge) at 100 μg via the IP route to CD-1 female mice (4–6-weeks old). EEEV-33 or EEEV-143 exhibited 27% or 80% therapeutic survival, respectively, against EEEV (FL93–939) aerosol challenge (1,631–1,825 PFU/mouse) compared to the control mAb (black; n=5).
(B) EEEV-143 administration at a higher inoculation dose of EEEV. EEEV-143 (orange; n=5) was administered as described in Figure 7A. EEEV-143 exhibited 20% therapeutic survival against EEEV (FL93–939) aerosol challenge (2,739 PFU/mouse).
(C) EEEV-143 administration at 200-μg dose after EEEV exposure. EEEV-143 (orange; n=5) was administered as described in Figure 7A except at a 200 μg dose. EEEV-143 conferred 20% therapeutic survival against EEEV (strain FL93–939) aerosol challenge (1,897 PFU/mouse) compared to the control mAb (black; n=5; 200 μg).
(D) In vivo imaging system (IVIS) images of CD-1 mice for EEEV-33 and EEEV-143 therapeutically treated group at days 4–5 after EEEV aerosol challenge. IVIS images for EEEV-33 (red) or EEEV-143 (orange). Four of the mice in the EEEV-33 group died prior to IVIS imaging on day 5 after virus challenge.
(E) Luminescence intensity of IVIS images. Total flux (photons/second) for the corresponding IVIS images in Figure 7D of the EEEV-33 and EEEV-143 groups are indicated. ~1 × 105 total flux is the background for uninfected mice.
Data A, B, D, E represent combined in vivo data for EEEV-33 (n=6, n=5) in two independent experiments (1,631 to 1,825 PFU/mouse). Data in A represent in vivo data for EEEV-143 (n=5) and rDENV-2D22 (n=5) in one independent experiment (1,631 to 1,825 PFU/mouse). Data in B, D, E represent in vivo data for EEEV-143 (n=5) in one independent experiment (2,739 PFU/mouse). Data in C represent in vivo data for EEEV-143 (n=5) and rDENV-2D22 (n=5) in one independent experiment (1,897 PFU/mouse). Data in A-C, the survival curves were compared using the log-rank test with Bonferroni multiple comparison correction. *p < 0.05, **p < 0.01, ns = not significant.
EEEV-143 protects and treats against WT EEEV aerosol challenge in mice.
The mucosal IgA response is suspected to play a role in protection because mice with low IgG serum titers following vaccination with EEEV live attenuated virus candidates still were protected against lethal EEEV aerosol challenge (Trobaugh et al., 2019). To assess this possibility, we studied the treatment efficacy of the IgA isotype form of the neutralizing mAb EEEV-143. EEEV-143 mediated 100% survival when a 100-μg dose was administered i.p. route 24 hours prior to virus exposure compared to rDENV-2D22 (Figure 6A). Remarkably, in one study (Figure 7A), 4 of 5 (80%) mice inoculated survived when EEEV-143 was administered 24 hours after exposure (1,825 PFU/mouse) compared to rDENV-2D22. In a replicate study, EEEV-143 also mediated 100% survival when a 100-μg dose was administered i.p. route 24 hours prior to virus exposure (2,379 PFU/mouse) compared to rDENV-2D22 (Figure 6B). However, in this cohort, only 20% of the EEEV-143-treated mice survived when mAbs were administered 24 hours after exposure (2,379 PFU/mouse) compared to rDENV-2D22 (Figure 7B). The difference in therapeutic efficacy between these two studies may be due to the variability of infection efficiency in the aerosol model, as slightly different virus titers were administered according to results from back-titration of the inocula on the days of challenge. To corroborate our findings, an additional study was performed. Here, 20% of the EEEV-143-treated mice survived when mAbs were administered a 200-μg dose 24 hours after exposure (1,897 PFU/mouse) (Figure 7C). Representative in vivo imaging (days 4 or 5) is shown for mice that survived infection in which EEEV replication was not observed in the brain as compared to rDENV-2D22 (Figures 6C and 7C). Survival was consistent with body weight of the mice over the course of 14 days. A reduction in body weight was observed for the mice that died (Figures S6A–B and S6E–F). For the mice that died, all clinical signs of disease were observed (Figures S6C–D and S6G–H).
DISCUSSION
In this study, we describe human mAbs isolated against EEEV from the B cells of an EEEV-immune individual with prior natural infection. Isolation of human anti-EEEV mAbs with moderate or potent neutralization activity against the chimeric virus Sindbis (SINV)/EEEV allowed for the characterization of the molecular and structural basis of neutralization against EEEV and addressed important questions relating to the human anti-EEEV antibody response. We observed diverse patterns of mAb reactivity and dependence on valency for binding and/or neutralization of SINV/EEEV. Potent mAbs neutralized SINV/EEEV as Fab molecules, suggesting bivalency or tetravalency is not necessary but may be required for optimal neutralization of SINV/EEEV. Two mAbs, EEEV-27 and EEEV-93, displayed reduced binding and neutralization potency as Fab molecules, indicating the requirement for bivalent interactions. Additionally, EEEV-7, −12, −21, −97, and −147 displayed reduced neutralization potency but not reduced binding as Fab molecules, indicating the neutralization mechanism for these mAbs may involve cross-linking of two E2 protomers as IgG molecules. Recognition of three antigenic determinants (domains A, B, and A/B) was observed, with a majority of neutralizing mAbs isolated from this individual recognizing the E2 structural domain B. Further characterization of EEEV-33 and EEEV-143 identified the molecular and structural basis of neutralization, which enabled these mAbs to exhibit in vivo efficacy against highly pathogenic EEEV in an aerosol challenge model.
EEEV-33 is a potently neutralizing human anti-EEEV mAb, with 3.1 pM or <37 pM IC50 values for neutralization against SINV/EEEV or EEEV, respectively. EEEV-33 preferentially binds SINV/EEEV particles compared to recombinant EEEV E2 glycoprotein, suggesting recognition of a quaternary epitope on the viral surface. A ~7.2 Å 3D image of SINV/EEEV bound to EEEV-33 Fab molecules elucidated the structural basis of neutralization by this antibody. The high neutralization potency of EEEV-33, which has similar potency as a Fab molecule, was consistent with the ability of three Fabs to bind each domain A binding site in the E2 trimer. The ability of three EEEV-33 Fabs to bind to each E2 trimer in the virus is unusual, compared to previous structural analyses of murine anti-EEEV mAbs in complex with SINV/EEEV particles (Hasan et al., 2018). From the murine antibody structural analysis, it was observed that steric clashes between Fab molecules, which target domain A of the E2 glycoprotein in a radial orientation, would limit the capacity for complete occupancy of the E2 trimer. However, EEEV-33 binds a unique epitope compared to the murine anti-EEEV mAbs, and the neutralization potency of EEEV-33 even as a Fab molecule is consistent with the occupancy observed in the cryo-EM model. Fab constant domain contacts were observed between Fabs bound within the trimeric spike, indicating that as an IgG molecule steric hindrance may limit occupancy. However, this feature may allow for intra-spike cross-linking to occur for neutralization. We also observed that the protomers of the E2 trimer do not change conformation upon EEEV-33 Fab binding. Together, these data suggest that EEEV-33 recognizes a conformational epitope present on domain A of the E2 trimer of SINV/EEEV particles. Binding to this epitope, EEEV-33 may stabilize or sterically hinder the E2 trimer, inhibiting viral entry or fusion.
A neutralizing VEEV-specific human mAb similar to EEEV-33, designated F5, was characterized previously (Hunt et al., 2010; Porta et al., 2014). Antibody F5 was isolated using phage display from B cells of VEEV-immune donors. This antibody binds to residues 115–119 in domain A of the E2 glycoprotein, analogous to those recognized by EEEV-33. In contrast to EEEV-33, F5 Fab molecules bind this region of VEEV in a radial orientation and occupy one third of the binding sites. F5 stabilized the E2 trimer via intra-spike cross-linking (Porta et al., 2014). The similarity in binding and neutralization activity for two human mAbs (EEEV or VEEV) suggests a conserved antigenic site (residues 115–120).
EEEV-143 is another potently neutralizing human anti-EEEV mAb with 2.8 pM or 315 pM IC50 values against SINV/EEEV or EEEV, respectively. The ~100-fold difference in IC50 values against SINV/EEEV and EEEV may be due to physical differences in the various virus preparations used, such as particle/PFU ratio or post-translational modifications of the structural proteins (Jose et al., 2010). EEEV-143 was isolated from a human B cell as an IgA1 antibody. In mice vaccinated with live-attenuated EEEV vaccine candidates, protection against aerosol challenge was observed even in some mice with low serum PRNT80 values, suggesting other immune responses may contribute to protection (Trobaugh et al., 2019). Strategies to prevent and/or treat EEEV through antibody-based methods that specifically target mucosal sites could be an important approach. Naturally occurring polymeric IgA (pIgA) molecules are transported actively across mucosal surfaces via the polymeric immunoglobulin receptor (pIgR) (Turula and Wobus, 2018). Transcytosis of neutralizing antibodies across the mucosa may block EEEV infection at or near the site of inoculation to prevent dissemination to the brain.
The structural basis of neutralization for EEEV-143 was determined using single particle cryo-EM of SINV/EEEV bound to EEEV-143 Fab molecules. The resulting ~8.3 Å structure showed that three Fabs also bound to each protomer in the E2 trimer. Fab constant domain contacts were observed around the 2-fold axis and ~11 Å distance between Fabs bound to the q3 and i3 spikes across the 3-fold axis. These contacts suggest that EEEV-143 forms inter-spike cross-links between adjacent E2 trimers as an IgG or IgA. This inter-E2 trimer cross-linking could stabilize or sterically hinder the trimer to inhibit viral entry or fusion. Again, the binding of three EEEV-143 Fab molecules to each E2 trimer is unusual when compared to studies of neutralizing antibodies generated in mice, where steric clashes between Fab molecules that target domain B of the E2 glycoprotein in a tangential orientation limited the capacity for complete occupancy of the E2 trimer. However, the neutralization potency of EEEV-143 as Fab molecules is consistent with our structural analysis. We suggest that steric clashes still will occur in the context of EEEV-143 expressed as a polymeric IgA molecule (dimeric IgA complex with joining [J] chain) as suggested by the reduction in the detected binding strength of this molecule compared to recombinant IgG1 or Fab molecules. Optimal in vitro neutralization of SINV/EEEV occurs as a polymeric IgA compared to Fab molecule, as there is >200-fold reduction in neutralization potency. Thus, the inter-spike cross-linking observed for EEEV-143 may require a bivalent (IgG) or tetravalent (IgA) antibody interaction for optimal neutralization of SINV/EEEV.
A neutralizing CHIKV mAb similar to EEEV-143, designated CHK-265, was characterized previously (Fox et al., 2015) CHK-265 is a broadly neutralizing and protective arthritogenic alphavirus mAb that inhibits viral entry and egress from cells. The ~16 Å cryo-EM reconstruction of CHIKV 181/25 particles in complex with CHKV-265 Fab molecules displays binding of three Fab molecules to each trimeric spike (q3 and i3). Inter-spike cross-linking was observed between two E2 protomers through recognition of domain B residues on one protomer and contacts domain A residues on an adjacent protomer. This binding leads to movement of domain B further over the E1 fusion loop and repositioning of domain A upon binding. CHK-265 Fab constant domain contacts were observed across the 2-fold axis further supporting the cross-liking mechanism of virus particles by CHK-265 as an IgG molecule. In contrast, EEEV-143 recognizes residues within domain B on one protomer and does not appear to induce conformational changes upon binding. Similarly, the constant domain Fab contacts observed for EEEV-143 around the 2-fold axis suggests that EEEV-143 may form inter-spike cross-links as an IgG or IgA molecule. The reduction in neutralization of EEEV-143 as a Fab molecule suggests that this mechanism is important for optimal neutralization of EEEV-143 at this site.
EEEV is classified as a USDA/CDC Select Agent due to potential for aerosolization and use as a bioterrorism agent. To mimic potential exposure of EEEV as a bioterrorism agent, aerosol challenge has been studied in mice and non-human primates (Phelps et al., 2019; Trobaugh et al., 2019). In our approach, mice were inoculated with EEEV via the aerosol route to assess the pre- or post-exposure treatment efficacy of EEEV-33 (IgG1) and EEEV-143 (IgA1). When given 24 hours before infection, EEEV-33 or EEEV-143 each protected mice with 91 or 100% survival compared to the control, respectively. When administered 24 hours after EEEV inoculation, EEEV-33 treated mice had a 27% survival rate whereas EEEV-143 treated mice showed 20 to 80% survival rates compared to the 0 to 20% for controls. This experimental animal aerosol challenge model is stringent, since the rapid kinetics of viral replication in the central nervous system quickly decreases the likelihood of antibody-mediated protection. There is need to study the in vivo efficacy in more detail, as multiple factors could affect effectiveness. As EEEV-33 and EEEV-143 recognize two different antigenic sites on the E2, a potential combination therapy with EEEV-33 and EEEV-143 might have greater efficacy and decrease the likelihood of viral escape mutant viruses generated in vivo.
Soon after EEEV aerosol challenge, virus-induced lesions are observed in the olfactory bulb. The spread and course of EEEV infection within the brain parenchyma corresponds with the severity of olfactory bulb lesions (Phelps et al., 2019). Once virus enters into the brain, it becomes challenging to neutralize the virus to allow for control of spread and survival. One of the difficulties in treatment of EEEV is the requirement for antibodies to cross the BBB (Morens et al., 2019), which limits transport of molecules >400 Da (Neves V, 2016). During viral infection, disruption of the BBB occurs, allowing for infiltration of immune cells and larger molecules, such as antibodies (Metcalf et al., 2013; Metcalf and Griffin, 2011). However, with alphaviruses in the mouse model, the breakdown of the BBB is a late event in the disease course (Salimi et al., 2020) and the inflammatory consequences of viral replication result in neurological deterioration, making recovery difficult after extensive infection (Griffin, 2016; Metcalf and Griffin, 2011). Developing molecules that cross this barrier more efficiently, such as through receptor-mediated transcytosis, might enhance the efficacy of therapeutic antibodies for EEEV infection (Pulgar, 2018).
Here we described the isolation and characterization of neutralizing human antibodies against EEEV. These studies provide molecular and structural bases of neutralization of EEEV by human mAbs and suggest future research directions that could provide treatment options for patients. These include defining the importance of mucosal IgA and avidity interactions for optimal neutralization of EEEV, the correlates of antibody-mediated protection, and mechanisms for enhancing antibody transport across the BBB in vivo. Our studies focused on binding, neutralization, and in vivo efficacy against the 1993 Florida FL93–939 isolate of EEEV. Genomic analysis of North American EEEV strains from 1934 to 2014 revealed a high level of conservation, with 99.76% average amino acid similarity (Tan et al., 2018), which suggests the FL93–939 strain may be representative of a majority of the EEEV strains. However, Madariaga virus (MADV), formally known as South American EEEV, is genetically distinct from EEEV (Lednicky et al., 2019). Analysis of neutralization activity by the mAbs described here against other EEEV strains circulating in North America, such as the 2019 strain and MADV, warrants further investigation. Overall, the studies described may help inform rationale structure-guided vaccine design and identify possible correlates of protection for lead therapeutic candidates against EEEV, and possibly other encephalitic alphaviruses.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents may be directed to and be fulfilled by the Lead Contact: Dr. James E. Crowe, Jr. (james.crowe@vumc.org).
Materials Availability
Materials described in this paper are available from the Lead Contact for distribution under the Uniform Biological Material Transfer Agreement, a master agreement that was developed by the NIH to simplify transfers of biological research materials.
Data and Code Availability
The cryo-EM reconstructions reported in this paper have been deposited to the Protein Data Bank or Electron Microscopy Data Bank under the accession numbers EMD-22223 (SINV/EEEV:rEEEV-33 Fab), EMD-22188 (SINV/EEEV:rEEEV-143 Fab), EMD-22276; PDB ID: 6XO4 (EEEV VLP), EMD-22277; PDB ID: 6XOB (EEEV VLP:rEEEV-143 Fab). All relevant data are included within the manuscript and are available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subject information.
The research subject was a 38-year-old otherwise healthy female subject who contracted EEEV infection naturally in September 2015 in New York state. The disease course was marked by systemic illness including acute meningoencephalitis and resolved with supportive therapy delivered in hospital. The New York State Department of Health (NYSDOH) Wadsworth Laboratory reported this was a laboratory-confirmed infection with EEEV. Peripheral blood from the subject was collected 13 months after infection, and peripheral blood mononuclear cells (PBMCs) were isolated by density gradient purification and cryopreserved until use. The subject gave written informed consent prior to participation, and the Institutional Review Board (IRB) at Vanderbilt University Medical Center approved the protocols for the recruitment and collection of blood samples used in this study.
Mouse model.
Mice were specific-pathogen-free CD-1 females of 4 to 6 weeks of age purchased from Charles River Laboratories. These studies were performed under University of Pittsburgh IACUC protocol #17121689 in accordance with the recommendations of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
Cell lines.
BHK-21 (hamster, male origin; ATCC) and Vero (monkey, female origin; ATCC) cells were maintained in DMEM (Gibco) and 10% heat-inactivated FBS (Thermo Fisher Scientific) at 37°C in a humidified atmosphere of 5% CO2. HMAA 2.5 cells (mouse-human, gender information not available; kindly provided by Lisa Cavacini) are a non-secreting myeloma cell line and were maintained in ClonaCell™-HY Medium A (Stem Cell Technologies) at 37°C in a humidified atmosphere of 7% CO2 as previously described (Yu et al., 2008a; Yu et al., 2008b). B95.8 (monkey, gender information not available; ATCC) cells were cultured in Medium A for collection of supernatants containing Epstein-Barr virus (EBV). Expi293F (human, female origin; Thermo Fisher Scientific) cells were maintained in Expi293 expression medium (Gibco) or Freestyle F17 medium (Gibco) supplemented with 4 mM L-glutamine (Gibco) and 0.1% pluronic F-68 (Gibco) at 125 rpm 37°C in a humidified atmosphere of 8% CO2. Expi293F cells were authenticated by the ATCC cell line authentication service using Short Tandem Repeat (STR) analysis. Cells were checked routinely for mycoplasma detection using a universal mycoplasma detection kit (ATCC).
Viruses.
The chimeric virus, Sindbis virus (SINV; TR339)/Eastern equine encephalitis virus (EEEV; FL93–939), and SINV/EEEV escape mutant viruses (M68T, G192R, and L227R) were described previously (Kim et al., 2019). EEEV FL93–939 was derived from the cDNA clone, as previously described (Gardner et al., 2011).
Plasmids.
Recombinant EEEV E1 ectodomain (strain FL93–939; amino acids Y1-S409) with the osteonectin leader sequence and a C-terminal 6x his-tag was codon-optimized, synthesized and cloned into the mammalian expression vector pcDNA3.1(+). Recombinant EEEV (strain FL93–939) structural protein genes (capsid-E3-E2–6K-1) were codon-optimized, synthesized and cloned into the mammalian expression vector pcDNA3.1(+) for expression of the WT EEEV structural proteins. Using the WT EEEV structural protein vector, residues D1-L267 of EEEV E2 structural protein were mutated to alanine or alanine residues to serine for expression of the EEEV E2 mutants for alanine-scanning mutagenesis library analyses. Recombinant human anti-EEEV variable genes were synthesized and cloned into a pTwist CMV Betaglobin WPRE Neo mammalian expression vector that was customized to contain isotype-specific constant regions (IgG1, IgA1, or Fab) (Twist Bioscience Inc.). Additionally, for expression of polymeric IgA1, the mouse J chain sequence (Uniprot: P01592) was synthesized a pTwist CMV Betaglobin WPRE Neo mammalian expression vector (Twist).
Recombinant proteins.
Recombinant EEEV E2 glycoprotein (strain v105) was purchased from IBT BioServices and contains a mixture of the p62 (E3E2) and E2 glycoproteins.
Recombinant CHIKV E1 was purchased from Meridian Life Science. EEEV virus-like particles (VLPs) were kindly provided by Dr. John Mascola at the NIH/NIAID Vaccine Research Center (Ko et al., 2019).
METHOD DETAILS
SINV/EEEV production.
BHK-21 cells were plated the day before using 3 × 107 cells per T-225 cm2 flask (Corning). The following day, cells were inoculated with SINV/EEEV at a MOI of 0.2 in DMEM/2% FBS. After incubation at 37°C in 5% CO2 for 48 hours, SINV/EEEV was harvested by clarification of infected BHK-21 cell supernatants through a 0.2-μm pore size filter (Nalgene). Virus then was used fresh or stored at −80°C until use. For cryo-EM studies, BHK-21 cells were inoculated with SINV/EEEV at a MOI of 5 in DMEM/2% FBS. After incubation at 37°C in 5% CO2 for 16 hours, SINV/EEEV was harvested by centrifugation at 2,000 × g at 4°C for 10 minutes. Virus supernatant was precipitated in 14% (w/v) PEG 6000 (Sigma-Aldrich) and 4.6% (w/v) NaCl (Corning) overnight at 4°C, followed by centrifugation at 2,500 × g at 4°C for 30 minutes. A linear, continuous 10 to 50% OptiPrep (Sigma-Aldrich) gradient was used to purify the SINV/EEEV particles further at 136,873 × g (rmax) for 1.5 hours at 4°C using an AH-650 swinging bucket rotor (Sorvall). SINV/EEEV particles were collected and buffer exchanged into TNE buffer (50 mM Tris-HCl pH 7.5 (Sigma), 100 mM NaCl, 0.1 mM EDTA (Corning)) to a concentration of ~0.1 mg/mL total protein content, as determined by a Bradford assay or BCA assay (Thermo Fisher Scientific). Virus then was used fresh or stored at 4°C until use.
Recombinant EEEV E1 ectodomain expression.
E1 was produced in Expi293F cells using the ExpiFectamine 293 transfection kit (Gibco). Briefly, 1 μg/mL of pcDNA3.1(+)-EEEV E1 ectodomain was diluted in Opti-MEM medium (Gibco) with ExpiFectamine 293 reagent for 15 to 20 minutes at room temperature before addition to Expi293F cells. Cells were incubated at 37°C in 8% CO2 and supernatant was harvested by centrifugation and subsequent filtering through a 0.45-μm pore size filter (Nalgene) 2 to 6 days after transfection. Cell supernatant was purified through a HisTrap excel column (GE Healthcare) according to the manufacturer’s protocol on an ÄKTA pure 25M chromatography system.
Human hybridoma generation.
Cryopreserved PBMCs were thawed and transformed with Epstein-Barr virus (EBV), as described (Yu et al., 2008a; Yu et al., 2008b). Briefly, in B cell growth medium (ClonaCell-HY Medium A [Stem Cell Technologies]), CpG (Invitrogen), Chk2 inhibitor (Sigma-Aldrich), cyclosporin A (Sigma-Aldrich), and EBV filtrate from the B95.8 cell line), 5 to 7 million PBMCs were added at 50 μL/well to 384-well plates (Thermo Fisher Scientific) and incubated at 37°C in 7% CO2. After 7–10 days, cells were expanded to 96-well plates in B cell expansion medium (Medium A, CpG, Chk2 inhibitor, and irradiated heterologous human PBMCs (Nashville Red Cross) at a density of 10 million cells/mL. The plates were incubated at 37°C in 7% CO2 for an additional 4–5 days prior to screening by ELISA, as below. Cells from wells containing reactive supernatants were fused with the myeloma cell line HMMA2.5 using an electrofusion protocol as described (Yu et al., 2008a). Fused hybridomas were selected by plating in HAT medium (Medium A, ClonaCell™-HY Medium E (Stem Cell Technologies), 50x HAT medium supplement (Sigma-Aldrich), ouabain octahydrate [Sigma-Aldrich]) at 50 μL/well in 384-well plates. The plates were incubated for 14 to 21 days at 37°C in a humidified atmosphere of 7% CO2 prior to screening by ELISA.
MAb generation.
Wells containing reactive hybridomas were cloned by single-cell fluorescence-activated cell sorting. These hybridoma clones were expanded in Medium E serially into 48-well plates, 12-well plates, and T-75 cm2 flasks, respectively. Hybridoma clones were expanded further into T-225 cm2 flasks or G-Rex® devices (Wilson Wolf) in serum-free medium (Hybridoma SFM [Gibco]). Supernatants were harvested after approximately 21 days, or in sets of 3 to 5 days, respectively, through a 0.2-μm pore size filter. Antibodies were purified from the filtrate using HiTrap Protein G (GE Healthcare Life Sciences), HiTrap MabSelect SuRe (GE Healthcare Life Sciences), HiTrap KappaSelect (GE Healthcare Life Sciences), HiTrap LambdaFabSelect (GE Healthcare Life Sciences), or CaptureSelect™ IgA affinity matrix (Thermo Fisher Scientific) columns on an ÄKTA pure 25M chromatography system. Antibodies were concentrated using 50K MWCO Amicon® Ultra centrifugal filter units (Millipore) followed by desalting and buffer exchange with 7K MWCO Zeba desalting columns (Thermo Fisher Scientific).
Hybridoma supernatant protein ELISA.
Recombinant EEEV E2 glycoprotein (E3E2) (strain V105; IBT Bioservices) was diluted to 0.5 μg/mL in 1x D-PBS to coat 384-well ELISA plates (Thermo Fisher Scientific) at 25 μL/well and incubated at 4°C overnight. The plates were washed 3x with D-PBS-T (1x D-PBS + 0.05% Tween 20 [Cell Signaling Technology]) and blocked for 1 hour at room temperature with 25 μL/well blocking solution (2% non-fat dry milk (Bio-Rad), 2% goat serum (Gibco) in D-PBS-T). After blocking, the plates then were washed 3x with D-PBS-T and a volume of 10 to 25 μL/well of supernatant from each well containing EBV-transformed B cells or hybridoma cell lines was added. Plates were incubated for 2 hours at room temperature or overnight at 4°C. Plates then were washed 3x with D-PBS-T and a suspension of secondary antibodies (goat anti-human IgG-AP (Meridian Life Science) and goat anti-human IgA-AP [Southern Biotech]) at a 1:4,000 dilution in 1% blocking solution (1% non-fat dry milk, 1% goat serum) was added at 25 μL/well for 1 hour at room temperature. Alkaline phosphatase substrate solution (phosphatase substrate tablets (Sigma-Aldrich) in AP substrate buffer (1M Tris aminomethane and 30 mM MgCl2) was added at 25 μL/well following plate washing 4x with D-PBS-T. Plates were incubated at room temperature in the dark for 1–2 hours and then read at an optical density of 405 nm with a plate reader.
Hybridoma supernatant SINV/EEEV ELISA.
A murine mAb (EEEV-66; MSD and ASK (Kim et al., 2019)) was diluted to 0.5 μg/mL in 1x D-PBS to coat 384-well ELISA plates at 25 μL/well and incubated at 4°C overnight. The remainder of the ELISA protocol follows as described above for the protein ELISA. However, after blocking, clarified SINV/EEEV supernatant diluted 1:10 in 1x D-PBS (approximately 1 × 106 to 1 × 107 FFU/mL as determined through focus reduction test (FRT) with BHK-21 cells) at 25 μL/well was added. After incubation for 1–2 hours at room temperature, the plates were washed 6x with D-PBS-T (the first 2–3 washes were conducted under BSL-2 conditions).
5′ RACE nucleotide sequence analysis.
Antibody heavy and light-chain variable region genes were sequenced from antigen-specific hybridoma lines that had been cloned biologically using single-cell flow cytometric sorting. Total RNA was extracted using the RNeasy Mini kit (Qiagen). A modified 5′ RACE (Rapid Amplification of cDNA Ends) approach was similar to that previously reported (Turchaninova et al., 2016). Briefly, 5 μL of total RNA was mixed with cDNA synthesis primer mix (10 μM each) and incubated for 2 min at 70°C and then the incubation temperature was decrease to 42°C to anneal the synthesis primers (1 to 3 min). After incubation, a mix containing 5x first-strand buffer (Clontech), 20 mM DTT, 5′ template switch oligo (10 μM), dNTP solution (10 mM each) and 10x SMARTScribe Reverse Transcriptase (Clontech) was added to the primer-annealed total RNA reaction and incubated for 60 min at 42°C. The first-strand synthesis reaction was purified using the AMPure Size Select Magnetic Bead Kit at a ratio of 0.6x (Beckman Coulter). Following, a single PCR amplification reaction containing 5 μL first-strand cDNA, 2x Q5 High Fidelity Mastermix (NEB), dNTP (10 mM each), forward universal primer (10 μM) and reverse primer mix (0.2 μM each in heavy-chain mix, 0.2 μM each in light-chain mix) were subjected to thermal cycling with the following conditions: initial denaturation for 1 min 30 s followed by 30 cycles of denaturation at 98°C for 10 s, annealing at 60°C for 20 s, and extension at 72°C for 40 s, followed by a final extension step at 72°C for 4 min. The first PCR reaction was purified using the AMPure Size Select Magnetic Bead Kit at a ratio of 0.6x (Beckman Coulter). Amplicon libraries then were prepared according to the Pacific Biosciences Multiplex SMRT Sequencing protocol and sequenced on a Pacific Biosciences Sequel system platform. Raw sequencing data was demultiplexed and circular consensus sequences (Cardoso et al.) were determined using the Pacific Biosciences SMRT Analysis tool suite. The identities of gene segments and mutations from germlines were determined by alignment using the ImMunoGenetics (IMGT) database (Brochet et al., 2008; Giudicelli and Lefranc, 2011).
Recombinant human Fab, IgG1, and IgA1 production.
Recombinant human anti-EEEV mAb or Fab molecules were produced in Expi293F cells using the ExpiFectamine 293 transfection kit according to the manufacturer’s protocol. Briefly, 1 μg/mL of DNA was diluted in Opti-MEM I medium with ExpiFectamine 293 reagent for 15 to 20 minutes at room temperature before addition to the Expi293F cells. Cells were incubated at 37°C in a humidified atmosphere of 8% CO2 and supernatant was harvested by centrifugation and subsequent filtering through a 0.45-μm pore size filter 6 to 7 days after transfection. For IgG1, IgA1, Fab molecules, cell supernatant was purified through a HiTrap MabSelect SuRe, CaptureSelect™ IgA affinity matrix, or CaptureSelect™ CH1-XL affinity column, respectively, according to the manufacturer’s protocol on an ÄKTA pure 25M chromatography system. Antibodies were concentrated using 30K or 50K MWCO Amicon® Ultra Centrifugal Filter Units followed by desalting and buffer exchange with 7K MWCO Zeba desalting columns.
Protein EC50 ELISA.
Recombinant EEEV E2 glycoprotein (E3E2) (strain V105; IBT BioServices), EEEV E1 ectodomain (strain FL93–939), or CHIKV E1 protein (Meridian Life Science) was diluted to 0.5, 2, or 2 μg/mL, respectively, in 1x D-PBS to coat 384-well ELISA plates at 25 μL/well and incubated at 4°C overnight. A protein screening ELISA was performed as previously described above. However, instead of hybridoma supernatant, purified mAb was diluted to 10 μg/mL in blocking solution (1% non-fat dry milk, 1% goat serum) and added at 25 μL/well for 2 hours at room temperature. Additionally, plates were incubated at room temperature in the dark for 2 hours and then read at an optical density of 405 nm with a BioTek™ plate reader. For recombinant anti-EEEV mAbs and Fab molecules, a suspension of secondary antibodies (goat anti-human kappa-HRP (Southern Biotech) and goat anti-human lambda-HRP (Southern Biotech)) at a 1:4,000 dilution in 1% blocking solution (1% non-fat dry milk, 1% goat serum) was added at 25 μL/well for 1 hour at room temperature. 1-Step Ultra TMB-ELISA substrate solution (Thermo Fisher Scientific) was added at 25 μL/well following plate washing 4x with D-PBS-T. The reaction was stopped after 10 minutes at room temperature by addition of 25 μL/well of 1N HCl (Fisher Scientific). The plates then were read at an optical density of 450 nm with a BioTek™ plate reader. EC50 values were determined after log transformation of concentration values and non-linear regression analysis using sigmoidal dose-response (variable slope) using GraphPad Prism software version 8.
SINV/EEEV EC50 ELISA.
A mouse anti-EEEV mAb (EEEV-66; MSD and ASK (Kim et al., 2019)) was coated onto 384-well plates and incubated at 4°C overnight, as previously described above. After the blocking step, SINV/EEEV was diluted in 1x D-PBS to a titer of approximately 2.2 × 107 FFU/mL as determined through FRT with BHK-21 cells. After incubation for 2 hours at room temperature, the plates then were washed 6x with D-PBS-T (the first 2–3 washes were conducted under BSL-2 conditions in a laminar flow biosafety cabinet).
Focus reduction test (FRT).
BHK-21 or Vero cells were plated at 2.5 × 106 cells/96-well plate in DMEM/5% FBS/10 mM HEPES (Corning) at 150 μL/well. Cells were incubated at 37°C in 5% CO2 overnight. Cells plated approximately 24 hours prior were washed 2x with 1x D-PBS.
Serial three-fold dilutions of SINV/EEEV were diluted in DMEM/2% FBS/10 mM HEPES and added at 100 μL/well to the cells. The virus and cells were incubated at 37°C in 5% CO2 for 1.5 hours. A 2% methylcellulose (Sigma-Aldrich):2x DMEM (Millipore):4% FBS:20 mM HEPES overlay then was added to the cells at 100 μL/well. Cells then were incubated at 37°C in 5% CO2 for 18 hours. Plates were fixed with 1% PFA (diluted in 1x D-PBS; Alfa Aesar) at 100 μL/well for 1 hour at room temperature. Plates then were washed 3x with 1x D-PBS followed by 1x Perm Wash (1x D-PBS, 0.1% saponin (Sigma-Aldrich), 0.1% BSA (Sigma-Aldrich)) at 200 μL/well. Immune EEEV ascites fluid (ATCC) at 1:6,000 dilution in 1x Perm Wash was then added at 50 μL/well. The plates were incubated either at room temperature for 2 hours with rocking or overnight at 4°C. Plates were washed 3x with 1x D-PBS-T followed by the addition of a suspension of secondary antibodies (goat anti-mouse IgG-Fc-specific-HRP (Jackson ImmunoResearch)) at 1:2,000 dilution in 1x Perm Wash. Plates were incubated for 1 hour at room temperature with rocking. Plates were washed 3x with 1x D-PBS-T followed by addition of TrueBlue™ peroxidase substrate solution (SeraCare) at 40 μL/well. Plates were incubated for ~15 minutes at room temperature followed by a rinse with MilliQ water. The plates then were air dried and imaged on an ImmunoSpot S6 Universal machine (CTL).
Focus reduction neutralization test (FRNT).
Vero cells were plated at 2.5 × 106 cells/96-well plate in DMEM/5% FBS/10 mM HEPES at 150 μL/well. Cells were incubated at 37°C in 5% CO2 overnight. Purified mAb was diluted to 20 μg/mL (final concentration 10 μg/mL) in DMEM/2% FBS/10 mM HEPES. Serial three-fold dilutions of the mAb were performed. MAb-only dilutions were separated to serve as a negative control. SINV/EEEV was diluted to ~100 focus-forming units (ffu)/well in DMEM/2% FBS/10 mM HEPES and added to the mAb serial dilutions. The mAb:virus mixture was incubated at 37°C in 5% CO2 for 1 hour. Vero cells plated approximately 24 hours prior were washed 2x with 1x D-PBS. The mAb:virus mixture then was added at 100 μL/well to the cells and incubated at 37°C in 5% CO2 for 1.5 hours. A 2% methylcellulose:2x DMEM/4% FBS/20 mM HEPES overlay then was added to the cells at 100 μL/well. Cells were incubated at 37°C in 5% CO2 for 18 hours. Plates were fixed and immunostained as described for FRT.
Post-attachment neutralization assay.
Vero cells were plated at 2.5 × 106 cells/96-well plate in DMEM/5% FBS/10 mM HEPES at 150 μL/well. Cells were incubated at 37°C in 5% CO2 overnight. Vero cells plated approximately 24 hours prior the media was replaced with chilled DMEM/2% FBS/10 mM HEPES and cells were chilled at 4°C for 15 minutes. SINV/EEEV was diluted to ~100 ffu per well in chilled DMEM/2% FBS/10 mM HEPES and added to the cells at 4°C for 1 hour. Purified mAb was diluted to 10 μg/mL in chilled DMEM/2% FBS/10 mM HEPES. Serial three-fold dilutions of the mAb were performed. MAb-only dilutions were separated to serve as a negative control. Cells were washed 3x with chilled DMEM/2% FBS/10 mM HEPES. mAb then was added at 100 μL/well to the cells and incubated at 4°C for 1 hour. Cells were washed 3x with DMEM/2% FBS/10 mM HEPES and incubated at 37°C for 15 minutes. A 2% methylcellulose:2x DMEM/4% FBS/20 mM HEPES overlay then was added to the cells at 100 μL/well. Cells were incubated at 37°C in 5% CO2 for 18 hours. Plates were fixed and immunostained as described for FRT.
Entry inhibition assay.
A FRNT was performed as previously described. However, prior to addition of overlay, the cells were washed 4x with DMEM/2% FBS/10 mM HEPES and incubated at 37°C in 5% CO2 for 15 minutes. A 2% methylcellulose:2x DMEM/4% FBS/20 mM HEPES overlay then was added to the cells at 100 μL/well. Cells were incubated at 37°C in 5% CO2 for 18 hours. Plates were fixed and immunostained as described for FRT.
EEEV plaque reduction neutralization test.
Antibody preparations were diluted serially (using 2-fold dilutions) and incubated with ~100 pfu of EEEV FL93–939 for 1 h at 37°C. Anti-EEEV ascites serum (ATCC) was used as a positive control. After incubation, Vero cell monolayer cultures in 6-well plates were inoculated and incubated for 1 h at 37°C. After overlay with agarose immunodiffusion grade (MP Biomedicals), plates were incubated for 2 days followed by overlay with neutral red for at least 6 h to count plaques. Percent neutralization was calculated based on the number of plaques in each mAb dilution compared to the number of plaques in untreated control wells inoculated with virus.
Competition-binding analysis using biolayer interferometry.
Anti-penta his (HIS1K) biosensor tips (FortéBio) on an Octet Red96 or HTX biolayer interferometry instrument (FortéBio) were soaked for 10 minutes in 1x kinetics buffer (FortéBio), followed by a baseline signal measurement for 60 seconds. Recombinant EEEV E2 glycoprotein (5 μg/mL; IBT BioServices) was immobilized onto the biosensor tips for 60 seconds. After a wash step in 1x kinetics buffer for 30 to 60 seconds, the first antibody (50 μg/mL) was incubated with the antigen-containing biosensor for 600 seconds. After a wash step in 1x kinetics buffer for 30 to 60 seconds, the biosensor tips then were immersed into the second antibody (50 μg/mL) for 180 seconds. Comparison between the maximal signal of each antibody compared to a buffer-only control was used to determine the percent binding of each antibody. A reduction in maximum signal to <33% of un-competed signal was considered full competition of binding for the second antibody in the presence of the first antibody. A reduction in maximum signal to between 33 to 67% of un-competed was considered intermediate competition of binding for the second antibody in the presence of the first antibody. Percent binding of the maximum signal >67% was considered absence of competition of binding for the second antibody in the presence of the first antibody.
Alanine-scanning mutagenesis analysis.
WT EEEV (strain FL93–939) structural proteins (capsid-E3-E2–6K-E1) and E2 mutants were expressed on the surface of Expi293F cells using the ExpiFectamine 293 transfection kit according to the manufacturer’s protocol as previously described. Cells were incubated at 37°C in a humidified atmosphere of 8% CO2, were harvested 24 hours after transfection, and fixed with 1% PFA/PBS. Cells were washed twice with 1x DPBS and stored at 4°C in FACS buffer (1x DPBS, 2% FBS, 2 mM EDTA) until use or used immediately. Cells were plated at 40,000–50,000 cells/well in 96-well V-bottom plates (Corning). Anti-EEEV mAbs or the irrelevant mAb negative control, rDENV-2D22, were diluted to 1 μg/mL in FACS buffer and incubated with the cells for 1 hour at 4°C. Cells were washed with FACS buffer and then incubated with secondary antibodies (anti-human IgG-PE (Southern Biotech) and anti-human IgA-PE (Southern Biotech) or anti-mouse IgG-PE (Southern Biotech)) diluted 1:1,000 in FACS buffer for 1 hour at 4°C. Cells were washed with FACS buffer and resuspended in 25 μL/well of FACS buffer. Number of events were collected on an IntelliCyt® iQue Screener PLUS flow cytometer (Sartorius). For analysis, mock transfected Expi293F cells were included as a negative control and subtracted as background. The percent binding of each mAb to the alanine mutants was compared to the WT EEEV structural protein control. An initial screen of residues D1-L267 was performed to identify residues with <25% binding and at least two mAbs with >70% binding to control for expression. These residues were further assessed for loss-of-binding phenotype for at least two additional biological replicates. Critical residues were defined as at least two mAbs with >70% binding to control for expression and <25% binding relative to WT protein.
Cryo-EM sample preparation and data acquisition.
Purified SINV/EEEV particles and recombinant anti-EEEV Fab (EEEV-33 or EEEV-143) (1:10 molar ratio) were incubated on ice for 30 minutes. 3 μL of mixture was applied on Lacy 400 mesh copper grids (TED PELLA) or carbon coated R2/2 copper Quantifoil holey grids (Electron Microscopy Sciences). The samples were vitrified in liquid ethane using a Vitrobot (Thermo Fisher Scientific) set at 4°C and 100% relative humidity under BSL-2 containment conditions. Images for SINV/EEEV:EEEV-33 Fab were collected on a Titan Krios electron microscope (Thermo Fisher Scientific) equipped with a K2 Summit Direct Electron Detector (Gatan) operated at 300 kV and having a nominal pixel size of 1.64 Å per pixel. Images for SINV/EEEV:EEEV-143 Fab were collected on a Glacios electron microscope (Thermo Fisher Scientific) equipped with a K2 Summit Direct Electron Detector operated at 200 kV and having a nominal pixel size of 2.0 Å per pixel. Micrographs were acquired automatically using Leginon software (Carragher et al., 2000). The total exposure time for both samples was 10 sec and frames were recorded every 0.2 sec. Defocus values ranged from 1.0 to 2.5 μm. The total accumulated does was ~50 e−/Å2 for SINV/EEEV:EEEV-33 Fab and ~25 e−/Å2 for and SINV/EEEV: EEEV-143 Fab samples.
Cryo-EM data processing.
Cryo-EM movies (50 frames, 200 msec exposure per frame) were corrected for beam-induced motion and dose-weighted using MotionCor2 (Zheng et al., 2017) resulting in global motion-corrected frame stacks and summed micrographs. Contrast transfer function (CTF) parameters was estimated using Gctf (Zhang, 2016). EEEV particles manually picked from selected micrographs in RELION 3.0 (Zivanov et al., 2018) using a box size of 800 pixels (1.64 Å) or 700 pixels (2.0 Å). Approximately 18,000 particles were picked from 2,111 micrographs of SINV/EEEV:EEEV-33 Fab and 10,000 particles were picked from 2,501 micrographs of SINV/EEEV:EEEV-143 Fab. The particles were subjected to reference-free 2D classification in RELION 3.0 and selected particles associated with good classes were exported to cisTEM (Grant et al., 2018). The de-novo initial model was generated without imposing any symmetry (C1 symmetry) which was subjected for 3D auto refinement (I1 symmetry) without mask in cisTEM. Further, the 3D model and associated particles obtained from cisTEM were exported to RELION 3.0 and used for the final masked 3D refinement (I1 symmetry) and postprocessing (Figure S7A). The resolution of the maps was evaluated using the “gold standard” Fourier shell correlation (FSC) at 0.143 criterion (Henderson et al., 2012; Scheres and Chen, 2012) (Figure S7B).
EEEV VLP negative stain grid preparation and imaging.
For screening and imaging of negatively stained (NS) EEEV VLP (Ko et al., 2019) or EEEV VLP:EEEV-143 Fab samples, ~3 μL of the sample at concentrations of 10–15 μg/mL was applied to glow discharged grid with continuous carbon film on 400 square mesh copper EM grids (Electron Microscopy Sciences). The grids were stained with 0.75% uranyl formate (Ohi et al., 2004). Images were recorded on a 4k × 4k CCD camera using an FEI TF20 transmission electron microscope (Thermo Fisher Scientific) operated at 200 keV and control with SerialEM (Mastronarde, 2005). All images were taken at 50,000× magnification with a pixel size of 2.18 A/pix in low-dose mode at a defocus of 1.5 to 1.8 μm. Image processing was performed using the Scipion software package (de la Rosa-Trevin et al., 2016). Images were import and particles were CTF estimated (Rohou and Grigorieff, 2015), then picked (Sorzano et al., 2013). 2D class averages were performed using Xmipp3.0 cl2d (de la Rosa-Trevin et al., 2013; Sorzano et al., 2013).
EEEV VLP cryo-EM sample preparation and data collection.
For the EEEV VLP:EEEV-143 Fab complex, EEEV VLP at concentration of 0.2 mg/mL was mixed with EEEV-143 Fab in a molar ratio of 720:1 (Fab:VLP) and incubated on ice for 1 hour. Then, 2.2 μL of either EEEV VLP or EEEV VLP/EEEV-143 Fab was applied 2x to a 300 mesh Lacey grid that was glow discharged for 25 s at 25 milliamperes. The grid was blotted for 2 s before being plunged into liquid ethane using a FEI Vitrobot Mark4 (Thermo Fisher Scientific) at 8° C and 100% humidity. The grids were imaged in Titan Krios (Thermo Fisher Scientific) operated at 300 keV equipped with Falcon 3EC Direct Electron Detector (Thermo Fisher Scientific) using counting mode. Movies collected at a nominal magnification of 96,000x, pixel size of 0.8608 A/pix for the EEEV VLP and at 75,000x, pixel size of 1.11 A/pix for the EEEV VLP:EEEV-143 Fab complex. Both data set were in a defocus range of 0.8 to 2.8 μm. Grids were exposed at 1e−/Å2/frame over 30 frames resulting in a total dose of ~30 e−/Å2 (see also Table S1).
EEEV VLP cryo-EM data processing.
Movies were pre-processed on-the fly (MotionCor2 (Zheng et al., 2017), Gctf (Zhang, 2016), using RELION (Scheres, 2012; Zivanov et al., 2018). Micrographs with low resolution, high astigmatism and defocus were removed from the data set. Further processing was done using RELION 3.1 beta. A small subset of micrographs were autopicked first by RELION LoG (Fernandez-Leiro and Scheres, 2017) and 2D class averages were determined. Representative classes were selected and used as templates for another round of autopicking. The particles were then subjected to multiple rounds of 2D class averages and 3D classification (with and without symmetry). The particles from the selected classes were re-extracted, 3D classified and subjected to 3D auto refinement. The data was processed further with Ctfrefine, polished and postprocessing was done (detailed statistics are provided in Table S1 and Figure S7).
EEEV VLP model building.
For the EEEV VLP, a homology model of SINV/EEEV (PDB: 6MX4) was used for docking to the cryoEM map with PHENIX (Adams et al., 2010). To improve coordinate fitting, the model was subjected to iterative refinement of manual building in Coot (Emsley and Cowtan, 2004) and PHENIX real-space refine (Adams et al., 2010). The model was validated with Molprobity (Chen et al., 2010). For the EEEV VLP:EEEV-143 Fab complex, the refined model of the VLP was used as starting model and was docked to the EM map with UCSF Chimera (Pettersen et al., 2004). The model was then refined in PHENIX (phenix real-space refine) by rigid body and a homology model of EEEV-143 Fab (PDB: 6MWX) was mutated to the EEEV-143 Fab sequence, docked, and rigid body refined to the EM map.
Mouse aerosol challenge with EEEV.
Mice were inoculated with EEEV strain FL93–939 by aerosol, as previously described (Trobaugh et al., 2019). Briefly, mice were challenged with ~10 LD50 (~2,500 PFU) of 20% sucrose-purified WT EEEV FL93-nLuc TaV using the AeroMP exposure system (Biaera Technologies, Hagerstown, MD) inside a class III biological safety cabinet and either an Aeroneb nebulizer (Aerogen) or 3-jet Collison nebulizer (CH Technologies). All mice were monitored twice daily for morbidity and mortality.
In vivo imaging
At different times after challenge, mice were injected with 10 μg of Nano-Glo substrate (Promega) subcutaneously in 500μL PBS, as previously described (Gardner et al., 2017). Four minutes after substrate injection, the mice were imaged using the IVIS Spectrum CT Instrument (PerkinElmer) using the autoexposure setting. The total flux (photons per second) in the head region was calculated for each animal using Living Image Software 4.5.1, with all images set to the same scale.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details can be found in the figure legends. EC50 values for binding and IC50 values for neutralization were determined after log transformation of concentration values and non-linear regression analysis using sigmoidal dose-response (variable slope). Direct comparison of differences in mAb binding to virion particle-specific epitopes versus sites in recombinant E2 glycoprotein cannot be performed on a molar basis. The precise total number of epitopes present on SINV/EEEV particles is unknown in an antibody-based capture ELISA format and may be greater than that on the recombinant EEEV E2 glycoprotein. To describe the differences in mAb binding, a virus/protein EC50 ratio was used. Survival curves were generated using the Kaplan-Meier method and curves compared the log-rank test with Bonferroni multiple comparison correction (n = number of mice, * = p<0.05, ** = p<0.01, ns = not statistically significant). A one-way ANOVA with Dunnett’s multiple comparison correction was used to compare luminescence intensity of IVIS images (* = p<0.01). All statistical analyses were performed using Prism software version 8 (GraphPad).
Supplementary Material
Figure S1, Related to Figure 2. Binding reactivity of neutralizing human anti-EEEV mAbs to SINV/EEEV particles or EEEV E2 glycoprotein.
(A) Binding ratio of neutralizing human anti-EEEV mAbs to SINV/EEEV particles vs. recombinant monomeric EEEV E2 glycoprotein. EEEV-33 is removed to display the binding reactivity groups for the rest of the panel of neutralizing human anti-EEEV mAbs at higher resolution. A dotted line indicates 32 pM half-maximal effective concentration (EC50) values for binding, revealing distinct binding patterns of human anti-EEEV mAbs to SINV/EEEV particles and EEEV E2 glycoprotein. Neutralizing human anti-EEEV mAbs are labeled with the anti-EEEV mAb name and colored according to binding group (Group 2 [green] = strong (SINV/EEEV EC50 = <32 pM) virus » protein binding; Group 3 [purple] = weak (SINV/EEEV EC50 = >32 pM) virus»protein binding; Group 4 [orange] = protein>virus binding).
(B) Representative binding curves of recombinant neutralizing human anti-EEEV IgG1, IgA1, or Fab molecules to three different antigens. Binding curves of recombinant neutralizing human anti-EEEV IgG1 (square), IgA1 (triangle), or Fab (circle) molecules to SINV/EEEV particles (green) or EEEV E2 glycoprotein (blue), with mAb concentration (nM) on the x-axis and optical density at 405 nm on the y-axis. Binding to EEEV E1 glycoprotein (purple) was not detected.
(C) EC50 values (pM) for binding of recombinant neutralizing human anti-EEEV IgG1, IgA1, or Fab molecules to SINV/EEEV particles or EEEV E2 glycoprotein. EC50 value in pM is indicated by blue fill color (<32 [dark blue], 32.01 to 100 [medium blue], 100.01 to 320 [light blue], <10,000 [lightest blue]). Ratio of binding to SINV/EEEV particles versus EEEV E2 glycoprotein is indicated as the ratio of EC50 values. Increasing depth of green color indicates lower ratios (<0.1 [dark green], 0.1 to 1.0 [medium green], 1.01 to 2.0 [light green], >2.0 [lightest green]), suggesting recognition of a quaternary epitope on virion particles. Ratio of binding of recombinant IgG1 versus Fab molecules to SINV/EEEV particles or EEEV E2 glycoprotein is indicated as the ratio of EC50 values. Increasing depth of purple color indicates lower ratios (<0.1 [dark purple], 0.1 to 1.0 [medium purple], 1.01 to 2.0 [light purple], >2.0 [lightest purple]), suggesting dependence on valency for binding. Antibody isotype is indicated as IgG1 or IgA1 for the heavy chain or k or l for the light chain. NA = not applicable.
Data in A to C represent mean ± SD of technical triplicates and are representative of two independent ELISA experiments. Each mAb is colored based on the binding group defined in Figure 2.
Figure S2, Related to Figure 3. Alanine-scanning mutagenesis library analysis. Bar graphs of neutralizing human anti-EEEV mAb binding to critical residues (<25% mAb binding with at least two other mAbs >70% to control for expression) or critical residues previously characterized for murine anti-EEEV mAbs (Kim et al., 2019) and the VEE-specific human mAb, F5 (Hunt et al., 2010; Porta et al., 2014). Each mAb is listed on the x-axis and percent binding relative to wildtype is on the y-axis. Residues are colored based on E2 domain (N-link - purple, Domain A - red, Arch 1 - magenta, Domain B - cyan, and Arch 2 - orange). Data represent mean ± SD of at least two independent experiments.
Figure S3, Related to Figure 3. Neutralization activity of human anti-EEEV mAbs to SINV/EEEV escape mutant viruses.
(A) Representative neutralization curves of potent neutralizing human anti-EEEV mAbs against SINV/EEEV WT, and escape mutant viruses M68T, G192R, and L227R. Neutralization curves of potent human anti-EEEV mAbs against SINV/EEEV WT (closed square), M68T (open triangle), G192R (open circle), and L227R (open diamond), with mAb concentration (nM) on the x-axis and % relative infectivity on the y-axis. mAbs are ordered based on IC50 values against SINV/EEEV WT. A positive control mouse mAb, mEEEV-3 (dark purple) (Kim et al., 2019), and a negative control mAb, rDENV-2D22 (black; B), were included.
(B) Representative neutralization curves of moderate neutralizing human anti-EEEV mAbs against SINV/EEEV WT and escape mutant viruses M68T, G192R, and L227R. Neutralization curves of moderate human anti-EEEV mAbs against SINV/EEEV WT (closed square), M68T (open triangle), G192R (open circle), and L227R (open diamond), with mAb concentration (nM) on the x-axis and % relative infectivity on the y-axis. A positive control mouse mAb, mEEEV-3 (dark purple; A) (Kim et al., 2019), and a negative control mAb, rDENV-2D22 (black), were included.
(C) Half-maximal inhibitory concentration (IC50) values (pM) for human anti-EEEV mAbs against SINV/EEEV WT and escape mutant viruses M68T, G192R, and L227R. IC50 values (pM) for neutralizing human anti-EEEV mAbs against SINV/EEEV WT, M68T, G192R, and L227R are indicated in the table. Neutralizing human anti-EEEV mAbs are listed in order of increasing IC50 value against SINV/EEEV WT. IC50 value in pM is indicated by the orange heat map (<33 [dark orange], 33 to 333 [medium orange], 333.01 to 3,333 [light orange], <10,000 [lightest orange]). Fold difference in IC50 value ratio of SINV/EEEV WT versus each escape mutant virus (M68T, G192R, or L227R) are indicated. Ratio of neutralization activity against SINV/EEEV WT versus each escape mutant virus (M68T, G192R, or L227R) is indicated as the fold difference of IC50 values. Increasing depth of maroon color indicates greater fold differences (>15 or > [dark maroon], 10–15 [medium maroon], 5–10 [light maroon], <5 [white]), suggesting reduction in neutralization activity of the mAb.
Data in A to C represent mean ± SD of technical triplicates and are representative of two independent focus reduction neutralization test (FRNT) experiments. mAbs are colored based on mAb legend in Figure 1.
Figure S4, Related to Figure 4 and Figure 5. EEEV-33 or EEEV-143 Fab binding to the E2 trimeric spike of SINV/EEEV particles.
Side view (A) or 40° top-view (B) of EEEV E2 trimeric spikes (in light blue) for side-by-side comparisons of native SINV/EEEV (left), SINV/EEEV:EEEV-33 Fab complex (right) and SINV/EEEV:EEEV-143 Fab complex (middle) showing binding of one Fab (EEEV-33 - red; EEEV-143 - orange) per E2 protomer within the trimeric spike. Low pass map of published native SINV/EEEV structure (EMD-9280) used for the comparison. Side view (C) or 40° top-view (D) of EEEV-33 Fab (red) and EEEV-143 Fab (orange) bound to SINV/EEEV or EEEV VLP, respectively. For the SINV/EEEV (PDB ID: 6MX4) and EEEV-143 Fab (mutated sequence of PDB ID: 6MWX) docked and rigid body refined model of SINV/EEEV:EEEV-33 Fab complex, the structural proteins are colored with E2 = dark green, E1 = salmon, and capsid = blue. For the EEEV VLP:EEEV-143 Fab complex (EMD-22277; PDB ID: 6XOB), the structural proteins are colored with E2 = light green, E1 = raspberry, and capsid = cyan.
Figure S5, related to Figure 5. Cryo-EM reconstruction of EEEV VLP in complex with EEEV-143 Fab molecules. Cryo-EM structure of the apo form of EEEV VLP (A–C) and of EEEV VLP:EEEV-143 Fab complex (D–F) showing radially colored surface representation of full (A, D) and cross-section (B, E) of the map. C, F. Roadmaps of the apo form of EEEV VLP and EEEV VLP:EEEV-143 Fab complex reconstructions to illustrate EEEV-143 Fab binding sites (in dark blue). EEEV-143 binds in a tangential orientation and makes contacts with neighboring spikes for inter-spike cross-linking of SINV/EEEV particles. G–H. Asymmetric unit view of EEEV structural proteins (E2 = green, E1 = red, capsid protein = cyan) of the apo form of EEEV VLP (G) or in complex with EEEV-143 Fab molecules (H; orange) bound in a tangential orientation. I. Alignment of EEEV structural proteins of the EEEV VLP apo form (dark blue) to the EEEV VLP:EEEV-143 Fab complex (pink) to show stabilization of the E2 glycoprotein upon EEEV-143 Fab binding.
Figure S6, Related to Figures 6 and 7. In vivo body weight change and clinical disease signs observed in EEEV-33 and EEEV-143 mAb administered lethal EEEV aerosol challenge model.
(A) Percent body weight change of EEEV-33, EEEV-143, and rDENV-2D22 prophylactically treated CD-1 mice over the course of 14 days. Percent change in weight (y-axis) for EEEV-33 (red; n=11), EEEV-143 (orange; n=5), and rDENV-2D22 (black; n=10) are indicated over the course of 14 days after EEEV challenge (1,631 to 1,825 PFU/mouse; x-axis).
(B) Percent body weight change of EEEV-143 and rDENV-2D22 prophylactically treated CD-1 mice over the course of 14 days. Percent change in weight (y-axis) for EEEV-143 (orange; n=5) and rDENV-2D22 (black; n=5) are indicated over the course of 14 days after EEEV challenge (2,739 PFU/mouse; x-axis).
(C) Clinical scores of EEEV-33, EEEV-143, and rDENV-2D22 prophylactically treated CD-1 mice over the course of 14 days. The number of mice (y-axis) for EEEV-33 (red; n=11), EEEV-143 (orange; n=5), and rDENV-2D22 (black; n=10) with defined clinical scores (dead (black), moribund (red), seizures/ataxia (yellow), hunched back/behavioral (blue), ruffled fur (green), healthy (white)) are indicated over the course of 14 days post EEEV challenge (1,631 to 1,825 PFU/mouse; x-axis).
(D) Clinical scores of EEEV-143 and rDENV-2D22 prophylactically treated CD-1 mice over the course of 14 days. The number of mice (y-axis) for EEEV-143 (orange; n=5) and rDENV-2D22 (black; n=5) with defined clinical scores (dead (black), moribund (red), seizures/ataxia (yellow), hunched back/behavioral (blue), ruffled fur (green), healthy (white)) are indicated over the course of 14 days after EEEV challenge (2,739 PFU/mouse; x-axis).
(E) Percent body weight change of EEEV-33, EEEV-143, and rDENV-2D22 therapeutically treated CD-1 mice over the course of 14 days. Percent change in weight (y-axis) for EEEV-33 (red; n=11), EEEV-143 (orange; n=5), and rDENV-2D22 (black; n=5) are indicated over the course of 14 days after EEEV challenge (1,631 to 1,825 PFU/mouse; x-axis).
(F) Percent body weight change of EEEV-143 therapeutically treated CD-1 and BALB/c mice, respectively, over the course of 14 days. Percent change in weight (y-axis) for EEEV-143 (orange; n=5) is indicated over the course of 14 days after EEEV challenge (2,739 PFU/mouse; x-axis).
(G) Clinical scores of EEEV-33, EEEV-143, and rDENV-2D22 therapeutically treated CD-1 mice over the course of 14 days. The number of mice (y-axis) for EEEV-33 (red; n=11), EEEV-143 (orange; n=5), and rDENV-2D22 (black; n=5) with defined clinical scores (dead (black), moribund (red), seizures/ataxia (yellow), hunched back/behavioral (blue), ruffled fur (green), healthy (white)) are indicated over the course of 14 days after EEEV challenge (1,631 to 1,825 PFU/mouse; x-axis).
(H) Clinical scores of EEEV-143 therapeutically treated CD-1 and BALB/c mice, respectively, over the course of 14 days. The number of mice (y-axis) for EEEV-143 (orange; n=5) with defined clinical scores (dead (black), moribund (red), seizures/ataxia (yellow), hunched back/behavioral (blue), ruffled fur (green), healthy (white)) are indicated over the course of 14 days after EEEV challenge (2,739 PFU/mouse; x-axis).
(I) Percent body weight change of CD-1 mice treated therapeutically with EEEV-143 or rDENV-2D22. Percent change in weight (y-axis) for EEEV-143 (orange; n=5) or rDENV-2D22 (black; n=5) treated animals is indicated over the course of 14 days after EEEV challenge (1,897 PFU/mouse; x-axis).
(J) Clinical scores of CD-1 mice treated therapeutically with EEEV-143 or rDENV-2D22. The number of mice (y-axis) treated with EEEV-143 (orange; n=5) or rDENV-2D22 (black; n=5) with defined clinical scores (dead (black), moribund (red), seizures/ataxia (yellow), hunched back/behavioral (blue), ruffled fur (green), or healthy (white)) are indicated over the course of 14 days after EEEV challenge (1,897 PFU/mouse; x-axis).
Figure S7, Related to STAR Methods. Cryo-EM processing of SINV/EEEV complexes with either EEEV-33 or EEEV-143 Fabs.
Flow chart of cryo-EM processing steps and Fourier shell correlation (FSCs) of the maps of SINV/EEEV:rEEEV-33 Fab complex (A), SINV/EEEV:rEEEV-143 Fab complex (B), EEEV VLP (C), and EEEV VLP:rEEEV-143 Fab complex (D). The FSCs of the refined models agree with each other, suggesting that the models are not over-refined.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| EEEV-7 (hybridoma-produced IgG1) | This paper | N/A |
| EEEV-12 (hybridoma-produced IgG1) | This paper | N/A |
| EEEV-21 (hybridoma-produced IgG1) | This paper | N/A |
| EEEV-27 (hybridoma-produced IgG1) | This paper | N/A |
| EEEV-33 (hybridoma-produced IgG1) | This paper | N/A |
| EEEV-93 (hybridoma-produced IgG1) | This paper | N/A |
| EEEV-94 (hybridoma-produced IgG1) | This paper | N/A |
| EEEV-97 (hybridoma-produced IgG1) | This paper | N/A |
| EEEV-106 (hybridoma-produced IgG1) | This paper | N/A |
| EEEV-129 (hybridoma-produced IgG1) | This paper | N/A |
| EEEV-143 (hybridoma-produced IgA) | This paper | N/A |
| EEEV-147 (hybridoma-produced IgG1) | This paper | N/A |
| rEEEV-7 IgG (recombinant expi293F-produced IgG1) | This paper | N/A |
| rEEEV-12 IgG (recombinant expi293F-produced IgG1) | This paper | N/A |
| rEEEV-21 IgG (recombinant expi293F-produced IgG1) | This paper | N/A |
| rEEEV-27 IgG (recombinant expi293F-produced IgG1) | This paper | N/A |
| rEEEV-33 IgG (recombinant expi293F-produced IgG1) | This paper | N/A |
| rEEEV-93 IgG (recombinant expi293F-produced IgG1) | This paper | N/A |
| rEEEV-94 IgG (recombinant expi293F-produced IgG1) | This paper | N/A |
| rEEEV-97 IgG (recombinant expi293F-produced IgG1) | This paper | N/A |
| rEEEV-106 IgG (recombinant expi293F-produced IgG1) | This paper | N/A |
| rEEEV-129 IgG (recombinant expi293F-produced IgG1) | This paper | N/A |
| rEEEV-143 IgG (recombinant expi293F-produced IgG1) | This paper | N/A |
| rEEEV-143 IgA (recombinant expi293F-produced IgA1 with mouse J chain) | This paper | N/A |
| rEEEV-147 IgG (recombinant expi293F-produced IgG1) | This paper | N/A |
| rEEEV-7 Fab (recombinant expi293F-produced Fab) | This paper | N/A |
| rEEEV-12 Fab (recombinant expi293F-produced Fab) | This paper | N/A |
| rEEEV-21 Fab (recombinant expi293F-produced Fab) | This paper | N/A |
| rEEEV-27 Fab (recombinant expi293F-produced Fab) | This paper | N/A |
| rEEEV-33 Fab (recombinant expi293F-produced Fab) | This paper | N/A |
| rEEEV-93 Fab (recombinant expi293F-produced Fab) | This paper | N/A |
| rEEEV-94 Fab (recombinant expi293F-produced Fab) | This paper | N/A |
| rEEEV-97 Fab (recombinant expi293F-produced Fab) | This paper | N/A |
| rEEEV-106 Fab (recombinant expi293F-produced Fab) | This paper | N/A |
| rEEEV-129 Fab (recombinant expi293F-produced Fab) | This paper | N/A |
| rEEEV-143 Fab (recombinant expi293F-produced Fab) | This paper | N/A |
| rEEEV-147 Fab (recombinant expi293F-produced Fab) | This paper | N/A |
| rDENV-2D22 IgG (recombinant expiCHO-produced IgG1) | Fibriansah et al., 2015 | N/A |
| Murine mAb: EEEV-10 | Michael S. Diamond | Kim et al., 2019 |
| Murine mAb: EEEV-66 | Michael S. Diamond | Kim et al., 2019 |
| Murine mAb: EEEV-69 | Michael S. Diamond | Kim et al., 2019 |
| Murine mAb: EEEV-86 | Michael S. Diamond | Kim et al., 2019 |
| Eastern equine encephalomyelitis immune ascites fluid | ATCC | Cat#VR-1242AF |
| Goat anti-human IgG-AP | Meridian Life Science | Cat#W90088A; RRID:AB_205090 |
| Goat anti-human IgA-AP | Southern Biotech | Cat#2050-04; RRID:AB_2795704 |
| Goat anti-human Kappa-HRP | Southern Biotech | Cat#2060-05; RRID:AB_2795720 |
| Goat anti-human Lambda-HRP | Southern Biotech | Cat#2070-05; RRID:AB_2795753 |
| Goat anti-mouse IgG-HRP | Jackson ImmunoResearch | Cat#115035008; RRID:AB_2313585 |
| Goat anti-human IgG-PE | Southern Biotech | Cat#2040-09; RRID:AB_2795648 |
| Goat anti-human IgA-PE | Southern Biotech | Cat#2050-09; RRID:AB_2795707 |
| Goat anti-mouse IgG-PE, human adsorbed | Southern Biotech | Cat#1030-09S; RRID:AB_2794298 |
| Bacterial and Virus Strains | ||
| Epstein-Barr virus (EBV) | This paper | N/A |
| Sindbis (TR339)/Eastern equine encephalitis virus (FL93-939) (SINV/EEEV) | Michael S. Diamond | Kim et al., 2019 |
| SINV/EEEV-M68T | Michael S. Diamond | Kim et al., 2019 |
| SINV/EEEV-G192R | Michael S. Diamond | Kim et al., 2019 |
| SINV/EEEV-L227R | Michael S. Diamond | Kim et al., 2019 |
| EEEV FL93-939 | William B. Klimstra | Gardner et al., 2011 |
| Biological Samples | ||
| Peripheral blood mononuclear cells (PBMCs) from EEEV survivor | This paper | N/A |
| Human PBMCs | Nashville Red Cross | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Poly(ethylene glycol) (PEG) - average Mn 6,000 | Sigma-Aldrich | Cat#81260 |
| OptiPrep | Sigma-Aldrich | Cat#D1556 |
| Uranyl formate | Electron Microscopy Sciences | Cat#22451 |
| Chk2 Inhibitor II Hydrate | Sigma-Aldrich | Cat#C3742 |
| Cyclosporin A | Sigma-Aldrich | Cat#C1832 |
| Ouabain Octahydrate | Sigma-Aldrich | Cat#O3125 |
| 50x HAT Medium Supplement | Sigma-Aldrich | Cat#H0262 |
| Paraformaldehyde | Alfa Aesar | Cat#J61899-AP |
| Saponin | Sigma-Aldrich | Cat#47036 |
| Methylcellulose | Sigma-Aldrich | Cat#M7027 |
| 2x Dulbecco’s Modified Eagle’s Medium (DMEM) | Millipore | Cat#SLM202B |
| TrueBlue™ Peroxidase Substrate Solution | SeraCare | Cat#5510-0030 |
| L-Glutamine (200mM) | Gibco | Cat#25030081 |
| Pluronic™ F-68 Non-ionic Surfactant (100X) | Gibco | Cat#24040032 |
| 10x Kinetics Buffer | FortéBio | Cat#181105 |
| Fetal Bovine Serum, ultra-low IgG | Thermo Fisher Scientific | Cat#16250078 |
| Expi293 Expression Medium | Gibco | Cat#A1435101 |
| Freestyle F17 Expression Medium | Gibco | Cat#A1383502 |
| Opti-MEM™ I Reduced Serum Medium | Gibco | Cat#31985088 |
| ExpiFectamine 293 Transfection Kit | Gibco | Cat#A14525 |
| ClonaCell™-HY Medium A | Stem Cell Technologies | Cat#03081 |
| ClonaCell™-HY Medium E | Stem Cell Technologies | Cat#03085 |
| Hybridoma SFM | Gibco | Cat#12045084 |
| 1-Step Ultra TMB-ELISA | Thermo Fisher Scientific | Cat#34029 |
| Recombinant EEEV E2 GP (E3E2) | IBT Bioservices | Cat#0560-001 |
| CHIKV E1 protein | Meridian Life Science | Cat#R01653 |
| EEEV E1 glycoprotein | This paper | N/A |
| EEEV virus-like particles (VLPs) | NIH Vaccine Research Center | Ko et al., 2019 |
| Critical Commercial Assays | ||
| Universal Mycoplasma Detection Kit | ATCC | Cat#30-1012K |
| Cell Authentication Service Short Tandem Repeat (STR) analysis | ATCC | N/A |
| Pierce™ Coomassie (Bradford) Protein Assay Kit | Thermo Fisher Scientific | Cat#PI23200 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat#23225 |
| Deposited Data | ||
| SINV/EEEV | EMDB: 9280; PDB ID: 6MX4 | Hasan et al., 2018 |
| Cryo-EM map of SINV/EEEV with rEEEV-33 Fab | This paper | EMD-22223 |
| Cryo-EM map of SINV/EEEV with rEEEV-143 Fab | This paper | EMD: 22188 |
| Cryo-EM map of EEEV VLP | This paper | PDB: 6XO4 EMD-22276 |
| Cryo-EM map of EEEV VLP with rEEEV-143 Fab | This paper | PDB: 6XOB EMD-22277 |
| Experimental Models: Cell Lines | ||
| Human: Expi293F | Thermo a1435101 Fisher Scientific |
Cat#A14527; RRID:CVCL_D615 |
| Monkey: Vero | ATCC | Cat#:CCL-81; RRID:CVCL_0059 |
| Monkey: B95.8 | ATCC | Cat#CRL-1612 (discontinued) RRID:CVCL_1953 |
| Hamster: BHK-21 | ATCC | Cat#:CCL-10; RRID:CVCL_1915 |
| Mouse-human HMMA 2.5 myeloma cell line | Dr. L. Cavacini | N/A |
| EEEV-7 hybridoma clone | This paper | N/A |
| EEEV-12 hybridoma clone | This paper | N/A |
| EEEV-21 hybridoma clone | This paper | N/A |
| EEEV-27 hybridoma clone | This paper | N/A |
| EEEV-33 hybridoma clone | This paper | N/A |
| EEEV-93 hybridoma clone | This paper | N/A |
| EEEV-94 hybridoma clone | This paper | N/A |
| EEEV-97 hybridoma clone | This paper | N/A |
| EEEV-106 hybridoma clone | This paper | N/A |
| EEEV-129 hybridoma clone | This paper | N/A |
| EEEV-143 hybridoma clone | This paper | N/A |
| EEEV-147 hybridoma clone | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: CD-1 female, 4-6-weeks old | Charles River Laboratories | N/A |
| Oligonucleotides | ||
| CpG (ZOEZOEZZZZZOEEZOEZZZT oligonucleotide) | Invitrogen | N/A |
| Recombinant DNA | ||
| Plasmid: pCDNA3.1(+)-EEEV (FL93-939) E1 ectodomain (Y1-S409) | This paper; Accession Number: AF159554 | GenScript Biotech |
| Plasmid: pCDNA3.1(+)-EEEV (FL93-939) structural protein (capsid-E3-E2-6K-E1) | This paper; Accession Number: AF159554 | GenScript Biotech |
| Plasmid: pCDNA3.1(+)-EEEV (FL93-939) structural protein (capsid-E3-E2-6K-E1) E2 mutants (D1-L267) | This paper; Accession Number: AF159554 | Twist Bioscience Inc. |
| Plasmid: pTwist-mouse J chain | This paper; UniProt: P01592 | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-7 rIgG1 heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-7 rFab heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-7 light chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-12 rIgG1 heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-12 rFab heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-12 light chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-21 rIgG1 heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-21 rFab heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-21 light chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-27 rIgG1 heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-27 rFab heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-27 light chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-33 rIgG1 heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-33 rFab heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-33 light chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-93 rIgG1 heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-93 rFab heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-93 light chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-94 rIgG1 heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-94 rFab heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-94 light chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-97 rIgG1 heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-97 rFab heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-97 light chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-106 rIgG1 heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-106 rFab heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-106 light chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-129 rIgG1 heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-129 rFab heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-129 light chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-143 rIgG1 heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-143 rIgA1 heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-143 rFab heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-143 light chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-147 rIgG1 heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-147 rFab heavy chain | This paper | Twist Bioscience Inc. |
| Plasmid (pTwist): EEEV-147 light chain | This paper | Twist Bioscience Inc. |
| Software and Algorithms | ||
| Prism | GraphPad | v8 |
| ImMunoGeneTics (IMGT) database | Brochet et al., 2008, Giudicelli et al., 2011 | N/A |
| Pymol | Schrödinger | v2.3.0 |
| Leginon software | Carragher et al., 2000 | N/A |
| MotionCor2 | Zheng et al., 2017 | N/A |
| Gctf | Zhang, 2016 | N/A |
| cisTEM | Grant et al., 2018 | N/A |
| SerialEM | Mastronarde, 2005 | N/A |
| Scipion software package | de la Rosa-Trevín et al., 2016 | N/A |
| Xmipp3.0 cl2d | Sorzano et al., 2013, de la Rosa-Trevín 2013 | N/A |
| RELION | Scheres, 2012, Zivanov et al., 2018 | v3.0; v3.1 beta |
| RELION LoG | Fernandez-Leiro et al., 2017 | N/A |
| PHENIX | Adams et al., 2010 | N/A |
| Coot | Emsley et al., 2004 | N/A |
| MolProbity | Chen et al., 2010 | N/A |
| UCSF Chimera | Pettersen et al., 2004 | N/A |
| Other | ||
| PacBio Sequel System | Pacific Biosciences | N/A |
| HIS1K Octet Sensor Tips | FortéBio | Cat#185121 |
| Octet RED96 System | FortéBio | N/A |
| Octet HTX System | FortéBio | N/A |
| BioStack™ 3 Microplate Stacker | BioTek™ | Cat#BIOSTACK3WR |
| EL406 Washer Dispenser | BioTek™ | Cat#406SUB3 |
| BioTek™ PowerWave™ Microplate Spectrophotometer | BioTek™ | Cat#BT-RPRWI |
| AH-650 Swinging Bucket Rotor | Sorvall | Cat#54294 |
| ThermoSorvall Model Discovery 90SE Ultracentrifuge | Sorvall | Cat#45905 |
| ImmunoSpot® S6 Universal | Cellular Technology Limited | Cat#S6UNV12 |
| IntelliCyt® iQue Screener PLUS | Sartorius | N/A |
| 6-well G-rex® plates | Wilson Wolf | Cat#80240M |
| ÄKTA Pure 25M Chromatography System | GE Healthcare Life Sciences | Cat#29018226 |
| HisTrap Excel | GE Healthcare Life Sciences | Cat#17371205 |
| HiTrap Protein G HP Columns | GE Healthcare Life Sciences | Cat#17040503 |
| HiTrap MabSelect SuRe | GE Healthcare Life Sciences | Cat#11003493 |
| HiTrap KappaSelect | GE Healthcare Life Sciences | Cat#17545812 |
| HiTrap LambdaFabSelect Column | GE Healthcare Life Sciences | Cat#17548212 |
| CaptureSelect™ IgA Affinity Matrix | Thermo Fisher Scientific | Cat#19428805 |
| CaptureSelect™ CH1-XL Pre-packed Column | Thermo Fisher Scientific | Cat#494346201 |
| Amicon® Ultra-4 Centrifugal Filter Unit 100K MWCO | Millipore | Cat#UFC810024 |
| Amicon® Ultra-15 Centrifugal Filter Unit 50K MWCO | Millipore | Cat#UFC905008 |
| Amicon® Ultra-15 Centrifugal Filter Unit 30K MWCO | Millipore | Cat#UFC903024 |
| Zeba™ Spin Desalting Columns, 7K MWCO | Thermo Fisher Scientific | Cat#89894 or 89890 |
| Lacy 400 Mesh Copper Grids | TED PELLA | Cat#01896-F |
| Carbon Coated R2/2 Copper Quantifoil Holey Grids | Electron Microscopy Sciences | Cat#Q250CR2-2nm |
| 300 Mesh Lacey Grids | TED PELLA | Cat#01883-F |
| 400 Square Mesh Copper EM Grids | Electron Microscopy Sciences | Cat#CF400-CU |
| Vitrobot | Thermo Fisher Scientific | N/A |
| FEI Vitrobot Mark4 | Thermo Fisher Scientific | N/A |
| FEI TF20 Transmission Electron Microscope | Thermo Fisher Scientific | N/A |
| Titan Krios G3i Electron Microscope | Thermo Fisher Scientific | N/A |
| Glacios Electron Microscope | Thermo Fisher Scientific | N/A |
| US4000 4k × 4k CCD camera | Gatan | N/A |
| K2 Summit Direct Electron Detector | Gatan | N/A |
| Falcon 3EC Direct Electron Detector | Thermo Fisher Scientific | N/A |
Highlights.
Potent human anti-EEEV monoclonal antibodies were isolated from an EEEV survivor
Fabs neutralized virus, but bivalent IgG was optimal for neutralization
Antibodies inhibit virus entry by stabilization of virus particles
EEEV-33 (IgG) and EEEV-143 (IgA) protected against aerosolized EEEV challenge
ACKNOWLEDGEMENTS
We thank J.D. Laughlin for performing Octet HTX experiments. We thank P. Gilchuk, C. Slaughter, M. Vogt, and S. Zost at Vanderbilt for helpful discussions and training. We thank D. Yousif, M. Jensen, N. Murphy, M. Goff, I. Kovach, J. Rodriguez, C. Gainza, W. Reichard, M. Majedi, J. Reidy, A. Trivette, and R. Troseth for technical support. We thank M. Leksell, M. Mayo, K. Moton, T. Martin, V. Morris, A. Bunnell, and A. Jordan for administrative assistance and R. Irving for lab management. The University of Michigan Cryo-EM Facility has received support from the U-M Life Sciences Institute and the U-M Biosciences Initiative. We acknowledge M. Su, A. Bondy, L. Keopping, C. Lilienthal, and B. Battey for U-M cryo-EM technical and IT support. Cryo-EM computation done at the LSI was supported by NIH S10OD020011. EM data collections of EEEV VLP and EEEV VLP:rEEEV-143 Fab were conducted at the Center for Structural Biology Cryo-EM facility at Vanderbilt University. L.E.W. was supported by NIH grants T32 HL069765 and F31 AI145189. This project received support from the U.S. Defense Threat Reduction Agency (HDTRA1-13-1-0034 J.E.C.; HDTRA1-15-1-0047 W.B.K.), grants from the NIH U19 AI142790 (J.E.C. and M.S.D.) and R01 AI095436 (W.B.K.), and by CTSA award UL1 TR002243 from the U.S. NCATS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
M.S.D. is consultant for Inbios, Vir Biotechnology, NGM Biopharmaceuticals, is on Scientific Advisory Boards of Moderna and Immunome, and a recipient of grants from Moderna, Vir Biotechnology, and Emergent BioSolutions. J.E.C. has served as consultant for Eli Lilly, Sanofi and is on Scientific Advisory Boards of CompuVax and Meissa Vaccines, is recipient of research grants from Moderna and Sanofi and is Founder of IDBiologics, Inc. Vanderbilt University has applied for patents that are related to this work.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agapov EV, Razumov IA, Frolov IV, Kolykhalov AA, Netesov SV, and Loktev VB (1994). Localization of four antigenic sites involved in Venezuelan equine encephalomyelitis virus protection. Arch Virol 139, 173–181. [DOI] [PubMed] [Google Scholar]
- Armstrong PM, and Andreadis TG (2010). Eastern equine encephalitis virus in mosquitoes and their role as bridge vectors. Emerg Infect Dis 16, 1869–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong PM, and Andreadis TG (2013). Eastern equine encephalitis virus--old enemy, new threat. N Engl J Med 368, 1670–1673. [DOI] [PubMed] [Google Scholar]
- Ayres JC, and Feemster RF (1949). The sequelae of eastern equine encephalomyelitis. N Engl J Med 240, 960–962. [DOI] [PubMed] [Google Scholar]
- Baxter VK, and Griffin DE (2016). Interferon gamma modulation of disease manifestation and the local antibody response to alphavirus encephalomyelitis. J Gen Virol 97, 2908–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard E, Solignat M, Gay B, Chazal N, Higgs S, Devaux C, and Briant L (2010). Endocytosis of chikungunya virus into mammalian cells: role of clathrin and early endosomal compartments. PLoS One 5, e11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochet X, Lefranc MP, and Giudicelli V (2008). IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res 36, W503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broeckel R, Fox JM, Haese N, Kreklywich CN, Sukulpovi-Petty S, Legasse A, Smith PP, Denton M, Corvey C, Krishnan S, et al. (2017). Therapeutic administration of a recombinant human monoclonal antibody reduces the severity of chikungunya virus disease in rhesus macaques. PLoS Negl Trop Dis 11, e0005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke CW, Froude JW, Miethe S, Hulseweh B, Hust M, and Glass PJ (2018). Human-Like Neutralizing Antibodies Protect Mice from Aerosol Exposure with Western Equine Encephalitis Virus. Viruses 10, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke CW, Froude JW, Rossi F, White CE, Moyer CL, Ennis J, Pitt ML, Streatfield S, Jones RM, Musiychuk K, et al. (2019). Therapeutic monoclonal antibody treatment protects nonhuman primates from severe Venezuelan equine encephalitis virus disease after aerosol exposure. PLoS Pathog 15, e1008157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso FD, Rezende IM, Barros ELT, Sacchetto L, Garces T, Silva NIO, Alves PA, Soares JO, Kroon EG, Pereira A, et al. (2019). Circulation of Chikungunya virus East-Central-South Africa genotype during an outbreak in 2016–17 in Piaui State, Northeast Brazil. Rev Inst Med Trop Sao Paulo 61, e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carragher B, Kisseberth N, Kriegman D, Milligan RA, Potter CS, Pulokas J, and Reilein A (2000). Leginon: an automated system for acquisition of images from vitreous ice specimens. J Struct Biol 132, 33–45. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (CDC) website, www.cdc.gov/easternequineencephalitis/tech/epi.html, accessed on July 5th, 2020.
- Centers for Disease Control and Prevention (CDC) website, https://www.cdc.gov/easternequineencephalitis/index.html, accessed on September 20th, 2020.
- Chen CL, Hasan SS, Klose T, Sun Y, Buda G, Sun C, Klimstra WB, and Rossmann MG (2020). Cryo-EM structure of eastern equine encephalitis virus in complex with heparan sulfate analogues. Proc Natl Acad Sci U S A 117, 8890–8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa-Trevin JM, Oton J, Marabini R, Zaldivar A, Vargas J, Carazo JM, and Sorzano CO (2013). Xmipp 3.0: an improved software suite for image processing in electron microscopy. J Struct Biol 184, 321–328. [DOI] [PubMed] [Google Scholar]
- de la Rosa-Trevin JM, Quintana A, Del Cano L, Zaldivar A, Foche I, Gutierrez J, Gomez-Blanco J, Burguet-Castell J, Cuenca-Alba J, Abrishami V, et al. (2016). Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J Struct Biol 195, 93–99. [DOI] [PubMed] [Google Scholar]
- DeTulleo L, and Kirchhausen T (1998). The clathrin endocytic pathway in viral infection. EMBO J 17, 4585–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnest JT, Basore K, Roy V, Bailey AL, Wang D, Alter G, Fremont DH, and Diamond MS (2019). Neutralizing antibodies against Mayaro virus require Fc effector functions for protective activity. J Exp Med 216, 2282–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- EnCheng S, Jing Z, Tao Y, QingYuan X, Yongli Q, WenShi W, Peng W, Liang S, Jing S, and DongLai W (2013). Analysis of murine B-cell epitopes on Eastern equine encephalitis virus glycoprotein E2. Appl Microbiol Biotechnol 97, 6359–6372. [DOI] [PubMed] [Google Scholar]
- Fernandez-Leiro R, and Scheres SHW (2017). A pipeline approach to single-particle processing in RELION. Acta Crystallogr D Struct Biol 73, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fibriansah G, Ibarra KD, Ng TS, Smith SA, Tan JL, Lim XN, Ooi JS, Kostyuchenko VA, Wang J, de Silva AM, et al. (2015). DENGUE VIRUS. Cryo-EM structure of an antibody that neutralizes dengue virus type 2 by locking E protein dimers. Science 349, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn DC, Meyer WJ, Mackenzie JM Jr., and Johnston RE (1990). A conformational change in Sindbis virus glycoproteins E1 and E2 is detected at the plasma membrane as a consequence of early virus-cell interaction. J Virol 64, 3643–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong RH, Banik SS, Mattia K, Barnes T, Tucker D, Liss N, Lu K, Selvarajah S, Srinivasan S, Mabila M, et al. (2014). Exposure of epitope residues on the outer face of the chikungunya virus envelope trimer determines antibody neutralizing efficacy. J Virol 88, 14364–14379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JM, Long F, Edeling MA, Lin H, van Duijl-Richter MKS, Fong RH, Kahle KM, Smit JM, Jin J, Simmons G, et al. (2015). Broadly Neutralizing Alphavirus Antibodies Bind an Epitope on E2 and Inhibit Entry and Egress. Cell 163, 1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JM, Roy V, Gunn BM, Huang L, Edeling MA, Mack M, Fremont DH, Doranz BJ, Johnson S, Alter G, et al. (2019). Optimal therapeutic activity of monoclonal antibodies against chikungunya virus requires Fc-FcgammaR interaction on monocytes. Sci Immunol 4, eaav5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner CL, Ebel GD, Ryman KD, and Klimstra WB (2011). Heparan sulfate binding by natural eastern equine encephalitis viruses promotes neurovirulence. Proc Natl Acad Sci U S A 108, 16026–16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner CL, Sun C, Luke T, Raviprakash K, Wu H, Jiao JA, Sullivan E, Reed DS, Ryman KD, and Klimstra WB (2017). Antibody Preparations from Human Transchromosomic Cows Exhibit Prophylactic and Therapeutic Efficacy against Venezuelan Equine Encephalitis Virus. J Virol 91, e00226–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudicelli V, and Lefranc MP (2011). IMGT/junctionanalysis: IMGT standardized analysis of the V-J and V-D-J junctions of the rearranged immunoglobulins (IG) and T cell receptors (TR). Cold Spring Harb Protoc 2011, 716–725. [DOI] [PubMed] [Google Scholar]
- Grant T, Rohou A, and Grigorieff N (2018). cisTEM, user-friendly software for single-particle image processing. Elife 7, e35383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin D (1995). Roles and reactivities of antibodies to alphaviruses. Seminars in Virology 6, 249–255. [Google Scholar]
- Griffin D, Levine B, Tyor W, Ubol S, and Despres P (1997). The role of antibody in recovery from alphavirus encephalitis. Immunol Rev 159, 155–161. [DOI] [PubMed] [Google Scholar]
- Griffin DE (2016). Alphavirus Encephalomyelitis: Mechanisms and Approaches to Prevention of Neuronal Damage. Neurotherapeutics 13, 455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan SS, Sun C, Kim AS, Watanabe Y, Chen CL, Klose T, Buda G, Crispin M, Diamond MS, Klimstra WB, et al. (2018). Cryo-EM Structures of Eastern Equine Encephalitis Virus Reveal Mechanisms of Virus Disassembly and Antibody Neutralization. Cell Rep 25, 3136–3147 e3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R, Sali A, Baker ML, Carragher B, Devkota B, Downing KH, Egelman EH, Feng Z, Frank J, Grigorieff N, et al. (2012). Outcome of the first electron microscopy validation task force meeting. Structure 20, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes AC, Basore K, Fremont DH, and Diamond MS (2020). A molecular understanding of alphavirus entry. PLoS Pathogens, In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongming M, Kim AS, Kafai NM, Earnest JT, Shah A, Case JB, Basore K, Gilliland TC, Sun C, Nelson CA, et al. (2020). LDLRAD3 is a receptor for Venezuelan equine encephalitis virus. Nature, In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulseweh B, Rulker T, Pelat T, Langermann C, Frenzel A, Schirrmann T, Dubel S, Thullier P, and Hust M (2014). Human-like antibodies neutralizing Western equine encephalitis virus. MAbs 6, 718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt AR, Bowen RA, Frederickson S, Maruyama T, Roehrig JT, and Blair CD (2011). Treatment of mice with human monoclonal antibody 24h after lethal aerosol challenge with virulent Venezuelan equine encephalitis virus prevents disease but not infection. Virology 414, 146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt AR, Frederickson S, Hinkel C, Bowdish KS, and Roehrig JT (2006). A humanized murine monoclonal antibody protects mice either before or after challenge with virulent Venezuelan equine encephalomyelitis virus. J Gen Virol 87, 2467–2476. [DOI] [PubMed] [Google Scholar]
- Hunt AR, Frederickson S, Maruyama T, Roehrig JT, and Blair CD (2010). The first human epitope map of the alphaviral E1 and E2 proteins reveals a new E2 epitope with significant virus neutralizing activity. PLoS Negl Trop Dis 4, e739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt AR, Johnson AJ, and Roehrig JT (1990). Synthetic peptides of Venezuelan equine encephalomyelitis virus E2 glycoprotein. I. Immunogenic analysis and identification of a protective peptide. Virology 179, 701–711. [DOI] [PubMed] [Google Scholar]
- Hunt AR, and Roehrig JT (1985). Biochemical and biological characteristics of epitopes on the E1 glycoprotein of western equine encephalitis virus. Virology 142, 334–346. [DOI] [PubMed] [Google Scholar]
- Hunt AR, and Roehrig JT (1995). Localization of a protective epitope on a Venezuelan equine encephalomyelitis (VEE) virus peptide that protects mice from both epizootic and enzootic VEE virus challenge and is immunogenic in horses. Vaccine 13, 281–288. [DOI] [PubMed] [Google Scholar]
- Hunt AR, Short WA, Johnson AJ, Bolin RA, and Roehrig JT (1991). Synthetic peptides of the E2 glycoprotein of Venezuelan equine encephalomyelitis virus. II. Antibody to the amino terminus protects animals by limiting viral replication. Virology 185, 281–290. [DOI] [PubMed] [Google Scholar]
- Jin J, Liss NM, Chen DH, Liao M, Fox JM, Shimak RM, Fong RH, Chafets D, Bakkour S, Keating S, et al. (2015). Neutralizing Monoclonal Antibodies Block Chikungunya Virus Entry and Release by Targeting an Epitope Critical to Viral Pathogenesis. Cell Rep 13, 2553–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose J, Snyder JE, and Kuhn RJ (2010). A structural and functional perspective of alphavirus replication and assembly. Future Microbiol 4, 837–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AS, Austin SK, Gardner CL, Zuiani A, Reed DS, Trobaugh DW, Sun C, Basore K, Williamson LE, Crowe JE Jr., et al. (2019). Protective antibodies against Eastern equine encephalitis virus bind to epitopes in domains A and B of the E2 glycoprotein. Nat Microbiol 4, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimstra WB, Nangle EM, Smith MS, Yurochko AD, and Ryman KD (2003). DC-SIGN and L-SIGN can act as attachment receptors for alphaviruses and distinguish between mosquito cell- and mammalian cell-derived viruses. J Virol 77, 12022–12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko SY, Akahata W, Yang ES, Kong WP, Burke CW, Honnold SP, Nichols DK, Huang YS, Schieber GL, Carlton K, et al. (2019). A virus-like particle vaccine prevents equine encephalitis virus infection in nonhuman primates. Sci Transl Med 11, eaav3113. [DOI] [PubMed] [Google Scholar]
- Kose N, Fox JM, Sapparapu G, Bombardi R, Tennekoon RN, de Silva AD, Elbashir SM, Theisen MA, Humphris-Narayanan E, Ciaramella G, et al. (2019). A lipid-encapsulated mRNA encoding a potently neutralizing human monoclonal antibody protects against chikungunya infection. Sci Immunol 4, eaaw6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Hardwick JM, Trapp BD, Crawford TO, Bollinger RC, and Griffin DE (1991). Antibody-mediated clearance of alphavirus infection from neurons. Science 254, 856–860. [DOI] [PubMed] [Google Scholar]
- Lindsey NP, Martin SW, Staples JE, and Fischer M (2020). Notes from the Field: Multistate Outbreak of Eastern Equine Encephalitis Virus - United States, 2019. MMWR Morb Mortal Wkly Rep 69, 50–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey NP, Staples JE, and Fischer M (2018). Eastern Equine Encephalitis Virus in the United States, 2003–2016. Am J Trop Med Hyg 98, 1472–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F, Fong RH, Austin SK, Chen Z, Klose T, Fokine A, Liu Y, Porta J, Sapparapu G, Akahata W, et al. (2015). Cryo-EM structures elucidate neutralizing mechanisms of anti-chikungunya human monoclonal antibodies with therapeutic activity. Proc Natl Acad Sci U S A 112, 13898–13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Metcalf TU, Baxter VK, Nilaratanakul V, and Griffin DE (2013). Recruitment and retention of B cells in the central nervous system in response to alphavirus encephalomyelitis. J Virol 87, 2420–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf TU, and Griffin DE (2011). Alphavirus-induced encephalomyelitis: antibody-secreting cells and viral clearance from the nervous system. J Virol 85, 11490–11501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer WJ, and Johnston RE (1993). Structural rearrangement of infecting Sindbis virions at the cell surface: mapping of newly accessible epitopes. J Virol 67, 5117–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell CJ, Niebylski ML, Smith GC, Karabatsos N, Martin D, Mutebi JP, Craig GB Jr., and Mahler MJ (1992). Isolation of eastern equine encephalitis virus from Aedes albopictus in Florida. Science 257, 526–527. [DOI] [PubMed] [Google Scholar]
- Morens DM, Folkers GK, and Fauci AS (2019). Eastern Equine Encephalitis Virus - Another Emergent Arbovirus in the United States. N Engl J Med 381, 1989–1992. [DOI] [PubMed] [Google Scholar]
- Neves V, A.-d.-S. F, Corte-Real S, Castanho MARB (2016). Antibody approaches to treat brain diseases. Trends in Biotechnology 34, 36–48. [Google Scholar]
- Nilaratanakul V, Chen J, Tran O, Baxter VK, Troisi EM, Yeh JX, and Griffin DE (2018). Germ Line IgM Is Sufficient, but Not Required, for Antibody-Mediated Alphavirus Clearance from the Central Nervous System. J Virol 92, e02081–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi M, Li Y, Cheng Y, and Walz T (2004). Negative Staining and Image Classification - Powerful Tools in Modern Electron Microscopy. Biol Proced Online 6, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal P, Dowd KA, Brien JD, Edeling MA, Gorlatov S, Johnson S, Lee I, Akahata W, Nabel GJ, Richter MK, et al. (2013). Development of a highly protective combination monoclonal antibody therapy against Chikungunya virus. PLoS Pathog 9, e1003312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereboev AV, Razumov IA, Svyatchenko VA, and Loktev VB (1996). Glycoproteins E2 of the Venezuelan and eastern equine encephalomyelitis viruses contain multiple cross-reactive epitopes. Arch Virol 141, 2191–2205. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Phelps AL, O’Brien LM, Eastaugh LS, Davies C, Lever MS, Ennis J, Zeitlin L, Nunez A, and Ulaeto DO (2019). Aerosol infection of Balb/c mice with eastern equine encephalitis virus; susceptibility and lethality. Virol J 16, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta J, Jose J, Roehrig JT, Blair CD, Kuhn RJ, and Rossmann MG (2014). Locking and blocking the viral landscape of an alphavirus with neutralizing antibodies. J Virol 88, 9616–9623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell LA, Fox JM, Kose N, Kim AS, Majedi M, Bombardi R, Carnahan RH, Slaughter JC, Morrison TE, Diamond MS, et al. (2020). Human monoclonal antibodies against Ross River virus target epitopes within the E2 protein and protect against disease. PLoS Pathog 16, e1008517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulgar VM (2018). Transcytosis to Cross the Blood Brain Barrier, New Advancements and Challenges. Front Neurosci 12, 1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroz JA, Malonis RJ, Thackray LB, Cohen CA, Pallesen J, Jangra RK, Brown RS, Hofmann D, Holtsberg FW, Shulenin S, et al. (2019). Human monoclonal antibodies against chikungunya virus target multiple distinct epitopes in the E1 and E2 glycoproteins. PLoS Pathog 15, e1008061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert E, Clase A, Bacetty A, and Larsen J (2009). Alphavirus antiviral drug development: scientific gap analysis and prospective research areas. Biosecur Bioterror 7, 413–427. [DOI] [PubMed] [Google Scholar]
- Roehrig JT, Day JW, and Kinney RM (1982). Antigenic analysis of the surface glycoproteins of a Venezuelan equine encephalomyelitis virus (TC-83) using monoclonal antibodies. Virology 118, 269–278. [DOI] [PubMed] [Google Scholar]
- Roehrig JT, Hunt AR, Kinney RM, and Mathews JH (1988). In vitro mechanisms of monoclonal antibody neutralization of alphaviruses. Virology 165, 66–73. [DOI] [PubMed] [Google Scholar]
- Roehrig JT, and Mathews JH (1985). The neutralization site on the E2 glycoprotein of Venezuelan equine encephalomyelitis (TC-83) virus is composed of multiple conformationally stable epitopes. Virology 142, 347–356. [DOI] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronca SE, Dineley KT, and Paessler S (2016). Neurological Sequelae Resulting from Encephalitic Alphavirus Infection. Front Microbiol 7, 959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose PP, Hanna SL, Spiridigliozzi A, Wannissorn N, Beiting DP, Ross SR, Hardy RW, Bambina SA, Heise MT, and Cherry S (2011). Natural resistance-associated macrophage protein is a cellular receptor for sindbis virus in both insect and mammalian hosts. Cell Host Microbe 10, 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salimi H, Cain MD, Jiang X, Roth RA, Beatty WL, Sun C, Klimstra WB, Hou J, and Klein RS (2020). Encephalitic Alphaviruses Exploit Caveola-Mediated Transcytosis at the Blood-Brain Barrier for Central Nervous System Entry. mBio 11, e02731–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH, and Chen S (2012). Prevention of overfitting in cryo-EM structure determination. Nat Methods 9, 853–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvarajah S, Sexton NR, Kahle KM, Fong RH, Mattia KA, Gardner J, Lu K, Liss NM, Salvador B, Tucker DF, et al. (2013). A neutralizing monoclonal antibody targeting the acid-sensitive region in chikungunya virus E2 protects from disease. PLoS Negl Trop Dis 7, e2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood JA, Stehman SV, Howard JJ, and Oliver J (2020). Cases of Eastern equine encephalitis in humans associated with Aedes canadensis, Coquillettidia perturbans and Culiseta melanura mosquitoes with the virus in New York State from 1971 to 2012 by analysis of aggregated published data. Epidemiol Infect 148, e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidwell RW, and Smee DF (2003). Viruses of the Bunya- and Togaviridae families: potential as bioterrorism agents and means of control. Antiviral Res 57, 101–111. [DOI] [PubMed] [Google Scholar]
- Smith SA, Silva LA, Fox JM, Flyak AI, Kose N, Sapparapu G, Khomandiak S, Ashbrook AW, Kahle KM, Fong RH, et al. (2015). Isolation and Characterization of Broad and Ultrapotent Human Monoclonal Antibodies with Therapeutic Activity against Chikungunya Virus. Cell Host Microbe 18, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorzano CO, de la Rosa Trevin JM, Oton J, Vega JJ, Cuenca J, Zaldivar-Peraza A, Gomez-Blanco J, Vargas J, Quintana A, Marabini R, et al. (2013). Semiautomatic, high-throughput, high-resolution protocol for three-dimensional reconstruction of single particles in electron microscopy. Methods Mol Biol 950, 171–193. [DOI] [PubMed] [Google Scholar]
- Sun E, Zhao J, Sun L, Xu Q, Yang T, Qin Y, Wang W, Wei P, Sun J, and Wu D (2013a). Comprehensive mapping of common immunodominant epitopes in the eastern equine encephalitis virus E2 protein recognized by avian antibody responses. PLoS One 8, e69349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Xiang Y, Akahata W, Holdaway H, Pal P, Zhang X, Diamond MS, Nabel GJ, and Rossmann MG (2013b). Structural analyses at pseudo atomic resolution of Chikungunya virus and antibodies show mechanisms of neutralization. Elife 2, e00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Gardner CL, Watson AM, Ryman KD, and Klimstra WB (2014). Stable, high-level expression of reporter proteins from improved alphavirus expression vectors to track replication and dissemination during encephalitic and arthritogenic disease. J Virol 88, 2035–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Lam TT-Y, Heberlein-Larson LA, Smole SC, Auguste AJ, Hennigan S, Halpin RA, Fedorova N, Puri V, Stockwell TB, et al. (2018). Large-scale complete-genome sequencing and phylodynamic analysis of eastern equine encephalitis virus reveals source-sink transmission dynamics in the United States. J Virol 92, e00074–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trobaugh DW, Sun C, Dunn MD, Reed DS, and Klimstra WB (2019). Rational design of a live-attenuated eastern equine encephalitis virus vaccine through informed mutation of virulence determinants. PLoS Pathog 15, e1007584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchaninova MA, Davydov A, Britanova OV, Shugay M, Bikos V, Egorov ES, Kirgizova VI, Merzlyak EM, Staroverov DB, Bolotin DA, et al. (2016). High-quality full-length immunoglobulin profiling with unique molecular barcoding. Nat Protoc 11, 1599–1616. [DOI] [PubMed] [Google Scholar]
- Turula H, and Wobus CE (2018). The Role of the Polymeric Immunoglobulin Receptor and Secretory Immunoglobulins during Mucosal Infection and Immunity. Viruses 10, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinuela-Berni V, Gomez-Gonzalez B, and Quintanar-Stephano A (2020). Blockade of Arginine Vasopressin receptors prevents blood-brain barrier breakdown in Experimental Autoimmune Encephalomyelitis. Sci Rep 10, 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss JE, Vaney MC, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, and Rey FA (2010). Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468, 709–712. [DOI] [PubMed] [Google Scholar]
- Yu X, McGraw PA, House FS, and Crowe JE Jr. (2008a). An optimized electrofusion-based protocol for generating virus-specific human monoclonal antibodies. J Immunol Methods 336, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Tsibane T, McGraw PA, House FS, Keefer CJ, Hicar MD, Tumpey TM, Pappas C, Perrone LA, Martinez O, et al. (2008b). Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature 455, 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Hryc CF, Cong Y, Liu X, Jakana J, Gorchakov R, Baker ML, Weaver SC, and Chiu W (2011). 4.4 A cryo-EM structure of an enveloped alphavirus Venezuelan equine encephalitis virus. EMBO J 30, 3854–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Kim AS, Fox JM, Nair S, Basore K, Klimstra WB, Rimkunas R, Fong RH, Lin H, Poddar S, et al. (2018). Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 557, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Sun EC, Liu NH, Yang T, Xu QY, Qin YL, Yang YH, and Wu DL (2012). Phage display identifies an Eastern equine encephalitis virus glycoprotein E2-specific B cell epitope. Vet Immunol Immunopathol 148, 364–368. [DOI] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, Related to Figure 2. Binding reactivity of neutralizing human anti-EEEV mAbs to SINV/EEEV particles or EEEV E2 glycoprotein.
(A) Binding ratio of neutralizing human anti-EEEV mAbs to SINV/EEEV particles vs. recombinant monomeric EEEV E2 glycoprotein. EEEV-33 is removed to display the binding reactivity groups for the rest of the panel of neutralizing human anti-EEEV mAbs at higher resolution. A dotted line indicates 32 pM half-maximal effective concentration (EC50) values for binding, revealing distinct binding patterns of human anti-EEEV mAbs to SINV/EEEV particles and EEEV E2 glycoprotein. Neutralizing human anti-EEEV mAbs are labeled with the anti-EEEV mAb name and colored according to binding group (Group 2 [green] = strong (SINV/EEEV EC50 = <32 pM) virus » protein binding; Group 3 [purple] = weak (SINV/EEEV EC50 = >32 pM) virus»protein binding; Group 4 [orange] = protein>virus binding).
(B) Representative binding curves of recombinant neutralizing human anti-EEEV IgG1, IgA1, or Fab molecules to three different antigens. Binding curves of recombinant neutralizing human anti-EEEV IgG1 (square), IgA1 (triangle), or Fab (circle) molecules to SINV/EEEV particles (green) or EEEV E2 glycoprotein (blue), with mAb concentration (nM) on the x-axis and optical density at 405 nm on the y-axis. Binding to EEEV E1 glycoprotein (purple) was not detected.
(C) EC50 values (pM) for binding of recombinant neutralizing human anti-EEEV IgG1, IgA1, or Fab molecules to SINV/EEEV particles or EEEV E2 glycoprotein. EC50 value in pM is indicated by blue fill color (<32 [dark blue], 32.01 to 100 [medium blue], 100.01 to 320 [light blue], <10,000 [lightest blue]). Ratio of binding to SINV/EEEV particles versus EEEV E2 glycoprotein is indicated as the ratio of EC50 values. Increasing depth of green color indicates lower ratios (<0.1 [dark green], 0.1 to 1.0 [medium green], 1.01 to 2.0 [light green], >2.0 [lightest green]), suggesting recognition of a quaternary epitope on virion particles. Ratio of binding of recombinant IgG1 versus Fab molecules to SINV/EEEV particles or EEEV E2 glycoprotein is indicated as the ratio of EC50 values. Increasing depth of purple color indicates lower ratios (<0.1 [dark purple], 0.1 to 1.0 [medium purple], 1.01 to 2.0 [light purple], >2.0 [lightest purple]), suggesting dependence on valency for binding. Antibody isotype is indicated as IgG1 or IgA1 for the heavy chain or k or l for the light chain. NA = not applicable.
Data in A to C represent mean ± SD of technical triplicates and are representative of two independent ELISA experiments. Each mAb is colored based on the binding group defined in Figure 2.
Figure S2, Related to Figure 3. Alanine-scanning mutagenesis library analysis. Bar graphs of neutralizing human anti-EEEV mAb binding to critical residues (<25% mAb binding with at least two other mAbs >70% to control for expression) or critical residues previously characterized for murine anti-EEEV mAbs (Kim et al., 2019) and the VEE-specific human mAb, F5 (Hunt et al., 2010; Porta et al., 2014). Each mAb is listed on the x-axis and percent binding relative to wildtype is on the y-axis. Residues are colored based on E2 domain (N-link - purple, Domain A - red, Arch 1 - magenta, Domain B - cyan, and Arch 2 - orange). Data represent mean ± SD of at least two independent experiments.
Figure S3, Related to Figure 3. Neutralization activity of human anti-EEEV mAbs to SINV/EEEV escape mutant viruses.
(A) Representative neutralization curves of potent neutralizing human anti-EEEV mAbs against SINV/EEEV WT, and escape mutant viruses M68T, G192R, and L227R. Neutralization curves of potent human anti-EEEV mAbs against SINV/EEEV WT (closed square), M68T (open triangle), G192R (open circle), and L227R (open diamond), with mAb concentration (nM) on the x-axis and % relative infectivity on the y-axis. mAbs are ordered based on IC50 values against SINV/EEEV WT. A positive control mouse mAb, mEEEV-3 (dark purple) (Kim et al., 2019), and a negative control mAb, rDENV-2D22 (black; B), were included.
(B) Representative neutralization curves of moderate neutralizing human anti-EEEV mAbs against SINV/EEEV WT and escape mutant viruses M68T, G192R, and L227R. Neutralization curves of moderate human anti-EEEV mAbs against SINV/EEEV WT (closed square), M68T (open triangle), G192R (open circle), and L227R (open diamond), with mAb concentration (nM) on the x-axis and % relative infectivity on the y-axis. A positive control mouse mAb, mEEEV-3 (dark purple; A) (Kim et al., 2019), and a negative control mAb, rDENV-2D22 (black), were included.
(C) Half-maximal inhibitory concentration (IC50) values (pM) for human anti-EEEV mAbs against SINV/EEEV WT and escape mutant viruses M68T, G192R, and L227R. IC50 values (pM) for neutralizing human anti-EEEV mAbs against SINV/EEEV WT, M68T, G192R, and L227R are indicated in the table. Neutralizing human anti-EEEV mAbs are listed in order of increasing IC50 value against SINV/EEEV WT. IC50 value in pM is indicated by the orange heat map (<33 [dark orange], 33 to 333 [medium orange], 333.01 to 3,333 [light orange], <10,000 [lightest orange]). Fold difference in IC50 value ratio of SINV/EEEV WT versus each escape mutant virus (M68T, G192R, or L227R) are indicated. Ratio of neutralization activity against SINV/EEEV WT versus each escape mutant virus (M68T, G192R, or L227R) is indicated as the fold difference of IC50 values. Increasing depth of maroon color indicates greater fold differences (>15 or > [dark maroon], 10–15 [medium maroon], 5–10 [light maroon], <5 [white]), suggesting reduction in neutralization activity of the mAb.
Data in A to C represent mean ± SD of technical triplicates and are representative of two independent focus reduction neutralization test (FRNT) experiments. mAbs are colored based on mAb legend in Figure 1.
Figure S4, Related to Figure 4 and Figure 5. EEEV-33 or EEEV-143 Fab binding to the E2 trimeric spike of SINV/EEEV particles.
Side view (A) or 40° top-view (B) of EEEV E2 trimeric spikes (in light blue) for side-by-side comparisons of native SINV/EEEV (left), SINV/EEEV:EEEV-33 Fab complex (right) and SINV/EEEV:EEEV-143 Fab complex (middle) showing binding of one Fab (EEEV-33 - red; EEEV-143 - orange) per E2 protomer within the trimeric spike. Low pass map of published native SINV/EEEV structure (EMD-9280) used for the comparison. Side view (C) or 40° top-view (D) of EEEV-33 Fab (red) and EEEV-143 Fab (orange) bound to SINV/EEEV or EEEV VLP, respectively. For the SINV/EEEV (PDB ID: 6MX4) and EEEV-143 Fab (mutated sequence of PDB ID: 6MWX) docked and rigid body refined model of SINV/EEEV:EEEV-33 Fab complex, the structural proteins are colored with E2 = dark green, E1 = salmon, and capsid = blue. For the EEEV VLP:EEEV-143 Fab complex (EMD-22277; PDB ID: 6XOB), the structural proteins are colored with E2 = light green, E1 = raspberry, and capsid = cyan.
Figure S5, related to Figure 5. Cryo-EM reconstruction of EEEV VLP in complex with EEEV-143 Fab molecules. Cryo-EM structure of the apo form of EEEV VLP (A–C) and of EEEV VLP:EEEV-143 Fab complex (D–F) showing radially colored surface representation of full (A, D) and cross-section (B, E) of the map. C, F. Roadmaps of the apo form of EEEV VLP and EEEV VLP:EEEV-143 Fab complex reconstructions to illustrate EEEV-143 Fab binding sites (in dark blue). EEEV-143 binds in a tangential orientation and makes contacts with neighboring spikes for inter-spike cross-linking of SINV/EEEV particles. G–H. Asymmetric unit view of EEEV structural proteins (E2 = green, E1 = red, capsid protein = cyan) of the apo form of EEEV VLP (G) or in complex with EEEV-143 Fab molecules (H; orange) bound in a tangential orientation. I. Alignment of EEEV structural proteins of the EEEV VLP apo form (dark blue) to the EEEV VLP:EEEV-143 Fab complex (pink) to show stabilization of the E2 glycoprotein upon EEEV-143 Fab binding.
Figure S6, Related to Figures 6 and 7. In vivo body weight change and clinical disease signs observed in EEEV-33 and EEEV-143 mAb administered lethal EEEV aerosol challenge model.
(A) Percent body weight change of EEEV-33, EEEV-143, and rDENV-2D22 prophylactically treated CD-1 mice over the course of 14 days. Percent change in weight (y-axis) for EEEV-33 (red; n=11), EEEV-143 (orange; n=5), and rDENV-2D22 (black; n=10) are indicated over the course of 14 days after EEEV challenge (1,631 to 1,825 PFU/mouse; x-axis).
(B) Percent body weight change of EEEV-143 and rDENV-2D22 prophylactically treated CD-1 mice over the course of 14 days. Percent change in weight (y-axis) for EEEV-143 (orange; n=5) and rDENV-2D22 (black; n=5) are indicated over the course of 14 days after EEEV challenge (2,739 PFU/mouse; x-axis).
(C) Clinical scores of EEEV-33, EEEV-143, and rDENV-2D22 prophylactically treated CD-1 mice over the course of 14 days. The number of mice (y-axis) for EEEV-33 (red; n=11), EEEV-143 (orange; n=5), and rDENV-2D22 (black; n=10) with defined clinical scores (dead (black), moribund (red), seizures/ataxia (yellow), hunched back/behavioral (blue), ruffled fur (green), healthy (white)) are indicated over the course of 14 days post EEEV challenge (1,631 to 1,825 PFU/mouse; x-axis).
(D) Clinical scores of EEEV-143 and rDENV-2D22 prophylactically treated CD-1 mice over the course of 14 days. The number of mice (y-axis) for EEEV-143 (orange; n=5) and rDENV-2D22 (black; n=5) with defined clinical scores (dead (black), moribund (red), seizures/ataxia (yellow), hunched back/behavioral (blue), ruffled fur (green), healthy (white)) are indicated over the course of 14 days after EEEV challenge (2,739 PFU/mouse; x-axis).
(E) Percent body weight change of EEEV-33, EEEV-143, and rDENV-2D22 therapeutically treated CD-1 mice over the course of 14 days. Percent change in weight (y-axis) for EEEV-33 (red; n=11), EEEV-143 (orange; n=5), and rDENV-2D22 (black; n=5) are indicated over the course of 14 days after EEEV challenge (1,631 to 1,825 PFU/mouse; x-axis).
(F) Percent body weight change of EEEV-143 therapeutically treated CD-1 and BALB/c mice, respectively, over the course of 14 days. Percent change in weight (y-axis) for EEEV-143 (orange; n=5) is indicated over the course of 14 days after EEEV challenge (2,739 PFU/mouse; x-axis).
(G) Clinical scores of EEEV-33, EEEV-143, and rDENV-2D22 therapeutically treated CD-1 mice over the course of 14 days. The number of mice (y-axis) for EEEV-33 (red; n=11), EEEV-143 (orange; n=5), and rDENV-2D22 (black; n=5) with defined clinical scores (dead (black), moribund (red), seizures/ataxia (yellow), hunched back/behavioral (blue), ruffled fur (green), healthy (white)) are indicated over the course of 14 days after EEEV challenge (1,631 to 1,825 PFU/mouse; x-axis).
(H) Clinical scores of EEEV-143 therapeutically treated CD-1 and BALB/c mice, respectively, over the course of 14 days. The number of mice (y-axis) for EEEV-143 (orange; n=5) with defined clinical scores (dead (black), moribund (red), seizures/ataxia (yellow), hunched back/behavioral (blue), ruffled fur (green), healthy (white)) are indicated over the course of 14 days after EEEV challenge (2,739 PFU/mouse; x-axis).
(I) Percent body weight change of CD-1 mice treated therapeutically with EEEV-143 or rDENV-2D22. Percent change in weight (y-axis) for EEEV-143 (orange; n=5) or rDENV-2D22 (black; n=5) treated animals is indicated over the course of 14 days after EEEV challenge (1,897 PFU/mouse; x-axis).
(J) Clinical scores of CD-1 mice treated therapeutically with EEEV-143 or rDENV-2D22. The number of mice (y-axis) treated with EEEV-143 (orange; n=5) or rDENV-2D22 (black; n=5) with defined clinical scores (dead (black), moribund (red), seizures/ataxia (yellow), hunched back/behavioral (blue), ruffled fur (green), or healthy (white)) are indicated over the course of 14 days after EEEV challenge (1,897 PFU/mouse; x-axis).
Figure S7, Related to STAR Methods. Cryo-EM processing of SINV/EEEV complexes with either EEEV-33 or EEEV-143 Fabs.
Flow chart of cryo-EM processing steps and Fourier shell correlation (FSCs) of the maps of SINV/EEEV:rEEEV-33 Fab complex (A), SINV/EEEV:rEEEV-143 Fab complex (B), EEEV VLP (C), and EEEV VLP:rEEEV-143 Fab complex (D). The FSCs of the refined models agree with each other, suggesting that the models are not over-refined.
Data Availability Statement
The cryo-EM reconstructions reported in this paper have been deposited to the Protein Data Bank or Electron Microscopy Data Bank under the accession numbers EMD-22223 (SINV/EEEV:rEEEV-33 Fab), EMD-22188 (SINV/EEEV:rEEEV-143 Fab), EMD-22276; PDB ID: 6XO4 (EEEV VLP), EMD-22277; PDB ID: 6XOB (EEEV VLP:rEEEV-143 Fab). All relevant data are included within the manuscript and are available from the Lead Contact upon request.