

Cells advance through G1 phase for 2 hours after DNA damage, and if not stopped in time, they will enter S phase with damaged DNA.
Abstract
The G1-S checkpoint is thought to prevent cells with damaged DNA from entering S phase and replicating their DNA and efficiently arrests cells at the G1-S transition. Here, using time-lapse imaging and single-cell tracking, we instead find that DNA damage leads to highly variable and divergent fate outcomes. Contrary to the textbook model that cells arrest at the G1-S transition, cells triggering the DNA damage checkpoint in G1 phase route back to quiescence, and this cellular rerouting can be initiated at any point in G1 phase. Furthermore, we find that most of the cells receiving damage in G1 phase actually fail to arrest and proceed through the G1-S transition due to persistent cyclin-dependent kinase (CDK) activity in the interval between DNA damage and induction of the CDK inhibitor p21. These observations necessitate a revised model of DNA damage response in G1 phase and indicate that cells have a G1 checkpoint.
INTRODUCTION
The cell cycle is controlled by a series of commitment points and checkpoints that ensure ordered progression through the phases of the cell cycle (1). In the absence of sufficient growth factors or mitogens, cells in early G1 may exit to quiescence, also referred to as G0, but later in G1 lose sensitivity to the presence of mitogens and remain committed to the cell cycle (2–4). Cell cycle checkpoints thought to be located at the G1-S and G2-M phase transitions are responsible for halting cell cycle progression if DNA damage is detected, thus preventing DNA damage from accumulating in subsequent generations (5). Due to the fundamental function of these checkpoints, many common oncogenes such as p53, BRCA1/2, and CHK1/2 are critically involved in checkpoint function and dysregulation (6). In G1 phase, mammalian cells with high levels of DNA damage fail to pass the G1-S checkpoint and arrest (7, 8). Thus, diverse environmental information is integrated in G1 phase and translated into two main mechanisms of proliferative regulation: the mitogen-regulated commitment point and the DNA damage–regulated G1-S checkpoint (Fig. 1A).
Fig. 1. Cells route to G0 rather than arresting at the G1-S transition after DNA damage.
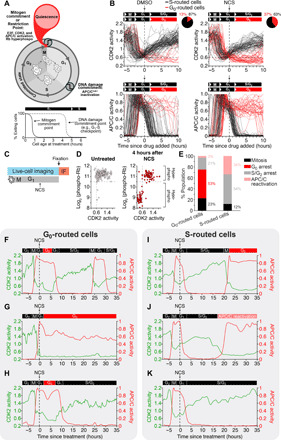
(A) Passage through the Restriction Point in early G1 phase is characterized by hyperphosphorylated Rb, E2F-mediated transcriptional activity, CDK2 activation, and high APC/C activity. Passage through the DNA damage commitment point is characterized by rapid APC/C inactivation at the end of G1 phase. (B) Single-cell traces of CDK2 (top) and APC/C (bottom) activity in MCF-10A cells treated with DMSO or NCS (200 ng/ml) during G1 phase, which was defined as CDK2 activity greater than 0.6 and APC/C activity greater than 0.3 at the time of treatment. Cells were colored black if APC/C activity fell below 0.3, indicating entry into S phase, and cells were colored red if APC/C activity remained above 0.3 6 hours after drug addition and CDK2 activity fell below 0.6, indicating rerouting to G0. Pie chart represents the percentage of cells that either routed to S or G0 phase after DNA damage. (C) Experimental setup for (D). Live-cell imaging was conducted to identify G1 cells at the time of treatment. Cells were treated with either DMSO or NCS (200 ng/ml) and then fixed 4 hours later. IF, immunofluorescence. (D) Scatterplot of single-cell CDK2 activity in the last frame of the movie versus the phospho-Rb levels after fixation. (E) Fate outcomes of G0- versus S phase–routed cells. Cells were tracked for 48 hours after NCS treatment, and fate outcomes were manually assigned. N > 2000 cells from n = 2 experiments. (F to H) Single-cell CDK2 and APC/C activity traces for cells that routed back to G0 after DNA damage. (I to K) Example single-cell CDK2 and APC/C activity traces for cells that continued on to S phase after DNA damage in G1.
The mitogen-regulated commitment point, called the Restriction Point, is located in early G1 phase (9). The Restriction Point has been defined mechanistically by the switch-like hyperphosphorylation of the retinoblastoma protein (Rb) by cyclin-dependent kinase (CDK) activity and the subsequent activation of the E2F family transcription factors (9–12). Cells that pass the Restriction Point continue on to mitosis even in the absence of mitogen signaling. Conversely, the DNA damage–regulated G1-S checkpoint is thought to be located at the end of G1 phase and arrests cells at the G1-S transition until the damage can be repaired. Mechanistically, the G1-S checkpoint is initiated through sensing of DNA damage by ataxia telangiectasia mutated (ATM) kinase (7), which phosphorylates and activates checkpoint kinase 1 (CHK1) and CHK2 (13). CHK1 and CHK2 are thought to regulate the cell cycle in two waves with a rapid response mediated by the degradation of the CDK-activating phosphatase Cdc25A (14) and a secondary response that involves CHK2-mediated phosphorylation of p53, a master tumor suppressor. P53, in turn, drives the transcriptional induction of p21, a CDK inhibitor (15). In addition, DNA damage has been shown to induce the degradation of cyclin D (16–19), which can also halt cell cycle progression through loss of CDK4/6 activity.
Incomplete or aberrant G1-S checkpoint passage, which occurs when cells transition from G1 to S phase despite the presence of DNA damage, has been reported, often in the context of oncogenesis (20, 21). Many cancer cells have missing or mutated checkpoint proteins, which can confer advantages in selection because of rapid proliferation (1, 22). However, aberrant checkpoint passage has also been observed even among cells with fully functional checkpoint proteins (23–25), suggesting that the G1-S checkpoint is inefficiently maintained (25). This inefficiency has been postulated to be due to inherent stochastic checkpoint error and the ability of signal transduction pathways to adapt to DNA damage (26, 27). Furthermore, different cell fates have been observed within populations of genetically identical cells, suggesting that additional factors may determine what fate an individual cell will choose (24, 25). Thus, despite decades of research on the G1-S checkpoint, recent studies have raised new questions about the molecular mechanisms underlying the G1-S checkpoint including which cell-intrinsic factors determine whether a cell will trigger the G1-S checkpoint.
Our study uses time-lapse imaging and single-cell tracking to follow cells after receiving DNA damage in G1 phase to determine the fate outcome of single cells. We find that the response to DNA damage at the single-cell level is highly variable. Rather than arresting at the G1-S transition, we observed a bifurcation in cellular fates with some cells rerouting back to G0 phase and other cells continuing on to S phase, despite the presence of considerable DNA damage. We show that routing back to G0 is mediated by induction of p21 after DNA damage, which can take place at any point in G1 phase, indicating that cells have an intra-G1 checkpoint rather than a G1-S checkpoint. Furthermore, we show that the cells that continue into S phase after DNA damage failed to trigger the G1 checkpoint due to persistent CDK2 activity during the 2 hours it takes for cells to induce a threshold level of p21 required to route cells back to G0. We have termed this phenomenon cell cycle inertia, which mediates continued forward progression through the G1 program even after DNA damage and, when combined with an irreversible G1-S phase transition, introduces divergent fate outcomes within a population of genetically identical cells. Collectively, our results indicate that an updated model of DNA damage checkpoints is required and demonstrate a nongenetic mechanism of checkpoint failure, which introduces substantial variability in cellular fate outcomes.
RESULTS
Cells route to G0 rather than arresting at the G1-S transition after DNA damage
To observe the fates of cells that experience DNA damage in G1 phase, we used live-cell imaging of both a CDK2 activity sensor, which undergoes nuclear-to-cytoplasm translocation in response to CDK2 phosphorylation (10, 28), and an anaphase-promoting complex/cyclosome (APC/C) activity sensor, which is based on a fluorescent substrate of the APC/C (29, 30). These sensors allowed us to follow the cell cycle trajectory of single cells before and after inducing DNA damage (Fig. 1A). Cells were considered to be past the Restriction Point and in G1 phase if they had a CDK2 activity greater than 0.6 and an APC/C activity greater than 0.3 (fig. S1, A to I; see also Materials and Methods), consistent with previously reported threshold values (10, 31). We treated asynchronously cycling cells with the radiomimetic drug neocarzinostatin (NCS), which induces DNA double-stranded breaks within 5 min of its addition to culture medium (32), and we only analyzed cells that were in G1 phase at the time of treatment. After inducing DNA damage, we observed that cells took divergent trajectories through the cell cycle. Rather than all cells arresting at the G1-S transition, we observed that approximately 37% of cells inactivated CDK2 activity back below the 0.6 threshold and maintained high APC/C activity, indicating that they routed back to a G0 state (Fig. 1B). The other 63% of cells unexpectedly continued to increase CDK2 activity and inactivated the APC/C, demonstrating that these cells entered into S phase despite the presence of damaged DNA (Fig. 1B and fig. S1J). To further support this conclusion, we fixed and stained cells 4 hours after NCS treatment and found that those cells that inactivated CDK2 also entered a hypophosphorylated Rb state (Fig. 1C), which is a marker of quiescence. Conversely, the cells that maintained high CDK2 activity and inactivated the APC/C had hyperphosphorylated Rb, which is a marker of the proliferative state.
To understand the significance of these divergent cell trajectories, we used long-term imaging and recorded the eventual fates of each single cell (Fig. 1E). Most of the cells that routed to G0 after DNA damage either recovered, reentered the cell cycle, and eventually went through mitosis (Fig. 1F) or remained in G0 for the duration of the imaging period (Fig. 1G). A small proportion of cells reentered the cell cycle but arrested at the G2-M transition (Fig. 1H). Conversely, only a small fraction of the cells that continued on to S phase successfully completed mitosis (Fig. 1I). The vast majority of these S phase–routed cells either arrested at the G2-M transition (Fig. 1J) or prematurely lost CDK2 activity and reactivated the APC/C in G2 phase (Fig. 1K), a state previously identified to be a precursor to senescence (33, 34). Thus, we find that cells that routed back to G0 are about twice as likely to subsequently undergo mitosis than cells that continued to S phase following DNA damage (Fig. 1E). Furthermore, considering that only 3% of G0-routed cells are destined for senescence compared to 34% of S phase–routed cells, cells that route back to G0 after DNA damage maintain greater proliferative potential in the long term, compared to cells that continue on to S phase, which are more likely to permanently exit the cell cycle.
Cells can reroute to G0 at any point in G1 phase
To determine when during G1 phase cells can route back to G0 after DNA damage, we treated asynchronously cycling cells with NCS and binned by cell age at the time of treatment (Fig. 2A). Notably, we found that cells that were between 0 and 2 hours old at the time of DNA damage were much more likely to route to G0 than cells that were 3 to 5 hours old (Fig. 2, B and C, and fig. S2, A and B), despite equal damage in all cells (Fig. 2D). In addition to chronological age, cell age can also be defined using CDK2 activity, which linearly increases over the course of G1 phase. Therefore, we also binned cells by CDK2 activity at the time of treatment and again found that cells with lower CDK2 activity at the time of DNA damage were much more likely to route to G0 than cells with higher CDK2 activity (Fig. 2E and fig. S2, C and D). Thus, contrary to a checkpoint at the end of G1 phase at the G1-S transition, we find that cells are much more likely to exit the cell cycle if they are damaged early in G1 phase rather than late. We observed the same pattern in cells damaged with γ-irradiation and in retinal pigment epithelial (RPE1) cells (fig. S2, E and F). Furthermore, while the probability of cell cycle exit decreases with cell age, we observed cells exiting the cell cycle at all points in G1 phase (Fig. 2B and fig. S2, A to C), indicating that cells can route to G0 at any point in G1 phase and arguing for an intra-G1 checkpoint rather than a G1-S checkpoint.
Fig. 2. Cells can reroute to G0 at any point in G1 phase.
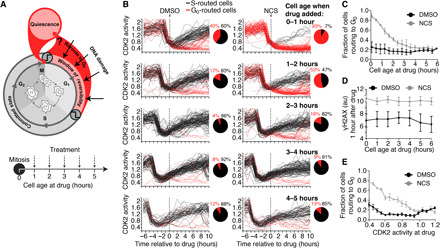
(A) Schematic diagram of the cell cycle and the experimental setup. Asynchronous cells were treated with NCS and binned by cell age (i.e., time since mitosis at the time of treatment), and each cell’s fate was recorded. (B) Single-cell CDK2 activity traces from cells treated with DMSO (left) or NCS (200 ng/ml; right) while in G1 phase, which was defined as CDK2 activity greater than 0.6 and APC/C activity greater than 0.3 at the time of treatment. Cells were binned by cell age at the time of treatment as indicated. Cells were colored black if APC/C activity fell below 0.3, indicating entry into S phase, and cells were colored red if APC/C activity remained above 0.3 6 hours after drug addition and CDK2 activity fell below 0.6, indicating rerouting to G0. (C) Quantification of the percentage of cells that routed to G0 after treatment with DMSO or NCS (200 ng/ml) as a function of cell age at the time of treatment as described in (B). Error bars are SEM from n = 3 independent experiments. (D) Median γH2AX staining 1 hour after treatment with DMSO or NCS (200 ng/ml) binned by cell age at the time of treatment. Error bars are SEM from n = 3 independent experiments. (E) Quantification of the percentage of cells that routed to G0 binned by CDK2 activity at the time of treatment. Error bars are SEM from n = 3 independent experiments.
The decision to route to G0 is largely deterministic
To understand why some cells route to G0 after DNA damage while other cells continue on to S phase (Fig. 3A), we tracked the fates of genetically identical sibling cells. Sibling cells have previously been shown to have highly similar intermitotic times (35, 36), and our own analysis shows that they have highly similar G1 lengths (fig. S3A), suggesting that they have similar amounts and stoichiometries of key cell cycle proteins that drive cell cycle progression. Consistent with this, we found that sibling cells have highly correlated CDK2 activity in G1 phase (fig. S3B) and highly correlated γH2AX levels 1 hour after NCS treatment (fig. S3C), suggesting that the amount of damage and the extent of the repair response are highly similar in sibling cells. We therefore reasoned that discordant fates in response to DNA damage between sibling cells would indicate that stochastic processes underlie the fate response to DNA damage while concordant fates would indicate that deterministic processes are involved. To this end, we measured the CDK2 activity of pairs of sibling cells before and after NCS treatment and found that sibling cells either both commit or both decommit from the cell cycle, as indicated by both sibling cells having a CDK2 activity above or below 0.6 (Fig. 3B). When we expanded this analysis to more than 750 pairs of sibling cells per condition, we found that the fate outcomes between sibling cells was concordant in about 80 to 85% of pairs compared to only 50% for pairs of random cells, 60% for pairs of age-matched random cells, and 55% for pairs of CDK2 activity–matched random cells (Fig. 3, C and D). Thus, if the fate outcome of one sibling cell is known, the fate outcome of the other sibling cell can be correctly predicted 85% of the time. While the fate outcomes are indeed correlated with cellular age (see Fig. 2, B, C, and E), cell age alone, as measured either chronologically or molecularly, is insufficient to predict which fate a cell will choose. Together, our data demonstrate that the decision to route to G0 after DNA damage is largely deterministic and correlates with, but is not determined by, cell age.
Fig. 3. The decision to route to G0 is largely deterministic.
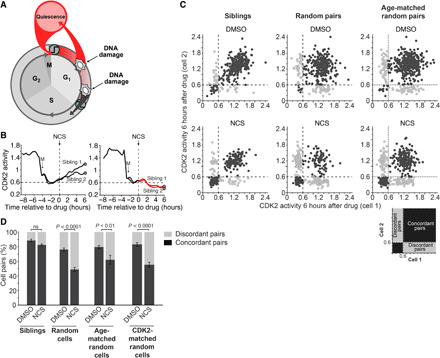
(A) Schematic diagram of the cell cycle showing divergent fate outcomes after DNA damage. (B) CDK2 activity from two pairs of sibling cells treated with NCS (200 ng/ml) at the indicated time. The time of mitosis is indicated as M. (C) Scatterplot of the CDK2 activity from pairs of sibling cells (left), pairs of random cells (middle), or pairs of age-matched random cells (right) 6 hours after treatment with either DMSO or NCS. Cells were considered concordant if both cells in the pair had CDK2 activity either above 0.6 or below 0.6. N > 700 cell pairs per condition. (D) Quantification of pairs of cells from (C) that were either concordant or discordant. Cells were considered age-matched if they were born within 12 min of each other and were considered CDK2-matched if they had CDK2 activity within 0.05 of each other at the time of treatment. Error bars are SEM from n = 3 independent experiments. P values were calculated using Fisher’s exact test. ns, not significant.
Persistent CDK2 activity after DNA damage leads to S phase entry if a cell is within 2 hours of S phase
To identify what molecular mechanism determines whether a cell routes to G0 after DNA damage or enters S phase, we looked at the CDK2 activity of single cells. Notably, we found that the CDK2 activity continued to increase in individual cells for up to 1 hour after DNA damage and did not fall back below the 0.6 threshold for Rb hyperphosphorylation until 2 to 4 hours later (Fig. 4, A to C, and fig. S4, A and B). These observations are again in stark contrast to the classical model of the G1-S checkpoint, where DNA damage signaling is thought to immediately halt cell cycle progression (14, 20, 37). Instead, we observed that after DNA damage, cells continued progressing toward S phase for 2 hours before reversing course and rerouting to G0. This intriguing observation may be best understood by borrowing from the concept of inertia, where an object in motion will remain in motion unless acted upon by a force. Analogously, a cell will continue progressing through the cell cycle program for 2 hours until a “force” generated by the DNA damage response builds to a level sufficient to stop cell cycle progression (Fig. 4D). Thus, it can be conceptualized that cell cycle progression displays a degree of inertia that prevents cells from immediately exiting the cell cycle after DNA damage.
Fig. 4. Persistent CDK2 activity after DNA damage leads to S phase entry if a cell is within 2 hours of S phase.
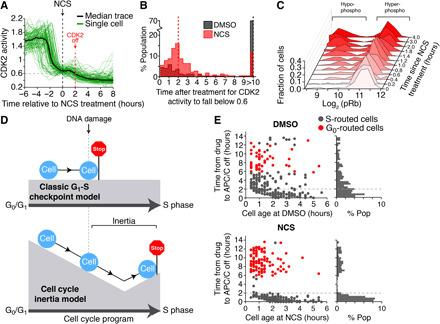
(A) Median and single-cell traces of CDK2 activity for cells treated with NCS during G1 phase. Small vertical dashed line indicates the time when median CDK2 activity falls below 0.6 after NCS treatment. N = 104 cells. (B) Histogram of time when CDK2 activity falls below 0.6 after the indicated treatment. Note that 70% of DMSO-treated cells did not fall below 0.6 during the imaging period. Single-cell data pooled from n = 3 independent experiments. (C) MCF-10A cells were preimaged using time-lapse microscopy to establish cell age and cell cycle phase. Cells were then treated with NCS (200 ng/ml) at various times before fixation. Cells were then immunostained for phospho-Rb (S807/S811). Only cells that were between 0 and 3 hours old at the time of treatment were analyzed. Data are histograms of the single-cell log2 phospho-Rb levels at each time point. Representative histograms from n = 3 independent experiments. (D) Schematic explaining the concept of cell cycle inertia and comparing it to the classical view of the G1-S checkpoint. (E) Scatterplot of single cells comparing cell age (i.e., time since mitosis) at the time of treatment versus the time after treatment when cells inactivate the APC/C. Cells were colored black if APC/C activity fell below 0.3, indicating entry into S phase, and cells were colored red if APC/C activity remained above 0.3 6 hours after drug addition and CDK2 activity fell below 0.6, indicating rerouting to G0. Right: Single-component histogram showing the distribution of the time after treatment when cells inactivate APC/C. Note that almost all cells that continued on to S phase after NCS treatment inactivated the APC/C within 2 hours of treatment.
We considered whether the 2-hour maintenance of CDK2 activity we observed may explain why some cells continue on to S phase despite the presence of DNA damage. We hypothesized that if a cell is within 2 hours of inactivating the APC/C and entering S phase when it receives DNA damage, then its cell cycle inertia will carry it through the G1-S transition and into S phase before the cell has enough time to route back to G0. To this end, we plotted each cell’s age and the time it takes each cell to inactivate the APC/C following DNA damage. We found that the only cells that entered S phase after treatment with NCS were within 2 hours of inactivating the APC/C at the time of damage, irrespective of their age at treatment (Fig. 4D). Cells that routed back to G0 took more than 6 hours to repair the damage, reenter the cell cycle, and eventually inactivate the APC/C after treatment with NCS. Notably, unlike in cells treated with dimethyl sulfoxide (DMSO), we failed to observe any cells that inactivated the APC/C between 2 and 6 hours after treatment with NCS. We repeated this analysis with RPE1 cells and found similar results (fig. S4C). These data indicate that if a cell is within 2 hours of inactivating the APC/C when it received DNA damage, then its uninterrupted CDK2 activity will cause it to pass through the G1-S transition, inactivate the APC/C, and enter S phase, whereas cells greater than 2 hours from S phase are able to reroute to G0. These data demonstrate a nongenetic origin for cells failing to trigger the DNA damage checkpoint in G1 phase and instead show that any cell can fail to trigger the checkpoint depending on how far from S phase a cell is when DNA damage occurs.
Persistence in CDK2 activity is mediated by a 2-hour delay between DNA damage and p21 induction above a threshold
We next aimed to identify the molecular mechanism underlying the observed cell cycle inertia after DNA damage. Given that the CDK inhibitor p21 links the DNA damage signaling network to cell cycle control (Fig. 5A and fig. S5, A and B), we reasoned that the 2-hour delay between DNA damage and CDK2 inhibition should be mediated by a delay between DNA damage and p21 induction. Therefore, we imaged and tracked MCF-10A cells expressing fluorescently tagged p21 from the endogenous CDKN1A locus (9) to investigate the dynamics of p21 induction in response to DNA damage in single cells. We observed, on average, a 1.5-hour delay between when DNA damage was induced and when p21 levels first started to rise above baseline (Fig. 5B and fig. S5C). To determine how much p21 is required to route cells back to G0, we measured both p21 levels and CDK2 activity in single cells. We found that cells needed to accumulate a threshold level of p21 to force cells to reroute to G0 (Fig. 5, C and D). To calculate in an unbiased way what threshold of p21 levels would best separate cells that rerouted to G0 from cells that route to S phase, we tested many different thresholds and plotted the percent of false positives and the percent of true positives for each possible threshold (receiver operating characteristic). The threshold that best separated these two populations was ~30 arbitrary units (au) or threefold over background in our experiments (Fig. 5, E and F). We observed a small proportion of cells in a spontaneous G0 state with low CDK2 activity, consistent with previous reports that described the same population (9, 10). We observed that the p21 levels in these cells also rose above the same threshold we observed in cells that rerouted to G0 after DNA damage (Fig. 5G).
Fig. 5. Persistence in CDK2 activity is mediated by a 2-hour delay between DNA damage and p21 induction above a threshold.
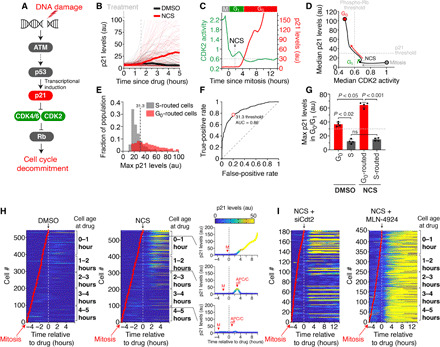
(A) DNA damage signaling diagram. (B) Single-cell p21 levels in MCF-10A cells expressing endogenously tagged mCitrine-p21. Median p21 traces are indicated by the thick lines. Dashed line indicates the median time for p21 induction after NCS. (C) Single-cell p21 levels, measured in arbitrary units (au), and CDK2 activity for a representative cell treated with NCS at the indicated time. The time CDK2 activity falls below 0.6 (G0) is indicated by the horizontal dashed line. (D) Phase plane diagram of the median CDK2 activity versus the median p21 levels. (E) Histogram of single-cell measurements of the maximum p21 levels reached during G0/G1 phase of cells treated with NCS. Data represent more than 1000 single cells in each population from five pooled experiments. Vertical dashed line is the optimal threshold for separating the two populations determined in (F). (F) Receiver operating characteristic of the data in (E). Red dot represents optimal p21 threshold. AUC, area under the curve. (G) Average maximum p21 levels achieved by each single cell during G0/G1 phase. Horizontal line indicates threshold level of p21 identified from (E) and (F). Error bars are SEM from n = 4 independent experiments. P values were obtained from one-way analysis of variance (ANOVA) with multiple comparisons test. (H) Heatmap of single-cell p21 levels after treatment with DMSO or NCS at the indicated time. Right: p21 levels from three representative cells. Time of APC/C inactivation is noted except for the top cell, which did not inactivate APC/C during the imaging period. (I) Heatmap of single-cell p21 levels after treatment with NCS at the indicated time and either Cdt2 siRNA or the pan-cullin inhibitor MLN-4924.
Having determined that p21 must reach a threshold level to inhibit CDK2 and force cells to route to G0, we measured the time it takes cells to induce p21 to the threshold after DNA damage. We found that it takes, on average, 2 hours (fig. S5D), which was slightly more than the average time of 1.5 hours it took to begin inducing p21 above background, but more consistent with the persistence in CDK2 activity we observed earlier (see Fig. 4, A and B).
Consistent with our previous observations that young cells are more likely to route back to G0 than old cells, we found that cells were more likely to induce p21 when they were 0 to 2 hours old than when they were 3 to 4 hours old at the time of DNA damage (Fig. 5H). Cells that were damaged in G1, before S phase, but continued on to S phase either failed to induce p21 above the necessary threshold or failed to induce p21 at all (Fig. 5H). Given that p21 is ubiquitinated and degraded once cells reach S phase by the E3 ubiquitin ligase CRL4Cdt2, we treated cells either with Cdt2 small interfering RNA (siRNA) (fig. S5E) or the pan-cullin inhibitor MLN-4924. We found that inhibition of CRL4Cdt2 by either method resulted in all cells inducing p21 independent of cell age or proximity to APC/C inactivation (Fig. 5I and fig. S5, F to H). These results show that the induction of p21 by itself is not sufficient to cause cell cycle exit, but rather, cells must accumulate and sustain p21 levels to inhibit CDK2 and trigger cell cycle exit (Fig. 6A). Furthermore, they demonstrate that all cells in G1 phase have the ability to induce p21 after DNA damage independent of cell age, but if their cell cycle inertia carries them into S phase where CRL4Cdt2 can degrade p21, then cells will fail to reroute to G0.
Fig. 6. Passage through G1 phase is controlled by p21 induction dynamics and G1 length.
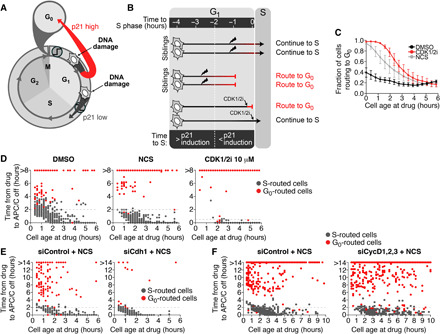
(A) Schematic diagram of the cell cycle. DNA damage induces high p21 accumulation in cells receiving the damage in early G1 but low p21 accumulation in cells receiving the damage in late G1. (B) Schematic diagram of our model of cell cycle inertia. If a damaged cell is more than 2 hours away from S phase, then it will have time to accumulate p21 (red bar) and decommit from the cell cycle. If a damaged cell is less than 2 hours away from S phase, then it will not have enough time to accumulate p21 and will commit to the cell cycle. Direct inhibition of CDK2 using a CDK1/2 inhibitor (CDK1/2i) can immediately force cells to reroute to G0. (C) Quantification of the percentage of cells that route to G0 after the indicated treatment as a function of cell age at the time of treatment. Error bars are SEM from n = 3 independent experiments. (D) Scatterplot of single cells comparing cell age at the time of treatment versus the time after treatment when cells inactivate the APC/C. Cells were treated with DMSO, NCS, or 10 μM CDK1/2 inhibitor. (E and F) Scatterplot of single cells comparing cell age at the time of treatment versus the time after treatment when cells inactivate the APC/C. Cells were treated with NCS plus either control siRNA, Cdh1 siRNA (E), or cyclins D1, D2, and D3 siRNA (F).
Passage through the G1 phase is controlled by p21 induction dynamics and G1 length
Our observations thus far have led us to a revised model of G1 checkpoint control. In this model, the slow induction of p21 after DNA damage allows for a 2-hour persistence in CDK2 activity that prevents cells within 2 hours of S phase when they received DNA damage from exiting the cell cycle and rerouting to G0 (Fig. 6B). To test this model, we simulated the effect of rapid p21 induction using a small-molecule inhibitor of CDK1 and CDK2, which has been shown to directly inhibit CDK2 activity within 15 min (10). Consistent with our model, we found that older cells routed back to G0 with a greater frequency in response to CDK2 inhibition than in response to DNA damage (Fig. 6C). The only cells that entered S phase after treatment with the CDK1/2 inhibitor were those that were within 30 min of APC/C inactivation at the time of treatment (Fig. 6D), which is significantly shorter than the 2-hour window observed with NCS treatment. This indicates that it is indeed the 2-hour delay in p21 induction that mediates cell cycle inertia during G1 phase and leads to ineffective G1 checkpoint control.
As a further test of our model, we altered the overall length of G1 phase by knocking down either Cdh1 (shorter G1 phase; fig. S6, A and B) or cyclins D1, D2, and D3 (longer G1 phase; fig. S6, B and C). Notably, at the population level, we found that a smaller percent of cells were able to route to G0 when G1 length was shorter than control (Fig. 6E and fig. S6D) while a greater percent of cells were able to route to G0 when G1 length was longer than control (Fig. 6F and fig. S6F). However, consistent with slow p21 induction mediating cell cycle inertia, we found that at the single-cell level, the probability of routing to G0 after DNA damage was independent of the overall G1 length (Fig. 6, E and F, and fig. S6, E to G). Rather, we found that cells with either a short or long G1 phase continued into S phase if they received DNA damage within 2 hours of APC/C inactivation and subsequent S phase entry. Thus, the probability that a cell will trigger the G1 checkpoint and route back to G0 is determined by the rate of p21 induction and the proximity of that cell to S phase at the time of damage and not necessarily by the presence of oncogenic mutations.
DISCUSSION
For many decades, the G1-S checkpoint has been viewed as a master regulator of cell cycle progression that rapidly and stringently prevents cells with damaged DNA from transitioning from G1 to S phase (37, 38). These early studies typically assessed the effect of DNA damage on the cell cycle by snapshot or population-level assays that lack temporal resolution and, by their nature, preclude the ability to observe the same cell before and after DNA damage. Studies that identified reduced efficacy of the G1-S checkpoint to arrest cells after DNA damage were unable to quantify the number of cells that still entered S phase despite damage and were moreover unable to identify the point of fate divergence due to lower temporal resolution specifically of G1 time points (25). To overcome these challenges, we used live-cell imaging and single-cell tracking to understand the cell cycle trajectories cells take following DNA damage. We found that cells bifurcate into two subpopulations after DNA damage in G1 phase, which eventually leads to heterogeneous fate outcomes at the single-cell level. Immediately after DNA damage, cells either route back to G0 phase where the damage can be repaired or continue on to S phase where the damage results in permanent cell cycle arrest. We showed that routing back to G0 phase requires cells to induce p21 to a threshold level, which, on average, takes approximately 2 hours. This 2-hour delay means that cells continue to progress through G1 phase and into S phase for 2 hours after DNA damage. This cell cycle inertia results in as many as 60% of cells with DNA damage continuing into S phase, demonstrating that the G1 checkpoint is highly ineffective even in cells without mutations in their checkpoint genes. Our finding that cells have cell cycle inertia provides a new conceptual framework for understanding previous observations of G1-S checkpoint failure.
The concept of cell cycle inertia is most likely not specific to DNA damage or stress signaling pathways. Cell cycle inertia will be observed in cases where the time constant of the inhibitory signaling pathway is longer than the time to transition between phases. An example of this was recently observed in mitogen signaling pathways (39). Removing mitogens during G1 phase resulted in a gradual loss of CDK4/6 activity, with some cells sustaining Rb hyperphosphorylation long enough to make it to S phase (12). It was also recently shown that persistence in CDK4/6 activity in response to stress increases as cells progress through G1 phase (39), which agrees with our findings that entry to S phase after damage is graded in relation to cellular age and may contribute to the inertia that we describe. These multiple observations suggest that cell cycle inertia may be a common feature of cell cycle phase transitions, a hypothesis that is consistent with a “brake model of cell cycle progression” recently proposed by Lemmens and Lindqvist (40). In this model, cell cycle progression is conceptualized by a ball rolling down a mountain with three break modules functioning to slow the ball’s movement. Within this conceptual framework, cell cycle inertia should be observed in each break module. In addition to delayed p21 induction during G1 phase, inertia during other phases could be mediated by additional factors such as mRNA turnover, protein turnover, phosphatase activity, etc.—all of which could play a role in maintaining forward progression through the cell cycle despite the induction of inhibitory signaling pathways.
Our data indicate that all cells in G1 phase are able to initiate activation of the ATM-p53-p21 signaling pathway in response to DNA damage, independently of where they lie in G1 phase. However, if cell cycle inertia carries a cell into S phase, the ubiquitin ligase CRL4Cdt2 degrades p21 and prevents its accumulation, giving the impression that the cell failed to initiate a DNA damage response via p21 in G1 phase. Consequently, failure to fully trigger cell cycle exit only occurs if p21 protein levels are not induced to high enough levels by the time the cell enters S phase. This relatively long time delay between initiating the checkpoint mechanism and fully engaging it results in a transient window of time during G1 phase when cells are able to fully trigger the DNA damage response checkpoint. Therefore, rather than a checkpoint that sits at the G1-to-S phase transition, we find that the checkpoint can be initiated at any point in G1 phase. This mechanism is practical for cells: If the checkpoint occurred in late G1, this would cause younger cells that were damaged to expend more energy on continuing to progress through the cell cycle, rather than allocating their resources to DNA repair as soon as possible. Previous studies have also suggested a G1-S checkpoint located temporally well before the G1-S transition. Our data describe that the mechanism for this checkpoint is primarily mediated by slow p21 induction combined with p21 degradation if a cell enters S phase (24). Thus, the classically defined G1-S checkpoint may be more parsimoniously described as a G1 checkpoint.
Our finding that the effectiveness of the G1 checkpoint is determined at the single-cell level by cell cycle inertia and at the population level by the length of G1 phase has broad implications for cell biology. Given that the molecular determinants of these features are inherent in all cells but variable between different cell types, failure to trigger the G1 checkpoint may be a significant contributor to genomic instability and ultimately oncogenesis. Cell-to-cell and tissue-specific variations in the ratio of p21 induction time to G1 length will give rise to G1 checkpoint control that is more, or less, effective. In the future, by making dynamic measurements, we can form a clearer understanding of why some cell populations have more stringent G1 checkpoint control than others. Such information could be used to predict the efficacy of G1 arrest after treatment with DNA-damaging agents.
MATERIALS AND METHODS
Cell culture
MCF-10A cells [American Type Culture Collection (ATCC), #CRL-10317] were cultured in the following full-growth media: phenol red–free Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Invitrogen) supplemented with 5% horse serum (ATCC, #30-2040), epidermal growth factor (20 ng/ml; PeproTech, #AF-100-15), insulin (10 μg/ml; Sigma-Aldrich, #I1882), hydrocortisone (500 μg/ml; Sigma-Aldrich, #H0888), cholera toxin (100 ng/ml; Sigma-Aldrich, #C8052), and 1% penicillin/streptomycin (Thermo Fisher Scientific, #15-140-163). hTERT-RPE1 human retinal epithelial cells (ATCC, #CRL-4000) were cultured in the following full-growth media: phenol red–free DMEM/F12 (Invitrogen) supplemented with 10% fetal bovine serum (ATCC, #30-2020) and hygromycin B (0.01 mg/ml). Cells were treated with NCS (200 ng/ml; Sigma-Aldrich, #N9162) unless otherwise specified. Media containing NCS was prepared 15 min before treating cells. Incubation was at 37°C and 5% CO2 for all cells. All cells were tested for mycoplasma.
Constructs and stable cell lines
CSII-pEF1a-H2B-mTurquoise, CSII-pEF1a-DHB(aa994-1087)-mVenus, and CSII-pEF1a-mCherry-Geminin(aa1-110) were described previously (10, 29). pLenti-PGK-Neo-PIP-NLS-mVenus was described previously (41) and obtained from Addgene (#118619). CSII-pEF1a-DHB(aa994-1087)-mCherry was cloned from CSII-pEF1a-DHB(aa994-1087)-mVenus swapping out mCherry for mVenus using the Gibson assembly method. MCF-10A cells expressing endogenously tagged mCitrine-p21 were a gift of S. L. Spencer’s laboratory and were described previously (9). Transduced cells were sorted on a BD Biosciences FACSAria Fusion to obtain pure populations expressing the desired fluorescent reporters.
Inhibitors
The inhibitors used in this study were MLN-4924 (3 μM; neddylation inhibitor, Active Biochem, #A-1139), Cdk1/2i III (3 μM; EMD Biosciences, #217714), PD0325901 [MEK1/2 (mitogen-activated protein kinase kinase 1/2) inhibitor, Sigma-Aldrich, #444968], and NCS (Sigma-Aldrich, N9162).
Immunofluorescence
Cells were fixed in 4% paraformaldehyde, washed three times in phosphate-buffered saline (PBS), permeabilized with 0.2% Triton X-100, and stained overnight at 4°C with anti–phospho-Rb (807/811) (Cell Signaling Technology, #8516). Primary antibodies were visualized using a secondary antibody conjugated to Alexa Fluor 647 and imaged with a FarRed filter.
siRNA transfection
MCF-10A cells were transfected using DharmaFECT 1 (Thermo Fisher Scientific) according to the manufacturer’s instructions. The following siRNAs from Dharmacon were used: On-Target plus control siRNA (nontargeting, D-001810-10-05); On-Target plus pooled set of 12 for cyclin D1, D2, and D3 (L-003210-00, L-003211-00, and L-003212-00); On-Target plus pooled set of four for Fzr1 (Cdh1, LQ-015377-00); On-Target plus pooled set of four siRNAs for CDKN1A (p21, LQ-003471-00); and On-Target plus pooled set of four for Cdt2 (L-020543-00) at final concentrations of 20 nM unless noted otherwise. Six hours after transfection, cells were washed with full growth medium, and then imaging was immediately started.
Time-lapse microscopy
Cells were plated >24 hours before imaging in full growth media in a 96-well dish (Ibidi) such that the density would remain subconfluent until the end of the imaging period. Time-lapse imaging was performed in 290-μl full growth media. Images were taken in cyan fluorescent protein, yellow fluorescent protein, and red fluorescent protein channels every 12 min on a Nikon Ti2-E inverted microscope (Nikon) with a 20× 0.45 numerical aperture objective. Total light exposure time was kept under 600 ms for each time point. Cells were imaged in a humidified, 37°C chamber in 5% CO2.
Immunoblotting
Cells were harvested and washed with PBS. Cell pellets were then incubated with whole-cell lysis buffer [50 mM tris (pH 7.4), 200 mM NaCl, 50 mM NaF, 1 mM Na3VO4, 0.5% Triton X-100, and protease inhibitor cocktail] in ice for 30 min. Lysates were centrifuged at high speed (16,000g), and the supernatants were collected. Protein concentration was measured by the Bradford method using bovine serum albumin as a standard. Samples were prepared in SDS sample buffer and resolved in SDS–polyacrylamide gel electrophoresis. Blots were developed by the chemiluminescence method. Antibodies used included anti-p21 (Cell Signaling Technologies, #2947S), anti-Cdh1 (Santa Cruz Biotechnology, DCS-266, SC-56312), anti-Cdt2 (Abcam, ab72264), and anti-vinculin (Sigma-Aldrich, V9131).
Image analysis
All image analyses were performed with custom MATLAB scripts as previously described (29). Briefly, optical illumination bias was empirically derived by sampling background areas across all wells in an imaging session and subsequently used to flatten all images. This enabled measurement and subtraction of a global background for each image. Cells were segmented for their nuclei based on either Hoechst staining (fixed-cell imaging) or H2B-mTurquoise (live-cell imaging). The time of mitosis was measured as the time of anaphase, which the analysis software identified as the time when chromosomes were separated into two objects. For DHB-mVenus measurements, cells were segmented for their cytoplasmic regions by spatially approximating a ring with an inner radius of 2 μm outside of the nuclear mask and an outer radius a maximum of 10 μm outside of the nuclear mask. Regions within 10 μm of another nucleus were excluded. Nuclear immunofluorescence, nuclear DHB-mVenus, nuclear DHB-mCherry, nuclear mCitrine-p21, and mCherry-Geminin signals were calculated as median nuclear intensity, as these signals were often excluded from the nucleoli. Cytoplasmic DHB-mVenus or DHB-mCherry signals were calculated as the median intensity within the cytoplasmic ring, excluding pixel intensities indistinguishable from background.
Identification of G1 cells
To identify G1 phase cells that had crossed the Restriction Point, we tracked cells that were treated with a small-molecule inhibitor of MEK1/2 for 16 hours to block mitogen signaling and classified each cell as either cycling or quiescent (fig. S1A). Using this as a truth set, we applied receiver operator characteristic analysis to determine a threshold level of CDK2 activity, measured at the time of the MEK1/2 inhibitor treatment, that optimally discriminated cycling cells (i.e., cells post–Restriction Point) (fig. S1, B to D). Similar to previous reports (10), we found that a CDK2 activity threshold of 0.6 minimized the false-positive rate and relative cost (fig. S1D). As another test of this threshold, we found that all cells with a CDK2 activity level greater than 0.6 also had hyperphosphorylated Rb (fig. S1E). To identify cells that had not yet entered S phase, we tracked cells that were treated with a small-molecule CDK1/2 inhibitor, which was previously shown to prevent APC/C inactivation (31), for 6 hours and classified them as either APC/C on or off (fig. S1F). Using similar methods as described above, we found that an APC/C activity threshold of 0.3 or 30% of maximal activity minimized the false-positive rate and relative cost (fig. S1, G to I). Thus, in this study, we considered single cells to be past the Restriction Point and in G1 phase if they have a CDK2 activity greater than 0.6 and an APC/C activity greater than 0.3.
Statistical analysis
Statistical analyses were performed in MATLAB (MathWorks), R, and Prism (GraphPad Software). Competing risks regression was performed as previously described using mets, timereg, and comp.risk package in R (42). Permutation tests were used to quantify the similarity in sibling G1 length. We calculated the mean difference in G1 length between sibling cell pairs and compared this with the mean difference in G1 length between randomly sampled cell pairs using 1000 random permutations of the dataset that were generated by randomly sampling cell pairs (with replacement). A two-tailed, unpaired Student’s t test (α = 0.05) was used to determine whether the observed mean difference in sibling G1 length was significantly different to that of random permutations. Further statistical details of experiments can be found in the figure legends.
Supplementary Material
Acknowledgments
We thank J. Luo and S. H. Yuspa for helpful discussions; K. Cappell, A. Jaimovich, S. L. Spencer, and M. Chung for critical reading of the manuscript; S. L. Spencer and A. Loewer for providing cell lines; S.-M. Jang for providing siRNA and antibodies; and the Flow Cytometry Core Facility of the Center for Cancer Research at the National Cancer Institute (NCI) for technical support. Funding: This research was supported by the Intramural Research Program of the NIH (grant ZIA BC 011830 to S.D.C.). Author contributions: J.F.N. and J.A.C. planned and executed most experiments, evaluated and interpreted results, and wrote the manuscript. M.M.A. performed the experiments with RPE1 cells. D.P. performed the validation of siRNA-mediated knockdown via Western blotting. S.D.C. conceived the study, planned experiments, interpreted the results, wrote the manuscript, and oversaw the research project. All authors contributed to the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. All unique/stable reagents generated in this study are available from the lead contact with a completed materials transfer agreement. The MATLAB code for the cell tracking and image analysis pipeline is available at https://github.com/scappell/Cell_tracking. Additional modified scripts for analysis of specific experiments are available from the corresponding author upon reasonable request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/3/eabe3882/DC1
REFERENCES AND NOTES
- 1.Hartwell L. H., Weinert T. A., Checkpoints: Controls that ensure the order of cell cycle events. Science 246, 629–634 (1989). [DOI] [PubMed] [Google Scholar]
- 2.Pardee A. B., A restriction point for control of normal animal cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 71, 1286–1290 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ekholm S. V., Zickert P., Reed S. I., Zetterberg A., Accumulation of cyclin E is not a prerequisite for passage through the restriction point. Mol. Cell. Biol. 21, 3256–3265 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zetterberg A., Larsson O., Kinetic analysis of regulatory events in G1 leading to proliferation or quiescence of Swiss 3T3 cells. Proc. Natl. Acad. Sci. U.S.A. 82, 5365–5369 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnum K. J., O’Connell M. J., Cell cycle regulation by checkpoints. Methods Mol. Biol. 1170, 29–40 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Medema R. H., Macůrek L., Checkpoint control and cancer. Oncogene 31, 2601–2613 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Bertoli C., Skotheim J. M., de Bruin R. A. M., Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 14, 518–528 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coller H. A., The essence of quiescence. Science 334, 1074–1075 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moser J., Miller I., Carter D., Spencer S. L., Control of the restriction point by Rb and p21. Proc. Natl. Acad. Sci. U.S.A. 115, E8219–E8227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spencer S. L., Cappell S. D., Tsai F.-C., Overton K. W., Wang C. L., Meyer T., The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 155, 369–383 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yao G., Lee T. J., Mori S., Nevins J. R., You L., A bistable Rb–E2F switch underlies the restriction point. Nat. Cell Biol. 10, 476–482 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Chung M., Liu C., Yang H. W., Köberlin M. S., Cappell S. D., Meyer T., Transient hysteresis in CDK4/6 activity underlies passage of the restriction point in G1. Mol. Cell 76, 562–573.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maréchal A., Zou L., DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 5, a012716 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mailand N., Falck J., Lukas C., Syljuåsen R. G., Welcker M., Bartek J., Lukas J., Rapid destruction of human Cdc25A in response to DNA damage. Science 288, 1425–1429 (2000). [DOI] [PubMed] [Google Scholar]
- 15.He G., Siddik Z. H., Huang Z., Wang R., Koomen J., Kobayashi R., Khokhar A. R., Kuang J., Induction of p21 by p53 following DNA damage inhibits both Cdk4 and Cdk2 activities. Oncogene 24, 2929–2943 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Agami R., Bernards R., Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell 102, 55–66 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Lin D. I., Barbash O., Kumar K. G. S., Weber J. D., Harper J. W., Klein-Szanto A. J. P., Rustgi A., Fuchs S. Y., Diehl J. A., Phosphorylation-dependent ubiquitination of cyclin D1 by the SCFFBX4-alphaB crystallin complex. Mol. Cell 24, 355–366 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okabe H., Lee S.-H., Phuchareon J., Albertson D. G., McCormick F., Tetsu O., A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PLOS ONE 1, e128 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santra M. K., Wajapeyee N., Green M. R., F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature 459, 722–725 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaltiel I. A., Krenning L., Bruinsma W., Medema R. H., The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 128, 607–620 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Paulovich A. G., Toczyski D. P., Hartwell L. H., When checkpoints fail. Cell 88, 315–321 (1997). [DOI] [PubMed] [Google Scholar]
- 22.Hartwell L. H., Kastan M. B., Cell cycle control and cancer. Science 266, 1821–1828 (1994). [DOI] [PubMed] [Google Scholar]
- 23.Heldt F. S., Barr A. R., Cooper S., Bakal C., Novák B., A comprehensive model for the proliferation–quiescence decision in response to endogenous DNA damage in human cells. Proc. Natl. Acad. Sci. U.S.A. 115, 2532–2537 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chao H. X., Poovey C. E., Privette A. A., Grant G. D., Chao H. Y., Cook J. G., Purvis J. E., Orchestration of DNA damage checkpoint dynamics across the human cell cycle. Cell Syst 5, 445–459.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deckbar D., Stiff T., Koch B., Reis C., Löbrich M., Jeggo P. A., The limitations of the G1-S checkpoint. Cancer Res. 70, 4412–4421 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Sandell L. L., Zakian V. A., Loss of a yeast telomere: Arrest, recovery, and chromosome loss. Cell 75, 729–739 (1993). [DOI] [PubMed] [Google Scholar]
- 27.Branzei D., Foiani M., The Rad53 signal transduction pathway: Replication fork stabilization, DNA repair, and adaptation. Exp. Cell Res. 312, 2654–2659 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Hahn A. T., Jones J. T., Meyer T., Quantitative analysis of cell cycle phase durations and PC12 differentiation using fluorescent biosensors. Cell Cycle 8, 1044–1052 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cappell S. D., Chung M., Jaimovich A., Spencer S. L., Meyer T., Irreversible APCCdh1 inactivation underlies the point of no return for cell-cycle entry. Cell 166, 167–180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakaue-Sawano A., Kurokawa H., Morimura T., Hanyu A., Hama H., Osawa H., Kashiwagi S., Fukami K., Miyata T., Miyoshi H., Imamura T., Ogawa M., Masai H., Miyawaki A., Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Cappell S. D., Mark K. G., Garbett D., Pack L. R., Rape M., Meyer T., Emi1 switches from being a substrate to an inhibitor of APC/CCDH1 to start the cell cycle. Nature 558, 313–317 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Batchelor E., Mock C. S., Bhan I., Loewer A., Lahav G., Recurrent initiation: A mechanism for triggering p53 pulses in response to DNA damage. Mol. Cell 30, 277–289 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johmura Y., Shimada M., Misaki T., Naiki-Ito A., Miyoshi H., Motoyama N., Ohtani N., Hara E., Nakamura M., Morita A., Takahashi S., Nakanishi M., Necessary and sufficient role for a mitosis skip in senescence induction. Mol. Cell 55, 73–84 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Wiebusch L., Hagemeier C., p53- and p21-dependent premature APC/C-Cdh1 activation in G2 is part of the long-term response to genotoxic stress. Oncogene 29, 3477–3489 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Chakrabarti S., Paek A. L., Reyes J., Lasick K. A., Lahav G., Michor F., Hidden heterogeneity and circadian-controlled cell fate inferred from single cell lineages. Nat. Commun. 9, 5372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sandler O., Mizrahi S. P., Weiss N., Agam O., Simon I., Balaban N. Q., Lineage correlations of single cell division time as a probe of cell-cycle dynamics. Nature 519, 468–471 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Bartek J., Lukas J., Pathways governing G1/S transition and their response to DNA damage. FEBS Lett. 490, 117–122 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Nyberg K. A., Michelson R. J., Putnam C. W., Weinert T. A., Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 36, 617–656 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Yang H. W., Cappell S. D., Jaimovich A., Liu C., Chung M., Daigh L. H., Pack L. R., Fan Y., Regot S., Covert M., Meyer T., Stress-mediated exit to quiescence restricted by increasing persistence in CDK4/6 activation. eLife 9, e44571 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lemmens B., Lindqvist A., DNA replication and mitotic entry: A brake model for cell cycle progression. J. Cell Biol. 218, 3892–3902 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grant G. D., Kedziora K. M., Limas J. C., Cook J. G., Purvis J. E., Accurate delineation of cell cycle phase transitions in living cells with PIP-FUCCI. Cell Cycle 17, 2496–2516 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cornwell J. A., Hallett R. M., der Mauer S. A., Motazedian A., Schroeder T., Draper J. S., Harvey R. P., Nordon R. E., Quantifying intrinsic and extrinsic control of single-cell fates in cancer and stem/progenitor cell pedigrees with competing risks analysis. Sci. Rep. 6, 27100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/3/eabe3882/DC1