

The EGR1 transcription factor blunts macrophage activation by coordinating epigenetic repression of inflammatory enhancers.
Abstract
Monocytes and monocyte-derived macrophages originate through a multistep differentiation process. First, hematopoietic stem cells generate lineage-restricted progenitors that eventually develop into peripheral, postmitotic monocytes. Second, blood-circulating monocytes undergo differentiation into macrophages, which are specialized phagocytic cells capable of tissue infiltration. While monocytes mediate some level of inflammation and cell toxicity, macrophages boast the widest set of defense mechanisms against pathogens and elicit robust inflammatory responses. Here, we analyze the molecular determinants of monocytic and macrophagic commitment by profiling the EGR1 transcription factor. EGR1 is essential for monopoiesis and binds enhancers that regulate monocytic developmental genes such as CSF1R. However, differentiating macrophages present a very different EGR1 binding pattern. We identify novel binding sites of EGR1 at a large set of inflammatory enhancers, even in the absence of its binding motif. We show that EGR1 repressive activity results in suppression of inflammatory genes and is mediated by the NuRD corepressor complex.
INTRODUCTION
Monocytes are postmitotic, mature myeloid cells that are generated through a stepwise differentiation process, beginning from CD34+ hematopoietic stem cells (HSCs). Monocytic commitment is orchestrated by lineage-determining transcription factors such as PU.1, CEBPA, and CEBPB (1–8). As in most developmental processes, transcription of protein-coding genes that specify and define cell identity is controlled by neighboring enhancer elements (1, 9–11). Upon commitment, a specific set of enhancers is coordinately activated to morph the transcriptome of the originating stem cell into that of a mature monocyte. We recently demonstrated that a critical set of monocytic enhancers are activated with the support of the early growth response-1 (EGR1) transcription factor (12). EGR1 and its paralog EGR2 have been previously implicated in promoting monocytic/macrophagic differentiation in human cell lines and primary myeloid precursors (13–16), but their function has been elusive. Our work demonstrated that pivotal lineage-specific enhancers, such as the FMS-intronic regulatory element (FIRE) element in the colony-stimulating factor 1 receptor (CSF1R) gene, rely on an EGR1-dependent axis for their activation (12).
Differentiated monocytes are short-lived cells that retain some ability to infiltrate tissues and secrete inflammatory cytokines and are predominantly found in the circulatory system, from where they can easily spread to sites of infection and inflammation. Monocytes can undergo an elaborate developmental program that generates macrophages, which are phagocytic cells that effectively promote inflammation and elicit an adaptive immune response via antigen presentation. In vivo, macrophagic differentiation occurs when monocytes begin infiltrating solid tissues. In vitro, blood- or bone marrow–derived monocytes can be differentiated into macrophages by administering granulocyte-macrophage CSF (GM-CSF) over the course of 5 to 7 days (17, 18). While the molecular underpinnings of monocyte-to-macrophage differentiation are not well defined, it is believed that this developmental step requires the same pool of transcription factors that govern earlier monocytic development (2, 5, 6). Notably, PU.1, CEBPA/B and activating protein 1 (AP1) are playing a major role in the process (4). In particular, PU.1 is believed to act as a priming, or pioneering, factor that allows additional cooperative transcription factor (TF) binding to occur and determine lineage fate (2, 5, 19). PU.1 also primes inflammatory enhancers in mature macrophages (19, 20). Here, we set out to systematically analyze the role of EGR1 in mono/macrophagic development in primary human cells. Strikingly, we find that EGR1 has very distinct chromatin-binding patterns in late-differentiating macrophages as opposed to monocytic-committed HSPCs, suggesting that the zinc finger TF may have distinct functional roles in these processes. We find that a large share of EGR1 target regions in macrophages are enhancers associated to the inflammatory response. Our data further suggest that EGR1 associates with the nucleosome remodeling and deacetylation (NuRD) chromatin remodeler and represses inflammatory enhancers in developing and mature macrophages, blunting the immune response. Together, our analysis supports a fundamental role for EGR1 in monocytic commitment and highlights an expanded repressive role for this transcription factor in macrophages.
RESULTS
EGR1 profiling in monocytic and macrophagic differentiation
We recently showed that EGR1 props enhancer activation during human monocyte differentiation via interaction with its cofactor NGFI-A Binding Protein 2 (NAB2) and the integrator protein complex (12). A recent study (16) suggested that EGR proteins (specifically EGR2) may also function in human monocyte-to-macrophage differentiation based on the evidence that this transcription factor is subjected to fine-tuned regulation during the differentiation of circulating monocytes into macrophages. To further investigate EGR1 contribution to macrophage differentiation, we analyzed Assay for Transposase-Accessible Chromatin (ATAC) sequencing data on monocytes and differentiated macrophages (day 5) and computed sequence-based motif analysis on the set of regions representing the top 5000 macrophage-specific peaks, ranking them based on the adjusted P value. This analysis retrieved an EGR1/EGR2-like consensus (GTGGGA/TG) as the second most enriched binding motif (e = 3.8 × 10−96), following the master regulator of hematopoiesis PU.1 (Fig. 1A and file S1) and consistent with previous reports (16) including our analysis of HL-60 cells (12).
Fig. 1. Late up-regulation of EGR1 during macrophage differentiation results in EGR1 binding at distal genomic regions.
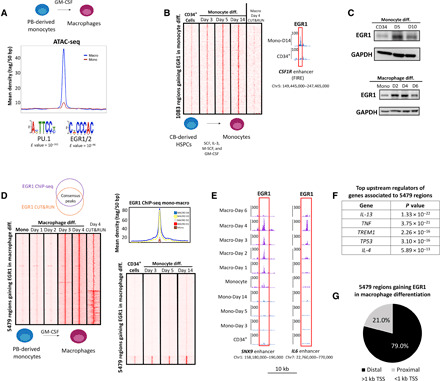
(A) ATAC-seq profile of top 5000 macrophage-specific peaks (ranked on the basis of adjusted P value) shows increased accessibility in macrophages after treatment with GM-CSF to promote differentiation from peripheral blood (PB)–derived monocytes. Motif analysis at these regions shows PU.1- and EGR1/EGR2-binding motifs as the most significantly enriched. (B) CUT&RUN heatmap shows that 1083 genomic sites gain EGR1 binding during monocyte differentiation from bone marrow–derived HSPCs. These sites include the FIRE enhancer of the CSF1R gene. (C) Immunoblot data reveal that EGR1 protein levels are up-regulated early during monocyte differentiation from HSPCs. Conversely, EGR1 is up-regulated after 3+ days of macrophage differentiation and quickly down-regulated past day 4. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (D) Genomic data support a late up-regulation of EGR1 during macrophage differentiation. On the basis of ChIP-seq– and CUT&RUN-replicated experiments, 5479 genomic regions gain EGR1 binding during macrophage differentiation. These 5479 regions are replicated across time ponts (day 3 and day 4) and between methodologies (ChIP-seq and CUT&RUN). Moreover, the 5479 regions specific to the macrophage lineage show limited overlap by EGR1 during monocyte differentiation from CD34+ cells. (E) CUT&RUN genome browser tracks show examples of enhancer regions occupied by EGR1 through macrophage differentiation. Shown are enhancers associated with SNX9 and the cytokine IL-6. (F) Ingenuity Pathway Analysis shows that genes associated to EGR1-gaining regions share upstream regulators involved in immune response pathways. (G) The very large majority of EGR1-gaining regions are distal (i.e., >1 kb from transcription start sites), suggesting that they are putative enhancers. All heatmaps displayed are from one replicate and are normalized by sequencing depth.
To delve into the function of EGR1 throughout the mono-macrophagic lineage, we set out to profile its genome-wide dynamics in primary human cells. First, we took advantage of CUT&RUN, which allows profiling of transcription factor binding with low cell numbers, to assess EGR1 recruitment over the course of a 2-week differentiation of cord blood–derived CD34+ cells (100,000 cells per time point). We assessed the progression of monocytic commitment by monitoring CD14 levels (fig. S1A). We and others showed that EGR1 is required by human stem and progenitor cells to form monocytic colonies in colony-forming unit (CFU) assays (12). Our data shows that EGR1 binds to a set of 1083 sites that are predominant enhancers and include the CSF1R FIRE element (Fig. 1B). EGR1 is likely required to prompt activation of most of these enhancer elements, as also supported by motif analysis retrieving EGR1 as the second most enriched motif after PU.1. Next, we profiled EGR1 binding in circulating monocytes differentiated to macrophages (cells were treated with GM-CSF over the course of 4 days; Fig. 1, B and C). While EGR1 is robustly activated during monocytic development (Fig. 1C), terminally differentiated monocytes show reduced levels of the TF by immunoblot (Fig. 1C and fig. S2) and lack of binding as evidenced by chromatin immunoprecipitation sequencing (ChIP-seq) and CUT&RUN (Fig. 1, D and E). EGR1 is significantly up-regulated after day 2 of macrophage development and recruited to thousands of genomic sites (Fig. 1D). We intersected ChIP-seq and CUT&RUN data at day 4 of differentiation to identify a high-confidence set of 5479 EGR1-binding sites, replicated across different techniques and time points (Fig. 1D). Strikingly, EGR1 binding in macrophages displays very limited overlap with the EGR1 sites in developing monocytes (Fig. 1, B and D), suggesting a different functional role for EGR1 in macrophages. We validated macrophagic commitment of monocytes with flow cytometry and excluded a dendritic cell bias in our protocol (fig. S1B).
Next, we linked each of the 5479 EGR1-gaining regions to the nearest gene (proximity to the closest transcription start site). We subjected EGR1-associated genes to Ingenuity Pathway Analysis and retrieved some of the best-known effectors/modulators of macrophage function as the top upstream regulators [e.g., interleukin-13 (IL-13), tumor necrosis factor (TNF), and IL-4; Fig. 1F]. We further characterized the 5479 regions based on distance from the closest transcription start site (TSS): Significantly, ~4300 of these sites (79.0%) are distal (i.e., distance from TSS > 1 kb; Fig. 1G), likely enhancer elements. Together, these data highlight a prominent role of EGR1 throughout the entire mono-macrophagic lineage and suggest that, in developing macrophages, EGR1 binds a large set of enhancer elements distinct from those bound at earlier developmental stages.
EGR1 binding at distal enhancers correlates with both transcriptional activation and repression
To delve into the scope of EGR1 binding at the ~4300 putative enhancer elements during macrophage differentiation, we leveraged our ATAC-seq data and compared them with publicly available ChIP-seq data for the active enhancer marker H3K27ac, generated in monocytes and GM-CSF–differentiated macrophages (16). This analysis revealed that the 4300 EGR1-bound distal sites can be clustered into three distinct groups: (i) activated sites, a group of 1693 regions poorly acetylated in monocytes, which gain significant acetylation and accessibility in macrophages following EGR1 binding (Fig. 2A); (ii) repressed sites, a group of 1,670 sites that are highly acetylated and highly accessible in monocytes but are repressed in developing macrophages following EGR1 binding (Fig. 2B); and (iii) unresponsive sites, a small set of 902 regions that appear inactive (i.e., not acetylated) either in monocytes or in macrophages and show unchanged accessibility before and after EGR1 binding (Fig. 2C). In all the shown comparisons, the overlap between EGR1 peaks and H3K27ac peaks is significantly higher than expected by chance (Fisher’s exact test, P < 2.2 × 10−16). The repressed, activated and unresponsive site clusters were obtained by performing a k-means clustering on the ~4300 enhancer elements. For differential H3K27ac and ATAC-seq analysis, we performed Wilcoxon’s rank sum test as done with our ChIP-seq data.
Fig. 2. EGR1 binding correlates with both enhancer activation and enhancer repression.
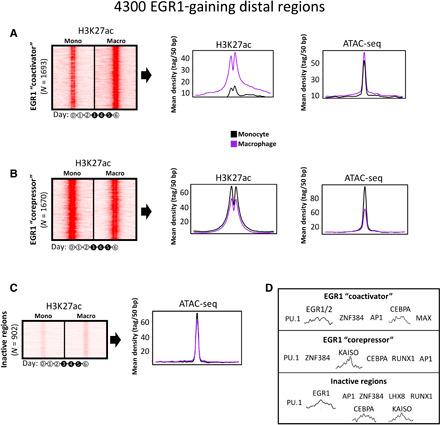
ChIP-seq for H3K27ac (21) and ATAC-seq data (this study) suggest that the EGR1-gaining distal regions can be divided into three main clusters: (A) regions not active in monocytes that gain acetylation and accessibility following EGR1 binding (ACTIVATED cluster), (B) regions active in monocytes that lose acetylation and accessibility following EGR1 binding (REPRESSED cluster), and (C) a small cluster of 900 regions not acetylated and with accessibility not affected by EGR1 binding. (D) Sequence-based motif analysis for the three clusters of EGR1-gaining distal regions. The activated cluster is enriched for the EGR1/2 motif, while the same motif is not present in the repressed cluster, suggesting that EGR1 may be recruited at these regions by other cofactors. All heatmaps depict one replicate per condition and are normalized by sequencing depth across conditions.
We searched these three clusters for motif enrichment and determined that the EGR1 motif was significantly enriched (e value = 10−32) in the activated site cluster and across the unresponsive sites, while it was not retrieved in the repressed site cluster (Fig. 2D and table S1). This latter cluster was the sole one enriched for motifs of transcriptional repressors that may cooperate to recruit EGR1 (i.e., Kaiso and ZNF384).
Collectively, these data suggest that EGR1 binding potentially underlies transcriptional outcomes at enhancer sites during macrophage differentiation. On one hand, EGR1 supports transcriptional activation via direct binding to its DNA motif, as previously indicated for developmental enhancers such as the CSF1R FIRE element. On the other hand, EGR1 seems to act as a corepressor TF at a different set of myeloid enhancers (enriched for key macrophage factors PU.1, AP1, and CEBPA), albeit recruited in the absence of a strong DNA motif.
EGR1 is required to repress enhancers in differentiating macrophages
Our data suggest that EGR1 binding correlates with both epigenetic activation and repression of enhancers. To determine whether EGR1 is functionally required for any of these transitions in developing macrophages, we transduced primary monocytes with lentiviral short hairpin RNAs against EGR1 (Fig. 3A) before stimulation with GM-CSF. Next, we performed ChIP-seq for H3K27ac and RNA sequencing (RNA-seq) on terminally differentiated macrophages (day 5) to assess the consequences of EGR1 depletion. Unlike the depletion of EGR1 from primary HSPCs, which blocks the development of monocytic colonies (12), the knockdown of EGR1 in monocytes did not prevent their differentiation into macrophages. At the genomic scale, EGR1 knockdown did not affect acetylation of enhancers bearing a coactivator signature (Fig. 3B). Conversely, the knockdown of EGR1 resulted in significant increase of acetylation at enhancers dynamically repressed during development (Wilcoxon’s rank sum test, P = 9.4 × 10−7; Fig. 3B and tables S2 to S3).
Fig. 3. EGR1 down-regulates enhancer activity and gene expression during macrophage differentiation.
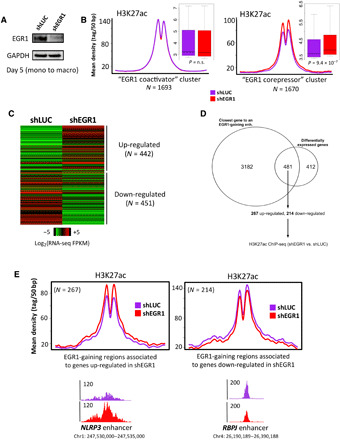
(A) Immunoblot confirms depletion of EGR1 protein levels upon treatment with short hairpin RNA (shRNA). (B) Average profiles and boxplots of ChIP-seq data for H3K27ac suggest that EGR1 depletion only affects acetylation in regions of the “repressed site” cluster. Loss of EGR1 produces increase of acetylation in these regions. P values were determined via Wilcoxon’s rank-sum test. n.s., not significant. (C) RNA-seq conducted in shEGR1 condition shows that depletion of EGR1 results in more than 900 differentially expressed genes (FDR < 5%). (D) 481 of the 893 genes differentially expressed upon EGR1-knockdown (EGR1-KD) are also the closest gene to an EGR1-gaining enhancer. FPKM, fragments per kilobase of exon per million fragments mapped. (E) Screening of H3K27ac levels in the EGR1-gaining enhancers associated to the 481 genes reveals that the enhancers associated to the up-regulated genes significantly gain acetylation upon EGR1-KD (Fisher’s exact test, P < 2.2 × 10−16).
Next, we interrogated the macrophage transcriptome to assess the transcriptional fallout of EGR1 loss. RNA-seq analysis resulted in 893 differentially expressed genes [false discovery rate (FDR) < 0.05], of which 442 up-regulated and 451 down-regulated (Fig. 3C and table S4). Four hundred eighty-one of 893 (48.1%) of the differentially expressed genes appear to be a direct target of EGR1 (they are the closest gene to an EGR1-binding region), this fraction being significantly higher than expected by chance (Fisher’s exact test, P < 2.2 × 10−16; Fig. 3D). Gene Ontology analysis showed that genes implicated in macrophage phagocytosis were the second most enriched pathway after cell cycle control/replication (file S2). EGR1 enhancers that are associated to genes up-regulated after EGR1 depletion also show an increase in H3K27ac levels (Wilcoxon’s rank sum test P = 6.3 × 10−4; Fig. 3E), whereas changes of H3K27ac occurring at regions associated to downregulated genes are not significant (Fig. 3E).
In summary, these data suggest that EGR1 is required for repression of ~1600 selected enhancers during macrophage differentiation. At these enhancers, EGR1 is not directly recruited by its DNA motif. Conversely, EGR1 seems dispensable for the activation of a second set of enhancers where it is recruited in a motif-dependent manner.
Restraining of inflammatory enhancers is mediated by the NuRD corepressor complex
While EGR1 is best characterized as a transcriptional activator, little is known about its repressive activity. To investigate how EGR1 restrains inflammatory enhancers, we carried out immunoprecipitation (IP) followed by mass spectrometry. These experiments were conducted in HL60 and human embryonic kidney (HEK) 293T cell lines using either an antibody targeting endogenous EGR1 (HL-60) or a FLAG antibody to target transduced flagged EGR1 (HEK293T). In both approaches, EGR1 coeluted with all subunits on the NuRD complex (Fig. 4A). NuRD is a ubiquitously expressed complex that carries histone lysine deacetylation (HDAC1/2) and chromodomain helicase DNA binding protein 4 (CHD4).
Fig. 4. EGR1 interacts with the NuRD chromatin remodeler complex.
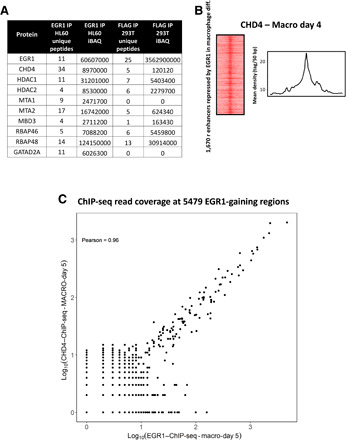
(A) Mass spectrometry data in both HL-60 and HEK293T cells show that EGR1 interacts with all NuRD subunits. (B) ChIP-seq data for NuRD’s catalytic subunit CHD4 support recruitment of NuRD at EGR1-binding sites at the repressed inflammatory enhancers. (C) Plot of Pearson correlation between CHD4 and EGR1 ChIP-seq read coverage at the 5479 EGR1-gaining regions in day 5 macrophages. A Pearson’s r of 0.96 indicates a high correlation of CHD4 and EGR1 binding at these sites (P = 2.2 × 10−16).
To assess whether EGR1 and NuRD functionally associate at chromatin in macrophages, we performed CHD4 ChIP-seq at day 4 of differentiation (peak of EGR1 expression and genome-wide binding) and observed broad overlap of the EGR1 and CHD4 signals at 1600 repressed inflammatory enhancers (Fig. 4B). Notably, EGR1 and CHD4 showed significant correlation even across the larger set of 5479 EGR1-binding regions (Pearson coefficient = 0.96; P = 2.2 × 10−16; Fig. 4C). In summary, our results suggest that EGR1 likely mediates the recruitment of NuRD at hundreds of inflammatory enhancers to retrain their activity during macrophage development.
EGR1 mitigates inflammatory enhancers and blunts the inflammatory response of macrophages
Our data show that EGR1 is linked to transcriptional repression of a set of ~1600 enhancers during a select window of macrophage development (days 3 to 4). We set out to determine the biological implications of EGR1 repressive activity. As previously noted, (Fig. 1E), genes associated to EGR1-binding regions during monocyte-to-macrophage differentiation are enriched for functions related to macrophage activation and inflammatory response. To determine whether the set of ~1600 repressed enhancers are implicated in macrophage activation, we mined publicly available ATAC-seq datasets (21) of lipopolysaccharide (LPS)–stimulated cells. We found that EGR1-repressed enhancers increase their accessibility in mature macrophages upon LPS activation (Fig. 5A). Next, we gauged the effect of sustained EGR1 expression on these cis-regulatory elements. EGR1 protein levels are rapidly induced by LPS induction (fig. S2). We forced expression of exogenous EGR1 in mature macrophages (Fig. 5B, flagEGR1) and performed ChIP–quantitative polymerase chain reaction (qPCR) analysis at selected candidate enhancers. H3K27ac levels decrease markedly at all enhancers upon LPS stimulation, suggesting that EGR1 overexpression prevents activation of inflammatory enhancers (Fig. 5C). To assess whether impaired enhancer activity results in bona fide attenuation of the inflammatory response, we performed RNA-seq on macrophages with and without ectopic EGR1 expression, before and after LPS treatment. We focused our analysis on a set of genes previously identified as bona fide LPS response genes in human macrophages (21). In particular, we identified 270 genes that were responsive to LPS in our system (fold change > 1.5; table S5) and were found in close proximity to enhancers bound by EGR1 during macrophage differentiation. Notably, our data demonstrate that EGR1 overexpression impairs the up-regulation of inflammatory genes upon LPS treatment (Wilcoxon’s rank sum test, P = 6.7 × 10−14; Fig. 5D). We extended our analysis genome wide and found that EGR1, in primary mature macrophages, coordinates a broad repressive effort, centered around inflammatory genes such as interferon-γ targets (Fig. 5E and file S3). Last, we sought to determine the extent to which EGR1-directed enhancer-repression blunts macrophage activation by examining cytokine release and immunophenotype of EGR1-transduced cells. We infected macrophages (4 days after differentiation) with lentivirus carrying exogenous EGR1 and stimulated cells with LPS for 24 hours before collecting their supernatant. Levels of TNFα and IL-12 were significantly reduced as compared to control cells (infected with the backbone vector), and IL-6 levels were moderately reduced (Fig. 6A). Cytokine levels secreted before LPS induction were comparable among wild-type and flagEGR1 samples, suggesting that EGR1-dependent repression dynamically antagonizes enhancer activation as opposed to preemptively shutting off the locus (Fig. 6A). We further assessed the activation status of macrophages by flow cytometry and found that surface expression of CD86 ligand was abolished in flagEGR1 cells (Fig. 6B). To further segregate between immunostimulatory or immunosuppressive abilities of EGR1, we probed the status of the programmed cell death protein 1 (PD-1) and CTLA4 checkpoints. A double-positive CD80/CD274 macrophage population was markedly enriched in flagEGR1 cells, further suggesting that EGR1 enacts a broad anti-inflammatory response (Fig. 6C).
Fig. 5. EGR1 represses activity of inflammatory enhancers and their target genes in mature macrophages.
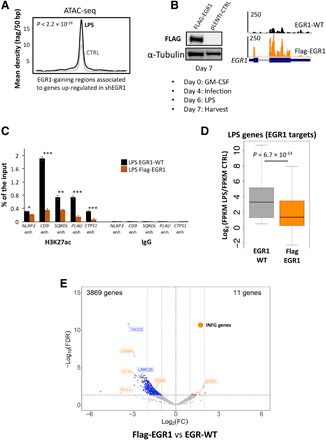
(A) ATAC-seq data (21) show that enhancers repressed by EGR1 during macrophage differentiation increase their accessibility upon mature macrophage activation with LPS. (B) Immunoblot and RNA-seq display EGR1 overexpression obtained by infecting macrophages with a FLAG-EGR1 pLenti vector after 4 days of differentiation. (C) ChIP-qPCR for H3K27ac: In condition of EGR1 overexpression and after LPS treatment, EGR1-gaining enhancers associated to inflammatory genes are significantly less acetylated than in the EGR1–wild-type (WT) condition (*P < 0.05, **P < 0.005, and ***P < 0.0005). (D) From the publicly available list of human LPS-responsive genes (21), we selected a subset of 271 genes that were LPS-responsive in our system (FPKM fold change of >1.5 between –LPS and +LPS) and targets of EGR1-gaining enhancers (i.e., representing the closest gene to an EGR1-gaining enhancer). RNA-seq indicates that, in EGR1 overexpression condition, the 271 genes are not properly up-regulated after LPS treatment when compared to EGR1-WT condition. (E) Volcano plot depicting the loss of gene expression in FLAG-EGR1 versus EGR1 wild-type macrophages as determined by RNA-seq in biological duplicates. The blue dots are down-regulated genes, and the red dots are up-regulated genes (fold change > 2, FDR < 0.1). Orange dots highlight either up-regulated or down-regulated genes that are regulated by interferon-γ (INF-γ genes) according to ingenuity pathway analysis. FC, fold change.
Fig. 6. EGR1 blunts the inflammatory response in human macrophages.
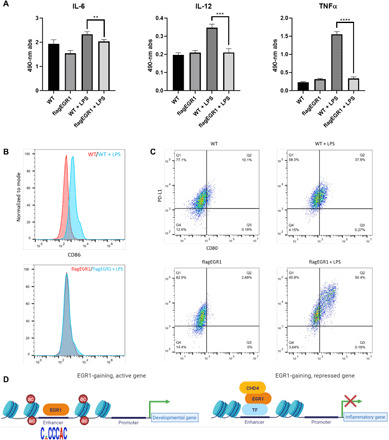
(A) Absorbance values of a cytokine panel ELISA show reduced secretion of select cytokines in EGR1-overexpressing macrophages upon stimulation with LPS. Of the cytokines assayed, IL-6 levels were significantly decreased by ~13%, while IL-12 and TNFα levels were decreased by ~40 and 80%, respectively, as compared to control macrophages after LPS stimulation (**P < 0.01, ***P < 0.001, and ****P < 0.0001). (B) Flow cytometry results of EGR1-overexpressing macrophages exhibit a lack of surface expression of the costimulatory signaling marker CD86 as compared to control macrophages after LPS stimulation. (C) Flow cytometry further shows a distinct population of PDL1(CD274)+- and CD80+-coexpressing cells in EGR1-overexpressing macrophages as compared to control macrophages. (D) Diagrammatic representation of the role of EGR1 in monomacrophagic cells. EGR1 associates with enhancers of both developmental and proinflammatory genes. At developmental enhancers, EGR1 is recruited by its DNA motif and promotes activation, especially during early monocytic commitment (12). Conversely, EGR1 is indirectly recruited at proinflammatory enhancers (likely via repressive TFs) during macrophage differentiation. EGR1 recruitment at inflammatory enhancers leads to deacetylation and loss of accessibility by way of the NuRD-repressive complex.
Together, our data suggest that EGR1 may target two distinct categories of regulatory elements with opposite outcomes (Fig. 6D): Near developmental genes, EGR1 binds enhancers via its DNA motif and supports activation, especially in stem and progenitor cells (Fig. 6D). Conversely, near inflammatory genes, EGR1 is recruited indirectly and associates with the NuRD complex to bring about deacetylation, decrease chromatin accessibility, and blunt the inflammatory response.
DISCUSSION
In this work, we investigate the epigenomic regulation of mono/macrophagic commitment in primary human cells and present the first comprehensive analysis of EGR1 in myelopoiesis. We characterize a novel function for the zinc-finger EGR1, demonstrating that this transcription factor attenuates the activity of inflammatory enhancers in developing and mature macrophages. The mono/macrophagic lineage undergoes a rather unique developmental route. HSCs generate a sequence of progenitor cells with the progressively restricted ability to differentiate into monocytes. This process requires several rounds of cell divisions and occurs over the span of few weeks. Mature monocytes are postmitotic cells, with a limited life span and are predominantly found in the bloodstream or in the spleen. Upon migrating to a variety of tissues, monocytes further differentiate into either dendritic cells or macrophages. This latter developmental step requires 4 to 6 days for completion and is not accompanied by further cell divisions. While morphologically different, monocytes and macrophages share some functional similarity: Both cell types have phagocytic ability and secrete an array of cytokines that elicit and control inflammation. In this light, development of monocytes and macrophages is thought to be orchestrated in a similar way by the same assortment of TFs. However, the actual enhancer dynamics underpinning myeloid commitment of human cells has not been previously investigated. Here, we take advantage of the recently developed CUT&RUN technique to track enhancer activation during HSPC-to-monocytes and monocyte-to-macrophages commitment, probing the transcription factor EGR1. Previous epigenomic studies have established a central role for the myeloid Ets family factor PU.1 in determining monocyte/macrophage cell identity. However, PU.1 is not sufficient to unambiguously address stem and progenitor cells; for instance, both macrophages and B cells depend on its activity. PU.1 operates as a pioneer factor, triggering accessibility at a large number of cell identity enhancers (2, 5). A combination of additional lineage-specific factors, such as AP1 and CEBP, tunes the activity of lineage-specific enhancers through cooperative binding with PU.1 (5, 22). Similarly, PU.1 sets the stage for enhancer activity in functional, differentiated macrophages (19), in which its binding is propaedeutic to recruitment of inflammatory TFs such as nuclear factor κB, signal transducers and activators of transcription 6, musculoaponeurotic fibrosarcoma transcription factor (MAF), and interferon regulatory factor 1 (6, 21, 23). Together, our data reveal that EGR1 is part of the PU.1 cistrome. First, unbiased analysis of DNA accessibility changes that occur during monocytic commitment of HSPC retrieves PU.1- and EGR1-like motifs as the first and second most enriched matrices, respectively. In addition, EGR1-centered analyses (from either CUT&RUN or ChIP-seq data) in stimulated HSPCs and monocytes identify PU.1 motifs as highly enriched throughout all EGR1-binding regions.
We recently demonstrated that EGR1 is essential to activate poised enhancer elements that drive monocytic lineage specification, such as the CSF1R-associated FIRE element (12). As a matter of fact, EGR1 is strictly required by CD34+ HSPCs to form monocytic colonies (13, 14). Here, we present the first time course analysis of EGR1 during monocytic differentiation of primary cells, further corroborating the central role of EGR1 in HSPC commitment. However, EGR1 depletion does not preclude macrophage differentiation from mature monocytes, and the same TF seems to be directed to a distinct set of enhancers. These data suggest that (i) macrophage commitment is not a mere extension of monocytic commitment but entails a distinct enhancer-promoter circuitry and (ii) that EGR1 is far more versatile than other lineage-determining TFs. Notably, unlike PU.1, EGR1 expression and binding ability are tightly regulated throughout myeloid development: While it is up-regulated during early HSPC commitment, EGR1 is lowly expressed in mature, circulating monocytes and disengaged from chromatin. Upon monocyte stimulation with GM-CSF, EGR1 undergoes a second “developmental” wave of up-regulation. Genome-wide recruitment of EGR1 is sustained during differentiation, tailing off at day 6 (loss of binding and reduced protein levels). During its transitory activation, the EGR1 macrophagic signature is strongly associated with repressed cis-regulatory elements comprising a large set of inflammatory enhancers. Notably, these enhancers are not bound in HSPCs and can be targeted in the absence of an EGR1 DNA motif. A repressive role for EGR1 has not been previously reported in hematopoietic cells, but it has been suggested in other cell types such as cardiomyocytes (24) and endothelial cells (25). Despite early evidence suggesting a proinflammatory role for EGR1 (26, 27), here, we show that EGR1 repressive activity in macrophages is likely mediated by the NuRD corepressor complex, which has been previously associated to EGR1 (28). NuRD activity has been shown to facilitate cell fate transitions in several developmental processes and across multiple species (29). Previous studies suggest that NuRD does not fully silence/dismantle enhancers but rather fine-tunes their accessibility and acetylation to limit their activity (30, 31). We posit that EGR1 restrains inflammatory enhancers during macrophage development, preventing their misfiring and subsequent unleash of inflammatory genes. To our knowledge, EGR1 is the first TF that counteracts the activation of inflammatory enhancers. Our data further suggests that the role of EGR factors in modulating inflammation may extend well beyond development. Overexpression of EGR1, in mature cells, blunts macrophage activation by reducing cytokine secretion, blocking stimulatory ligands such as CD86, and increasing CTLA4 and PD-1 ligands. Albeit reduced, EGR1 is expressed in mature macrophages, even during LPS stimulation, similar to other EGR family members such as EGR2. Beyond their developmental role, the EGR TFs may also constitute a group of dynamically regulated anti-inflammatory transcription factors across all mature myeloid cells.
MATERIALS AND METHODS
Macrophage differentiation
Circulating monocytes were obtained by the University of Pennsylvania Human Immunology Primary Cell Core Facility and differentiated into adherent macrophages in Iscove’s Modified Dulbecco’s Media (IMDM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and human recombinant GM-CSF (25 ng/ml; PeproTech) for 5 days.
Monocyte differentiation
Deidentified human cord blood was obtained from volunteers with informed consent at Helen F. Graham Cancer Center and Research Institute at Christiana Hospital. The protocol was approved by the ChristianaCare Institutional Review Board. Mononucleated cells were separated with Ficoll-Hystopaque Plus (GE Healthcare). CD34+ cells were then isolated using the human CD34 MicroBeads Kit (Miltenyi Biotec) following the manufacturer’s instructions. CD34+ cells were differentiated into monocytes in serum-free expansion medium (SFEM) supplemented with stem cell factor (SCF; 100 ng/ml), IL-3 (10 ng/ml), M-SCF (50 ng/ml), and GM-CSF (25 ng/ml; PeproTech).
ChIP-seq and ChIP-qPCR sample processing
For each experiment, two biological replicates were used. Samples from different conditions were processed together to prevent batch effects. For each replicate, 10 million cells were cross-linked with 1% formaldehyde for 5 min at room temperature, harvested, and washed twice with 1× phosphate-buffered saline (PBS). The pellet was resuspended in ChIP lysis buffer [150 mM NaCl, 1% Triton X-100, 0.7% SDS, 500 mM dithiothreitol (DTT), 10 mM tris-HCl, and 5 mM EDTA], and chromatin was sheared to an average length of 200 to 400 base pairs (bp) using the Covaris M220 ultrasonicator. The chromatin lysate was diluted with SDS-free ChIP lysis buffer. For ChIP-seq, 10 μg of antibody (5 μg for histone modifications) was added to the 10 million lysated cells along with Protein A magnetic beads (Invitrogen) and incubated at 4°C overnight. On day 2, beads were washed twice with each of the following buffers: mixed micelle buffer (150 mM NaCl, 1% Triton X-100, 0.2% SDS, 20 mM tris-HCl, 5 mM EDTA, and 65% sucrose), buffer 500 (500 mM NaCl, 1% Triton X-100, 0.1% Na deoxycholate, 25 mM Hepes, 10 mM tris-HCl, and 1 mM EDTA), LiCl/detergent wash (250 mM LiCl, 0.5% Na deoxycholate, 0.5% NP-40, 10 mM tris-HCl, and 1 mM EDTA). A final wash was performed with 1× Tris-EDTA (TE) buffer. Beads were resuspended twice with 1/3 of a final volume of TE containing 1% SDS and incubated at 65°C for 10 min to elute immunocomplexes. Elution was repeated twice, and the samples were further incubated overnight at 65°C to reverse cross-linking, along with the untreated input (5% of the starting material). On day 3, after treatment with proteinase K (0.5 mg/ml) for 3 hours, DNA was purified using the Zymo ChIP DNA Clear Concentrator Kit (Zymo research) and quantified with Qubit. Barcoded libraries were made with the NEB Ultra II DNA Library Prep Kit for Illumina and sequenced on Illumina NextSeq 500. For ChIP-qPCR samples, on day 3, DNA was purified with Wizard SV Gel and PCR Clean-Up system (Promega) and resuspended in 200 and 5 μl were for each PCR reaction.
CUT&RUN
For each experiment, two biological replicates were used. Cells per sample (400,000) were used to perform CUT&RUN as described in (32) with no modifications. Barcoded libraries were made with the NEB Ultra II DNA Library Prep Kit for Illumina and sequenced on Illumina NextSeq 500.
ChIP-seq and CUT&RUN analyses
Sequences were aligned to the reference hg19 using Burrows-Wheeler Alignment (BWA) tool, with the maximal exact matches (MEM) algorithm (33). Aligned reads were filtered on the basis of mapping quality (>10) to restrict our analysis to higher quality and likely uniquely mapped reads, and PCR duplicates were removed. We called peaks for each individual using Model-Based Analysis of ChIP-Seq (MACS2) at 5% FDR, with default parameters (34). To identify EGR1-gained regions, we took advantage of the fact that EGR1 is not expressed until day 3, so there are no peaks at all until the day 3. Therefore, we considered as “gained” all the peaks that were called at day 3. Notably, >90% of the peaks were replicated at days 3 and 4. We acknowledge that the standard pipeline for analysis of CUT&RUN data include the dovetail parameter for the alignment with bowtie and the use of sparse enrichment analysis for CUT&RUN (SEACR) for peak calling. However, we find that there is strong correlation of the aligned reads with the BWA method we used and bowtie with dovetail.
RNA-seq sample processing
For each experiment, two biological replicates were used. Samples from different conditions were processed together to prevent batch effects. Total RNA was extracted using the Zymo Direct-Zol RNA MiniPrep Kit (Zymo research) and the miRNeasy Kit with TRIzol protocol and in-column deoxyribonuclease treatment (QIAGEN), respectively. Quality of total RNA was assessed by the RNA integrity number (RIN) using Agilent Bioanalyzer. All retained RNA samples had a RIN > 8. Ribodepletion was performed with the QIaseq FastSelect RNA Removal Kit. Stranded libraries were produced with the KAPA RNA HyperPrep Kit and sequenced on Illumina NextSeq 500.
RNA-seq analyses
Reads were aligned to hg19 using STAR v2.5 (35), in two-pass mode with the following parameters: quantMode TranscriptomeSAM–outFilterMultimapNmax 10–outFilterMismatchNmax 10–outFilterMismatchNoverLmax 0.3–alignIntronMin 21–alignIntronMax 0–alignMatesGapMax 0–alignSJoverhangMin 5–runThreadN 12–twopassMode Basic–twopass1readsN 60000000–sjdbOverhang 100. We filtered bam files based on alignment quality (q = 10) using SAMtools v0.1.19 (36). We used the latest annotations obtained from Ensembl to build reference indexes for the spliced transcripts alignment to a reference (STAR) alignment. FeatureCounts (37) was used to count reads mapping to each gene. RNA-seq by expectation maximization (RSEM) (38) was instead used to obtain fragments per kilobase of exon per million fragments mapped. We analyzed differential gene expression levels using read counts, normalized by feature length with DESeq2 (39), with the following model: design = $condition.
ATAC-seq sample processing and analysis
For each sample/replicate, 50,000 cells per condition were processed as described in the original ATAC-seq protocol paper (40). Data were processed using the same pipeline described for ChIP-seq, with one modification: All mapped reads were offset by +4 bp for the forward strand and 5 bp for the reverse strand.
Western blot
Cells were harvested and washed three times in 1× PBS and lysed in radioimmunoprecipitation assay buffer [50 mM tris-HCl (pH 7.5), 150 mM NaCl, 1% IGEPAL, 0.5% sodium deoxycholate, 0.1% SDS, and 500 mM DTT] supplemented with aprotinin (1 mg/ml), leupeptin (1 mg/ml; Sigma-Aldrich), and pepstatin (1 mg/ml; BMB). Whole-cell lysate (50 mg) was loaded in Bolt 4 to 12% Bis-Tris Plus gel (Invitrogen) or Novex WedgeWell 10% tris-glycine gel (Invitrogen) and separated through gel electrophoresis [SDS–polyacrylamide gel electrophoresis (SDS-PAGE)] in Bolt MES running buffer (Invitrogen) or tris-glycine-SDS buffer (Bio-Rad), respectively. Separated proteins were transferred to ImmunBlot polyvinylidene difluoride membranes (Bio-Rad) for antibody probing. Membranes were incubated with 10% bovine serum albumin (BSA) in tris-buffered saline containing 0.1% tween (TBST) for 30 min at room temperature, then incubated for 2 hours at room temperature (RT) or overnight at 4°C with the suitable antibodies diluted in 5% BSA in 1× TBST, washed with TBST, and incubated with a dilution of 1:10,000 of horseradish peroxidase–linked anti-mouse or anti-rabbit secondary antibody (Cell Signaling Technology) for 1 hour at RT. Antibodies were then visualized using Clarity Western ECL substrate (Bio-Rad) and imaged using Fujifilm LAS-3000 Imager (Fujifilm).
Immunoprecipitation
HL-60 cells were washed twice with ice-cold PBS before resuspension in BC100 [20 mM tris (pH 8.0), 0.1 M KCl, 0.1% NP-40, 10% glycerol, 0.2 mM EDTA, 0.5 mM DTT, and protease inhibitors aprotinin, leupeptin, and pepstatin (1 mg/ml each)] and incubated at 4°C for 5 min. The pellet was resuspended in buffer C [20 mM tris (pH 8.0), 1.5 mM MgCl2, 0.42 M NaCl, 25% glycerol, 0.2 mM EDTA, 0.5 mM DTT, and protease inhibitors] and incubated at 4°C for 30 min. For chromatin-enriched nuclear extracts, Benzonase nuclease (Sigma-Aldrich) was added after 15 min and incubated for an additional 30 min at 4°C. The resulting extract was spun down at 12,000 rpm for 30 min. The pellet was discarded, and the supernatant was kept as nuclear extract. The nuclear extract was dialyzed overnight in BC80 [20 mM tris (pH 8.0), 80 mM KCl, 0.2 mM EDTA, 10% glycerol, 1 mM β-mercaptoethanol, and 0.2 mM phenylmethylsulfonyl fluoride], cleared, and stored at 80°C. A 500 mg (for Western blot) or 2 to 4 mg (for mass spectrometry) of nuclear extract were diluted in co-IP buffer [20 mM tris (pH 7.9), 100 mM NaCl, 0.1% NP-40, and protease inhibitors). EGR1 antibody–cross-linked Dynabeads Protein A (Invitrogen) were incubated with nuclear extract at 4°C for 4 or 2 hours, respectively. The supernatant was kept as flow through. Beads were washed three times with co-IP buffer, followed by one wash with 0.05% NP-40 in PBS. Glycine elution was performed with agitation in 0.1 M glycine (pH 3.0) for 1 min, and 1 M tris base (pH 11.0) was added to neutralize the pH of the eluate.
Mass spectrometry
After co-IP, eluates were prepared for SDS-PAGE as described previously. The eluates were run into a 10% tris-glycine gel at 110 V for 10 min. The gel was stained with the Colloidal Blue Staining Kit (Invitrogen) and further processed at the proteomics facility at the Wistar Institute. Briefly, the gel lanes were excised, digested with trypsin, and analyzed by liquid chromatography–tandem mass spectrometry on the Q Exactive HF mass spectrometer. The data were searched against the UniProt human database (September 2016) and provided sequences using MaxQuant 1.5.2.8 (41). FDRs for protein and peptide identifications were set at 1%.
EGR1 overexpression
To generate the EGR1 overexpression, a FLAG-EGR1 was synthesized and cloned into a pLenti-PURO vector. Macrophages were lentivirally transduced at day 4 of differentiation, adding polybrene (5 μg/ml). Cells were selected with puromycin (1 μg/ml; InvivoGen) for 48 hours. At day 6, LPS solution was added at a concentration of 500 ng/ml. Cells were harvested after 24 hours of LPS treatment. A pLENTI against luciferase was used as control.
EGR1 knockdown
shEGR1 was designed with the Broad Institute algorithm (https://portals.broadinstitute.org/gpp/public/) and subsequently cloned in either pLKO.1 (12). A pLENTI against luciferase was used as a control. Monocytes were lentivirally transduced at day 0 of differentiation through one round of spin inoculation (1800 rpm, 45 min at 30°C, brake off) in IMDM medium without serum. Inactivated FBS and GM-CSF (25 ng/ml) were added right after spin inoculation. Puromycin (1 μg/ml; InvivoGen) was added the next day. Cells were harvested at day 5 of differentiation.
Real-time qPCR
Cells were lysed in Tri-reagent, and RNA was extracted using the Direct-zol RNA MiniPrep Kit (Zymo Research). Template RNA (1 mg) was retrotranscribed into complementary DNA (cDNA) using random primers and the Revertaid first-strand cDNA Synthesis Kit (Thermo Fisher Scientific) according to the manufacturer’s directions. The cDNA (50 ng) was used for each real-time qPCR reaction with 0.4 mM of each primer and 10 ml of iQ SYBR Green Supermix (Bio-Rad) in a final volume of 20 ml using the CFX96 Real-Time System (Bio-Rad). Thermal cycling parameters were as follows: 3 min at 95°C, followed by 40 cycles of 10 s at 95°C, 20 s at 63°C, followed by 30 s at 72°C. Each sample was run in triplicate. 18S ribosomal RNA was used as normalizer. Primer sequences are reported in table S7.
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay (ELISA) was carried out according to the manufacturer’s specifications for the cytokine ELISA Kit (Signosis). Media were gathered from differentiated macrophages after 72 hours in culture and loaded into precoated cytokine ELISA assay well strips. Absorbance values were measured via absorbance microplate reader at 490 nm.
Antibodies
EGR1 (ChIP, IP, Western blotting and CUT&RUN) was from Bethyl (A303-390A). CHD4 was from Abcam (ab72418). H3K27ac was from Abcam (ab4279). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was from Cell Signaling Technology (2118). PU.1 was from Santa Cruz Biotechnology (T-21).
Statistical and computational analyses
All statistical analyses were performed using R v3.3.1. Figures were made with the package ggplot2+. BEDtools v2.25.0 (42) was used for genomic analyses. Pathway analysis was performed with Ingenuity Pathway Analysis Suite (QIAGEN, www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis). Motif analyses were performed with Meme-ChIP (43). In Fig. 2D, we showed all of the significant motifs found by Meme-ChIP. Average profiles were generated with seqMINER version 1.3.4 (44). The E value, as defined by MEME-ChIP, is the statistical significance of the motif. It is an estimate of the expected number of motifs with the given log likelihood ratio (or higher) and with the same width and site count that one would find in a similarly sized set of random sequences. In Fig. 2, the k-means clustering of the 4300 EGR1 enhancers was performed with seqMINER (44).
Supplementary Material
Acknowledgments
We would like to thank S. Welsh, C. Hill, S. Offley, and P. Porazzi for insightful discussions. All genome-wide data were generated and processed at the Wistar Institute with the outstanding technical support from all members of the Genomics Facility, especially S, Bala (NIH-NCI funded CCSG: P30-CA010815). Funding: This work was supported by grant R01 HL141326 (A.G.) from the NIH, grant RSG-18-157-01-DMC from the American Cancer Society (A.G.), and a grant from the G. Harold & Leyla Y. Mathers Foundation (A.G.). A.Z. is supported by an NIH training grant T32-CA009171. Author contributions: M.T. and A.G. designed the project. M.T., A.Z., and E.B. performed the experiments. M.T. and S.D. analyzed the data. F.V., F.W., and D.G. supervised experiments and analyses and contributed to the conceptual development of the manuscript. M.T. and A.G. wrote the paper. All authors read and approved the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The original genome-wide data described in this paper are deposited at the GEO (accession number GSE136216). Previously published datasets that were reanalyzed in this paper are found at GEO: GSE31621. A full list of experiments and replicates is available in table S6. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/3/eaaz8836/DC1
REFERENCES AND NOTES
- 1.Álvarez-Errico D., Vento-Tormo R., Sieweke M., Ballestar E., Epigenetic control of myeloid cell differentiation, identity and function. Nat. Rev. Immunol. 15, 7–17 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Barozzi I., Simonatto M., Bonifacio S., Yang L., Rohs R., Ghisletti S., Natoli G., Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol. Cell 54, 844–857 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman A. D., Transcriptional control of granulocyte and monocyte development. Oncogene 26, 6816–6828 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Fonseca G. J., Tao J., Westin E. M., Duttke S. H., Spann N. J., Strid T., Shen Z., Stender J. D., Sakai M., Link V. M., Benner C., Glass C. K., Diverse motif ensembles specify non-redundant DNA binding activities of AP-1 family members in macrophages. Nat. Commun. 10, 414 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ostuni R., Piccolo V., Barozzi I., Polletti S., Termanini A., Bonifacio S., Curina A., Prosperini E., Ghisletti S., Natoli G., Latent enhancers activated by stimulation in differentiated cells. Cell 152, 157–171 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Scott E. W., Simon M. C., Anastasi J., Singh H., Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science 265, 1573–1577 (1994). [DOI] [PubMed] [Google Scholar]
- 8.Link V. M., Duttke S. H., Chun H. B., Holtman I. R., Westin E., Hoeksema M. A., Abe Y., Skola D., Romanoski C. E., Tao J., Fonseca G. J., Troutman T. D., Spann N. J., Strid T., Sakai M., Yu M., Hu R., Fang R., Metzler D., Ren B., Glass C. K., Analysis of genetically diverse macrophages reveals local and domain-wide mechanisms that control transcription factor binding and function. Cell 173, 1796–809.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang J., Liu X., Li D., Shao Z., Cao H., Zhang Y., Trompouki E., Bowman T. V., Zon L. I., Yuan G.-C., Orkin S. H., Xu J., Dynamic control of enhancer repertoires drives lineage and stage-specific transcription during hematopoiesis. Dev. Cell 36, 9–23 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heinz S., Romanoski C. E., Benner C., Glass C. K., The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 16, 144–154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang C., Lee J.-E., Lai B., Macfarlan T. S., Xu S., Zhuang L., Liu C., Peng W., Ge K., Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc. Natl. Acad. Sci. U.S.A. 113, 11871–11876 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barbieri E., Trizzino M., Welsh S. A., Owens T. A., Calabretta B., Carroll M., Sarma K., Gardini A., Targeted enhancer activation by a subunit of the integrator complex. Mol. Cell 71, 103–116.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krishnaraju K., Hoffman B., Liebermann D. A., Early growth response gene 1 stimulates development of hematopoietic progenitor cells along the macrophage lineage at the expense of the granulocyte and erythroid lineages. Blood 97, 1298–1305 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Laslo P., Spooner C. J., Warmflash A., Lancki D. W., Lee H.-J., Sciammas R., Gantner B. N., Dinner A. R., Singh H., Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell 126, 755–766 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Nguyen H. Q., Hoffman-Liebermann B., Liebermann D. A., The zinc finger transcription factor Egr-1 is essential for and restricts differentiation along the macrophage lineage. Cell 72, 197–209 (1993). [DOI] [PubMed] [Google Scholar]
- 16.Pham T.-H., Benner C., Lichtinger M., Schwarzfischer L., Hu Y., Andreesen R., Chen W., Rehli M., Dynamic epigenetic enhancer signatures reveal key transcription factors associated with monocytic differentiation states. Blood 119, e161–e171 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Lacey D. C., Achuthan A., Fleetwood A. J., Dinh H., Roiniotis J., Scholz G. M., Chang M. W., Beckman S. K., Cook A. D., Hamilton J. A., Defining GM-CSF– and macrophage-CSF–dependent macrophage responses by in vitro models. J. Immunol. 188, 5752–5765 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Burgess A. W., Metcalf D., The nature and action of granulocyte-macrophage colony stimulating factors. Blood 56, 947–958 (1980). [PubMed] [Google Scholar]
- 19.Ghisletti S., Barozzi I., Mietton F., Polletti S., De Santa F., Venturini E., Gregory L., Lonie L., Chew A., Wei C.-L., Ragoussis J., Natoli G., Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity 32, 317–328 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Tagore M., McAndrew M. J., Gjidoda A., Floer M., The lineage-specific transcription factor PU.1 prevents polycomb-mediated heterochromatin formation at macrophage-specific genes. Mol. Cell. Biol. 35, 2610–2625 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park S. H., Kang K., Giannopoulou E., Qiao Y., Kang K., Kim G., Park-Min K.-H., Ivashkiv L. B., Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat. Immunol. 18, 1104–1116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gosselin D., Link V. M., Romanoski C. E., Fonseca G. J., Eichenfield D. Z., Spann N. J., Stender J. D., Chun H. B., Garner H., Geissmann F., Glass C. K., Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang K., Park S. H., Chen J., Qiao Y., Giannopoulou E., Berg K., Hanidu A., Li J., Nabozny G., Kang K., Park-Min K.-H., Ivashkiv L. B., Interferon-γ represses M2 gene expression in human macrophages by disassembling enhancers bound by the transcription factor MAF. Immunity 47, 235–250.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng Y., Desjardins C. A., Cooper O., Kontor A., Nocco S. E., Naya F. J., EGR1 functions as a potent repressor of MEF2 transcriptional activity. PLOS ONE 10, e0127641 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu M.-Y., Wu X.-Y., Li Q.-S., Zheng R.-M., Expression of Egr-1 gene and its correlation with the oncogene proteins in non-irradiated and irradiated esophageal squamous cell carcinoma. Dis. Esophagus 19, 267–272 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Coleman D. L., Bartiss A. H., Sukhatme V. P., Liu J., Rupprecht H. D., Lipopolysaccharide induces Egr-1 mRNA and protein in murine peritoneal macrophages. J. Immunol. 149, 3045–3051 (1992). [PubMed] [Google Scholar]
- 27.Mostecki J., Showalter B. M., Rothman P. B., Early growth response-1 regulates lipopolysaccharide-induced suppressor of cytokine signaling-1 transcription. J. Biol. Chem. 280, 2596–2605 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Srinivasan R., Mager G. M., Ward R. M., Mayer J., Svaren J., NAB2 represses transcription by interacting with the CHD4 subunit of the nucleosome remodeling and deacetylase (NuRD) complex. J. Biol. Chem. 281, 15129–15137 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Signolet J., Hendrich B., The function of chromatin modifiers in lineage commitment and cell fate specification. FEBS J. 282, 1692–1702 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynolds N., Latos P., Hynes-Allen A., Loos R., Leaford D., O’Shaughnessy A., Mosaku O., Signolet J., Brennecke P., Kalkan T., Costello I., Humphreys P., Mansfield W., Nakagawa K., Strouboulis J., Behrens A., Bertone P., Hendrich B., NuRD suppresses pluripotency gene expression to promote transcriptional heterogeneity and lineage commitment. Cell Stem Cell 10, 583–594 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bornelov S., Reynolds N., Xenophontos M., Gharbi S., Johnstone E., Floyd R., Ralser M., Signolet J., Loos R., Dietmann S., Bertone P., Hendrich B., The nucleosome remodeling and deacetylation complex modulates chromatin structure at sites of active transcription to fine-tune gene expression. Mol. Cell 71, 56–72.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skene P. J., Henikoff J. G., Henikoff S., Targeted in-situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 13, 1006–1019 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Li H., Durbin R., Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nussbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buenrostro J. D., Giresi P. G., Zaba L. C., Chang H. Y., Greenleaf W. J., Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cox J., Mann M., MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Machanick P., Bailey T. L., MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics 2712, 1696–1697 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhan X., Liu D. J., SEQMINER: An R-package to facilitate the functional interpretation of sequence-based associations. Genet. Epidemiol. 39, 619–623 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/3/eaaz8836/DC1