

Increased O-GlcNAcylation can confer neuroprotective effects against necroptosis in the context of Alzheimer’s disease.
Abstract
O-GlcNAcylation (O-linked β-N-acetylglucosaminylation) is notably decreased in Alzheimer’s disease (AD) brain. Necroptosis is activated in AD brain and is positively correlated with neuroinflammation and tau pathology. However, the links among altered O-GlcNAcylation, β-amyloid (Aβ) accumulation, and necroptosis are unclear. Here, we found that O-GlcNAcylation plays a protective role in AD by inhibiting necroptosis. Necroptosis was increased in AD patients and AD mouse model compared with controls; however, decreased necroptosis due to O-GlcNAcylation of RIPK3 (receptor-interacting serine/threonine protein kinase 3) was observed in 5xFAD mice with insufficient O-linked β-N-acetylglucosaminase. O-GlcNAcylation of RIPK3 suppresses phosphorylation of RIPK3 and its interaction with RIPK1. Moreover, increased O-GlcNAcylation ameliorated AD pathology, including Aβ burden, neuronal loss, neuroinflammation, and damaged mitochondria and recovered the M2 phenotype and phagocytic activity of microglia. Thus, our data establish the influence of O-GlcNAcylation on Aβ accumulation and neurodegeneration, suggesting O-GlcNAcylation–based treatments as potential interventions for AD.
INTRODUCTION
The incidence of Alzheimer’s disease (AD), a degenerative brain disorder and the most common type of dementia (1, 2), has increased in aged populations, and therefore, the impact of AD on society is increasing with the aging of the population (3). The pathological hallmarks of AD include β-amyloid (Aβ) accumulation, neuroinflammation, and neuronal death, which manifests as decreased brain volume and is directly related to memory loss (1, 3). Therefore, patients with AD present several symptoms associated with the loss of brain function, such as deficits in memory retention, reasoning, abstraction, and language skills (3). Aβ accumulation adversely affects the brain environment surrounding the neuronal cells. Aβ further disrupts the surrounding environment by activating astrocytes and microglia, which play important roles in maintaining brain homeostasis, and ultimately induces neuronal cell death and cognitive impairment (4). Microglia change according to the surrounding environment and stimulatory signals. Activated microglia can be classified as exhibiting a proinflammatory state (M1) or those exhibiting an anti-inflammatory state (M2) (4). M2-type signals alter the properties of microglia and change their profile from proinflammatory to anti-inflammatory, which increases Aβ phagocytosis and maintains brain homeostasis (4). Although considerable effort has been dedicated to the understanding of AD pathology, the mechanism of these phenomena in AD is unclear. Thus, active research focusing on the identification of the mechanisms in AD is ongoing.
Necroptosis or programmed necrosis, one of the possible cellular mechanisms underlying AD, has been found to be activated in humans’ brains with AD and is positively correlated with the pathological manifestations of AD, such as neuronal death, neuroinflammation, and Braak stage (5, 6). In addition to cell death induction, necroptosis is involved in proinflammatory responses (7, 8). Inhibition of necroptosis in AD effectively suppresses neuroinflammation (7). Necroptosis blockade in mouse models of AD inhibits Aβ accumulation and improves cognitive function (7, 9). In necroptosis, three proteins, receptor-interacting serine/threonine protein kinase 1 (RIPK1), RIPK3, and mixed lineage kinase domain–like pseudo-kinase (MLKL), are sequentially phosphorylated and interact with one another to form the necrosome (8). This complex migrates to the membrane and causes cell membrane breakdown, resulting in the release of intracellular organelles (6). Constitutively active MLKL induces a higher degree of neuronal death and exacerbates cognitive deficits in AD mice (5). Conversely, the pharmacological or genetic inhibition of RIPK1 attenuates biochemical pathology and behavioral deficits in the amyloid precursor protein (APP)/presenilin 1 (PS1) mice (7). In addition to AD, inhibition of necroptosis elicits a positive effect on brain function in several neurodegenerative diseases including multiple sclerosis, amyotrophic lateral sclerosis, and Parkinson’s disease, making it an important target for the treatment of these diseases (10–12).
O-linked β-N-acetylglucosaminylation (O-GlcNAcylation), a posttranslational modification in which O-linked β-N-acetylglucosamine (O-GlcNAc) is attached to serine/threonine residues, is a tightly regulated process that plays an integral role in fundamental cell signaling including transcription, protein-protein interaction, and sensing of the metabolic status (13). O-GlcNAcylation is enriched in brain tissues, and a number of O-GlcNAcylated brain proteins, such as tau and α-synuclein, are associated with neurodegenerative diseases (13). O-GlcNAcylation inhibits aggregation of proteins related to neurodegenerative diseases (14–16). O-linked β-N-acetylglucosaminase (O-GlcNAcase or OGA), an enzyme that specifically removes O-GlcNAc from proteins, and O-GlcNAc transferase (OGT), an enzyme that attaches O-GlcNAc to proteins, are distributed throughout the brain and play important roles in regulating O-GlcNAcylation (13, 17). Notably, OGA nullizygosity is embryonically lethal in mice, underscoring the developmental requirement for O-GlcNAcylation (18). In the brain of patients with AD, low levels of O-GlcNAcylated proteins are present (19, 20), and decreased O-GlcNAc levels in the mouse brain induce neurodegeneration and inflammatory response (21). In a few pharmacological studies, OGA inhibitors effectively decreased the amount of Aβ plaques and neurofibrillary tangles in the brain of AD mouse models (15, 22). In tauopathy mice, an increase in tau O-GlcNAcylation with OGA inhibition rendered tau less susceptible to aggregation and decreased neurodegeneration (16). In addition, O-GlcNAcylation alters the mitochondrial bioenergetic capacity (23), and decreased O-GlcNAcylation impairs mitochondrial activity in patients with AD and in a mouse model of AD (24). Despite the potential of O-GlcNAcylation as a therapeutic target in AD, the relationship between neuronal death and O-GlcNAcylation in AD is unclear.
As both O-GlcNAcylation and necroptosis have been separately shown to be targets for alleviating AD pathology, we further evaluated their association in AD. In this study, we generated 5xFAD with OGA haploinsufficiency and evaluated the changes in the pathologies of AD. We found that O-GlcNAcylation suppressed the activation of necroptosis in a mouse model of AD. Accordingly, an increase in O-GlcNAcylation induced by insufficient OGA expression in the brain of AD mice reduced AD pathological manifestations, including Aβ accumulation, neuronal death, cognitive decline, neuroinflammation, and dysfunctional mitochondria. Thus, O-GlcNAcylation appears to act as a major regulator of AD pathology through the inhibition of necroptosis.
RESULTS
Decreased O-GlcNAcylation and increased necroptosis in the AD brain
To investigate the association of O-GlcNAcylation and necroptosis in AD, we measured the expression of O-GlcNAcylation and necroptosis-related proteins in inferior parietal lobule samples from patients with AD and age-matched control patients with no symptoms of dementia. The global O-GlcNAcylated protein level was significantly higher in the individuals with no dementia symptoms than in the patients with AD, whereas no significant differences in the expression of OGT and OGA were observed (Fig. 1, A and C). Regarding necroptosis, the levels of RIPK1 and MLKL were significantly increased in the patients with AD (Fig. 1, A and D). In addition, the patients with AD exhibited elevated levels of phosphorylated RIPK1 (P-RIPK1), RIPK3 (P-RIPK3), and MLKL (P-MLKL) compared with those in the non-AD group (Fig. 1, A and E). The levels of synaptophysin, a synaptic marker, were significantly reduced in the brain tissues of patients with AD compared with the corresponding control patients (Fig. 1, A and F). These results suggest that O-GlcNAcylation decreases with an increase in necroptosis in the brain of patients with AD compared with control subjects.
Fig. 1. Association of O-GlcNAcylation and necroptosis in AD.
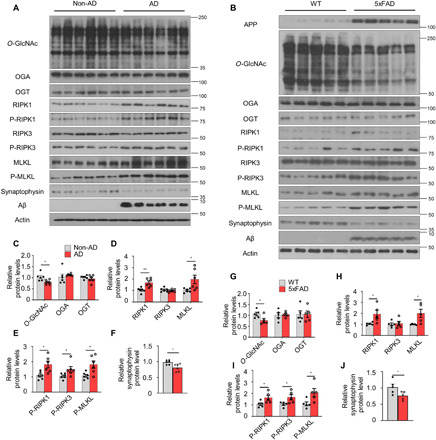
(A and B) Western blot analysis of brain tissues of patients with AD and nondemented controls (non-AD) (n = 6) (A) and 5xFAD and WT mice (n = 5; 12 months of age) (B). (C to J) Quantification of O-GlcNAcylation–related proteins (C and G), necroptosis-related proteins (D and H), phosphorylated necroptosis–related proteins (E and I), and synaptophysin (F and J) in (A) and (B). The levels of phosphorylated necroptosis–related proteins were normalized to the levels of the corresponding total proteins. Values are presented as means ± SEM. *P < 0.05 and **P < 0.01 versus non-AD (C to F) or WT (G to J); two-tailed Student’s t test.
Next, markers of O-GlcNAcylation and necroptosis in the brain were compared between the mouse model of AD (5xFAD) and wild-type (WT) control mice of the same age. The global O-GlcNAcylated protein level was significantly lower in the brains of 5xFAD mice, but no significant differences in OGA or OGT protein expression were observed (Fig. 1, B and G). Although the protein expression of OGA did not differ between WT and 5xFAD mice, OGA activity in the brain tissue was significantly higher in 5xFAD mice than in WT mice of the same age (fig. S1A). However, the activity of OGT did not differ between WT and 5xFAD mice (fig. S1B). Unlike in vivo, the activity of O-GlcNAc–related enzymes decreased in rat primary neurons after Aβ exposure (fig. S1C). The expression of all necroptosis-related factors, except total RIPK3, was higher in 5xFAD mice than in WT mice (Fig. 1, B, H, and I). The loss of synapses is a key process in the pathological progression of AD. The level of synaptophysin was decreased in the brain tissues of 5xFAD mice compared with WT mice (Fig. 1, B and J). We also found age-dependent changes in the levels of these proteins in 5xFAD mice (fig. S1, D and E). We further evaluated O-GlcNAcylation in rat primary neurons by immunostaining for O-GlcNAc and found that treatment with Aβ significantly attenuated the level of O-GlcNAcylation. Moreover, this effect could be reversed by treatment with the OGA inhibitor, thiamet-G (fig. S1, F to H).
Upon activation, necroptosis markers form a multimeric, insoluble, amyloid-like structure (25). The levels of necroptosis-related factors in the insoluble pellet of brain lysates were found to be significantly elevated in patients with AD and 5xFAD mice compared with age-matched control patients and WT mice (fig. S1, I to N). Overall, these data suggest that necroptosis is activated when O-GlcNAcylation decreases in the brains of patients with AD and AD mouse models.
OGA insufficiency increased global O-GlcNAc levels and inhibited necroptosis-related factors
To elucidate whether increased O-GlcNAc levels regulate the activity of necroptosis factors in the context of AD, we generated 5xFAD mice with OGA haploinsufficiency by crossing 5xFAD mice with OGA+/− mice. Genotyping was performed to divide the progeny into four groups (fig. S2A): WT, WT;OGA+/−, 5xFAD, and 5xFAD;OGA+/− mice. The levels of O-GlcNAcylation and OGA expression in the brain varied among these groups (Fig. 2, A to D, and fig. S2, B and D). Consistently, OGA activity in the brain significantly decreased with OGA haploinsufficiency (fig. S2E). Despite the enhanced level of O-GlcNAcylation, glucose tolerance and body weight were not changed by OGA haploinsufficiency (fig. S2, F to H), which was consistent with the results from previous reports (26, 27). Thus, OGA haploinsufficiency effectively decreased OGA activity and increased O-GlcNAcylation in the mouse model of AD.
Fig. 2. OGA haploinsufficiency increased global O-GlcNAc levels in the brain and decreased activation of necroptosis.
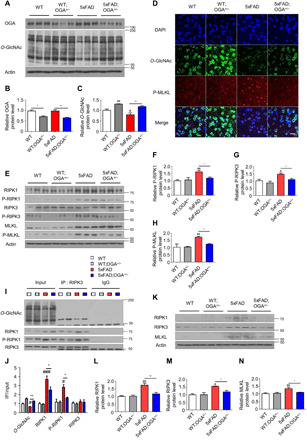
(A) Western blot analysis of OGA and O-GlcNAcylated proteins in brain samples of indicated mouse genotypes (n = 3). (B and C) Quantification of OGA protein (B) and O-GlcNAc protein (C) level in (A). (D) Immunostaining of O-GlcNAc and P-MLKL in the cortical region of mice samples (n = 3 to 4). DAPI, 4′,6-diamidino-2-phenylindole. Scale bar, 20 μm. (E) Necroptosis-related proteins in the brain of indicated genotypes of mice (n = 3). (F to H) Quantification of P-RIPK1 (F), P-RIPK3 (G), and P-MLKL (H) in (E). The levels of phosphorylated necroptosis–related proteins were normalized to the levels of the corresponding total proteins. (I) Western blot analysis of necroptosis-related proteins in RIPK3 immunoprecipitates from mouse brain samples. The precipitated immunoglobulin G (IgG) heavy chain is marked with an asterisk. IP, immunoprecipitation. (J) Quantification of O-GlcNAc, RIPK1, P-RIPK1, and RIPK3 binding to RIPK3 in (I). (K) Necroptosis-related proteins in insoluble fractions of brain samples collected from the indicated mouse genotypes (n = 3). (L to N) Quantification of necroptosis-related proteins RIPK1 (L), RIPK3 (M), and MLKL (N) in (K). Three slices of each sample were used to normalize each sample. Values are presented as means ± SEM. #P < 0.05, ##P < 0.01, and ###P < 0.001 versus WT; *P < 0.05 and **P < 0.01 versus 5xFAD; one-way analysis of variance (ANOVA) with Tukey’s test.
To evaluate whether O-GlcNAcylation affects the activation of necroptosis in the brain, we performed dual immunostaining of the cortex. The number of P-MLKL–positive cells was significantly reduced in 5xFAD;OGA+/− mice compared with 5xFAD mice (Fig. 2D and fig. S2I). While 5xFAD;OGA+/− mice and 5xFAD mice showed no differences in the total levels of necroptosis-related proteins, the levels of activated phosphorylated necroptosis factors were significantly lower in 5xFAD;OGA+/− mice (Fig. 2, E to H). O-GlcNAcylation competes with phosphorylation for target proteins (13); therefore, we determined whether RIPK3 in the brain is modified by O-GlcNAcylation. The amount of O-GlcNAcylated RIPK3 was significantly reduced in 5xFAD mice compared to that of WT mice, and increased O-GlcNAcylation of RIPK3 was found with OGA haploinsufficiency (Fig. 2, I and J). Since necroptosis factors interact during the necroptosis process, we used immunoprecipitation to examine the interactions between these factors in different genotypes. The amount of RIPK1 bound to RIPK3 was significantly higher in 5xFAD mice than in WT mice and significantly lower in 5xFAD;OGA+/− mice than in 5xFAD mice. Unlike 5xFAD mice, OGA haploinsufficiency did not cause similar effects in WT mice (Fig. 2, I and J).
Thereafter, we evaluated whether O-GlcNAcylation is involved in necrosome complex formation. While high levels of necroptosis factors were detected in the insoluble pellets of brain lysates from 5xFAD mice, the levels of these proteins were significantly decreased in those from 5xFAD;OGA+/− mice (Fig. 2, K to N). In summary, these data show that OGA haploinsufficiency increases O-GlcNAc levels in the brain of mouse model of AD and that O-GlcNAcylation inhibits the necroptosis pathway.
Increased O-GlcNAc levels in the brain inhibited neuronal cell death
As we showed that enhanced O-GlcNAc levels inhibited the necroptosis pathway in 5xFAD mice, we next aimed to determine whether O-GlcNAcylation affects neuronal loss in AD by performing Nissl staining of AD mouse brain samples. Previous reports showed that the number of neurons was visibly reduced in 5xFAD mice (28). As expected, 5xFAD mice exhibited higher neuronal loss in the cortical layer 5 than that of WT mice. However, 5xFAD;OGA+/− mice showed less neuronal loss in this region than 5xFAD mice (Fig. 3, A and B). The difference in neuronal density between 5xFAD mice and 5xFAD;OGA+/− mice was confirmed by evaluating the expression of NeuN, a neuronal marker (Fig. 3, C and D). Thereafter, we examined the coexpression of NeuN and P-MLKL by immunostaining. Unlike in WT mice, P-MLKL was highly expressed in 5xFAD mice; however, it was expressed at significantly lower levels in 5xFAD;OGA+/− mice (Fig. 3, C and E). Sites positive for Fluoro-Jade, a staining reagent for detecting neurodegeneration, were observed in 5xFAD mice, unlike in 5xFAD;OGA+/− mice (Fig. 3, F and G). Because neuronal death destroys the synaptic network and is one of the symptoms of AD (1), we evaluated synaptic density by measuring synaptophysin expression. Immunostaining revealed that the overall synaptophysin level in the cortical region was lower in 5xFAD mice than in WT mice; however, no difference was observed in the synaptophysin level in the cortical region between 5xFAD;OGA+/− and WT mice (Fig. 3, H and J). In addition, Western blot analysis revealed similar differences in synaptophysin expression among the mouse strains (Fig. 3, I and K). These findings suggest that an increase in the O-GlcNAc level due to OGA insufficiency suppresses neuronal death related to necroptosis and maintains synaptic density in the mouse model of AD.
Fig. 3. Increased O-GlcNAcylation reduced neuronal loss in the brain tissue of 5xFAD.
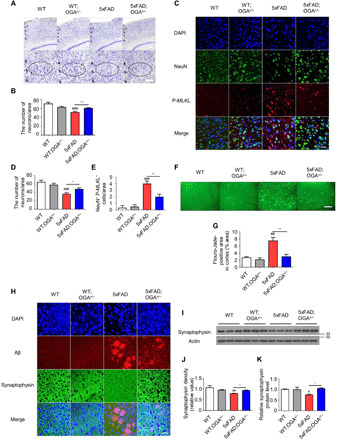
(A) Nissl staining in the cortical region of indicated mouse genotypes (n = 3 to 5). Scale bars, 100 μm. (B) The number of neurons in the cortical region of mice in (A). (C) Immunostaining of NeuN and P-MLKL in the cortical region of mice samples (n = 3 to 4). Scale bar, 20 μm. (D and E) The number of NeuN-positive cells (D) and NeuN/P-MLKL double-positive cells (E) in (C). (F) Flouro-Jade C staining in the cortical region of mice samples (n = 3). Scale bar, 100 μm. (G) Quantification of Flouro-Jade C–positive area in (F). (H) Immunostaining of Aβ and synaptophysin in the cortical region of mice samples (n = 3 to 4). Scale bar, 20 μm. (I) Synaptophysin protein level in the cortical region of mice samples (n = 3). (J) Quantification of synaptophysin density in (H). (K) Quantification of synaptophysin proteins levels in (I). Three to five slices of each sample were used to normalize each sample. Values are presented as means ± SEM. #P < 0.05, ##P < 0.01, and ###P < 0.001 versus WT; *P < 0.05 and **P < 0.01 versus 5xFAD; one-way ANOVA with Tukey’s test.
Increased O-GlcNAc levels ameliorated cognitive decline in 5xFAD mice
To determine whether O-GlcNAcylation is associated with cognitive function, we subjected mice in each genotype group to cognitive function testing. When the experiments were performed in these mice at the age of 3 months, 5xFAD mice did not show any cognitive impairment and Aβ accumulation (fig. S2, J to L). Following an additional period of aging, cognitive testing was conducted in the experimental animals at 8 months of age (Fig. 4A). In the cognitive function tests, the movements of the mice were recorded in the absence of behavioral impediments that could affect the measurements (Fig. 4, B to D). At 8 months of age, the decreased spontaneous alternation behavior of 5xFAD mice in the Y-maze, which evaluates short-term memory, was recovered to the level of WT mice when a copy of the OGA gene was deleted (Fig. 4E). Similarly, in the novel object recognition test, 5xFAD mice showed a decreased ability to recognize novel objects compared with WT mice, but 5xFAD;OGA+/− mice maintained the cognitive ability to explore and sniff around a new object (Fig. 4F). In the passive avoidance test, which measures fear memory, 5xFAD;OGA+/− mice were able to remember having been received an electric shock in a dark room and showed a tendency not to enter the darkroom (Fig. 4G). In addition, in the Morris water maze, 5xFAD;OGA+/− mice found the hidden platform faster than 5xFAD mice, exhibited significantly decreased escape latency, and found the hidden platform more frequently after training (Fig. 4, H and I). In the probe test, 5xFAD;OGA+/− mice stayed in the quadrant where the platform had previously been located as much as the WT mice did, while 5xFAD mice swam randomly (Fig. 4, J to L). The cognitive testing results indicate that the memory-related function of 5xFAD;OGA+/− mice is superior to that of 5xFAD mice.
Fig. 4. Increased O-GlcNAc levels ameliorate cognitive deficits in 5xFAD mice.
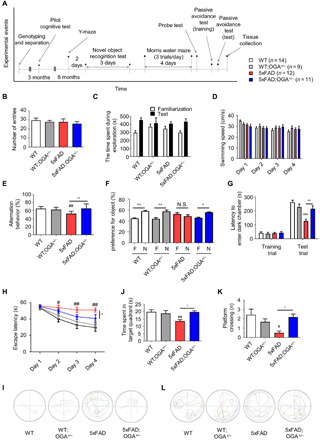
(A) Schematic drawing of the schedule of cognitive tests in indicated mouse genotypes (n = 9 to 14). (B) The number of entries in the Y-maze. (C) Time spent to the exploration of indicated mouse genotypes in novel object test. (D) Swimming speed of indicated mouse genotypes in the Morris water maze. (E) The Y-maze spontaneous alteration. (F) Preference for familiar and novel objects in the novel object recognition test. F, familiar object; N, novel object; N.S., not significant. (G) Duration of memory retention in the passive avoidance test. (H) The escape latency measured during the Morris water maze training trials. (I) Swimming traces obtained during training trials. (J) Time spent in the target quadrant where the hidden platform was previously placed during probe trial. (K) The number of times mice passed through the area in which the hidden platform was previously located during probe trial. (L) Swimming traces obtained during probe test. Values are presented as means ± SEM. #P < 0.05, ##P < 0.01, and ###P < 0.001 versus WT; *P < 0.05 and **P < 0.01 versus 5xFAD except familiar object (F); one-way ANOVA with Tukey’s test.
In addition, we examined whether cognitive function could be recovered with an OGA inhibitor in another model of Aβ-induced cognitive decline (fig. S3A). We determined changes in O-GlcNAc levels following the injection of the oligomer of Aβ or Aβ with thiamet-G. We found that the Aβ-mediated reduction in O-GlcNAc levels in the brain was recovered by treatment with the OGA inhibitor (fig. S3B). In the Y-maze test, no difference in the movement was observed among the control, Aβ, and Aβ with thiamet-G groups (fig. S3C). While the Aβ-injected mice showed a significant reduction in spontaneous alteration behavior compared with the control mice, treatment with thiamet-G rescued the impairment in spontaneous alternation behavior in the Aβ-injected mice (fig. S3D). In summary, O-GlcNAcylation restores cognitive abilities in 5xFAD or Aβ-injected model.
OGA insufficiency significantly reduced the amount of Aβ in the brain of 5xFAD mice
Since the cognitive ability of 5xFAD mice was restored with haploinsufficiency of the OGA gene, we next investigated the factors that influence the cognitive ability in 5xFAD mice. As the accumulation of Aβ is the most common pathological manifestation of AD and abnormal accumulation of Aβ leads to neuronal death (1), we examined the changes of factors related to Aβ production. First, we found that O-GlcNAcylation of APP was not detected in the brain tissues of this model (fig. S4A). Next, we measured the expression of proteins related to the amyloidogenic process (APP, β-site APP cleaving enzyme 1, and PS1). While the levels of the APP processing proteins were increased in 5xFAD mice compared with those of WT mice, the levels of these proteins were not significantly different between 5xFAD and 5xFAD;OGA+/− mice (fig. S4, B and C). In addition, the levels of APP–C-terminal fragments (CTFs), C99, and C83 were not changed by OGA haploinsufficiency (fig. S4, B and C). However, the amount of Aβ plaques that accumulated in the brain was significantly decreased in the brain tissue of 5xFAD;OGA+/− mice compared with that of 5xFAD mice (Fig. 5, A and B). To evaluate whether O-GlcNAcylation affects Aβ accumulation in brain regions related to cognitive functions, we performed diaminobenzidine tetrahydrochloride (DAB) staining of the cortex and hippocampus, followed by staining with an anti-Aβ antibody. Notably, Aβ accumulation in the two examined regions was significantly reduced in 5xFAD;OGA+/− mice compared with 5xFAD mice (Fig. 5, C to F). The same result was confirmed by Aβ staining using other antibodies (fig. S4, D to G). These results were quantitatively confirmed by enzyme-linked immunosorbent assay (ELISA), with high levels of both Aβ40 and Aβ42 proteins that were detected in the brain lysates of 5xFAD mice, while Aβ protein levels were significantly lower in 5xFAD;OGA+/− mice (Fig. 5, G to J). The level of Aβ protein was lower in the brains of 5xFAD;OGA+/− mice than in those of 5xFAD mice in Western blotting (fig. S4, H to J). In addition, staining was performed using thioflavin S, which can label proteins that form β sheet structures, such as Aβ plaques. The number of observed thioflavin S–positive sites was lower in 5xFAD;OGA+/− mice than in 5xFAD mice (Fig. 5, K and L). Together, these data suggest that the amount of Aβ is significantly reduced when O-GlcNAcylation is enhanced in 5xFAD mice.
Fig. 5. Increased levels of O-GlcNAc reduce Aβ accumulation.
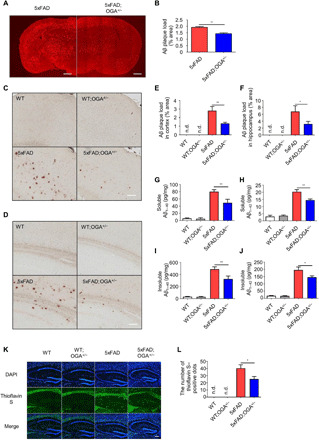
(A) Immunostaining of Aβ plaques in the brain of 5xFAD and 5xFAD;OGA+/− mice (n = 4). Scale bars, 1 mm. (B) Quantification of Aβ plaque loads in (A). (C and D) Images of Aβ immunoreactivity with DAB staining in the cortex (C) and hippocampus (D) tissue of mice samples (n = 3 to 5). Scale bars, 100 μm. (E and F) Quantification of Aβ immunoreactivity detected with DAB staining in the cortex (E) and hippocampus (F) tissue of mice samples. (G to J) The levels of soluble (G and H) and insoluble (I and J) Aβ40 and Aβ42 measured by ELISA in mouse cerebral cortex samples (n = 4 to 6). (K) Staining of Aβ plaques with thioflavin S in the hippocampus of mice samples (n = 3 to 5). Scale bar, 150 μm. (L) The number of thioflavin S–positive dots in (K). Three slices of each sample were used to normalize each sample. Values are presented as means ± SEM. *P < 0.05 and **P < 0.01 versus 5xFAD; one-way ANOVA with Tukey’s test (G to J) and two-tailed Student’s t test (B, E, F, and L). n.d., not detectable.
Increased O-GlcNAcylation suppressed the overactivation of astrocytes and microglia
To evaluate whether O-GlcNAcylation affects the neuroinflammatory response, we examined mouse brain tissue sections via immunostaining. Brain sections of 5xFAD mice contained many reactive astrocytes, and this tendency was notably less in the brain sections of 5xFAD;OGA+/− mice (Fig. 6, A and C). The neuroinflammatory response was also evaluated by microglial immunostaining, which showed that the regions positive for the microglial marker Iba1 (Ionized calcium binding adaptor molecule 1) were significantly decreased in the brain sections of 5xFAD;OGA+/− mice compared with those of 5xFAD mice (Fig. 6, B and D). Western blotting revealed that the expression levels of glial fibrillary acidic protein (GFAP) and Iba1 proteins in the brains of 5xFAD mice were significantly lower than those in the brains of 5xFAD;OGA+/− mice (Fig. 6, E to H).
Fig. 6. Increasing O-GlcNAcylation alters the inflammatory response in the brain of AD mice.
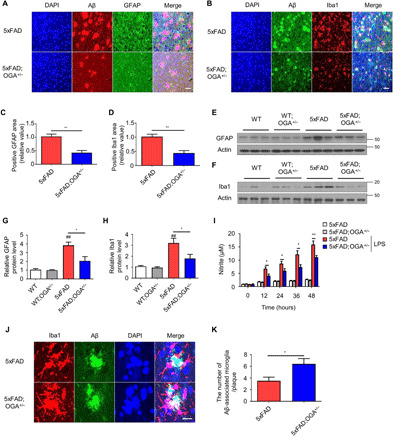
(A) Aβ plaques and GFAP in the cortical region of 5xFAD and 5xFAD;OGA+/− mice (n = 4). Scale bar, 20 μm. (B) Aβ plaques and Iba1 in the cortical region of 5xFAD and 5xFAD;OGA+/− mice (n = 4). Scale bar, 20 μm. (C) Quantification of the GFAP area in (A). (D) Quantification of the Iba1 area in (B). (E and F) Western blot analysis of GFAP (E) and Iba1 (F) in mice brain samples (n = 3). (G and H) Quantification of GFAP (G) and Iba1 (H) proteins in mice samples. (I) Nitrite concentrations in conditioned media of 5xFAD and 5xFAD;OGA+/− primary microglia stimulated by LPS (n = 4). (J) Aβ-associated microglia in cerebral cortex of 5xFAD (n = 4) and 5xFAD;OGA+/− mice (n = 5). Scale bar, 20 μm. (K) The number of Aβ-associated microglia in (J). Three slices of each sample were used to normalize each sample. Values are presented as means ± SEM. ##P < 0.01 versus WT; *P < 0.05 and **P < 0.01 versus 5xFAD; except for 5xFAD microglia stimulated by LPS (I); two-tailed Student’s t test.
Continuous stimulation of microglia by Aβ contributes to neurotoxicity through the activation of several proinflammatory cascades (29). The mRNA levels of cytokines known to participate in proinflammatory responses, such as tumor necrosis factor–α (TNF-α), interleukin-1β (IL-1β), IL-6, and inducible nitric oxide synthase (iNOS), were significantly increased in 5xFAD mice compared with 5xFAD;OGA+/− and WT mice (fig. S5, A to D). The changes in the TNF-α and IL-1β protein levels in brain tissues corresponded to the measured levels of the corresponding mRNAs (fig. S5, E and F). The alteration of iNOS related to the production of nitrite was reconfirmed by nitrite assay. The measurement of nitrite production in primary microglia in response to lipopolysaccharide (LPS) stimulation demonstrated reduced nitrite production in stimulated 5xFAD;OGA+/− microglia compared with stimulated 5xFAD microglia (Fig. 6I). These results demonstrate that the reduction in Aβ accumulation due to O-GlcNAcylation decreases the hyperactivation of astrocytes and microglia, which, in turn, prevents excessive inflammation.
Enhanced O-GlcNAcylation altered the polarization and phagocytic activity of microglia
Since we observed that proinflammatory protein levels were decreased by OGA haploinsufficiency in 5xFAD mice, we measured the levels of M2-related genes to evaluate whether an increase in O-GlcNAc levels are associated with microglial polarization into the M2 state. Arginase 1 (Arg1) expression was decreased in 5xFAD mice compared with WT mice and was restored to WT levels in 5xFAD;OGA+/− mice (fig. S6A). The levels of the found in inflammatory zone 1 protein were not different between 5xFAD and WT mice and were higher in 5xFAD;OGA+/− mice than in 5xFAD mice (fig. S6B). The mRNA levels of another M2 marker, YM1 (Chitinase-like protein-1), were higher in 5xFAD mice than in WT mice. Moreover, YM1 expression was increased in both WT;OGA+/− and 5xFAD;OGA+/− mice compared with WT mice (fig. S6C). These data indicate that O-GlcNAcylation affects microglial polarization.
To measure the phagocytic function of microglia in the brain tissues of 5xFAD mice, we assessed Aβ-associated microglial recruitment in the Aβ lesions of comparable size by fluorescence staining. We observed a greater degree of Aβ-associated microglia in the cortical region of 5xFAD;OGA+/− mice than in those of 5xFAD mice (Fig. 6, J and K). This observation was subsequently confirmed in vitro by measuring the phagocytic activity of primary microglia. The amount of Aβ remaining in the culture medium of the microglial cultures over 4 hours showed that OGA+/− microglia removed Aβ more rapidly than WT microglia (fig. S6D). This finding was quantitatively confirmed through Western blotting for extracellular and intracellular Aβ42 in primary microglia, which showed the advanced phagocytic activity of OGA insufficient microglia (fig. S6, E to G). These results suggest that an increase in O-GlcNAcylation enhances the phagocytic activity of microglia, thereby eliminating Aβ.
Abnormal mitochondrial function was attenuated by OGA insufficiency
Damaged mitochondria are an important indicator of abnormalities in neuronal functions and are found in the brain tissue of patients with AD (30). Mitochondria damaged by Aβ are shorter than normal mitochondria, and restoration of normal mitochondrial shape is necessary to mitigate AD symptoms (1). Since we found that Aβ levels were decreased in the brain tissues of 5xFAD;OGA+/− mice compared with 5xFAD mice, we further characterized the structural changes in the mitochondria in brain tissue samples from WT, WT;OGA+/−, 5xFAD, and 5xFAD;OGA+/− mice using high-resolution transmission electron microscopy (TEM). The mitochondria in the brain tissues of WT mice were normal in shape and length, whereas those in the brain tissues of 5xFAD mice were shorter (fig. S7, A to C). Conversely, the mitochondria in the brain tissues of 5xFAD;OGA+/− mice were more similar to those in the brain tissues of WT mice compared with those in the brain tissues of 5xFAD mice (fig. S7, A to C). In addition, the number of the mitochondria was increased in the brain tissues of 5xFAD mice compared with WT mice and was restored to the baseline in the brain tissues of 5xFAD;OGA+/− mice (fig. S7D). Data were analyzed by segmenting mitochondrial size. Fewer fragmented mitochondria, shorter than 200 nm, were found in 5xFAD;OGA+/− mice compared with 5xFAD mice (fig. S7, B and E), indicating that O-GlcNAcylation restored mitochondrial morphology.
Subsequently, we evaluated whether these morphological changes are related to mitochondrial function. By comparing the adenosine triphosphate (ATP) levels in the brain tissues of WT mice with those of 5xFAD mice of the same age, we found that the ATP level in the brain tissue was decreased in 5xFAD mice compared to WT mice, and the brain ATP level in 5xFAD mice was decreased in an age-dependent manner (fig. S7F). In addition, the activity of complex VI was reduced in 5xFAD mice compared with WT mice but recovered up to the WT level in 5xFAD;OGA+/− mice (fig. S7G). Moreover, the ATP levels observed in brain tissue samples at this time point were in accordance with these findings (fig. S7H). The levels of reactive oxygen species (ROS) were significantly elevated in the damaged mitochondria in the brain tissue. The ROS levels were higher in 5xFAD than in WT mice and lower in 5xFAD;OGA+/− mice than in WT mice (fig. S7I).
The maintenance of the mitochondrial membrane potential (ΔΨm) is critical for the generation of ATP. A significantly decreased ΔΨm was previously observed in a mouse model of AD (30). We evaluated whether O-GlcNAcylation rescues the ΔΨm in the primary neuron. In vitro data demonstrated a decreased ΔΨm in neurons exposed to Aβ manifested as a decrease in the tetramethylrhodamine (TMRM) positive signal, whereas OGA insufficiency or treatment with thiamet-G restored the ΔΨm to nontreated group levels (fig. S7, J to M). Overall, these findings indicated that an increase in O-GlcNAcylation can rescue mitochondrial abnormalities in a mouse model of AD and neuronal cultures.
DISCUSSION
In this study, we investigated the pleiotropic neuroprotective effects of O-GlcNAcylation on multiple models of AD (Fig. 7). O-GlcNAcylation is involved in a variety of cellular processes essential for the maintenance of intracellular homeostasis (13). The maintenance of O-GlcNAc levels is associated with decreased tissue and cell damage, leading to a decrease in stress-induced apoptosis (13). Normally, O-GlcNAcylated proteins are enriched in the brain (17). Moreover, various neurodegenerative disorders have been shown to be associated with decreased O-GlcNAc levels (13), and relatively low O-GlcNAc levels in patients with AD suggest that AD pathology is associated with a failure to maintain appropriate O-GlcNAcylation (19, 20). However, two studies have demonstrated the presence of increased O-GlcNAcylation in the brains of patients with AD (31, 32). Therefore, it is important to consider many critical factors in the analysis of O-GlcNAcylation in AD. These factors include age, gender, genetic heterogeneity, disease progression, cause of death, brain region, and postmortem interval (PMI). In this study, we further evaluated the effect of attenuating the decrease in O-GlcNAcylation in a mouse model of AD with a model of OGA gene haploinsufficiency. Beyond the original role of O-GlcNAcylation, we assessed the effects of O-GlcNAcylation on neuronal loss and necroptosis, the hallmarks of AD. Notably, we showed that an increase in global O-GlcNAc levels did not influence the total protein expression of necroptosis factors (RIPK1, RIPK3, and MLKL) in a mouse model of AD but reduced the levels of phosphorylated active necroptosis factors, which induce cell death and neuroinflammation. OGT suppresses RIPK3 activation in response to sepsis and inflammation (33). Decreased O-GlcNAc levels by OGT deletion lead to an enhanced immune response and inflammation in sepsis (33). O-GlcNAcylation of RIPK3 prevents the formation of the necrosome and downstream necroptosis signaling (33). Considering liver disease, decreased O-GlcNAc levels have been detected in patients with liver fibrosis and in mice with liver injury. Liver-specific OGT-deficient mice exhibit liver fibrosis and portal inflammation (34). In addition, these indicators of necroptosis exhibit reduced binding activity with one another under the conditions of increased global O-GlcNAcylation and, therefore, are unable to form the necrosome complex to activate necroptosis (33, 34). Before this study, to our knowledge, O-GlcNAc was not shown to regulate the activation of necroptosis in AD, although it does in other disease processes. In this study, we first found that RIPK3 is the target of O-GlcNAcylation in the brain, and the activity of necroptosis factors decreases with an increase in the amount of O-GlcNAcylated RIPK3. Reduction in activated necroptosis factors mediated by enhanced O-GlcNAc levels was clearly associated with decreased neuronal cell death and improved cognitive function in 5xFAD mice. Together, these data suggest that necroptosis activation was suppressed by the increased O-GlcNAcylation of RIPK3, which subsequently alleviated the pathological manifestations of AD. Therefore, on the basis of our findings, a therapeutic approach that promotes O-GlcNAcylation offers a new opportunity for the treatment of AD, including neuronal loss and cognitive impairments associated with this disease.
Fig. 7. Diagram showing the mechanism of how O-GlcNAcylation ameliorates AD pathology.
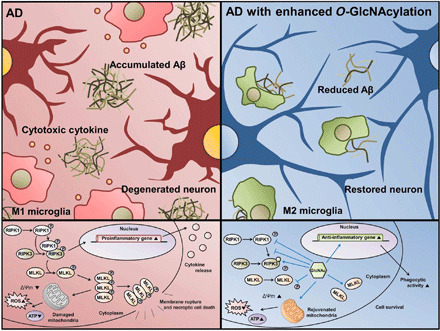
RIPK3 is directly O-GlcNAcylated, which suppresses the phosphorylation of RIPK3 and interaction between RIPK1 and RIPK3. O-GlcNAcylation reduces the activation of necroptosis factors, formation of necrosome, and necroptotic cell death. In addition, O-GlcNAcylation restores phagocytic activity of microglia and mitochondria function. Therefore, O-GlcNAcylation ameliorates AD pathologies including accumulated Aβ, neuroinflammation, and neuronal loss.
In addition to the positive effect of O-GlcNAcylation on neuronal survival, we observed that O-GlcNAcylation reduces the severity of cellular stresses in the brain, specifically the increased Aβ burden and neuroinflammation, which are the hallmarks of AD progression (4). Aβ initiates a cascade of inflammatory events that results in neuronal death and, eventually, dementia (29). As a protective role of O-GlcNAcylation in AD, treatment with an OGA inhibitor was reported to successfully inhibit Aβ accumulation in the 5xFAD model (35). Although Aβ plaques accumulated with age in 5xFAD mice, 5xFAD;OGA+/− mice exhibited significantly decreased amounts of Aβ plaques. We further examined this effect from two perspectives: a reduction in Aβ production and improvement in Aβ clearance. A previous report showed that a treatment with an OGA inhibitor activated non-amyloidogenic pathways in vitro (36). Conversely, pharmacological inhibition of OGA did not alter the APP processing–related protein including APP-CTFs in primary hippocampal neurons (22). We observed that 5xFAD;OGA+/− and 5xFAD mice did not show any difference in the levels of proteins involved in the amyloidogenic pathway or APP-CTFs. Further detailed studies elucidating the O-GlcNAcylation effects on the amyloidogenic pathway would strengthen the association between amyloid pathology and O-GlcNAcylation.
Although O-GlcNAcylation in the brain did not directly affect Aβ production, it initiated a number of changes in microglia affected by Aβ plaques. Microglia are the major cell type that regulate neuroinflammation in the brain (4). Activated microglia trigger a variety of inflammatory cascades, which, in turn, result in cytokine secretion (29). These secreted cytokines can elicit neurotoxic effects, that is, continuous cytokine stimulation results in neuronal degeneration (29). We found that an increase in O-GlcNAc levels due to OGA haploinsufficiency reduced the activation of microglia and astrocytes, which resulted in attenuating the expression levels of proinflammatory cytokines in 5xFAD mice. Microglia undergo dynamic changes in response to environmental stimuli and are classified as M1 (proinflammatory) or M2 (anti-inflammatory role) cells, which have either a neurotoxic or a neuroprotective function (4). In AD, Aβ plaques and proinflammatory cytokines, such as IL-1β and TNF-α, skew microglia into the M1 state and diminish their phagocytic capability (37). Conversely, M2-type microglia exhibit enhanced Aβ clearance and secrete cytokines associated with neuroprotection (4). Intracerebral injection of IL-4 and IL-13 have been reported to attenuate Aβ plaques and cognitive impairment with the up-regulation of Arg1- and YM1-positive M2 cells in APP23 mice (38). Regardless of the rate of Aβ production, improvement in Aβ clearance effectively alleviates the symptoms of AD (39). In this study, we demonstrated that enhanced O-GlcNAcylation successfully eliminates Aβ accumulation. A few reports have shown the effect of OGA inhibitors on Aβ accumulation (22, 35), but the mechanism of reduction of Aβ accumulation by O-GlcNAcylation is unclear. Although increased O-GlcNAcylation in the brain tissue of 5xFAD mice did not inhibit the amyloidogenic pathways, it stimulated the M2 polarization of microglia and thereby improved phagocytic activity to increase Aβ clearance. We have unraveled the mechanism by which O-GlcNAcylation ameliorates the Aβ pathology; however, the reason for the altered level of O-GlcNAc in AD is unclear. Some reports have suggested that exposing neuronal cells to Aβ triggered the reduction of O-GlcNAcylation (19, 40). In the brain of 3xTg-AD mice, the level of O-GlcNAcylation appeared to decrease with an increase in OGA activity (41). We found that the activity of O-GlcNAc–related enzymes in neurons exposed to Aβ is significantly affected, indicating that Aβ interferes with the homeostasis of O-GlcNAcylation. Further studies are needed to elucidate the detailed mechanisms underlying the regulation of O-GlcNAcylation in AD.
In addition, the mitochondrion, an organelle that supplies energy to cells, is known to be sensitive to stress signals. Although there are conflicting opinions on whether mitochondrial abnormalities are the cause or result of AD, it is generally accepted that the mitochondria are damaged in AD (30). As AD progresses, the mitochondria are morphologically fragmented, resulting in abnormal ROS production and reduced cellular respiration by dysfunctional mitochondria (30). We found that increased O-GlcNAc levels in the brain are associated with the recovery of mitochondrial function and normal mitochondrial morphology. The rescue of mitochondrial dysfunction was previously shown to be closely associated with the alleviated symptoms of AD (30), and our results support this association.
Since O-GlcNAcylation is an important posttranslational modification involved in neurodegenerative diseases, several studies have examined the small molecules that modulate O-GlcNAcylation. Two small-molecule inhibitors of OGA, namely, MK-8719 (42) and ASN120290 (43), are currently in phase 1 clinical trial for progressive supranuclear palsy and related tauopathies. Thus, our study provides a robust logical and experimental basis for OGA as a promising therapeutic target for altering disease progression in AD, not limited to tauopathy.
In conclusion, our findings suggest that O-GlcNAc up-regulation can ameliorate neuronal death and neuroinflammation in the brain and, subsequently, cognitive dysfunction in AD models. An increase in O-GlcNAc levels due to OGA insufficiency inhibited the activation of necroptosis factors and decreased neuronal death, thereby rescuing AD-related cognitive deficits. In addition, Aβ accumulation was reduced in 5xFAD mice due to the conversion of proinflammatory microglia into anti-inflammatory microglia by the increased global O-GlcNAcylation. In addition, the function of damaged mitochondria in the brain of an AD mouse model was restored to normal levels. Therefore, the regulation of O-GlcNAcylation represents a promising novel approach for AD treatment.
MATERIALS AND METHODS
Human brain specimens
Inferior parietal lobule specimens from the brains of patients with AD and individuals without dementia collected by the University of Kentucky Alzheimer’s Disease Center Autopsy Program were used for this study. All patients with AD met the clinical and neuropathological diagnostic criteria for AD (44–46). Control subjects had no history or neuropathological symptoms of brain disorder. Sex, age, PMI, Braak stage, and plaque density of the subjects are shown in table S1. Tissue specimens were rapidly removed at autopsy and were frozen and stored at −80°C.
Mice
B5SJL-Tg (APPSwFlLon,PSEN1*M146L*L286V)6799Vas/Mmjax (5xFAD) mice were purchased from the Jackson laboratory. The 5xFAD mice were backcrossed to the C57BL/6J mice for eight generations. The OGA+/− mice were obtained from Suh’s laboratory (18). The 5xFAD-heterozygous mice were maintained on C57BL6/SJL background. Thus, 5xFAD mice were backcrossed to C57BL/6J mice for at least eight generations (45). The backcrossed 5xFAD mice were used to produce 5xFAD;OGA+/− mice. Genotyping of all mice was performed by polymerase chain reaction (PCR) using DNA extracted from a tail snip. Genotyping was performed as previously described (47). Three to four same-sex littermate mice were housed in a single cage until they were age appropriate for the experiment. This study was reviewed and approved by the Institutional Animal Care and Use Committee of Sungkyunkwan University (approval number: 2018-02-01-1).
Primary neuronal cell culture
Primary neuronal cell cultures were prepared from Sprague-Dawley rat embryos at day 18.5 and genotyped mouse pups on postnatal day 1, as described previously (30). Brains were dissected from rat embryos and mouse pups, and the blood vessels and meninges were removed. Dissected brains were treated with trypsin for 10 min at 37°C and were gently triturated with a pipette. Trypsinized tissues were passed through a 100-μm cell strainer to isolate individual cells. Isolated cells were plated on a poly-l-lysine–coated cover glass (Sigma-Aldrich). Cells were maintained in a neurobasal medium (Thermo Fisher Scientific) containing the B-27 supplement (Thermo Fisher Scientific) at 37°C in a humidified atmosphere containing 5% CO2. After 10 days of culturing, primary neurons were evaluated according to the experimental schedule.
Primary microglia culture
Cerebral cortical and hippocampal tissues were dissected from WT and OGA+/− mice at postnatal day 1 or 2 and subjected to trypsinization and homogenization by triturating with pipettes. Homogenized suspensions were passed through a 70-μm cell strainer to separate individual cells. The cells from each homogenized brain were plated in two T-75 flasks containing a growth medium [10% fetal bovine serum (Capricorn), Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Thermo Fisher Scientific), 1% penicillin/streptomycin (Thermo Fisher Scientific), 1% l-glutamine (Thermo Fisher Scientific), 1% sodium pyruvate (Thermo Fisher Scientific), and 1% nonessential amino acids (Thermo Fisher Scientific)] and cultured at 37°C in a humidified atmosphere containing 5% CO2. Mixed microglia and astrocytes were grown until isolation on day 16. After 16 days, microglia were isolated according to the manual of a cell isolation kit (STEMCELL Technologies) and cultured according to the experimental schedule.
Y-maze
The Y-maze is a behavioral test that uses the rodent’s tendency to move to a new arm rather than to a previously existing one. The Y-maze consisted of three arms inclined at an angle of 120°. All arms were 5 cm wide and 30 cm long and surrounded by 15-cm-high walls made of dark gray plastic. The mice were randomly placed in one of the three arms and were free to explore the maze for 10 min. During this phase, the arm exploration sequence was recorded, and the number of times the three arms were entered at once was measured. At the end of each test, the mouse was moved to its customary cage, and the maze was cleaned. Short-term memory was evaluated by calculating the ratio of the total number of explorations to the number of three arms explored at once.
Novel object recognition test
Novel object recognition test is a common behavioral experiment evaluating the cognitive abilities of experimental animals with less stress (45). This test was conducted in three stages: habituation, training, and test sessions. The experiment was carried out in an opaque dark-colored plastic box (length × width × height, 50 cm × 50 cm × 50 cm). On the first day of the test, mice were placed in an empty open-field box for 10 min to adapt to the experimental conditions. After the habituation step was completed, the open field box was cleaned, and the next mouse was exposed to the same conditions. The next day, a training session was performed, in which two similar objects were placed in a box, and mice were allowed to explore for 10 min. The two objects observed by the mice had a similar color and texture and were slightly larger in size than the mice, making it impossible for the mice to climb. After the training session, the mice were rested for 24 hours and then were subjected to the test session. In the test session, one of the two objects was replaced with a new object, and the exploration pattern was recorded. Meaningful exploration pattern was defined as mice sniffing the objects with their noses or their faces touching its surface or within 2 cm of the objects. After the recording was finished, the box and objects were wiped off. The exploration record was quantified by dividing the time spent exploring each object by the total time spent in approaching the object. Tests were performed by an operator blinded to the mouse type involved in the test.
Morris water maze
The Morris water maze measures long-term spatial acquisition and memory capacity. It was performed as described previously (30). The test consisted of a 4-day training trial and a 1-day probe trial. The Morris water maze consisted of a pool with a diameter of 150 cm surrounded by 60-cm-high walls. Before the experiment, the pool was filled with water containing a nontoxic white pigment, and the temperature was set to 22°C. The pool was divided into four quarters, one of which included a transparent platform with a diameter of 9 cm and a height of 25 cm. The pool was filled with water to a level of 1.5 cm above the platform; hence, the platform could not be identified from the perspective of the experimental animal. Several visual cues were placed around the pool, which remained in the same position throughout the test. In the training trial, mice were subjected to this experiment three times a day, with 1-hour intervals between the experimental sessions. The escape latency was recorded three times a day during training sessions. The average of these three records was calculated and compared between the groups. During the test, the location of the mice in the pool changed in each trial, whereas the position of the hidden platform was unchanged. Each individual trial required 60 s. During this time, if the mouse could not find the platform, then the experimenter placed the mouse on the platform for 10 s to observe the surroundings. However, the experimenter would retreat quickly behind the curtain to remain hidden to avoid mice being distracted from observing the surroundings. The probe test was carried out 24 hours after the last test trial. All the other conditions were kept the same as the test trial, except for the removal of the hidden platform. Under this condition, mice were allowed to swim in the pool for 60 s. During the 60-s period, the time spent in the area where the hidden platform was located was recorded, and the number of times the mouse passed the location of the platform was counted.
Passive avoidance task
The passive avoidance task measures how well rodents with neurodegenerative diseases acquire and maintain fear-based memories. This test is based on the tendency of rodents to seek darker areas. The test was performed as described previously (30). The passive avoidance task appratus consisted of a transparent clear room and a dark room connected, with a barrier wall between them. The bottom of the apparatus had metal bars, with bars in the dark room designed to conduct electricity. This experiment was divided into two sessions: a training session, during which the mice were subjected to an electrical shock, and a test session, in which the memory of shock was evaluated after 24 hours. For the training session, mice were initially placed in the transparent room. After some exploration, the mouse entered the dark room and closed the door in between. After the mouse entered the dark room, an electric shock of 0.4-mA intensity was applied for 2 s. Because mice excreted a lot of feces after receiving an electric shock, the dark room was cleaned between the experiments. A test session was conducted 24 hours after training. Mice subjected to electric shock were placed in the transparent room, and the time until they entered the darkroom was measured. The test session lasted for up to 300 s. The experiment proceeded in a random order, and the experimenter was unaware of which mice were being tested.
Intracerebroventricular injection
Intracerebroventricular injection was performed using a previously described method (48). A 28-gauge stainless steel needle (3.0 mm long) attached to a microsyringe was used for all experiments. The 12-week-old Institute of Cancer Research (ICR) mice were anesthetized, and the needle was inserted 1 mm to the right of the midline point equidistant from each eye, at an equal distance between the eyes and the ears. Aβ peptide (10 μM) or saline was delivered gradually within 5 s. The injected volume of Aβ was 5 μl, and the final amount of injected Aβ was 0.2257 μg. Neither the insertion of the needle nor the injection of the peptide or saline had a significant influence on survival.
Tissue sample preparation
All brain tissue samples were prepared after the completion of behavioral tests. Tissue sample preparation was performed as described previously (45). Briefly, mice were anesthetized with Zoletil (Virbac) and Rompun (Bayer). Once anesthetized, mice were perfused with phosphate-buffered saline (PBS; P3813) with 0.9% NaCl concentration. Samples for Western blotting analysis were dissected to separate the hippocampus and cortex. The dissected samples were flash-frozen in liquid nitrogen and stored at −80°C until analysis. For immunohistochemistry, brains were perfused with 4% paraformaldehyde (PFA) after the complete removal of the blood. Perfused samples were fixed with 4% PFA at 4°C overnight. Brain tissues were embedded in paraffin blocks and serially sectioned at 5 μm in thickness. The hippocampus series of sections was used for immunohistochemistry.
Aβ preparation and treatment
Aβ1–42 peptide (ANYGEN) was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP; Sigma-Aldrich). Aβ-HFIP solution was incubated at room temperature until the solution became colorless and clear. The prepared solution was aliquoted and lyophilized using a SpeedVac concentrator. The lyophilized Aβ peptide was sealed and stored at −70°C until the experiment. Before the experiment, the lyophilized Aβ peptide was thawed to room temperature, dissolved in dimethyl sulfoxide (Merck), and adjusted to a final concentration of 5 μM using the DMEM/F12 media. To investigate the effect of Aβ peptide, primary neurons were incubated with Aβ peptide for 24 hours.
Thiamet-G treatment
Thiamet-G (Sigma-Aldrich) was dissolved in distilled water. Cells were incubated with 2 μM thiamet-G for 24 hours. For the intracerebroventricular Aβ injection model, 12-week-old ICR mice were intraperitoneally injected with thiamet-G (50 mg/kg) for 3 days.
Nitrite assay
Measurement of nitrite levels in microglia cultured media was performed according to the manufacturer’s protocol (Cell Biolabs). Microglia were seeded in plates and stimulated with LPS (100 ng/ml) for 12 hours. Culture media were collected over time and added into a 96-well plate, mixed with kit reagents, and incubated for 10 min. The nitrite concentration in the media was determined on the basis of standard values.
Western blot analysis
The samples were prepared and processed on the basis of previously described standard procedures (46). Brain tissue samples were lysed in a tissue protein extraction buffer (Thermo Fisher Scientific) containing phosphatase and protease inhibitors and subsequently homogenized. After incubation for 1 hour on ice, the supernatant and pellet were separated by centrifugation at 10,000g for 30 min at 4°C. The supernatant was used for the subsequent analysis of the changes in protein levels in the tissue, whereas the pellet was used to measure the Aβ amount. The insoluble pellet was dissolved in a buffer containing 5% SDS (Thermo Fisher Scientific) and 8 M urea (Merck). The amount of protein in each sample was quantified using the Pierce BCA assay (Thermo Fisher Scientific). Equal amounts of proteins were mixed with a 4× sample buffer (Thermo Fisher Scientific) containing 5% mercaptoethanol (Sigma-Aldrich) and boiled for 10 min at 95°C. Samples were prepared just before Western blotting. Approximately 10 to 20 μg of quantified samples were loaded onto 8 to 16% SDS–polyacrylamide gels and run until the proteins were completely separated. After the completion of the running step, separated proteins were transferred to a 0.45 μM polyvinylidene difluoride membrane (Merck) at 85 V and 180 to 220 mA for 90 min. The membrane with transferred proteins was blocked with 5% nonfat dry milk for 1 hour at room temperature and subsequently incubated with a primary antibody at 4°C with gentle shaking overnight. The next day, the membrane was washed three times with tris-buffered saline containing 0.1% Tween 20 (Merck) for 10 min per wash and incubated with peroxidase-conjugated anti-mouse or anti-secondary antibodies for 1 hour at room temperature. The final immunoreactive band was visualized using enhanced chemiluminesence solution (DONGIN) after the second washing step. The used membrane was reprobed for β-actin, which was used as a loading control. Detailed information about antibody procedures is shown in table S2.
Immunoprecipitation
Immunoprecipitation was performed using a previously described method (49). For immunoprecipitation, mouse brain tissues were lysed using a lysis buffer [30 mM Hepes (pH 7.5), 150 mM NaCl, 1 mM EDTA, 0.01% SDS, and 0.5% NP-40] containing phosphatase (BioVision) and protease inhibitors (BioVision). The homogenized brain lysates were precleared by incubation with protein G agarose beads (Santa Cruz Biotechnology) at 4°C for 6 hours. An anti-RIPK3 antibody (1:200 dilution) was coupled to protein G agarose beads in the lysis buffer for 4 hours at 4°C. Each lysate was subsequently mixed with the antibody-coupled beads and incubated overnight at 4°C. The following day, the beads were washed three times in cold lysis buffer and boiled with the sample buffer for 10 min at 95°C.
Quantitative real-time PCR
Total RNA was extracted from brain tissue samples after homogenization with the RNAiso plus solution and total RNA extraction reagent (TaKaRa). RNA was prepared according to the manufacturer’s protocol. The isolated RNA was treated with ribonuclease-free deoxyribonuclease to remove genomic DNA contamination. RNA aliquots (500 ng) of each sample were used for complementary DNA (cDNA) synthesis using a cDNA synthesis kit (TaKaRa). Subsequently, we performed real-time reverse transcription PCR (RT-PCR) using a CFX Connect (Bio-Rad, USA). PCR amplification was performed using the SYBR Premix Ex Taq II (TaKaRa). The PCR conditions for all genes were as follows: 95°C for 30 s, 40 cycles at 95°C for 5 s, and 60°C for 30 s. We calculated the differences between the Ct values for the experimental and reference genes (glyceraldehyde-3-phosphate dehydrogenase) as ΔCt. The quantitative RT-PCR primer sequences are listed in table S3.
Immunohistochemistry
Histological samples were stained using a previously published standard procedure (45). Serially sectioned brain slices were deparaffinized and hydrated using a clearing agent (VWR Life Science) and serial concentrations of ethyl alcohol. For antigen retrieval, slices were placed at 100°C in an incubator for 10 min and blocked at room temperature for 1 hour in PBS with Tween 20 solution containing 5% bovine serum albumin (BSA; Bovogen). Following blocking, the primary antibodies were added, and slices were kept overnight at 4°C. The slices were sufficiently washed the next day and incubated with secondary fluorescent antibodies at room temperature for 2 hours. Upon the completion of all incubation steps, the slices were rinsed and mounted. Images were acquired using a confocal microscope (TCS SP8 HyVolution, Korea), and immunostaining data were evaluated using the ImageJ 1.48v software. To quantify the degree of staining between each group, the value of the activated area of each sample without nonspecific signals and with same threshold values was used. Detailed antibody information is shown in table S2.
DAB staining
DAB staining was performed in accordance to the manufacturer’s protocol (Abcam), with slight modifications. Briefly, brain slices were subjected to progressive staining using methods similar to those used in the preparation of immunostaining samples until the antigen retrieval step. All slices were free-floating and treated with an oxidase blocker for 10 min and subsequently washed. Slices were blocked with 5% BSA for 1 hour, followed by overnight incubation with a primary Aβ antibody at 4°C. The next day, slices were washed and incubated with the biotinylated goat anti-mouse antibody at room temperature for 20 min. Thereafter, the slides were rinsed, and the slices were incubated with streptavidin peroxidase for 10 min. A mixture of DAB chromogen and substrates was applied to the slides. The color change resulting from staining was observed using a microscope, and washing was performed three times immediately after the appropriate state was reached. Last, the slices were dried and mounted.
Nissl staining
Following deparaffinization of paraffin-embedded tissue sections, they were incubated for 10 min in 0.1% cresyl violet solution (Sigma-Aldrich). During this process, light was blocked, and the staining solution was preheated. The sections were washed with distilled water and visually checked under a microscope to determine the clearest state, while incubating in 95% ethyl alcohol. After clearing the xylene twice for 5 min, sections were mounted using a permanent mounting medium (D.P.X., Sigma-Aldrich).
Thioflavin S staining
Thioflavin S staining was performed using a method based on standard procedures. Deparaffinized tissue sections were incubated for 10 min with 1% filtered aqueous thioflavin S (Sigma-Aldrich). After serial washing with ethyl alcohol, the last wash was performed with distilled water. The sections were then mounted with aqueous mounting media.
Fluoro-Jade C staining
Fluoro-Jade C staining was performed according to the manufacturer’s protocol (Merck). Deparaffinized tissue sections were incubated for 10 min in 0.06% potassium permanganate solution (Merck). After washing with distilled water, samples were incubated in 0.0001% Fluoro-Jade solution in distilled water with 0.1% acetic acid (Merck) for 20 min. Sections were washed with distilled water, cleared with xylene, and mounted with D.P.X. Ambient light was blocked as much as possible during the entire procedure.
Measurement of Δψm
To measure mitochondrial membrane potential, primary neurons were stained with TMRM (Thermo Fisher Scientific). Staining was performed according to the manufacturer’s protocol with slight modifications. TMRM solution was diluted in a cell culture medium to a final concentration of 100 nM, and the cells were incubated for 30 min. Ten minutes before TMRM staining, Hoechst dye (1 μg/ml; Thermo Fisher Scientific) was applied to stain the nuclei of neurons, followed by two washes with PBS.
ROS assay
The ROS assay kit (DoGenBio) was used to measure the degree of oxidative stress according to the manufacturer’s protocol. To reduce the variations in ROS between the analyzed samples, the entire process (from sampling to measurement) was conducted within 1 day. The prepared standard and samples were added to a 96-well plate, and the samples were incubated with the probe provided in the kit for 30 min. After the completion of the reaction, the absorbance of the plated samples was measured at 560 nm using a microplate reader (BioTek, Synergy HTX, USA).
OGT and OGA activity assay
The OGT activity test was performed using a previously described method with some modifications (50). This method is a coupled enzyme method based on the production of uridine diphosphate (UDP) by OGT. Briefly, the synthesized peptide (YSDSPSTST, AB Frontier), UDP-GlcNAc (Merck), and brain lysate were incubated to separate UDP from UDP-GlcNAc. After that, phosphoenolpyruvate (Merck) and pyruvate kinase (Merck) were added to the mixed samples to catalyze pyruvate. Last, the level of pyruvate was measured using the pyruvate assay kit (BioVision) to confirm OGT activity in the brain tissue. The measurements of OGA activity were performed using the β-N-acetylglucosaminidase assay kit (Sigma-Aldrich) according to the manufacturer’s instructions. Equilibrated substrates and standard solutions were incubated with the reaction components for 30 min. After treatment with the stop solution, activity was quantified by measuring the absorbance of samples at 405 nm using a microplate reader.
Complex IV activity test
To measure mitochondrial activity, a complex IV activity test was conducted using a commercial kit (Abcam). All procedures were carried out according to the manufacturer’s protocol with slight modifications. Brain tissues were lysed using the detergent provided in the kit and placed into a 96-well plate precoated with a complex IV–specific antibody. Following 3 hours of incubation, the plate was rinsed and incubated with the same amount of assay solution. The absorbance, corresponding to the enzyme activity, was measured using a microplate reader. Plate measurements were taken at 1-min intervals for 30 min, and the slope of the generated graph was quantified to calculate the activity of each sample.
Measurement of ATP
ATP levels were measured using the ATP Determination Kit (Molecular Probes) according to the manufacturer’s instructions. A total of 90 μl of the prepared reaction mix was added to 10 μl of tissue homogenate and ATP standard solutions. Then, a microplate reader was used to measure the luminescence at 560 nm. Concentrations were calculated on the basis of the values measured using the standards provided in the kit.
Oral glucose tolerance test
Mice fasted overnight were used in the oral glucose tolerance test. Before injecting the glucose, resting blood glucose levels were measured. To initiate the experiment, glucose was orally injected to mice (1 mg/g of body weight). Blood glucose levels were measured at 0, 30, 60, and 120 min after injection. The concentration of glucose in the blood was measured using a glucometer (Accu-Check, Roche).
Enzyme-linked immunosorbent assay
The levels of Aβ40 (Thermo Fisher Scientific), Aβ42 (Thermo Fisher Scientific), and cytokines, IL-1β (Abcam) and TNF-α (R&D Systems), in mouse brain tissue samples were analyzed using ELISA kits according to the manufacturer’s instructions. The samples were prepared to obtain tissue lysate supernatants for analysis using procedures described in the ELISA kit manual. To quantify the concentration of the target molecule, samples were added on a precoated plate and incubated with a horseradish peroxidase–conjugated detection antibody. Then, a microplate reader was used to measure the absorbance. Concentrations were calculated on the basis of the values measured using the standards provided in the kit.
Transmission electron microscopy
Mitochondrial morphology was analyzed in brain samples collected from the four genotyped mice used in the behavioral experiments using TEM. Samples were fixed with a solution containing 4% PFA and 1.5% glutaraldehyde and incubated overnight at 4°C. Next, the samples were rinsed, dehydrated using acetone and serial concentrations of ethanol, and passed through a transition solvent. The samples were subsequently embedded in liquid resin. The embedded samples were sectioned into semithin sections (1 μm) with a glass knife and stained with toluidine blue to select a small area for ultrathin sectioning. Then, the selected regions were subjected to ultrathin sectioning using a diamond knife to generate samples with a thickness of 50 to 70 nm. The sections were placed on a metal grid, stained with lead–uranyl acetate, and analyzed using a transmission electron microscope (Hitachi, HT7800, Japan). At least seven random images were collected for each independent mouse sample preparation. The shape and length of mitochondria were analyzed by a researcher blinded to the experimental groups using the ImageJ software.
Statistical analysis
All histological and biochemical data analyses were performed by a researcher blinded to the experimental groups. All statistical analyses, namely, two-tailed Student’s t test, one-way analysis of variance (ANOVA) with Tukey’s test, and two-way ANOVA, were performed using GraphPad Prism 8 (GraphPad Software). All data are expressed as means ± SEM from three independent experiments. Differences between groups were considered significant at P < 0.05 (*P < 0.05, **P < 0.01, and ***P < 0.001).
Supplementary Material
Acknowledgments
Funding: This study was supported by the National Research Foundation of Korea (NRF-2019R1A2C3011422, NRF-2019R1A5A2027340, NRF-2018M3C7A1021851, and NRF-2017M3C7A1048268) and a grant from the Korea Polar Research Institute (PE20010). Author contributions: J.P. and D.-G.J. conceived the study. J.P., H.-J.H., S.W.S., and D.-G.J. designed the research. E.S.C. and S.H.B. performed cognitive testing in animal models. Y.C. and J.L. performed immunostaining. H.K.K. and J.H. conducted the animal work and performed data analyses. E.K., J.H.S., and J.K. conducted the imaging experiments and analyzed the imaging data. Y.R.Y., M.P., S.H.K., T.V.A., H.J., S.W.S., P.-G.S., and D.-G.J. conducted data analysis and contributed tools and reagents. J.P. and D.-G.J. wrote the manuscript. All authors approved and contributed to the final version of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The OGA+/− mice can be provided by P.-G.S. pending scientific review and a completed material transfer agreement. Requests for the OGA+/− mice should be submitted to P.-G.S. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/3/eabd3207/DC1
REFERENCES AND NOTES
- 1.Wang J., Gu B. J., Masters C. L., Wang Y.-J., A systemic view of Alzheimer disease—Insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 13, 612–623 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Lee H., Kim D., Lee W., Kim H. Y., Kim Y., Preventive approach for overcoming dementia. Arch. Pharm. Res. 42, 647–657 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Livingston G., Sommerlad A., Orgeta V., Costafreda S. G., Huntley J., Ames D., Ballard C., Banerjee S., Burns A., Cohen-Mansfield J., Cooper C., Fox N., Gitlin L. N., Howard R., Kales H. C., Larson E. B., Ritchie K., Rockwood K., Sampson E. L., Samus Q., Schneider L. S., Selbæk G., Teri L., Mukadam N., Dementia prevention, intervention, and care. Lancet 390, 2673–2734 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Li Q., Barres B. A., Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 18, 225–242 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Caccamo A., Branca C., Piras I. S., Ferreira E., Huentelman M. J., Liang W. S., Readhead B., Dudley J. T., Spangenberg E. E., Green K. N., Belfiore R., Winslow W., Oddo S., Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 20, 1236–1246 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Galluzzi L., Kroemer G., Necroptosis: A specialized pathway of programmed necrosis. Cell 135, 1161–1163 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Rubinsztein D. C., RIPK1 promotes inflammation and β-amyloid accumulation in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 114, 10813–10814 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan J., Amin P., Ofengeim D., Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 20, 19–33 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang S.-H., Lee D. K., Shin J., Lee S., Baek S., Kim J., Jung H., Hah J.-M., Kim Y., Nec-1 alleviates cognitive impairment with reduction of Aβ and tau abnormalities in APP/PS1 mice. EMBO Mol. Med. 9, 61–77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ito Y., Ofengeim D., Najafov A., Das S., Saberi S., Li Y., Hitomi J., Zhu H., Chen H., Mayo L., Geng J., Amin P., DeWitt J. P., Mookhtiar A. K., Florez M., Ouchida A. T., Fan J.-b., Pasparakis M., Kelliher M. A., Ravits J., Yuan J., RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353, 603–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ofengeim D., Ito Y., Najafov A., Zhang Y., Shan B., DeWitt J. P., Ye J., Zhang X., Chang A., Vakifahmetoglu-Norberg H., Geng J., Py B., Zhou W., Amin P., Berlink Lima J., Qi C., Yu Q., Trapp B., Yuan J., Activation of necroptosis in multiple sclerosis. Cell Rep. 10, 1836–1849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iannielli A., Bido S., Folladori L., Segnali A., Cancellieri C., Maresca A., Massimino L., Rubio A., Morabito G., Caporali L., Tagliavini F., Musumeci O., Gregato G., Bezard E., Carelli V., Tiranti V., Broccoli V., Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in Parkinson’s disease models. Cell Rep. 22, 2066–2079 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park J., Lai M. K. P., Arumugam T. V., Jo D.-G., O-GlcNAcylation as a therapeutic target for Alzheimer’s disease. Neuromol. Med. 22, 171–193 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Lewis Y. E., Galesic A., Levine P. M., De Leon C. A., Lamiri N., Brennan C. K., Pratt M. R., O-GlcNAcylation of α-synuclein at serine 87 reduces aggregation without affecting membrane binding. ACS Chem. Biol. 12, 1020–1027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hastings N. B., Wang X., Song L., Butts B. D., Grotz D., Hargreaves R., Hess J. F., Hong K.-L. K., Huang C. R.-R., Hyde L., Laverty M., Lee J., Levitan D., Lu S. X., Maguire M., Mahadomrongkul V., McEachern E. J., Ouyang X., Rosahl T. W., Selnick H., Stanton M., Terracina G., Vocadlo D. J., Wang G., Duffy J. L., Parker E. M., Zhang L., Inhibition of O-GlcNAcase leads to elevation of O-GlcNAc tau and reduction of taupathy and cerebrospinal fluid tau in rTg4510 mice. Mol. Neurodegener. 12, 39 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuzwa S. A., Shan X., Macauley M. S., Clark T., Skorobogatko Y., Vosseller K., Vocadlo D. J., Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 8, 393–399 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Gao Y., Wells L., Comer F. I., Parker G. J., Hart G. W., Dynamic O–glycosylation of nuclear and cytosolic proteins: Cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J. Biol. Chem. 276, 9838–9845 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Yang Y. R., Song M., Lee H., Jeon Y., Choi E.-J., Jang H.-J., Moon H. Y., Byun H.-Y., Kim E.-K., Kim D. H., Lee M. N., Koh A., Ghim J., Choi J. H., Lee-Kwon W., Kim K. T., Ryu S. H., Suh P.-G., O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging Cell 11, 439–448 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Pinho T. S., Correia S. C., Perry G., Ambrósio A. F., Moreira P. I., Diminished O-GlcNAcylation in Alzheimer’s disease is strongly correlated with mitochondrial anomalies. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 2048–2059 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Liu F., Shi J., Tanimukai H., Gu J., Gu J., Grundke-Iqbal I., Iqbal K., Gong C.-X., Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain 132, 1820–1832 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang A. C., Jensen E. H., Rexach J. E., Vinters H. V., Hsieh-Wilson L. C., Loss of O-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 113, 15120–15125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuzwa S. A., Shan X., Jones B. A., Zhao G., Woodward M. L., Li X., Zhu Y., McEachern E. J., Silverman M. A., Watson N. V., Gong C.-X., Vocadlo D. J., Pharmacological inhibition of O-GlcNAcase (OGA) prevents cognitive decline and amyloid plaque formation in bigenic tau/APP mutant mice. Mol. Neurodegener. 9, 42 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan E. P., McGreal S. R., Graw S., Tessman R., Koppel S. J., Dhakal P., Zhang Z., Machacek M., Zachara N. E., Koestler D. C., Peterson K. R., Thyfault J. P., Swerdlow R. H., Krishnamurthy P., DiTacchio L., Apte U., Slawson C., Sustained O-GlcNAcylation reprograms mitochondrial function to regulate energy metabolism. J. Biol. Chem. 292, 14940–14962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cha M.-Y., Cho H. J., Kim C., Jung Y. O., Kang M. J., Murray M. E., Hong H. S., Choi Y.-J., Choi H., Kim D. K., Choi H., Kim J., Dickson D. W., Song H. K., Cho J. W., Yi E. C., Kim J., Jin S. M., Mook-Jung I., Mitochondrial ATP synthase activity is impaired by suppressed O-GlcNAcylation in Alzheimer’s disease. Hum. Mol. Genet. 24, 6492–6504 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J., McQuade T., Siemer A. B., Napetschnig J., Moriwaki K., Hsiao Y.-S., Damko E., Moquin D., Walz T., McDermott A., Chan F. K.-M., Wu H., The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macauley M. S., Shan X., Yuzwa S. A., Gloster T. M., Vocadlo D. J., Elevation of global O-GlcNAc in rodents using a selective O-GlcNAcase inhibitor does not cause insulin resistance or perturb glucohomeostasis. Chem. Biol. 17, 949–958 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keembiyehetty C., Love D. C., Harwood K. R., Gavrilova O., Comly M. E., Hanover J. A., Conditional knock-out reveals a requirement for O-linked N-acetylglucosaminase (O-GlcNAcase) in metabolic homeostasis. J. Biol. Chem. 290, 7097–7113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oakley H., Cole S. L., Logan S., Maus E., Shao P., Craft J., Guillozet-Bongaarts A., Ohno M., Disterhoft J., Eldik L. V., Berry R., Vassar R., Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butovsky O., Weiner H. L., Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 19, 622–635 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baek S. H., Park S. J., Jeong J. I., Kim S. H., Han J., Kyung J. W., Baik S.-H., Choi Y., Choi B. Y., Park J. S., Bahn G., Shin J. H., Jo D. S., Lee J.-Y., Jang C.-G., Arumugam T. V., Kim J., Han J.-W., Koh J.-Y., Cho D.-Y., Jo D.-G., Inhibition of Drp1 ameliorates synaptic depression, Aβ deposition, and cognitive impairment in an Alzheimer’s disease model. J. Neurosci. 37, 5099–5110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griffith L. S., Schmitz B., O-linked N-acetylglucosamine is upregulated in Alzheimer brains. Biochem. Biophys. Res. Commun. 213, 424–431 (1995). [DOI] [PubMed] [Google Scholar]
- 32.Förster S., Welleford A. S., Triplett J. C., Sultana R., Schmitz B., Butterfield D. A., Increased O-GlcNAc levels correlate with decreased O-GlcNAcase levels in Alzheimer disease brain. Biochim. Biophys. Acta 1842, 1333–1339 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X., Gong W., Wang H., Li T., Attri K. S., Lewis R. E., Kalil A. C., Bhinderwala F., Powers R., Yin G., Herring L. E., Asara J. M., Lei Y. L., Yang X., Rodriguez D. A., Yang M., Green D. R., Singh P. K., Wen H., O-GlcNAc transferase suppresses inflammation and necroptosis by targeting receptor-interacting serine/threonine-protein kinase 3. Immunity 50, 576–590.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang B., Li M.-D., Yin R., Liu Y., Yang Y., Mitchell-Richards K. A., Nam J. H., Li R., Wang L., Iwakiri Y., Chung D., Robert M. E., Ehrlich B. E., Bennett A. M., Yu J., Nathanson M. H., Yang X., O-GlcNAc transferase suppresses necroptosis and liver fibrosis. JCI Insight 4, 127709 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim C., Nam D. W., Park S. Y., Hong H. S., Boo J. H., Jung E. S., Kim Y., Baek J. U., Kim K. S., Cho J. W., Mook-Jung I., O-linked β-N-acetylglucosaminidase inhibitor attenuates β-amyloid plaque and rescues memory impairment. Neurobiol. Aging 34, 275–285 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Jacobsen K. T., Iverfeldt K., O-GlcNAcylation increases non-amyloidogenic processing of the amyloid-β precursor protein (APP). Biochem. Biophys. Res. Commun. 404, 882–886 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Jimenez S., Baglietto-Vargas D., Caballero C., Moreno-Gonzalez I., Torres M., Sanchez-Varo R., Ruano D., Vizuete M., Gutierrez A., Vitorica J., Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: Age-dependent switch in the microglial phenotype from alternative to classic. J. Neurosci. 28, 11650–11661 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawahara K., Suenobu M., Yoshida A., Koga K., Hyodo A., Ohtsuka H., Kuniyasu A., Tamamaki N., Sugimoto Y., Nakayama H., Intracerebral microinjection of interleukin-4/interleukin-13 reduces β-amyloid accumulation in the ipsilateral side and improves cognitive deficits in young amyloid precursor protein 23 mice. Neuroscience 207, 243–260 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Pan R.-Y., Ma J., Kong X.-X., Wang X.-F., Li S.-S., Qi X.-L., Yan Y.-H., Cheng J., Liu Q., Jin W., Tan C.-H., Yuan Z., Sodium rutin ameliorates Alzheimer’s disease–like pathology by enhancing microglial amyloid-β clearance. Sci. Adv. 5, eaau6328 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryu I.-H., Lee K.-Y., Do S.-I., Aβ-affected pathogenic induction of S-nitrosylation of OGT and identification of Cys-NO linkage triplet. Biochim. Biophys. Acta 1864, 609–621 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Tramutola A., Sharma N., Barone E., Lanzillotta C., Castellani A., Lavarone F., Vincenzoni F., Castagnola M., Butterfield D. A., Gaetani S., Cassano T., Perluigi M., Di Domenico F., Proteomic identification of altered protein O-GlcNAcylation in a triple transgenic mouse model of Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 3309–3321 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Sandhu P., Lee J., Ballard J., Walker B., Ellis J., Marcus J., Toolan D., Dreyer D., McAvoy T., Duffy J., Michener M., Valiathan C., Trainor N., Savage M., McEachern E., Vocadlo D., Smith S. M., Struyk A., Pharmacokinetics and pharmacodynamics to support clinical studies of MK-8719: An O-GlcNAcase inhibitor for progressive supranuclear palsy. Alzheimers Dement. 12, P1028 (2016). [Google Scholar]
- 43.Ryan J. M., Quattropani A., Abd-Elaziz K., den Daas I., Schneider M., Ousson S., Neny M., Sand A., Hantson J., Permanne B., Wiessner C., Beher D., O1-12-05: Phase 1 study in healthy volunteers of the O-GlcNAcase inhibitor ASN120290 as a novel therapy for progressive supranuclear palsy and related tauopathies. Alzheimers Dement. 14, P251 (2018). [Google Scholar]
- 44.Hyman B. T., Trojanowski J. Q., Consensus recommendations for the post-mortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J. Neuropathol. Exp. Neurol. 56, 1095–1097 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Bahn G., Park J.-S., Yun U. J., Lee Y. J., Choi Y., Park J. S., Baek S. H., Choi B. Y., Cho Y. S., Kim H. K., Han J., Sul J. H., Baik S.-H., Lim J., Wakabayashi N., Bae S. H., Han J.-W., Arumugam T. V., Mattson M. P., Jo D.-G., NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer’s models. Proc. Natl. Acad. Sci. U.S.A. 116, 12516–12523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gwon A.-R., Park J.-S., Arumugam T. V., Kwon Y.-K., Chan S. L., Kim S.-H., Baik S.-H., Yang S., Yun Y.-K., Choi Y., Kim S., Tang S.-C., Hyun D.-H., Cheng A., Dann C. E. III, Bernier M., Lee J., Markesbery W. R., Mattson M. P., Jo D.-G., Oxidative lipid modification of nicastrin enhances amyloidogenic γ-secretase activity in Alzheimer’s disease. Aging Cell 11, 559–568 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baik S.-H., Fane M., Park J. H., Cheng Y.-L., Fann D. Y.-W., Yun U. J., Choi Y., Park J.-S., Chai B. H., Park J. S., Back S. H., Jeong J. I., Jang Y. J., Bahn G., Lee J.-Y., Li Y.-I., Sobey C. G., Uchida T., Park J. H., Kim H. T., Tang S.-C., Arumugam T. V., Jo D.-G., Pin1 promotes neuronal death in stroke by stabilizing Notch intracellular domain. Ann. Neurol. 77, 504–516 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Kim H. Y., Lee D. K., Chung B.-R., Kim H. V., Kim Y., Intracerebroventricular injection of amyloid-β peptides in normal mice to acutely induce Alzheimer-like cognitive deficits. J. Vis. Exp. 109, e53308 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park J., Madan M., Chigurupati S., Baek S. H., Cho Y., Mughal M. R., Yu A., Chan S. L., Pattisapu J. V., Mattson M. P., Jo D.-G., Neuronal Aquaporin 1 inhibits amyloidogenesis by suppressing the interaction between beta-secretase and amyloid precursor protein. J. Gerontol. A Biol. Sci. Med. Sci. , glaa068 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L., Ren F., Li J., Ma X., Wang P., A modified coupled enzyme method for O-linked GlcNAc transferase activity assay. Biol. Proced. Online 11, 170–183 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He Y., Ma X., Li D., Hao J., Thiamet G mediates neuroprotection in experimental stroke by modulating microglia/macrophage polarization and inhibiting NF-κB p65 signaling. J. Cereb. Blood Flow Metab. 37, 2938–2951 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rossi C., Cusimano M., Zambito M., Finardi A., Captondo A., Garcia-Manteiga J. M., Comi G., Furlan R., Martino G., Muzio L., Interleukin 4 modulates microglia homeostasis and attenuates the early slowly progressive phase of amyotrophic lateral sclerosis. Cell Death Dis. 9, 250 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/3/eabd3207/DC1