

CXCL2 was highly expressed in ACPA+ RA than in ACPA− RA. CXCL2 promoted osteoclastogenesis and was related to bone erosion in RA, which suggest that the blockade of CXCL2 might be a novel strategy for the treatment of RA.

Keywords: ACPA, CXCL2, osteoclast, osteoclastogenesis, rheumatoid arthritis
Summary
Anti‐citrullinated protein/peptide antibodies (ACPA) play important roles in the pathogenesis of rheumatoid arthritis (RA). ACPA‐positive (ACPA+) and ACPA‐negative (ACPA−) RA were suggested to be different disease subsets, with distinct differences in genetic variation and clinical outcomes. The aims of the present study were to compare gene expression profiles in ACPA+ and ACPA− RA, and to identify novel candidate gene signatures that might serve as therapeutic targets. Comprehensive transcriptome analysis of peripheral blood mononuclear cells (PBMCs) from ACPA+ and ACPA− RA patients and healthy controls was performed via RNA sequencing. A validation cohort was used to further investigate differentially expressed genes via polymerase chain reaction (PCR) and enzyme‐linked immunosorbent assay (ELISA). Spearman’s correlation test was used to evaluate the correlation of differentially expressed genes and the clinical and laboratory data of the patients. The role of differentially expressed genes in osteoclastogenesis was further investigated. Expression of C‐X‐C motif chemokine ligand 2 (CXCL2) was significantly increased in ACPA+ RA than in ACPA− RA, which was validated in PBMCs and serum. CXCL2 promoted the migration of CD14+ monocytes and increased osteoclastogenesis in RA patients. RAW264.7 macrophages were used to investigate specific mechanisms, and the results suggested that CXCL2 stimulated osteoclastogenesis via extracellular receptor kinase (ERK) mitogen‐activated protein kinase (MAPK) and nuclear factor kappa B pathways. In conclusion, CXCL2 was highly expressed in ACPA+ RA than in ACPA− RA. CXCL2 promoted osteoclastogenesis and was related to bone erosion in RA, which suggests that the blockade of CXCL2 might be a novel strategy for the treatment of RA.
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease that involves activation of inflammatory cells, synovial hyperplasia and destruction of cartilage and bone [1]. In recent years, anti‐citrullinated protein/peptide antibodies (ACPAs) have been used as biomarkers in the diagnosis of RA and to predict clinical outcomes [2, 3, 4]. Based on the presence or absence of ACPA, RA can be classified into two subtypes as ACPA‐positive (ACPA+) or ACPA‐negative (ACPA−). It has been reported that ACPAs are detectable years before the onset of arthritis, and they are strong predictors of progression to classic RA in patients with undifferentiated arthritis [5, 6]. Moreover, ACPAs are associated with RA severity. RA patients with ACPAs exhibit more severe radiographic damage than those without ACPAs [7, 8, 9, 10]. Many studies have suggested that ACPAs play a pathophysiological role in RA [11]. In recent years, a number of candidate genes have been shown to be differentially associated with susceptibility in ACPA+ and ACPA− RA [12, 13, 14]. With the advent of next‐generation sequencing technologies, such as RNA sequencing, more comprehensive and accurate transcriptome analysis has become feasible and affordable. In RNA sequencing, short fragments of complementary DNA (cDNA) are sequenced (‘reads’) then mapped onto the reference genome. RNA sequencing facilitates both the identification of differentially expressed genes and precise quantitative determination of exon and isoform (alternative splicing) expression, along with the characterization of transcription initiation sites and new splicing variants [15, 16].
In the present study, comprehensive transcriptome analysis of peripheral blood mononuclear cells (PBMCs) from ACPA+ and ACPA− RA patients was performed to identify novel candidate gene signatures that might be involved in the pathogenesis of different subsets of disease.
Materials and methods
Patients
Two ACPA+ and two ACPA− RA patients from Peking University Third Hospital who fulfilled the 2010 European League Against Rheumatism/American College of Rheumatology classification criteria for RA [2] were enrolled into the study. All four patients were newly diagnosed, treatment‐naive, menopausal and non‐smoker women from the northern Han population with symptom duration of fewer than 6 months, and none had any other chronic diseases. Three matched healthy individuals were included into the study.
PBMC collected from 40 ACPA+, 40 ACPA− RA patients and 40 healthy controls were applied for quantitative polymerase chain reaction (qPCR) to validate candidate genes expression. Serum C‐X‐C motif chemokine ligand 2 (CXCL2) levels were detected by enzyme‐linked immunosorbent assay (ELISA) in a cohort consisting of 70 ACPA+, 37 ACPA− RA patients and 40 healthy controls. The clinical and laboratory data of the patients were collected from electronic medical records. In all comparisons mentioned, the groups were age‐ and sex‐matched. The present study was conducted in accordance with the Declaration of Helsinki. The Ethics Committee of Peking University Third Hospital approved the study. All procedures involving specimens obtained from humans were performed with informed consent from each patient.
RNA extraction, sequencing and analysis
PBMCs were isolated from the blood of RA patients and healthy donors via centrifugation over Ficoll‐Paque Plus (GE Healthcare, Danderyd, Sweden). RNA was extracted from PBMCs using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s protocol. Sample quality was assessed using Nanodrop. Total RNA was enriched for mRNA using poly‐A selection. In brief, mRNA was fragmented, reverse‐transcribed, adapted with sequencing primers and sample barcodes, size‐selected and polymerase chain reaction (PCR)‐enriched. Libraries were sequenced on the HiSeq 2000 platform. RNA sequence reads were aligned to the human reference genome (NCBI, hg19) using TopHat2 aligners. Normalized gene counts were calculated using HTSeq, and differential gene expression between samples was identified via edgeR. Gene expression [reads per kilobase million (RPKM)] values were calculated using Cufflinks version 2.0.2. Gene Ontology (http://geneontology.org/) associations were determined using Gorilla. Network construction was based on the Ingenuity Pathway Analysis (Qiagen, Valencia, CA, USA) experimental evidence database.
Cell culture
PBMCs were isolated from the blood of RA patients and healthy donors, as described above. CD14+ monocytes were positively selected via magnetic‐activated cell sorting (Miltenyi Biotec, Bergisch Gladbach, Germany) and an anti‐CD14 antibody. The isolated CD14+ monocytes (1·5 × 105 cells per well in 48‐well plates or 6 × 105 cells per well in 12‐well plates) were incubated in RPMI‐1640 media containing 10% fetal bovine serum (FBS). Osteoclast differentiation was induced from CD14+ monocytes treated with macrophage colony‐stimulating factor (M‐CSF, 50 ng/ml) and receptor activator of nuclear factor kappa‐Β ligand (RANKL, 100 ng/ml). CXCL2 (100 ng/ml or otherwise as indicated) were added to the cultures. The medium was replaced every 3 days.
RAW264.7 macrophages (1.0 × 104 cells per well in 48‐well plates or 4·5 × 104 cells per well in 12‐well plates) were incubated in Dulbecco’s modified Eagle’s medium (DMEM) media containing 10% FBS. Osteoclast differentiation was induced by M‐CSF (10 ng/ml) and RANKL (50 ng/ml) with or without CXCL2.
Flow cytometry
To identify the expression of C‐X‐C motif chemokine receptor 2 (CXCR2), CD14+ monocytes were incubated for 45 min at 4°C with anti‐human CXCR2 in the dark. Cells were washed twice with staining wash buffer and centrifuged at 300 g for 5 min to pellet the cells. Stained cells were resuspended in 200 µl of phosphate‐buffered saline (PBS) then analyzed via flow cytometry. In each case, staining was compared with that of the appropriately labeled isotype control antibody.
Cell migration
Monocyte migration testing was performed with 5 μm pore‐size polycarbonate membrane Transwell tissue culture inserts (Corning, NY, USA). Cells (5 × 104 per well) were seeded into the wells in the top chamber in 10% FBS medium, and CXCL2 was added to the lower chamber in 10% FBS medium. After incubation for 1 h at 37°C the medium was removed and the filters were fixed with 4% paraformaldehyde for 20 min. The filters were then stained with 0·1% crystal violet for 15 min. The number of cells that had migrated from the upper surface of the filter to the lower surface of the filter was counted.
qPCR
Total RNA was extracted using TRIzol reagent (Invitrogen), and 1 μg of total RNA was reverse‐transcribed into cDNA using a FastQuant RT Kit (with gDNase) (Tiangen Biotech, Beijing, China). Reverse transcription–PCR (RT–PCR) was carried out using the QuantStudio™ 5 real‐time PCR system (Thermo Fisher Scientific, Fremont, CA, USA) with Talent qPCR PreMix (SYBR green) (Tiangen Biotech). Data were normalized to the expression of glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). The primer nucleotide sequences for PCR were obtained from GenBank database and synthesized by Sangon Biotech (Shanhai, China). The primer sequence information used in the study is shown in the Supporting information, Table S1.
ELISA
The isolated CD14+ monocytes (1·5 × 105 cells per well in 48‐well plates) from RA patients were treated with M‐CSF (50 ng/ml) for 72 h, then the supernatants were collected.
CXCL2 levels in serum from ACPA+ RA, ACPA− RA and healthy controls, as well as levels in the supernatants of CD14+ monocyte cultures, were measured using human CXCL2 enzyme‐linked immunosorbent assay kits, in accordance with the manufacturer’s instructions.
Tartrate‐resistant acid phosphatase (TRAP) staining
TRAP staining of CD14+ monocytes and RAW264.7 macrophages was performed after culturing with or without CXCL2 in the presence of M‐CSF and RANKL. TRAP staining was conducted using the TRAP kit (387A‐1KT; Sigma, St Louis, MO, USA), in accordance with the manufacturer’s instructions. TRAP‐positive multi‐nucleated cells containing more than three nuclei were identified as osteoclasts and counted under a microscope.
F‐actin ring immunofluorescence
Cells were fixed with 4% paraformaldehyde and incubated with fluorescein isothiocyanate (FITC)‐conjugated anti‐actin antibody for 1 h at 25°C. After a PBS wash the cells were incubated with 4′,6‐diamidino‐2‐phenylindole (DAPI) for 5 min at 25°C, then analyzed using an Olympus BX51 microscope (Tokyo, Japan).
Scanning electron microscopy
Cells on bone slices were fixed in 4% glutaraldehyde, dehydrated via graded alcohol solutions and hexamethyldisilazane solutions (Sigma‐Aldrich, MO, USA). Bone slices were then mounted onto aluminum stubs, sputtered with gold and examined using a scanning electron microscope (JSM7900F; JEOL, Tokyo, Japan).
Bone resorption assay
Functional evidence of osteoclast formation was observed via a lacunar resorption assay system using cell cultures on bone slices [17]. Cells were removed from the bone slices by treatment with 0·1 M ammonium hydroxide. The bone slices were washed in distilled water and ultrasonicated to remove adherent cells, then fixed in 4% glutaraldehyde to reveal areas of lacunar resorption, as mentioned above in scanning electron microscopy.
Western blotting
Total protein was extracted using radioimmunoprecipitation assay (RIPA) lysis buffer (Applygen, Beijing, China). A total of 40 μg of protein was loaded into a 10% sodium dodecyl sulphate‐polyacrylamide gel and electrophoretically transferred to polyvinylidene fluoride membranes (Merck Millipore, Billerica, MA, USA). After blocking with 5% milk for 1 h, the membranes were incubated with specific antibodies at the indicated dilutions for 12–16 h at 4°C. The membranes were scanned using an Odyssey Sa Imaging System (LICOR Biosciences, Lincoln, NE, USA). The antibody information used in this study is shown in the Supporting information, Table S2.
Statistical analysis
Statistical analysis of RNA sequencing count data was conducted using edgeR with a 5% false discovery rate (FDR). All experiments were repeated at least three times and are presented as the mean ± standard deviation (s.d.) and the median with interquartile range. Statistical analyses were assessed via Spearman’s correlation, Mann–Whitney test and one‐way analysis of variance followed by Tukey’s test for multiple comparisons. P‐values < 0.05 were considered statistically significant.
Results
Selection of candidate genes using RNA sequencing
Further analysis was conducted on RNA sequencing data from seven samples; two ACPA+ RA patients, two ACPA− RA patients and three healthy controls. Differentially expressed genes (DEGs) were analyzed in the two different sample types, and three groups of DEGs were identified (ACPA+ versus control, ACPA+ versus ACPA−, ACPA− versus control). We identified 1007 DEGs (522 up‐regulated and 485 down‐regulated) between ACPA+ and control, 628 DEGs (344 up‐regulated and 284 down‐regulated) between ACPA+ and ACPA−, and 1097 DEGs (549 up‐regulated and 548 down‐regulated) between ACPA− and control (Fig. 1a,b). All these DEGs exhibited distinct expression patterns in the three sample types (Fig. 1c).
Fig. 1.
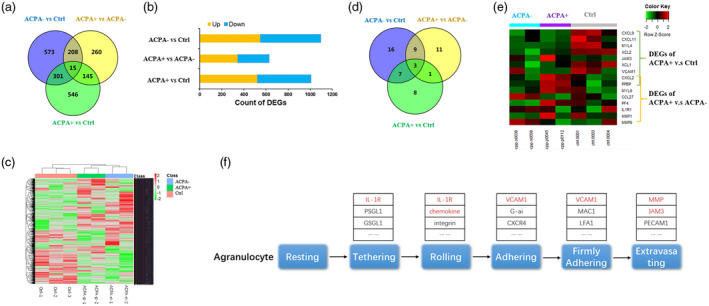
Transcriptome analysis of anti‐citrullinated protein/peptide antibodies (ACPA)− rheumatoid arthritis (RA) patients (n = 2), ACPA+ RA patients (n = 2) and control (n = 3) samples revealed the global gene expression pattern and identified agranulocyte adhesion and diapedesis as one key pathway that particularizes ACPA+ RA. (a) Three groups of differentially expressed genes (DEGs) were identified using DEseq2 at a threshold of P‐value < 0·05, fold change > 2, the Venn diagram shows the overlap of three groups of DEGs. (b) Count of up‐ and down‐regulated genes. (c) Heat‐map of expression pattern of all 2048 DEGs in patient and control samples. (d) Each group of DEGs were submitted to pathway enrichment analysis using ingenuity pathway analysis (IPA), the Venn diagram shows the overlap of significantly enriched canonical pathways. Agranulocyte adhesion and diapedesis pathway significantly enriched in both ACPA+ versus control group and ACPA+ versus ACPA− group, but not in ACPA− versus control group. (e) The heat‐map presents the expression pattern of DEGs that involve in the agranulocyte adhesion and diapedesis pathway. (f) A simplified schematic of the agranulocyte adhesion and diapedesis pathway and some involved genes (red‐marked genes) are differentially expressed in ACPA+ RA patients.
Pathway enrichment analysis/ingenuity pathway analysis of DEGs yielded 19 canonical pathways that were significantly enriched between ACPA+ and control, 24 between ACPA+ and ACPA− and 35 between ACPA− and control. Some of these pathways were enriched in more than one comparison (Fig. 1d). The agranulocyte adhesion and diapedesis pathway was particularly enriched in ACPA+ versus control and ACPA+ versus ACPA− (Fig. 1d). Different DEGs were involved in the agranulocyte adhesion and diapedesis pathway in two comparisons (Fig. 1e,f), and of these, CXCL2 and C‐X‐C motif chemokine ligand 7 (CXCL7) were exclusively up‐regulated in ACPA+ samples (Fig. 1e).
Validation of candidate genes expression in ACPA+ RA patients
To validate the differential expression levels of candidate genes in ACPA+ and ACPA− RA patients, the expression levels of CXCL2 and CXCL7 were measured using RT–PCR. CXCL2 levels were higher in ACPA+ RA patients (n = 40) than in ACPA− RA patients (n = 40) and healthy controls (n = 40) (Fig. 2a), confirming that CXCL2 was differentially expressed in ACPA+ RA patients and ACPA− RA patients. However, there was no significant difference in CXCL7 expression between ACPA+ RA patients and ACPA− RA patients.
Fig. 2.
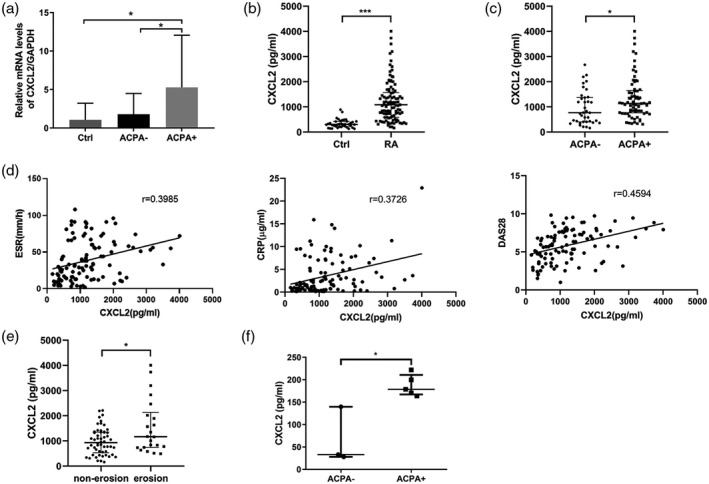
The expression of C‐X‐C motif chemokine ligand 2 (CXCL2) in rheumatoid arthritis (RA) patients (ACPA+, ACPA−) and control samples revealed higher CXCL2 expression in PBMC, serum and monocytes from ACPA+ RA patients. (a) Validation of CXCL2 expression in PBMC from ACPA+ (n = 40) RA patients, ACPA− (n = 40) RA patients and normal controls (n = 40) by real‐time–polymerase chain reaction (RT–PCR). (b) CXCL2 levels in serum of RA patients (n = 107) and normal controls (n = 40). (c) The serum CXCL2 in ACPA+ (n = 70) and ACPA− (n = 37) RA patients. (d) Correlations between CXCL2 and clinical parameters. (e) CXCL2 levels in serum of RA patients without bone erosion (n = 54) and patients with bone erosion (n = 22). (f) CXCL2 in the supernatant of CD14+ monocytes derived from ACPA+ (n = 5) and ACPA− (n = 3) RA patients. Data are expressed as mean ± standard deviation (s.d.) (a) and median with interquartile range (b,c,e,f). *P < 0·05, **P < 0·01, ***P < 0·001.
CXCL2 levels in serum and CD14+ cell supernatant
CXCL2 levels in serum were detected in 70 ACPA+ RA patients, 37 ACPA− RA patients and 40 healthy controls. Age and sex were balanced between the groups, disease activity score of 28 joints (DAS28) and treatment were similar between ACPA+ RA patients and ACPA− RA patients, whereas the disease duration, rheumatoid factor (RF)‐positive rate and bone erosion‐positive rate were significantly different among ACPA+ RA patients and ACPA− RA patients (Table 1).
Table 1.
Baseline characteristics of the enrolled rheumatoid arthritis (RA) patients
| Total (n = 107) | ACPA+ (n = 70) | ACPA− (n = 37) | P‐value | |
|---|---|---|---|---|
| Age (mean years) | 55·94 ± 14·11 | 55·13 ± 13·64 | 57·49 ± 15·02 | 0·414 |
| Gender (male/female) | 26/81 | 16/54 | 10/27 | 0·632 |
| Duration (months) | 24 (12, 108) | 60 (17, 183) | 13 (5, 36) | 0·000 |
| RF‐positive | 57·94% | 82·86% | 10·81% | 0·000 |
| ESR (mm/h) | 32 (14, 60) | 33 (14·5, 60) | 29 (13·5, 57·5) | 0·692 |
| CRP (mg/dl) | 1·86 (0·51, 5·22) | 1·565 (0·5, 4·37) | 2·46 (0·61, 7·72) | 0·250 |
| DAS28 | 5·84 (4·41, 7·59) | 6·62 (4·13, 7·77) | 5·64 (4·5, 6·71) | 0·187 |
| Bone erosion | 28·95% (n = 22/76) | 38% (n = 19/50) | 11·54% (n = 3/26) | 0·016 |
| Treatment | ||||
| DMARDS | 99·07% | 100% | 97·30% | 0·346 |
| Methotrexate | 43·93% | 47·14% | 37·84% | 0·416 |
| Hydroxychloroquine | 73·83% | 74·29% | 72·97% | 1·000 |
| Leflunomide | 42·06% | 42·86% | 40·54% | 0·840 |
| Sulfasalazine | 21·50% | 24·29% | 16·22% | 0·459 |
| Glucocorticoids | 42·06% | 37·14% | 51·35% | 0·217 |
| Biologicals | 6·54% | 5·41% | 7·14% | 1·000 |
Data are represented as mean ± standard deviation (s.d.) and median with interquartile range.
ACPA= anti‐citrullinated protein/peptide antibodies; ESR = erythrocyte sedimentation rate; RF = rheumatoid factor; CRP = C‐reactive protein; DAS28 = Disease Activity Score of 28 joints; DMARDs = disease‐modifying anti‐rheumatic drugs.
Median serum CXCL2 in RA patients [1083·43 (586·94, 1569·50) pg/ml] was significantly higher than in healthy controls [301·44 (217·62, 422·26) pg/ml] (P < 0·001; Fig. 2b). Median serum CXCL2 was significantly higher in ACPA+ RA patients than in ACPA− RA patients (P < 0·05; Fig. 2c). Correlation analysis showed that CXCL2 was positively correlated with DAS28 (r = 0·4594, P < 0·001), estimated sedimentation rate (ESR) (r = 0·3985, P < 0·001) and C‐reactive protein (CRP) (r = 0·3726, P < 0·001) (Fig. 2d). However, the CXCL2 level was not related to disease duration (r = −0·084, P = 0·391) and RF titer (r = 0·1358, P = 0·309). Serum CXCL2 was significantly higher in RA patients with bone erosion than in RA patients without bone erosion (P < 0·05; Fig. 2e).
CXCL2 expression was significantly higher in the supernatant of CD14+ monocyte cultures derived from ACPA+ RA patients than from the ACPA− RA patients (Fig. 2f).
CXCL2 promoted migration of CD14+ monocytes from RA patients
Flow cytometry analysis using anti‐CXCR2 antibody revealed that CD14+ monocytes from ACPA+ patients expressed CXCR2 at a higher level than those from ACPA− patients, but the difference was not statistically significant (Fig. 3a). In the Transwell experiments, CXCL2 significantly increased the number of migrating monocytes in cultures derived from RA patients (Fig. 3b,c).
Fig. 3.
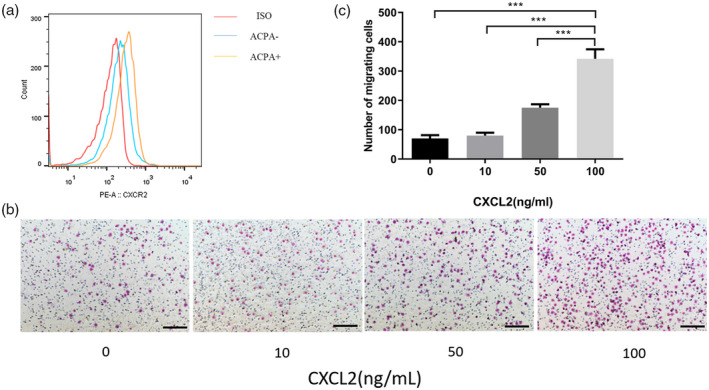
Migration of CD14+ monocytes from rheumatoid arthritis (RA) patients by C‐X‐C motif chemokine ligand 2 (CXCL2). (a) The expression of CXCR2 on the surface of CD14+ monocytes isolated from ACPA+ rheumatoid arthritis (RA) patients and ACPA− RA patients was analyzed by flow cytometry. (b) A migration assay of CD14+ monocytes from RA patients was performed using the Transwell system. (c) The number of cells that migrated through to the lower surface was counted; n = 3 per group. Data are expressed as mean ± standard deviation (s.d.); ***P < 0·001. Scale bar in (b) is 100 μm.
CXCL2 increased osteoclast differentiation and enhanced osteoclastic bone resorption in RA patients and healthy donors
In experiments conducted to evaluate the effects of CXCL2 on osteoclastogenesis of monocytes from RA patients, monocytes formed osteoclasts on plastic and the average numbers of osteoclasts were similar in the control cultures and cultures exposed to a low concentration of CXCL2 (10 ng/ml and 50 ng/ml) (Supporting information, Fig. S1). However, high doses of CXCL2 (100 ng/ml) tended to promote the formation of osteoclasts (Fig. 4a,f). Formation of the F‐actin ring by osteoclasts is a necessary step in bone resorption. Immunofluorescence microscopy of CD14+ monocytes cultured for 14 days treated with CXCL2 (100 ng/ml) in the presence of M‐CSF and RANKL revealed that F‐actin rings were significantly increased in number (Fig. 4b). CXCL2 stimulation also increased cell fusion, resulting in multi‐nucleated cells (Fig. 4c), which suggested that CXCL2 promoted osteoclast formation.
Fig. 4.
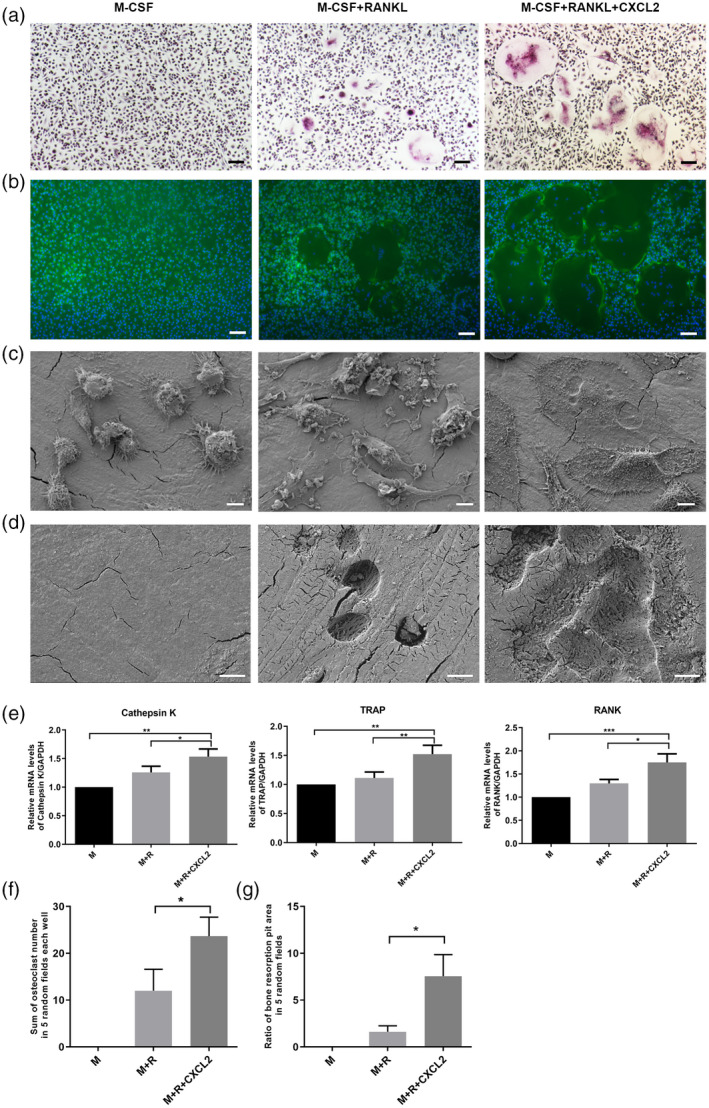
C‐X‐C motif chemokine ligand 2 (CXCL2)‐induced osteoclast differentiation and bone resorption in rheumatoid arthritis (RA) patients. CD14+ monocytes isolated from rheumatoid arthritis (RA) patients were cultured in the presence of macrophage colony‐stimulating factor (M‐CSF) (50 ng/ml) and receptor activator of nuclear factor kappa‐Β ligand (RANKL) (100 ng/ml) with CXCL2 (100 ng/ml) during the osteoclastogenesis. (a) Cells were then fixed with 4% paraformaldehyde and stained for tartrate‐resistant acid phosphatase (TRAP). (b) Cells were fixed and stained for F‐actin. (c) Representative scanning electron microscopy images of cell fusion. (d) Representative scanning electron microscopy images of bone resorption pits. (e) CXCL2 increased the mRNA expression of cathepsin K, TRAP and RANK. (f) The sum of osteoclast number in five random fields each well counted in TRAP staining. (g). The ratio of resorption pit area in five random fields per group measured in scanning electron microscopy. Three separate experiments were performed with similar results. Data are expressed as mean ± standard deviation (s.d.); *P < 0·05, **P < 0·01, ***P < 0·001. Scale bar in (a) and (b) is 100 μm. Scale bar in (c) and (d) is 10 μm.
Functional evidence of osteoclast formation was obtained via the lacunar resorption assay system using cell cultures on bone slices. The result showed that the resorption area was increased in bone slices exposed to CXCL2 (P < 0·05, Fig. 4d,g). Expression of the osteoclast markers RANK, cathepsin K and TRAP was significantly increased after 3 days of stimulation with the CXCL2 (100 ng/ml) in the presence of M‐CSF (50 ng/ml) and RANKL (100 ng/ml) (P < 0·05; Fig. 4e).
In experiments of monocytes from healthy donors, CXCL2 increased osteoclast differentiation and enhanced osteoclastic bone resorption (Fig. 5) consistent with the results in RA patients (Fig. 4a–e).
Fig. 5.
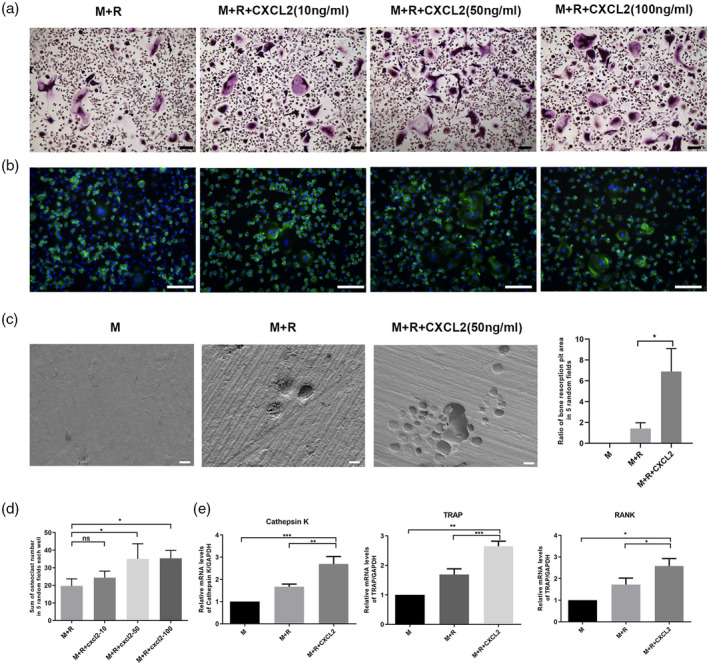
C‐X‐C motif chemokine ligand 2 (CXCL2)‐induced osteoclast differentiation and bone resorption in healthy donors. CD14+ monocytes isolated from healthy donors were cultured in the presence of macrophage colony‐stimulating factor (M‐CSF) (50 ng/ml) and receptor activator of nuclear factor kappa‐Β ligand (RANKL) (100 ng/ml) with CXCL2 (10, 50, 100 ng/ml) during the osteoclastogenesis. (a) Cells were then fixed with 4% paraformaldehyde and stained for tartrate‐resistant acid phosphatase (TRAP). (b) Cells were fixed and stained for F‐actin. (c) Representative scanning electron microscopy images of bone resorption pits and the ratio of pit area in five random fields per group. (d) The sum of osteoclast number in five random fields each well, counted in TRAP staining. (e) CXCL2 (50 ng/ml) increased the mRNA expression of cathepsin K, TRAP and RANK. Three separate experiments were performed with similar results. Data are expressed as mean ± standard deviation (s.d.); *P < 0·05, **P < 0·01, ***P < 0·001. Scale bar in (a) and (b) is 100 μm. Scale bar in (c) is 20 μm.
CXCL2 promoted osteoclast differentiation in RAW264.7 cells
Consistent with the results obtained in CD14+ monocytes, the effects of exogenous CXCL2 on osteoclast formation were also observed in RAW264.7 cells. Compared with the control group, much larger multi‐nucleated cells were evident in the CXCL2 (100 ng/ml) groups (Fig. 6a). Expression of osteoclast markers such as cathepsin K and TRAP was also significantly increased in the CXCL2 group after 3 days of stimulation in the presence of M‐CSF and RANKL (P < 0·05; Fig. 6b). These results suggest that CXCL2 enhances osteoclast formation and bone resorption. In addition, CXCL2 significantly increased the expression of nuclear factor of activated T cells 1 (NFATc1) and c‐Fos by RAW264.7 cells after 3 days of stimulation with M‐CSF and RANKL (P < 0·05; Fig. 6b).
Fig. 6.
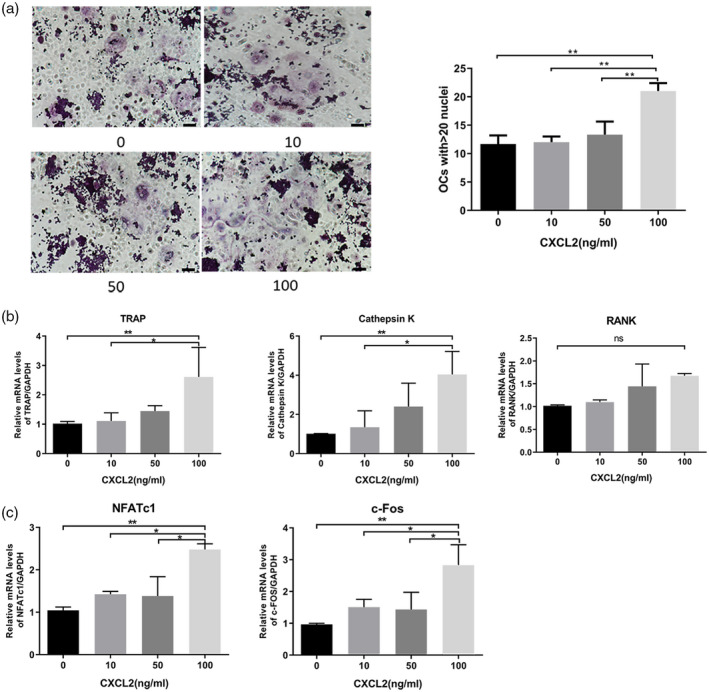
C‐X‐C motif chemokine ligand 2 (CXCL2)‐induced osteoclast differentiation in RAW264.7 cells. (a) RAW264.7 cells were used to induce osteoclast formation for 5 days in the presence of macrophage colony‐stimulating factor (M‐CSF) (10 ng/ml) and receptor activator of nuclear factor kappa‐Β ligand (RANKL) (50 ng/ml) with CXCL2. Large osteoclasts with more than 20 nuclei were counted. (b) CXCL2 increased the mRNA expression of cathepsin K and tartrate‐resistant acid phosphatase (TRAP) at 3 days in RAW264.7 cells. C. CXCL2 induced the mRNA expression of NFATc1 and c‐Fos in RAW264.7 cells at 3 days in the presence of M‐CSF and RANKL. Values shown were the mean ± standard deviation (s.d.) for three separate experiments; *P < 0·05, **P < 0.01. Scale bar is 100 μm.
CXCL2‐stimulated osteoclastogenesis via extracellular receptor kinase (ERK) mitogen‐activated protein kinase (MAPK) and nuclear factor kappa B (NFκB) pathways
To explore the signal pathways involved in the osteoclastogenesis with the stimulation of CXCL2 (100 ng/ml), RAW264.7 cells were incubated in osteoclast‐promoting medium with or without CXCL2. Western blotting at different time‐points was conducted to detect the phosphorylation of nuclear factor of kappa light polypeptide gene enhancer in B cell inhibitor alpha (IκBα), p65, ERK1/2 and c‐Jun N‐terminal kinase (JNK). The addition of CXCL2 dramatically promoted phosphorylation of p65 and ERK1/2 (P < 0·05, Fig. 7a,b), with no effect on the phosphorylation of IκBα or JNK (Fig. 7c,d).
Fig. 7.
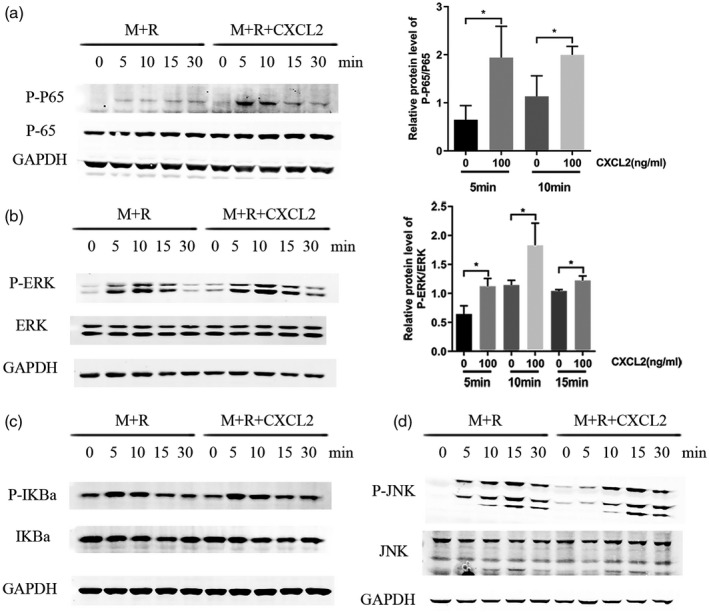
C‐X‐C motif chemokine ligand 2 (CXCL2)‐stimulated osteoclastogenesis through nuclear factor kappa B (NF‐kB) p65 and mitogen‐activated protein kinase (MAPK) extracellular‐signal‐regulated kinase (ERK1/2) pathways in RAW264.7 cells. RAW264.7 cells were incubated in macrophage colony‐stimulating factor (M‐CSF) and receptor activator of nuclear factor kappa‐Β ligand (RANKL) with or without CXCL2 (100 ng/ml). (a) CXCL2 increased the activation of the p‐p65 at 5 and 10 min. (b) CXCL2 increased the activation of the p‐ERK at 5, 10 and 15 min. (c) CXCL2 had no effect on the phosphorylation of nuclear factor of kappa light polypeptide gene enhancer in B‐cells inhibitor alpha (IkBa). (d) CXCL2 had no effect on the phosphorylation of c‐Jun N‐terminal kinase (JNK). Values shown are the mean ± standard deviation (s.d.) for three separate experiments; *P < 0·05.
Discussion
RNA sequencing technology has been used to detect differences in gene expression profiles in RA patients for the last few years. Several gene expression profiling studies of synovial tissues and PBMCs from RA patients have revealed marked variation in gene expression profiles that have facilitated the identification of distinct molecular disease mechanisms involved in RA pathology [18, 19, 20, 21]. The heterogeneity of RA was demonstrated by the presence of distinct autoantibody specificities such as ACPA in serum, differential responsiveness to treatment and variability in clinical presentation. RA patients can be stratified into two subgroups defined by the presence or absence of ACPA, and ACPA+ patients exhibit more severe inflammation and radiographic damage than ACPA− patients [5, 6, 7]. In the present study, various genes were compared in ACPA+ and ACPA− patients via RNA sequencing, and two significantly increased chemokines, CXCL2 and CXCL7, were identified in ACPA+ patients. The next, more focused, analysis using PCR technology on many more patients found that only CXCL2 was differentially expressed in ACPA+ RA patients and ACPA− RA patients. There was no significant difference in CXCL7 expression between ACPA+ RA patients and ACPA− RA patients.
CXCL2 was first identified as a major chemokine produced by endotoxin‐treated macrophages [22], which bind to the G‐protein coupled receptor CXCR2 expressed on macrophages, neutrophils and epithelial cells [23]. In previous studies, CXCL2 level was found to be more highly expressed in RA patients than in normal controls [24]. Xiaokun et al. downloaded microarray data sets of GSE1919, GSE12021 and GSE21959 [35 RA samples and 32 normal controls] from the Gene Expression Omnibus database [https://www.ncbi.nlm.nih.gov/geo/] and identified DEGs in RA samples. They found that CXCL2 was strongly associated with DEGs involved in RA progression [24]. Jacobs et al. [25] performed a microarray analysis to characterize the molecular events underlying pathology in autoantibody‐mediated arthritis, and reported that CXCL2 was up‐regulated in parallel with the disease. Jeongim et al. [26] reported that CXCL2 was significantly higher in synovial fluid and serum from RA patients compared with corresponding samples from osteoarthritis patients. In the present study, serum CXCL2 was significantly increased in RA patients compared with healthy controls, and serum CXCL2 was higher in ACPA+ RA patients than in ACPA− RA patients. Compared with ACPA− patients, ACPA+ patients had a longer disease duration and a higher positive rate of RF. To exclude the impact of these two factors on CXCL2 level, correlation analysis was applied and showed that CXCL2 was irrelevant to disease duration and RF titer.
CXCL2 was involved in various biological progresses, such as angiogenesis, inflammation and cancer biological behaviors [23, 27, 28, 29, 30]. CXCL2 was also considered to be a proinflammatory factor in RA [24]. In line with this, our study showed that CXCL2 was positively correlated with DAS28, ESR and CRP. Moreover, recent studies revealed CXCL2 was also involved in the process of osteoclastogenesis [26, 31]. In the present study, we also found serum CXCL2 was significantly higher in RA patients with bone erosion than in RA patients without bone erosion. Thus, we hypothesized that CXCL2 could be one of the key molecules up‐regulated in RA progression, especially in ACPA+ RA, which exhibit more radiographic damage, and focused on the CXCL2 for subsequent analyses.
As the most important osteoclast precursors, CD14+ monocytes were derived from ACPA+ patients and exhibited elevated CXCL2 secretion compared with those derived from ACPA− patients. There may be various mechanisms for elevated CXCL2 secretion in CD14+ monocytes from ACPA+ patients. First, different genetic background in ACPA+ and ACPA− patients may result in different immune cell activity and gene expression patterns [32]. Secondly, ACPA can activate complement via the classical pathway and the alternative pathway [33], which might activate immune cells to increase the expression of CXCL2 in monocyte indirectly. Immune complexes containing ACPA and citrullinated fibrinogen have been shown to trigger tumor necrosis factor (TNF) secretion through engagement of FcγR on human macrophages [34]. Furthermore, CXCL1 and CXCL2 mRNA levels were elevated in ankle joints from ACPA intra‐articular‐injected mice [35]. In parallel work in one of their laboratories, they found that CXCL8/interleukin (IL)‐8, another member of the CXC chemokine family, was released by human osteoclasts in response to ACPA stimulation and CXCL8/IL‐8 is an essential mediator of ACPA‐driven osteoclast [36]. We speculate that ACPA tends to affect the CXCL2 levels in monocytes/macrophages. However, how ACPA affects the CXCL2 levels in monocytes/macrophages and the possible specific mechanism still needs to be clarified.
CXCR2 is known to be the major receptor for CXCL2 [37]. It has been suggested that CXCL2 acts as a chemoattractant for various types of cells by binding to CXCR2, and monocytes constitutively express CXCR2 [37]. However, whether or not there is a difference in the expression of cell surface CXCR2 in CD14+ monocytes from ACPA+ and ACPA− RA patients remains unclear. In the present study, we found that CXCR2 expression was higher on the surfaces of CD14+ monocytes derived from ACPA+ patients than those from ACPA− patients, but the difference was not statistically significant.
ACPA+ RA patients usually develop more severe radiological damage, and enhanced osteoclastogenesis may be involved in the bone erosion [7]. Osteoclasts are polykaryocytes formed via the fusion of mononuclear monocytic precursors such as monocytes in peripheral blood. We surmise that elevated CXCL2 may recruit more monocytes to joint sites and augment the formation of osteoclasts. The present study showed that CXCL2 promoted the migration, differentiation and function of osteoclasts in experiments using CD14+ monocytes isolated from RA patients.
Jeongim et al. [26] reported that RANKL significantly increased the expression of CXCL2 in bone marrow‐derived macrophages (BMMs) and that CXCL2 mediated RANKL‐dependent differentiation of osteoclasts from BMMs in mice. The osteoclastogenesis‐enhancing effects of CXCL2 were further corroborated by their investigation with an in‐vivo bone resorption model. Notably, CXCL2 alone induced significant bone loss in mice calvarial similar to that induced by RANKL. They also reported that CXCL2 mediated lipopolysaccharide (LPS)‐induced osteoclastogenesis in RANKL‐primed precursors (i.e. BMMs) [31]. LPS stimulated the production of CXCL2 in BMMs, and the conditioned medium from LPS‐treated BMMs could enhance the migration of osteoclast precursors, which was blocked by treatment with CXCL2‐neutralizing antibody or CXCR2 receptor antagonist. Blocking CXCL2 also reduced LPS‐induced osteoclastogenesis, and in addition CXCL2 neutralization prevented bone destruction in mice treated with LPS, suggesting a critical role of CXCL2 in the process of osteoclastogenesis. In the present study, CXCL2 was also evidently involved in the process of osteoclast differentiation in RA patients and healthy donors.
The effects of CXCL2 on activating the signaling pathway during osteoclast differentiation were examined in RAW 264.7 cells to explore the molecular mechanisms of the observed enhancement effect on osteoclastogenesis. The addition of CXCL2 resulted in dramatically increased phosphorylation of p65 and ERK1/2, while it did not affect the phosphorylation of IκBα or JNK. NFATc1 and c‐Fos are critical transcription factors in the regulation of osteoclast differentiation [38]. In the current study, NFATc1 and c‐fos were increased significantly with stimulation of CXCL2 in the presence of M‐CSF and RANKL. NFATc1 is an important regulatory factor in the process of osteoclast differentiation mediated by RANKL‐activated MAPK and nuclear factor kappa B (NFκB) signaling pathways [39]. Therefore, it suggests that CXCL2 may promote osteoclast differentiation and bone resorption by regulating NFATc1 expression via interfering with the phosphorylation of NFκB p65 and MAPK ERK1/2.
Therapies targeting CXCL2/CXCR2 have been tested in animal models of arthritis with concomitant reduction in neutrophil recruitment and TNF‐α production [40, 41], and immunization against CXCL2 was reportedly efficient in delaying the onset of arthritis and reducing disease severity in a murine collagen‐induced arthritis model [42]. The novel orally active non‐competitive allosteric inhibitor of CXCR2 known as DF 2162 significantly ameliorates adjuvant‐induced arthritis in rats, an effect that is quantitatively and qualitatively similar to that of anti‐TNF antibody treatment [40]. In addition, the CXCR2 antagonist SCH563705 led to a dose‐dependent decrease in clinical disease scores and paw thickness measurements, and clearly reduced inflammation and bone and cartilage degradation in a mouse model of arthritis [41]. The results of our study imply that targeting CXCL2/CXCR2 as a strategy to treat RA may contribute to protection from bone destruction by directly inhibiting bone resorption, in addition to the already suggested anti‐inflammatory effects.
In conclusion, we identified novel pathways associated with ACPA+ RA patients using RNA sequencing, and detected higher CXCL2 expression in ACPA+ RA patients than in ACPA− RA patients. Increased levels of CXCL2 promoted osteoclastogenic process in CD14+ monocytes from RA patients. These results suggest a previously unreported role of CXCL2 during osteoclastogenesis in RA patients, and indicate that CXCL2 blockade might be a novel therapeutic strategy in RA.
Disclosures
The authors declare no conflicts of interest. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author contributions
X. W., L. S., N. H., Z. A., R. Y., C. L., Y. L., Y. L. and X. L. carried out the molecular biochemical studies. X. W., L. S., and N. H. drafted the manuscript and performed the statistical analysis. X. F. and J. Z. participated in the design of the study and helped to draft manuscript. All authors have read and approved the final manuscript.
Supporting information
Fig S1. CXCL2 induced osteoclast differentiation in RA patients. CD14+ monocytes isolated from RA patients were cultured in the presence of M‐CSF (50 ng/ml) and RANKL (100 ng/ml) with CXCL2 (10, 50, 100 ng/ml) during the osteoclastogenesis. Cells were then fixed with 4% paraformaldehyde and stained for TRAP. The sum of osteoclast number in 5 random fields each well were counted. Three separate experiments were performed with similar results. Data are expressed as mean ± standard deviation (s.d.). *P < 0.05. Scale bar is 100 μm.
Table S1. Information of the primer sequence.
Table S2. Information of the antibodies.
Acknowledgements
We gratefully thank the Medical Research Center of Peking University Third Hospital for providing experimental equipment and technical support. This work was supported by the National Natural Science Foundation of China (no. 81571573 and no. 81771744).
Contributor Information
X. Fang, Email: fangxd@big.ac.cn.
J. Zhao, Email: zhao-jinxia@163.com.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Li R, Sun J, Ren LM et al Epidemiology of eight common rheumatic diseases in China: a large‐scale cross‐sectional survey in Beijing. Rheumatology 2012; 51:721–9. [DOI] [PubMed] [Google Scholar]
- 2. Aletaha D, Neogi T, Silman AJ et al 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010; 62:2569–81. [DOI] [PubMed] [Google Scholar]
- 3. Zhao J, Su Y, Li R et al Classification criteria of early rheumatoid arthritis and validation of its performance in a multi‐centre cohort. Clin Exp Rheumatol 2014; 32:667–73. [PubMed] [Google Scholar]
- 4. Burgers LE, van Steenbergen HW, Ten Brinck RM, Huizinga TW, van der Helm‐van Mil AH. Differences in the symptomatic phase preceding ACPA‐positive and ACPA‐negative RA: a longitudinal study in arthralgia during progression to clinical arthritis. Ann Rheum Dis 2017; 76 1751–4. [DOI] [PubMed] [Google Scholar]
- 5. Ding B, Padyukov L, Lundstrom E et al Different patterns of associations with anti‐citrullinated protein antibody‐positive and anti‐citrullinated protein antibody‐negative rheumatoid arthritis in the extended major histocompatibility complex region. Arthritis Rheum 2009; 60:30–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Belakova G, Manka V, Zanova E, Racay P. Benefits of anticitrullinated peptides examination in rheumatoid arthritis. Nigerian J Clin Pract 2018; 21:1380–3. [DOI] [PubMed] [Google Scholar]
- 7. Im CH, Kang EH, Ryu HJ et al Anti‐cyclic citrullinated peptide antibody is associated with radiographic erosion in rheumatoid arthritis independently of shared epitope status. Rheumatol Int 2009; 29:251–6. [DOI] [PubMed] [Google Scholar]
- 8. Cheng TT, Yu SF, Su FM et al Anti‐CCP‐positive patients with RA have a higher 10‐year probability of fracture evaluated by FRAX(R): a registry study of RA with osteoporosis/fracture. Arthritis Res Ther 2018; 20:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hecht C, Englbrecht M, Rech J et al Additive effect of anti‐citrullinated protein antibodies and rheumatoid factor on bone erosions in patients with RA. Ann Rheum Dis 2015; 74:2151–6. [DOI] [PubMed] [Google Scholar]
- 10. Schett G. The role of ACPAs in at‐risk individuals: Early targeting of the bone and joints. Best Pract Res Clin Rheumatol 2017; 31:53–8. [DOI] [PubMed] [Google Scholar]
- 11. England BR, Thiele GM, Mikuls TR. Anticitrullinated protein antibodies: origin and role in the pathogenesis of rheumatoid arthritis. Curr Opin Rheumatol 2017; 29:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Padyukov L, Seielstad M, Ong RT et al A genome‐wide association study suggests contrasting associations in ACPA‐positive versus ACPA‐negative rheumatoid arthritis. Ann Rheum Dis 2011; 70:259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Viatte S, Plant D, Bowes J et al Genetic markers of rheumatoid arthritis susceptibility in anti‐citrullinated peptide antibody negative patients. Ann Rheum Dis 2012; 71:1984–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yarwood A, Huizinga TW, Worthington J. The genetics of rheumatoid arthritis: risk and protection in different stages of the evolution of RA. Rheumatology 2016; 55:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA‐seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 2008; 18:1509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heruth DP, Gibson M, Grigoryev DN, Zhang LQ, Ye SQ. RNA‐seq analysis of synovial fibroblasts brings new insights into rheumatoid arthritis. Cell Biosci 2012; 2:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adamopoulos IE, Chao CC, Geissler R et al Interleukin‐17A upregulates receptor activator of NF‐kappaB on osteoclast precursors. Arthritis Res Ther 2010; 12:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pratt AG, Swan DC, Richardson S et al A CD4 T cell gene signature for early rheumatoid arthritis implicates interleukin 6‐mediated STAT3 signalling, particularly in anti‐citrullinated peptide antibody‐negative disease. Ann Rheum Dis 2012; 71:1374–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wright HL, Cox T, Moots RJ, Edwards SW. Neutrophil biomarkers predict response to therapy with tumor necrosis factor inhibitors in rheumatoid arthritis. J Leuk Biol 2017; 101:785–95. [DOI] [PubMed] [Google Scholar]
- 20. Yu F, Duan C, Zhang X et al RNA‐seq analysis reveals different gene ontologies and pathways in rheumatoid arthritis and Kashin–Beck disease. Int J Rheum Dis 2018; 21:1686–94. [DOI] [PubMed] [Google Scholar]
- 21. Sumitomo S, Nagafuchi Y, Tsuchida Y et al Transcriptome analysis of peripheral blood from patients with rheumatoid arthritis: a systematic review. Inflamm Regen 2018; 38:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wolpe SD, Cerami A. Macrophage inflammatory proteins 1 and 2: members of a novel superfamily of cytokines. FASEB J 1989; 3:2565–73. [DOI] [PubMed] [Google Scholar]
- 23. De Filippo K, Dudeck A, Hasenberg M et al Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 2013; 121:4930–7. [DOI] [PubMed] [Google Scholar]
- 24. Gang X, Sun Y, Li F et al Identification of key genes associated with rheumatoid arthritis with bioinformatics approach. Medicine 2017; 96:e7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jacobs JP, Ortiz‐Lopez A, Campbell JJ, Gerard CJ, Mathis D, Benoist C. Deficiency of CXCR2, but not other chemokine receptors, attenuates autoantibody‐mediated arthritis in a murine model. Arthritis Rheum 2010; 62:1921–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ha J, Choi HS, Lee Y, Kwon HJ, Song YW, Kim HH. CXC chemokine ligand 2 induced by receptor activator of NF‐kappa B ligand enhances osteoclastogenesis. J Immunol 2010; 184:4717–24. [DOI] [PubMed] [Google Scholar]
- 27. Boro M, Balaji KN. CXCL1 and CXCL2 regulate NLRP3 inflammasome activation via G‐protein‐coupled receptor CXCR2. J Immunol 2017; 199:1660–71. [DOI] [PubMed] [Google Scholar]
- 28. Henn D, Abu‐Halima M, Wermke D et al MicroRNA‐regulated pathways of flow‐stimulated angiogenesis and vascular remodeling in vivo . J Translat Med 2019; 17:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Subat S, Mogushi K, Yasen M, Kohda T, Ishikawa Y, Tanaka H. Identification of genes and pathways, including the CXCL2 axis, altered by DNA methylation in hepatocellular carcinoma. J Cancer Res Clin Oncol 2019; 145:675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu J, Xu W, Qian J et al Transcriptome profiling analysis reveals that CXCL2 is involved in anlotinib resistance in human lung cancer cells. BMC Med Genom 2019; 12(Suppl 2):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ha J, Lee Y, Kim HH. CXCL2 mediates lipopolysaccharide‐induced osteoclastogenesis in RANKL‐primed precursors. Cytokine 2011; 55:48–55. [DOI] [PubMed] [Google Scholar]
- 32. Willemze A, Trouw LA, Toes RE, Huizinga TW. The influence of ACPA status and characteristics on the course of RA. Nat Rev Rheumatol 2012; 8:144–52. [DOI] [PubMed] [Google Scholar]
- 33. Trouw LA, Haisma EM, Levarht EW et al Anti‐cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum 2009; 60:1923–31. [DOI] [PubMed] [Google Scholar]
- 34. Clavel C, Nogueira L, Laurent L et al Induction of macrophage secretion of tumor necrosis factor alpha through Fcgamma receptor IIa engagement by rheumatoid arthritis‐specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Rheum 2008; 58:678–88. [DOI] [PubMed] [Google Scholar]
- 35. Wigerblad G, Bas DB, Fernades‐Cerqueira C et al Autoantibodies to citrullinated proteins induce joint pain independent of inflammation via a chemokine‐dependent mechanism. Ann Rheum Dis 2016; 75:730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krishnamurthy A, Joshua V, Haj Hensvold A et al Identification of a novel chemokine‐dependent molecular mechanism underlying rheumatoid arthritis‐associated autoantibody‐mediated bone loss. Ann Rheum Dis 2016; 75:721–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheng Y, Ma XL, Wei YQ, Wei XW. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim Biophys Acta Rev Cancer 2019; 1871:289–312. [DOI] [PubMed] [Google Scholar]
- 38. Park JH, Lee NK, Lee SY. Current understanding of RANK signaling in osteoclast differentiation and maturation. Mol Cells 2017; 40:706–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Song F, Zhou L, Zhao J et al Eriodictyol inhibits RANKL‐induced osteoclast formation and function via inhibition of NFATc1 activity. J Cell Physiol 2016; 231:1983–93. [DOI] [PubMed] [Google Scholar]
- 40. Barsante MM, Cunha TM, Allegretti M et al Blockade of the chemokine receptor CXCR2 ameliorates adjuvant‐induced arthritis in rats. Br J Pharmacol 2008; 153:992–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Min SH, Wang Y, Gonsiorek W et al Pharmacological targeting reveals distinct roles for CXCR2/CXCR1 and CCR2 in a mouse model of arthritis. Biochem Biophys Res Communi 2010; 391:1080–6. [DOI] [PubMed] [Google Scholar]
- 42. Kim SJ, Chen Z, Essani AB et al Differential impact of obesity on the pathogenesis of RA or preclinical models is contingent on the disease status. Ann RheumDis 2017; 76:731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. CXCL2 induced osteoclast differentiation in RA patients. CD14+ monocytes isolated from RA patients were cultured in the presence of M‐CSF (50 ng/ml) and RANKL (100 ng/ml) with CXCL2 (10, 50, 100 ng/ml) during the osteoclastogenesis. Cells were then fixed with 4% paraformaldehyde and stained for TRAP. The sum of osteoclast number in 5 random fields each well were counted. Three separate experiments were performed with similar results. Data are expressed as mean ± standard deviation (s.d.). *P < 0.05. Scale bar is 100 μm.
Table S1. Information of the primer sequence.
Table S2. Information of the antibodies.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.