

Sixteen CGD cases were clinically, functionally and genetically characterized and classified as suffering from different variant forms (X910, X91− or X91+) according to NOX2 expression and NADPH oxidase activity in their neutrophils. The pathological impact of each mutation of the new and extremely rare double missense mutation Thr208Arg‐Thr503Ile in CYBB was investigated using stable transfection in the NOX2 knock‐out PLB‐985 cell line. Low NADPH oxidase activity found in X91−‐CGD patients correlates with mild clinical forms, whereas X910‐CGD and X91+‐CGD cases remain the most clinically severes.

Keywords: clinical severity, NADPH oxidase, NOX, X‐linked CGD variants
Summary
Chronic granulomatous disease (CGD) is a rare inherited disorder in which phagocytes lack nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity. The most common form is the X‐linked CGD (X91‐CGD), caused by mutations in the CYBB gene. Clinical, functional and genetic characterizations of 16 CGD cases of male patients and their relatives were performed. We classified them as suffering from different variants of CGD (X910, X91− or X91+), according to NADPH oxidase 2 (NOX2) expression and NADPH oxidase activity in neutrophils. Eleven mutations were novel (nine X910‐CGD and two X91−‐CGD). One X910‐CGD was due to a new and extremely rare double missense mutation Thr208Arg‐Thr503Ile. We investigated the pathological impact of each single mutation using stable transfection of each mutated cDNA in the NOX2 knock‐out PLB‐985 cell line. Both mutations leading to X91−‐CGD were also novel; one deletion, c.‐67delT, was localized in the promoter region of CYBB; the second c.253‐1879A>G mutation activates a splicing donor site, which unveils a cryptic acceptor site leading to the inclusion of a 124‐nucleotide pseudo‐exon between exons 3 and 4 and responsible for the partial loss of NOX2 expression. Both X91−‐CGD mutations were characterized by a low cytochrome b 558 expression and a faint NADPH oxidase activity. The functional impact of new missense mutations is discussed in the context of a new three‐dimensional model of the dehydrogenase domain of NOX2. Our study demonstrates that low NADPH oxidase activity found in both X91−‐CGD patients correlates with mild clinical forms of CGD, whereas X910‐CGD and X91+‐CGD cases remain the most clinically severe forms.
Introduction
Chronic granulomatous disease (CGD) is a rare genetic disorder belonging to innate immunodeficiency syndromes [1]. CGD is due to a defect in the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex of phagocytes, highly expressed in phagocytic cells such as neutrophils, monocytes and macrophages. The pathophysiological consequence is a defect in reactive oxygen species (ROS) production normally responsible for the killing of bacteria and fungi during an infection. Thus, CGD is generally diagnosed early, before the age of 2 years by recurrent and severe infections, although some rare cases may remain undiagnosed until later childhood or even adult life [2]. The incidence of CGD is approximately one in 250 000 newborn individuals. CGD is a genetically heterogeneous disease, with all ethnic groups equally affected. The NADPH oxidase complex is composed of a membrane‐bound flavocytochrome b 558 (cytb 558), the redox center of the enzyme, consisting of an α subunit, p22phox, and a β subunit, NOX2 (also named gp91phox) and three cytosolic components: p47phox, p67phox and p40phox. CGD is caused by mutations located in any of these five subunits [3, 4, 5]. NOX2 is the flavin‐ and heme‐containing oxidase element capable of transferring electrons from NADPH in the cytosol to molecular oxygen in the extracellular or intraphagosomal compartment, while p22phox stabilizes the expression of this component in phagocytic cells [6]. In neutrophils, the defect in expression of one of these two subunits compromises the expression of the other [7]. Recently it was demonstrated that mutations in EROS/CYBC1 are responsible for a decrease in NADPH oxidase activity of phagocytes, leading to chronic granulomatous disease [8]. A small G protein, Rac2, is also involved in regulating NADPH oxidase activity and can be mutated in rare cases, leading to an innate immunodeficiency [9, 10, 11, 12, 13, 14, 15]. In resting cells, membrane and cytosolic components of the NADPH oxidase complex are dissociated, while in stimulated phagocytes they become associated at the membrane, leading to an active oxidase complex able to produce superoxide anions [16].
On the basis of the mode of inheritance, two classical forms of the CGD are known: an autosomal form (AR‐CGD) with mutations in CYBA (OMIM number 233690), NCF1 (OMIM number 233700), NCF2 (OMIM number 233710) or NCF4 (OMIM number 613960), encoding p22phox, p47phox, p67phox and p40phox proteins, respectively. In the X‐linked form of CGD (X91‐CGD), mutations are present in CYBB (OMIM number 306400) encoding NOX2, which accounts for more than 60% of all CGD cases. Clear information on the severity of CGD according to the genetic forms is often difficult to establish. However, the majority of mutations affecting the membrane cytb 558 are mainly associated with severe clinical features of CGD, when no measurable NADPH oxidase activity is found [17, 18]. Indeed, X‐CGD patients with mutations in CYBB can be classified as having different variant forms (X910, X91− or X91+), according to the level of cytb 558 expression and NADPH oxidase activity in their phagocytes [19]. The CYBB gene, encoding NOX2, encompasses 13 exons spanning approximately 30 kb of the human X chromosome DNA [20, 21]. X910‐CGD, which represents more than 90% of X‐CGD cases, is characterized by an absence of cytb 558 expression and NADPH oxidase activity and is related to the most severe clinical form. A few numbers of cases with ‘variant’ forms of the disease, called X91−‐CGD, have also been described, in which low levels of cytb 558 expression can be accompanied by a proportionally decreased NADPH oxidase activity [22]. Mutations associated with this phenotype are usually located in the coding region of CYBB. These variants are of interest because they cause a structural disorganization, leading either to an incomplete loss of protein or to a partial dysfunction or both [23]. The severity of the clinical forms of these variants varies considerably, relating to the level of NADPH oxidase activity [24]. Very rare mutations (five published cases) in the upstream promoter region of CYBB leading to the X91−‐CGD phenotype have also been described [25, 26, 27, 28]. These mutations are located between the ‘CCAAT’ and the ‘TATA’ boxes in a consensus binding site for the erythroblast transformation‐specific (ETS) family of transcription factors in the NOX2 promoter region, and are responsible for defects in CYBB transcription [29]. A striking point is that, in most of the X91−‐CGD cases characterized by a mutation in the CYBB promoter, the CGD diagnosis is made in adolescents (< 10 years) or in adults, and usually the clinical form is mild. In the last rare cases of variants, named X91+‐CGD, mutated NOX2 is normally expressed (also membrane cytb 558), but no NADPH oxidase activity can be detected. To date, approximately 25 X91+‐CGD mutations have been reported [4]. Most of them are missense mutations or small deletions, and are primarily located in the C‐terminal cytosolic tail of NOX2, confirming the importance of this region in catalytic activity but not structural stability. This feature can be used to obtain insight into the importance of certain regions, but due to the scarcity of patient material, model systems are usually employed for detailed studies. Indeed 18 X91+‐CGD mutations have been reproduced in the PLB‐985 cell line by mutagenesis, stable transfection and clonal selection for functional studies [30, 31, 32, 33, 34, 35, 36].
We investigated 16 male patients suspected of suffering from X‐linked CGD and their families. Clinical, genotypical and phenotypical data are reported. These X‐CGD patients were classified as having different variant forms of CGD (X910, X91− or X91+), according to their NOX2 expression and NADPH oxidase activity. Eleven mutations in CYBB were novel. Two rare novel mutations (−67delT and a c.253‐1879A>G mutation activating a splicing donor site) leading to X91−‐CGD were fully characterized. The impact of an intriguing double missense mutation, Thr208Arg‐Thr503Ile, was also deciphered after stable transfection of Thr208Arg‐NOX2 and Thr503Ile‐NOX2 in the NOX2 knock‐out PLB‐985 cell line. The functional importance of novel missense mutations is discussed in the context of a new three‐dimensional model of dehydrogenase domain of NOX2 [37].
Materials and methods
Ethical considerations
Blood samples were collected from healthy volunteers, patients and relatives after obtaining their signed informed consent. Written consent for DNA analysis of samples from patients, parents and relatives was also obtained.
Patients
Patients from Italy, France, Croatia, Lithuania and Finland were diagnosed as having CGD on the basis of their clinical history, examination and the inability of their phagocytes to generate ROS species (Tables 1 and 2). The results of the mosaic pattern by nitro blue tetrazolium (NBT) assay or dihydrorhodamine‐1, 2, 3 (DHR) flow cytometry from the mothers’ peripheral neutrophils were consistent with an X‐linked inheritance of their sons’ disease. X‐CGD subtypes were determined according to the nomenclature X910, X91−, X91+, where the superscript denotes whether the level of NOX2 is undetectable (0), low (−) or normal (+), as determined by immunoblot or flow cytometry analysis. A summary of the clinical history of the patients is given in Table 1. All patients are currently in good health under prophylactic treatment and are under constant care and follow‐up in a specialized medical center.
Table 1.
Clinical data of the 16 X91‐CGD patients
| Patients | X‐CGD variants | Date of birth | Date of diagnosis | Severe clinical signs | Minor clinical signs | Actual treatments |
|---|---|---|---|---|---|---|
| P1 | X91– | 12/22/13 | 01/20/15 | – | Multiple abscessed skin lesions, splenomegaly | Prophylactic a |
| P2 | X910 | 11/18/12 | 09/24/14 | Prolonged fever | Inflammatory syndrome, xanthogranuloma (Serratia marcescens), tonsil adenophlegmon (Staphylococcus aureus), microcytic anemia due to martial deficiency, diarrhea, vomiting, rotavirus gastroenteritis | Prophylactic a |
| P3 | X910 | 03/10/09 | 02/01/2012 | Sepsis, hepato‐splenomegaly hepatic granuloma | Pallor, pustulosis, inflammatory syndrome, bloody diarrhea (Enterococcus faecium), multiple necrotic adenopathies (S. marcescens), hepatic cytolysis and cholestasis, failure to thrive | Prophylactic a |
| P4a b | X91– | 02/04/86 | 10/18/2012 | – | Mild childhood asthma, at teenage recurrent perianal abscesses, treated for Crohn disease, nose infection (S. aureus) | Prophylactic a |
| P4b a | 1988 | 2012 postmortem | Aspergillus pneumonitis | – | – | |
| P5 | X910 | 07/29/12 | 04/01/14 | Pulmonary abscesses (S. constellatus) | Neonatal pustular melanosis, eosinophilic pancolitis, maculo‐papular skin rash | Engraftment at 3 years of age. Joint chronic graft‐versus‐host disease. Complete chimerism |
| P6 | X910 | 03/13/09 | 04/28/09 | Severe sepsis, three outbreaks of pneumonia | – | Prophylactic a |
| P7 | X910 | 06/20/16 | 06/22/17 | – | Cervical adenitis, febrile (S. aureus), papular lesions, fungal infections in the penis, hypothermia, cyanosis | Prophylactic a |
| P8 | X910 | 12/08/15 | 05/19/16 | Hepatic abcesses | Fever, inflammatory syndrome, rotavirus gastroenteritis, cervical adenopathies, granuloma of the tonsil, perforated acute otitis, ileitis | Recent engraftment (HLA id.) with infectious complications and viral reactivation, persistent diarrhea 6 months post‐transplant |
| P9 | X910 | 10/28/12 | 06/02/14 | Prolonged fever, liver abscesses | – | Engraftment at 3 years of age, complete chimerism |
| In good health | ||||||
| P10 | X910 | 09/02/18 | 12/18/18 | Bacteremia (S. aureus) | Reccurent suppurative inguinal (S. aureus) | Prophylactic a |
| P11 | X910 | 10/14/14 | 03/15/17 | Mycobacterium bovis infection | Recurrent suppurative lymphadenitis, pneumonia, other pulmonary bacterial infections | Prophylactic a |
| P12 | X910 | 01/30/86 | 06/24/87 | Sepsis (S. typhimurium), pulmonary aspergillosis | Suppurative inguinal adenopathies (Candida albicans), furonculosis (Klebsiella pneumoniae), cervical adenopathy (Streptoccocus), balano‐prepucial fistula, recurrent cold sores | Prophylactic a |
| P13 | X910 | 09/07/13 | 07/20/16 | Abscess of the mediastinum, bacteremia, bacterial pneumonia, suspected Noonan syndrome | Cervical lymphadenitis, BCGitis | Prophylactic a |
| P14 | X910 | 02/16/83 | 12/02/10 | Bone infection (Aspergillus sp.) treated by laminectomy, permanent respiratory failure (restrictive and obstructive) | Peritonsillar abcess | Prophylactic a corticotherapy, hydroxychloroquine, amethopterine, permanent oxygenotherapy |
| P15 | X91+ | 12/19/03 | 09/20/09 | Prolonged fever, pulmonary abcess | Cervical adenopathy, BCGititis, abscess of the anal margin, acute appendicitis, diarrhea, chronic inflammation syndrome | Prophylactic a |
| P16 | X910 | 01/21/13 | 06/30/16 | Liver abcesses | Cervical adenitis | Engraftment at 16 months years of age, acute intestinal graft versus host disease. Bronchiolitis obliterans syndrome, complete chimerism |
Trimethoprim‐sulfamethoxazole and itraconazole
P4a and P4b are brothers. P4b died aged 24 years. At that time CGD diagnosis was performed for him and his brother P4a.
Dead patient.
Patients with new mutations are shown in bold type.
HLA = human leucocyte antigen; CGD = chronic granulomatous disease; – = absence of clinical signs.
Table 2.
Phenotypical and genotypical profiles of the 16 X91‐CGD patients
| Patients | NBT test % of positive cells | Cyt c Red O2 ‐ nmol/min/106cells a | Resorufin H2O2 nmol/min/106cells b | DHR, index (% of cells) c | Protein expression flow cytometry index (% of cells) or WB | Cyt b 558 picomol/mg proteins | cDNA nucleotide change | Gene location | Amino‐acid change | CGD‐type |
|---|---|---|---|---|---|---|---|---|---|---|
| Standard values (n=100) | 90 ± 6 | 11.3 ± 3.3 | 25.7 ± 10.3 | > 5 | 40 ± 13 | 166 ± 30 | – | – | – | – |
| P1 | – | – | – | 2.3 (98)–277 (2) | 1.0 (98)–25·1 (2) | – | –67 delT | Promoter | No | X91– |
| M1 | – | – | – | 4.3 (36)–22.3 (63) | 1.0 (33)–17·1 (67) | – | Carrier | |||
| P2 | – | – | – | 1.1 (100) | 1.1 (100) | – | c.39delT | Exon 1 | p.Phe13LeufsX21 | X910 |
| M2 | – | – | – | 2.6 (47)–47.0 (53) | 1.0 (46)–18.9 (54) | – | Carrier | |||
| Gm2 | – | – | – | – | – | – | Mutation not found | Not carrier | ||
| P3 | 1.0 | – | 0 | 1.0 (100) | Abs NOX2 | – | c.201dupT | Exon 3 | p.Leu68SerfsX34 | X910 |
| P4a | 21.0 | – | – | 10.4 (100) | 2.3 (100) | 36 | c.253–1879A>G | Intron 3 | Skip exon 3, p.Cys85LeufsX32 | X91– |
| M4 | 80 | – | – | 3.4 (16)–13.4 (84) | 2.8 (10)–30.0 (90) | 342 | Carrier | |||
| S4 | 58 | – | – | 1.2 (47)–12.4 (53) | 2.2 (37)–30.0 (63) | 315 | Carrier | |||
| P5 | 0 | – | – | 1.0 (100) | 1.0 (100) | – | c.600_603dupTTAC | Exon 6 | p.Phe202LeufsX2 | X910 |
| M5 | 30 | – | – | 8.4 (78)–49.8 (22) | 1.0 (65)–55.0 (35) | – | Carrier | |||
| Am5 | 72 | – | – | 2.1 (24)–6.9 (76) | 1.0 (17)–9.3 (83) | – | Carrier | |||
| Gam5 | – | – | – | – | – | – | Mutation not found | Not carrier | ||
| P6 | 0 | 0 | 0 | – | – | 0 | c.623 C>G and c.1508 C>T | Exon 6 and Exon 12 | p.Thr208Arg and Thr503Ile | X910 |
| M6 | 39 | 5.5 | 10.4 | – | – | 76 | Carrier | |||
| Am6 | – | – | – | – | – | – | Mutation not found | Not carrier | ||
| P7 | 0 | 1.3 (100) | 1.3 (100) | c.674+3G>T | Intron 6 | Deletion of exon 6 or exon 5 and 6, p.Asp162ThrfsX15 | X910 | |||
| M7 | 94 | – | – | 57.8 (100) | 44.7 (100) | – | Not carrier | |||
| P8 | 0 | – | – | 1.0 (100) | 1.2 (100) | – | c.676C>T | Exon 7 | p.Arg226X | X910 |
| M8 | 49 | – | – | 39.0 (48)–154 (52) | 1.2 (49)–19.0 (51) | – | Carrier | |||
| S8 d | 94 | – | – | 15.5 (100) | 8.3 (100) | – | – | ND d | ||
| P9 | 0 | – | – | 1.0 (100) | 1.0 (100) | – | c.816G>A | Exon 8 | p.Trp272X | X910 |
| M9 | 36 | – | – | 2.8 (78)–25.0 (22) | 1.0 (60)–23.0 (40) | – | Carrier | |||
| Am9 | 90 | – | – | 158 (100) | 12 (100) | – | Mutation not found | Not carrier | ||
| Am9' | 96 | – | – | 97 (100) | 20 (100) | – | Mutation not found | Not carrier | ||
| Gam9 | – | – | – | 63 (100) | 10 (100) | – | Mutation not found | Not carrier | ||
| P10 | – | – | – | 1.0 (100) | 1.0 (100) | – | c.980T>A | Exon 9 | Val327Asp | X910 |
| M10 | – | – | – | 46.0 (58)–244 (42) | 1.0 (64)–23.0 (46) | – | Carrier | |||
| P11 | 0 | – | – | 1.0 (100) | 1.0 (100) | – | c.1061_1065delATATC | Exon 9 | p.His354ProfsX16 | X910 |
| M11 | 12 | – | – | 3.5 (93)–217 (7) | 1.3 (90)–44.7 (10) | – | Carrier | |||
| P12 | 0 | 0 | – | – | Abs NOX2 | 0 | c.897+1258_1151+129del (del of 1657bp) | Intron 9–exon 9 | Skip exon 9, p.Val300AspfsX4 | X910 |
| M12 | 57 | 1.9 | – | – | NOX2 diminished | 39 | Carrier | |||
| S12 | 73 | 5.0 | – | – | NOX2 diminished | 55 | Carrier | |||
| B12 d | 80 | 10.0 | – | – | 103 | ND d | ||||
| P13 | 2 | – | – | – | Abs NOX2 | – | c. 1166G>A | Exon 10 | p.Gly389Glu | X910 |
| P14 | 4 | – | 0 | – | Abs NOX2 | – | c.1167_1179delGCCCTTTGGCACT | Exon 10 | p.Phe391ValfsX9 | X910 |
| D14 | 48 | – | – | – | – | Carrier | ||||
| P15 | 0 | 0 | – | – | Pres NOX2 | – | c.1235G>A | Exon 10 | p.Gly412Glu | X91+ |
| M15 | 88 | 14.6 | – | – | – | – | Carrier | |||
| Am15 | – | – | – | – | – | – | Mutation not found | Not carrier | ||
| P16 | 4 | – | – | 1.1 (100) | 1.2 (100) | – | c.1546T>C | Exon 12 | p.Trp516Arg | X910 |
| M16 | 36 | – | – | 42 (68)–189 (32) | 2.0 (69)–35.9 (31) | – | Carrier |
P = patient; M = mother; Gm = grandmother; S = sister; B = brother; D = daughter; Am = maternal aunt; Gam = maternal grand‐aunt.
Activation with PMA;
activation with PMA;
MFI of activated cells/MFI resting cells (% of cells having index) or Western blot results; Abs = absence; Pres = presence; Decr = decrease;
genetic analysis not performed because they were safe and not of age.
Patients with new mutations are shown in bold type.
DHR = dihydrorhodamine; CGD = chronic granulomatous disease; NOX2 = nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2; – = not determined; PMA = phorbol myristate acetate; MFI = mean fluorescence intensity.
Cell preparation
Human neutrophils and mononuclear cells (lymphocytes plus monocytes) were isolated from citrated blood from patients, their relatives and healthy volunteers. Lymphocytes purified by Ficoll‐Hypaque density gradient centrifugation were infected with the B95‐8 strain of Epstein–Barr virus (EBV) and cultured as previously described [38, 39].
Wild‐type (WT), X‐CGD (lacking NOX2 expression and/or NADPH oxidase activity) and transfected X‐CGD PLB‐985 cell lines were grown in RPMI‐1640 supplemented with 10% (v/v) fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin and 2 mM L‐glutamine at 37°C in a humidified 5% CO2 atmosphere. Geneticin (0·5 mg/ml) was added to maintain the selection pressure of the plasmid in the transfected cells. For granulocytic differentiation, PLB‐985 cells were exposed to 0·5% (v/v) dimethylformamide for 6 days [40].
ROS measurements in phagocytic cells
ROS production in human neutrophils was measured by superoxide dismutase (SOD)‐sensitive cytochrome c reduction, resorufine fluorescence measurement (Amplex Red® hydrogen peroxide⁄peroxidase assay kit; Invitrogen Life Technologies, Villebon sur Yvette, France), flow cytometry with dihydrorodamine‐1, 2, 3 (DHR) (TebuBio, Le Perray‐en‐Yvelines, France) or NBT slide test after opsonized latex bead phagocytosis, as previously described [41]. In flow cytometry experiments with DHR, 5 µg/ml of catalase were added to the mother’s neutrophils to avoid passive diffusion of ROS from oxidase‐positive to oxidase‐negative cells. ROS production in PLB‐985 cell lines was measured by chemiluminescence in the presence of luminol and horseradish peroxidase (HRP) [33]. Relative light units (RLU) were recorded at 37°C, over a time–course of 60 min in a Luminoscan luminometer (Labsystems, Helsinki, Finland). In some experiment cells were labeled with 2·5 μg/ml Alexa Fluor 647 rat anti‐human CD294 (CRTH2; BD Biosciences, Pont de Claix, France) as described below, prior to incubation with DHR and phorbol myristate acetate (PMA) stimulation [28].
NADPH oxidase subunits and CD294 expression in phagocytic cells
Flow cytometry – single staining
Monoclonal antibody (mAb) 7D5 directed against external epitopes of NOX2 (D162‐3; Clinisciences, Nanterre, France), monoclonal anti‐p22phox (SC‐130550; Santa Cruz Biotechnologies Inc., Heidelberg, Germany), with secondary antibody conjugated with Alexa Fluor 488 (A11070; Invitrogen Life Technologies) or phycoerythrin (PE) (A10543; Invitrogen Life Technologies) were used for analysis of NADPH oxidase subunit expression in phagocytic cells. Control staining with appropriate isotype‐matched control antibodies was included to establish thresholds for positive staining. Cell fluorescence was quantified using a fluorescence activated cell sorter (FACS) Canto II (BD Biosciences). Data were collected and analyzed with the FACS DIVA software and FlowJo software, Tree Star (BD Biosciences).
Expression of NOX2 and CD294 in neutrophils and eosinophils by flow cytometry
Granulocytes (106 cells) were suspended in 0·1 m of Hepes‐buffered saline/bovine serum albumin (HBS‐BSA) and incubated with 5 μg/ml of mAb 7D5 (anti‐gp91phox) or control isotype‐matched immunoglobulin (Ig)G1 mAb (02‐6100; ThermoFisher Scientific, Illkirsh, France) for 30 min on ice. The cells were washed twice and incubated with Alexa Fluor 488 goat‐F(ab)2 anti‐mouse IgG1 (A11070; Invitrogen, Villebon sur Yvette, France) for 30 min on ice. After two washes as above, the cells were incubated with 2·5 μg/ml Alexa Fluor 647 rat anti‐human CD294 (CRTH2; BD Biosciences) for 30 min on ice and washed [28]. Cell‐associated fluorescence (FL1 and FL4) was measured on 100 000 events. Data were collected and analysed with the FACS DIVA software and FlowJo software‐Tree Star (BD Biosciences).
Western blot
Expression of NOX2, p22phox, p47phox, p67phox and p40phox in a 1% Triton X100 soluble extract prepared from human neutrophils was examined by Western blot analysis using mAbs anti‐NOX2 48 and anti‐p22phox 449 [42, 43, 44] polyclonal antibodies anti‐p67phox (C19, SC7662; Santa Cruz), anti‐p47phox and anti‐p40phox [45, 46]. Polyclonal anti‐gG‐HRP was used as a second antibody and the immune complexes were detected by chemiluminescence using an ECL kit Femtomax (Rockland Immunochemicals, Limerick, PA, USA). Protein concentration was determined by Pierce BCA Protein AssayTM Kit (ThermoFisher) [47].
Molecular analysis
Preparation of RNA and DNA
Total RNA was isolated from either mononuclear cells or EBV‐transformed B lymphocytes of both CGD patients and healthy individuals, using a modified single‐step method [48]. Genomic DNA was purified with a purification kit (ref. 51206 Flexigene DNA kit; Qiagen, Hilden, Germany).
Sequencing
When possible, first‐strand cDNA was synthesized from total RNA by reverse transcriptase reaction according to the manufacturer’s instructions (ref. 11EMAMV203; MP Biomedicals Santa Ana, CA, USA). Total cDNA was immediately amplified by polymerase chain reaction (PCR) in three overlapping PCR fragments and separated by gel electrophoresis [49]. The bands were photographed under UV (GelDoc XR+Biorad; Biorad, Marnes‐la‐Coquette, France). All PCR products were sequenced using an ABI 3730 XL 96 capillary sequencer (Perkin Elmer, Foster City, CA, USA). More than 500 sequences of control cDNA of NOX2 were analyzed to rule out the possibility of polymorphisms. In all cases, location of mutations found in cDNA was verified in CYBB genomic DNA after PCR amplification of each exon and flanking intron regions using appropriate forward and backward primers [[49]] followed by Sanger sequencing. This was performed for all patients except for P1, P4a and P12. For patient P1 we used primers to amplify the promoter region of CYBB, as previously described [28]. For patients P4a and P12 new primers were designed to amplify a partial sequence of intron 3 (I3 forward: GAG AGT CTG AGT CAT TGC TCA G; I3 reverse: CTA TCC GGA AGG TGG TCA TAG) and introns 8 and 9 (I8 forward: GCG ACA GAG GCA TTA TTT GGT T; I9 reverse CCT ATG TCT TGC CAG GAC ATC) of CYBB, respectively. Aliquot of all PCR products in bromophenol blue solution were run together with a DNA ladder (ref. R0211, ThermoFisher Scientific) on 1·5% (wt/vol) agarose containing Gel RedTM nucleic acid stain (ref. 41003; Biotium, Inc., Fremont, CA, USA) in parallel with a negative control (PCR products amplified without DNA) and a positive control (PCR amplification of control DNA) to analyze size and purity. In some cases, PCR products were purified from agarose gel according to the manufacturer’s instructions (QIAquick Gel Extraction kit, ref. 28704; Qiagen, Courtaboeuf, France).
Directed mutagenesis and stable transfection into X‐CGD PLB‐985 cells
Thr208Arg and Thr503Ile point mutations were introduced into the wild‐type (WT) NOX2 cDNA in pBluescript II KS(+) vector using the QuikChange site‐directed mutagenesis kit (Stratagene, San Diego, CA, USA), according to the manufacturer’s instructions. The sequence of WT and the mutated NOX2 cDNA were verified by dideoxynucleotide sequencing. The WT or mutant NOX2 cDNAs were subcloned into the mammalian expression vector pEF‐PGKneo and electroporated into X‐CGD PLB‐985 cells in which the CYBB gene was disrupted by gene targeting, resulting in the absence of NOX2 expression and NADPH oxidase activity, as previously described [33]. Clones expressing WT or mutated NOX2 were selected by limiting dilution in 1·5 mg/ml geneticin.
Results
Summary of clinical data
X‐CGD patients were diagnosed from the age of 1 month (P6) to 26 years (P4a), but 10 of 16 patients were diagnosed before the age of 2 years. The most frequent feature was prolonged fever, skin abscesses, adenopathies and pulmonary infections (Table 1). Some patients suffered from an inflammatory syndrome in the gastrointestinal tract (P2, P3, P4a, and P8). Patients P3, P8, P9 and P16 suffered from liver abscesses. The severity criteria were defined according to the presence of severe infections in deep organs associated with sepsis and the age of first symptoms. Tissue examination of some patients (P2, P3 and P8) showed granuloma in skin, liver or tonsils. Patients P1, P4a, P7 and P10 suffered from skin infections and gastroenteritis only without infections in deep organs. Patient P1 was diagnosed at the age of 2 when he suffered from mild infections in the skin. A splenomegaly was detected at the time of diagnosis. He is now in good health under prophylactic treatment. Patient P4a had no susceptibility to infections during childhood and adolescence. He suffered from pollen allergy and mild asthma, but he received no regular medication (Table 1). At the age of 13 he had recurrent perianal abscesses and fistulas. Crohn disease was suspected. In 2012 he had mild Staphylococcus aureus infection around his nose. He was diagnosed at 26 years of age at the time of the death of his younger brother P4b from sudden‐onset Aspergillus pneumonitis. P4a has been symptom‐free for years and he is now in good health under prophylactic treatment. Patient P7 had only superficial adenopathies and infections, but was febrile and suffered from cyanosis and hypothermia. Six of 16 patients suffered from sepsis or prolonged fever (P2, P6, P9, P10, P12 and P15) and one had a permanent hyperthermia (P2). S. aureus was the most frequently encountered bacterium (P2, P4a, P7, P10 and P14), Serratia marcescens being also found two times (P2, P3). Other bacteria were detected often at the origin of infections in CGD patients, such as S. typhimurium and Klebsiella pneumonia, which were found only in patient P12, Mycobacterium bovis in patient P11 and Streptococcus spp. in patients P5 and P12. Pulmonary aspergillosis was found in patients P4b and P12, and suppurative inguinal adenopathies with Candida albicans in P12 and at S. aureus in P10. Regarding the oldest patients P4a, P12 and P14, two were diagnosed early (P12 and P14), while patient P4a was diagnosed at the age of 26. This late diagnosis correlates perfectly with the level of severity of the disease (Table 1). This will be discussed according to the patients’ CGD subtypes.
With respect to the treatment of CGD patients, all had a prophylactic lifelong treatment (trimethoprim/sulfamethoxazole/itraconazole). Low‐dose permanent corticosteroid therapy was given to patients P2 and P14 because of permanent hyperthermia and inflammatory syndrome (P2), and permanent respiratory failure required permanent oxygen therapy (P14). Four patients (P5, P8 P9 and P16) had bone marrow precursor cell transplantation at the age of 16 months to 4 years. Three patients have complete chimerism (P5, P9 and P16). Patient P5 suffered from chronic joint GvH, P16 had an acute digestive graft‐versus‐host disease (GVHD) and patient P8 had persistent diarrhea 6 months post‐transplant. Patient P9 is currently in good health.
Phenotypical and genotypical characterization of X‐CGD patients
The NADPH oxidase activity of purified neutrophils from the 16 X‐CGD patients and their relatives was measured by several different methods (as described in Materials and methods) and appeared to be totally abolished, except for patients P1 and P4a (Table 2). We emphasize that the NADPH oxidase activities measured by the NBT reduction test correlate perfectly with those obtained by flow cytometry with the DHR probe in all the patients’ neutrophils. Sometimes the amount of catalase added to the tube before to test the DHR oxidation in mothers’ neutrophils is not sufficient to prevent passive diffusion of ROS from NOX2‐positive to NOX2‐negative cells (M8, M10 and M16 in Table 2). In addition, for the mothers (M1‐2, M4, M5, M8, M10‐11 and M16) the percentage of ‘oxidase‐positive’ neutrophils correlates perfectly with the percentage of neutrophils presenting a normal expression of NOX2, measured by flow cytometry (Table 2, Fig. 1a). We confirmed the large variation in the maternal phenotype due to different X inactivation patterns. Only one mother was not a carrier of her son’s disease (M7) (Fig. 1a), but we did not investigate the mothers of patients P13 and P14. The daughter of patient P14 was also diagnosed as a carrier (Table 2). In some cases, the absence of NADPH oxidase activity found in patients was measured by SOD‐sensitive cytochrome c reduction (P6, P12 and P15) or resorufin oxidation (P3, P6, P14), which measures external production of superoxide () or hydrogen peroxide (H2O2), respectively (Table 2). However, these methods are less useful to detect the carrier status of the mothers, because the two populations of cells (oxidase‐positive and ‐negative populations) cannot be distinguished by these methods. We also investigated the carrier status of maternal aunts, great‐aunts and grandmothers by looking for the genetic mutation found in the index X‐CGD patients (Gm2, Gam5, Am6, Am9, Am9′, Gam9 and Am15). None were X‐linked carriers. Some sisters of X‐CGD patients (S4 and S12) were carriers, whereas S8 seemed not to be a carrier regarding the NADPH oxidase activity. However, S8 was not fully investigated (no genetic analysis) because she was healthy and under the age for genetic diagnosis (to sign informed consent). For B12, the brother of P12, we tested only the NADPH oxidase activity in his neutrophils for the same reason as S8. Patient P12 was classified as an X910‐CGD patient because of the total absence of NOX2 and p22phox expression by Western blotting (Fig. 1b). However, normal NOX2 and p22phox expression was found in patient P15’s neutrophils even in the absence of NADPH oxidase activity (Fig. 1c). The equal expression of cytosolic factors p40phox, p47phox and p67phox in all the samples can be considered as an internal control of protein loading. This is a typical profile of an X91+‐CGD subtype. In addition, for patients P1 and P4a, faint NADPH oxidase activity and low NOX2 and p22phox expression were found, which is typical for X91−‐CGD subtypes (Table 2). These cases will be discussed according to their specific genetic mutation.
Fig. 1.
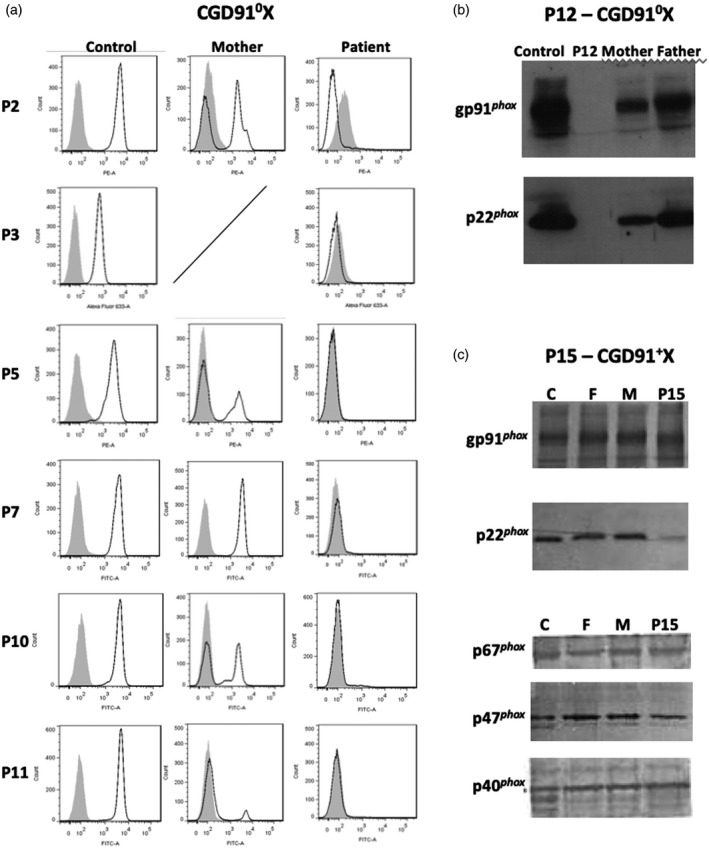
Phenotypical analysis of the neutrophils from X‐linked chronic granulomatous disease (CGD) patients having new CYBB mutations. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2) expression of patients and their mothers’ neutrophils was measured by flow cytometry, as described in Materials and methods. Solid grey curve corresponds to dihydrorhodamine (DHR)‐loaded resting neutrophils, empty black curve corresponds to DHR‐loaded neutrophils after phorbol myristate acetate (PMA) stimulation (a). For patient P12, the absence of gp91phox (or NOX2) and p22phox expression was shown by Western blotting using the primary specific antibodies 48, 449, respectively (b). Note that all the subunits of the NADPH oxidase complex are expressed in patient P15’s neutrophils (X91+CGD) by Western blotting using specific antibodies, as described in Material and methods (c).
X910‐CGD patients
Thirteen of 16 X‐linked CGD patients suffered from X910‐CGD, except patients P1, P4a (X91−‐CGD) and P15 (X91+‐CGD). Indeed, neither NADPH oxidase activity nor NOX2 and p22phox expression were found in the neutrophils of these 13 X0 CGD patients (Table 2). Among these 13 patients we found four missense mutations, three small deletions, two non‐sense mutations, one small insertion, one large deletion, one duplication and one splice mutation. The new double missense mutation c.637 C>G (exon 6) and c.1522 C>T (exon 12), leading p.Thr208Arg and p.Thr503Ile mutations in NOX2, respectively (patient P6), are responsible for a X910‐CGD phenotype (Fig. 2a,b,c). This case is extremely rare in CGD [4]. In order to discard a possible polymorphism, we studied the functional impact of each mutation in the NOX2 knock‐out PLB‐985 cell line, as we performed previously. Using directed mutagenesis and stable transfection in this cellular model, we found that each mutation (p.Thr208Arg and p.Thr503Ile) was deleterious for the NADPH oxidase activity (Fig. 2e), whereas only p.Thr208Arg mutation leads to the absence of NOX2 expression (Fig. 2d). However, we can conclude that neither mutation is a polymorphism. His mother is heterozygous for each mutation. The structural impact of each mutation will be discussed in the context of a three‐dimensional dehydrogenase model of NOX2 (Fig. 3a,b) [37].
Fig. 2.
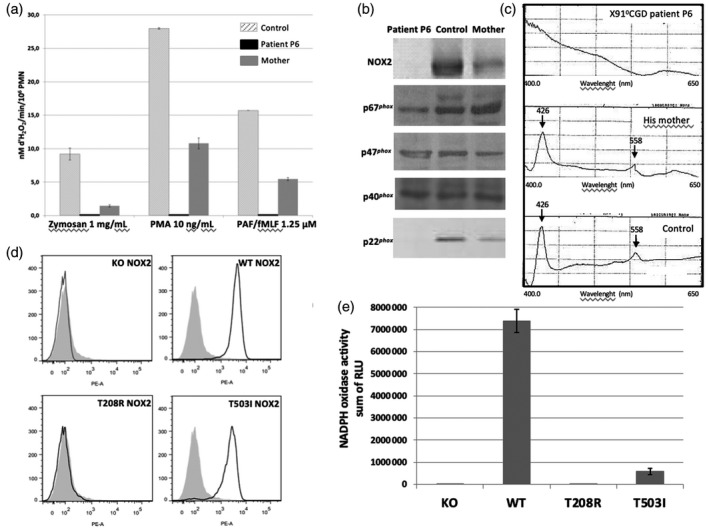
Functional analysis of patient P6’s T208R and T503I substitutions in nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2) by means of transgenic PLB‐985 cells. Measurement of the NOX2 oxidase activity of neutrophils of patient P6 and of his mother by resorufine oxidation (Amplex RedR) after opsonized zymosan, phorbol myristate acetate (PMA) and platelet‐activating factor/N‐formyl‐Met‐Leu‐Phe (PAF/fMLF) activation, as described in Materials and methods (a). Expression of NADPH oxidase subunits NOX2, p67phox and p47phox p40phox and p22phox in patient P6 and his mother’s neutrophils by Western blot analysis (b). Cytochrome b 558 differential spectra of 1% Triton X100 soluble extract from patient P6 and his mother’s neutrophils (c). NOX2 expression in differentiated T208R–NOX2 and T503I–NOX2 transgenic PLB‐985 cells by flow cytometry using the 7D5 monoclonal antibody against NOX2 and phycoerythrin (PE)‐coupled polyclonal antibody anti‐IgG1 (empty black curve). Solid grey curve corresponds to resting neutrophils labeled with monoclonal mouse IgG1 isotype as an irrelevant antibody (d). NADPH oxidase activity of differentiated T208R–NOX2 and T503I–NOX2 transgenic PLB‐985 cells measured by chemiluminescence in presence of luminol, horseradish peroxidase (HRP) after 80 ng/ml PMA stimulation (e).
Fig. 3.
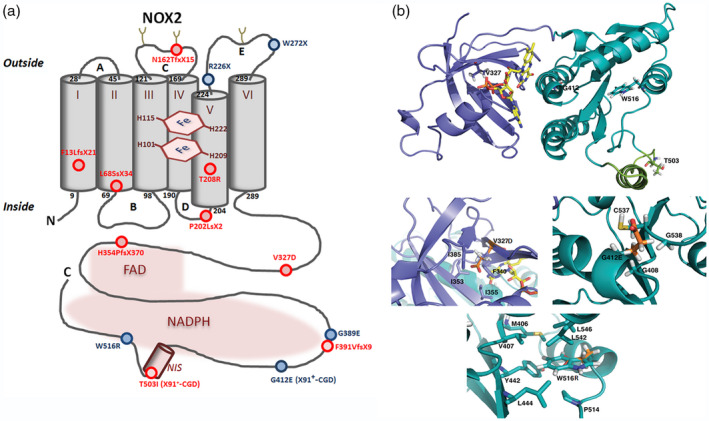
(a) Schematic representation of X‐linked chronic granulomatous disease (CGD) point mutations within the NOX2 protein – new mutations are shown in red and known mutations in blue. New mutations in the promoter (patient P1) or in intron sequences (patients P4a and P12) of CYBB are not represented. (b) NOX2 dehydrogenase model and modeling of CGD mutations. The DH model is represented as ribbons, dark green for the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2) binding domain – corresponding to the 3A1F Protein Data Bank (db) structure – and purple‐blue for the modelled FAD binding domain ([37]). NOX2 NIS, in light green, is an approximate structural model but is partly disordered, flexible as suggested by the absence of density in the 3A1F structure for this sequence. Some of the residues corresponding to X910 and X91+ mutations are highlighted as shown in stick (V327, G412, W516 and T503). Three zooms highlight the V327, G412 and W516 residues with emphasis on the neighboring residues around the considered mutations. In each case, the mutated position is presented with both the wild‐type side chain and the corresponding mutation, in orange, superimposed. Molecular graphic images were produced using PyMOL software
X91+‐CGD patients
Among 681 X‐linked CGD cases reported in 2010 [4], only 38 patients with 28 different mutations were identified as having an X91+‐CGD subtype. Often, these CGD variants are due to missense mutations [19]. The phenotype of patient P15’s neutrophils fitted perfectly with this type of variant (Table 2). Indeed, all the NADPH oxidase subunits were normally expressed in P15’s neutrophils (Fig. 1c), whereas the NADPH oxidase activity was totally abolished (Table 2). A point mutation in exon 10 of CYBB (c.1235G>A) was responsible for the change of glycine‐412 to glutamic acid in NOX2 (p.Gly412Glu) (Fig. 3b). The total absence of NADPH oxidase activity correlates with the severe clinical profile found in this patient (Table 1). This mutation was reported in Roos et al. [4], but not fully characterized. Gly412 is in the dehydrogenase domain of NOX2 close to the NADPH binding site (Fig. 3). This will be discussed in the context of a three‐dimensional dehydrogenase model of NOX2 (Fig. 3b) [37].
X91−‐CGD patients
A faint but significant NADPH oxidase activity was detected in the neutrophils of patients P1 and P4a (Table 2). The pattern of NOX2 expression was different in both patients. In patient P1 98% of the neutrophils exhibited no NADPH oxidase activity, whereas approximately 2% of cells had a normal oxidase activity related to a normal expression of NOX2 (Table 2, Fig. 4a). This small population of cells was identified as eosinophils, owing to CD294 double‐staining. However, a CD294‐positive and oxidase‐negative population of cells was also highlighted (Fig. 4a). CD294, also named CRTH2, is a seven‐transmembrane G‐protein‐coupled receptor known as the chemoattractant receptor‐homologous molecule expressed also in T helper type 2 (Th2) cells. Thus, perhaps these cells were lymphocytes. The level of NOX2 expression is also in accordance with the percentage of both populations of cells (neutrophils and eosinophils) (Fig. 4b). NOX2 expression and the cytochrome b 558 spectrum of patient P1’s cells are probably due to eosinophilic contamination of the neutrophils after Ficoll purification (Fig. 4c,d). Using the same amount of proteins for each sample (control, mother and patient P1) (20 µg) loaded on the sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS‐PAGE), a faint NOX2 and p22phox expression in patient P1’s granulocytes was seen by Western blot analysis (Fig.4c). We verified that NOX2 was correctly glycosylated by performing a Western blot with 200 µg of protein for patient P1 (data not shown). A novel −67delT mutation was found in the promoter region of CYBB (Fig. 4e). This point mutation is located in a region between the CCAAT and the TATA boxes in a consensus binding region of transcription factors (TFs) of the ETS family (Elf1 and PU.1) [29, 50, 51] (Fig. 4f). We and others have previously demonstrated that mutations located in this region disturb the binding of these TFs, which are necessary to trigger NOX2 expression [25, 26, 28]. However, in eosinophils, NOX2 expression is not under the control of these TFs [52]. This explains the NOX2 expression and the NADPH oxidase activity seen in the eosinophils of patient P1 (Fig. 4a,b).
Fig. 4.
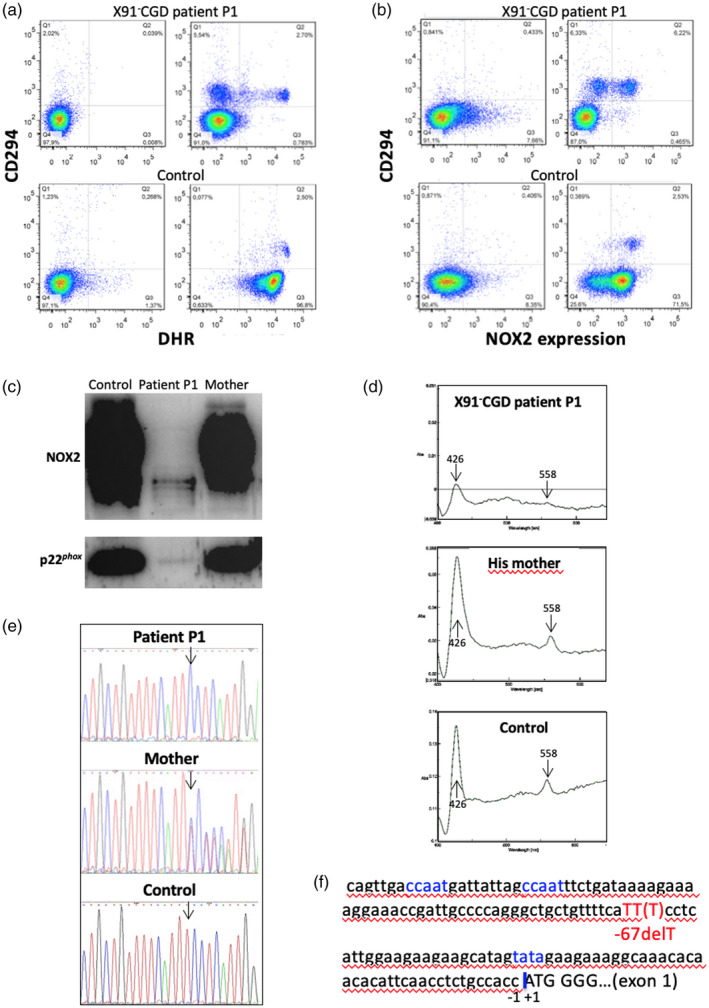
Phenotypical and genotypical characterization of the X91− ‐chronic granulomatous disease (CGD) of patient P1 due to a new point mutation in the CYBB promoter. Flow cytometry dot‐plots of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2) oxidase activity [dihydrorhodamine (DHR)] and CD294 expression (eosinophil marker) in neutrophils and eosinophils of granulocyte preparations from a healthy subject (bottom panels) and patient P1 (top panels) at rest (left panels) or after stimulation with phorbol myristate acetate (PMA) (right panels). Left panels correspond to dot‐plots of cells labeled with isotype control antibodies and loaded with DHR (a). Flow cytometry dot‐plots of NOX2 and CD294 expression (eosinophil marker) in neutrophils and eosinophils of granulocyte preparations from a healthy subject (bottom panels) and patient P1 (top panels) using 7D5 monoclonal antibody and anti‐CD294 antibody (right panels) compared to dot‐plots of isotype controls (left panels) (b). NOX2 and p22phox expression in patient P1 and his mother’s granulocytes by Western blot analysis. A faint NOX2 and p22phox expression are visible probably due to the presence of eosinophils in the granulocyte preparation (c). Cytochrome b 558 differential spectra of 1% Triton X100 soluble extract from patient P1 and his mother’s neutrophils (d). Thymidine deletion at position −67 detected in the CYBB promoter region of patient P1 and his mother as a carrier, compared to a healthy donor sequence (e). Schematic representation of part of the CYBB promoter region, with localization of the ‘CCAAT’ and ‘TATA’ boxes, the ATG starting codon and the point mutation described in panel (e) (f).
For patient P4a, we found that the whole population of neutrophils exhibited a faint NADPH oxidase activity (Fig. 5a). This result correlates with the faint formazan staining of patient P4a’s neutrophils in the NBT reduction test (Fig. 5b). A low NOX2 expression was also found in patient P4a’s neutrophils by flow cytometry and Western blot analysis, which correlates with the level of NADPH oxidase activity found in these cells (Fig. 5c,d). In addition, a substantial p22phox expression was found in patient P4a’s neutrophils, but in fewer amounts than in control neutrophils (Fig. 5e). His mother and sister seemed to be carriers, because they both possessed two populations of neutrophils, one oxidase‐positive and an oxidase‐negative population. This distribution was also seen in the NBT reduction test (Fig. 5b). However, the percentage of each population differs in the mother and sister of patient P4a (Fig. 5a). His mother has a major population of neutrophils with a normal oxidase activity (80% of the neutrophils) and the remaining neutrophils have a slight oxidase activity, as seen in her son (20% of neutrophils), even in the presence of catalase. In contrast, his sister has approximately 50% of neutrophils with a normal oxidase activity and 50% of cells with no oxidase activity (Table 2, Fig. 5a). This distribution is confirmed by the NOX2 and p22phox expression profiles in their neutrophils (Fig. 5d,e). These differences are probably caused by different X inactivation patterns in both carriers.
Fig. 5.
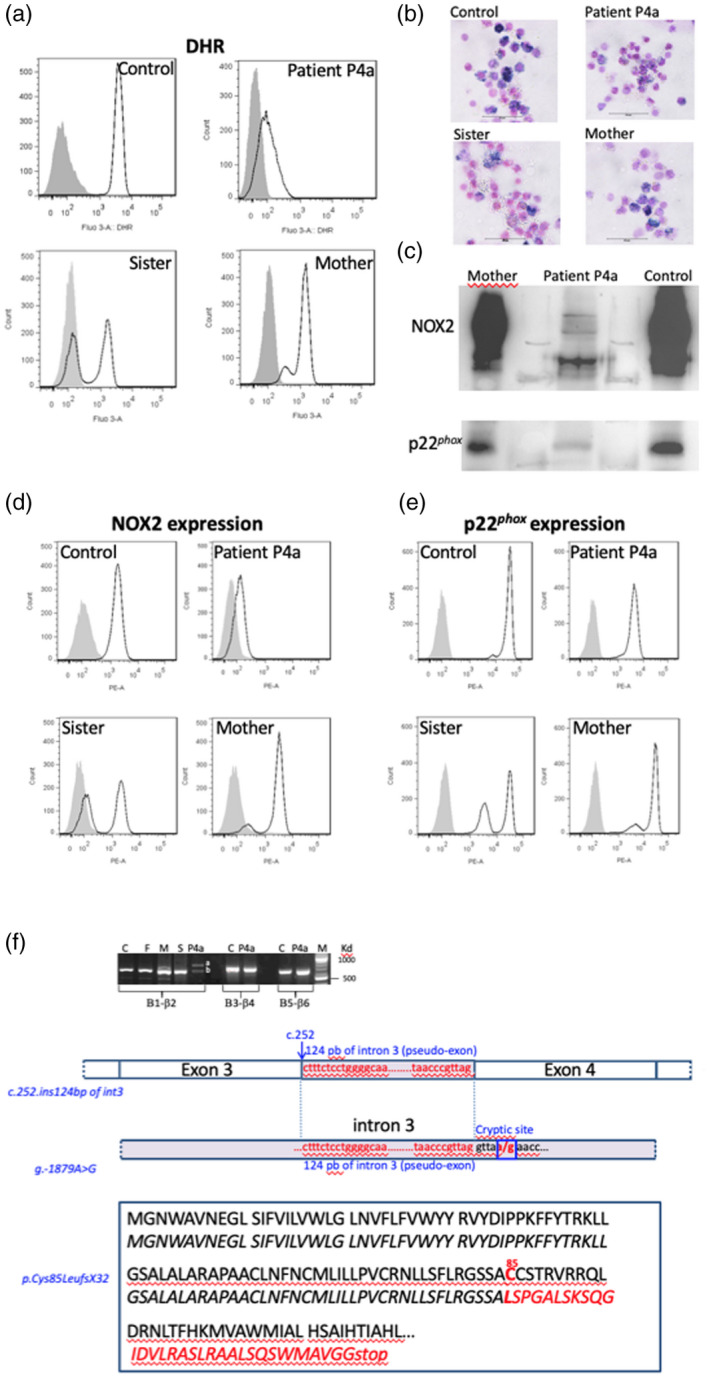
Phenotypical analysis of the neutrophils of patient P4a suffering from an X91−‐chronic granulomatous disease (CGD) and having a new CYBB mutation. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2) oxidase activity of neutrophils from patient P4a, his sister and his mother was measured by flow cytometry after stimulation during 15 min with 200 ng/ml phorbol myristate acetate (PMA) in the presence of the dihydrorodamine (DHR) probe, as described in Materials and methods. Solid grey curve corresponds to DHR‐loaded resting neutrophils, empty black curve corresponds to DHR‐loaded neutrophils after PMA stimulation (a). The NADPH oxidase activity of neutrophils from patient P4a, his sister and his mother was also measured using the nitro blue‐tetrazolium (NBT) reduction test (b). NOX2 and p22phox expression was shown by Western blot using the primary specific antibodies 48 and 449, respectively (c). Nox2 (d) and p22phox expression (e) was shown by flow cytometry using the primary specific antibodies 7D5 and 449, respectively. Solid grey curve corresponds to resting neutrophils labeled with irrelevant antibodies; empty black curve corresponds to neutrophils labeled with the 7D5 (anti‐NOX2) or 449 antibodies (p22phox) and secondary antibodies coupled with phycoerythrin (PE). Genetic analysis of the mutation of patient P4a in CYBB gene, leading to an X91−‐CGD. NOX2 cDNA was amplified in three overlapping fragments, as described in Materials and methods. A 124 base pair (bp) of intron 3 was inserted between exons 3 and 4 leading to the creation of a pseudoexon. Intronic regions surrounding the inserted region of intron 3 in the NOX2 cDNA was amplified and sequenced. A c.253‐1879A>G hemizygous mutation was found creating a splicing donor site, which unveils a cryptic acceptor site leading the inclusion of 124 nucleotides as a pseudo‐exon responsible for the partial loss of the NOX2 expression. The consequence in the NOX2 protein is a missense mutation at Cys85→Leu and a frameshift leading to the generation of a stop codon located 32 amino acids further on (p.Cys85LeufsX32) (f).
To find the mutation in CYBB of patient P4a, NOX2 cDNA was amplified in three overlapping fragments from total mRNA by reverse transcription–polymerase chain reaction (RT–PCR). Two bands were found in the first amplified fragment (β1–β2), one having a normal size [621 base pairs (bp)] corresponding to exons 1 to 6, the other having an abnormal higher size. Each band was excised, and cDNA was purified and sequenced. No mutation was found in the normal‐sized band (exons 1 to 6). The sequence of the upper band showed an insertion of 124 bp of intron 3, as described in Table 2 and Fig. 5f. No mutations were found in the two other amplified fragments of cDNA (β3–β4 and β5‐β6). We sequenced all exons of CYBB with intronic flanking regions after PCR amplification from the genomic DNA of patient P4a and his relatives and no mutation was found. Intronic regions surrounding the inserted region of intron 3 were then amplified and sequenced. A novel c.253‐1879A>G hemizygous mutation was found that activates a splicing donor site. The inclusion of the 124 bp fragment into the mRNA unveils an active cryptic acceptor site at the start of a pseudo‐exon, and this inclusion is responsible for the partial loss of the NOX2 expression (Fig. 5f). Bioinformatic analysis allows the estimation of the splice donor sequence strength. The c.253‐1879A>G mutation induces an increase of the splice donor strength from 6·02 to 9·14 with the MaxEntScan matrix and from 83·52 to 95·58 with the Human Splicing Finder (HSF) matrix (http://www.umd.be/HSF/technicaltips.html). The inserted sequence in the NOX2 cDNA creates a frameshift and introduction of a stop codon at position 85, explaining the loss of NOX2 expression (Fig. 5f). However, the neutrophils of patient P4a exhibited a low NOX2 expression related to a low NADPH oxidase activity by flow cytometry; it seems that a faint amount of normal NOX2 was expressed in his cells. The c.253‐1879A>G mutation was also found in the genomic DNA of his mother and sister in one allele, confirming their carrier status.
In conclusion, patients P1 and P4a suffer from an X91−‐CGD but the genetic mutation is very different for both patients, which can explain the phenotypical differences in their neutrophils. According to their clinical data, both patients suffer from a mild form of CGD.
Discussion
We report here 16 patients with X‐linked CGD (X91‐CGD, OMIM #306400) presenting mutations in the CYBB gene. Most of the patients were from France, except patients P1, P4a, P11 and P13, who came from Italy, Finland, Lithuania and Croatia, respectively. Most of the patients suffered from an X910‐CGD, except patient P15, who had an X91+‐CGD and patients P1 and P4a who exhibited an X91−‐CGD. This classification is based on the level of NOX2 expression, as previously demonstrated. All types of mutations were found leading to X910‐CGD type, and nine of 13 were new and spread over the whole coding region. Two of these nine new X910‐CGD mutations were a point mutation in intron 6 (patient P7) and a large deletion (intron 9/exon 9) (patient P12) leading to the deletion of exons 5/6 and exon 9, respectively. Regarding their clinical signs, the X910‐CGD cases were by far the most severe forms, probably because of the absence of NOX2 expression and NADPH oxidase activity. The severity of the impact of missense mutations on the NADPH oxidase expression and activity can be highlighted regarding their effect on the structure of the enzyme. Indeed, we analyzed an extremely rare case of X910‐CGD due to a double missense mutation (p.Thr208Arg and p.Thr503Ile). To our knowledge, it is the first listed case of a double missense mutation causing a X910‐CGD. We previously published a unique case of a similar double missense mutation p.His303Asn/Pro304Arg leading to an X91+‐CGD subtype ([53, 54]). Each missense mutation was deleterious for the NADPH oxidase activity, but not for its expression, ruling out the possibility of a polymorphism. However, polymorphisms are rare in CYBB [4]. In the present case, and regarding their respective position in the NOX2 model, each mutation acts through different mechanisms. Thr208 is located in the transmembrane segment V just upstream of His209, which is one of the proximal heme ligands (Fig. 3a). Thus, the mutation in the transmembrane domain of this Thr208 to an arginine, cumulating in higher steric properties and a positive charge, may be deleterious to the structure of the transmembrane domain and at least locally may impair proper orientation of His209. This may lead in cascade to the absence of proximal heme, and thus the impossibility to mature the final protein, explaining a X910 phenotype for this modeled single mutant. Regarding Thr503ILe, the second cumulative missense mutation in this patient, its modelling in PLB985 leads to a normal expression but the loss of activity and thus a X91+ phenotype. This Thr503 is located in a hot‐spot for X91+‐CGD mutations in the NIS (NOX insertion sequence), which is a sequence specific of NOX dehydrogenase domains but absent in other structurally related dehydrogenase domains (e.g. ferredoxin NADP+ reductase) (Fig. 3b). Previous mutations reported in the NIS have been shown to impair assembly with cytosolic factor and/or activation or the initial electron transfer from NADPH to FAD [33, 34, 35]. Regarding its location, the Thr503ILe mutation is likely to induce similar defects to those already studied and characterized in this NIS [37]. Among missense mutations in CYBB (Fig. 3), the p.Val327Asp mutation leading to a X910‐CGD in NOX2 was new. This Val residue is located in the FAD binding subdomain of the NOX2 dehydrogenase domain (Fig. 3b) and is at the center of a hydrophobic cluster within the domain. Introducing an aspartic acid in that position is expected to disrupt the structure of the domain. This is fully compatible with the X910 phenotype within patient P10. A similar analysis can be drawn for the Trp516Arg mutant, where the Trp516 side chain is at the center of a hydrophobic cluster within the NADPH binding subdomain (Fig. 3b). Its replacement by an Arg residue does not allow the correct folding.
Replacement of Gly412 by a glutamic acid leads to a rare X91+ phenotype. At this position in the structure a glycine residue is strictly conserved, as well in the neighboring loops containing G408 and G538 (Fig. 3b). Addition of a Glu in this position may distort the proper packing of the Rossman fold and thus an efficient NADPH binding site. In addition, a Glu in this position adds a negative charge in the heart of the NADPH binding site which is already a strongly negatively charged coenzyme. These various elements explain a defective recognition of NADPH in the Gly412Glu mutant [37]. However, the clinical feature of this X91+‐CGD was as severe as X910‐CGD cases because of the total absence of NADPH oxidase activity in phagocytes. The clinical severity of X91−‐CGD is variable and depends upon the localization of the mutations in CYBB related to the level of NADPH oxidase activity found in patients’ phagocytes [19, 22, 24]. This X91−‐CGD variant is often due to missense mutations, small deletions or insertions in the encoding region of NOX2 (52 cases of X91−‐CGD) listed in [4], leading to a decrease of its expression in phagocytes, the NADPH oxidase activity being slightly or totally abolished. We previously demonstrated that there are two categories of X91−‐CGD mutations, one in which the NADPH oxidase activity is proportional to the NOX2 expression level and another showing an absence of NADPH oxidase activity associated with variable levels of NOX2 expression, the last one being associated with more severe clinical cases [22]. This work also allowed us to determine potential binding regions of NOX2 with p22phox, essential for normal synthesis of cytochrome b 558. In this work, the impact of the point mutation in intron 3 of CYBB on NOX2 expression and NADPH oxidase activity in phagocytes of patient P4a was quite unusual. Indeed, a unique population of neutrophils was found by flow cytometry characterized by a faint NOX2 expression associated with a low NADPH oxidase activity, suggesting that normal mRNA was present in phagocytes. Even if mutated mRNA can be removed by non‐sense‐mediated mRNA decay, the normal splice manages to remain in the patient P4a’s neutrophils. In addition, p22phox expression was faintly impacted by this mutation. Thus, the clinical form of this X91−‐CGD case was mild. The faint but substantial NADPH oxidase activity found in the neutrophils’ population of patient P4a could partially protect him against infections. However, we must also emphasize that patient P4a had a younger brother, P4b, who died at 24 years of age of sudden‐onset Aspergillus pneumonitis. CGD was suspected post mortem for patient P4b and no biological investigation was done for him. Patient P4a was diagnosed after his brother’s death. He is in good health and is now the father of two children. Patient P1 suffered from a mild clinical form, as was the case for the other patients with this type of mutation [25, 26, 28]. Only five different mutations in the CYBB promoter leading to X91−‐CGD were documented in the literature (for review see [23]). Location of these mutations is situated in a region between the CCAAT and the TATA boxes in a consensus‐binding region of transcription factors of the ETS family (Elf1 and PU.1) that regulate NOX2 expression in neutrophils and monocytes [29, 50, 51]. However, NOX2 expression is regulated differently in eosinophils [52], explaining that patient P1 can partially be protected against infections because of the normal NADPH oxidase activity in his eosinophils.
In conclusion, in addition to rapid and efficient diagnosis provided by the new generation of high‐throughput sequencing, certain rare monogenic diseases such as CGDs require complete functional and molecular investigation to more clearly understand the impact of each genetic mutation on the enzyme expression and activity related to the clinical severity of the disease. This refined characterization helps understanding of the molecular mechanisms of the disease and to some extent may assist in the care and follow‐up of these patients. In addition, owing to the three‐dimensional model of the dehydrogenase domain of NOX2 and the functional study of single mutation in the NOX2 knock‐out PLB‐985 cell line, analysis of the impact of certain CGD missense mutations on NOX2 activity and synthesis permits us to improve our knowledge of the functioning of this enzyme.
Disclosures
On behalf of all authors, the corresponding author states that there are no conflicts of interest.
Author contributions
M. M., S. B., B. V. and J. B. performed the functional and molecular analysis; N. R‐B., J. R. and J. F. performed the DNA sequencing, V. B., G. C., C. D., F. F., V. G., M. R., J‐P. B., C. B‐B, E. J., L. E., C. J., S. D. H., M H‐C., M. H., G. M., Y. B., D. P., J. K., R. T. and L. K., performed the clinical diagnosis and collected the clinical data; D. M. collected some functional data; F. F. contributed to analysis of the data, drew Fig. 1b and revised the article; M. J. S. carried out the conception and design of the study, acquisition, analysis and interpretation of data and the drafting of the article.
Data sharing statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Acknowledgements
The authors would like to pay tribute to Cécile Martel, who was largely involved in the diagnosis of CGD patients for more than 30 years and who passed away in 2016. A warm thank you to Professor Dirk Roos, the Netherlands, for his careful re‐reading, corrections and his judicious advices. M. J. S. is grateful for support from the University Grenoble Alpes (UGA) (AGIR program 2014); the Faculty of Medicine Grenoble, Interreg France‐Suisse [Programme de Cooperation Territoriale Européenne, Fond Européen de Développement Regional (FEDER), 2017–2019]. This work was also supported by the Delegation for Clinical Research and Innovations, University Hospital Grenoble Alpes (CHUGA) (DRCI, Rementips project 2014). The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1. Mahlaoui N, Picard C, Bach P et al Genetic diagnosis of primary immunodeficiencies: a survey of the French national registry. J Allergy Clin Immunol 2019; 143:1646–1649.e1610. [DOI] [PubMed] [Google Scholar]
- 2. Roos D. Chronic granulomatous disease. Br Med Bull 2016; 118:50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roos D, Kuhns DB, Maddalena A et al Hematologically important mutations: the autosomal recessive forms of chronic granulomatous disease (second update). Blood Cells Mol Dis 2010; 44:291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roos D, Kuhns DB, Maddalena A et al Hematologically important mutations: X‐linked chronic granulomatous disease (third update). Blood Cells Mol Dis 2010; 45:246–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van de Geer A, Nieto‐Patlan A, Kuhns DB et al Inherited p40phox deficiency differs from classic chronic granulomatous disease. J Clin Invest 2018; 128:3957–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu L, DeLeo FR, Biberstine‐Kinkade KJ, Renee J, Nauseef WM, Dinauer MC. Biosynthesis of flavocytochrome b558. gp91(phox) is synthesized as a 65‐kDa precursor (p65) in the endoplasmic reticulum. J Biol Chem 1999; 274:4364–9. [DOI] [PubMed] [Google Scholar]
- 7. Nauseef WM, Clark RA. Intersecting stories of the phagocyte NADPH oxidase and chronic granulomatous disease. Methods Mol Biol 2019; 1982:3–16. [DOI] [PubMed] [Google Scholar]
- 8. Thomas DC, Charbonnier LM, Schejtman A et al EROS/CYBC1 mutations: decreased NADPH oxidase function and chronic granulomatous disease. J Allergy Clin Immunol 2019; 143:782–785.e781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gu Y, Williams DA. RAC2 GTPase deficiency and myeloid cell dysfunction in human and mouse. J Pediatr Hematol Oncol 2002; 24:791–4. [DOI] [PubMed] [Google Scholar]
- 10. Yu H, Leitenberg D, Li B, Flavell RA. Deficiency of small GTPase Rac2 affects T cell activation. J Exp Med 2001; 194:915–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roberts AW, Kim C, Zhen L et al Deficiency of the hematopoietic cell‐specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity 1999; 10:183–96. [DOI] [PubMed] [Google Scholar]
- 12. Sharapova SO, Haapaniemi E, Sakovich IS et al Heterozygous activating mutation in RAC2 causes infantile‐onset combined immunodeficiency with susceptibility to viral infections. Clin Immunol 2019; 205:1–5. [DOI] [PubMed] [Google Scholar]
- 13. Hsu AP, Donko A, Arrington ME et al Dominant activating RAC2 mutation with lymphopenia, immunodeficiency, and cytoskeletal defects. Blood 2019; 133:1977–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Williams DA, Tao W, Yang F et al Dominant negative mutation of the hematopoietic‐specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood 2000; 96:1646–54. [PubMed] [Google Scholar]
- 15. Ambruso DR, Knall C, Abell AN et al Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Natl Acad Sci USA 2000; 97:4654–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sumimoto H. Structure, regulation and evolution of Nox‐family NADPH oxidases that produce reactive oxygen species. FEBS J 2008; 275:3249–77. [DOI] [PubMed] [Google Scholar]
- 17. van den Berg JM, van Koppen E, Ahlin A et al Chronic granulomatous disease: the European experience. PLOS ONE 2009; 4:e5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Winkelstein JA, Marino MC, Johnston RB Jr et al Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine 2000; 79:155–69. [DOI] [PubMed] [Google Scholar]
- 19. O’Neill S, Brault J, Stasia MJ, Knaus UG. Genetic disorders coupled to ROS deficiency. Redox Biol 2015; 6:135–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Royer‐Pokora B, Kunkel LM, Monaco AP et al Cloning the gene for the inherited disorder chronic granulomatous disease on the basis of its chromosomal location. Cold Spring Harb Symp Quant Biol 1986; 51:177–83. [DOI] [PubMed] [Google Scholar]
- 21. Teahan C, Rowe P, Parker P, Totty N, Segal AW. The X‐linked chronic granulomatous disease gene codes for the beta‐chain of cytochrome b‐245. Nature 1987; 327:720–1. [DOI] [PubMed] [Google Scholar]
- 22. Beaumel S, Grunwald D, Fieschi F, Stasia MJ. Identification of NOX2 regions for normal biosynthesis of cytochrome b558 in phagocytes highlighting essential residues for p22phox binding. Biochem J 2014; 464:425–37. [DOI] [PubMed] [Google Scholar]
- 23. Stasia MJ, Li XJ. Genetics and immunopathology of chronic granulomatous disease. Semin Immunopathol 2008; 30:209–35. [DOI] [PubMed] [Google Scholar]
- 24. Kuhns DB, Alvord WG, Heller T et al Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med 2010; 363:2600–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Newburger PE, Skalnik DG, Hopkins PJ, Eklund EA, Curnutte JT. Mutations in the promoter region of the gene for gp91‐phox in X‐linked chronic granulomatous disease with decreased expression of cytochrome b558. J Clin Invest 1994; 94:1205–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weening RS, De Boer M, Kuijpers TW, Neefjes VM, Hack WW, Roos D. Point mutations in the promoter region of the CYBB gene leading to mild chronic granulomatous disease. Clin Exp Immunol 2000; 122:410–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stasia MJ, Brion JP, Boutonnat J, Morel F. Severe clinical forms of cytochrome b‐negative chronic granulomatous disease (X91‐) in 3 brothers with a point mutation in the promoter region of CYBB. J Infect Dis 2003; 188:1593–604. [DOI] [PubMed] [Google Scholar]
- 28. Defendi F, Decleva E, Martel C, Dri P, Stasia MJ. A novel point mutation in the CYBB gene promoter leading to a rare X minus chronic granulomatous disease variant – impact on the microbicidal activity of neutrophils. Biochim Biophys Acta 2009; 1792:201–210. [DOI] [PubMed] [Google Scholar]
- 29. Voo KS, Skalnik DG. Elf‐1 and PU.1 induce expression of gp91(phox) via a promoter element mutated in a subset of chronic granulomatous disease patients. Blood 1999; 93:3512–20. [PubMed] [Google Scholar]
- 30. Zhen L, King AA, Xiao Y, Chanock SJ, Orkin SH, Dinauer MC. Gene targeting of X chromosome‐linked chronic granulomatous disease locus in a human myeloid leukemia cell line and rescue by expression of recombinant gp91phox. Proc Natl Acad Sci USA 1993; 90:9832–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu L, Cross AR, Zhen L, Dinauer MC. Functional analysis of NADPH oxidase in granulocytic cells expressing a delta488‐497 gp91(phox) deletion mutant. Blood 1999; 94:2497–504. [PubMed] [Google Scholar]
- 32. Zhen L, Yu L, Dinauer MC. Probing the role of the carboxyl terminus of the gp91phox subunit of neutrophil flavocytochrome b558 using site‐directed mutagenesis. J Biol Chem 1998; 273:6575–81. [DOI] [PubMed] [Google Scholar]
- 33. Li XJ, Grunwald D, Mathieu J, Morel F, Stasia MJ. Crucial role of two potential cytosolic regions of Nox 2, 191TSSTKTIRRS200 and 484DESQANHFAVHHDEEKD500, on NADPH oxidase activation. J Biol Chem 2005; 280:14962–73. [DOI] [PubMed] [Google Scholar]
- 34. Li XJ, Fieschi F, Paclet MH et al Leu505 of Nox2 is crucial for optimal p67phox‐dependent activation of the flavocytochrome b558 during phagocytic NADPH oxidase assembly. J Leukoc Biol 2007; 81:238–49. [DOI] [PubMed] [Google Scholar]
- 35. Debeurme F, Picciocchi A, Dagher MC et al Regulation of NADPH oxidase activity in phagocytes: relationship between FAD/NADPH binding and oxidase complex assembly. J Biol Chem 2010; 285:33197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Picciocchi A, Debeurme F, Beaumel S et al Role of putative second transmembrane region of Nox2 protein in the structural stability and electron transfer of the phagocytic NADPH oxidase. J Biol Chem 2011; 286:28357–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beaumel S, Picciocchi A, Debeurme F et al Down‐regulation of NOX2 activity in phagocytes mediated by ATM‐kinase dependent phosphorylation. Free Radic Biol Med 2017; 113:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boyum A. A one‐stage procedure for isolation of granulocytes and lymphocytes from human blood. General sedimentation properties of white blood cells in a 1g gravity field. Scand J Clin Lab Invest Suppl 1968; 97:51–76. [PubMed] [Google Scholar]
- 39. Batot G, Paclet MH, Doussiere J et al Biochemical and immunochemical properties of B lymphocyte cytochrome b558. Biochim Biophys Acta 1998; 1406:188–202. [DOI] [PubMed] [Google Scholar]
- 40. Tucker KA, Lilly MB, Heck L Jr, Rado TA. Characterization of a new human diploid myeloid leukemia cell line (PLB‐985) with granulocytic and monocytic differentiating capacity. Blood 1987; 70:372–8. [PubMed] [Google Scholar]
- 41. Martel C, Mollin M, Beaumel S et al Clinical, functional and genetic analysis of twenty‐four patients with chronic granulomatous disease – identification of eight novel mutations in CYBB and NCF2 genes. J Clin Immunol 2012; 32:942–58. [DOI] [PubMed] [Google Scholar]
- 42. Verhoeven AJ, Bolscher BG, Meerhof LJ et al Characterization of two monoclonal antibodies against cytochrome b558 of human neutrophils. Blood 1989; 73:1686–94. [PubMed] [Google Scholar]
- 43. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227:680–5. [DOI] [PubMed] [Google Scholar]
- 44. Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 1979; 76:4350–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vergnaud S, Paclet MH, El Benna J, Pocidalo MA, Morel F. Complementation of NADPH oxidase in p67‐phox‐deficient CGD patients p67‐phox/p40‐phox interaction. Eur J Biochem 2000; 267:1059–67. [DOI] [PubMed] [Google Scholar]
- 46. Stasia MJ, Mollin M, Martel C et al Functional and genetic characterization of two extremely rare cases of Williams–Beuren syndrome associated with chronic granulomatous disease. Eur J Hum Genet 2013; 21:1079–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smith PK, Krohn RI, Hermanson GT et al Measurement of protein using bicinchoninic acid. Anal Biochem 1985; 150:76–85. [DOI] [PubMed] [Google Scholar]
- 48. Chomczynski P, Sacchi N. Single‐step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal Biochem 1987; 162:156–9. [DOI] [PubMed] [Google Scholar]
- 49. Bakri FG, Martel C, Khuri‐Bulos N et al First report of clinical, functional, and molecular investigation of chronic granulomatous disease in nine Jordanian families. J Clin Immunol 2009; 29:215–30. [DOI] [PubMed] [Google Scholar]
- 50. Eklund EA, Luo W, Skalnik DG. Characterization of three promoter elements and cognate DNA binding protein(s) necessary for IFN‐gamma induction of gp91‐phox transcription. J Immunol 1996; 157:2418–29. [PubMed] [Google Scholar]
- 51. Suzuki S, Kumatori A, Haagen IA et al PU.1 as an essential activator for the expression of gp91(phox) gene in human peripheral neutrophils, monocytes, and B lymphocytes. Proc Natl Acad Sci USA 1998; 95:6085–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang D, Suzuki S, Hao LJ et al Eosinophil‐specific regulation of gp91(phox) gene expression by transcription factors GATA‐1 and GATA‐2. J Biol Chem 2000; 275:9425–32. [DOI] [PubMed] [Google Scholar]
- 53. Stasia MJ, Lardy B, Maturana A et al Molecular and functional characterization of a new X‐linked chronic granulomatous disease variant (X91+) case with a double missense mutation in the cytosolic gp91phox C‐terminal tail. Biochim Biophys Acta 2002; 1586:316–30. [DOI] [PubMed] [Google Scholar]
- 54. Bionda C, Li XJ, van Bruggen R et al Functional analysis of two‐amino acid substitutions in gp91 phox in a patient with X‐linked flavocytochrome b558‐positive chronic granulomatous disease by means of transgenic PLB‐985 cells. Hum Genet 2004; 115:418–27. [DOI] [PubMed] [Google Scholar]