

Abstract
Nevoid basal cell carcinoma syndrome (NBCCS) is mainly characterised by multiple basal cell carcinomas (BCCs) caused by PTCH1, PTCH2, and SUFU. However, clinical and genetic data on Asian NBCCS patients are limited. We aimed to analyse the clinical phenotypes and genetic spectrum of Korean patients with NBCCS. Fifteen patients with NBCCS at Seoul National University Hospital were included, and their clinical data were analysed. Whole-exome sequencing and/or multiplex ligation-dependent probe amplification using peripheral blood were performed to identify genetic causes. Genetic analysis revealed that 73.3% (11/15) of the patients carried 9 pathogenic variants, only in the PTCH1 gene. Variants of uncertain significance (VUS) and likely benign were also detected in 2 (13.3%) and 2 (13.3%) patients, respectively. BCCs were found in the majority of the cases (93.3%) and the number of BCCs increased with age (ρ = 0.595, P = 0.019). This study revealed that PTCH1 pathogenic variants were the main cause of NBCCS in Korean patients. As BCCs are commonly detected, a periodic dermatologic examination is recommended. Finally, our results support the addition of genetic screening to the existing criteria for NBCCS diagnosis.
Subject terms: Skin cancer, Next-generation sequencing
Introduction
Nevoid basal cell carcinoma syndrome (NBCCS, MIM 109400), also known as Gorlin syndrome, is a rare autosomal dominant disorder predisposing to multiple basal cell carcinomas (BCCs)1. It is characterised by BCCs, odontogenic keratocysts, palmoplantar pits, falx cerebri calcification, and other developmental anomalies2. The diagnostic criteria for NBCCS proposed by Kimonis et al. in 1997 are still widely used3. When either two major, or one major and two minor criteria are fulfiled, the diagnosis of NBCCS can be confirmed.
NBCCS is mainly caused by variants in PTCH1—the human homolog of the Drosophila patched gene4. It is a tumour suppressor gene of the Sonic hedgehog (SHH) signalling pathway which encodes the transmembrane glycoprotein, patched-1. Loss-of-function variants of PTCH1 cause abnormal constitutive upregulation of the pathway and development of BCC. Additionally, PTCH2 and SUFU variants are known to cause NBCCS in rare cases5. The detection rate of PTCH1 variants in clinically diagnosed NBCCS was reported to range from 40 to 85%, while SUFU variants were found in 5.3% of all NBCCS patients6. However, the genomic characteristics of NBCCS in Asian descents remain largely unknown.
Although the prevalence of NBCCS is approximately 1 in 56,000–256,000, the incidence of the syndrome in Asia is uncertain7,8. In previous studies, the prevalence of NBCCS was reported to be 1 in 235,800 in Japan and 1 in 13,939,393 in Korea9,10. A systematic review of NBCCS literature revealed that significant differences exist between ethnic groups. In Northern Europe, patients showed significantly higher frequencies of BCCs, falx cerebri calcification, palmar and plantar pits, and family history. East Asians displayed significantly higher frequencies of keratocystic odontogenic tumours, cleft lips and palates, and hypertelorism11. A previous study describing a pooled analysis of Korean patients with NBCCS reported a high frequency of odontogenic keratocysts (90.9%), but a low BCC detection rate (15.2%)10.
In this study, we aimed to analyse clinical phenotypes and characterise the genetic profiles of Korean patients with NBCCS by whole-exome sequencing (WES) and multiplex ligation-dependent probe amplification (MLPA).
Results
Clinical characteristics of patients with NBCCS
A total of 15 patients were included. The age distribution of patients with NBCCS was 9–66 years (median 34 years) and the age of onset was 8–51 years (median 19 years). The male to female ratio was 1:2.75, although NBCCS is known to present equally in both males and females12.
Unlike previous studies which showed that BCCs accounted for 15.2–37.8% of NBCCS cases in Asian patients, our data demonstrated that most patients (93.3%) had at least one BCC10,13,14. Specifically, more than 2 BCCs were found in 86.7% of patients, while BCC onset before the age of 20 was observed in 46.7% of patients. The number of BCCs increased with age (ρ = 0.595, P = 0.019). In our cohort, BCCs were generally managed successfully with surgical excision. For small BCCs, punch biopsies were used to minimise normal skin-loss. CO2 laser ablation was used for multiple superficial BCCs. BCC went undetected in 1 patient only (case 14). She was diagnosed with NBCCS with multiple odontogenic cysts and palmoplantar pits at 12 years old. Follow-up examinations did not reveal any evidence of BCC until the age of 19 years.
Palmar or plantar fits were found in majority of the cases (93.3%), followed by macrocephaly (66.7%), odontogenic keratocysts (60.0%), lamellar calcification of the falx cerebri (60.0%), congenital malformations (46.7%), first degree relative with NBCCS (40.0%), bifid, fused or markedly splayed ribs (33.3%), ovarian fibroma (6.7%) and radiologic abnormalities (6.7%; Table 1 and Fig. 1). Medulloblastoma was detected in 3 patients (20.0%). Case 3 had a meningioma, which was not included in the diagnostic criteria but often presents in NBCCS15.
Table 1.
Diagnostic criteria for NBCCS and clinical phenotype of 15 Korean patients.
| Diagnostic criteria for NBCCS | N (%) |
|---|---|
| Major criteria | |
| 1. More than two BCCs or one under the age of 20 years | 14 (93.3) |
| 2. Odontogenic keratocysts of the jaw | 9 (60.0) |
| 3. Three or more palmar or plantar pits | 14 (93.3) |
| 4. Lamellar calcification of the falx cerebri | 9 (60.0) |
| 5. Bifid, fused or markedly splayed ribs | 5 (33.3) |
| 6. First degree relative with NBCCS | 6 (40.0) |
| Minor criteria | |
| 1. Macrocephaly determined after adjustment for height | 10 (66.7) |
| 2. Congenital malformations | 7 (46.7) |
| 3. Other skeletal abnormalities | 3 (20.0) |
| 4. Radiological abnormalities | 1 (6.7) |
| 5. Ovarian fibroma | 1 (6.7) |
| 6. Medulloblastoma | 3 (20.0) |
| Total | 15 (100.0) |
NBCCS nevoid basal cell carcinoma syndrome, BCC basal cell carcinoma.
Figure 1.
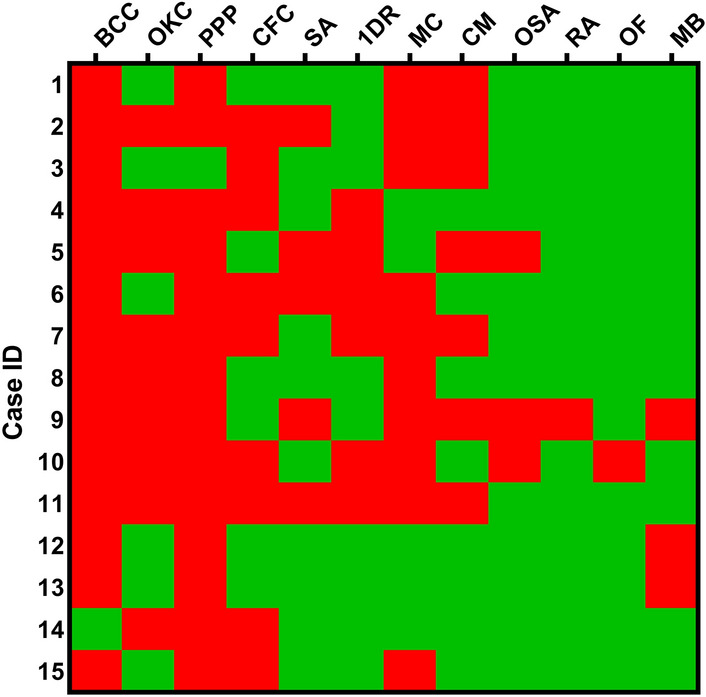
Heatmap of 15 NBCCS cases versus clinical features according to the criteria set by Kimonis et al.3. Clinical features are indicated in red (present) and green (absent). NBCCS nevoid basal cell carcinoma syndrome, BCC more than two basal cell carcinomas or one under the age of 20 years, OKC odontogenic keratocysts, PPP three or more palmar or plantar pits, CFC lamellar calcification of the falx cerebri, SA bifid, fused, or markedly splayed ribs, 1DR first-degree relative with nevoid basal cell carcinoma syndrome, MC macrocephaly, CM congenital malformations, OSA other skeletal abnormalities, RA radiological abnormalities, OF ovarian fibroma, MB medulloblastoma.
All patients fulfiled the criteria proposed by Kimonis et al.3 during clinical examination, except for one child (case 1). This patient fulfiled one major and one minor criterion (palmoplantar pits and macrocephaly) at the first visit (age 8 years), which was insufficient to meet the diagnostic criteria. Due to clinical suspicion, genetic analysis was performed. The results revealed whole gene deletion of PTCH1. Therefore, we performed detailed physical and dermatologic examinations using dermoscopy. The patient displayed multiple, 1–2 mm-sized brown papules on the abdomen. Dermoscopic examination revealed patterns associated with BCC. A subsequent punch biopsy confirmed the presence of BCCs. In addition, hypertelorism was detected by measuring the interpupillary distance.
Genetic profiles of patients with NBCCS
Of the 15 NBCCS patients, we found 4 nonsense, 4 splice site, 3 missense variant, 2 frameshift, 1 partial deletion, and 1 gross deletion. When we categorized them according to the American College of Medical Genetics and the Association for Molecular Pathology (ACMG/AMP) guidelines16, 9 PTCH1 pathogenic variants (including likely pathogenic variants) were found in 11 patients (73.3%). Variants of uncertain significance (VUS) and likely benign were also detected in 2 (13.3%) and 2 (13.3%) patients, respectively (Table 2). All variants were located along PTCH1 and scattered without mutational hotspot (Fig. 2). However, none of the patients had PTCH2 or SUFU pathogenic variants.
Table 2.
PTCH1 variants of 15 NBCCS patients.
| Case | Sex/age | DNA variants | Protein alteration | Location | Variant type | Classification | ACMG criteria | References |
|---|---|---|---|---|---|---|---|---|
| 1 | F/9 | del 9q22.31–q22.33 | Whole gene | Gross deletion | Pathogenic | Matsudate et al.17 | ||
| 2 | M/37 | Exon 13–15 deletion | Exon 13–15 | Partial deletion | Pathogenic | Novel | ||
| 3 | M/56 | c.290delA | p.Asn97Thrfs*20 | Exon 2 | Frameshift | Pathogenic | PVS1, PM2, PP5 | Wilson et al.18 |
| 4 | F/62 | c.403C > T | p.Arg135* | Exon 3 | Nonsense | Pathogenic | PVS1, PM2, PP5 | Wicking et al.19 |
| 5 | F/22 | c.403C > T | p.Arg135* | Exon 3 | Nonsense | Pathogenic | PVS1, PM2, PP5 | Wicking et al.19 |
| 6 | F/66 | c.1347 + 1G > A | p.? | Intron 9 | Splice site | Pathogenic | PVS1, PM2, PP5 | Reinders et al.20 |
| 7 | F/38 | c.1347 + 1G > A | p.? | Intron 9 | Splice site | Pathogenic | PVS1, PM2, PP5 | Reinders et al.20 |
| 8 | F/24 | c.1847G > A | p.Ser616Asn | Exon 13 | Missense | VUS | PM2, PP3 | Novel |
| 9 | M/13 | c.2251-3C > G | p.? | Intron 14 | Splice site | VUS | PM2, PP3, BS3 | Sun JS et al.21 |
| 10 | F/35 | c.2415dup | p.Val806Serfs*23 | Exon 15 | Frameshift | Likely pathogenic | PVS1, PM2 | Novel |
| 11 | F/20 | c.2422C > T | p.Gln808* | Exon 15 | Nonsense | Likely pathogenic | PVS1, PM2 | Waszak et al.22 |
| 12 | F/34 | c.2560 + 7C > T | p.? | Intron 15 | Splice region | Likely benign | BS1, BP6 | rs75576651 |
| 13 | F/19 | c.2678G > A | p.Arg893His | Exon 16 | Missense | Likely benign | BS1, BP6 | Tate et al.23 |
| 14 | F/19 | c.2802T > A | p.Tyr934* | Exon 17 | Nonsense | Likely pathogenic | PVS1, PM2 | Novel |
| 15 | M/60 | c.3467T > G | p.Leu1156Arg | Exon 21 | Missense | Likely pathogenic | PM2, PM6, PP3, PP5 | Gianferante et al.24 |
VUS variant of uncertain significance, PVS pathogenic very strong, PM pathogenic moderate, PP pathogenic supporting, BS benign strong, BP benign supporting.
Figure 2.

PTCH1 variant distribution of 15 Korean patients with NBCCS along with the patched-1 protein. DEL deletion, red nonsense variant, yellow frameshift variant, green large deletion, dark green splice site variant, blue missense variant, purple splice region variant.
Of the 15 patients, 6 (40.0%) had a family history of NBCCS and their variants were confirmed as familial variants with pedigree analysis. Cases 4, 5 and 6, 7 had a mother–daughter relationship and the same pathogenic variants. There were 3 nonsense, 2 frameshift, 1 splice site and 1 missense variant among the pathogenic sequence variants, which is in line with a recent review article demonstrating the most common variant form is frameshift, followed by nonsense variant25. Among these, two pathogenic variants (c.2415dup and c.2802T > A) were considered novel, and one variant (c.2422C > T) was not reported in NBCCS, but in medulloblastoma22. Although copy number alterations have been reported as rare, 2 patients (13.3%) in our cohort showed gross or partial deletion of PTCH124.
Two variants (c.1847G > A and c.2251-3C > T) were classified as VUS (Table 2). Both were not observed in the normal population (PM2) and in silico tools predicted them to be pathogenic (PP3). However, Sun et al. previously showed that c.2251-3C > T had no splicing effect, using RT-PCR (BS3)21. As no further information on the other variant (c.1847G > A) was available, we classified it as VUS.
Discussion
In this study, we described the clinical phenotypes and genetic profiles of 15 Korean patients with NBCCS. While the genetic background of NBCCS was first revealed in 1992, the genetic spectrum in Asian patients remains unknown26. Because PTCH1 has 24 exons, WES can be used for variant analysis, making it possible to analyse additional genes, like PTCH2 and SUFU. Therefore, we used WES for the genetic analysis of NBCCS. Additionally, we utilised MLPA for the detection of PTCH1 copy number variants.
Previous studies have shown that the frequency of BCC ranges from 15.2 to 51.4% in Asians9,10,13. However, we detected BCC in all except one NBCCS case (93.3%). The difference between our data and those of previous studies is that this study was conducted in clinics by experienced dermatosurgeons with expertise in skin cancers. Previous studies were conducted in dental clinics10,27. Selection and observation bias may therefore exist in previous literature.
We found 9 patients had PTCH1 pathogenic single nucleotide variants and 2 patients had large deletions, so exonal deletions of PTCH1 accounted for 13.3% of our cohort. According to previous Japanese data, 3 out of 20 patients (13.6%) were reported to have PTCH1 gross deletions with chromosomal microarray analysis17. Also, large genomic deletions and duplications were found in 8% of the 110 new variants in European cohorts20. Therefore, copy number analysis of PTCH1 as well as sequence analysis are necessary for the genetic consultation of patients with NBCCS.
Since NBCCS was first described by Gorlin and Goltz in 1960, several groups have recommended guidelines for the diagnosis of NBCCS2. Among these, the criteria proposed by Kimonis et al.3 are most widely used. Based on these criteria, we successfully diagnosed patients with NBCCS, except for one young patient (case 1) who initially did not fulfil the criteria. Based on clinical suspicion of the syndrome, a molecular test was performed and PTCH1 deletion was detected. According to the consensus statement from the first international colloquium on NBCCS, genetic confirmation with one major criterion can be used to make NBCCS diagnosis28. Because some features tend to occur in older NBCCS patients, this criterion would be particularly helpful for young patients, as in our case29. Since early detection and timely management of BCCs can result in the prevention of deformities with minimal scarring, genetic testing might be essential in younger patients.
Genotype–phenotype correlations in NBCCS have not been evident in previous studies19,30,31. In a previous study, the risk for medulloblastoma was higher in SUFU-related NBCCS cases than in PTCH1-related NBCCS patients6. Specifically, medulloblastoma was found in 3 out of 9 individuals (33.3%) with SUFU variants, but in only 2 out of 115 patients (1.7%) with PTCH1 variants. In our study, 20% of patients had medulloblastoma, but no PTCH1 pathogenic variants were detected. Our results, therefore, supported that PTCH1-related NBCCS patients have a relatively low risk for medulloblastoma.
The management of NBCCS should be tailored according to each patient’s condition. Various specialists should participate in treating the patients, including dermatologists, oral or dental surgeons, paediatricians, plastic surgeons and medical geneticists. As BCC has a prevalence of 47–96%, early detection and treatment is essential to minimise cosmetic disfigurement32. Although surgeries like wide excision or Mohs micrographic surgery are used for high-risk BCCs or destructing/disfiguring lesions, they are often not suitable for patients with NBCCS with multiple and extensive lesions as they can be time-consuming and deforming1. In patients in our study, BCCs were generally well managed with surgery, except for one case (case 6). This patient displayed multifocal recurrence where she received a full-thickness skin graft after BCC removal. Hence, we suggest that skin grafting be avoided as it makes it difficult to detect recurrence in patients with NBCCS.
A limitation of our study is the small sample size because of the low prevalence of NBCCS in Korea. Only patients of Korean descent were included. Patients presenting with similar and different features from the other population need to be investigated and a larger prospective multinational cohort study should be conducted. Besides, the correlation of molecular profiles between cancer tissues and germline variants requires further investigation. Lastly, the overall detection rate of pathologic variants was 73.3%. In our study, we did not analyse copy number variation of other genes except PTCH1. In addition, there is a possibility of involvement of unknown candidate genes that account for Gorlin syndrome. Further studies about the SHH pathway-related genes and copy number analysis might be helpful for understanding the pathogenesis of those patients33.
In summary, we explored the clinical and genetic profiles of Korean patients with NBCCS and demonstrated the diagnostic utility of genetic testing by WES and MLPA. Additionally, we detected a high rate of BCC in Korean patients with NBCCS, as opposed to previous knowledge. This suggests the necessity of regular screening for BCC in Korean patients with NBCCS. Finally, our results underline the need for genetic diagnosis, particularly in younger patients. We, therefore, suggest the addition of genetic criterion to the existing diagnostic criteria for NBCCS.
Methods
Study population
Fifteen patients who fulfiled the diagnostic criteria for NBCCS set by Kimonis et al.3 were recruited for this study, which was conducted from 2017 to 2019 at Seoul National University Hospital. Among them, 4 patients from 2 families (cases 4, 5, and 6, 7) were included. Clinical information, including radiological examination and family history was collected from the hospital medical records. Written informed consent was obtained from all patients. This study was performed in accordance with the Declaration of Helsinki and was approved by the Institutional Review Board of Seoul National University Hospital (No. 1910-170-1074). This study was supported by the National Supporting Program for Genetic Diagnosis of Rare Diseases of the Korea Centers for Disease Control & Prevention.
Whole-exome sequencing
Peripheral blood from patients was collected and stored in EDTA bottles and genomic DNA was extracted from each sample using the Gentra Puregene Blood kit (Qiagen, Hilden, Germany), according to the manufacturer’s protocol. The DNA concentration and purity were quantified using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). DNA was sonicated using Covaris (Covaris, Inc., Woburn, MA, USA). Target enrichment was performed on the extracted DNA with SureSelect Human All Exon (version 5; Agilent, Santa Clara, CA, USA), for exome capture. Paired-end sequencing of all captured libraries was performed with the Illumina HiSeq 2000 or 2500 platform (Illumina, San Diego, CA, USA), as previously described34.
Variant analysis
Bioinformatics analyses were performed by aligning to the human reference sequence, GRCh37/hg19, using a lab-developed pipeline based on NextGENe software version 2.4.0.1 (SoftGenetics, State College, PA, USA; https://softgenetics.com). The reference sequences of PTCH1 (NM_000264.4), PTCH2 (NM_003738.4), and SUFU (NM_016169.3) were used. The cut-off of variant allele frequency was 20% and the minimum read count was 10×. The Genome Aggregation Database and Korean Reference Genome Database were used to filter out benign variants. The Human Gene Mutation Database and ClinVar database were screened for previously reported variants. For in silico prediction, SIFT, Mutation Taster and PolyPhen2 were used as described previously35. Variants were classified as benign, likely benign, VUS, likely pathogenic and pathogenic, according to the ACMG/AMP guidelines16. The distribution of variants was analysed using ProteinPaint36.
Deletion/duplication analysis
Gene dosage analysis of PTCH1 was performed using samples that showed no pathogenic variants by WES analysis. MLPA was performed using SALSA MLPA Probemix P067-B3 (MRC-Holland, Amsterdam, The Netherlands), according to the manufacturer’s guidelines. In brief, 100 ng of genomic DNA was denatured for 5 min at 98 °C and hybridised with probes for 16 h at 60 °C. Ligation was done by ligase for 15 min at 54 °C and polymerase chain reaction of the ligated probes was done for 35 cycles using the Veriti 96-Well Thermal Cycler (Thermo Fisher Scientific). Each reaction was quantified using capillary electrophoresis with an ABI PRISM 3130xl Genetic Analyzer (Thermo Fisher Scientific). The results were analysed with the Coffalyser.Net version 140721.1958 (MRC-Holland; https://www.mrcholland.com/technology/software/coffalyser-net). We defined the normal range of the gene as 0.7–1.3, and deletion was identified when normalised peak ratio value was < 0.7 and duplication when > 1.3.
Statistical analysis
Spearman’s correlation coefficient was used for correlation analysis, and data were analysed with SPSS version 25.0 (IBM, Armonk, NY, USA). All statistical analyses were two-tailed and P values < 0.05 were considered statistically significant.
Acknowledgements
This work was supported by the National Supporting Program for Genetic Diagnosis of Rare Diseases of the Korea Centers for Disease Control & Prevention. We also thank the patients and their families for their participation in the study.
Author contributions
B.K. performed data interpretation, statistical analysis and wrote the main manuscript text; M.J.K. collected the results and analysed the genetic profile; K.H., S.J.J., J.M.K. and J.-H.M. recruited the patients and did the physical exams of patients; S.S.P., M.-W.S. and J.-H.M. conceived the study design and reviewed the cases. M.-W.S. and J.-H.M. wrote the main manuscript text and contributed equally to this work as corresponding authors. All authors revised, reviewed and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Moon-Woo Seong, Email: mwseong@snu.ac.kr.
Je-Ho Mun, Email: jehomun@snu.ac.kr.
References
- 1.John AM, Schwartz RA. Basal cell naevus syndrome: An update on genetics and treatment. Br. J. Dermatol. 2016;174:68–76. doi: 10.1111/bjd.14206. [DOI] [PubMed] [Google Scholar]
- 2.Gorlin RJ, Goltz RW. Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. N. Engl. J. Med. 1960;262:908–912. doi: 10.1056/NEJM196005052621803. [DOI] [PubMed] [Google Scholar]
- 3.Kimonis VE, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am. J. Med. Genet. 1997;69:299–308. doi: 10.1002/(SICI)1096-8628(19970331)69:3<299::AID-AJMG16>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 4.Johnson RL, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 5.Peris K, et al. Diagnosis and treatment of basal cell carcinoma: European consensus-based interdisciplinary guidelines. Eur. J. Cancer. 2019;118:10–34. doi: 10.1016/j.ejca.2019.06.003. [DOI] [PubMed] [Google Scholar]
- 6.Smith MJ, et al. Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J. Clin. Oncol. 2014;32:4155–4161. doi: 10.1200/JCO.2014.58.2569. [DOI] [PubMed] [Google Scholar]
- 7.Evans DG, et al. Complications of the naevoid basal cell carcinoma syndrome: Results of a population based study. J. Med. Genet. 1993;30:460–464. doi: 10.1136/jmg.30.6.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lo Muzio L, et al. Nevoid basal cell carcinoma syndrome Clinical findings in 37 Italian affected individuals. Clin. Genet. 1999;55:34–40. doi: 10.1034/j.1399-0004.1999.550106.x. [DOI] [PubMed] [Google Scholar]
- 9.Endo M, et al. Nationwide survey of nevoid basal cell carcinoma syndrome in Japan revealing the low frequency of basal cell carcinoma. Am. J. Med. Genet. A. 2012;158A:351–357. doi: 10.1002/ajmg.a.34421. [DOI] [PubMed] [Google Scholar]
- 10.Ahn SG, et al. Nevoid basal cell carcinoma syndrome: A retrospective analysis of 33 affected Korean individuals. Int. J. Oral. Maxillofac. Surg. 2004;33:458–462. doi: 10.1016/j.ijom.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 11.MacDonald DS. A systematic review of the literature of nevoid basal cell carcinoma syndrome affecting East Asians and North Europeans. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. 2015;120:396–407. doi: 10.1016/j.oooo.2015.05.024. [DOI] [PubMed] [Google Scholar]
- 12.Pino LC, et al. Basal cell nevus syndrome: Clinical and molecular review and case report. Int. J. Dermatol. 2016;55:367–375. doi: 10.1111/ijd.12993. [DOI] [PubMed] [Google Scholar]
- 13.Sim YC, et al. Novel PTCH1 gene mutation in nevoid basal cell carcinoma syndrome. J. Craniofac. Surg. 2018;29:e252–e255. doi: 10.1097/SCS.0000000000004274. [DOI] [PubMed] [Google Scholar]
- 14.Guo YY, et al. PTCH1 gene mutations in keratocystic odontogenic tumors: A study of 43 Chinese patients and a systematic review. PLoS ONE. 2013;8:e77305. doi: 10.1371/journal.pone.0077305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lo Muzio L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome) Orphanet. J. Rare Dis. 2008;3:32. doi: 10.1186/1750-1172-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–423. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsudate Y, et al. Targeted exome sequencing and chromosomal microarray for the molecular diagnosis of nevoid basal cell carcinoma syndrome. J. Dermatol. Sci. 2017;86:206–211. doi: 10.1016/j.jdermsci.2017.02.282. [DOI] [PubMed] [Google Scholar]
- 18.Wilson LC, et al. Patched mutations and hairy skin patches: A new sign in Gorlin syndrome. Am. J. Med. Genet. A. 2006;140A:2625–2630. doi: 10.1002/ajmg.a.31374. [DOI] [PubMed] [Google Scholar]
- 19.Wicking C, et al. Most germ-line mutations in the nevoid basal cell carcinoma syndrome lead to a premature termination of the PATCHED protein, and no genotype-phenotype correlations are evident. Am. J. Hum. Genet. 1997;60:21–26. [PMC free article] [PubMed] [Google Scholar]
- 20.Reinders MG, et al. New mutations and an updated database for the patched-1 (PTCH1) gene. Mol. Genet. Genom. Med. 2018;6:409–415. doi: 10.1002/mgg3.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun LS, Li XF, Li TJ. PTCH1 and SMO gene alterations in keratocystic odontogenic tumors. J. Dent. Res. 2008;87:575–579. doi: 10.1177/154405910808700616. [DOI] [PubMed] [Google Scholar]
- 22.Waszak SM, et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: A retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018;19:785–798. doi: 10.1016/S1470-2045(18)30242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tate G, Kishimoto K, Mitsuya T. Biallelic disruption of the PTCH1 gene in multiple basal cell carcinomas in Japanese patients with nevoid basal cell carcinoma syndrome. Acta Med. Okayama. 2014;68(3):163–170. doi: 10.18926/AMO/52657. [DOI] [PubMed] [Google Scholar]
- 24.Gianferante DM, et al. Whole-exome sequencing of nevoid basal cell carcinoma syndrome families and review of Human Gene Mutation Database PTCH1 mutation data. Mol. Genet. Genom. Med. 2018;6:1168–1180. doi: 10.1002/mgg3.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shoko O, Yuriko N, Toshifumi A. Gorlin syndrome: Recent advances in genetic testing and molecular and cellular research. Int. J. Mol. Sci. 2020;21(20):E7559. doi: 10.3390/ijms21207559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farndon PA, et al. Location of gene for Gorlin syndrome. Lancet. 1992;339:581–582. doi: 10.1016/0140-6736(92)90868-4. [DOI] [PubMed] [Google Scholar]
- 27.MacDonald DS, Li TK, Goto TK. A consecutive case series of nevoid basal cell carcinoma syndrome affecting the Hong Kong Chinese. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. 2015;120:408–415. doi: 10.1016/j.oooo.2015.05.025. [DOI] [PubMed] [Google Scholar]
- 28.Bree AF, Shah MR, BNCS Colloquium Group Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS) Am. J. Med. Genet. A. 2011;155:2091–2097. doi: 10.1002/ajmg.a.34128. [DOI] [PubMed] [Google Scholar]
- 29.Klein RD, Dykas DJ, Bale AE. Clinical testing for the nevoid basal cell carcinoma syndrome in a DNA diagnostic laboratory. Genet. Med. 2005;7:611–619. doi: 10.1097/01.gim.0000182879.57182.b4. [DOI] [PubMed] [Google Scholar]
- 30.Bale AE. Variable expressivity of patched mutations in flies and humans. Am. J. Hum. Genet. 1997;60:10–12. [PMC free article] [PubMed] [Google Scholar]
- 31.Cosgun B, et al. Lack of genotype–phenotype correlation in basal cell nevus syndrome: A Dutch multicenter retrospective cohort study. J. Am. Acad. Dermatol. 2019;S0190–9622(19):32463–32466. doi: 10.1016/j.jaad.2019.07.072. [DOI] [PubMed] [Google Scholar]
- 32.Solis DC, et al. Risk factors for basal cell carcinoma among patients with basal cell nevus syndrome: Development of a basal cell nevus syndrome patient registry. JAMA Dermatol. 2017;153:189–192. doi: 10.1001/jamadermatol.2016.4347. [DOI] [PubMed] [Google Scholar]
- 33.Kei-ichi M, et al. Simultaneous detection of both single nucleotide variations and copy number alterations by next-generation sequencing in Gorlin syndrome. PLoS ONE. 2015;10(11):e0140480. doi: 10.1371/journal.pone.0140480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joo K, et al. Genotypic profile and phenotype correlations of ABCA4-associated retinopathy in Koreans. Mol. Vis. 2019;25:679–690. [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S, et al. Spectrum of MNX1 pathogenic variants and associated clinical features in Korean patients with Currarino syndrome. Ann. Lab. Med. 2018;38:242–248. doi: 10.3343/alm.2018.38.3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou X, et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat. Genet. 2016;48:4–6. doi: 10.1038/ng.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]