

Abstract
Background
Altered lipid metabolism in early life has been associated with subsequent weight gain and predicting this could aid in obesity prevention and risk management. Here, a lipidomic approach was used to identify circulating markers for future obesity risk in translational murine models and validate in a human infant cohort.
Methods
Lipidomics was performed on the plasma of APOE*3 Leiden, Ldlr-/-.Leiden, and the wild-type C57BL/6J mice to capture candidate biomarkers predicting subsequent obesity parameters after exposure to high-fat diet. The identified candidate biomarkers were mapped onto corresponding lipid metabolism pathways and were investigated in the Cambridge Baby Growth Study. Infants’ growth and adiposity were measured at 0-24 months. Capillary dried blood spots were sampled at 3 months for lipid profiling analysis.
Findings
From the mouse models, cholesteryl esters were correlated with subsequent weight gain and other obesity parameters after HFD period (Spearman's r≥0.5, FDR p values <0.05) among APOE*3 Leiden and Ldlr-/-.Leiden mice, but not among the wild-type C57BL/6J. Pathway analysis showed that those identified cholesteryl esters were educts or products of desaturases activities: stearoyl-CoA desaturase-1 (SCD1) and fatty acid desaturase (FADS) 1 and 2. In the human cohort, lipid ratios affected by SCD1 at 3 months was inversely associated with 3-12 months weight gain (B±SE=-0.31±0.14, p=0.027), but positively with 12-24 months weight and adiposity gains (0.17±0.07, p=0.02 and 0.17±0.07, 0.53±0.26, p=0.04, respectively). Lipid ratios affected by SCD1 and FADS2 were inversely associated with adiposity gain but positively with height gain between 3-12 months.
Interpretation
From murine models to human setting, the ratios of circulating lipid species indicating key desaturase activities in lipid metabolism were associated with subsequent body size increase, providing a potential tool to predict early life weight gain.
Keywords: Lipid ratio, SCD1, FADS, Infant growth, Adiposity, Desaturase
1. INTRODUCTION
Early life growth and nutrition significantly contribute to the risk of developing metabolic diseases later in life, especially obesity, type 2 diabetes (T2D) and cardiovascular diseases [1]. The Avon Longitudinal Study of Pregnancy and Childhood (ALSPAC) showed that infants with rapid weight gain between 0-24 months had higher weight and body fat percentage at the age of 5 years [2]. Further projected into later life, excessive weight-for-length gain during the first 3 months postnatally has been associated with increased metabolic risks in early adult life (21 years of age) [3].
While weight gain can be attributed to the imbalance between energy consumption and energy expenditure [4], there are still many other unexplained metabolic pathways affecting basal energy expenditure and/or satiety, leading to variations in weight gain. Better understanding of individual susceptibility to excessive weight gain, especially in early life, can provide a window of opportunity for better weight management and reducing the risk of metabolic disease [5].
In human, variation in lipid metabolism during infancy has been associated with subsequent weight gain [6]. Specific lipid species as well as key enzymes influencing their metabolism are involved, although the underlying mechanisms are still unclear.
In the present study, we used an unbiased lipidomics approach to identify candidate lipid biomarkers associated with early weight gain and other obesity-related parameters. First, we performed diet-induced obesity experiments in mice to identify circulating lipids in early-life plasma that could potentially predict future obesity risk. We used mouse models with established translational characteristics of human lipid metabolism and dyslipidaemia, including ApoE*3Leiden (E3L) and Ldlr-/-.Leiden mice [7,8]. The findings from the mouse models would then be verified in an infant population from the Cambridge Baby Growth Study (CBGS), a large birth cohort in the UK setting [9], by associating candidate lipid biomarkers to weight gain in infants up to 24 months.
2. METHODS
2.1. Mouse studies
2.1.1. Experimental design
Three animal experiments were included in this study. First, inbred male mice of the heterozygous E3L strain were kept under specific-pathogen-free conditions (TNO, Leiden, The Netherlands; n=40). E3L mouse is the wildtype C57BL/6J mouse with a knocked-in mutated human apolipoprotein E3 gene (APOE3). This mutation caused reduced protein binding capacity to the low-density lipoprotein receptor (LDLr) leading to slower cholesterol clearance [10]. Animals were housed in Makrolon cages at a relative humidity of 50-60% and temperature at around 21°C under a 12:12 light/dark cycle with the light on at 7 am. Food and acidified tap water were available ad libitum. All mice were fed with standard lab chow (Ssniff-R, Uden, The Netherlands) until 12 weeks of age. Ten mice were sacrificed at 12 weeks of age to serve as a baseline reference group. Following that, the remaining 30 mice received 24% w/w lard and sucrose-containing energy-dense high-fat diet (HFD; #D12451, Research Diets, New Brunswick, NJ) between 12-20 weeks of age to induce obesity as illustrated in Figure 1. Diet and macronutrient composition are described in detail elsewhere [11]. Bodyweight and caloric intake were monitored regularly. Tail blood was sampled after 4 hours of fasting at 12 weeks (before HFD intervention) and 16 and 20 weeks for lipidomic analyses. At 20 weeks, the remaining mice were sacrificed and the subcutaneous, epidydimal, and mesenteric adipose tissue depots were isolated.
Fig. 1.

Schematic overview of the ApoE3L mice experiment
In the second study, the findings from the E3L experiment were subsequently validated in 12-week-old Ldlr-/-.Leiden and C57BL/6J mice. The Ldlr-/-.Leiden mouse is the wildtype C57BL/6J with a knocked-out LDLr gene to mimic humans with LDLR-defective mutations. These two types of mice are 94% genetically similar and the wildtype C57BL/6J is the most common strain used to study obesity. To induce obesity, the same HFD was fed to the Ldlr-/-.Leiden mice and wildtype C57BL/6J8 for 21 and 24 weeks, respectively.
Finally, to evaluate whether the identified candidate biomarkers were apparent at earlier ages, another lipidomic analysis of plasma of E3L mice collected at the age of 4 weeks was performed. In this study, animals were supplemented with arachidonic acid/docosahexaenoic acid (ARA/DHA) from 4-12 weeks of age. Lipid concentrations reflecting desaturase activities were analysed against 4-12 weeks weight gain between mice treated with ARA/DHA or placebo (n=15 each) [12].
All animal care and experimental procedures were approved by the Ethical Committee on Animal Care and Experimentation (Zeist, the Netherlands; approval reference numbers DEC-3117, -3076, -3260 and -3277) and were performed in compliance with ARRIVE guidelines and the European Community specifications regarding the use of laboratory animals.
2.1.2. Obesity endpoints and adipose tissue analyses
During the HFD feeding period, mice were weighed regularly to record delta weight gain as the primary obesity endpoint. Additional obesity endpoints included final absolute body weight after completing HFD, overall adiposity (i.e. the total mass of all adipose tissue depots), and absolute mass of specific adipose tissue depots. Histological analysis of subcutaneous, epidydimal, and mesenteric adipose tissue depots allowed determination of the average adipocyte cell size, a determinant of white adipose tissue (WAT) quality [13].
2.1.3. Lipidomics on mouse plasma
Lipids were analysed as described elsewhere [14] with electrospray and atmospheric pressure chemical ionization (APCI) ultra-high performance liquid chromatography/high-resolution mass spectrometry (ESI-UHPLC-HRMS; Thermo Accela and LTQ OrbitrapTM). The method is explained in more detail in the supplementary materials.
2.2. Human study
The lipid ratios that were associated with the desaturases activities in animal study were also examined in lipidomics data from an infant cohort, in order to verify the associations between lipid ratios associated with desaturase enzyme activity and weight gain in humans.
2.2.1. Study population
The experiment built upon the methodology employed in our previous studies [6,15,16]. The Cambridge Baby Growth Cohort Study (CBGS) is a UK prospective observational cohort, with longitudinal infancy anthropometric measures and collection of detailed demographics and infant feeding [9]. Mother-infant pairs were recruited from a single maternity unit in Cambridge, UK. Inclusion criteria included singleton, term, and healthy infants with normal birth weight and no significant comorbidities during pregnancy. Infants with a genetic or syndromal disease were excluded from the analyses. Studies were approved by National Research Ethics Service Cambridgeshire 2 Research Ethics Committee (REC) with REC reference 00/325, and all mothers gave informed written consent. This study involved a subgroup of CBGS (total n=201) with available dried capillary blood spots samples. Subjects were recruited between 02/10/2003 and 29/08/2009.
2.2.2. Growth and adiposity measurement
Birth weight was obtained from hospital records. Other birth and subsequent (3, 12, and 24 months) anthropometry measures were performed by three trained paediatric research nurses. Weight was measured to the nearest 1 g using a Seca 757 electronic baby scale and length was measured to the nearest 0.1 cm using an Infantometer (SECA 416). Skinfold thickness was measured in triplicate at four sites (triceps, subscapular, flank, and quadriceps) on the left side of the body using a Holtain Tanner/Whitehouse Skinfold Caliper (Holtain Ltd).
2.2.3. Sample collection
Blood samples from capillary heel-prick sampling were collected at 3 months of age. Blood was dropped onto filtered paper cards, air-dried at ambient room temperature overnight, and stored in Ziploc bags at -80°C. A single spot with diameter 3.2 mm was punched from the card for analysis.
2.2.4. Lipidomics on human dried blood spots
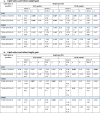
Blood spots samples were extracted by adapting a protocol described previously [6]. Bloods/analytes were placed in the wells of glass coated 2.4 ml deep well plate and added with 100 μl of MilliQ H2O, 250 μl of methanol, and 500 μl of methyl tertiary butyl ether to partition the lipids. The plates were then centrifuged for 10 minutes at 6,000 rpm after being shaken for 10 minutes at 600 rpm. The organic layer on top of the aqueous phase was transferred, dried down, reconstituted, and used for lipid analysis by direct infusion high-resolution untargeted mass spectrometry (HRMS), as previously described [6,15]. Additional lipid identification was done by LC-MS/MS. Selected masses were isolated, and all spectra were recorded [6]. Only the signals of (CE(16:1), CE(16:0), PC(32:1), PC(32:0) PC(38:4), PC(38:3) TG(54:4), TG(54:3), PC(36:3), PC(36:2), TG(50:3) and TG(50:2) were used to calculate the desaturase activities yielding 6 values of lipid ratios (Table 3).
Table 3.
Regression models associating lipid ratios at 3 months and subsequent growth
 |
2.2.5. Statistical analyses
In the mouse models the natural variation in response to HFD feeding was used to establish correlations between plasma lipids and specified obesity endpoints as was reported in Wopereis et al [14]. Correlations and corresponding p-values were calculated in R version 3.6.3 using Spearman's tests. The resulting p-values were corrected for multiple testing by calculating the false discovery rate (FDR) using the Benjamini Hochberg method. Graphs were produced using GraphPad Prism version 8.4.2.
Infancy age- and sex-appropriate standard deviation scores (SDS) were calculated for weight and length measurements, (with adjustment for gestational age at birth and 3 months), by comparison to the UK 1990 growth reference using LMSgrowth software [17]. Internally-derived SDS were calculated for each individual skinfolds, adjusted for infant sex, GA, and exact age at visit. Growth gains were derived from delta weight, height, and mean skinfolds SDS between 3-12 and 12-24 months. Maternal BMI was derived from self-reported pre-pregnancy weight divided by the square of measured height (kg/m2).
Unless otherwise stated, all descriptive data are presented as means±standard deviations (SD) for continuous variables or as a percentage (%) for categorical variables. Multiple linear regression was used to investigate associations between lipid ratios separately at 3 months of age with weight, height, and skinfolds (reflecting adiposity) gains during both 3-12 and 12-24 months. For weight and height SDS, models were adjusted for maternal parity, maternal pre-pregnancy BMI, and infant feeding history. For mean skinfold, models were adjusted for infant sex, gestational age, postnatal age at measurement, and feeding history, as well as maternal pre-pregnancy BMI and parity. For linear regression models, assumptions of normality, linearity, homoscedasticity, and absence of multicollinearity had been evaluated and none were violated. Since there were only 6 variables/lipid ratios in the regression models, statistical significance was achieved if p values<0.05. Statistical analyses were carried out using SPSS version 25.0 (IBM) and R version 1.0.136.
2.3. Role of the funding source
The funders of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
3. RESULTS
3.1. Identification of candidate obesity biomarkers in mouse cohorts
Obesity was induced in standard chow-fed E3L mice at age 12 weeks by exposing them to HFD for 8 weeks (n=30). The average body weight at baseline was 26.0±1.7 g and mice weight gain ranged between 3.1 and 11.8 g (Supplementary Figure 1A-C). A similar inter-individual variation was observed for other obesity endpoints, including body fat percentage; epidydimal, mesenteric and subcutaneous WAT mass; and adipocyte cell size of major WAT depots (Supplementary Figure 1D-J).
Lipidomic analysis was performed on plasma collected prior to HFD feeding, involving more than 200 lipids of all major lipid classes. This allowed for identification of specific circulating lipids that correlated with weight-gain as our primary obesity endpoint and other obesity-related parameters after HFD. At 12 weeks, the circulating relative concentration of four lipids, the cholesteryl esters (CEs), CE(20:2), CE(20:3), CE(22:4), and CE(22:5), was significantly correlated with weight-gain during HFD feeding (FDR<0.05; Figure 2; complete analyte results in Supplementary Table 1). Moreover, when measured at 20 weeks, all four CEs showed significant and stronger correlations with delta weight gain during the HFD period (Figure 2E).
Fig. 2.

(a-d) Correlations between plasma concentrations of four CEs at 12 weeks with subsequent weight gain after 8 weeks of HFD feeding (1 mouse was excluded due to technical error, total n=29, r=Spearman‘s correlation coefficient; FDR-corrected p <0.05). (e) Correlations between mice body weight gain from 12-20 weeks during HFD feeding and each CE level over time (each cell denotes Spearman's correlation coefficient, increased red intensity indicates stronger correlation (FDR-corrected p <0.05).
An overview of all lipids that significantly correlated with one or more obesity endpoints is displayed in Table 1, showing the number of correlations and which obesity-related endpoints were predicted by each analyte. Most lipids that correlated with 3 or more obesity endpoints were CEs. Half of the remaining lipids that correlated with ≤2 obesity endpoints belonged to the phosphatidylcholine (PC) group. Of note, CEs are components of lipoproteins, while PCs are essential elements of biological membranes. If significant correlations were only observed between lipids and 1-2 obesity endpoints, the endpoint would usually reflect the quality of adipose tissue, e.g. mass and size of specific WAT depots.
Table 1.
Correlation between each lipid species and obesity-related endpoints (Spearman's r≥0.50, FDR-corrected p<0.05) in ApoE3L mice
| Lipid species | Number of correlations | Correlation coefficient (r) per obesity endpoint | |
|---|---|---|---|
| 1 | CE(20:2) | 8 | BW gain (0.68), omental fat mass (0.56), subcutaneous fat mass (0.69), perirenal fat mass (0.64), total fat mass (0.64), body fat percentage (0.63), subcutaneous adipocyte size (0.53) |
| 2 | CE(20:3) | 6 | BW gain (0.59), omental fat mass (0.58), subcutaneous fat mass (0.65), body fat percentage (0.61), omental adipocyte size (0.53), subcutaneous adipocyte size (0.66) |
| 3 | CE(22:5) | 6 | BW gain (0.57), omental fat mass (0.57), subcutaneous fat mass (0.63), body fat percentage (0.61), omental adipocyte size (0.57), subcutaneous adipocyte size (0.66) |
| 4 | CE(20:4) | 5 | Omental fat mass (0.57), subcutaneous fat mass (0.63), body fat percentage (0.6), omental adipocyte size (0.5), subcutaneous adipocyte size (0.66) |
| 5 | CE(22:4) | 4 | BW gain (0.66), subcutaneous fat mass (0.64), body fat percentage (0.59), subcutaneous adipocyte size (0.6) |
| 6 | CE(16:1) | 4 | Omental fat mass (0.65), subcutaneous fat mass (0.57), omental adipocyte size (0.52), subcutaneous adipocyte size (0.53) |
| 7 | SM(16:1) | 4 | Omental fat mass (0.57), subcutaneous fat mass (0.57), omental adipocyte size (0.55), subcutaneous adipocyte size (0.5) |
| 8 | CE(18:2) | 3 | Omental fat mass (0.63), subcutaneous fat mass (0.59), subcutaneous adipocyte size (0.51) |
| 9 | PC(32:2) | 3 | Omental fat mass (0.58), omental adipocyte size (0.61), subcutaneous adipocyte size (0.56) |
| 10 | PC(34:3) | 3 | Omental fat mass (0.58), omental adipocyte size (0.6), subcutaneous adipocyte size (0.54) |
| 11 | CE(22:6) | 3 | Omental fat mass (0.54), subcutaneous fat mass (0.56), subcutaneous adipocyte size (0.61) |
| 12 | PC(32:1) | 2 | Omental fat mass (0.58), omental adipocyte size (0.55) |
| 13 | PC(36:5) | 2 | Omental adipocyte size (0.62), subcutaneous adipocyte size (0.61) |
| 14 | C20:5n3 | 2 | Omental adipocyte size (0.56), subcutaneous adipocyte size (0.61) |
| 15 | C20:4n3 | 2 | Omental adipocyte size (0.55), subcutaneous adipocyte size (0.53) |
| 16 | C22:6n3 | 2 | Omental adipocyte size (0.56), subcutaneous adipocyte size (0.57) |
| 17 | CE(20:5) | 2 | Omental adipocyte size (0.56), subcutaneous adipocyte size (0.63) |
| 18 | PC(38:3) | 2 | Omental adipocyte size (0.55), subcutaneous adipocyte size (0.5) |
| 19 | PC(38:6) | 2 | Omental adipocyte size (0.54), subcutaneous adipocyte size (0.52) |
| 21 | C20:4n6 | 2 | Omental adipocyte size (0.53), subcutaneous adipocyte size (0.55) |
| 22 | PC(40:7) | 2 | Omental adipocyte size (0.53), subcutaneous adipocyte size (0.5) |
| 23 | PC(36:4) | 2 | Omental adipocyte size (0.52), subcutaneous adipocyte size (0.57) |
| 24 | PC(38:5) | 2 | Omental adipocyte size (0.52), subcutaneous adipocyte size (0.54) |
| 25 | CE(14:0) | 1 | Omental fat mass (0.6) |
| 26 | SM(23:1) | 1 | Omental fat mass (0.54) |
| 27 | C24:0 | 1 | Omental fat mass (0.53) |
| 28 | LPC sn2(18:0) | 1 | Subcutaneous adipocyte size (0.52) |
| 29 | PC(38:4) | 1 | Subcutaneous adipocyte size (0.5) |
| 30 | PC(38:3) | 1 | Subcutaneous adipocyte size (0.5) |
| 31 | C22:5n3 | 1 | Omental adipocyte size (0.55) |
| 32 | LPC sn1(16:1) | 1 | Omental adipocyte size (0.52) |
| 33 | PC(36:3) | 1 | Omental adipocyte size (0.53) |
| 34 | C20:3n9 | 1 | Omental adipocyte size (0.5) |
| 35 | C20:3n3 | 1 | Omental adipocyte size (0.5) |
| 36 | PC(36:5) | 1 | Omental adipocyte size (0.5) |
BW=body weight
The four identified CEs that correlated with weight gain are highlighted in grey (no 1, 2, 3, and 5).
CE=cholesteryl esters, PC=phosphatidylcholine, LPC=lysophosphatidylcholine, SM=sphingomyelins, TG=triglycerides
To verify if the identified 4 CEs could be predictive for obesity endpoints, lipidomics analyses were performed in two different independent HFD-induced obesity mice experiments. The first experiment was carried out with mice that displayed a human-like lipoprotein profile with elevated relative concentration of apolipoprotein B-containing lipoproteins (Ldlr-/-.Leiden mice). The second experiment was conducted on mice in which those lipoproteins are rapidly cleared from the circulation (C57BL/6J mice). In Ldlr-/-.Leiden mice, plasma concentrations of all 4 CEs at 9 weeks of age strongly correlated with mice body weight after completing 21 weeks of HFD (data not shown): CE(20:3) (Spearman's r=0.727), CE(20:4) (r=0.735), CE(22:4) (r=0.817), and CE(22:5) (r=0.787). On the contrary, in C57BL/6J mice, a model without a humanised lipoprotein profile, those CEs did not correlate with weight gain, except for CE(16:1). This particular CE also predicted WAT weight and adipocyte size in the E3L cohort (data not shown).
In an attempt to delineate putative mechanistic relationships between the identified lipid biomarkers, their fatty acids (FA) moieties were plotted in the lipid pathways relevant for FA elongation and desaturation detailing n-3, n-6 and n-9 FA pathways (Figure 3). Analysing this mechanistic framework provided evidence that the FA moieties of the identified CEs are educts or products of desaturase activity and their ratios are often used as biomarkers for these enzyme acitivities. This suggests that three key enzyme activities in early-life of mice, FADS1/Δ5-FA desaturase, FADS2/Δ6-FA desaturase, and SCD-1/Δ9-FA desaturase, are critical for later body weight gain and obesity endpoints. Among the E3L mice in the first animal study, C16:1/C16:0 ratio (a marker of SCD-1 activity) was positively correlated with body weight gain (r=0.59, FDR<0.05) and subcutaneous adipocyte cell size (r=0.54; FDR<0.05). Interestingly, among the lipids that correlated with obesity endpoints in mice (Table 1), there were several lipids typically used to estimate FADS1, FADS2, and SCD-1 activity in human, including in our infant cohort in the following section (Table 4), e.g. CE(16:1), PC(32:1), PC(38:3), PC(38:4), and PC(36:3).
Fig. 3.

Schematic diagram of identified lipid biomarkers and their relation to the activities of desaturase enzymes, showing FA of the n-3, n-6 and n-9 pathways that are converted by elongases and desaturases. The reactions catalysed by desaturases FADS1, FADS2 and SCD-1 are shown with each corresponding coloured line. The FA moieties of the biomarkers shown in Table 1 are highlighted in green. Only lipids with single distinct FA moiety are displayed whereas complex lipids (e.g. PC with multiple FA) are omitted. SCD= stearoyl Co-A desaturase, FADS= fatty acid desaturase.
Table 4.
Summary of results of the association analyses between infant growth/adiposity outcomes and desaturase activities
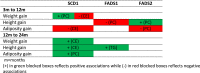 |
m=months
(+) in green blocked boxes reflects positive associations while (-) in red blocked boxes reflects negative associations
To test if the mapping of lipid ratios to corresponding desaturase activities within this mechanistic framework already applies early in life, lipidomics was conducted on the plasma of 4-week-old E3L mice. Lipid concentrations were used to calculate product/educt ratios of desaturase-mediated enzymatic reactions or desaturase indices/activities. As shown in Figure 4, the calculated SCD1 and FADS1 activities correlated with the subsequent weight gain from 4-12 weeks after being exposed to HFD. As opposed to the previous experiment (Figure 2 and Table 1), SCD-1 was inversely associated with subsequent weight gain. In addition, this experiment also demonstrated that ARA/DHA supplementation could modulate CE circulating levels and desaturase activities corresponding to obesity endpoints (Supplementary Figure 2, obesity endpoints have been reported elsewhere [12]).
Fig. 4.

Correlation between desaturases activities measured at 4 weeks and subsequent body weight gain between 4-12 weeks among HFD-fed E3L mice (n=10). The SCD-1, FADS1, and FADS2 activities were estimated using lipid concentrations from lipidomics as those enzymes mediate specific FA conversions as shown in Figure 3.
3.2. Validation of candidate obesity biomarkers in human infants
To translate the findings from murine experiments to the human context, lipidomics data from a subgroup of the CBGS cohort were analysed. In particular, associations between lipid ratios affected by desaturase enzyme activities identified from the animal models and growth gains in infancy were explored. The baseline characteristics of the cohort are shown in Table 2. All infants were born at term from healthy mothers, had normal birth weight, were mostly of Caucasian descent, with equal proportion in sex and birth order. The characteristics shown in Table 2 did not differ significantly with the whole group of normal birth weight infants in the CBGS (data not displayed).
Table 2.
CBGS baseline characteristics
| Characteristics | N | Mean±SD or N (%) |
|---|---|---|
| Maternal characteristics | ||
| Age at delivery, year | 201 | 33.42±4.5 |
| Pre-pregnancy BMI, kg/m2 | 168 | 23.5±4.37 |
| Height, cm | 176 | 166.37±6.99 |
| Ethnicity (N (%) of European descent) | 179 | 168(93.9%) |
| Parity (N (%) of primiparous) | 201 | 91(45.3%) |
| Smoking history (N (%) of smoking mothers) | 201 | 8(4%) |
| Birth and infantcharacteristics | ||
| Sex (N(%) of male infants) | 201 | 88(43.8%) |
| Gestational age at delivery, weeks | 201 | 39.85±1.44 |
| Delivery methods (N(%) of vaginal delivery) | 194 | 140(72.2%) |
| Birth weighta, kg | 201 | 3.42±0.55 |
| Birth weight-SDSa | 201 | -0.05±1.04 |
| Birth lengthb, cm | 195 | 51.07±2.3 |
| Birth length-SDSb | 195 | -0.02±0.97 |
| Birth head circumferenceb, cm | 196 | 34.86±1.48 |
| Birth head circumference-SDS | 196 | -0.24±1.05 |
| BMI at birthb, kg/m2 | 195 | 13.15±1.88 |
| BMI-SDSb | 195 | -0.15±1.65 |
| Mean skinfolds at birthb,c, cm | 196 | 5.88±1.42 |
| LGA, % | 201 | 7(3.5%) |
| Exclusively breastfed until 3 months, % | 199 | 87(43.7%) |
| 0-3 months catch-up weight gaind, % | 200 | 53(26.5%) |
| 3-12 months catch-up weight gaind, % | 200 | 42(21%) |
| 12-24 months catch-up weight gaind, % | 200 | 31(15.5%) |
SDS values are based on UK 1990 growth reference, adjusted for sex, gestational (only for birth measurement) and postnatal age at measurement. LGA=large-for-gestational age, if birth weight SDS≥2 SD.
Values represent mean±SD or %.
aData taken from hospital records
bMeasured in ≤8 days after birth
cTaken from 4 sites: triceps, subscapular, flank, and quadriceps
dCatch-up is defined if delta growth between 2 times ≥0.67 SD
The desaturases activities have been classically assessed by the ratios of specific FA, measured by gas chromatography [18,19]. From the lipid profiling data on dried blood spots in our previous studies, hydrolysed FA were unavailable, rather, the data exhibited intact lipids containing those specific acids. From our detailed lipid analysis published previously [6,15], particular lipids could be identified as suitable proxies for the FA necessary to assess the desaturase activities. Lipid ratios at age 3 months indicating SCD1, FADS1, and FADS2 activities that were examined in this study are listed in Table 3 and each corresponding molecular mass in Supplementary table 1.
Table 3.
(Continued)
 |
B: unstandardized beta, 95% CI: 95% confidence interval (lower bound, upper bound) for the unstandardized beta
All lipid ratios are log-transformed
Weight, height, BMI SDS values are based on UK 1990 growth reference, adjusted for sex, gestational age (at 3 months only), and postnatal age at measurement. Skinfolds SDS values are internally-derived, adjusted for sex, gestational age, and postnatal age at measurement.
Age at visit is limited to 70-112 days for 3 months, 337-393 days for 12 months, and 702-758 days for 24 months visits
aModel 2 is adjusted for maternal parity (primiparous vs non), maternal pre-pregnancy BMI, and infant feeding history at 3 months
Linear regression models were employed to demonstrate the associations between lipid ratios representing enzyme activities with growth gains during infancy, with early (3-12 months) and late infancy (12-24 months) being analysed separately. In these analyses, first models were unadjusted while the second models were adjusted for significant confounding factors observed from our previous studies [6,20], including maternal parity, maternal pre-pregnancy BMI, and 0-3 months infant feeding history.
In the fully adjusted models, PC ratio reflecting SCD1 activity was positively associated with weight gain between 3-12 months of age (p=0.012, Table 3a). Meanwhile, CE ratio reflecting SCD1 activity showed negative associations with 3-12 months weight and BMI gains (p=0.027 and 0.033, respectively, Table 3a and c). The association between SCD1 activity represented by CEs and growth gains became positive at 12-24 months (fully adjusted, p=0.02 with weight and p=0.009 with height, Table 3a and b). Similarly, SCD1 PC ratio was also positively associated with 12-24 month-skinfolds gain reflecting adiposity (Table 3d). FADS1 PC ratio was inversely-, while FADS2 PC ratio was positively associated with subsequent 3-12 months height gain. The latter lipid ratio was inversely associated with adiposity gain from 3-12 months.
4. DISCUSSION
Here, we present a translational framework for the identification of lipid species, lipid ratios and potentially related enzyme activities associated with growth and adiposity outcomes during early life. The identified lipids and corresponding ratios might serve as predictive biomarkers to provide insights into individual susceptibility for obesity risk. The enzyme activities, as suggested by the lipid ratios, may offer opportunities for potential dietary interventions to modulate this risk, highlighting a window of opportunity early in life.
In our previous study, we reported that breastfeeding-related circulating lipids in infants could predict subsequent weight gain early in life [6]. However, that study was strictly observational and therefore had limited scope to provide direct insight into the mechanisms driving these associations. In light of this limitation, animal experiments can help in identifying robust candidate lipids to predict excessive weight-gain susceptibility. This is mainly because animal models allow for rigid control of diet and handling procedures, exclusion of genetic and environmental confounding factors, and accessibility of tissues that is impossible in humans. Extrapolation of results from animal studies has to take species-unique lipid metabolism and relative nutrient density per body weight into consideration [21] but can also benefit from additional insights based on specific lipid metabolism characteristics compared in particular strains. However, animal studies with translatable information on the link between lipid metabolism and growth are still scarce, with only a few genetically modified transgenic mouse models to mimic human lipid metabolism [7].
In this study, we used mouse models with established translational characteristics of human lipid metabolism and dyslipidaemia including E3L and Ldlr-/-.Leiden mice [7,8]. Independent diet-induced obesity experiments involving large cohorts of mice were set out to identify detectable circulating lipids in early-life plasma that could potentially predict future obesity risk. In these experiments, standardisation of genetic and environmental factors was implemented to minimise their confounding effects. This included (1) using male-only mice with identically-modified genetic background and (2) applying uniform nutrients provision, fasting period, age when commencing the energy-dense diet, and plasma sampling methods. Obesity was determined by various endpoints, such as body weight, adiposity, visceral and subcutaneous fat depots.
From the first E3L experiment, several lipids were noted to correlate with subsequent weight gain and other obesity parameters after 8 weeks of HFD exposure, mainly classified as CEs and PCs. The 4 best correlating CEs also displayed significant correlations with subsequent weight gain after HFD among Ldlr-/- mice, but not among the wild-type C57BL/6J. From lipid metabolism pathways, those CEs were identified to be educts or products of desaturases activities: SCD1, FADS1, and FADS2. Finally, the circulating level of these identified CEs at 4 weeks correlated with subsequent weight gain among E3L mice, suggesting their potentials to be early life candidate biomarkers (CBM) to predict later obesity in mice. Interestingly, circulating levels could be modulated by early life dietary intervention with ARA/DHA. We then attempted to translate the findings from our animal studies into the human setting via measuring lipid abundance from infants at 3 months of age.
Our findings on lipid ratios potentially related to SCD1, FADS1 and FADS2 activities are in line with recent human studies that have associated polyunsaturated fatty acids (PUFA) intake and the related enzyme activities, SCD1, FADS1, and FADS2, with childhood metabolic parameters [22]. For example, Wolters et al reported that a FADS1 polymorphism influenced blood pressure and body mass index (BMI) in a multinational European study involving children aged 2-10 years [23]. Another study from the same group reported positive associations between fatty acid ratios representing SCD1 and FADS2 activities at baseline with BMI and TG levels both at baseline and 2 years later [24]. In contrast, FADS1 activity measured by fatty acid ratios at baseline was inversely associated with both measures at both time points [24]. Similar to this particular study, we also discovered a positive association between SCD1 indices level at 3 months with later adiposity gain between 12-24 months, as well as with weight and height gains (Table 4). There was also a contemporaneous positive association between FADS2 activity and infant BMI at 3 months (B±SE=3.74±1.61, p=0.021, data not shown), and a positive association with height gain between 3-12 months (Table 3b). Meanwhile, a negative association between FADS1 was not observed with adiposity but with height gain between 3-12 months with PC ratio. As shown in the summary table 4, from all lipid ratios employed as proxies of enzyme activities, SCD1 activity appeared to be the most significant marker of growth and adiposity development during early life.
SCD1 plays a pivotal role in fat storage, lipid homeostasis, and energy metabolism. This enzyme is involved in monounsaturated fatty acids (MUFA) biosynthesis via introduction of a double bond into saturated fatty acids that can come from lipogenesis or from the diet. Increased hepatic SCD1 activity, measured as fatty acid ratio has been linked to obesity and its comorbidities, both in animals and humans [25,26]. In a population study involving more than 1800 elderly participants, Vinkness reported positive associations between plasma SCD1 indices, CE 16:1/16:0 and 18:1/18:0, and adiposity parameters, including BMI and body fat mass measured by dual X-ray absorptiometry (DXA). In contrast, those markers showed inverse associations with polyunsaturated fatty acids (PUFA) and exercise [25].
The rationale for translating results on lipid-related outcomes from animal models to humans is supported by specific similarities in lipid physiology observed across mammals. This includes comparable roles of lipids in supporting cellular, nuclear, and organelles membranes, lipoprotein, and the use of triglycerides (TGs) as storage molecules. Consistent with this, the potential biomarkers identified in this study belong to lipid classes that exert roles in storage and transport (cholesteryl esters) or as membrane lipids (phosphatidylcholine). Of note, lipid clearance and packaging, which influence lipid homeostasis, differ between mammals. Different lipid molecules might possibly be observed in the circulation due to species-dependent characteristics such as, e.g. diet, and lipid processing, e.g. enzymes involved in very-low-density lipoprotein (VLDL) assembly and secretion, lipolysis, lipoprotein clearance. Nonetheless, it is plausible to hypothesise that the pathways and enzymatic processes involved in lipid metabolism affecting an individual's susceptibility to rapid weight gain are analogous across species. The main rationale for this is that lipid metabolism orchestrating enzymes and their transcriptional regulators are evolutionarily conserved [27].
The predominant lipoproteins in human are apolipoprotein B-containing (V)LDL particles that are formed and released by the liver to transport lipids to the peripheral organs. In wild-type mice such as C57BL/6J mice, the (V)LDL concentrations in the circulation are extremely low because of rapid LDL receptor-mediated clearance of (V)LDL particles by the liver. Hence, in this study, HDL particles prevailed in the circulation of wild-type C57BL/6J mice whereas in E3L mice and LDLr-/-.Leiden mice, VLDL/LDL particles were abundantly present, similar to what would be observed in humans [28,29]. Many of the biomarkers identified in E3L mice that were confirmed in the independent experiment in LDLr-/-.Leiden mice belong to the class of CEs, which are constituents of (V)LDL particles. Of these CEs, only the lipid CE(16:1) could be recapitulated in C57BL/6J, possibly because oleic acid (C16:1) is one of the most abundant FA in the circulation, even among the wild-type mice [13]. The observation that all identified potential biomarkers with exception of CE(16:1) could be confirmed in LDLr-/-.Leiden mice but not in C57BL/6J mice indicates that the potential biomarkers are confined to lipoproteins.
Figure 3 displays the relationship between the identified lipid biomarkers and desaturases activities. While elongases mediate the elongation of the FA backbones, the fatty acid desaturases catalyse the synthesis of (poly)unsaturated fatty acids (PUFA) by introducing double carbon bonds. The ratio between saturated FA and PUFA influences the composition of phospholipids in cell membranes and affects membrane fluidity. Differences in lipid ratios reflecting desaturase activity have previously been associated with obesity and the metabolic syndrome [26].
It is interesting to speculate on the origin of the identified lipids and the underlying mechanism for their predictive value for obesity later in life. In infants, the demand for PUFA is different from adults. Moreover, the genetic makeup may contribute to differences in plasma lipid profiles, since single nucleotide polymorphisms (SNPs) in FADS genes can modulate desaturase activity [30]. Furthermore, pre- (maternal) and postnatal feeding could influence circulating desaturase activity markers. In mice, genetic and environmental factors can be excluded as confounding factors. Therefore, the predictive value of desaturase activity markers for obesity development in genetically identical mice suggests that the markers reflect a metabolic set point, possibly established via epigenetic mechanisms [31].
Gene expression of the desaturase enzymes FADS1, FADS2 and SCD1 is regulated by the lipogenic transcription factors SREBP1c and peroxisome proliferator-activated receptors (PPARs), predominantly in the liver [32], but also in adipose tissue [33]. Interestingly, studies focusing on mechanisms of metabolic programming have implicated PPARs as candidate gatekeeper pathway of developmental programming [34] and adipose tissue expansion during obesity development [8]. Notably, the PPARα gene has shown sensitivity for epigenetic changes [35], which may account for long-term changes in PPARα and its target genes. Future research may shed light on whether modulation of PPARs in early life (e.g. by PUFAs as endogenous ligands), could be an interesting opportunity to modulate the metabolic setpoint, measured by lipid ratios reflecting desaturase activity, as shown in our previously reported animal study (Supplementary Figure 2) [12].
In our mouse studies, ratios of CE lipids as proxies for FADS1, FADS2 or SCD1 activity were associated with subsequent weight gain. However, the identified CE lipids were not present in the existing CBGS lipidomics database. This is mainly because these particular CEs are of low abundance and not consistently detected in all of the studied infants. The lipidomics data were obtained from only a 3.2 mm disc of blood spot material from heel prick sample per infant, which limited the depth of lipid profiling analysis. Moreover, as lipids are apolar and can stick to other non-charged molecules, large particles such as VLDL and LDL are relatively difficult to dissolve once the blood is dried. Moreover, the method of detection is also less sensitive for apolar lipids. Therefore, desaturase activities were derived from analyses of other lipids in the CBGS lipidomics database.
From the CBGS data, of all 3 enzymes activities measured as lipid ratios, only SCD1 differed between 0-3 months infant feeding history, with breastfed infants had higher levels of CE and PC ratios (both p values<0.0005). The differences in direction of the associations between lipid ratios and growth parameters in the early (3-12 months) versus late infancy period (12-24 months) demonstrate how dynamic lipid metabolism is and thus affecting its potential use as candidate biomarkers. This difference could also be observed in the mice study; SCD1 (CE16:1)/CE(16:0) ratio level was inversely associated with subsequent weight gain between 4-12 weeks (Figure 4), but positively with 12-20 weeks weight gain, both after HFD exposure (Figure 2 and Table 1). Of note, studies with adult participants have shown that the activity of those enzymes is strongly driven by FA composition in diet [36]. Therefore, the effects of FA enzymatic activity on growth and metabolism might also change with the large variations in diet over time in infants.
A major strength of our work is the ability to align the lipid and weight gain results of gender- and diet-controlled E3L and LDLr-/- mouse models with lipid data from a human infant cohort. There are very few birth cohorts that have both detailed weight, growth and body composition data combined with lipid profiling data, which makes this approach unique. The regression models were also controlled for feeding history, therefore, the associations between the identified lipid ratios and growth parameters were independent on how the infants were fed. Furthermore, lipid profiling approaches were independent between the model system and the human birth cohort study, which emphasises the robustness of translated results. The translational approach also allowed us to use a hypothesis based analysis of specific lipid ratios, rather than a data-driven multivariate approach, which at best could only generate hypotheses.
There are important limitations to note. First, lipid metabolism in mice is inherently different to lipid metabolism of humans. The use of the E3L and LDLr-/-.Leiden mouse models allowed us to study lipid metabolism that is closer to humans then it would be the case if wild type mice were used. This is because apolipoprotein B-containing particles are the predominant circulating lipoproteins in both transgenic mice models. Despite similarities in lipid handling and metabolome [7,37], mice do not fully represent human lipid metabolism because they lack, for instance, cholesterylester transfer protein. Secondly, the dietary context differed between the mouse studies and the early life situation in humans: at three months infants are only consuming breast milk or formula, and it is possible that we would have identified other lipids or lipid ratios if the dietary condition could have been mimicked better. Third, the use of lipid ratios is only a proxy to estimate an enzyme activity. Desaturases are expressed in multiple tissues among which liver and adipose tissue, and lipids measured in the plasma compartment may reflect the net activity of desaturases in one or more of these tissues. Assessing the molecular causes that underlie the observed lipid differences between infants requires a lot of futher work, including mRNA and protein expression studies as well as lipid profiling of tissue biopsies and lipid flux studies. Fourth, the lipid profiling technology used between the mouse models and the human cohorts limited our ability to directly compare the exact same lipids, or very specific lipid ratios that could have improved the sensitivity to pick up relationships between desaturase activities and weight gain. Despite rapid development of lipidomics technologies, it is not ideal to repeat lipidomic analysis on precious and hard-to-obtain biological samples, such as infant's blood. Hence, re-analyse the samples using more recent technology was not feasible.
In conclusion, lipid ratios potentially reflecting SCD1, FADS1, and FADS2 activities were identified in this study and they were associated with subsequent body size, first in murine models and subsequently validated in an infant human cohort. While these lipids are promising as candidate biomarkers, further longitudinal investigations are required to confirm these findings.
5. Contributors
AK and RK had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. RK, IBP, TK, WvD, and PYW were responsible for animal studies and data analyses. DBD, KKO, IAH, and PMP were involved in the conduction of the CBGS. LO and PMP were responsible for infant recruitment, clinic visit, and blood sampling in the CBGS. AK, SSG, and SF conducted the lipidomics experiments on the infant DBS. LO and AK analysed the infant lipidomics data and LO performed the statistical analyses. LO, JAvD, AK, and RK drafted the manuscript. GG, IBP, TK, IAH, KKO, DBD, and PYW helped to improve the manuscript. All authors contributed to interpretation of data, critically revised the article for important intellectual content, and approved the final version.
6. Funding
The animal study and respective lipidomics analyses were supported by a grant from a research program of the Dutch government (Eli-Co project) to IBP, WvD, TK, and RK, and the TNO research program ‘BMH-PMC-13 Functional Biomarkers’.
The CBGS has been funded by the Medical Research Council [7500001180, G1001995], European Union Framework 5 [QLK4-1999–01422], the Mothercare Charitable Foundation [RG54608], Newlife Foundation for Disabled Children (07/20), and the World Cancer Research Fund International (2004/03). This research was also supported by the National Institute for Health Research/Wellcome Trust Clinical Research Facility at Cambridge University Hospitals NHS Foundation Trust and the NIHR Cambridge Comprehensive Biomedical Research Centre. KKO is supported by the Medical Research Council (Unit programmes: MC_UU_12015/2 and MC_UU_00006/2).
RK acknowledges funding from the Early Research Program ‘Body Brain Interactions’ (ERP020). AK, SF, and SGS are supported by the Biotechnology and Biological Sciences Research Council (BBSRC) (BB/M027252/1 for AK and SF, BB/M027252/2 for SGS). AK also gratefully acknowledges funding from the NIHR Biomedical Research Centre Cambridge (IS-BRC-1215-20014).
All sponsors had no role in the study design, collection, analysis or interpretation of the data, the writing of the manuscript or the decision to submit it for publication.
7. Data sharing
The data that support the findings of this study are available on request from Dr Albert Koulman (for human study, ak675@medschl.cam.ac.uk) and Dr Robert Kleeman (for animal study, Robert.kleemann@tno.nl).
Declaration of Competing Interest
Both animal and human studies received funding support from Mead Johnson Nutrition. JAvD and GG are employees of Mead Johnson Nutrition/Reckitt Benckiser, MHS and EAFvT were employees of Mead Johnson Nutrition at the time of the study. No other authors declare a conflict of interest.
Acknowledgements
The authors thank Drs Elly de Wit†, Dr Lars Verschuren, and Drs Erik Offerman for technical assistance and advice provided in the animal studies. The authors also acknowledge the CBGS research nurses Suzanne Smith, Anne-Marie Wardell, and Karen Forbes, all the families who contributed to the study, the staff at the National Institute for Health Research (NIHR) Cambridge/Wellcome Trust Clinical Research Facility, the NIHR Cambridge Comprehensive Biomedical Research Centre, and the midwives at the Rosie Maternity Hospital, Cambridge, UK.
Footnotes
Funding: The list of funding sources is available at the end of the manuscript (see Acknowledgments).
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.ebiom.2020.103198.
Appendix. Supplementary materials
References
- 1.Barker DJP. The developmental origins of chronic adult disease. Acta Paediatr Int J Paediatr Suppl. 2004;93(446):26–33. doi: 10.1080/08035320410022730. [DOI] [PubMed] [Google Scholar]
- 2.Ong KK, Ahmed ML, Emmett PM, Preece M.A., Dunger D.B. the ALSPAC Study Team. Association Between Postnatal Catch-Up Growth and Obesity in Childhood: Prospective Cohort Study. BMJ. 2000;320:967–971. doi: 10.1177/000992280003901210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kerkhof GF, Hokken-Koelega ACS. Rate of neonatal weight gain and effects on adult metabolic health. Nat Rev Endocrinol. 2012;8:689–692. doi: 10.1038/nrendo.2012.168. [DOI] [PubMed] [Google Scholar]
- 4.Piaggi P. Metabolic Determinants of Weight Gain in Humans. Obesity. 2019;27(5):691–699. doi: 10.1002/oby.22456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanson MA, Gluckman PD. Early developmental conditioning of later health and disease: physiology or pathophysiology? Physiol Rev. 2014;94:1027–1076. doi: 10.1152/physrev.00029.2013. Table 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prentice P, Koulman A, Matthews L, Acerini CL, Ong KK, Dunger DB. Lipidomic analyses, breast- and formula-feeding, and growth in infants. J Pediatr. 2015;166(2):276–281. doi: 10.1016/j.jpeds.2014.10.021. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zadelaar S, Kleemann R, Verschuren L. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol. 2007;27(8):1706–1721. doi: 10.1161/ATVBAHA.107.142570. [DOI] [PubMed] [Google Scholar]
- 8.Zwamborn RA, Slieker RC, Mulder PC. Prolonged high-fat diet induces gradual and fat depot-specific DNA methylation changes in adult mice. Sci Rep. 2017;7(43261):1–9. doi: 10.1038/srep43261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prentice P, Acerini CL, Eleftheriou A, Hughes IA, Ong KK, Dunger DB. Cohort profile: The Cambridge Baby Growth Study (CBGS) Int J Epidemiol. 2016;45(1) doi: 10.1093/ije/dyv318. 35-35g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pouwer MG, Heinonen SE, Behrendt M. The APoE∗3-Leiden heterozygous glucokinase knockout mouse as novel translational disease model for type 2 diabetes, dyslipidemia, and diabetic atherosclerosis. J Diabetes Res. 2019 doi: 10.1155/2019/9727952. 2019(Cvd) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gart E, Lima ES, Schuren F. Diet-independent correlations between bacteria and dysfunction of gut, adipose tissue, and liver: A comprehensive microbiota analysis in feces and mucosa of the ileum and colon in obese mice with NAFLD. Int J Mol Sci. 2019;20(1):1–20. doi: 10.3390/ijms20010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wielinga PY, Harthoorn LF, Verschuren L. Arachidonic acid/docosahexaenoic acid-supplemented diet in early life reduces body weight gain, plasma lipids, and adiposity in later life in ApoE*3Leiden mice. Mol Nutr Food Res. 2012;00:1–10. doi: 10.1002/mnfr.201100762. [DOI] [PubMed] [Google Scholar]
- 13.Mulder P, Morrison MC, Wielinga PY, Van Duyvenvoorde W, Kooistra T, Kleemann R. Surgical removal of inflamed epididymal white adipose tissue attenuates the development of non-alcoholic steatohepatitis in obesity. Int J Obes. 2016;40(4):675–684. doi: 10.1038/ijo.2015.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wopereis S, Radonjic M, Rubingh C. Identification of prognostic and diagnostic biomarkers of glucose intolerance in ApoE3Leiden mice. Physiol Genomics. 2012;44:293–304. doi: 10.1152/physiolgenomics.00072.2011. [DOI] [PubMed] [Google Scholar]
- 15.Koulman A, Prentice P, Wong MCY. The development and validation of a fast and robust dried blood spot based lipid profiling method to study infant metabolism. Metabolomics. 2014:1–8. doi: 10.1007/s11306-014-0628-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Acharjee A, Prentice P, Acerini C. The translation of lipid profiles to nutritional biomarkers in the study of infant metabolism. Metabolomics. 2017;13(3):1–9. doi: 10.1007/s11306-017-1166-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan H, Cole T. a Microsoft Excel add-in to access growth references based on the LMS method. 2012. LMSgrowth.http://www.healthforallchildren.co.uk/ [Google Scholar]
- 18.Steffen LM, Vessby B, Jacobs DR. Serum phospholipid and cholesteryl ester fatty acids and estimated desaturase activities are related to overweight and cardiovascular risk factors in adolescents. Int J Obes. 2008;32(8):1297–1304. doi: 10.1038/ijo.2008.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hodge AM, English DR, O'Dea K. Plasma phospholipid and dietary fatty acids as predictors of type 2 diabetes: Interpreting the role of linoleic acid. Am J Clin Nutr. 2007;86(1):189–197. doi: 10.1093/ajcn/86.1.189. [DOI] [PubMed] [Google Scholar]
- 20.Prentice PM, Olga L, Petry CJ. Reduced size at birth and persisting reductions in adiposity in recent, compared with earlier, cohorts of infants born to mothers with gestational diabetes mellitus. Diabetologia. 2019;62(11):1977–1987. doi: 10.1007/s00125-019-4970-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Innis SM. Essential fatty acids in infant nutrition: Lessons and limitations from animal studies in relation to studies on infant fatty acid requirements. Am J Clin Nutr. 2000;71:238–244. doi: 10.1093/ajcn/71.1.238s. [DOI] [PubMed] [Google Scholar]
- 22.Andersen KR, Harslof LBS, Schnurr TM. A study of associations between early DHA status and fatty acid desaturase (FADS) SNP and developmental outcomes in children of obese mothers. Br J Nutr. 2017;117(2):278–286. doi: 10.1017/S0007114516004645. [DOI] [PubMed] [Google Scholar]
- 23.Wolters M, Dering C, Siani A. The role of a FADS1 polymorphism in the association of fatty acid blood levels, BMI and blood pressure in young children—Analyses based on path models. PLoS One. 2017;12(7):1–17. doi: 10.1371/journal.pone.0181485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolters M, Schlenz H, Börnhorst C. Desaturase activity is associated with weight status and metabolic risk markers in young children. J Clin Endocrinol Metab. 2015;100(10):3760–3769. doi: 10.1210/jc.2015-2693. [DOI] [PubMed] [Google Scholar]
- 25.Vinknes KJ, Elshorbagy AK, Nurk E. Plasma stearoyl-CoA desaturase indices: Association with lifestyle, diet, and body composition. Obesity. 2013;21(3):294–302. doi: 10.1002/oby.20011. [DOI] [PubMed] [Google Scholar]
- 26.Warensjö E, Risérus U, Vessby B. Fatty acid composition of serum lipids predicts the development of the metabolic syndrome in men. Diabetologia. 2005;48(10):1999–2005. doi: 10.1007/s00125-005-1897-x. [DOI] [PubMed] [Google Scholar]
- 27.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 28.Verschuren L, Kleemann R, Offerman EH. Effect of low dose atorvastatin versus diet-induced cholesterol lowering on atherosclerotic lesion progression and inflammation in apolipoprotein E*3-Leiden transgenic mice. Arter Thromb Vasc Biol. 2005;25(1):161–167. doi: 10.1161/01.ATV.0000148866.29829.19. [DOI] [PubMed] [Google Scholar]
- 29.Verschuren L, Kooistra T, Bernhagen J. MIF deficiency reduces chronic inflammation in white adipose tissue and impairs the development of insulin resistance, glucose intolerance, and associated atherosclerotic disease. Circ Res. 2009;105(1):99–107. doi: 10.1161/CIRCRESAHA.109.199166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Czumaj A, Śledziński T. Biological role of unsaturated fatty acid desaturases in health and disease. Nutrients. 2020;12(2) doi: 10.3390/nu12020356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burdge GC, Lillycrop KA. Fatty acids and epigenetics. Curr Opin Clin Nutr Metab Care. 2014;17(2):156–161. doi: 10.1097/MCO.0000000000000023. [DOI] [PubMed] [Google Scholar]
- 32.Rakhshandehroo M, Knoch B, Müller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010;2010 doi: 10.1155/2010/612089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Al-Hasani H, Joost HG. Nutrition-/diet-induced changes in gene expression in white adipose tissue. Best Pract Res Clin Endocrinol Metab. 2005;19(4):589–603. doi: 10.1016/j.beem.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Tain YL, Hsu CN, Chan JYH. PPARs link early life nutritional insults to later programmed hypertension and metabolic syndrome. Int J Mol Sci. 2016;17(20):1–11. doi: 10.3390/ijms17010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. Dietary Protein Restriction of Pregnant Rats Induces and Folic Acid Supplementation Prevents Epigenetic Modification of Hepatic Gene Expression in the Offspring. J Nutr. 2005;135(6):1382–1386. doi: 10.1093/jn/135.6.1382. [DOI] [PubMed] [Google Scholar]
- 36.Warensjö E, Rosell M, Hellenius ML, Vessby B, De Faire U, Risérus U. Associations between estimated fatty acid desaturase activities in serum lipids and adipose tissue in humans: Links to obesity and insulin resistance. Lipids Health Dis. 2009;8:6–11. doi: 10.1186/1476-511X-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morrison MC, Kleemann R, van Koppen A, Hanemaaijer R, Verschuren L. Key inflammatory processes in human NASH are reflected in Ldlr-/-.Leiden mice: A translational gene profiling study. Front Physiol. 2018;9(FEB):1–13. doi: 10.3389/fphys.2018.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.