

Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is caused by incomplete silencing of the disease locus, leading to pathogenic misexpression of DUX4 in skeletal muscle. Previously, we showed that CRISPR inhibition could successfully target and repress DUX4 in FSHD myocytes. However, an effective therapy will require both efficient delivery of therapeutic components to skeletal muscles and long-term repression of the disease locus. Thus, we re-engineered our platform to allow in vivo delivery of more potent epigenetic repressors. We designed an FSHD-optimized regulatory cassette to drive skeletal muscle-specific expression of dCas9 from Staphylococcus aureus fused to HP1α, HP1γ, the MeCP2 transcriptional repression domain, or the SUV39H1 SET domain. Targeting each regulator to the DUX4 promoter/exon 1 increased chromatin repression at the locus, specifically suppressing DUX4 and its target genes in FSHD myocytes and in a mouse model of the disease. Importantly, minimizing the regulatory cassette and using the smaller Cas9 ortholog allowed our therapeutic cassettes to be effectively packaged into adeno-associated virus (AAV) vectors for in vivo delivery. By engineering a muscle-specific epigenetic CRISPR platform compatible with AAV vectors for gene therapy, we have laid the groundwork for clinical use of dCas9-based chromatin effectors in skeletal muscle disorders.
Keywords: FSHD, facioscapulohumeral muscular dystrophy, DUX4, CRISPR inhibition, dCas9, gene therapy, gene regulation, epigenetic repression, muscular dystrophy, AAV
Graphical Abstract
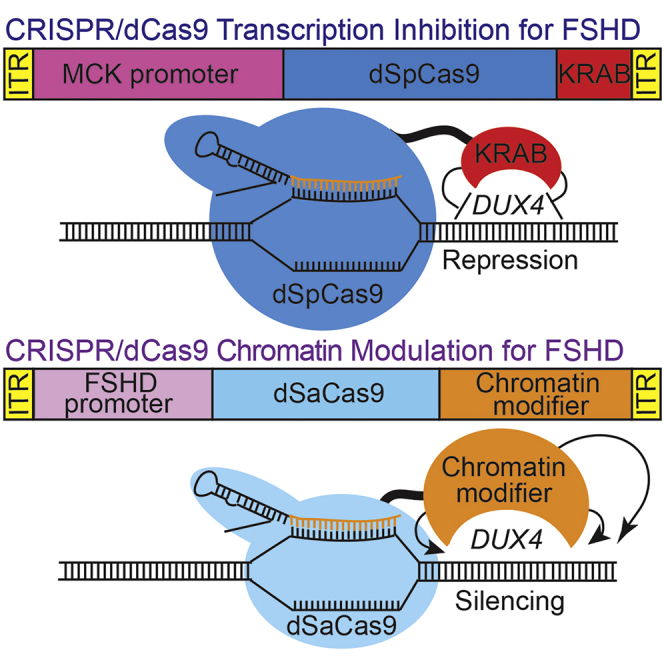
FSHD is caused by incomplete epigenetic silencing, leading to pathogenic misexpression of DUX4 in skeletal muscles. We designed an FSHD-optimized cassette to express dCas9 fused to each of four epigenetic repressors. This epigenetic CRISPR inhibition effectively and specifically represses DUX4-fl and its targets in primary FSHD myocytes and in vivo.
Introduction
Facioscapulohumeral muscular dystrophy (FSHD) (MIM: 158900 and 158901) is the third most common muscular dystrophy,1,2 characterized by progressive weakness and atrophy of specific muscle groups. Both forms of the disease are caused by epigenetic dysregulation of the D4Z4 macrosatellite repeat array at chromosome 4q35.3,4 FSHD1, the most common form of the disease, is linked to contractions at this array,5, 6, 7 resulting in relaxation of chromatin that is normally repressed. FSHD2 is caused by mutations in proteins that maintain epigenetic silencing of the D4Z4 array, leading to a similar chromatin relaxation.8,9 In both forms of the disease, loss of epigenetic repression leads to the aberrant expression of the DUX4 retrogene in skeletal muscle.10 The DUX4 protein, in turn, activates a host of genes normally expressed in early development, which cause pathology when misexpressed in adult skeletal muscle.4,11,12 Although an intact DUX4 open reading frame resides in every D4Z4 repeat unit in the macrosatellite array, only the full-length DUX4 mRNA (DUX4-fl) encoded by the distal-most repeat is stably expressed and translated due to the polyadenylation signal residing in an exon distal to the array in disease-permissive alleles.10,13 Thus, FSHD is a toxic gain-of-function disease.
There are no cures or ameliorative treatments for FSHD, so an effective therapy is critically needed. Since the discovery that FSHD pathogenesis is caused by aberrant expression of DUX4 in skeletal muscles,10,13,14 numerous therapeutic approaches targeting DUX4 and its downstream pathways are being developed.15, 16, 17, 18, 19, 20, 21 The most direct path to an FSHD therapy is eliminating expression of DUX4 mRNA. While the amount of DUX4 inhibition required for effective therapy is unknown, data from clinically affected and asymptomatic FSHD subjects support the idea that any reduction in DUX4 expression will have therapeutic benefit.22, 23, 24 Small molecules targeting DUX4 expression, independently identified from highly similar indirect expression screens, are promising; however, their discovery is limited by the chemical libraries screened, dosing, and modes of action. Despite the clear overlap in libraries, two published screens with similar approaches identified different molecules, targets, and pathways for DUX4 inhibition, even to the exclusion of other targets,20,21 which is cause for concern. Long-term, harmful side effects are also a concern, as many potential FSHD drug targets play key roles within the target tissue16 (e.g., the p38 inhibitor losmapimod, currently being tested in a clinical trial for FSHD25, 26, 27).
With these issues in mind, correcting the fundamental epigenetic dysregulation that leads to FSHD may prove to be the most efficacious and cost-effective avenue of therapy.28 As with many repetitive elements in the human genome, the D4Z4 macrosatellite array that encodes DUX4 is normally under strong epigenetic repression in adult somatic cells. The loss of repression seen in FSHD patients is marked by reduced enrichment of the repressive H3K9me3 mark, HP1γ, and cohesin at D4Z4 arrays29,30 and DNA hypomethylation at the 4q D4Z4 array.3,23,31, 32, 33, 34 Thus, returning the chromatin at D4Z4 to its normal state of repression in skeletal myocytes is a viable and attractive therapeutic avenue. CRISPR technology provides a means for effectively modifying specific regions of the human genome and, thus, the ability to target the root cause of a disease. Therefore, we are developing CRISPR-based approaches to correct the epigenetic dysregulation in FSHD and therapeutically reduce pathogenic DUX4 expression.
CRISPR gene targeting consists of two components working together: (1) the Cas9 nuclease and (2) a single guide RNA (sgRNA), which directs Cas9 to a specific site in the genome. There are essentially two approaches one could take toward CRISPR-based silencing of DUX4: editing (CRISPRe), which utilizes a functional Cas9 to alter the genomic sequence, and inhibition (CRISPRi), which uses an enzymatically inactive “dead” Cas9 (dCas9) fused to a transcriptional or chromatin repressor. CRISPRi platforms can effectively modulate endogenous gene expression in mammalian cells,35 and for an approach to FSHD with a path to the clinic, CRISPRi has significant advantages over CRISPRe. (1) It does not damage the genome. (2) Any CRISPR approach targeting DUX4 will have unavoidable specific “off targets,” because D4Z4/ DUX4 is present in many copies on the non-contracted 4q and both 10q chromosomes, in addition to polymorphic D4Z4s at other loci. However, as these regions are normally heterochromatic, they would simply remain repressed by CRISPRi—not cut, which could result in numerous double-strand breaks and likely apoptosis. (3) CRISPRi, using an appropriate epigenetic regulator, can potentially switch the epigenetic state of a region to make the effect heritable and essentially permanent.
Previously, we demonstrated that CRISPRi using dCas9 from Streptococcus pyogenes (dSpCas9) fused to the KRAB transcriptional repression domain (TRD) can successfully target the pathogenic D4Z4 repeat array to repress DUX-fl expression in FSHD myocytes.15 However, repression mediated by dCas9-KRAB requires its continuous expression,36, 37, 38 which is not ideal for therapeutic applications. Here we extend the utility of CRISPRi to epigenetic regulators capable of mediating stable repression for long-term silencing, yet small enough to be packaged in adeno-associated viruses (AAVs). Key to this advance was minimizing the gene regulatory cassette while maintaining high activity in skeletal muscle, creating more space for delivery of larger epigenetic regulators fused to the smaller dCas9 from Staphylococcus aureus (dSaCas9). We demonstrate that targeting the DUX4 promoter or exon 1 with dSaCas9 fused to any one of four epigenetic repressors (the SUV39H1 SET domain, the MeCP2 TRD, HP1α, or HP1γ) increases chromatin repression at the disease locus, leading to suppression of DUX4 and its target genes in FSHD myocytes and in a DUX4-based, FSHD-like transgenic mouse model. Targeting each dSaCas9-repressor to DUX4 has no significant effects on the closest-matching off-target genes expressed in skeletal muscle and minimal effects on global gene expression. Importantly, by reducing the size of the regulatory cassette, our therapeutic cassettes can be effectively packaged into AAV vectors for in vivo delivery.
Results
Design of an FSHD-specific CRISPRi platform for epigenetic repression of DUX4
While we have demonstrated proof of principle that CRISPRi for FSHD is feasible,15 an effective therapy will require both efficient delivery of therapeutic components to skeletal muscles and long-term repression of the disease locus. To address these needs, we re-engineered our CRISPRi platform. Our initial study used dSpCas9 fused to the KRAB TRD (Figure 1A), which was sufficient for short-term inhibition in cultured cells but not ideal for long-term silencing. Stable silencing might be directly accomplished by targeting DNA methyltransferases (DNMTs)42 to DUX4; however, the catalytic domains of these enzymes are much too large to fit the packaging constraints (∼4.4 kb) of current AAV vectors needed for in vivo delivery. Thus, we turned our attention to smaller epigenetic regulators and repressive domains that can also effect stable silencing.
Figure 1.

CRISPRi constructs for epigenetic repression of DUX4
(A) Original two-vector system: (1) dSpCas9 fused to the KRAB TRD under control of the CK8 regulatory cassette,39,40 and (2) a DUX4-targeting sgRNA with dSpCas9-compatible scaffold under control of the U6 promoter. (B) Optimized two-vector system: (1) the smaller dSaCas9 ortholog fused to one of four epigenetic repressors (HP1α, HP1γ, the MeCP2 TRD, or the SUV39H1 pre-SET, SET, and post-SET domains) under control of a minimized skeletal muscle regulatory cassette, and (2) a DUX4-targeting sgRNA with dSaCas9-compatible scaffold incorporating modifications that remove a putative Pol III terminator and improve assembly with dCas941 under control of the U6 promoter. (C) Schematic diagram of the FSHD locus at chromosome 4q35. Distances are shown relative to the DUX4 MAL start codon (∗). For simplicity, only the distal D4Z4 repeat unit of the macrosatellite array (dark gray) is depicted. DUX4 exons 1 and 2 are located within the D4Z4 repeat, and exon 3 lies in the distal subtelomeric sequence. The locations of sgRNA target sequences (#1–6) are indicated. Positions of ChIP amplicons are shown as unlabeled red bars (in order from 5′ to 3′: DUX4 promoter, exon 1, and exon 3). Refer to text for additional details. ITR, inverted terminal repeat; NLS, nuclear localization signal; HGA, hemagglutinin; bGH, bovine growth hormone; PAS, polyadenylation signal; cPPT, central polypurine tract; CTS, central termination sequence.
Of these, HP1 proteins are key mediators of heterochromatin formation. HP1α predominantly localizes to heterochromatin,43, 44, 45 and HP1γ is enriched at the D4Z4 macrosatellite array in healthy myocytes and lost in FSHD.29 SUV39H1 is a histone methyltransferase that establishes constitutive heterochromatin at pericentric and telomeric regions.46 The SET domain of SUV39H1 participates in stable binding to heterochromatin and mediates H3K9 trimethylation, a repressive mark that recruits HP1.46 Although the SET domain contains the active site of enzymatic activity, both pre-SET and post-SET domains are required for methyltransferase activity and are included in our SET cassette. The methyl-CpG-binding protein MeCP2 plays diverse roles in chromatin regulation, but its TRD is all that is required for binding repressive histone marks and co-repressor complexes to enable efficient gene silencing.47, 48, 49
To accommodate dCas9 fused to even these relatively small repressors and repressive domains in AAV vectors required minimizing current regulatory cassettes. Building on key work from the Hauschka lab39,40 and taking into account that cardiac muscle is not involved in FSHD,50,51 we designed a minimized skeletal muscle regulatory cassette to allow larger therapeutic components to be delivered in vivo. Starting with the CK8 cassette, which is a modified version of three muscle creatine kinase (CKM) enhancers upstream of the CKM promoter,39 we removed additional space between elements and deleted the CarG and AP2 sites, which are dispensable for expression in skeletal muscle.52,53 This reduced the size of the regulatory cassette to 378 bp, allowing us to build constructs containing the smaller dSaCas9 ortholog fused to epigenetic repressors that were previously too large to fit into AAV vectors. Thus, our optimized CRISPRi platform consists of: (1) dSaCas9 fused to one of four epigenetic repressors (HP1α, HP1γ, the MeCP2 TRD, or the SUV39H1 pre-SET, SET, and post-SET domains) under control of our FSHD-optimized regulatory cassette, and (2) an sgRNA targeting the DUX4 locus under control of the U6 promoter (Figure 1B). These components were expressed in lentiviral (LV) vectors for transduction of cultured myocytes and in AAV vectors for in vivo use.
dSaCas9-mediated recruitment of epigenetic repressors to the DUX4 promoter or exon 1 represses DUX4-fl and DUX4-FL targets in FSHD myocytes
To target our dSaCas9-repressors, we designed sgRNAs compatible with the Sa protospacer adjacent motif (PAM) (NNGRRT) across the DUX4 locus (Materials and methods; Figure 1C). For all experiments, we performed four serial coinfections of FSHD myogenic cultures, as described.15 Cells were transduced with combinations of LV supernatants expressing either dSaCas9 fused to each epigenetic regulator or individual sgRNAs. Cells were harvested 3 days following the final round of infection for analysis of gene expression by quantitative reverse-transcriptase PCR (qRT-PCR).
Although targeting DUX4 exon 3 or the D4Z4 upstream enhancers54 had no effect, targeting each dSaCas9-repressor to the DUX4 promoter or exon 1 significantly reduced levels of DUX4-fl mRNA to ∼30%–50% of endogenous levels (Figure 2; Figure S1). As levels of DUX4-FL protein are low and difficult to assess in FSHD myocytes, we routinely assess DUX4-FL target gene expression as the more reliable assay and relevant functional readout of DUX4 activity. Importantly, expression levels of DUX4-FL targets thought to have pathogenic consequences55 are significantly decreased in parallel with the reduction in DUX4-fl mRNA (Figure 2; Figure S1).
Figure 2.

dSaCas9-mediated recruitment of epigenetic repressors to the DUX4 promoter or exon 1 represses DUX4-fl and DUX4-FL targets in FSHD myocytes
(A–D) FSHD myocytes were transduced with dSaCas9 fused to either: (A) the SUV39H1 pre-SET, SET, and post-SET domains (SET); (B) the MeCP2 TRD; (C) HP1γ; or (D) HP1α, with or without sgRNAs targeting DUX4 (#1–6) or non-targeting sgRNAs (NT). Expression levels of DUX4-fl and DUX4-FL target genes TRIM43 and MBD3L2 were assessed by qRT-PCR. Data are plotted as the mean + SD value of at least four independent experiments, with relative mRNA expression for cells expressing each dCas9-epigenetic regulator alone set to 1. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 are from comparing to NT.
To verify that enzymatic activity of the SET domain is required for the effect on DUX4-fl, we targeted dSaCas9-SET containing a mutation (C326A) within the SET domain that abolishes enzymatic activity56 to the DUX4 promoter/exon 1. Although the effect was highly variable, the inactive SET domain did not significantly affect levels of DUX4-fl (Figure S2), indicating that enzymatic activity of this region is required for DUX4-fl repression.
Targeting dSaCas9-repressors to DUX4 has no effect on Myosin heavy chain 1 or D4Z4 proximal genes
To rule out a nonspecific effect of dSaCas9-repressors on muscle differentiation, we assessed levels of Myosin heavy chain 1 (MYH1), a marker of terminal muscle differentiation, by qRT-PCR in the cells described above. Importantly, MYH1 levels were equivalent in all cultures (Figures 3A–3D; Figure S3), indicating that lower levels of DUX4-fl are not due to impairment of differentiation. We also measured expression of FRG1 and FRG2, which lie proximal to the D4Z4 macrosatellite. Recruitment of each dSaCas9-repressor to the DUX4 promoter/exon 1 did not reduce expression of these D4Z4 proximal genes (Figures 3A–3D; Figure S3).
Figure 3.

Targeting dSaCas9-repressors to DUX4 has no effect on Myosin heavy chain 1, D4Z4 proximal genes, or closest-match off-target genes expressed in skeletal muscle
(A–D) Expression levels of the terminal muscle differentiation marker Myosin heavy chain 1 (MYH1) and D4Z4 proximal genes FRG1 and FRG2 were assessed by qRT-PCR in the FSHD myocyte cultures described in Figure 2. (E and F) Expression levels of (E) Lysosomal amino acid transporter 1 homolog (LAAT1), (F) Ribosome biogenesis regulatory protein homolog (RRS1), or Guanine nucleotide-binding protein G(i) subunit alpha-1 isoform 1 (GNAI1) were assessed by qRT-PCR in the relevant FSHD myocyte cultures described in Figure 2. Intron 1 of LAAT1 contains a potential off-target match to sgRNA #1. The single exon of RRS1 and the downstream flanking sequence of GNAI1 contain potential off-target matches to sgRNA #5. Data are plotted as the mean + SD value of at least four independent experiments, with relative mRNA expression for cells expressing each dCas9-epigenetic regulator alone set to 1.
Targeting dSaCas9-repressors to DUX4 has minimal off-target effects in FSHD myocytes
A major concern of CRISPR-based approaches for gene therapy is the prospect of deleterious off-target effects. While many versions of Cas9 are being engineered for increased specificity,57, 58, 59, 60 for preclinical studies it is always necessary to determine the off-target effects of each potential treatment. For the sgRNAs that worked best in combination with each dSaCas9-repressor, we used the publicly available Cas-OFFinder tool (http://www.rgenome.net/cas-offinder/) to search for the closest-matching off-target sequences in the human genome. Only sgRNAs #1 and #5 had close-matching predicted off-targets in or near genes expressed in skeletal muscle (Table S1). Intron 1 of Lysosomal amino acid transporter 1 homolog (LAAT1) contains a potential off-target match to sgRNA #1. The single exon of Ribosome biogenesis regulatory protein homolog (RRS1) and the sequence 283 bp downstream of Guanine nucleotide-binding protein G(i) subunit alpha-1 isoform 1 (GNAI1) contain potential off-target matches to sgRNA #5. However, in contrast to the striking reduction in DUX4-fl, targeting dSaCas9-SET with sgRNA #1 had no effect on LAAT1 expression (Figure 3E; Figure S4). Similarly, targeting dSaCas9-HP1γ with sgRNA #5 had no effect on levels of RRS1 or GNAI1 (Figure 3F; Figure S4).
Since an analysis of off-target DNA binding (by chromatin immunoprecipitation sequencing [ChIP-seq]) sheds no light on the more critical off-target gene expression profiles, we performed RNA sequencing (RNA-seq) to assess the global effects of targeting each dSaCas9-repressor to DUX4 with the most effective sgRNAs. Primary FSHD myocytes were transduced with each combination of vectors (described in Figure 4) or with dSaCas9-KRAB + sgRNA #6 for comparison. Gene Ontology (GO) analysis shows that the majority of misregulated cellular responses are likely due to the LV transduction or possibly dCas9 expression and not due to off-target repression mediated by the dCas9 effectors (Figures S5–S9). The fact that targeting with four different sgRNAs yields very similar profiles of differentially expressed genes (DEGs), consistent with an innate immune response, strongly supports this conclusion (Table S2. Significant DEGs following targeting of dSaCas9-repressors to DUX4, Table S3. Comparison of DEGs following targeting of dSaCas9-repressors to DUX4, Table S4. Changes in expression among developmental and myogenic DEGs following targeting of dSaCas9-repressors to DUX4), although some immune-related DEGs may represent a correction of DUX4-mediated dysregulation, since DUX4 targets include immune mediators. After removing DEGs consistent with a response to virus, the vast majority remaining are part of embryonic programs or developmental pathways that are deregulated by DUX4 misexpression (Table 1; Table S4).61, 62, 63, 64, 65 Many of these genes are common to multiple treatments, and their differential expression represents a return to a more normal pattern of gene expression. For example, DUX4 expression reduces levels of TRIM14, KREMEN2, LY6E, and PARP14 in multiple independent studies (compared in Jagannathan et al.61); consistent with these studies, we found that all four dSaCas9-epigenetic repressor treatments led to an increase in expression of these genes (Table 1; Table S4). Conversely, TM6SF1 and ITGA8, which are upregulated following DUX4 overexpression,61,63 were both decreased following treatment with every dSaCas9-epigenetic repressor (Table 1; Table S4).
Figure 4.

Targeting dSaCas9-repressors to DUX4 has minimal effects on global gene expression in FSHD myocytes
(A–E) FSHD myocytes were transduced with: (A) dSaCas9-KRAB + sgRNA #6, (B) dSaCas9-HP1α + sgRNA #2, (C) dSaCas9-HP1γ + sgRNA #5, (D) dSaCas9-SET + sgRNA #1, or (E) dSaCas9-TRD + sgRNA #6. For each treatment, five independent experiments were analyzed by RNA-seq using the Illumina HiSeq 2 × 100 bp platform. Adjusted volcano scatterplots show the global transcriptional changes between each treatment versus mock-infected cells. Each data point represents a gene. Upregulated genes (p < 0.05 and a log2 fold change > 1) are indicated by red dots. Downregulated genes (p < 0.05 and a log2 fold change < −1) are indicated by blue dots. Unique differentially expressed genes (summarized in F) are indicated by yellow dots.
Table 1.
Changes in DUX4-dependent gene expression following targeting of dSaCas9-repressors to DUX4
| dSaCas9-TRD | dSaCas9-SET | dSaCas9-HP1γ | dSaCas9-HP1α | |
|---|---|---|---|---|
| KREMEN2 | 1.33 | 1.57 | 1.64 | 1.26 |
| FRAS1 | 1.08 | 1.15 | 1.05 | NS |
| TRIM14 | 1.05 | 1.10 | 1.16 | 1.03 |
| FRZB | NS | NS | 1.02 | 1.09 |
| COL9A2 | NS | NS | 1.15 | NS |
| TYMP | 1.53 | 1.63 | 1.70 | 1.30 |
| CMPK2 | 4.47 | 4.80 | 4.82 | 4.12 |
| SPTBN5 | 1.14 | 1.13 | 1.23 | 1.02 |
| GRIA1 | 1.09 | 1.18 | 1.39 | 1.33 |
| TPPP3 | 1.07 | 1.28 | 1.20 | 1.01 |
| LY6E | 1.69 | 1.96 | 1.92 | 1.54 |
| PARP14 | 1.11 | 1.33 | 1.21 | 1.05 |
| ACSM5 | 1.13 | 1.45 | 1.35 | NS |
| PRELP | 1.01 | 1.02 | 1.05 | NS |
| TM6SF1 | −1.10 | −1.35 | −1.16 | −1.22 |
| ITGA8 | −1.24 | −1.36 | −1.37 | −1.19 |
| COL10A1 | NS | −1.02 | NS | −1.13 |
Shown are log2 fold changes in DUX4 target genes,61, 62, 63, 64, 65 whose expression in FSHD myocytes is altered following transduction with each dSaCas9-repressor + DUX4-targeting sgRNA. These genes are part of developmental pathways dysregulated by DUX4, and their differential expression following CRISPRi treatment represents a return to a more normal pattern of gene expression. NS, not significant.
Expression levels of myogenic genes that were assessed by qRT-PCR (MYOD1, MYOG, and MYH1) also showed no changes by RNA-seq analysis (Table S4). The only muscle genes with differential expression are CKM, which is increased ∼2-fold following treatment with dSaCas9-SET, -HP1γ, or -TRD, an antisense transcript of MEF2C, and MYBPC2, which are increased ∼2-fold following treatment with dSaCas9-SET (Table S4). Since DUX4 expression is reported to inhibit myogenesis, these changes also likely represent a beneficial correction of DUX4-mediated transcriptional dysregulation.
Importantly, the number of detectable off-target responses to each treatment was extremely low. Treatment with dSaCas9-TRD or -HP1α yielded no significant unique DEGs, while treatment with dSaCas9-SET and -HP1γ yielded only 7 and 8 unique DEGs, respectively (Figure 4; Table S3). Thus, as predicted by the in silico search of sgRNA targets, our system for CRISPRi is highly specific in human myocytes. In contrast, treatment with dSaCas9-KRAB, which was targeted using the same sgRNA (#6) as dSaCas9-TRD, yielded 37 unique DEGs (versus 0 for dSaCas9-TRD). This result was surprising, considering the high specificity reported for dSpCas9-KRAB;66 however, it suggests that, at least in myocytes, the KRAB repressor is recruited to genomic locations independent of sgRNA targeting and is a more promiscuous repressor than the MeCP2 TRD.
Targeting dSaCas9-repressors to DUX4 increases chromatin repression at the locus
Since targeting each epigenetic repressor to the DUX4 promoter/exon 1 reduces levels of DUX4-fl, we expected that each would mediate direct changes in chromatin structure at the locus. Using ChIP assays, we analyzed several marks of repressive chromatin across D4Z4 following CRISPRi treatment in FSHD myocytes. Increases in repressive marks are difficult to assess across the FSHD1 DUX4 locus, because three of the four 4q/10q alleles, which are comprised of many more copies of D4Z4, are already in a compacted, heterochromatic state. Thus, any attempt to assess an increase in repression at the contracted allele is dampened by the presence of the other three. Unsurprisingly, changes in overall levels of the repressive H3K9me3 histone mark were undetectable across the D4Z4 repeats; however, other repressive marks were detectably and significantly elevated, overcoming the high background. Recruitment of HP1α to DUX4 led to a ∼30%–40% enrichment of this factor across the locus, as well as increased occupancy of the KAP1 co-repressor (Figures 5A and 5B; Figures S10A and S10B). Recruitment of HP1γ led to an increase in both HP1α and KAP1 at DUX4 exon 3, and recruitment of the MeCP2 TRD led to an increase in HP1α across the locus (Figures 5A and 5B; Figures S10A and S10B). Recruitment of each of the four factors also led to a ∼40%–60% decrease in the elongating form of RNA Pol II (phospho-serine 2) at the pathogenic repeat (Figure 5C; Figure S10C), consistent with the lower levels of DUX4-fl mRNA observed (Figure 2). Taken together, these results indicate that treatment with dSaCas9-repressors returns the chromatin at the disease locus to a more normal state of repression.
Figure 5.

Targeting dSaCas9-repressors to DUX4 increases chromatin repression at the locus
ChIP assays were performed using FSHD myocytes transduced with each dSaCas9-epigenetic regulator + individual optimal sgRNA targeting the DUX4 promoter or exon 1. (A–C) Chromatin was immunoprecipitated using antibodies specific for (A) HP1α or (B) KAP1 and analyzed by qPCR using primers to the promoter (Pro), transcription start site (TSS), or exon 3 of DUX4 or to MYOD1, or (C) antibodies specific for the elongating form of RNA Pol II (phospho-serine 2) and analyzed by qPCR using primers specific to DUX4 exon1/intron1 on chromosome 4 or to MYOD1. MYOD1 was used as a negative control for an active gene that should not be affected by CRISPRi targeted to DUX4. Locations of DUX4 primers are shown in Figure 1C. Data are presented as fold enrichment of the target region by each specific antibody normalized to α-histone H3, with enrichment for mock-infected cells set to 1. For all panels, each bar represents the average of at least three independent ChIP experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 are from comparing to enrichment at MYOD1.
The FSHD-optimized regulatory cassette is only active in skeletal muscles
Following development of our optimized cassette, it was important to confirm that our smaller CKM-based regulatory cassette retains high activity in skeletal muscles with low to no activity in other tissues. Thus, in vivo expression of the FSHD-optimized regulatory cassette was analyzed using AAV9-mediated transgene delivery to wild-type mice. Viral particles were delivered by systemic retro-orbital injection (2.8 × 1014 genome copy [GC]/kg body weight), and the mCherry reporter signal was visualized at 12 weeks post-injection. As previously reported,67 AAV9 strongly transduced skeletal muscles, cardiac muscle, and liver (Figure S11). Nonetheless, mCherry expression was detected only in skeletal muscles and was undetectable in the heart (Figure 6), even at a higher exposure (Figure S12), and in non-muscle tissues (Figure S13), indicating that our FSHD-optimized regulatory cassette is only active in the critical target tissue. The lack of tropism/activity in testis is particularly important, because DUX4 is normally expressed in this tissue in healthy individuals.10,68
Figure 6.

The FSHD-optimized regulatory cassette is active in skeletal muscles
In vivo AAV9-mediated mCherry expression under control of the FSHD-optimized regulatory cassette (A), (Figure 1B) was visualized in the indicated tissues at 12 weeks post-injection with the same exposure time (0.5 s). (B–H and L) For two-tissue panels, tissues from uninjected mice are shown on the left. (I’–K) Single-tissue panels are AAV injected, and (I) is uninjected. All injected tissues are indicated by an asterisk. Expression of mCherry was detected in skeletal muscles (tibialis anterior [TA], gastrocnemius [GA], extensor digitorum longus [EDL], and quadriceps [QUA], as well as pectoral, abdominal, and facial muscles) and was undetectable in soleus (SOL), heart, and testis.
In vivo targeting of dSaCas9-repressors to DUX4 exon 1 represses DUX4-fl and DUX4-FL targets in ACTA1-MCM;FLExDUX4 bi-transgenic mice
To test the ability of our CRISPRi platform to repress DUX4-fl in vivo, we utilized our lab’s ACTA1-MCM;FLExDUX4 (FLExD) FSHD-like bi-transgenic mouse model, which can be induced to express DUX4-fl and develop a moderate pathology in response to a low dose of tamoxifen.69,70 These mice carry one human D4Z4 repeat from which DUX4-fl is expressed and can be targeted by our sgRNAs to exon 1. Mice were injected intramuscularly with AAV9 vectors encoding dSaCas9-TRD or -KRAB and sgRNAs targeting DUX4 exon 1 at different ratios, followed 3.5 weeks later by intraperitoneal injection of tamoxifen to induce mosaic DUX4-fl expression in skeletal muscles. Two weeks post-induction, expression of DUX4-fl and the mouse homologs of two direct target genes that are robustly induced by DUX4-FL were assessed by qRT-PCR in the injected TAs. Although transcript levels of the DUX4-fl transgene are difficult to assess in this model,69,70 targeting either dSaCas9-TRD or -KRAB to DUX4 exon 1 led to a ∼30% decrease in expression of DUX4-fl at the higher ratio of sgRNA to effector (Figure 7). Transcript levels of DUX4-FL targets Wfdc3 and Slc34a2 were also reduced, although the reduction was only significant for dSaCas9-TRD at the lower ratio of sgRNA to effector (Figure 7). Although these effects are modest, they provide proof of principle that our epigenetic CRISPRi platform is a viable strategy for ongoing preclinical development.
Figure 7.

In vivo targeting of dSaCas9-repressors to DUX4 exon 1 represses DUX4-fl and DUX4-FL targets in ACTA1-MCM;FLExD bi-transgenic mice
(A–F) dSaCas9-TRD or -KRAB ± sgRNAs were delivered intramuscularly using AAV9 to the ACTA1-MCM;FLExD moderate pathology FSHD-like transgenic mouse model, which carries one human D4Z4 repeat.70 Expression of DUX4-fl and DUX4-FL downstream markers Wfdc3 and Slc34a2 were assessed by qRT-PCR and normalized to levels of Rpl37. Copy-number ratios of dSaCas9-TRD or -KRAB to sgRNA are indicated. ∗p < 0.05, ∗∗p < 0.01 are from comparing to dSaCas9-TRD or -KRAB control.
Discussion
CRISPR/Cas9 technology has been used extensively to target and modify specific genomic regions, offering the potential for permanent correction of many diseases.15,71, 72, 73 While the dangers associated with standard CRISPR editing are a concern for any locus, they are of particular concern in a highly repetitive region such as the FSHD locus. However, the use of CRISPR to repress gene expression is ideally suited to FSHD. Unfortunately, CRISPRi platforms for human gene therapy are limited by the large size of Cas9 targeting proteins, which take up most of the available space in AAV vectors, leaving little room for effectors. Not surprisingly, most proof-of-principle studies have utilized dSpCas9 in LV vectors, which have a larger genome capacity and are convenient for expression in cultured cells but not useful for clinical gene delivery. The smaller dSaCas9 ortholog has been shown to work well with a fused effector,74 but its coding sequence is still over 3 kb, leaving little room for a chromatin modulator and regulatory sequences within the 4.4 kb packaging capacity of AAVs. It is worth emphasizing that the packaging limitation of AAV vectors continues to be a major hurdle for gene therapy of FSHD and many other diseases. To bring a CRISPRi platform for FSHD to the clinic, it was imperative to find stable repressors small enough to be included in dCas9 therapeutic cassettes and to reduce the size of current muscle-specific regulatory cassettes.
Studies from many labs, including ours, have utilized dCas9-KRAB to repress target genes; however, repression mediated by this effector requires its continuous expression. While dCas9-effectors may be continuously expressed from stable episomal AAV vectors, this is not guaranteed. From a clinical standpoint, it seems far more desirable to achieve stable repression that does not rely on continuous, lifelong expression of the transgene. Thus, we minimized the widely used CK8 cassette39 while maintaining high activity and specificity for skeletal muscles and used this FSHD-optimized cassette to drive expression of dSaCas9 fused to each of four small epigenetic repressors capable of mediating stable silencing. Here we demonstrate that dSaCas9-mediated targeting of these repressors returns the chromatin at the FSHD locus to a more normal state of repression and reduces expression of DUX4-fl and its targets in FSHD myocytes and in a DUX4-based transgenic mouse model, with minimal effects on the muscle transcriptome.
We observed more robust repression of DUX4-fl and its targets in primary FSHD myocytes than in ACTA1-MCM;FLExD bi-transgenic mice, likely due to limitations of the mouse model, which contains only a single D4Z4 repeat that may not be enough for efficient epigenetic silencing. Thus, ongoing studies will also test our CRISPRi platform in a human xenograft model containing mature FSHD myofibers.75,76 These mice are immunocompromised and, thus, not useful for assessing the effects of CRISPRi on DUX4-mediated immune pathologies. However, because they contain a full D4Z4 array from an FSHD patient, a xenograft model may be ideal for assessing long-term epigenetic changes at the disease locus. Determining the stability of DUX4 repression mediated by CRISPRi is a critical goal, since current AAV vectors for gene therapy can only be administered once.
A major concern of Cas9 editing is the potential for off-target cutting leading to deleterious mutations, something that was not considered to be a problem for dCas9 effectors. However, it was recently demonstrated in yeast that R-loops formed by dCas9 binding to DNA can cause mutagenesis at both on- and off-target sites,77 although the frequency is several orders of magnitude lower than that induced by Cas9. Consistent with this very low rate, mutations induced by dCas9 have not been detected in mammalian cells.78 Additionally, this concern is ameliorated when targeting the D4Z4 region, which is normally silent. Fortunately, for CRISPRi of FSHD, both the nature of the targeted region and the type of modulation employed tend to mitigate the general concerns related to CRISPR platforms.
As CRISPR and other gene-targeting systems continue to evolve, it is important that the results of this study can be adapted to a changing platform. The identification of sgRNAs that successfully target the DUX4 locus with minimal off-target effects should prove useful with engineered Cas9 variants and dCas9 fused to other effectors. In addition, we have identified the DUX4 promoter and exon 1 as targets for epigenetic modulation, and these regions contain numerous sgRNA targets compatible with different orthologs of Cas9. Once these orthologs are better characterized, smaller and less immunogenic versions should become available, rendering fusions with larger epigenetic regulators more amenable to in vivo delivery. We are currently making further improvements in vector and cassette design that allow all therapeutic components to be included on single AAV vectors, a clinically important milestone for improving efficiency, lowering the high cost of therapy, and reducing the immunotoxicity associated with high viral doses.
Here we demonstrate the successful use of dCas9-mediated epigenetic repression in a muscle disorder, thus laying the groundwork for subsequent, ongoing studies to assess the functional efficacy and stability of this approach in vivo. Ultimately, we are attempting to correct the underlying pathogenic mechanism in FSHD using a therapeutically relevant platform. In addition, our successful use of dCas9-based chromatin effectors should be applicable to other diseases of aberrant gene regulation.
Materials and methods
Study design
The primary goal of this study was to design and test a CRISPRi platform that will ultimately allow therapeutic delivery of epigenetic repressors capable of mediating stable repression of DUX4 in FSHD patients. Thus, we designed our therapeutic cassettes (minimized skeletal muscle-specific regulatory sequences driving dSaCas9 fused to different epigenetic repressors) for use in AAV gene therapy vectors. Two experimental systems were used: primary FSHD myocytes, which contain endogenous patient D4Z4 arrays, and ACTA1-MCM;FLExD bi-transgenic mice,69,70 which can be induced to express DUX4-fl at controllable levels in skeletal muscles and exhibit pathologies consistent with FSHD. For experiments in primary cell culture, therapeutic cassettes were delivered via LV, but all in vivo experiments utilized therapeutic cassettes in AAV9 for efficient delivery to skeletal muscles. For in vivo experiments, we tested increasing ratios of sgRNA to dSaCas9-repressor, since dual-vector CRISPR editing has been shown to be more effective using a higher ratio of sgRNA to Cas9.79 Since our AAV-sgRNA contains three sgRNAs in one vector, we delivered AAV-dSaCas9-repressor:AAV-sgRNA at ratios of 1:0, 1:1, and 1:4, which is equivalent to dSaCas9:sgRNA copy number ratios of 1:0, 1:3, and 1:12. All experiments (biological replicates) in primary cells were performed at least four times (for gene expression analysis by qRT-PCR), at least three times (for ChIP analysis), and five times (for global transcriptome analysis by RNA-seq). AAV transductions of dSaCas9-TRD or -KRAB + sgRNAs utilized nine mice for each dSaCas9-repressor.
Antibodies and plasmids
The ChIP-grade antibodies used in this study, α-KAP1 (ab3831), α-HP1α (ab77256), α-RNA Pol II CTD repeat (phospho S2) (ab5095), and α-histone H3 (ab1791) were purchased from Abcam (Cambridge, MA, USA). dSaCas9 constructs were designed with a skeletal muscle-specific regulatory cassette consisting of three modified CKM enhancers in tandem, upstream of a modified CKM promoter. Enhancer modifications are as follows: (1) Left E-box mutated to Right E-box,80 (2) enhancer CArG and AP2 sites removed, (3) 63 bp between Right E-box and MEF2 site removed,39 (4) sequence between TF binding motifs minimized, (5) +1 to +50 promoter sequence used,39 and (6) consensus Inr site added.39 The sequence is available upon request. This regulatory cassette was designed upstream of the SV40 bipartite nuclear localization signal flanking dSaCas9, which was fused in-frame to one of four epigenetic repressors (the SUV39H1 pre-SET, SET, and post-SET domains, the MeCP2 TRD, HP1α, or HP1γ) and HA tag, followed by the SV40 late pA signal. sgRNA constructs were designed with the U6 promoter upstream of BfuAI cloning sites, followed by an SaCas9-optimized scaffold and cPPT/CTS. Constructs were synthesized in pUC57 and sequenced by GenScript Biotech, then cloned into a pRRLSIN LV vector for transduction of primary FSHD myocytes. pRRLSIN.cPPT.PGK-GFP.WPRE was a gift from Didier Trono (Addgene plasmid #12252 ; http://addgene.org/12252; RRID:Addgene_12252). For evaluation of activity in vivo, the FSHD-optimized regulatory cassette driving mCherry (Figure 6, top) was cloned between the AAV2 ITRs (using MluI and RsrII) of the pAAV-CA plasmid,81 a gift from Naoshige Uchida (Addgene plasmid #69616; http://addgene.org/69616; RRID:Addgene_69616). All AAV9 vectors used this study were produced by Vector Biolabs (Malvern, PA, USA).
sgRNA design and plasmid construction
We used the publicly available sgRNA design tool from the Broad Institute (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design) to design dSaCas9-compatible sgRNAs targeting across the DUX4 locus. sgRNAs were cloned individually into BfuAI sites in the parent construct and sequence verified. We used the publicly available Cas-OFFinder tool (http://www.rgenome.net/cas-offinder/) to search for the closest-matching off-target sequences in genes expressed in skeletal muscle. Refer to Table S1 for additional details.
Cell culture, transient transfection, and LV transduction
Myogenic cells derived from biceps muscle of an FSHD1 patient (17Abic) were obtained from the Wellstone FSHD cell repository22,23 and grown as described.15 LV particles were generated using 293T packaging cells, as described.15 At ∼70%–80% confluency, 17Abic myoblasts were subjected to four serial infections, as described,15 and allowed to self-differentiate. Cells were harvested ∼72 h following the last round of infection.
qRT-PCR
Total RNAs were extracted using TRIzol (Invitrogen) and purified using the RNeasy Mini kit (QIAGEN) after on-column DNase I digestion. From cultured myocytes, total RNA (2 μg) was used for cDNA synthesis using oligo dT and Superscript III Reverse Transcriptase (Invitrogen); DUX4-fl was amplified by qPCR using 200 ng of cDNA, and other genes were amplified using 10–20 ng of cDNA, as described.23 From mouse TAs, DUX4-fl was amplified using 100 ng of cDNA, and downstream targets were amplified using 5–20 ng of iScript cDNA. Oligonucleotide primer sequences are provided in Table S5.15,16,54,82, 83, 84, 85, 86
RNA-seq
FSHD myocytes (17Abic) were subjected to four serial co-infections with LV supernatants expressing dSaCas9 fused to either: (1) the SUV39H1 pre-SET, SET, and post-SET domains (SET); (2) the MeCP2 TRD; (3) HP1γ; (4) HP1α; or (5) the KRAB TRD, each in combination with LV expressing an sgRNA targeting DUX4. Cells were harvested ∼72 h following the last round of infection. For all treatments, five separate experiments were performed, and reduction of DUX4-fl and DUX4-FL targets was confirmed by qRT-PCR prior to submitting samples for sequencing. RNA-seq analysis was performed by GeneWiz using the Illumina HiSeq 2 × 100 bp platform, which is ideal for identifying gene expression levels, splice variant expression, and the de novo transcriptome assembly (including un-annotated sequences). The rRNA depletion, library construction, sequencing, and initial analysis (mapping all sequence reads to the human genome, reading hit count measurements, and differential gene expression comparisons) were performed by GeneWiz. Sequence reads were trimmed to remove possible adaptor sequences and nucleotides with poor quality using Trimmomatic v.0.36. The trimmed reads were mapped to the Homo sapiens GRCh38 reference genome available on ENSEMBL using the STAR aligner v.2.5.2b. The STAR aligner is a splice aligner that detects splice junctions and incorporates them to help align the entire read sequences. Unique gene hit counts were calculated by using featureCounts from the Subread package v.1.5.2. Only unique reads that fell within exon regions were counted. Since a strand-specific library preparation was performed, the reads were strand-specifically counted. Comparisons of gene expression between groups of samples were performed using DESeq2, described below. A GO analysis was performed on the statistically significant set of genes by implementing the software GeneSCF v.1.1-p2. The goa_human GO list was used to cluster the set of genes based on their biological processes and determine their statistical significance. To estimate the expression levels of alternatively spliced transcripts, the splice variant hit counts were extracted from the RNA-seq reads mapped to the genome. Differentially spliced genes were identified for groups with more than one sample by testing for significant differences in read counts on exons (and junctions) of the genes using DEXSeq. Volcano plots of differentially expressed genes were generated using Prism 7 (GraphPad).
ChIP
ChIP assays were performed with LV-transduced 17Abic differentiated myocytes using the Fast ChIP method87 as described.15 Chromatin was immunoprecipitated using 2 μg of specific antibodies. SYBR green quantitative PCR assays were performed as described.15 Oligonucleotide primer sequences are provided in Table S5.
In vivo characterization of FSHD-optimized regulatory cassette
All animal experiments were approved by the institutional Animal Care and Use Committee of the University of Nevada, Reno. All mice were anesthetized prior to injection. Systemic AAV9-mediated delivery of the FSHD-optimized regulatory cassette driving mCherry (100 μL of 3.2 × e13 GC/mL) was performed via retro-orbital injection (ROI) to wild-type C57BL/6J mice (2 males and 2 females) at 3.5 weeks of age with an average dose of 2.8 × 1011 GC/g body weight. At 12 weeks post-ROI, mCherry signals in all tissues were captured with Leica MZ9.5/DFC-7000T fluorescent imaging system and Leica LAS X software, using the same exposure unless indicated. Images were assembled with Adobe Photoshop CS6, and exposures were adjusted equally. For assessment of systemic AAV9-mediated infection, genomic DNA was isolated from individual tissues, and viral genomes were quantified by qPCR (50 ng genomic DNA) using primers to the bovine growth hormone polyadenylation signal (PAS) and normalized to the endogenous single copy Rosa26 gene. Oligonucleotide primer sequences are provided in Table S5.
AAV transduction of dSaCas9-TRD or -KRAB in ACTA1-MCM;FLExD bi-transgenic mice
Tibialis anterior (TA) muscles of 4-week-old male ACTA1-MCM;FLExD bi-transgenic animals were injected with various ratios of AAV9-dSaCas9-TRD or -KRAB and AAV9-sgRNAs. AAV-dSaCas9-TRD or -KRAB was injected at 5 × 105 GC/TA for all experiments. At 3.5 weeks post-AAV injection, mice were subjected to intraperitoneal injections of 5 mg/kg of tamoxifen (TMX) to induce DUX4-fl expression in skeletal muscles. TA muscles were sampled at 14 days post-TMX injection for gene expression analysis.
Statistical analysis
Experiments in primary cells were performed using at least four biological replicates (for qRT-PCR analysis) and at least three biological replicates (for ChIP analysis), and data were analyzed using an unpaired, two-tailed Student’s t test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). RNA-seq analysis was performed by GeneWiz using five biological replicates, and comparisons of gene expression between groups of samples were performed using DESeq2. The Wald test was used to generate p values and log2 fold changes. Genes with a p value < 0.05 and absolute log2 fold change > 1 were called as DEGs for each comparison. Enrichment of GO terms was tested using Fisher exact test (GeneSCF v1.1-p2). Significantly enriched GO terms had an adjusted p value <0.05 in the differentially expressed gene sets. For AAV transductions in mice, gene expression was analyzed using an unpaired, two-tailed Student’s t test.
Acknowledgments
We thank Ms. Monique Ramirez and Dr. Andreia Marcelino Nunes for help with mouse experiments, and Dr. Jeffrey B. Miller for critical reading of the manuscript. This work was funded by an award from the FSHD Canada Foundation to C.L.H., an award from the FSHD Global Research Foundation to P.L.J., a grant from the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (award number R01AR062587) to P.L.J., Chip and Shannon Wilson, and the William R. Lewis Family.
Author contributions
C.L.H., T.I.J., and P.L.J. designed the study. C.L.H. and P.L.J. wrote the manuscript. C.L.H. designed therapeutic cassettes, performed all experiments using FSHD myocytes, and analyzed data. T.I.J. performed all experiments using mice and analyzed data. All authors edited and approved the final version of the manuscript.
Declaration of interests
C.L.H., P.L.J., and T.I.J. are listed as inventors on two US patent applications (nos. 62/398801 and 63/011476) pertaining to the use of CRISPR inhibition for FSHD.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.12.001.
Supplemental information
References
- 1.Padberg G.W. Leiden University; 1982. Facioscapulohumeral Disease. Thesis; p. 243. [Google Scholar]
- 2.Orphanet . 2018. Prevalence and incidence of rare diseases: Bibliographic data.https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf [Google Scholar]
- 3.van Overveld P.G., Lemmers R.J., Sandkuijl L.A., Enthoven L., Winokur S.T., Bakels F., Padberg G.W., van Ommen G.J., Frants R.R., van der Maarel S.M. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 2003;35:315–317. doi: 10.1038/ng1262. [DOI] [PubMed] [Google Scholar]
- 4.Himeda C.L., Jones P.L. The Genetics and Epigenetics of Facioscapulohumeral Muscular Dystrophy. Annu. Rev. Genomics Hum. Genet. 2019;20:265–291. doi: 10.1146/annurev-genom-083118-014933. [DOI] [PubMed] [Google Scholar]
- 5.Wijmenga C., Frants R.R., Brouwer O.F., Moerer P., Weber J.L., Padberg G.W. Location of facioscapulohumeral muscular dystrophy gene on chromosome 4. Lancet. 1990;336:651–653. doi: 10.1016/0140-6736(90)92148-b. [DOI] [PubMed] [Google Scholar]
- 6.Wijmenga C., Hewitt J.E., Sandkuijl L.A., Clark L.N., Wright T.J., Dauwerse H.G., Gruter A.M., Hofker M.H., Moerer P., Williamson R. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat. Genet. 1992;2:26–30. doi: 10.1038/ng0992-26. [DOI] [PubMed] [Google Scholar]
- 7.van Deutekom J.C., Wijmenga C., van Tienhoven E.A., Gruter A.M., Hewitt J.E., Padberg G.W., van Ommen G.J., Hofker M.H., Frants R.R. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 1993;2:2037–2042. doi: 10.1093/hmg/2.12.2037. [DOI] [PubMed] [Google Scholar]
- 8.Lemmers R.J., Tawil R., Petek L.M., Balog J., Block G.J., Santen G.W., Amell A.M., van der Vliet P.J., Almomani R., Straasheijm K.R. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012;44:1370–1374. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van den Boogaard M.L., Lemmers R.J.L.F., Balog J., Wohlgemuth M., Auranen M., Mitsuhashi S., van der Vliet P.J., Straasheijm K.R., van den Akker R.F.P., Kriek M. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016;98:1020–1029. doi: 10.1016/j.ajhg.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snider L., Geng L.N., Lemmers R.J., Kyba M., Ware C.B., Nelson A.M., Tawil R., Filippova G.N., van der Maarel S.M., Tapscott S.J., Miller D.G. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6:e1001181. doi: 10.1371/journal.pgen.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tassin A., Laoudj-Chenivesse D., Vanderplanck C., Barro M., Charron S., Ansseau E., Chen Y.W., Mercier J., Coppée F., Belayew A. DUX4 expression in FSHD muscle cells: how could such a rare protein cause a myopathy? J. Cell. Mol. Med. 2013;17:76–89. doi: 10.1111/j.1582-4934.2012.01647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campbell A.E., Belleville A.E., Resnick R., Shadle S.C., Tapscott S.J. Facioscapulohumeral dystrophy: activating an early embryonic transcriptional program in human skeletal muscle. Hum. Mol. Genet. 2018;27(R2):R153–R162. doi: 10.1093/hmg/ddy162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lemmers R.J., van der Vliet P.J., Klooster R., Sacconi S., Camaño P., Dauwerse J.G., Snider L., Straasheijm K.R., van Ommen G.J., Padberg G.W. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao Z., Snider L., Balog J., Lemmers R.J., Van Der Maarel S.M., Tawil R., Tapscott S.J. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum. Mol. Genet. 2014;23:5342–5352. doi: 10.1093/hmg/ddu251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Himeda C.L., Jones T.I., Jones P.L. CRISPR/dCas9-mediated Transcriptional Inhibition Ameliorates the Epigenetic Dysregulation at D4Z4 and Represses DUX4-fl in FSH Muscular Dystrophy. Mol. Ther. 2016;24:527–535. doi: 10.1038/mt.2015.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Himeda C.L., Jones T.I., Virbasius C.M., Zhu L.J., Green M.R., Jones P.L. Identification of Epigenetic Regulators of DUX4-fl for Targeted Therapy of Facioscapulohumeral Muscular Dystrophy. Mol. Ther. 2018;26:1797–1807. doi: 10.1016/j.ymthe.2018.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wallace L.M., Liu J., Domire J.S., Garwick-Coppens S.E., Guckes S.M., Mendell J.R., Flanigan K.M., Harper S.Q. RNA interference inhibits DUX4-induced muscle toxicity in vivo: implications for a targeted FSHD therapy. Mol. Ther. 2012;20:1417–1423. doi: 10.1038/mt.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marsollier A.C., Ciszewski L., Mariot V., Popplewell L., Voit T., Dickson G., Dumonceaux J. Antisense targeting of 3′ end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: a new gene-silencing approach. Hum. Mol. Genet. 2016;25:1468–1478. doi: 10.1093/hmg/ddw015. [DOI] [PubMed] [Google Scholar]
- 19.Choi S.H., Bosnakovski D., Strasser J.M., Toso E.A., Walters M.A., Kyba M. Transcriptional Inhibitors Identified in a 160,000-Compound Small-Molecule DUX4 Viability Screen. J. Biomol. Screen. 2016;21:680–688. doi: 10.1177/1087057116651868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cruz J.M., Jr., Hupper N., Wilson L.S., Concannon J.B., Wang Y., Oberhauser B., Patora-Komisarska K., Zhang Y., Glass D.J., Trendelenburg A.U., Clarke B.A. Protein kinase A activation inhibits DUX4 gene expression in myotubes from patients with facioscapulohumeral muscular dystrophy. J. Biol. Chem. 2018;293:11837–11849. doi: 10.1074/jbc.RA118.002633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campbell A.E., Oliva J., Yates M.P., Zhong J.W., Shadle S.C., Snider L., Singh N., Tai S., Hiramuki Y., Tawil R. BET bromodomain inhibitors and agonists of the beta-2 adrenergic receptor identified in screens for compounds that inhibit DUX4 expression in FSHD muscle cells. Skelet. Muscle. 2017;7:16. doi: 10.1186/s13395-017-0134-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones T.I., Chen J.C., Rahimov F., Homma S., Arashiro P., Beermann M.L., King O.D., Miller J.B., Kunkel L.M., Emerson C.P., Jr. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis. Hum. Mol. Genet. 2012;21:4419–4430. doi: 10.1093/hmg/dds284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones T.I., King O.D., Himeda C.L., Homma S., Chen J.C., Beermann M.L., Yan C., Emerson C.P., Jr., Miller J.B., Wagner K.R., Jones P.L. Individual epigenetic status of the pathogenic D4Z4 macrosatellite correlates with disease in facioscapulohumeral muscular dystrophy. Clin. Epigenetics. 2015;7:37. doi: 10.1186/s13148-015-0072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L.H., Friedman S.D., Shaw D., Snider L., Wong C.J., Budech C.B., Poliachik S.L., Gove N.E., Lewis L.M., Campbell A.E. MRI-informed muscle biopsies correlate MRI with pathology and DUX4 target gene expression in FSHD. Hum. Mol. Genet. 2019;28:476–486. doi: 10.1093/hmg/ddy364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oliva J., Galasinski S., Richey A., Campbell A.E., Meyers M.J., Modi N., Zhong J.W., Tawil R., Tapscott S.J., Sverdrup F.M. Clinically Advanced p38 Inhibitors Suppress DUX4 Expression in Cellular and Animal Models of Facioscapulohumeral Muscular Dystrophy. J. Pharmacol. Exp. Ther. 2019;370:219–230. doi: 10.1124/jpet.119.259663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rojas L.A., Valentine E., Accorsi A., Maglio J., Shen N., Robertson A., Kazmirski S., Rahl P., Tawil R., Cadavid D. p38α Regulates Expression of DUX4 in a Model of Facioscapulohumeral Muscular Dystrophy. J. Pharmacol. Exp. Ther. 2020;374:489–498. doi: 10.1124/jpet.119.264689. [DOI] [PubMed] [Google Scholar]
- 27.Segalés J., Perdiguero E., Muñoz-Cánoves P. Regulation of Muscle Stem Cell Functions: A Focus on the p38 MAPK Signaling Pathway. Front. Cell Dev. Biol. 2016;4:91. doi: 10.3389/fcell.2016.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darrow J.J., Sharma M., Shroff M., Wagner A.K. Efficacy and costs of spinal muscular atrophy drugs. Sci. Transl. Med. 2020;12:eaay9648. doi: 10.1126/scitranslmed.aay9648. [DOI] [PubMed] [Google Scholar]
- 29.Zeng W., de Greef J.C., Chen Y.Y., Chien R., Kong X., Gregson H.C., Winokur S.T., Pyle A., Robertson K.D., Schmiesing J.A. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD) PLoS Genet. 2009;5:e1000559. doi: 10.1371/journal.pgen.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balog J., Thijssen P.E., de Greef J.C., Shah B., van Engelen B.G., Yokomori K., Tapscott S.J., Tawil R., van der Maarel S.M. Correlation analysis of clinical parameters with epigenetic modifications in the DUX4 promoter in FSHD. Epigenetics. 2012;7:579–584. doi: 10.4161/epi.20001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Overveld P.G., Enthoven L., Ricci E., Rossi M., Felicetti L., Jeanpierre M., Winokur S.T., Frants R.R., Padberg G.W., van der Maarel S.M. Variable hypomethylation of D4Z4 in facioscapulohumeral muscular dystrophy. Ann. Neurol. 2005;58:569–576. doi: 10.1002/ana.20625. [DOI] [PubMed] [Google Scholar]
- 32.de Greef J.C., Lemmers R.J., van Engelen B.G., Sacconi S., Venance S.L., Frants R.R., Tawil R., van der Maarel S.M. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum. Mutat. 2009;30:1449–1459. doi: 10.1002/humu.21091. [DOI] [PubMed] [Google Scholar]
- 33.Jones T.I., Yan C., Sapp P.C., McKenna-Yasek D., Kang P.B., Quinn C., Salameh J.S., King O.D., Jones P.L. Identifying diagnostic DNA methylation profiles for facioscapulohumeral muscular dystrophy in blood and saliva using bisulfite sequencing. Clin. Epigenetics. 2014;6:23. doi: 10.1186/1868-7083-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lemmers R.J., Goeman J.J., van der Vliet P.J., van Nieuwenhuizen M.P., Balog J., Vos-Versteeg M., Camano P., Ramos Arroyo M.A., Jerico I., Rogers M.T. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum. Mol. Genet. 2015;24:659–669. doi: 10.1093/hmg/ddu486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brezgin S., Kostyusheva A., Kostyushev D., Chulanov V. Dead Cas Systems: Types, Principles, and Applications. Int. J. Mol. Sci. 2019;20:6041. doi: 10.3390/ijms20236041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amabile A., Migliara A., Capasso P., Biffi M., Cittaro D., Naldini L., Lombardo A. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell. 2016;167:219–232.e14. doi: 10.1016/j.cell.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Geen H., Ren C., Nicolet C.M., Perez A.A., Halmai J., Le V.M., Mackay J.P., Farnham P.J., Segal D.J. dCas9-based epigenome editing suggests acquisition of histone methylation is not sufficient for target gene repression. Nucleic Acids Res. 2017;45:9901–9916. doi: 10.1093/nar/gkx578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tarjan D.R., Flavahan W.A., Bernstein B.E. Epigenome editing strategies for the functional annotation of CTCF insulators. Nat. Commun. 2019;10:4258. doi: 10.1038/s41467-019-12166-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salva M.Z., Himeda C.L., Tai P.W., Nishiuchi E., Gregorevic P., Allen J.M., Finn E.E., Nguyen Q.G., Blankinship M.J., Meuse L. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol. Ther. 2007;15:320–329. doi: 10.1038/sj.mt.6300027. [DOI] [PubMed] [Google Scholar]
- 40.Himeda C.L., Chen X., Hauschka S.D. Design and testing of regulatory cassettes for optimal activity in skeletal and cardiac muscles. Methods Mol. Biol. 2011;709:3–19. doi: 10.1007/978-1-61737-982-6_1. [DOI] [PubMed] [Google Scholar]
- 41.Tabebordbar M., Zhu K., Cheng J.K.W., Chew W.L., Widrick J.J., Yan W.X., Maesner C., Wu E.Y., Xiao R., Ran F.A. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stepper P., Kungulovski G., Jurkowska R.Z., Chandra T., Krueger F., Reinhardt R., Reik W., Jeltsch A., Jurkowski T.P. Efficient targeted DNA methylation with chimeric dCas9-Dnmt3a-Dnmt3L methyltransferase. Nucleic Acids Res. 2017;45:1703–1713. doi: 10.1093/nar/gkw1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.James T.C., Elgin S.C. Identification of a nonhistone chromosomal protein associated with heterochromatin in Drosophila melanogaster and its gene. Mol. Cell. Biol. 1986;6:3862–3872. doi: 10.1128/mcb.6.11.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smothers J.F., Henikoff S. The hinge and chromo shadow domain impart distinct targeting of HP1-like proteins. Mol. Cell. Biol. 2001;21:2555–2569. doi: 10.1128/MCB.21.7.2555-2569.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Canzio D., Larson A., Narlikar G.J. Mechanisms of functional promiscuity by HP1 proteins. Trends Cell Biol. 2014;24:377–386. doi: 10.1016/j.tcb.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schotta G., Ebert A., Reuter G. SU(VAR)3-9 is a conserved key function in heterochromatic gene silencing. Genetica. 2003;117:149–158. doi: 10.1023/a:1022923508198. [DOI] [PubMed] [Google Scholar]
- 47.Jones P.L., Veenstra G.J., Wade P.A., Vermaak D., Kass S.U., Landsberger N., Strouboulis J., Wolffe A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 48.Nan X., Ng H.H., Johnson C.A., Laherty C.D., Turner B.M., Eisenman R.N., Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 49.Hite K.C., Adams V.H., Hansen J.C. Recent advances in MeCP2 structure and function. Biochem. Cell Biol. 2009;87:219–227. doi: 10.1139/o08-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trevisan C.P., Pastorello E., Armani M., Angelini C., Nante G., Tomelleri G., Tonin P., Mongini T., Palmucci L., Galluzzi G. Facioscapulohumeral muscular dystrophy and occurrence of heart arrhythmia. Eur. Neurol. 2006;56:1–5. doi: 10.1159/000094248. [DOI] [PubMed] [Google Scholar]
- 51.Galetta F., Franzoni F., Sposito R., Plantinga Y., Femia F.R., Galluzzi F., Rocchi A., Santoro G., Siciliano G. Subclinical cardiac involvement in patients with facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2005;15:403–408. doi: 10.1016/j.nmd.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 52.Amacher S.L., Buskin J.N., Hauschka S.D. Multiple regulatory elements contribute differentially to muscle creatine kinase enhancer activity in skeletal and cardiac muscle. Mol. Cell. Biol. 1993;13:2753–2764. doi: 10.1128/mcb.13.5.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Donoviel D.B., Shield M.A., Buskin J.N., Haugen H.S., Clegg C.H., Hauschka S.D. Analysis of muscle creatine kinase gene regulatory elements in skeletal and cardiac muscles of transgenic mice. Mol. Cell. Biol. 1996;16:1649–1658. doi: 10.1128/mcb.16.4.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Himeda C.L., Debarnot C., Homma S., Beermann M.L., Miller J.B., Jones P.L., Jones T.I. Myogenic enhancers regulate expression of the facioscapulohumeral muscular dystrophy-associated DUX4 gene. Mol. Cell. Biol. 2014;34:1942–1955. doi: 10.1128/MCB.00149-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Geng L.N., Yao Z., Snider L., Fong A.P., Cech J.N., Young J.M., van der Maarel S.M., Ruzzo W.L., Gentleman R.C., Tawil R., Tapscott S.J. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev. Cell. 2012;22:38–51. doi: 10.1016/j.devcel.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rea S., Eisenhaber F., O’Carroll D., Strahl B.D., Sun Z.W., Schmid M., Opravil S., Mechtler K., Ponting C.P., Allis C.D., Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 57.Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen J.S., Dagdas Y.S., Kleinstiver B.P., Welch M.M., Sousa A.A., Harrington L.B., Sternberg S.H., Joung J.K., Yildiz A., Doudna J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature. 2017;550:407–410. doi: 10.1038/nature24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hu J.H., Miller S.M., Geurts M.H., Tang W., Chen L., Sun N., Zeina C.M., Gao X., Rees H.A., Lin Z., Liu D.R. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556:57–63. doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tan Y., Chu A.H.Y., Bao S., Hoang D.A., Kebede F.T., Xiong W., Ji M., Shi J., Zheng Z. Rationally engineered Staphylococcus aureus Cas9 nucleases with high genome-wide specificity. Proc. Natl. Acad. Sci. USA. 2019;116:20969–20976. doi: 10.1073/pnas.1906843116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jagannathan S., Shadle S.C., Resnick R., Snider L., Tawil R.N., van der Maarel S.M., Bradley R.K., Tapscott S.J. Model systems of DUX4 expression recapitulate the transcriptional profile of FSHD cells. Hum. Mol. Genet. 2016;25:4419–4431. doi: 10.1093/hmg/ddw271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tanaka Y., Kawazu M., Yasuda T., Tamura M., Hayakawa F., Kojima S., Ueno T., Kiyoi H., Naoe T., Mano H. Transcriptional activities of DUX4 fusions in B-cell acute lymphoblastic leukemia. Haematologica. 2018;103:e522–e526. doi: 10.3324/haematol.2017.183152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Preussner J., Zhong J., Sreenivasan K., Gunther S., Engleitner T., Kunne C., Glatzel M., Rad R., Looso M., Braun T. Oncogenic Amplification of Zygotic Dux Factors in Regenerating p53-Deficient Muscle Stem Cells Defines a Molecular Cancer Subtype. Cell Stem Cell. 2018;23:794–805.e4. doi: 10.1016/j.stem.2018.10.011. [DOI] [PubMed] [Google Scholar]
- 64.Caron L., Kher D., Lee K.L., McKernan R., Dumevska B., Hidalgo A., Li J., Yang H., Main H., Ferri G. A Human Pluripotent Stem Cell Model of Facioscapulohumeral Muscular Dystrophy-Affected Skeletal Muscles. Stem Cells Transl. Med. 2016;5:1145–1161. doi: 10.5966/sctm.2015-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Okimoto R.A., Wu W., Nanjo S., Olivas V., Lin Y.K., Ponce R.K., Oyama R., Kondo T., Bivona T.G. CIC-DUX4 oncoprotein drives sarcoma metastasis and tumorigenesis via distinct regulatory programs. J. Clin. Invest. 2019;129:3401–3406. doi: 10.1172/JCI126366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thakore P.I., D’Ippolito A.M., Song L., Safi A., Shivakumar N.K., Kabadi A.M., Reddy T.E., Crawford G.E., Gersbach C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods. 2015;12:1143–1149. doi: 10.1038/nmeth.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Inagaki K., Fuess S., Storm T.A., Gibson G.A., Mctiernan C.F., Kay M.A., Nakai H. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther. 2006;14:45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Young J.M., Whiddon J.L., Yao Z., Kasinathan B., Snider L., Geng L.N., Balog J., Tawil R., van der Maarel S.M., Tapscott S.J. DUX4 binding to retroelements creates promoters that are active in FSHD muscle and testis. PLoS Genet. 2013;9:e1003947. doi: 10.1371/journal.pgen.1003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jones T., Jones P.L. A cre-inducible DUX4 transgenic mouse model for investigating facioscapulohumeral muscular dystrophy. PLoS ONE. 2018;13:e0192657. doi: 10.1371/journal.pone.0192657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jones T.I., Chew G.L., Barraza-Flores P., Schreier S., Ramirez M., Wuebbles R.D., Burkin D.J., Bradley R.K., Jones P.L. Transgenic mice expressing tunable levels of DUX4 develop characteristic facioscapulohumeral muscular dystrophy-like pathophysiology ranging in severity. Skelet. Muscle. 2020;10:8. doi: 10.1186/s13395-020-00227-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wright A.V., Nuñez J.K., Doudna J.A. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell. 2016;164:29–44. doi: 10.1016/j.cell.2015.12.035. [DOI] [PubMed] [Google Scholar]
- 72.Himeda C.L., Jones T.I., Jones P.L. Scalpel or Straitjacket: CRISPR/Cas9 Approaches for Muscular Dystrophies. Trends Pharmacol. Sci. 2016;37:249–251. doi: 10.1016/j.tips.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu X., Qi L.S. A CRISPR-dCas Toolbox for Genetic Engineering and Synthetic Biology. J. Mol. Biol. 2019;431:34–47. doi: 10.1016/j.jmb.2018.06.037. [DOI] [PubMed] [Google Scholar]
- 74.Josipović G., Zoldoš V., Vojta A. Active fusions of Cas9 orthologs. J. Biotechnol. 2019;301:18–23. doi: 10.1016/j.jbiotec.2019.05.306. [DOI] [PubMed] [Google Scholar]
- 75.Mueller A.L., O’Neill A., Jones T.I., Llach A., Rojas L.A., Sakellariou P., Stadler G., Wright W.E., Eyerman D., Jones P.L., Bloch R.J. Muscle xenografts reproduce key molecular features of facioscapulohumeral muscular dystrophy. Exp. Neurol. 2019;320:113011. doi: 10.1016/j.expneurol.2019.113011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sakellariou P., O’Neill A., Mueller A.L., Stadler G., Wright W.E., Roche J.A., Bloch R.J. Neuromuscular electrical stimulation promotes development in mice of mature human muscle from immortalized human myoblasts. Skelet. Muscle. 2016;6:4. doi: 10.1186/s13395-016-0078-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Laughery M.F., Mayes H.C., Pedroza I.K., Wyrick J.J. R-loop formation by dCas9 is mutagenic in Saccharomyces cerevisiae. Nucleic Acids Res. 2019;47:2389–2401. doi: 10.1093/nar/gky1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lei L., Chen H., Xue W., Yang B., Hu B., Wei J., Wang L., Cui Y., Li W., Wang J. APOBEC3 induces mutations during repair of CRISPR-Cas9-generated DNA breaks. Nat. Struct. Mol. Biol. 2018;25:45–52. doi: 10.1038/s41594-017-0004-6. [DOI] [PubMed] [Google Scholar]
- 79.Min Y.L., Li H., Rodriguez-Caycedo C., Mireault A.A., Huang J., Shelton J.M., McAnally J.R., Amoasii L., Mammen P.P.A., Bassel-Duby R., Olson E.N. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019;5:eaav4324. doi: 10.1126/sciadv.aav4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nguyen Q.G., Buskin J.N., Himeda C.L., Shield M.A., Hauschka S.D. Differences in the function of three conserved E-boxes of the muscle creatine kinase gene in cultured myocytes and in transgenic mouse skeletal and cardiac muscle. J. Biol. Chem. 2003;278:46494–46505. doi: 10.1074/jbc.M308194200. [DOI] [PubMed] [Google Scholar]
- 81.Menegas W., Bergan J.F., Ogawa S.K., Isogai Y., Umadevi Venkataraju K., Osten P., Uchida N., Watabe-Uchida M. Dopamine neurons projecting to the posterior striatum form an anatomically distinct subclass. eLife. 2015;4:e10032. doi: 10.7554/eLife.10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bodega B., Ramirez G.D., Grasser F., Cheli S., Brunelli S., Mora M., Meneveri R., Marozzi A., Mueller S., Battaglioli E., Ginelli E. Remodeling of the chromatin structure of the facioscapulohumeral muscular dystrophy (FSHD) locus and upregulation of FSHD-related gene 1 (FRG1) expression during human myogenic differentiation. BMC Biol. 2009;7:41. doi: 10.1186/1741-7007-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Klooster R., Straasheijm K., Shah B., Sowden J., Frants R., Thornton C., Tawil R., van der Maarel S. Comprehensive expression analysis of FSHD candidate genes at the mRNA and protein level. Eur. J. Hum. Genet. 2009;17:1615–1624. doi: 10.1038/ejhg.2009.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krom Y.D., Thijssen P.E., Young J.M., den Hamer B., Balog J., Yao Z., Maves L., Snider L., Knopp P., Zammit P.S. Intrinsic epigenetic regulation of the D4Z4 macrosatellite repeat in a transgenic mouse model for FSHD. PLoS Genet. 2013;9:e1003415. doi: 10.1371/journal.pgen.1003415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Taberlay P.C., Kelly T.K., Liu C.C., You J.S., De Carvalho D.D., Miranda T.B., Zhou X.J., Liang G., Jones P.A. Polycomb-repressed genes have permissive enhancers that initiate reprogramming. Cell. 2011;147:1283–1294. doi: 10.1016/j.cell.2011.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nelson J.D., Denisenko O., Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 2006;1:179–185. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.