

Abstract
NLRP3 (NACHT, LRR and PYD domains-containing protein 3) is an intracellular sensor that detects a broad range of microbial motifs, endogenous danger signals and environmental irritants, resulting in the formation and activation of the NLRP3 inflammasome. Assembly of the NLRP3 inflammasome leads to caspase-1-dependent release of the proinflammatory cytokines, IL-1β and IL-18, as well as to gasdermin D-mediated pyroptotic cell death. Recent studies have revealed new regulators of the NLRP3 inflammasome, including new interacting or regulatory proteins, metabolic pathways and a regulatory mitochondrial hub. In this Review, we present the molecular, cell biological and biochemical basis of NLRP3 activation and regulation, and describe how this mechanistic understanding is leading to potential therapeutics that target the NLRP3 inflammasome.
Introduction
The discovery that NLRP3 gain-of-function mutations cause the dominantly inherited autoinflammatory disease known as cryopyrin-associated periodic syndrome (CAPS) represented a major advance in inflammation research1, 2, 3. CAPS belongs to a group of diseases with varied severity that includes familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS) and chronic infantile neurological cutaneous and articular syndrome (CINCA; also known as neonatal-onset multisystem inflammatory disorder (NOMID)). Early studies linked NLRP3 to inflammation mediated by the cytokine IL-1 and revealed the involvement of NLRP3 in autoinflammatory diseases, which has been reviewed in depth elsewhere4, 5. In parallel, the discovery that NLRP1 can form a complex with activated caspase-1 termed the inflammasome6, followed by the revelation that NLRP3 can perform a similar function, provided a molecular basis to explain the CAPS phenotype7. Beyond the monogenic autoinflammatory diseases in humans, NLRP3 was shown to affect a wide range of disease models in mice, highlighting the potential application of NLRP3-targeted therapies for these diseases.
The past decade has witnessed a burgeoning appreciation of inflammasomes as critical innate immune components that orchestrate host immune homeostasis. This Review focuses on the recent advances in our understanding of the activation and intrinsic regulation of the NLRP3 inflammasome machinery, as well as the emerging pharmacological approaches that target the NLRP3 inflammasome and show potential for clinical translation.
The NLRP3 inflammasome: an overview
The NLRP3 inflammasome consists of a sensor (NLRP3), an adaptor (ASC; also known as PYCARD) and an effector (caspase-1). NLRP3 is a tripartite protein that contains an amino-terminal pyrin domain (PYD), a central NACHT domain (domain present in NAIP, CIITA, HETE and TP1) and a carboxy-terminal leucine-rich repeat (LRR) domain [G]. The NACHT domain has ATPase activity that is vital for NLRP3 self-association and function8, whereas the LRR domain is thought to induce autoinhibition by folding back onto the NACHT domain. The adaptor ASC has two protein interaction domains, an N-terminal PYD and a C-terminal caspase-recruitment domain (CARD). Full-length caspase-1 has an N-terminal CARD, a central large catalytic domain (p20) and a C-terminal small catalytic subunit domain (p10). Upon stimulation, NLRP3 oligomerizes through homotypic interactions between NACHT domains (Figure 1). Oligomerized NLRP3 recruits ASC through homotypic PYD-PYD interactions and nucleates helical ASC filament formation, also through PYD-PYD interactions. Multiple ASC filaments coalesce into a single macromolecular focus, known as an ASC speck9, 10, 11. Assembled ASC recruits caspase-1 through CARD-CARD interactions, and enables proximity-induced caspase-1 self-cleavage and activation. Caspase-1 clustered on ASC self-cleaves at the linker between p20 and p10 to generate a complex of p33 (comprising the CARD and p20) and p10, which remains bound to ASC and is proteolytically active12. Further processing between the CARD and p20 releases p20-p10 from ASC. The released p20-p10 heterotetramer is unstable in cells, hence terminating its protease activity12. Recently NIMA-related kinase 7 (NEK7), a serine-threonine kinase that is known to be involved in mitosis, was found to be essential for NLRP3 inflammasome activation13, 14, 15. NEK7 specifically interacts with NLRP3, but not the other inflammasome sensors NLRC4 or interferon-inducible protein AIM2 [G].Upon inflammasome activation, the NEK7-NLRP3 interaction increases, and NEK7 oligomerizes with NLRP3 into a complex that is essential for ASC speck formation and caspase-1 activation14, 15. Thus, NEK7 appears to be a core component specific to the NLRP3 inflammasome.
Figure 1 |. NLRP3 inflammasome priming and activation.
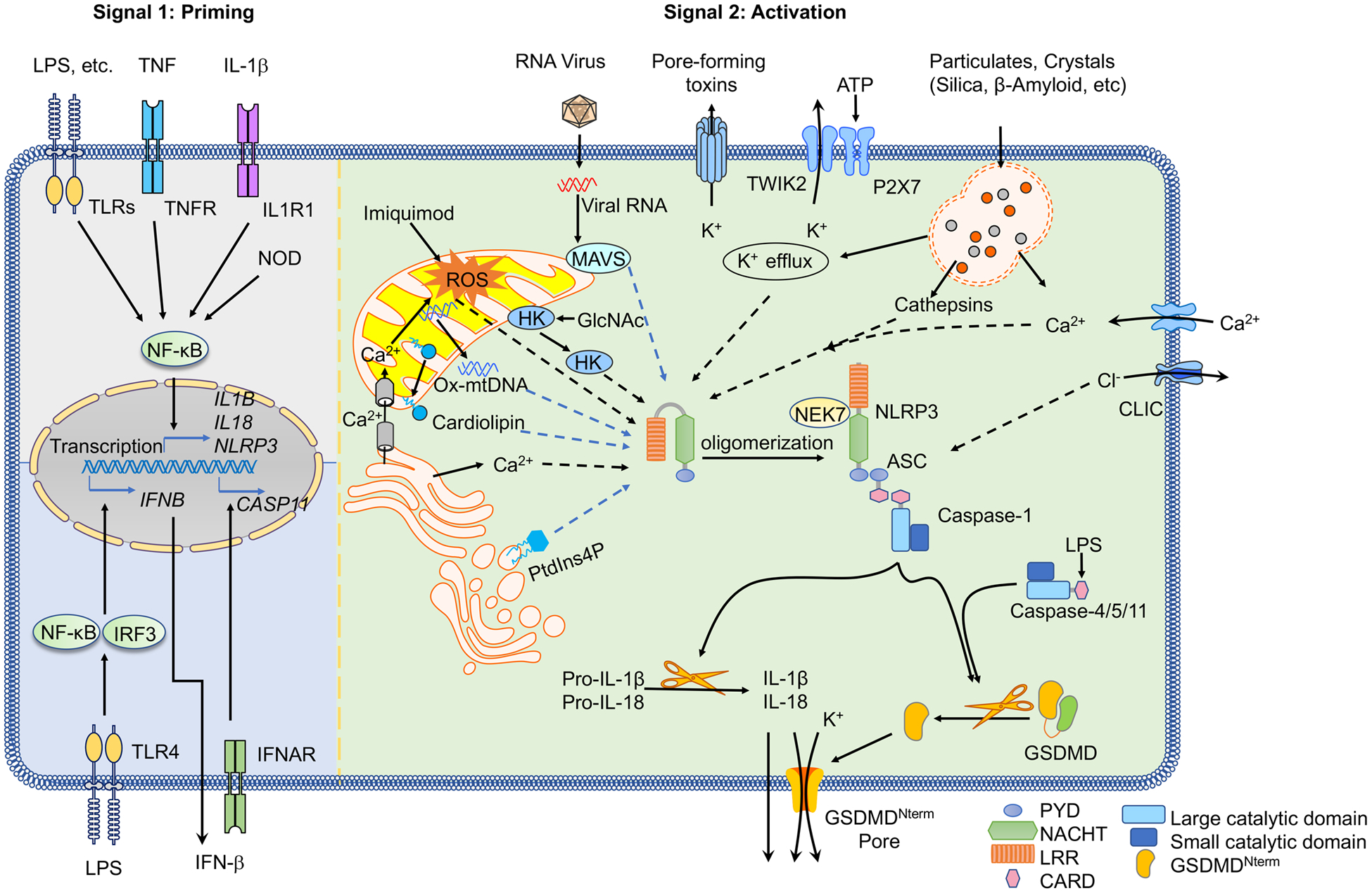
The signal 1 (priming, left) is provided by activation of cytokines or pathogen-associated molecular patterns (PAMPs), leading to the transcriptional upregulation of canonical and non-canonical NLRP3 inflammasome components. Signal 2 (activation; right) is provided by any of numerous PAMPs or damage-associated molecular patterns (DAMPs), such as particulates, crystals and ATP, that activate multiple upstream signaling events. These include K+ efflux, Ca+ flux, lysosomal disruption, mitochondrial reactive oxygen species (mtROS) production, the relocalization of cardiolipin to the outer mitochondrial membrane and the release of oxidized mtDNA (Ox-mtDNA), followed by Cl− efflux. RNA viruses activate NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) through mitochondrial antiviral signaling protein (MAVS) on the mitochondrial outer membrane. Formation of the inflammasome activates caspase 1, which in turn cleaves pro-IL-1β and pro-IL-18. Gasdermin D (GSDMD) is also cleaved and inserts into the membrane, forming pores and inducing pyroptosis. Upon detection of cytosolic lipopolysaccharide (LPS), caspases 4, 5 and 11 are activated and cleave GSDMD, triggering pyroptosis. CARD, caspase recruitment domain; CLIC, chloride intracellular channel protein; GlcNAc, N-acetylglucosamine; GSDMDNterm, GSDMD amino-terminal cell death domain; HK , hexokinase; IFNAR , IFNα/β receptor ; IL-1R1, IL-1 receptor type 1; LRR , leucine-rich repeat; MDP, muramyl dipeptide; NEK7 , NIMA-related kinase 7; NF-κB, nuclear factor-κB; P2X7 , P2X purinoceptor 7; PtdIns4P, phosphatidylinositol-4-phosphate; PYD, pyrin domain; ROS, reactive oxygen species; TLR , Toll-like receptor ; TNF, tumor necrosis factor ; TNFR , tumor necrosis factor receptor ; TWIK2, two-pore domain weak inwardly rectifying K+ channel 2.
Priming the NLRP3 inflammasome
Because inflammasome activation is an inflammatory process, its activation must be highly regulated. With few exceptions, inflammasome activation is considered to be a two-step process — first it must be ‘primed’ and then it can be activated (Figure 1). Priming serves at least two functions. The first function is to upregulate the expression of the inflammasome components NLRP3, caspase-1 and pro-IL-1β. This transcriptional upregulation can be induced through the recognition of various pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) that engage pattern-recognition receptors (PRRs) such as Toll-like receptors (TLRs) or nucleotide-binding oligomerization domain-containing protein 2 (NOD2), or through cytokines such as tumor necrosis factor (TNF) and IL-1β that lead to nuclear factor-κB (NF-κB) activation and gene transcription16, 17, 18. In addition, priming with the TLR4 ligand lipopolysaccharide (LPS) shifts macrophage metabolism from oxidative phosphorylation to glycolysis, indirectly causing the stabilization of hypoxia-inducible factor 1α (HIF1α) and an increase in IL1B gene transcription19. The second function of priming is the induction of posttranslational modifications (PTMs) of NLRP3 (Figure 2), which stabilize NLRP3 in an auto-suppressed inactive, but signal-competent, state. Multiple PTMs have been described for NLRP3, including ubiquitylation, phosphorylation and sumoylation. PTM of NLRP3 can occur at the unstimulated, priming, activation and resolution phases. PTM of NLRP3 is further discussed in a later section.
Figure 2 |. Post-transcriptional modifications of NLRP3.
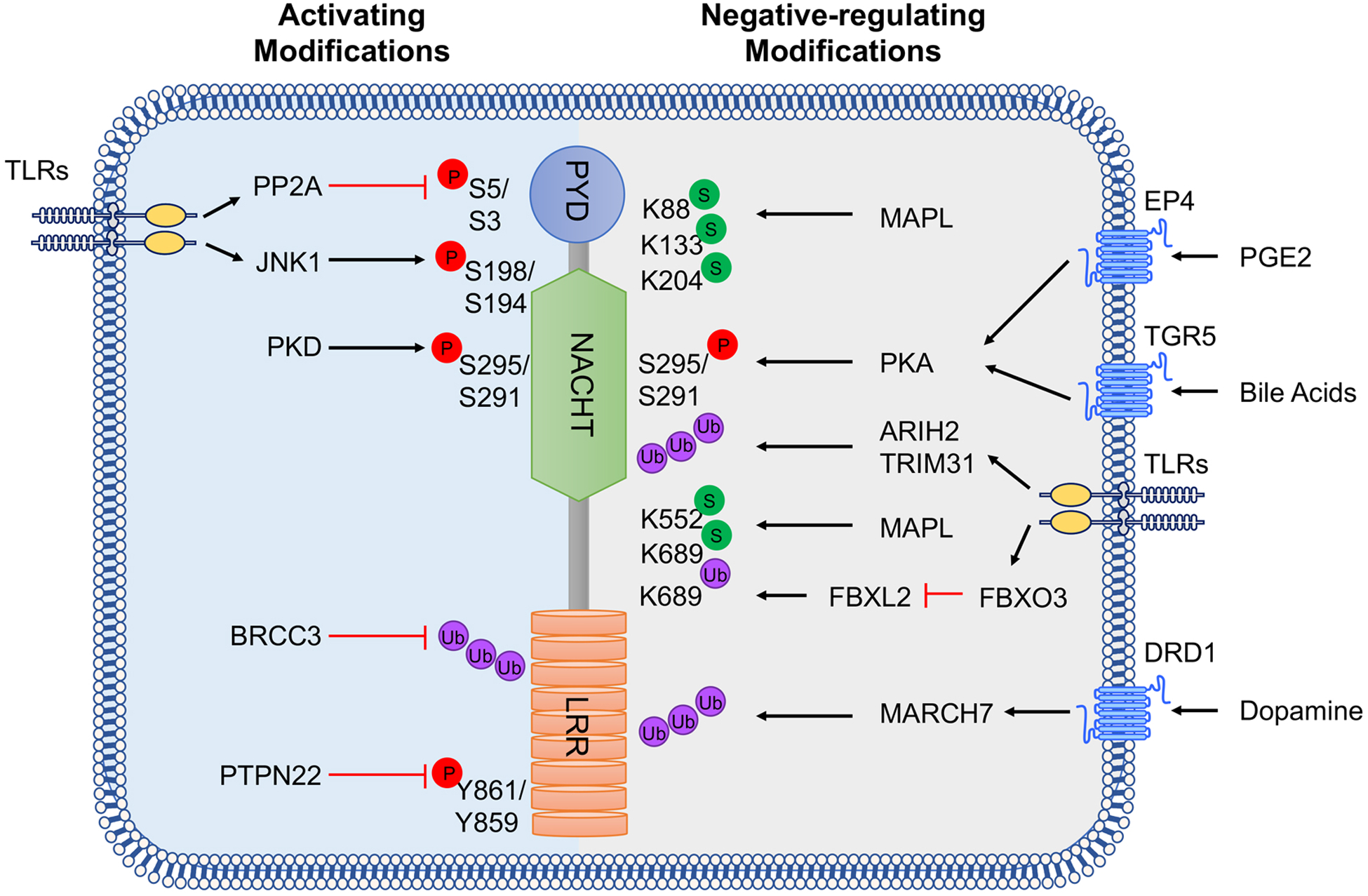
NLRP3 is regulated by phosphorylation (P), ubiquitylation (Ub) and sumoylation (S). Activating regulatory modifications are listed on the left and those that inhibit NLRP3 activation are on the right. Amino acid residues in both human and mouse NLRP3 are listed for phosphorylation, whereas ubiquitylation and sumoylation refer only to the murine protein residues. PP2A, protein phosphatase 2A; JNK1, JUN N-terminal kinase 1; PKD, protein kinase D; BRCC3, BRCA1/BRCA2-containing complex subunit 3; PTPN22, tyrosine-protein phosphatase non-receptor type 22; MUL1, E3 SUMO-protein ligase MUL1; PKA, protein kinase A; ARIH2, E3 ubiquitin-protein ligase ARIH2; TRIM31, tripartite motif-containing protein 31; FBXL2, F-box/LRR-repeat protein 2; MARCH7, membrane-associated RING finger protein 7; PYD, pyrin domain; NACHT, NAIP, CIITA, HET-E and TP1 shared domain; LRR, leucine-rich repeat domain.
NLRP3 activation
The priming step of inflammasome activation licenses the cell, whereas a second step occurs following the recognition of an NLRP3 activator and induces full activation and inflammasome formation. Although most PRRs have limited specificity for one or a few related PAMPs or DAMPs, NLRP3 is unique in that it is activated by a wide variety of unrelated stimuli (Table 1, Figure 1). NLRP3 is activated in bacterial, viral and fungal infections, as well as in sterile inflammation mediated by endogenous DAMPs and on exposure to environmental irritants. The unifying factor of these activators is that they all induce cellular stress; cellular stress is then sensed by NLRP3. Exactly how NLRP3 senses cellular stress and which pathways are induced to culminate in NLRP3 activation and inflammasome formation remain to be fully elucidated. It is thought to include multiple upstream signals, most of which are not mutually exclusive, including efflux of potassium ions (K+) or chloride ions (Cl−), flux of calcium ions (Ca2+), lysosomal disruption, mitochondrial dysfunction, metabolic changes and trans-Golgi disassembly. Although there are an abundance of data describing the upstream signaling events, many pathways are interrelated and overlapping and the data are sometimes conflicting. Therefore, as yet there is no consensus model of NLRP3 activation. Hence, we discuss the various upstream signals — which may act in tandem or independently — that have been mooted to have a role in NLRP3 inflammasome activation.
Table 1 |.
NLRP3 inflammasome activators
| Activator | Source | Examples | Refs |
|---|---|---|---|
| DAMP | Self-derived | ATP, cholesterol crystals, monosodium urate crystals, calcium pyrophosphate dihydrate crystals, calcium oxalate crystals, soluble uric acid, neutrophil extracellular traps, cathelicidin, α-synuclein, amyloid-β, serum amyloid A, prion protein, biglycan, hyaluronan, islet amyloid polypeptide, hydroxyapatite, haeme, oxidized mitochondrial DNA, membrane attack complex, cyclic GMP-AMP, lysophosphatidylcholine, ceramides, oxidized phospholipid 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine, sphingosine | 20, 25, 26, 57, 99, 111, 156, 157, 158, 159, 160, 161, 162, 163 |
| Foreign-derived | Alum, silica, aluminium hydroxide, nanoparticles, carbon nanotubes, chitosan, palmitate (also self-derived), UVB, imiquimod (R837)/CL097, resiquimod (R848) | 28, 40, 164 | |
| PAMP | Bacterial | Lipolysaccharide, peptidoglycan, muramyl dipeptide, trehalose-6,6’-dibehenate, c-di-GMP-c-di-AMP, bacterial RNA, RNA/DNA hybrid | 31, 165, 166, 167, 168, 169 |
| Toxins: nigericin (Streptomyces hygroscopicus), gramicidin (Bacillus brevis), valinomycin (Streptomyces fulvissimus, Streptomyces tsusimaensis), β-hemolysin (Group B Streptococcus), α-hemolysin (Staphylococcus aureus), M protein (Group A Streptococcus), leucocidin (Staphylococcus aureus), tetanolysin O (Clostridium tetani), pneumolysin (Streptococcus pneumoniae), listeriolysin O (Listeria monocytogenes), aerolysin (Aeromonas hydrophila), streptolysin O (Streptococcus pyogenes), enterohaemolysin (Escherichia coli O157:H7), haemolysin BL (Bacillus cereus), adenylate cyclase toxin (Bordetella pertussis), M protein (Group A Streptococcus), maitotoxin (Marina dinoflagellates) | 170, 171, 172, 173 | ||
| Viral | Double-strand RNA, single-strand RNA | 62, 66, 67 | |
| Fungal | β-glucans, hyphae, mannan, zymosan | 174, 175 |
DAMP, damage-associated molecular pattern; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; PAMP, pathogen-associated molecular pattern; UVB, ultraviolet B.
K+ efflux.
With few exceptions, K+ efflux is a necessary upstream event in NLRP3 activation. It has been known for decades that nigericin, a K+/H+ ionophore, and ATP activation of P2X7 [G] , a ligand-gated ion channel of the purinergic receptor family, promote IL-1β maturation via K+ efflux20, 21, 22. Although the P2X family receptors are membrane non-selective cation channels for Na+, K+ and Ca2+ ions, it was only recently appreciated that P2X7 does not act as the ion channel for K+ efflux23. After ATP stimulation, P2X7 promotes Ca2+ and Na+ influx and coordinates with the K+ channel TWIK2 (two-pore domain weak inwardly rectifying K+ channel 2), which mediates K+ efflux24. In addition, NLRP3 inflammasome activation induced either by LPS treatment or by caecal ligation and puncture [G] depend on TWIK224. LPS activation of NLRP3 in vivo is attributed to its activation of the complement cascade. The complement cascade component, membrane attack complex activates NLRP325, 26, while C3a engagement of its receptor on monocytes enhances inflammasome activation via the release of ATP27. In addition to ATP and pore-forming toxins, the particulate stimuli alum, silica and calcium pyrophosphate crystals all induce K+ efflux that is critical to NLRP3 inflammasome activation28. Importantly, low extracellular concentrations of K+ are sufficient to activate the NLRP3 inflammasome in THP1 cell-free lysates, as well as in cultured bone marrow-derived macrophages, whereas high extracellular concentrations of K+ prevent its activation28, 29. Although it has been proposed that K+ efflux is the common denominator upstream of all NLRP3 activators, there are several reports of NLRP3 inflammasome activation independent of K+ efflux30, 31, 32.
Ca2+ flux.
Ca2+ mobilization is also reported to be a critical upstream event in NLRP3 activation33, 34. Mobilization of Ca2+ occurs by either the opening of plasma membrane channels or the release of endoplasmic reticulum (ER)-linked intracellular Ca2+ stores to allow the flux of Ca2+ to the cytosol. Both pathways are linked, in that when either plasma Ca2+ channels open or ER-linked stores are released, the other pathway typically follows. Furthermore, K+ efflux can regulate Ca2+ flux by acting as a counter ion at the plasma membrane for Ca2+ influx. Thus, it is often found that Ca2+ flux and K+ efflux are coordinated in NLRP3 activation. For example, ATP mobilizes Ca2+ by inducing a weak Ca2+ influx via its receptor P2X7 and coordinating K+ efflux24. K+ efflux promotes the release of ER-linked Ca2+ stores followed by the opening of plasma Ca2+ channels33, 35. Additionally, NLRP3 activation induced by nigericin, alum, monosodium urate crystals and the membrane-attack complex have been shown to depend on Ca2+ flux in addition to K+ efflux, as noted above25, 33. Importantly, there is contradictory evidence showing that for some stimuli, the Ca2+ flux occurs downstream of both NLRP3 and caspase-1 activation and, therefore, is not critical for NLRP3 activation36. Thus, the role of Ca2+ flux in NLRP3 activation remains debated.
Cl− efflux.
Low extracellular Cl− levels are known to enhance ATP-induced IL-1β secretion. Accordingly, Cl− channel blockers and high extracellular Cl− levels inhibit NLRP3 activation37, 38, suggesting a role for Cl− efflux in NLRP3 activation. Although there is a report that nigericin-induced activation of NLRP3 does not result in changes in intracellular Cl− levels28, two groups have recently shown that chloride intracellular channel proteins (CLICs) are necessary for NLRP3 activation by several stimuli, including nigericin, but dispensable for AIM2 and NLRC4 inflammasome activation37, 38. While deficiency of CLIC4 impedes nigericin-induced IL-1β release, knockdown of Clic1 and Clic5 in Clic4−/− bone marrow-derived macrophages further impedes IL-1β release, suggesting that CLICs might have redundant roles in NLRP3 activation38. CLICs are present in both the cytosol and plasma membrane, where they form anion channels. Translocation of CLIC1, CLIC4 and CLIC5 to the plasma membrane depends on the release of mitochondrial reactive oxygen species (mtROS), whereas Cl− efflux occurs downstream of K+ efflux38. Another recent study separated the contribution of K+ efflux from Cl− efflux by inhibiting K+ or Cl− efflux independently during NLRP3 activation. They showed that the role of K+ efflux is to drive NLRP3 oligomerization, whereas the role of Cl− efflux is to promote ASC polymerization during NLRP3 inflammasome formation39.
Lysosomal disruption.
Phagocytosis of particulates, either self-derived particulates such as uric acid and cholesterol crystals, or foreign-derived particulates such as alum, silica and asbestos, causes lysosomal rupture and release of the particulates into the cytoplasm40. Lysosomal acidification precedes lysosomal swelling and damage, and is crucial for NLRP3 activation. Additionally, lysosomal rupture induced by the lysosomotropic dipeptide Leu-Leu-OMe is sufficient to activate the NLRP3 inflammasome40. The use of broad spectrum cathepsin inhibitors suggested that cathepsins, which reside in the lysosomes, are important for NLRP3 activation in response to particulate stimuli. However, genetic deletion of cathepsins B, X, L or S, individually, or knock-down of cathepsin X individually, had little effect on NLRP3 activation41, suggesting that cathepsins might have redundant roles in NLRP3 activation. Importantly, lysosomal damage by Leu-Leu-OMe and NLRP3 particulate stimuli activate K+ efflux and Ca2+ influx, suggesting that many NLRP3 activation pathways converge on either K+ and/or Ca2+ flux28, 33, 42.
Mitochondria.
Mitochondrial dysfunction, and release of mtROS and mtDNA into the cytosol, are additional key upstream events implicated in NLRP3 activation. Mitochondria continually produce ROS as a by-product of oxidative phosphorylation, although during cellular stress mtROS levels are greatly increased. Mitophagy [G] is therefore an important regulator of NLRP3 activation as it removes damaged and dysfunctional mitochondria and reduces mtROS. Increases in damaged and dysfunctional mitochondria following treatment with inhibitors of mitophagy enhance NLRP3 inflammasome activation43. Early studies show that ATP and particulate stimulation induce ROS production that is necessary for NLRP3 inflammasome activation44, 45. Imiquimod, a small-molecule adenine derivative, and the related compound CL097, were recently shown to activate the NLRP3 inflammasome via inhibition of the quinone oxidoreductases and mitochondrial complex I31, 46. Consistent with previous reports showing that disruptors of complex I and III induce mtROS and NLRP3 activation43, 47, imiquimod-induced NLRP3 activation was independent of K+ flux and lysosomal disruption, but dependent on mtROS production31. Importantly, this finding suggests that K+ efflux is not necessary as long as mitochondrial disruption and mtROS production occur. However, because most inhibitors are known to have off-target effects, the studies that use mtROS inhibitors to show a role for ROS should be verified with other approaches. Indeed, at least one study showed that ROS inhibitors block the priming step, but not activation of the NLRP3 inflammasome48.
In addition to ROS, a role for nuclear factor erythroid 2-related factor 2 (NRF2) has also been reported in inflammasome regulation. NRF2 regulates both basal and induced levels of antioxidant genes to support cell survival during oxidative stress [G]. By limiting ROS levels, NRF2 inhibits NLRP3 activation49. In addition, NRF2-driven gene transcription attenuates NF-κB activation and downregulates expression of the inflammasome components NLRP3, CASP1, IL1B and IL18, thus limiting NLRP3 inflammasome activity50. However, others showed that NRF2 is also necessary for NLRP3 activation51, 52, 53, 54 and is enriched in ASC specks53.
In addition to mitochondrial dysfunction and mtROS release, circulating mtDNA was first found to act as a DAMP for NLRP3 activation55 and is also a critical component of the NLRP3 activation pathway47, 56, 57. During oxidative stress, mtROS and Ca2+ work together to open mitochondrial permeability transition (MPT) pores58. The release of mtDNA into the cytoplasm was shown to depend on MPT pores and mtROS47. Shimada et al.57 found that mtDNA was rapidly released into the cytoplasm after stimulation with various NLRP3 activators, and importantly was oxidized. Oxidized mtDNA specifically activated the NLRP3 inflammasome, whereas non-oxidized mtDNA preferentially stimulated the AIM2 inflammasome57. Multiple studies show that oxidized mtDNA localized with NLRP3 and co-immunoprecipitated with NLRP3 after activator stimulation, although these studies did not differentiate between direct or indirect interaction56, 57. Recently, it was found that priming with the TLR2, TLR3 or TLR4 agonists results in IRF1 activation, which in turn upregulates cytidine/uridine monophosphate kinase 2 (CMPK2); CMPK2 controls the pool of mitochondrial dNTPs, thus increasing mtDNA synthesis56. Surprisingly, this stands in contrast to earlier reports showing that IRF1 is dispensable for NLRP3 activation59, 60. A report using a genetic deletion approach found no role for MPT pores or mitophagy in NLRP3 activation61. Whereas another study showed that mtDNA was not released into the cytosol in the absence of NLRP347. Thus further studies are needed to resolve the conflicting data and better understand the role of mitochondria in NLRP3 activation.
Besides the potential role of mitochondria in NLRP3 activation, there is increasing evidence that mitochondria act as docking sites for inflammasome assembly. In unstimulated cells, NLRP3 is a cytoplasmic protein that associates with the ER, but upon activation it becomes associated with both mitochondria and the mitochondria-associated membrane (MAM)43, 62. At least three proteins have been implicated as the connection point between NLRP3 and mitochondria. One such protein is cardiolipin. Cardiolipin is a mitochondrial phospholipid of the inner membrane that, upon mitochondrial stress, is exposed on the outer membrane where it serves as a binding site for molecules associated with autophagy [G] and apoptosis63. Cardiolipin also binds NLRP3 and full-length caspase-1 independently, and these interactions were shown to be necessary for inflammasome activation64, 65. A second mitochondrial protein, mitochondrial antiviral signaling protein (MAVS), which is an adaptor protein in RNA sensing pathways, has been shown to be important for NLRP3 inflammasome activation during RNA viral infections and after stimulation with the synthetic RNA polyinosinic–polycytidylic acid62, 66, 67, 68. MAVS recruits NLRP3, directing its location to the mitochondria for inflammasome activation62, 67. Although MAVS was required for NLRP3 inflammasome activation after RNA viral infections, it appears to be nonessential for NLRP3 inflammation induced by other NLRP3 stimuli67. Lastly, mitofusin 2, which is found on the outer mitochondrial membrane, the ER and contact sites at the MAM, was found to be important for NLRP3 activation during RNA viral infections68. During viral infection, mitofusin 2 formed a complex with MAVS to support localization of NLRP3 to the mitochondria68.
Metabolic changes.
A major role of mitochondria is energy production via ATP and regulation of metabolism. As such, mitochondria are poised to detect cellular cues and metabolites to tune metabolic flux. Priming of dendritic cells (DCs) with LPS results in their switch from oxidative phosphorylation to aerobic glycolysis69. Phosphorylation of glucose is the first step in glycolysis and is mediated by hexokinase. During bacterial infection, degradation of the bacterial cell wall component peptidoglycan in lysosomes releases N-acetylglucosamine (GlcNAc). Hexokinase, which localizes at the mitochondrial surface, then binds to GlcNAc, promoting its relocalization into the cytosol32. GlcNAc-induced hexokinase relocalization promoted NLRP3 inflammasome activation independent of K+ efflux. A peptide inhibitor of hexokinase was sufficient to induce hexokinase localization to the cytosol and drive inflammasome activation32. Importantly, during this process, although no mitochondrial membrane disruption was observed, mtDNA was detected in the cytosol. Similarly, another study reported that chemical disruption of glycolysis after the priming step activates NLRP3 inflammasome70. However, the interpretation of this observation is complicated by the fact that inhibition of glycolysis during priming inhibits LPS-induced IL1B gene transcription19.
Free fatty acids (FAs) that are liberated through diet or by upregulation of FA synthesis activate the NLPR3 inflammasome71, 72, 73. The anti-inflammatory AMP-activated protein kinase (AMPK) is an essential mediator of FA metabolism, while suppressing FA-induced inflammation. One of the ways in which AMPK suppresses inflammation is by limiting ROS production and activating autophagy, thereby inhibiting NLRP3 inflammasome activation74. The saturated FA palmitate however, was found to suppress AMPK activation and in turn increase ROS production and NLRP3 activation71.
In addition to promoting NLRP3 activation, metabolic changes also negatively regulate NLRP3. Fasting and caloric restriction are known to reduce inflammation. During periods of low blood glucose, such as during caloric restriction, fasting and uncontrolled type I diabetes, metabolism switches to fatty acid oxidation leading to the production of ketone bodies. One of these ketone bodies, β-hydroxybutyrate (BHB), inhibits NLRP3 activation induced by a wide variety of stimuli75. BHB also suppresses caspase-1 activation and IL-1β release in murine models of CAPS and in the urate crystal model [G] of inflammation. Interestingly, BHB regulation of NLRP3 was not due to the starvation mechanisms of AMPK activation and autophagy, ROS decreases or glycolysis inhibition, but rather was due to inhibition of K+ efflux by an unknown mechanism.
Short-chain fatty acids (SCFAs) are fermentation products of the gut microbiota that are incorporated during FA synthesis and are reported to have anti-inflammatory effects. Butyrate, which is a SCFA that is structurally related to BHB, had no effect on ATP- or particulate-induced NLRP3 activation75. Conversely, a recent report shows that butyrate and propionate inhibit NLPR3 priming and activation by palmitate, with a major effect on reducing pro-IL-1β levels and a modest effect on inflammasome activation76. More investigation into whether SCFAs have a role in NLRP3 activation are warranted since metabolic regulation of NLRP3 is becoming an important subject for many diseases77.
Trans-Golgi disassembly.
Until recently, the role of the Golgi apparatus in NLRP3 activation had been underappreciated. Using a cellular reconstitution system, NLRP3 stimuli were found to promote trans-Golgi network disassembly into vesicles called dispersed trans-Golgi network (dTGN)78. The phospholipid phosphatidylinositol-4-phosphate on dTGN recruits NLRP3 and promotes NLRP3 aggregation that is essential for downstream ASC oligomerization and caspase-1 activation78. Although both K+ efflux-dependent stimuli (nigericin) and K+ efflux-independent stimuli (imiquimod) induce dTGN formation and NLRP3 recruitment, K+ efflux is only necessary for NLRP3 recruitment and not for dTGN formation78. This suggests that K+ efflux-dependent and mitochondria-dependent NLRP3 activation may be separate pathways that converge on Golgi disassembly. Another study found that, upon activation, NLRP3 translocates to the Golgi apparatus in a complex with sterol regulatory element-binding protein 2 (SREBP2) and SREBP cleavage-activating protein (SCAP) and couples activation with SREBP2 maturation and cholesterol biosynthesis79. The SCAP and SREBP2 complex is required for NLRP3 activation.
Although there has been a lot of effort in understanding the upstream events during NLRP3 activation, there is still no single unifying model. Much of the evidence reported so far relies on pharmacological inhibition rather than genetic approaches. This makes it difficult to parse out indirect and off-target effects. Indeed, because most stimuli that activate NLPR3 also induce ion fluxes and organelle damage, it is hard to know whether mitochondrial dysfunction and mtROS are crucially involved or just associated with NLRP3 activation. The recent suggestion that K+ efflux and mitochondrial dysfunction are separable events and that they both independently converge on trans-Golgi disassembly78 is intriguing, but requires further investigation.
NLRP3 and pyroptosis
Besides inflammatory cytokine production, NLRP3 inflammasome activation leads to pyroptosis [G] , which is a rapid, inflammatory form of lytic programmed cell death (Figure 1). Recent key studies have identified that gasdermin D (GSDMD) is the executioner of pyroptosis. GSDMD has an N-terminal cell death domain (GSDMDNterm), a central short linker region and a C-terminal autoinhibition domain. Caspase-1 cleaves GSDMD, removing its C-terminus and releasing it from intramolecular inhibition80, 81. GSDMDNterm then binds phosphatidylinositol phosphates and phosphatidylserine in the cell membrane inner leaflet, oligomerizes and inserts into the plasma membrane forming a 10–14 nm pore containing 16 symmetric protomers82, thus killing cells from within. In addition, GSDMDNterm demonstrates bactericidal activity in vitro by binding to cardiolipin present in both outer and inner bacterial membranes83. How this direct bactericidal activity functions during infections remains elusive. Cardiolipin is also present in the inner and outer mitochondrial membranes after NLRP3 activation, but it is not clear whether GSDMDNterm can permeabilize mitochondria by binding to mitochondrial cardiolipin. Another feature of GSDMD-dependent pyroptosis is that it facilitates the release of IL-1β and IL-18 via non-conventional secretion84, 85. Pyroptosis induces the secretion of IL-1α in both full-length and calpain-processed forms, although processing does not determine its bioactivity86. Together, these findings reveal GSDMD biochemically determines pyroptosis downstream of inflammasome activation. In addition to the important role played by caspase-1 in pro-IL-1β processing and pyroptosis, the apical activator caspase, caspase-8, also plays a role in inducing a non-canonical pathway of IL-1β and IL-18 maturation and cell death87, 88, 89. Although several groups have shown caspase-8 mediated maturation of pro-IL1β and IL-18, this can occur through both NLRP3 dependent or independent pathways.
Non-canonical NLRP3 activation
Cytosolic LPS during Gram-negative bacterial infection is sensed by human caspase-4 and caspase-5, and murine caspase-11, to induce non-canonical NLRP3 inflammasome activation90, 91, 92, 93. In this pathway, extracellular LPS activates TLR4, and the induced type I interferon response together with the complement C3-C3aR axis upregulate caspase-11 expression94. Type I interferons also induce the expression of a cluster of small GTPases, guanylate-binding proteins (GBPs) and their phylogenetic relative IRGB1095, 96. GBPs and IRGB10 are recruited to intracellular bacteria, where they lyse bacteria and liberate LPS into the cytosol95, 96. Direct recognition of cytosolic LPS by caspase-11 triggers its oligomerization and activation by auto-proteolytic cleavage93, 97. Subsequently, GSDMD is cleaved by activated caspases 4, 5 or 11 and this induces pyroptosis80, 98. The K+ efflux caused by pyroptosis activates NLRP3-caspase-1 dependent IL-1β secretion. The oxidized phospholipid 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (oxPAPC) also binds to caspase-11 and activates this non-canonical pathway99. Recently, a report showed that in mouse models of age-related macular degeneration, the non-canonical caspase-11-NLRP3 inflammasome pathway drives disease pathology and suggested that oxPAPC is important for inflammasome activation during disease100. Interestingly, although mtDNA was important in these models, it did not directly activate the canonical NLRP3 pathway, but rather induced type I interferon production via the cyclic GMP-AMP synthase (cGAS)-STING DNA sensing pathway. Conversely, a recent study suggests that oxPAPC suppresses rather than activates the caspase-11-dependent inflammasome101. The discrepancy between these studies requires further investigation.
In neutrophils, the detection of cytosolic LPS triggers non-canonical inflammasome activation and GSDMD cleavage by caspase-11 and also activates the release of neutrophil extracellular traps [G] (NETs; also known as NETosis)102. Of interest, NETosis can engage a feedforward loop that augments inflammation. NETs and the NET-associated antimicrobial peptide cathelicidin can induce NLRP3 inflammasome activation, and reciprocally, IL-18 released after inflammasome assembly can induce NETosis103. Together, the data reveal the critical role of the caspase-11-NLRP3 axis in intracellular bacteria surveillance.
One-step NLRP3 inflammasome activation
NLRP3 inflammasome activation does not always follow the two-step activation model. One exception is the species-specific observation that TLR4 stimulation alone is sufficient to induce the secretion of IL-1β from human and porcine monocytes, but not from murine monocytes30, 104. Given the observation that LPS induces endogenous ATP release from human monocytes104, 105, it was initially thought that LPS-induced inflammasome activation was mediated by the binding of extracellular ATP to P2X7 and the induction of K+ efflux105. But a second study revealed that in fact this pathway signals via a TRIF-RIPK1-FADD-caspase-8 pathway upstream of NLRP3 and is not associated with the usual hallmarks of inflammasome activation, including ASC speck formation, pyroptosis and K+ efflux; it is therefore referred to as alternative NLRP3 inflammasome activation30. A second exception to the two-step activation model is observed in mouse bone marrow derived DCs (BMDCs) and mouse splenic DCs, in which LPS alone is also sufficient to activate the NLRP3 inflammasome by an unknown mechanism106. This one-step rapid response might contribute to maintaining sterility of blood and have implications during bacterial sepsis.
Another one-step activation pathway has been observed in mouse bone marrow-derived macrophages in which simultaneous TLR and NLRP3 stimulation leads to rapid inflammasome activation. Stimulation of TLR2, TLR4, TLR7 or TLR9, together with extracellular ATP, rapidly activates NLRP3. Although this type of NLRP3 activation does not promote IL-1β secretion and pyroptosis, it does however lead to IL-18 secretion, which occurs independent of de novo transcription107. The transcription-independent inflammasome activation provides a fast response mechanism in tissue inflammation, as microbial infection might simultaneously engage both TLRs and NLRP3. Early pyroptosis and release of IL-18 may be important in decreasing intracellular pathogen burden.
Cooperation with other inflammasome sensors
There are numerous reports of inflammasome activation involving more than one sensor108, 109, 110, 111, 112. Most of these reports include NLRP3 as one of the sensors. Salmonella species activate both NLRP3 and NLRC4108, and the colocalization of NLRP3 and NLRC4 during infection has been shown by microscopic analysis113. The inflammasome response to lysophosphatidylcholine (also known as lysolecithin) in macrophages, microglia and astrocytes is also dependent on both of these sensors111. Malaria and Aspergillus species can activate both the NLRP3 and AIM2 inflammasomes109, 110. During malaria infection, inflammasome activation depends on plasmodium gDNA (sensed by AIM2) and haemozoin (sensed by NLRP3)109. In mouse macrophages and DCs, cytosolic DNA and the DNA virus murine cytomegalovirus (MCMV) are sensed by AIM2 to form an inflammasome114. Cytosolic DNA is also sensed by the cGAS-STING pathway, which produces the secondary messenger cGAMP, which further promotes activation of the NLRP3 and AIM2 inflammasomes and is important for control of MCMV112. In contrast to murine macrophages and DCs and the human monocyte-like cell line THP-1, AIM2 is dispensable for DNA-induced inflammasome activation in the human monocyte-like cell line BlaER1; in BlaER1 cells, DNA activates inflammasomes solely through a cGAS-STING-NLRP3 pathway115. It is currently unclear whether dual sensing reflects two sensors activated in different cells, two sensors within the same cell each forming independent inflammasomes, or two sensors cooperating to form a single inflammasome in the same cell.
PTM regulation of NLRP3
PTM by covalent addition of functional groups is important for the regulation of protein folding, localization and functional activities (Figure 2). Various PTMs have been shown to regulate innate immunity through their effects on the activation, survival, proliferation, differentiation and migration of immune cells116. There is an emerging appreciation that priming not only upregulates NLRP3 expression, albeit in an inactive configuration, but also licenses NLRP3 to rapidly respond to activating stimuli. This non-transcriptional role of priming, as well as the subsequent activation phase, is at least partly regulated by PTMs.
Ubiquitylation.
NLRP3 is ubiquitylated in resting macrophages and is deubiquitylated upon priming and activation117. FBXL2, a SCF (SKP1-cullin-F-box) E3 ligase complex subunit, recognizes Trp73 of NLRP3 and targets it for ubiquitylation and proteasomal degradation118. LPS priming extends the half-life of NLRP3 by inducing another SCF subunit FBOX3, which degrades FBXL2118. In resting cells, the E3 ubiquitin ligase TRIM31 interacts with the PYD of NLRP3 and induces Lys48-linked ubiquitylation and proteasomal degradation of NLRP3119. LPS or IL-1β treatment further increase TRIM31 expression, suggesting that TRIM31 is a feedback suppressor of the NLRP3 inflammasome119. Dopamine signaling via dopamine D1 receptor increases intracellular levels of cAMP, which then binds to NLRP3. This promotes MARCH7-dependent Lys48-linked ubiquitylation of the LRR domain of NLRP3 promoting its autophagic degradation120. Finally, Lys63-specific deubiquitinase BRCC3 deubiquitylates the LRR domain of NLRP3 and is required for NLRP3 oligomerization and activation121. NLRP3 is not the only protein in the inflammasome that undergoes ubiquitylation. Linear ubiquitylation of ASC by the linear ubiquitin assembly complex is specifically required for NLRP3 inflammasome activation122.
Phosphorylation.
TLR priming induces the phosphorylation of NLRP3 on Ser198 (in human NLRP3; Ser194 in mouse NLRP3) by JUN N-terminal kinase 1 (JNK1; also known as MAPK8), and promotes NLRP3 self-association and activation123. Mechanistically, Ser198 (or Ser194) phosphorylation promotes BRCC3 binding and subsequent deubiquitylation123. After NLRP3 activator stimulation, protein kinase D (PKD) at the Golgi phosphorylates NLRP3 on Ser295 (in human NLRP3; Ser291 in mouse NLRP3) and promotes NLRP3 activation124. However, phosphorylation also can suppress NLRP3 activation. Phosphorylation at NLRP3 Ser5 (in human NLRP3; Ser3 in mouse NLRP3) within the PYD prevents inflammasome activation owing to electrostatic repulsion of the ASC PYD125, whereas protein phosphatase 2A dephosphorylates Ser5 (or Ser3) to enhance NLRP3 nucleation with ASC125. In resting cells, NLRP3 is phosphorylated at Tyr861 (in human NLRP3; Tyr859 in mouse NLRP3)126. Upon activation, protein tyrosine phosphatase, non-receptor type 22 (PTPN22) interacts with NLRP3 in an ASC-dependent manner, and dephosphorylates NLRP3 at Tyr861 (in human NLRP3; Tyr859 in mouse NLRP3) to allow efficient NLRP3 activation126. Bile acids and prostaglandin E2 can suppress the NLRP3 inflammasome by inducing protein kinase A (PKA)-mediated phosphorylation on NLRP3 Ser295 (in human NLRP3; Ser291 in mouse NLRP3)127, 128, which can occur both prior to and after NLRP3 activation127. Ser295 (or Ser291) phosphorylation attenuates NLRP3 ATPase activity and blocks NLRP3 activation127. It is currently unclear why PKA and PKD phosphorylate NLRP3 at the same site but have opposing effects, although Ser295 (or Ser291) phosphorylation in combination with other NLRP3 PTMs may determine the final effect and further studies are warranted to clarify the effect.
Sumoylation.
In resting cells, NLRP3 is sumoylated at multiple sites by the E3 SUMO protein ligase MUL1 (also known as MAPL), which restrains NLRP3 activation. Upon activation, NLRP3 becomes desumoylated by sentrin-specific protease 6 (SENP6) and SENP7, promoting inflammasome activation129. Notably, a defect in sumoylation of NLRP3 due to a Lys689Arg mutation causes hyperactivation of NLRP3, which phenocopies the reported mutations found in patients with CAPS129. In summary, these studies highlight the delicate control of NLRP3 inflammasome activity by this dynamic landscape of PTMs.
NLRP3-interacting proteins or regulators
In addition to the proteins that form the inflammasome complex, such as ASC or pro-IL-1β, there are other proteins that interact with NLRP3. Pyrin-only proteins (POPs; also known as PYDC proteins) and CARD-only proteins (COPs) are small cytoplasmic decoy proteins that contain single homotypic protein-binding domains130. They function in the downregulation of inflammation. Both POPs and COPs are only found in primates. Of the four POPs (POP1–POP4), POP1 and POP2 regulate NLRP3 activation and are upregulated in response to NF-κB during priming. The PYD of POP1 and POP2 have 64% and 37% identity, respectively, with the PYD of ASC, and in in vitro overexpression systems both POP1 and POP2 bind to ASC and inhibit the NLRP3-ASC interaction130, 131. Similar to NF-κB, IL-1β also upregulates the expression of POP1 and POP2, thus forming a feedback loop to prevent excessive NLRP3 activation. Furthermore, POP2 negatively regulates NF-κB activation by regulating the non-canonical pathway during LPS priming132. Transgenic expression of POP1 and POP2 in mice reveals that they have functions beyond dampening of excessive inflammatory responses, as POP2-expressing mice are more resistant to bacterial infection than wild-type mice133. Mice in which monocytes, macrophages and DCs express transgenic POP1 are protected from death in an otherwise lethal LPS sepsis model and in a model of the autoinflammatory disease MWS134.
There are five human COPs that function to regulate caspase-1 activation. Three of them, COP1 (also known as CARD16), ICEBERG (also known as CARD18) and INCA (also known as CARD17), have 97%, 81% and 52% identity, respectively, to the CARD of caspase-1 and can bind to full-length caspase-1, thereby preventing its autoactivation and limiting inflammasome activation130. Because POPs and COPs are not expressed in mice, we are only beginning to understand their role during infections and diseases that activate NLRP3. With the recent development of transgenic POP mice, further studies towards the understanding of their role and regulation in diseases are expected.
Therapeutic targeting of NLRP3
An important focus of future research is to use our understanding of the molecular mechanisms of NLRP3 inflammasome activation to identify effective NLRP3 inhibitors (Table 2) or inhibitory pathways (Figure 2) and assess their therapeutic potential. Excitingly, a number of NLRP3 inhibitors have been reported to date, including those that either directly inhibit NLRP3 or indirectly inhibit inflammasome components or related signaling events. A major caveat for using these inhibitors is that the inhibitory mechanism or precise target is not fully elucidated, thus representing a potential risk due to off-target effects.
Table 2 |.
NLRP3 inhibitors
| Inhibitor | ~IC50a | Inhibition mechanism | Specificity | Primingb | Clinical status | Ref |
|---|---|---|---|---|---|---|
| MCC950 (CP-456,773) | 8 nM | Unknown | NLRP3 | Phase II | 139 | |
| CY-09 | 5 μM | Binds Walker A motif, NACHT ATPase inhibitor | NLRP3 | 148 | ||
| Oridonin | 0.5 μM | Irreversibly binds NLRP3 Cys279, inhibits the NLRP3-NEK7 interaction | NLRP3 | 153 | ||
| Tranilast | 25–50 μM | Binds NACHT Inhibits the NLRP3-NLRP3 interaction | NLRP3 | + | Approved | 151 |
| MNS | 2 μM | NACHT ATPase inhibitor | NLRP3 | 176 | ||
| OLT1177 Dapansutrile | 1–100 nM (mouse) 1 μM (human) | NACHT ATPase inhibitor | NLRP3 | Phase II | 177 | |
| Bay 11–7082 | 5 μM | NACHT ATPase inhibitor | NLRP3 NLRC4 | + | 148, 178 | |
| BOT-4-one | 0.59–1.28 μM | Alkylation, NACHT ATPase inhibitor | NLRP3 NLRC4 | + | 179 | |
| Parthenolide | 5 μM | NACHT ATPase inhibitor, caspase-1 inhibitor | NLRP3 AIM2 NLRC4 NLRP1 | + | 148, 178 | |
| INF39 | 10 μM | NACHT ATPase inhibitor | NLRP3 | + | 149 |
Approximate IC50 of inhibitor in vitro.
+ denotes inhibition on priming.
IC50, median inhibitory concentration; NLRP, NOD-, LRR- and pyrin domain-containing protein; NEK7 , NIMA-related kinase 7.
IL-1 signaling blockade is an example of successful clinical translation of basic immunology research and is currently being used in the treatment of NLRP3-driven immunopathologies. Three biologicals are approved by the US Food and Drug Administration for multiple inflammatory diseases: canakinumab, an IL-1β neutralizing antibody; anakinra, a recombinant IL-1 receptor antagonist; and rilonacept, a decoy receptor that binds IL-1β and IL-1α135. Two similar agents, including GSK1070806, an IL-18 blocking antibody, and MABp1, an IL-1α neutralizing antibody, are under early development136. Given the initial remarkable therapeutic benefits of these approved biologicals in patients with CAPS, the clinical indications of IL-1 blockade are expanding continuously. However, it is important to note that IL-1β blockade or IL-1R1 deficiency failed to rescue the lethality in a mouse model of CAPS, suggesting that other inflammatory mediators released during NLRP3 activation may be important for disease progression137.
Directly targeting NLRP3 by small molecules is specific, cost-effective and less invasive than cytokine blockade. Several such inhibitors have been discovered to date (Table 2). The diarylsulfonylurea compound MCC950 (originally reported as CRID3/CP-456773) is the most potent and specific NLRP3 inhibitor138. MCC950 specifically inhibits canonical and non-canonical NLRP3 inflammasome activation in both mouse and human macrophages in vitro, without impairing the NLRP1, NLRC4 and AIM2 inflammasomes or TLR-mediated priming signals139. Of note, MCC950 is not able to inhibit the release of IL-1α and prevent cell death in response to particulate stimuli and transfected LPS, because these events are NLRP3 independent. Strikingly, MCC950 demonstrated therapeutic efficacy against a variety of preclinical immunopathological models, including CAPS, experimental autoimmune encephalomyelitis139, Alzheimer’s disease140, traumatic brain injury141, atherosclerosis142, cardiac arrhythmias143, myocardial infarction144, diabetes145, steatohepatitis146 and colitis147. Although these studies provide compelling rationale for targeting NLRP3, a Phase II clinical trial of MCC950 for rheumatoid arthritis was suspended due to hepatic toxicity4. C172, an inhibitor for cystic fibrosis transmembrane conductance regulator channel (CFTR), and its analogue CY-09, were recently recognized as NLRP3 inhibitors in screening libraries of bioactive compounds148. Because CY-09 does not have CFTR-inhibitory activity, thus reducing its risks of off-target effects, it can move forward to preclinical trials148. CY-09 specifically inhibits the NLRP3 inflammasome, but not the NLRC4 and AIM2 inflammasomes nor TLR-mediated priming signals148. Mechanistically, CY-09 binds to the ATP-binding Walker A motif of NLRP3 and inhibits its ATPase activity and oligomerization149. Tranilast, an analogue of a tryptophan metabolite, has been approved for use in treating allergy, asthma and hypertrophic scars in Korea and Japan since 1982150. Only recently was its mechanism uncovered, showing that Tranilast binds the NACHT domain of NLRP3 abolishing the NLRP3-NLRP3 interaction and oligomerization without affecting its ATPase activity. Thus, it specifically inhibits NLRP3 inflammasomes, but not AIM2 or NLRC4 inflammasomes151. Tranilast also inhibits LPS-induced pro-IL-1β and IL-6 release147, suggesting that Tranilast suppresses inflammation via a multi-target mechanism.
Rabdosia rubescens is an over-the-counter herbal medicine that is used for the treatment of inflammatory diseases in China152. Oridonin, an ent-kaurane diterpenoid, is the main ingredient in Rabdosia rubescens and it has been shown to specifically inhibit the NLRP3 inflammasome, but not AIM2 and NLRC4 inflammasomes153. Oridonin irreversibly binds NLRP3 Cys279 via a covalent carbon-carbon bond, and blocks the NLRP3-NEK7 interaction140. Although Oridonin has NF-κB and MAPK inhibitory effects, the dose required for this was ten times higher than that needed for NLRP3 inhibition154. CY-09, Tranilast and Oridonin also showed remarkable therapeutic effects in mouse models including CAPS, peritonitis and type 2 diabetes. Many other direct inhibitors including, MNS, OLT1177, Bay 11–7082, BOT-4-one, parthenolide and INF39, target NLRP3 by inhibiting its ATPase activity, however, they have not been tested in vivo in NLRP3-dependent models148. Additionally, Bay 11–7082, BOT-4-one, parthenolide and INF39 have multiple biological activities, and are thus unlikely to serve as specific NLRP3 inhibitors.
Several inhibitors have been shown to be ineffective in the treatment of NLRP3-associated diseases. The P2X7 inhibitors AZD9056, CE-224535 and GSK1482169 were unsuccessful in the treatment of rheumatoid arthritis135. Glyburide is commonly used in the treatment of type 2 diabetes and acts by inhibiting ATP-sensitive K+ channels in pancreatic β-cells and stimulating insulin release. Although glyburide potently inhibits NLRP3 activation in vitro, it requires high doses in vivo and the mechanism of action is unknown. Additionally, the associated hypoglycemia and perturbation of glucose metabolism limits the use of this inhibitor beyond diabetes136. Two caspase-1-inhibiting peptidomimetic pro-drugs, VX-740 and VX-765, reached Phase II clinical trials for arthritis, epilepsy and psoriasis, but were discontinued due to hepatic toxicity155.
Conclusions and future directions
The study NLRP3 inflammasome activation is a rich field in immunology, with rapidly emerging insight into its mechanism of action and regulation. The discoveries of the NEK7-NLRP3 interaction, GSDMD as the pyroptosis executor, and roles for ionic flux, mitochondrial dysfunction, metabolic alteration and Golgi disassembly in NLRP3 activation, represent major advances in this field.
While refinement of our understanding of NLRP3 activation continues, targeting of NLRP3 as a therapeutic for multiple diseases is rapidly progressing. Current treatment for NLRP3 pathologies focuses on inhibition of the inflammasome-derived cytokine IL-1β. Although this has been highly effective, it isn’t without problems. Inflammasome activation is critical for immune control of numerous pathogens, and so loss of IL-1β can have detrimental effects on immune defense. Many of the new therapeutics advancing to clinical trials are specific for NLRP3 activation and do not affect the function of other inflammasome sensors. With increases in the number of individuals affected by inflammatory conditions, because of the Western lifestyle and ageing population, it is likely that there will be a greater need for NLRP3-specific therapeutics. Biochemical studies on structure and phase separation will unravel the molecular principles underlying inflammasome assembly. Continued profiling, refinement and repurposing of direct and specific NLRP3 inhibitors will boost future clinical translation, epitomizing the use of precision medicine in inflammasome related disorders.
Acknowledgements
This review was supported by the National Center for Advancing Translational Sciences, US National Institutes of Health (NIH), through grant KL2TR002490 awarded to K.V.S., and by the NIH, through grants AI029564, CA156330, DK094779, AI109965 and AI067798 awarded to J.P.T. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
GLOSSARY
- Leucine-rich repeat (LRR) domain
A domain that mediates the detection of ligands derived from microbial components, which is found in TLRs and may serve a similar role in certain NLRs. The LRR domain of NLRs (NACHT-LRR proteins) is structurally similar to that of Toll-like receptors. It consists of leucine-rich amino-acid strands forming a peptide loop. The loops occur as tandem repeats that, together, form a coil or ‘solenoid’ and contain constant sequences, as well as unique ‘insertions’ or variable residues for each ligand.
- Urate crystal model
A mouse model of crystal-induced peritonitis that activates the NLRP3 inflammasome
- Autophagy
A cytoplasmic bulk degradation system in which cytoplasmic cargo is targeted and is sequestered typically in double-membrane vesicles, leading to subsequent fusion with the lysosome. This process is essential for the response to starvation because it facilitates the recycling of cellular components. In addition, autophagy can be targeted to intracellular bacteria to restrict their growth.
- Mitophagy
A term referring to the selective removal of mitochondria by macroautophagy under conditions of nutrient starvation or mitochondrial stress.
- AIM2
(Also known as absent in melanoma 2) A sensor that combines with the adaptor ASC and the protease caspase-1 to form the AIM2 inflammasome. It senses cytosolic double-strand DNA from bacterial or viruses, or from mislocalized self-DNA and contributes to infection defense.
- Pyroptosis
A lytic, inflammatory form of programmed cell death that is triggered by cleavage of gasdermin D by the inflammatory caspases 1, 4, 5 or 11. It is characterized by cytoplasmic swelling, early plasma membrane rupture and nuclear condensation. The cytoplasmic content is released into the extracellular space, and this is thought to augment inflammatory and repair responses.
- Neutrophil extracellular traps
Fibrous networks that are released into the extracellular environment by neutrophils. They are composed mainly of DNA, but also contain proteins from neutrophil granules. NETs act as a mesh that traps microorganisms and exposes them to neutrophil-derived effector molecules.
- P2X7
An ATP-gated cation channel that is expressed by haematopoietic cells and participates in cell proliferation and apoptosis. It belongs to the family of purinoceptors for ATP and is responsible for the ATP-dependent activation of NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3).
- Caecal ligation and puncture
An experimental model of peritonitis in rodents, in which the caecum is ligated and then punctured, thereby forming a small hole. This leads to leakage of intestinal bacteria into the peritoneal cavity and subsequent peritoneal infection.
- Oxidative stress
Cells continuously produce reactive oxygen species (ROS) such as hydrogen peroxide or superoxide anions. Under physiological conditions, mitochondria are the main source, and cellular antioxidants ensure that the redox equilibrium is maintained. During inflammatory responses (and also in cancer), excessive production of ROS leads to a metabolic condition known as oxidative stress, which can lead to apoptosis and necrosis.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Hoffman HM, Mueller JL, Broide DH, Wanderer AA & Kolodner RD Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 29, 301–305 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aganna E et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum 46, 2445–2452 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Aksentijevich I et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum 46, 3340–3348 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]; References 1, 2 and 3 report that NLRP3 gain-of-function mutations promote human autoinflammatory diseases.
- 4.Mangan MS et al. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov 17, 588 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Guo H, Callaway JB & Ting JP Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21, 677–687 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinon F, Burns K & Tschopp J The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10, 417–426 (2002). [DOI] [PubMed] [Google Scholar]; This is the first report describing an inflammasome complex that mediates cleavage of IL-1β.
- 7.Agostini L et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20, 319–325 (2004). [DOI] [PubMed] [Google Scholar]; This is the first report showing NLRP3 assembles an inflammasome complex that mediates cleavage of IL-1β by caspase-1.
- 8.Duncan JA et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci U S A 104, 8041–8046 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that ATP binding is essential for NLRP3 function, suggesting a therapeutic target for treating NLRP3 related diseases.
- 9.Cai X et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 156, 1207–1222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu A et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193. [DOI] [PMC free article] [PubMed] [Google Scholar]; References 9 and 10 show that the pyrin domains from inflammasome sensors nucleates the polymerization of ASC.
- 11.Schmidt FI et al. A single domain antibody fragment that recognizes the adaptor ASC defines the role of ASC domains in inflammasome assembly. J Exp Med 213, 771–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boucher D et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med 215, 827–840 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmid-Burgk JL et al. A genome-wide CRISPR screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J Biol Chem 291, 103–109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He Y, Zeng MY, Yang D, Motro B & Núñez G NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi H et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 17, 250–258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; References 13, 14 and 15 identify NEK7 as an integral component of the NLRP3 inflammasome.
- 16.Bauernfeind FG et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183, 787–791 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franchi L, Eigenbrod T & Núñez G Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol 183, 792–796 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xing Y et al. Cutting Edge: TRAF6 Mediates TLR/IL-1R Signaling-Induced Nontranscriptional Priming of the NLRP3 Inflammasome. J Immunol 199, 1561–1566 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Tannahill GM et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perregaux D & Gabel CA Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem 269, 15195–15203 (1994). [PubMed] [Google Scholar]
- 21.Walev I, Reske K, Palmer M, Valeva A & Bhakdi S Potassium-inhibited processing of IL-1 beta in human monocytes. EMBO J 14, 1607–1614 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Surprenant A, Rassendren F, Kawashima E, North RA & Buell G The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272, 735–738 (1996). [DOI] [PubMed] [Google Scholar]
- 23.Samways DS, Li Z & Egan TM Principles and properties of ion flow in P2X receptors. Front Cell Neurosci 8, 6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Di A et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity 49, 56–65.e54 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Triantafilou K, Hughes TR, Triantafilou M & Morgan BP The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci 126, 2903–2913 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Laudisi F et al. Cutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1β release. J Immunol 191, 1006–1010 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Asgari E et al. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood 122, 3473–3481 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Muñoz-Planillo R et al. K⁺ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pétrilli V et al. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14, 1583–1589 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Gaidt MM et al. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 44, 833–846 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Groß CJ et al. K + Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 45, 761–773 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Wolf AJ et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell 166, 624–636 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murakami T et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A 109, 11282–11287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee GS et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492, 123–127 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yaron JR et al. K(+) regulates Ca(2+) to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis 6, e1954 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katsnelson MA, Rucker LG, Russo HM & Dubyak GR K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J Immunol 194, 3937–3952 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Domingo-Fernández R, Coll RC, Kearney J, Breit S & O’Neill LAJ The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1β transcription and activate the NLRP3 inflammasome. J Biol Chem 292, 12077–12087 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang T et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun 8, 202 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Green JP et al. Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc Natl Acad Sci U S A 115, E9371–E9380 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hornung V et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9, 847–856 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orlowski GM et al. Multiple Cathepsins Promote Pro-IL-1β Synthesis and NLRP3-Mediated IL-1β Activation. J Immunol 195, 1685–1697 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Katsnelson MA, Lozada-Soto KM, Russo HM, Miller BA & Dubyak GR NLRP3 inflammasome signaling is activated by low-level lysosome disruption but inhibited by extensive lysosome disruption: roles for K+ efflux and Ca2+ influx. Am J Physiol Cell Physiol 311, C83–C100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou R, Yazdi AS, Menu P & Tschopp J A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Cruz CM et al. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem 282, 2871–2879 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dostert C et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Courbet A et al. Imidazoquinoxaline anticancer derivatives and imiquimod interact with tubulin: Characterization of molecular microtubule inhibiting mechanisms in correlation with cytotoxicity. PLoS One 12, e0182022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakahira K et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12, 222–230 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bauernfeind F et al. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol 187, 613–617 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu X et al. Nuclear Factor E2-Related Factor-2 Negatively Regulates NLRP3 Inflammasome Activity by Inhibiting Reactive Oxygen Species-Induced NLRP3 Priming. Antioxid Redox Signal 26, 28–43 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W et al. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem Pharmacol 76, 1485–1489 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sussan TE et al. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS One 3, e3791 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Freigang S et al. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur J Immunol 41, 2040–2051 (2011). [DOI] [PubMed] [Google Scholar]
- 53.Zhao C, Gillette DD, Li X, Zhang Z & Wen H Nuclear factor E2-related factor-2 (Nrf2) is required for NLRP3 and AIM2 inflammasome activation. J Biol Chem 289, 17020–17029 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sogawa Y et al. Infiltration of M1, but not M2, macrophages is impaired after unilateral ureter obstruction in Nrf2-deficient mice. Sci Rep 7, 8801 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Q et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhong Z et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shimada K et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lemasters JJ, Theruvath TP, Zhong Z & Nieminen AL Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta 1787, 1395–1401 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Man SM et al. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat Immunol 16, 467–475 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuriakose T, Zheng M, Neale G & Kanneganti TD IRF1 Is a Transcriptional Regulator of ZBP1 Promoting NLRP3 Inflammasome Activation and Cell Death during Influenza Virus Infection. J Immunol 200, 1489–1495 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Allam R et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep 15, 982–990 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Subramanian N, Natarajan K, Clatworthy MR, Wang Z & Germain RN The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dudek J Role of Cardiolipin in Mitochondrial Signaling Pathways. Front Cell Dev Biol 5, 90 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iyer SS et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Elliott EI et al. Cutting Edge: Mitochondrial Assembly of the NLRP3 Inflammasome Complex Is Initiated at Priming. J Immunol 200, 3047–3052 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Franchi L et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J Immunol 193, 4214–4222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Park S et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol 191, 4358–4366 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ichinohe T, Yamazaki T, Koshiba T & Yanagi Y Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc Natl Acad Sci U S A 110, 17963–17968 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krawczyk CM et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sanman LE et al. Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. Elife 5, e13663 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wen H et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 12, 408–415 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moon JS et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest 125, 665–680 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 73.Moon JS et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat Med 22, 1002–1012 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 74.Li XN et al. Activation of the AMPK-FOXO3 pathway reduces fatty acid-induced increase in intracellular reactive oxygen species by upregulating thioredoxin. Diabetes 58, 2246–2257 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Youm YH et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med 21, 263–269 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that an endogenous molecule produced during fasting potently inhibits NLRP3 activation.
- 76.Truax AD et al. The Inhibitory Innate Immune Sensor NLRP12 Maintains a Threshold against Obesity by Regulating Gut Microbiota Homeostasis. Cell Host Microbe 24, 364–378.e366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hughes MM & O’Neill LAJ Metabolic regulation of NLRP3. Immunol Rev 281, 88–98 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Chen J & Chen ZJ PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 564, 71–76 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that disassembly of the trans-Golgi network serves as a scaffold for NLRP3 aggregation and activation.
- 79.Guo C et al. Cholesterol Homeostatic Regulator SCAP-SREBP2 Integrates NLRP3 Inflammasome Activation and Cholesterol Biosynthetic Signaling in Macrophages. Immunity 49, 842–856.e847 (2018). [DOI] [PubMed] [Google Scholar]
- 80.Shi J et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660 (2015). [DOI] [PubMed] [Google Scholar]
- 81.He W. t. et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell research 25, 1285 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ding J et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111 (2016). [DOI] [PubMed] [Google Scholar]
- 83.Liu X et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Evavold CL et al. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Monteleone M et al. Interleukin-1β Maturation Triggers Its Relocation to the Plasma Membrane for Gasdermin-D-Dependent and -Independent Secretion. Cell Rep 24, 1425–1433 (2018). [DOI] [PubMed] [Google Scholar]
- 86.Groß O et al. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 36, 388–400 (2012). [DOI] [PubMed] [Google Scholar]
- 87.Antonopoulos C et al. Caspase-8 as an Effector and Regulator of NLRP3 Inflammasome Signaling. J Biol Chem 290, 20167–20184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Antonopoulos C, El Sanadi C, Kaiser WJ, Mocarski ES & Dubyak GR Proapoptotic chemotherapeutic drugs induce noncanonical processing and release of IL-1β via caspase-8 in dendritic cells. J Immunol 191, 4789–4803 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bossaller L et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J Immunol 189, 5508–5512 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kayagaki N et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011). [DOI] [PubMed] [Google Scholar]; This study identifies non-canonical NLRP3 activation via LPS-mediated caspase-11 activation and shows it is an important immune mediator during sepsis.
- 91.Aachoui Y et al. Caspase-11 protects against bacteria that escape the vacuole. Science 339, 975–978 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kayagaki N et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249 (2013). [DOI] [PubMed] [Google Scholar]
- 93.Shi J et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 (2014). [DOI] [PubMed] [Google Scholar]; This study shows that caspases 4, 5 and 11 are intracellular receptors for LPS, whose activation induce non-canonical inflammsome activation.
- 94.Napier BA et al. Complement pathway amplifies caspase-11-dependent cell death and endotoxin-induced sepsis severity. J Exp Med 213, 2365–2382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Man SM et al. IRGB10 liberates bacterial ligands for sensing by the AIM2 and caspase-11-NLRP3 inflammasomes. Cell 167, 382–396. e317 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Meunier E et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 509, 366 (2014). [DOI] [PubMed] [Google Scholar]
- 97.Lee BL et al. Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. J Exp Med 215, 2279–2288 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kayagaki N et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature 526, 666 (2015). [DOI] [PubMed] [Google Scholar]; References 80, 81 and 98 show that cleavage of gasdermin D by caspases 1, 4, 5, or 11 during inflammasome activation causes pyroptosis.
- 99.Zanoni I et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352, 1232–1236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kerur N et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat Med 24, 50–61 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chu LH et al. The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nat Commun 9, 996 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen KW et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol 3, eaar6676 (2018). [DOI] [PubMed] [Google Scholar]
- 103.Kahlenberg JM, Carmona-Rivera C, Smith CK & Kaplan MJ Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. The journal of immunology, 1202388 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Netea MG et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 113, 2324–2335 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Piccini A et al. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A 105, 8067–8072 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.He Y, Franchi L & Núñez G TLR agonists stimulate Nlrp3-dependent IL-1β production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J Immunol 190, 334–339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lin K-M et al. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc Natl Acad Sci U S A 111, 775–780 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Broz P et al. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med 207, 1745–1755 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kalantari P et al. Dual engagement of the NLRP3 and AIM2 inflammasomes by plasmodium-derived hemozoin and DNA during malaria. Cell Rep 6, 196–210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Karki R et al. Concerted activation of the AIM2 and NLRP3 inflammasomes orchestrates host protection against Aspergillus infection. Cell Host Microbe 17, 357–368 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Freeman L et al. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med 214, 1351–1370 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Swanson KV et al. A noncanonical function of cGAMP in inflammasome priming and activation. J Exp Med 214, 3611–3626 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Man SM et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A 111, 7403–7408 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rathinam VA et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 11, 395–402 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gaidt MM et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 171, 1110–1124.e1118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liu J, Qian C & Cao X Post-translational modification control of innate immunity. Immunity 45, 15–30 (2016). [DOI] [PubMed] [Google Scholar]
- 117.Juliana C et al. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem, 287, 36617–36622 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Han S et al. Lipopolysaccharide primes the NALP3 inflammasome by inhibiting its ubiquitination and degradation mediated by the SCFFBXL2 E3 ligase. J Biol Chem 290, 18124–18133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Song H et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat Commun 7, 13727 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yan Y et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160, 62–73 (2015). [DOI] [PubMed] [Google Scholar]
- 121.Py BF, Kim M-S, Vakifahmetoglu-Norberg H & Yuan J Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 49, 331–338 (2013). [DOI] [PubMed] [Google Scholar]
- 122.Rodgers MA et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med 211, 1333–1347 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Song N et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol Cell 68, 185–197. e186 (2017). [DOI] [PubMed] [Google Scholar]
- 124.Zhang Z et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J Exp Med 214, 2671–2693 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Stutz A et al. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J Exp Med 214, 1725–1736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Spalinger MR et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J Clin Invest 126, 1783–1800 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mortimer L, Moreau F, MacDonald JA & Chadee K NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat. Immunol 17, 1176 (2016). [DOI] [PubMed] [Google Scholar]
- 128.Guo C et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 45, 802–816 (2016). [DOI] [PubMed] [Google Scholar]
- 129.Barry R et al. SUMO-mediated regulation of NLRP3 modulates inflammasome activity. Nat Commun 9, 3001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Indramohan M, Stehlik C & Dorfleutner A COPs and POPs Patrol Inflammasome Activation. J Mol Biol 430, 153–173 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bedoya F, Sandler LL & Harton JA Pyrin-only protein 2 modulates NF-kappaB and disrupts ASC:CLR interactions. J Immunol 178, 3837–3845 (2007). [DOI] [PubMed] [Google Scholar]
- 132.Ratsimandresy RA et al. The PYRIN domain-only protein POP2 inhibits inflammasome priming and activation. Nat Commun 8, 15556 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Periasamy S et al. Pyrin-only protein 2 limits inflammation but improves protection against bacteria. Nat Commun 8, 15564 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.de Almeida L et al. The PYRIN Domain-only Protein POP1 Inhibits Inflammasome Assembly and Ameliorates Inflammatory Disease. Immunity 43, 264–276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dinarello CA, Simon A & Van Der Meer JW Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nature reviews Drug discovery 11, 633 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ozaki E, Campbell M & Doyle SL Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflamm Res 8, 15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Brydges SD et al. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity 30, 875–887 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Laliberte RE et al. Glutathione S-transferase omega 1–1 is a target of cytokine release inhibitory drugs and may be responsible for their effect on interleukin-1β posttranslational processing. Journal of Biological Chemistry (2003). [DOI] [PubMed] [Google Scholar]
- 139.Coll RC et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21, 248–255 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that the small-molecule MCC950 specifically inhibits NLRP3 inflammasome activation and is effective in NLRP3-activated mouse disease models.
- 140.Dempsey C et al. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav Immun 61, 306–316 (2017). [DOI] [PubMed] [Google Scholar]
- 141.Ismael S, Nasoohi S & Ishrat T MCC950, the selective NLRP3 inflammasome inhibitor protects mice against traumatic brain injury. J. Neurotrauma 35, 1294–1303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.van der Heijden T et al. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E–Deficient Mice—Brief Report. Arterioscler Thromb Vasc Biol 37, 1457–1461 (2017). [DOI] [PubMed] [Google Scholar]
- 143.Monnerat G et al. Macrophage-dependent IL-1β production induces cardiac arrhythmias in diabetic mice. Nat Commun 7, 13344 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Van Hout GP et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Euro Heart J 38, 828–836 (2016). [DOI] [PubMed] [Google Scholar]
- 145.Zhai Y et al. Inhibiting the NLRP3 Inflammasome Activation with MCC950 Ameliorates Diabetic Encephalopathy in db/db Mice. Molecules 23, 522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mridha AR et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol 66, 1037–1046 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Perera AP et al. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci Rep 8, 8618 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jiang H et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med 214, 3219–3238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cocco M et al. Development of an acrylate derivative targeting the NLRP3 inflammasome for the treatment of inflammatory bowel disease. J Med Chem 60, 3656–3671 (2017). [DOI] [PubMed] [Google Scholar]
- 150.Darakhshan S & Pour AB Tranilast: a review of its therapeutic applications. Pharmacol Res 91, 15–28 (2015). [DOI] [PubMed] [Google Scholar]
- 151.Huang Y et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med 10, e8689 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ma Z, Hu C & Zhang Y Therapeutic effect of Rabdosia rubescens aqueous extract on chronic pharyngitis and its safety. Zhong Nan Da Xue Xue Ban 36, 170–173 (2011). [DOI] [PubMed] [Google Scholar]
- 153.He H et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Baldwin AG et al. Boron-based inhibitors of the NLRP3 inflammasome. Cell Chem Biol 24, 1321–1335. e1325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.MacKenzie SH, Schipper JL & Clark AC The potential for caspases in drug discovery. Curr Opin Drug Discov Develop 13, 568–576 13, 568 (2010). [PMC free article] [PubMed] [Google Scholar]
- 156.Duewell P et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Martinon F, Pétrilli V, Mayor A, Tardivel A & Tschopp J Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 (2006). [DOI] [PubMed] [Google Scholar]
- 158.Masters SL et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol 11, 897–904 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mulay SR et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J Clin Invest 123, 236–246 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lai M et al. The NLRP3-Caspase 1 Inflammasome Negatively Regulates Autophagy via TLR4-TRIF in Prion Peptide-Infected Microglia. Front Aging Neurosci 10, 116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Niemi K et al. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J Immunol 186, 6119–6128 (2011). [DOI] [PubMed] [Google Scholar]
- 162.Babelova A et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem 284, 24035–24048 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yamasaki K et al. NLRP3/cryopyrin is necessary for interleukin-1beta (IL-1beta) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J Biol Chem 284, 12762–12771 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Baron L et al. The NLRP3 inflammasome is activated by nanoparticles through ATP, ADP and adenosine. Cell Death Dis 6, e1629 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Martinon F, Agostini L, Meylan E & Tschopp J Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol 14, 1929–1934 (2004). [DOI] [PubMed] [Google Scholar]
- 166.Abdul-Sater AA et al. Cyclic-di-GMP and cyclic-di-AMP activate the NLRP3 inflammasome. EMBO Rep 14, 900–906 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sha W et al. Human NLRP3 inflammasome senses multiple types of bacterial RNAs. Proc Natl Acad Sci U S A 111, 16059–16064 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kailasan Vanaja S et al. Bacterial RNA:DNA hybrids are activators of the NLRP3 inflammasome. Proc Natl Acad Sci U S A 111, 7765–7770 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Schweneker K et al. The mycobacterial cord factor adjuvant analogue trehalose-6,6’-dibehenate (TDB) activates the Nlrp3 inflammasome. Immunobiology 218, 664–673 (2013). [DOI] [PubMed] [Google Scholar]
- 170.Greaney AJ, Leppla SH & Moayeri M Bacterial Exotoxins and the Inflammasome. Front Immunol 6, 570 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gurcel L, Abrami L, Girardin S, Tschopp J & van der Goot FG Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 126, 1135–1145 (2006). [DOI] [PubMed] [Google Scholar]
- 172.Mariathasan S et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 (2006). [DOI] [PubMed] [Google Scholar]
- 173.Mathur A et al. A multicomponent toxin from Bacillus cereus incites inflammation and shapes host outcome via the NLRP3 inflammasome. Nat Microbiol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lamkanfi M, Malireddi RK & Kanneganti TD Fungal zymosan and mannan activate the cryopyrin inflammasome. J Biol Chem 284, 20574–20581 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kankkunen P et al. (1,3)-beta-glucans activate both dectin-1 and NLRP3 inflammasome in human macrophages. J Immunol 184, 6335–6342 (2010). [DOI] [PubMed] [Google Scholar]
- 176.He Y et al. 3, 4-Methylenedioxy-β-nitrostyrene inhibits NLRP3 activation by blocking assembly of the inflammasome. J Biol Chem 289, 1142–1150 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Marchetti C et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci U S A 115, E1530–E1539 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Juliana C et al. Anti-inflammatory compounds parthenolide and Bay 11–7082 are direct inhibitors of the inflammasome. J Biol Chem 285, 9792–9802 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Shim D-W et al. BOT-4-one attenuates NLRP3 inflammasome activation: NLRP3 alkylation leading to the regulation of its ATPase activity and ubiquitination. Sci Rep 7, 15020 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]