

Abstract
BACKGROUND
Type 2 diabetes mellitus (T2DM) is significantly increasing worldwide, and the incidence of its complications is also on the rise. One of the main complications of T2DM is diabetic kidney disease (DKD). The glomerular filtration rate (GFR) and urinary albumin creatinine ratio (UACR) increase in the early stage. As the disease progresses, UACR continue to rise and GFR begins to decline until end-stage renal disease appears. At the same time, DKD will also increase the incidence and mortality of cardiovascular and cerebrovascular diseases. At present, the pathogenesis of DKD is not very clear. Therefore, exploration of the pathogenesis of DKD to find a treatment approach, so as to delay the development of DKD, is essential to improve the prognosis of DKD.
AIM
To detect the expression of tenascin-C (TNC) in the serum of T2DM patients, observe the content of TNC in the glomerulus of DKD rats, and detect the expression of TNC on inflammatory and fibrotic factors in rat mesangial cells (RMCs) cultured under high glucose condition, in order to explore the specific molecular mechanism of TNC in DKD and bring a new direction for the treatment of DKD.
METHODS
The expression level of TNC in the serum of diabetic patients was detected by enzyme-linked immunosorbent assay (ELISA), the protein expression level of TNC in the glomerular area of DKD rats was detected by immunohistochemistry, and the expression level of TNC in the rat serum was detected by ELISA. Rat glomerular mesangial cells were cultured. Following high glucose stimulation, the expression levels of related proteins and mRNA were detected by Western blot and polymerase chain reaction, respectively.
RESULTS
ELISA results revealed an increase in the serum TNC level in patients with T2DM. Increasing UACR and hypertension significantly increased the expression of TNC (P < 0.05). TNC expression was positively correlated with glycosylated haemoglobin (HbA1c) level, body mass index, systolic blood pressure, and UACR (P < 0.05). Immunohistochemical staining showed that TNC expression in the glomeruli of rats with streptozotocin-induced diabetes was significantly increased compared with normal controls (P < 0.05). Compared with normal rats, serum level of TNC in diabetic rats was significantly increased (P < 0.05), which was positively correlated with urea nitrogen and urinary creatinine (P < 0.05). The levels of TNC, Toll-like receptor-4 (TLR4), phosphorylated nuclear factor-κB p65 protein (Ser536) (p-NF-κB p65), and miR-155-5p were increased in RMCs treated with high glucose (P < 0.05). The level of TNC protein peaked 24 h after high glucose stimulation (P < 0.05). After TNC knockdown, the levels of TLR4, p-NF-κB p65, miR-155-5p, connective tissue growth factor (CTGF), and fibronectin (FN) were decreased, revealing that TNC regulated miR-155-5p expression through the TLR4/NF-κB p65 pathway, thereby regulating inflammation (NF-κB p65) and fibrosis (CTGF and FN) in individuals with DKD. In addition, metformin treatment may relive the processes of inflammation and fibrosis in individuals with DKD by reducing the levels of the TNC, p-NF-κB p65, CTGF, and FN proteins.
CONCLUSION
TNC can promote the occurrence and development of DKD. Interfering with the TNC/TLR4/NF-κB p65/miR-155-5p pathway may become a new target for DKD treatment.
Keywords: Tenascin-C, miR-155-5p, Metformin, Type 2 diabetes mellitus, Diabetic kidney disease, Toll-like receptor 4
Core Tip: We investigated the correlation between serum tenascin-C (TNC) levels and urine albumin clearance ratios in a total of 380 patients with type 2 diabetes mellitus, TNC can regulate miR-155-5p through Toll-like receptor 4/nuclear factor κB signaling in rat mesangial cells under hyperglycaemia condition, and metformin can inhibit inflammation and fibrosis in diabetic kidney disease through TNC.
INTRODUCTION
Diabetes has now developed into the most common metabolic disease and affects the health of approximately 6.4% of the global population. Among patients with diabetes, diabetic kidney disease (DKD) is one of the most important causes of poor prognosis[1,2]. Approximately 40% of patients with type 2 diabetic mellitus (T2DM) will eventually progress to DKD. The combined actions of high glucose levels, oxidative stress, inflammatory chemokines, and multiple signalling pathways lead to endothelial cell injury, haemodynamic changes, and increased microvascular permeability, all of which may recruit inflammatory cells to the site of the diseased kidney, leading to the occurrence and development of DKD[3]. Therefore, studies of the mechanism of DKD are particularly important for developing new treatments, particularly treatments specific for DKD[4].
Tenascins are a family of extracellular matrix (ECM) glycoproteins comprised of tenascin-C (TNC), tenascin-X, tenascin-R, and tenascin-W[5]. Tenascin-R is mainly expressed in the central nervous system, tenascin-W is mainly expressed in stem cells, and tenascin-X is mainly expressed in loose connective tissue[6]. TNC is the most critical tenascin. TNC is expressed at very low levels in normal adult tissues, and its levels are only increased during embryonic development, in tumours, during injury repair and in inflammation[7]. TNC is the main regulator of early fibrosis, and an increase in TNC expression may reflect the development of fibrosis as a useful biomarker of matrix reconstruction[8]. Ishizaki et al[9] identified TNC as a potentially important indicator of disease severity. In patients with various chronic kidney diseases, plasma TNC levels are increased and negatively correlate with the glomerular filtration rate; additionally, urine TNC levels are negatively correlated with creatinine clearance[10]. In patients with DKD, TNC is widely expressed in renal tissues and positively correlated with ECM expansion[11]. However, the precise pathway by which TNC affects the development of DKD has not been reported.
Toll-like receptors (TLRs) are type І membrane glycoproteins that can identify pathogen-related molecular patterns, such as lipopolysaccharide (LPS), and endogenous damage-associated molecular pattern molecules, such as high mobility group box 1 (HMGB1)[12]. HMGB1 is an endogenous activator of oxidative stress in individuals with DKD[13]. After high-glucose treatment, TLR4 activates HMGB1 and further promotes the development of tubulointerstitial inflammation in individuals with DKD[14]. In addition, TLR4 stimulates the generation of IL-1 receptor associated kinase (IRAK) and tumor necrosis factor receptor associated factor (TRAF) through myeloid differentiation factor 88 (MyD88) and finally produces nuclear factor-κB (NF-κB), which promotes the occurrence and development of DKD[15].
MicroRNAs are small non-coding RNAs of 21-25 nucleotides in length that regulate the development of diseases by regulating the expression of target genes[16]. As shown in the study by Guay et al[17], miR-155 in exosomes released by T lymphocytes is converted into its activated form to promote the apoptosis of insulin beta cells, which leads to the occurrence of type 1 diabetes. Beltrami et al[18] also observed increased expression of miR-155 in the urine of patients with DKD. Although studies have shown the presence of miR-155 in individuals with DKD, the pathway by which miR-155 modulates the development of DKD has not been reported.
As a first-line drug for the treatment of diabetes, metformin has been widely explored for the treatment of DKD. In recent years, metformin has been shown to inhibit the occurrence and development of inflammation through NF-κB[19]. However, the mechanism by which it relieves inflammation through NF-κB and delays the development of DKD requires further investigation.
MATERIALS AND METHODS
Human studies
Subjects: Three hundred and eighty patients who were newly diagnosed with T2DM at our hospital from January 2018 to December 2018 were selected as an experimental group. Members of a healthy normal control group (n = 60) were selected from The Physical Examination Center of The First Affiliated Hospital of China Medical University. The exclusion criteria were as follows: (1) Patients under the age of 18 years old or who experienced acute complications of diabetes, such as hyperosmolar coma or diabetic ketoacidosis, within the last 3 mo; (2) Patients with cardiovascular, cerebrovascular, liver, kidney, infectious, autoimmune, or other systemic diseases or malignant tumours; (3) Patients with a recent infection, patients who underwent surgery or experienced trauma, patients under other stressful conditions, patients who were pregnant or breast feeding, and patients in other special physical states; and (4) Patients treated with angiotensin-converting enzyme inhibitors or angiotensin receptor blockers over the last 3 mo.
Patients with T2DM were divided into a normal albuminuria group [n = 70, urinary albumin creatinine ratio (UACR) < 30 mg/g], microalbuminuria group (n = 105, UACR of 30-300 mg/g), microalbuminuria with hypertension group (n = 75, UACR of 30-300 mg/g, with hypertension), macroalbuminuria group (n = 65, UACR > 300 mg/g), and macroalbuminuria with hypertension group (n = 65, UACR > 300 mg/g, with hypertension).
Data collection: Patient age, height, weight, systolic blood pressure (SBP), and diastolic blood pressure were collected. Fasting plasma glucose, fasting plasma insulin, glycosylated hemoglobin (HbA1c), high-density lipoprotein-cholesterol (HDL-C), low-density lipoprotein-cholesterol (LDL-C), total serum cholesterol, triglyceride, and uric acid levels were determined. Five millilitres of morning urine was collected, and urinary albumin and creatinine levels were detected. Body mass index (BMI) and UACR were calculated. Homeostasis model assessment of insulin resistance levels were calculated to evaluate insulin resistance.
Enzyme-linked immunosorbent assay: Five millilitres of venous blood was used for the enzyme-linked immunosorbent assay (ELISA). The blood was incubated at room temperature for 30 min, an anticoagulant was added, and the blood was centrifuged for 15 min (3000 r/min, 4 °C). The supernatant was collected and stored at -80 °C in aliquots. An ELISA was used to determine serum TNC levels (SEB975Hu, Cloud-Clon Corp) among patients within the group (variable < 8%, variance < 10%).
Animal experiments
Animal feeding: Eight-week-old male Sprague-Dawley rats (Beijing Vital River Laboratory Animal Technology Co., Ltd.) were fed in a standard specific pathogen-free laboratory on a 12 h light/dark cycle, housed in groups of three rats/cage, and provided free access to food and water. All experiments were approved by The Institutional Animal Care and Use Committee (IACUC) of The China Medical University Animal Experiment Department (Approval No. 2017112).
We used a well-established low-dose streptozotocin (STZ, S0130, Sigma-Aldrich, United States)-induced diabetic rat model. The rats were fasted for 12-16 h and injected with a low dose of STZ (35 mg/kg, dissolved in cold 0.1 mol/L citrate buffer pH 4.5). A One Touch Ultra glucometer and test paper (Johnson & Johnson, United States) were used to measure the blood glucose levels of diabetic rats in blood samples collected from the tail vein when the condition of diabetes was stable at 72 h after the STZ injection. Rats with significant hyperglycaemia (blood glucose > 16.7 mmol/L) accompanied by polydipsia were considered the diabetic rat model.
Histological analysis: Renal cortices were fixed with polyoxymethylene and embedded in paraffin. The sections (3 um) were stained with hematoxylin-eosin (HE) and periodic acid-Schiff (PAS). The pathological changes were observed using a Leica microscope.
Immunohistochemistry: The deparaffinized and rehydrated sections (3 um) of paraffin-embedded renal cortices were subjected to heat-mediated antigen retrieval and incubated with 3% H2O2 for 10 min. Sections were then incubated overnight at 4 °C with primary antibody against TNC (CST, 12221, 1:400), and the pathology score of micrographs was blindly evaluated by more than two experienced pathologists. At least ten images from each kidney section were counted.
ELISA and biochemical index determination: Five millilitres of venous blood of rats was used for ELISA. The blood was incubated at room temperature for 30 min, an anticoagulant was added, and the blood was centrifuged for 15 min (3000 r/min, 4 °C). The supernatant was collected and stored at -80 °C in aliquots. An ELISA was used to determine serum TNC levels (SEB975Ra, Cloud-Clon Corp) among rats within the group. The levels of urea nitrogen (C013-2-1, Nanjing Jiancheng Bioengineering Institute) and creatinine (C011-2-1, Nanjing Jiancheng Bioengineering Institute) in serum of rats were measured with commercial kits.
Cell-based experiments
Reagents and antibodies: The antibodies used in this study were rabbit polyclonal antibodies against fibronectin (FN) (sc-8422, purchased from Santa), connective tissue growth factor (CTGF) (sc-365970, purchased from Santa), TNC (12221, purchased from CST), TLR4 (sc-30002, purchased from Santa), NF-κB p65 (8242, purchased from CST), p-p65 (3033, purchased from CST), and GAPDH (Zhongshan Jinqiao). In addition, miR-155-5p was detected using a kit and primer purchased from Gema (Suzhou, China).
Cell culture: Rat mesangial cells (RMCs) were purchased from the Wuhan Cell Bank (Wuhan Collection Center, China). The cells were cultured in minimal essential medium (Life Technologies, Carlsbad, CA, United States) supplemented with 10% foetal bovine serum (Abgent, San Diego, CA, United States) at a density of 5 × 105 cells/2 mL in a 6-well plate at 37 °C in an atmosphere containing 5% CO2 and 95% air. Cells were pre-treated with serum-free medium overnight before subsequent experiments.
High-glucose screening experiment. After RMCs had grown to a confluence of approximately 50%, the cells were selected and divided into a normal glucose group, high mannitol osmotic control group (osmotic pressure control group), and hyperglycaemic group and cultured as described below: (1) The normal glucose control group (NG, 5.5 mmol/L glucose) was cultured for 24 h, and cells were then collected; (2) The high mannitol osmotic control group (5.5 mmol/L glucose + 24.5 mmol/L mannitol; the osmotic pressure was the same as the high glucose group) was cultured for 24 h, and cells were then collected; and (3) The high glucose group (30 mmol/L glucose) was cultured for 24 h, and cells were then collected. The collected cells and supernatants were stored in a freezer at -80 °C. Real-time polymerase chain reaction (PCR) was conducted to detect the expression of miR-155-5p in RMCs, Western blot was used to detect the levels of the TNC, TLR4, p-p65, NF-κB p65, CTGF, and FN proteins in RMCs, and ELISA was used to detect the levels of TNC expression in supernatants.
Transient transfection. Small interfering RNAs (siRNAs) targeting TNC, TLR4, and miR-155-5p were designed and synthesized by Gema (Shanghai, China). All of the sequences are listed in Table 1. Lipofectamine 2000 (Life Technologies, Carlsbad, CA, United States) was placed into four plates and used for transient transfection according to the manufacturer’s instructions. The cells were plated in 6-well plates, and the siRNAs were then added. After transfection for 6 h, the media were replaced with normal- or high-glucose medium.
Table 1.
Sequences of siRNAs used for transfection
|
Gene
|
Sequence
|
|
| TNC-siRNA | siRNA-TNC-T1 sense | 5’-GCA UCU GUG AUG AUG ACU ATT-3’ |
| Anti-sense | 5’-UAG UCA UCA UCA CAG AUG CTT-3’ | |
| siRNA-TNC-T2 sense | 5’-GCU GAG AAG GGC AGA CAU ATT-3’ | |
| Anti-sense | 5’-UAU GUC UGC CCU UCU CAG CTT-3’ | |
| siRNA-TNC-T3 sense | 5’-GCA UUU GUG AGG AUG GUU UTT-3’ | |
| Anti-sense | 5’-AAA CCA UCC UCA CAA AUG CTT-3’ | |
| NC-siRNA-T | 5’-UUC UCC GAA CGU GUC ACG UTT-3’ | |
| Anti-sense | 5’-ACG UGA CAC GUU CGG AGA ATT-3’ | |
| TLR4-siRNA | siRNA-TLR4 | 5’-CGA GCU GGU AAA GAA UUU ATT-3’ |
| Anti-sense | 5’-UAA AUU CUU UAC CAG CUC GTT-3’ | |
| NC-siRNA-TLR4 sense | 5’-UUC UCC GAA CGU GUC ACG UTT-3’ | |
| Anti-sense | 5’-ACG UGA CAC GUU CGG AGA ATT-3’ | |
| miR-155-5p inhibitor | Inhibitor | 5’-ACC CCU AUC ACA AUU AGC AUU AA-3’ |
| Inhibitor-NC | 5’-CAG UAC UUU UGU GUA GUA CAA-3’ |
TNC: Tenascin-C; TLR4: Toll-like receptor-4; NC: Normal control.
TNC screening experiment. The siRNAs designed by Gema were screened. Appropriate fragments for subsequent experiments were selected according to their transfection efficiencies.
Signaling pathway experiment. TNC blocking peptide (3628BP-50, Biovision) was used to block TNC. TN-C blocking peptide (0.5 mg/mL) was incubated with RMCs at 37 °C for 1 h and then high glucose stimulation was performed. Western blot was subsequently performed as above. RMCs were treated with 2.5 μg/mL recombinant TNC (r-TNC; CC065, Sigma). All RMCs were treated for 24 h and collected for subsequent experiments.
Metformin treatment. Metformin powder was purchased from Beyotime (s1741). A metformin stock solution was prepared and diluted to 1, 5, 10, 20, and 50 μmol/L metformin solutions. RMCs were divided into MET (5.5 mmol/L glucose + 10 μmol/L metformin), 24H (30 mmol/L glucose), H+1 (30 mmol/L glucose + 1 μmol/L metformin), H+5 (30 mmol/L glucose + 5 μmol/L metformin), H+10 (30 mmol/L glucose + 10 μmol/L metformin), H+20 (30 mmol/L glucose + 20 μmol/L metformin), and H+50 ( 30 mmol/L glucose + 50 μmol/L metformin) groups. All RMCs were treated for 24 h and collected for subsequent experiments.
MiRNA extraction and detection of miR-155-5p expression using real-time quantitative PCR
The miRNAs were isolated using a miRcute miRNA extraction kit (Gema, Shanghai, China) according to the manufacturer’s instructions. Five microliters of miRNAs were used for reverse transcription using the miRcute miRNA first strand cDNA synthesis kit (Gema, Shanghai, China). A miRcute miRNA qPCR kit (SYBR Green; Gema, Shanghai, China) was used for PCR with 2.5 μL of cDNA templates and the following protocol: 40 cycles of 94 °C for 2 min, 94 °C for 20 s, and 60 °C for 34 s. The expression in each sample was standardized to the corresponding U6 expression level. The experimental data were analysed using the 2--Δ-ΔCt method. Each sample was tested three times. Experiments were repeated independently six times under the same experimental conditions.
Protein extraction and Western blot analysis
Radioimmunoprecipitation buffer containing phenylmethanesulfonyl fluoride as a protease inhibitor and phosphatase inhibitors was used for cell lysis. The protein concentration was detected with a BCA kit (Beyotime Institute of Biotechnology, China), and a standard curve was generated with bovine serum albumin (BSA). Samples were mixed with 5 × loading buffer and heated in a water bath at 100 °C for 5 min. Equivalent amounts of proteins (50 μg for each sample) were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis with an 8%-12% separating gel and then transferred to a methanol-activated polyvinylidene fluoride membrane using a constant current of 200 mA. BSA was diluted with Tris-buffered saline-Tween 20 (pH 7.6). The membrane was incubated with a 5% BSA solution at room temperature for 2 h and then incubated with the primary antibody at 4 °C overnight. Antibodies against TNC, TLR4, NF-κB p65, p-NF-κB p65 (Ser536), FN, and CTGF were diluted 1:500, while the antibody against GAPDH was diluted 1:1000. After the incubation with the primary antibody and washing, the membrane was sequentially incubated with anti-rabbit and anti-mouse IgG secondary antibodies (1:500). Then, horseradish peroxidase was detected with enhanced chemiluminescence reagent (Beyotime Institute of Biotechnology, China), and a MicroChemi 4.2 Bio-imaging system was used to detect luminescence. Gelpro32 software was used to analyse the grey values of the bands. The densities of phosphorylated proteins were compared with the corresponding total protein densities to detect protein activation. The experiment was repeated independently six times under the same experimental conditions.
Statistical analysis
Analysis of ELISA data: The obtained data were statistically analysed using SPSS statistical analysis software (version 19.0, IBM Corp., United States). The normality of the distribution of each variable was assessed, and normally distributed measured data are reported as the mean ± SE, while measured data with a non-normal distribution are reported as medians (interquartile intervals). For inter-group comparisons of normally distributed measured data, one-way ANOVA was used to determine the significance of differences in the indicators among three or more groups, and the LSD method was used for pairwise comparisons between groups. Indicator data that did not conform to a normal distribution were statistically analysed after logarithmic transformation. The correlation between TNC levels and the UACR was analysed with a ridge regression model. P < 0.05 indicated statistical significance.
Cytological data analysis: The obtained data were statistically analysed using SPSS statistical analysis software (version 19.0, IBM Corp., United States). All data were obtained from six independent experiments and are reported as the mean ± SE. ANOVA was used to analyze differences among three or more groups, and the LSD method was used to analyse normally distributed data. All P values were double-tailed, where P < 0.05 indicated a statistically significant difference and P < 0.001 indicated a highly statistically significant difference.
RESULTS
Serum TNC levels in patients with T2DM are significantly increased, and the expression levels of TNC are positively correlated with diabetes-related indicators
In the present study, the serum TNC level in patients with T2DM was measured using an ELISA. With an increasing UACR, the TNC level was significantly increased, and the TNC level was further increased in the hypertensive group compared to the control group (P < 0.05) (Table 2). According to the statistical analysis, serum TNC levels in patients with T2DM were positively correlated with HbA1c levels, BMI, SBP, and UACR (Table 3). The microalbuminuria and macroalbuminuria groups presented significantly higher levels of TNC than the normal albuminuria group (P < 0.05) (Table 2). The serum TNC level in the macroalbuminuria group was obviously higher than that in the microalbuminuria group (P < 0.05) (Table 2).
Table 2.
Comparison of clinical characteristics and biochemical data among groups [mean ± SEM, M (quartile)]
|
Group
|
n
|
Age (yr)
|
TG (mmol/L)
|
TC (mmol/L)
|
LDL-C (mmol/L)
|
HDL-C (mmol/L)
|
UA (μmol/L) |
HbA1C (%)
|
TNC (pg/mL)
|
UACR (mg/g)
|
SBP (mmHg)
|
DBP (mmHg)
|
HOMA-IR
|
| A | 60 | 50 ± 1.29 | 1.17 (0.92-1.48) | 4.58 ± 0.11 | 2.80 ± 0.11 | 1.44 ± 0.05 | 279.54 ± 8.43 | 5.40 (5.25-5.70) | 15.20 (14.08-15.95) | 9.92 (7.58-11.23) | 119 ± 1.55 | 72 ± 1.29 | 1.02 ± 0.08 |
| B | 70 | 52 ± 1.79 | 0.91 (0.71-1.59) | 5.13 ± 0.15 | 3.40 ± 0.14 | 1.29 ± 0.04 | 265.77 ± 7.08 | 7.30 (6.05-8.70)a | 15.07 (12.10-17.95) | 16.76 (13.58-24.26)a | 118 ± 1.79 | 74 ± 0.84 | 2.53 ± 0.12a |
| C | 105 | 56 ± 1.07 | 2.07 (1.32-3.01)a, b | 4.62 ± 0.07 | 2.88 ± 0.06 | 1.12 ± 0.03a | 340.24 ± 8.17a, b | 8.40 (7.50-9.80)a | 17.07 (14.70-19.88)a, b | 44.85 (33.91-59.59)a, b | 126 ± 0.68 | 77 ± 0.68 | 4.68 ± 0.26a, b |
| D | 75 | 57 ± 1.5 | 1.84 (1.55-3.87)a, b | 4.49 ± 0.13 | 2.89 ± 0.11 | 1.00 ± 0.02a, b | 348.87 ± 10.83a, b | 7.30 (6.60-8.50)a | 18.96 (14.70-21.40)a, b, c | 56.17 (45.80-159.74)a,b, c | 152 ± 1.04a, b, c | 92 ± 1.5a, b, c | 4.85 ± 0.34a, b |
| E | 65 | 52 ± 0.99 | 2.08 (1.76-2.88)a, b | 5.61 ± 0.16a, c, d | 3.68 ± 0.13a, c, d | 1.21 ± 0.03 | 381.11 ± 10.47a, b | 7.80 (7.00-9.25)a | 18.71 (16.40-19.95)a, b, c | 871.58 (390.94-871.58)a, b, c, d | 127 ± 1.36 | 79 ± 0.99 | 5.65 ± 0.45a, b |
| F | 65 | 59 ± 1.36 | 1.99 (1.38-4.07)a, b | 4.84 ± 0.15 | 2.84 ± 0.12e | 1.11 ± 0.04a | 358.38 ± 8.27a, b | 9.10 (7.10-9.70)a | 21.16 (16.92-21.35)a, b, c, d, e | 1094.62 (790.00-5246.40)a, b, c, d, e | 160 ± 1.49a, b, c, e | 96 ± 1.86a, b, c, e | 5.75 ± 0.4a, b |
P < 0.05 compared with group A.
P < 0.05 compared with group B.
P < 0.05 compared with group C.
P < 0.05 compared with group D.
P < 0.05 compared with group E. 1 mmHg = 0.133 kPa; A: Healthy group; B: Normal albuminuria group; C: Microalbuminuria group; D: Microalbuminuria combined with hypertension group; E: Macroalbuminuria group; F: Macroalbuminuria combined with hypertension group. BMI: Body mass index; SBP: Systolic blood pressure; DBP: Diastolic blood pressure; UACR: Urinary albumin to creatinine ratio; HOMA-IR: Homeostasis model assessment-insulin resistance; HOMA-IR: Homeostasis model assessment of insulin resistance, = FRG × FINS / 22.5; HDL-C: High-density lipoprotein-cholesterol; LDL-C: Low-density lipoprotein-cholesterol; TC: Total cholesterol; TG: Triglycerides; UA: Uric acid; TNC: Tenascin-C.
Table 3.
Correlation between tenascin-C levels and urinary albumin excretion rate
|
Index
|
lgTNC
|
||
|
β
|
t
|
P
value
|
|
| Constant | 0.0001 | -31.965 | 0.001 |
| lgHbA1c | 0.405 | 15.893 | 0.001 |
| BMI | 0.110 | 4.090 | 0.002 |
| SBP | 0.341 | 13.379 | 0.002 |
| lgUACR | 0.131 | 4.621 | 0.001 |
BMI: Body mass index; SBP: Systolic blood pressure; 1 mmHg = 0.133 kPa; UACR: Urinary albumin to creatinine ratio; TNC: Tenascin-C.
The prevention and treatment of DKD primarily include the control of blood glucose levels and the use of antihypertensive drugs, and an increase in blood pressure will further increase the rate of urinary albumin excretion[20-22]. Therefore, we assayed the hypertensive group to observe the effect of hypertension on the serum TNC level. Serum TNC levels in the microalbuminuria hypertension and macroalbuminuria hypertension groups were significantly higher than those in the microalbuminuria and macroalbuminuria groups, respectively (P < 0.05) (Table 2). Moreover, the serum TNC level in the macroalbuminuria hypertension group was significantly higher than that in the microalbuminuria hypertension group (P < 0.05) (Table 2).
The serum TNC level was used as the dependent variable, while the HbA1c level, BMI, SBP, and UACR were used as independent variables for the collinearity analysis. The tolerance values revealed by the statistical analysis of collinearity were all less than 0.05, and the variance inflation factor values were all greater than 30, indicating a collinearity between the dependent variable and the independent variables and suggesting that these variables could be included in the ridge regression analysis.
A ridge regression analysis was conducted with the serum TNC level as the dependent variable and the HbA1c level, BMI, SBP, and UACR as independent variables. The following ridge regression equation was obtained: YlgTNC = 0.0001 + 0.405X1 + 0.110X2 + 0.341X3 + 0.131X4 (P < 0.05, X1 = lgHbA1c, X2 = BMI, X3 = SBP, and X4 = lgUACR). From this equation, the HbA1c, BMI, SBP, and UACR coefficients are positive and P < 0.05, indicating the significance of this equation; namely, the TNC level was positively correlated with the HbA1c level, BMI, SBP and UACR. As the HbA1c level, BMI, SBP and UACR increased, the TNC level also increased (Figure 1).
Figure 1.

Relationship between the determination coefficient (R2) and K value, as shown by the ridge regression analysis. RSQ: R-SQUARE; SBP: Systolic blood pressure; BMI: Body mass index; HBA: Glycosylated hemoglobin; UAC: Urinary albumin creatinine ratio.
The expression level of TNC is significantly increased in the kidney tissue and serum of diabetic rats, and is positively correlated with impaired renal function
Each kidney section had at least ten images, and the pathology score of micrographs was blindly evaluated by more than two experienced pathologists. The HE and PAS staining results showed an increase in mesangial dilation and ECM deposition in DKD rats. Further immunohistochemical staining was performed using kidney tissues from rats with DKD. We stained the glomeruli of DKD rats and observed a significant increase in the expression of TNC in the glomeruli of DKD rats compared to the normal rat kidney tissue (P < 0.05) (Figure 2), which suggested that TNC may be involved in the occurrence and development of DKD.
Figure 2.

Histological changes and tenascin-C expression in the kidneys of diabetic rats. A: Hematoxylin-eosin staining; B: Periodic acid-Schiff staining (400 ×); C: Immunohistochemical staining; D: Pathological scoring of tenascin-C (TNC). Scale bar: 20 μm. Representative immunohistochemical images and immunohistochemical scores for TNC in renal section are shown. aP < 0.05 compared with the NC control group. NC, n = 6, DN, n = 6. NC: Normal rat renal section; DN: Diabetic rat renal section; HE: Hematoxylin-eosin; PAS: Periodic acid-Schiff; TNC: Tenascin-C.
In clinical practice, serum creatinine is the most commonly used indicator to determine whether the kidney is damaged. When the glomerular damage is severe and the filtration rate decreases, the serum creatinine increases, which can accurately reflect the degree of kidney damage. As the main component of low-molecular-weight nitrogen-containing substances excreted by the kidneys, urea nitrogen is of great significance for the diagnosis of the course and prognosis of renal diseases. After the diagnosis of diabetes, a kidney examination found that serum creatinine and urea nitrogen increased, indicating that the patient’s kidneys were damaged. Therefore, we detected the expression of TNC, serum creatinine, and urea nitrogen in the serum of diabetic rats and normal rats. We found that the expression levels of serum TNC, urea nitrogen, and creatinine in diabetic rats were significantly higher than those of the normal group (P < 0.05) (Table 4). Pearson correlation analysis showed that TNC levels were positively correlated with creatinine and urea nitrogen (r = 0.796 and 0.958, respectively, P < 0.01).
Table 4.
Comparison of expression of tenascin-C, creatinine, and urea nitrogen in serum of rats [mean ± SD, M (quartile)]
|
Group
|
n
|
TNC (ng/mL)
|
Creatinine (mmol/L)
|
Urea nitrogen (mmol/L)
|
| N | 6 | 40.85 ± 2.99 | 37.27 ± 6.41 | 6.02 ± 1.57 |
| D | 6 | 59.25 ± 3.46a | 129.08 ± 39.88a | 22.48 ± 2.71 |
P < 0.05 compared with group. N, n = 6 for each group. N: Normal rat serum; D: Diabetic rat serum; TNC: Tenascin-C.
High-glucose stimulation increases the levels of TNC protein in RMCs
The mechanism underlying the correlation between TNC levels and the UACR has not been reported. Cell-based experiments were conducted to further explore this mechanism. We used RMCs (HBZY-1 cells) for the experiment designed to analyse the effect of high-glucose stimulation on TNC expression. RMCs were stimulated with normal glucose (5.5 mmol/L glucose) or hypertonic glucose (5.5 mmol/L glucose + 24.5 mmol/L mannitol) for 72 h and with high glucose (30 mmol/L glucose) for 24, 48, and 72 h. Western blot analysis showed that high-glucose stimulation increased the level of TNC protein, which peaked at 24 h (P < 0.05) (Figure 3). Therefore, we applied high-glucose stimulation for 24 h in subsequent experiments. In addition, 24.5 mmol/L mannitol was added to the normal glucose group each time to control for permeability, and the addition of mannitol did not significantly increase the level of TNC protein (Figure 3). Thus, the increase in the levels of the TNC protein in RMCs treated with high glucose was not caused by an alteration in the osmotic pressure.
Figure 3.

Changes in the levels of the tenascin-C and toll-like receptor-4 proteins and nuclear factor-κB p65 protein (Ser536) phosphorylation in cells stimulated with high glucose. A and B: Protein bands and protein expression of tenascin-C (TNC). Rat mesangial cells (RMCs) were cultured under normal (NG, 5.5 mmol/L glucose) and hypertonic (HM, 5.5 mmol/L glucose + 24.5 mmol/L mannitol) conditions for 72 h, or high-glucose (HG, 30 mmol/L glucose) conditions for 24, 48, and 72 h. aP < 0.05 compared with the 72 h control group; C-G: Protein bands and protein expression of TNC, Toll-like receptor-4, and phosphorylated nuclear factor-κB p65 protein (Ser536). RMCs were cultured under normal (NG, 5.5 mmol/L glucose), hypertonic (HM, 5.5 mmol/L glucose + 24.5 mmol/L mannitol), and high-glucose (HG, 30 mmol/L glucose) conditions for 24 h. aP < 0.05 compared with the N control group. Protein levels were detected using Western blot. The results are presented as the mean ± SD of six independent experiments after normalization to GAPDH levels. TNC: Tenascin-C; TLR4: Toll-like receptor-4; p-NF-κB p65: Phosphorylated nuclear factor-κB p65 protein (Ser536); N: Normal glucose; M: Hypertonic; H: High-glucose.
Moreover, the TNC expression level in the high glucose treated RMC supernatant was significantly higher than that in the normal glucose group (P < 0.05) (Table 5).
Table 5.
Comparison of expression of tenascin-C in the supernatant of rat mesangial cell culture [mean ± SD, M (quartile)]
|
Group
|
n
|
TNC (ng/mL)
|
| Ns | 6 | 57.43 ± 5.94 |
| Hs | 6 | 70.13 ± 2.84a |
P < 0.05 compared with group Ns, n = 6 for each group. Ns: Normal condition supernatant (NG, 5.5 mmol/L glucose). Hs: High-glucose condition supernatant (HG, 30 mmol/L glucose).
High-glucose stimulation of RMCs increases TLR4 expression through TNC
As shown in previous studies, TNC and TLR4 jointly promote the inflammatory response through a loop in macrophages[6,23-25], monocytes[26], dendritic cells[27], bone marrow stem cells[26,28], adipocytes[29,30], astrocytes[31], and liver cancer cells[32]. RMCs were separately cultured under normal glucose, hypertonic, and hyperglycaemic conditions to explore the role of TLR4 in RMCs cultured with high glucose concentrations. Compared with RMCs cultured with normal glucose levels, high-glucose-cultured RMCs exhibited significantly increased levels of TLR4 protein (P < 0.05) (Figure 3). However, a significant difference in the level of TLR4 protein was not observed between RMCs cultured under normal-glucose and hyperosmotic conditions (Figure 3), excluding an effect of osmotic pressure.
We used siRNA technology to silence the expression of TNC and further confirm its effect on TLR4 expression in RMCs cultured with high glucose concentrations. First, three different siRNAs for TNC (siRNA-TNC-T1, siRNA-TNC-T2, and siRNA-TNC-T3) were used to silence TNC expression, and non-specific siRNA (NC-siRNA-T) was used as a control (Table 1). Cells were transfected for 6 h and cultured with normal glucose concentrations for 24 h. Compared with the normal control (N) group, transfection with the three different siRNAs targeting TNC reduced the expression of TNC (P < 0.05) (Figure 4). Among the three siRNAs, siRNA-TNC-T2 displayed the highest transfection efficiency (P < 0.05) (Figure 4). Therefore, siRNA-TNC-T2 was used in subsequent experiments. A significant difference in TNC expression was not observed between the N and NC-siRNA-T groups (P > 0.05) (Figure 4). Therefore, the effect of the transfection on the experimental results was eliminated. Next, siRNA-TNC-T2 was used to silence the expression of the TNC gene, and NC-siRNA-T was used as a blank control. After transfection for 6 h, RMCs were cultured with normal glucose or high glucose concentrations for 24 h. Compared with RMCs cultured with normal glucose concentrations, those cultured with high glucose concentrations exhibited significantly increased TLR4 expression (P < 0.05) (Figure 5). However, this effect was blocked by siRNA-TNC-T2 (P < 0.05) (Figure 5). In addition, the expression of TLR4 in cells cultured with normal glucose concentrations was not affected by siRNA-TNC-T2 (P > 0.05) (Figure 5).
Figure 4.

Screening siRNAs to silence the expression of tenascin-C protein. A: Protein bands; B: Protein expression of tenascin-C (TNC). Rat mesangial cells (RMCs) were transfected with siRNA-TNC-T1, siRNA-TNC-T2, siRNA-TNC-T3, and siRNA-NC. RMCs were cultured in normal glucose (NG, 5.5 mmol/L glucose) medium for 24 h. aP < 0.05 compared with untransfected RMCs cultured with normal glucose concentrations. The level of the tenascin-C protein was detected using Western blot. The results are presented as the mean ± SD of six independent experiments after normalization to GAPDH levels. TNC: Tenascin-C; NC: Negative control; N: Normal glucose.
Figure 5.

Silencing of tenascin-C protein expression inhibits the expression of Toll-like receptor-4 and fibrosis factors (connective tissue growth factor and fibronectin), as well as the phosphorylation of p65. A: Protein bands; B-F: Protein expression of tenascin-C (TNC), Toll-like receptor-4 (TLR4), phosphorylated nuclear factor-κB p65 (Ser536) (p-NF-κB p65), connective tissue growth factor (CTGF), and fibronectin (FN). Rat mesangial cells (RMCs) were transfected with siRNA-TNC-T2 for 6 h, and the media were then replaced with normal-glucose (NG, 5.5 mmol/L glucose) or high-glucose (HG, 30 mmol/L glucose) medium for 24 h (N, 5.5 mmol/L glucose). RMCs were transfected with siRNA-TNC-T2 and siRNA-NC for 6 h, and the media were then replaced with normal-glucose (NG, 5.5 mmol/L glucose) or high-glucose (HG, 30 mmol/L glucose) medium for 24 h. aP < 0.05 compared with RMCs cultured with normal glucose concentrations; cP < 0.05 compared with RMCs cultured with high glucose concentrations. TNC, TLR4, p-NF-κB p65, NF-κB p65, CTGF, and FN levels were all detected using Western blot. The results are presented as the mean ± SD of six independent experiments after normalization to GAPDH levels. TNC: Tenascin-C; TLR4: Toll-like receptor-4; p-p65: Phosphorylated nuclear factor-κB p65 (Ser536); p65: Nuclear factor-κB p65; CTGF: Connective tissue growth factor; FN: Fibronectin; N: Normal control; NC: Negative control; H: High glucose.
Therefore, the inhibition of TNC expression decreases the level of TLR4 in high-glucose-cultured RMCs.
TNC expressed in high-glucose-cultured RMCs induces the expression of NF-κB through TLR4
The inhibition of NF-κB alters the expression of TNC in primary human monocytes[28]. After TNC stimulation, the expression of the TLR4 mRNA is significantly increased in Huh 7.0 (hepatocellular carcinoma) cells, and NF-κB fluorescence in the nucleus is increased[1]. In subsequent experiments, we decided to verify the effect of TNC on TLR4/NF-κB in high-glucose-cultured RMCs.
First, we cultured RMCs under normal-glucose, hyperosmotic, and hyperglycaemic conditions. Compared with the levels measured after culture with normal glucose concentrations, NF-κB p65 phosphorylation was significantly increased after culture with high glucose concentrations for 24 h (P < 0.05) (Figure 3). However, a significant difference in the level of NF-κB p65 phosphorylation was not observed between cells cultured under normal-glucose and hyperosmotic conditions (P > 0.05) (Figure 3). Therefore, we eliminated the potential effect of osmotic pressure on the experimental results.
Based on the results from previous studies by our research group, siRNA-TLR4 was used to silence the expression of the TLR4 gene (Table 1), and a non-specific siRNA (NC-siRNA-TL) was used as a control. RMCs were transfected with the siRNAs for 6 h and cultured with normal glucose or high glucose concentrations for 24 h. Notably, siRNA-TLR4 significantly reduced the level of NF-κB p65 phosphorylation upon high-glucose stimulation (P < 0.05) (Figure 6) but had no effect on NF-κB p65 phosphorylation in cells cultured with normal glucose concentrations. In addition, the silencing of TLR4 expression inhibited not only NF-κB p65 phosphorylation but also the expression of the upstream protein TNC (P < 0.05) (Figure 6). Based on these results, TLR4 silencing decreases the activation of the NF-κB p65 signalling pathway induced by high glucose concentrations. Furthermore, transfection with siRNA-TNC-T2 not only inhibited the expression of the TLR4 protein (P < 0.05) (Figure 5) but also decreased the phosphorylation of NF-κB p65 in high-glucose-cultured RMCs (P < 0.05) (Figure 5).
Figure 6.

Silencing Toll-like receptor-4 protein expression inhibits the expression of tenascin-C and fibrosis factors (connective tissue growth factor and fibronectin), as well as nuclear factor-κB p65 phosphorylation. A: Protein bands; B-F: Protein expression of tenascin-C (TNC), Toll-like receptor-4 (TLR4), phosphorylated nuclear factor-κB p65 (Ser536) (p-NF-κB p65), connective tissue growth factor (CTGF), and fibronectin (FN). Rat mesangial cells (RMCs) were transfected with siRNA-TLR4 for 6 h, and the media were then replaced with normal-glucose (NG, 5.5 mmol/L glucose) or high-glucose (HG, 30 mmol/L glucose) medium for 24 h (NG, 5.5 mmol/L glucose), (HG, 30 mmol/L glucose). RMCs were transfected with siRNA-TLR4 and siRNA-NC for 6 h, and the media were then replaced with normal-glucose (NG, 5.5 mmol/L glucose) or high-glucose (HG, 30 mmol/L glucose) medium for 24 h. aP < 0.05 compared with RMCs cultured with normal glucose concentrations; cP < 0.05 compared with RMCs cultured with high glucose concentrations. TNC, TLR4, p-NF-κB p65, NF-κB p65, CTGF, and FN levels were detected using Western blot. The results are presented as the mean ± SD of six independent experiments after normalization to GAPDH levels. TNC: Tenascin-C; TLR4: Toll-like receptor-4; p-p65: Phosphorylated nuclear factor-κB p65 (Ser536); p65: Nuclear factor-κB p65; CTGF: Connective tissue growth factor; FN: Fibronectin; N: Normal control; NC: Negative control; H: High glucose.
High glucose induces the expression of miR-155-5p in RMCs
Pre-miR-155 adopts two mature forms, miR-155-3p and miR-155-5p, of which miR-155-5p is the most abundant[33]. Most studies of DKD have focused on miR-155-5p. Therefore, in this experiment, we investigated the effect of high glucose concentrations on miR-155-5p levels in RMCs. RMCs were treated with high glucose or normal glucose concentrations (as a control) for 0.25, 0.5, 1, 2, 4, 8, 12, 24, 36, and 48 h. Real-time PCR results revealed an increase in the expression of miR-155-5p in cells cultured with high-glucose medium; its expression peaked at 4 h and was 24.03-fold higher than miR-155-5p expression in cells cultured in normal-glucose medium (P < 0.05) (Figure 7).
Figure 7.
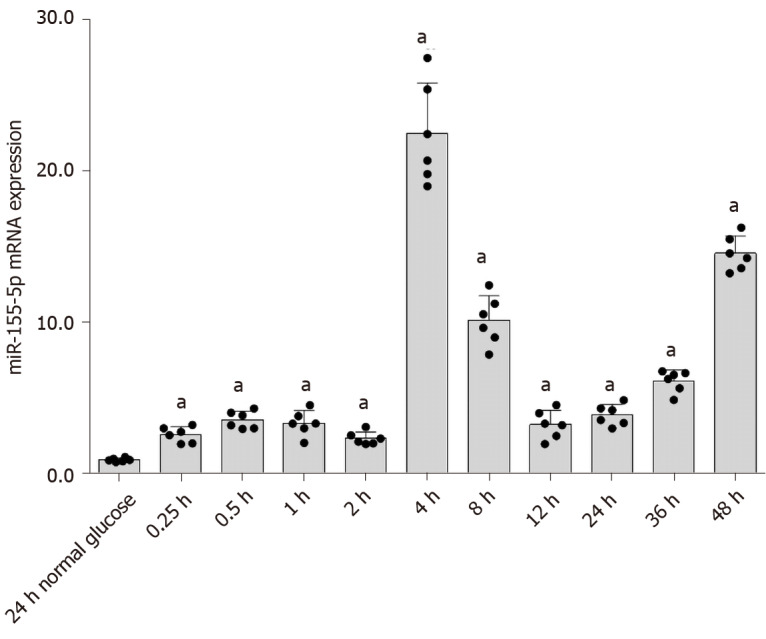
Expression of miR-155-5p induced by high glucose treatment for different durations. Rat mesangial cells (RMCs) cultured under normal-glucose (NG, 5.5 mmol/L glucose) and high-glucose (HG, 30 mmol/L glucose) conditions were treated with high glucose concentrations for 0.25, 0.5, 1, 2, 4, 8, 12, 24, 36, and 48 h. The expression of miR-155-5p increased beginning at 0.25 h and peaked after 4 h. aP < 0.05 compared with RMCs cultured with normal glucose concentrations. The expression of miR-155-5p was quantified using real-time polymerase chain reaction. The results were normalized to the expression of the U6 mRNA and are presented as the mean ± SD of six independent experiments.
TNC regulates the expression of miR-155-5p in high-glucose-cultured RMCs through the TLR4/NF-κB pathway
In bone marrow-derived macrophages, LPS stimulation significantly increases the expression of miR-155, which is significantly inhibited in TNC knockout (TNKO) mice[33]. In addition, in a liver fibrosis study, increased expression of miR-155 was observed in Kupffer cells, along with increased expression of TLR4 protein[34,35]. Furthermore, an NF-κB inhibitor inhibits the expression of miR-155 in RAW264.7 macrophages[36]. Thus, TNC may regulate the expression of miR-155 through the TLR4/NF-κB pathway, but the specific mechanism still requires experimental verification.
In the present study, high-glucose stimulation significantly increased the expression of miR-155-5p in RMCs by 5.38-fold (P < 0.05; Figure 8). We transfected cells with siRNA-TLR4 to silence the expression of the TLR4 gene and further confirm the role of TLR4 in increased miR-155-5p levels in RMCs cultured under high-glucose conditions. Real-time PCR showed a significant decrease in the expression of miR-155-5p after TLR4 silencing, and miR-155-5p was expressed at 2.29-fold lower levels than in cells cultured under high-glucose conditions (P < 0.05; Figure 8). Furthermore, transfection of RMCs with siRNA-TNC-T2 not only inhibited the activity of TLR4 and phosphorylation of NF-κB p65 (P < 0.05; Figure 5) but also downregulated the expression of miR-155-5p by 2.12-fold (P < 0.05; Figure 9) in high-glucose-cultured RMCs.
Figure 8.
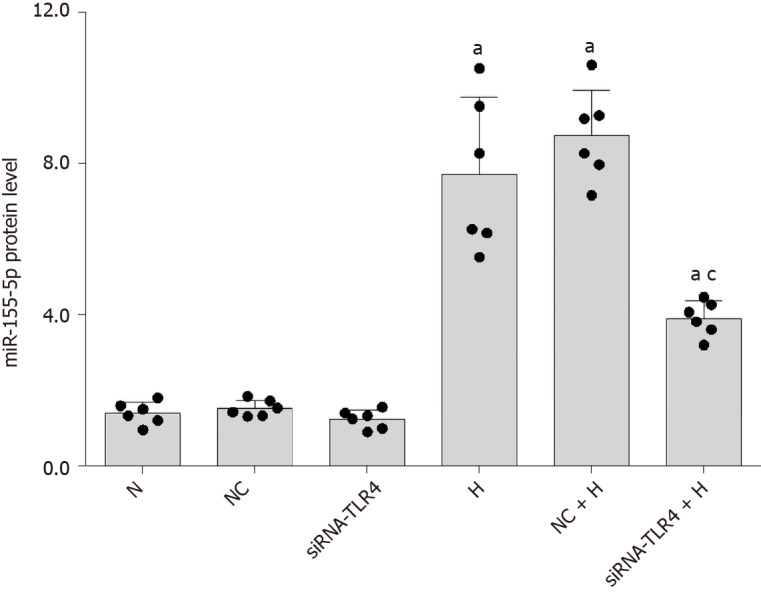
Silencing of Toll-like receptor-4 expression inhibits miR-155-5p expression. Rat mesangial cells (RMCs) were transfected with siRNA-TLR4 to silence Toll-like receptor-4 (TLR4) expression and siRNA-NC for 6 h, and the media were then replaced with normal-glucose (NG, 5.5 mmol/L glucose) or high-glucose (HG, 30 mmol/L glucose) medium for 24 h. aP < 0.05 compared with RMCs cultured with normal glucose concentrations; cP < 0.05 compared with RMCs cultured with high glucose concentrations. The expression of miR-155-5p was quantified using real-time polymerase chain reaction. The results were normalized to the expression of the U6 mRNA and are presented as the mean ± SD of six independent experiments. TLR4: Toll-like receptor-4; N: Normal control; NC: Negative control; H: High-glucose.
Figure 9.
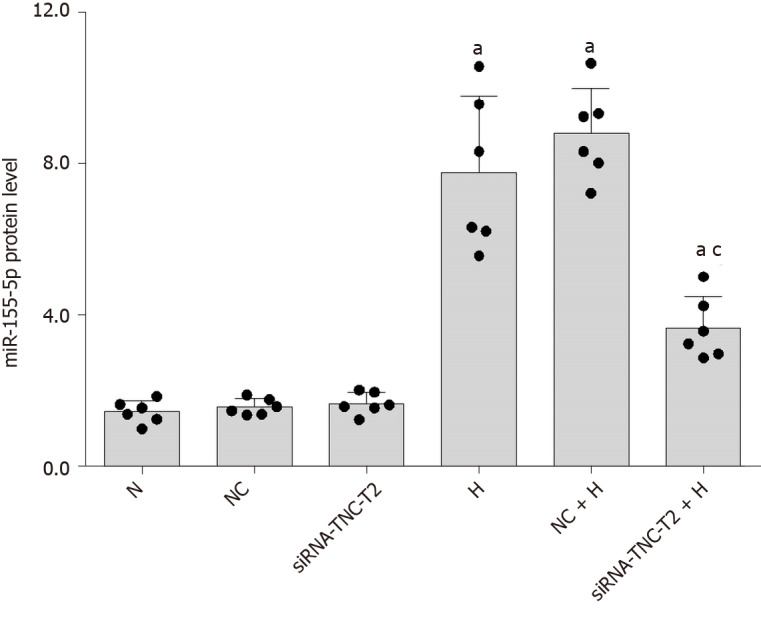
Silencing of tenascin-C expression inhibits miR-155-5p expression. Rat mesangial cells (RMCs) were transfected with siRNA-TNC-T2 and siRNA-NC for 6 h, and the media were then replaced with normal-glucose (NG, 5.5 mmol/L glucose) or high-glucose (HG, 30 mmol/L glucose) medium for 24 h. aP < 0.05 compared with RMCs cultured with normal glucose concentrations; cP < 0.05 compared with RMCs cultured with high glucose concentrations. The expression of miR-155-5p was quantified using real-time polymerase chain reaction. The results were normalized to the expression of the U6 mRNA and are presented as the mean ± SD of six independent experiments. N: Normal control; NC: Negative control; H: High-glucose.
TNC regulates the expression of inflammatory and fibrogenic factors in high-glucose-treated RMCs through the TLR4/NF-κB/miR-155-5p pathway
In the unilateral ureteral obstruction (UUO) model, the level of TNC protein was significantly increased in day 7, while PCR showed that the level of the TNC mRNA started to increase on day 1 after UUO, indicating that TNC was produced before the development of renal fibrosis. The stimulation of rat stromal fibroblasts (NRK-49F cells) with the recombinant human TNC protein significantly increased the number of NRK-49F cells, suggesting that TNC promotes the proliferation of renal fibroblasts. TNC, a secreted protein that is part of the ECM, is involved in formation of the ECM microenvironment. Overexpression of TNC promotes the expansion of fibroblasts. After decellularization of the UUO kidney, TNC still promoted the proliferation and expansion of fibroblasts, indicating that TNC was concentrated in the matrix. A TNC-specific siRNA was injected into UUO mice, and TNKO significantly reduced the renal expression of the FN and alpha smooth muscle actin proteins, as well as fibrotic injury, as evidenced by Masson’s trichrome staining of collagen deposition[37].
The silencing of TNC expression might inhibit the proliferation of fibroblasts and alleviate the development of renal fibrosis, indicating an important role for TNC in the development of renal fibrosis. Based on the conclusions drawn from the aforementioned experiments, we conducted the next experiment.
First, we used an miR-155-5p inhibitor to specifically inhibit the expression of miR-155-5p. After transfection with the miR-155-5p inhibitor for 6 h, the medium was changed, and cells were cultured with normal or high glucose concentrations for 24 h. The levels of the fibrogenic factors FN and CTGF were decreased (P < 0.05) (Figure 10). Further use of siRNA-TLR4 to inhibit the expression of TLR4 revealed that the silencing of TLR4 expression decreased not only the phosphorylation of the inflammatory factor NF-κB p65 (P < 0.05) (Figure 6) but also the expression of miR-155-5p (P < 0.05) (Figure 8) and the fibrosis factors CTGF and FN (P < 0.05) (Figure 6) in high-glucose-treated RMCs. Then, siRNA-TNC-T2 was used to specifically silence the expression of TNC. Following the specific silencing of TNC expression, TLR4 expression and phosphorylation of the inflammatory factor NF-κB p65 were decreased (P < 0.05) (Figure 5), miR-155-5p expression was decreased (P < 0.05) (Figure 9), and the expression of the fibrosis factors CTGF and FN was decreased (P < 0.05) (Figure 5) in high-glucose-treated RMCs.
Figure 10.

Inhibition of miR-155-5p expression. A: Protein bands; B-F: Protein expression of tenascin-C (TNC), Toll-like receptor-4 (TLR4), phosphorylated nuclear factor-κB p65 (Ser536) (p-NF-κB p65), connective tissue growth factor (CTGF), and fibronectin (FN). Rat mesangial cells (RMCs) were transfected with a miR-155-5p inhibitor to inhibit the expression of miR-155-5p (IN) or non-specific inhibitor (IN-NC) for 6 h and further cultured with normal glucose or high glucose concentrations for 24 h. The inhibition of miR-155-5p reduced the levels of TNC, TLR4, fibrosis factors (CTGF and FN), and p-NF-κB p65. RMCs were transfected with siRNA-IN to inhibit miR-155-5p and siRNA-NC for 6 h, and the media were then replaced with normal-glucose (NG, 5.5 mmol/L glucose) or high-glucose (HG, 30 mmol/L glucose) medium for 24 h. aP < 0.05 compared with RMCs cultured with normal glucose concentrations; cP < 0.05 compared with RMCs cultured with high glucose concentrations. TNC, TLR4, p-NF-κB p65, NF-κB p65, CTGF, and FN levels were all detected using Western blot. The results are presented as the mean ± SD of six independent experiments after normalization to GAPDH levels. TNC: Tenascin-C; TLR4: Toll-like receptor-4; p-NF-κB p65: Phosphorylated nuclear factor-κB p65 (Ser536); NF-κB p65: Nuclear factor-κB p65; CTGF: Connective tissue growth factor; FN; Fibronectin; N: Normal control; NC: Negative control; H: High-glucose.
Inhibition of miR-155-5p expression reduces the expression of TNC, TLR4, and NF-κB in RMCs cultured under high-glucose conductions
We transfected RMCs with the miR-155-5p inhibitor to further confirm the effect of miR-155-5p on TNC, TLR4, and NF-κB p65 levels (Table 6). When the expression of miR-155-5p was inhibited, the levels of the TNC, TLR4, and phosphorylated NF-κB p65 proteins in RMCs were significantly reduced in cells stimulated with hyperglycaemia (P < 0.05) (Figure 10).
Table 6.
Real-time polymerase chain reaction primer sequences
|
Primer
|
Sequence
|
|
| miR-155-5p | Forward | 5’-GGT GCG GTT AAT GCT AAT TGT G-3’ |
| Reverse | 5’-CAG AGC AGG GTC CGA GGTA-3’ | |
| U6 | Forward | 5’-CGC TTC GGC AGC ACA TAT AC-3’ |
| Reverse | 5’-TTC ACG AAT TTG CGT GTC ATC-3’ |
Signaling pathway experiment
TNC inhibitory antibodies and recombinant proteins were used to stimulate rat mesangial cells, respectively, and it was found that inhibitory antibodies significantly reduced the expression of TLR4, CTGF, and FN proteins under high glucose stimulation, while the expression of TLR4, CTGF and FN was significantly increased after the addition of r-TNC protein; the expression of CTGF and FN could be restored when silencing TLR4 under r-TNC stimulation. These results suggested that TNC downregulated inflammatory and fibrosis factors through TLR4/NF-κB (P < 0.05) (Figure 11).
Figure 11.

Signaling pathway experiment. A: Protein bands; B-F: Protein expression of Toll-like receptor-4 (TLR4), phosphorylated nuclear factor-κB p65 (Ser536) (p-NF-κB p65), connective tissue growth factor (CTGF), and fibronectin (FN). Rat mesangial cells (RMCs) were treated with TNC inhibitory antibody or TNC recombinant protein with or without transfected with siRNA-TLR4. RMCs cultured with normal glucose concentrations (N, 5.5 mmol/L glucose) or high-glucose (H, 30 mmol/L glucose) medium for 24 h, RMCs were pretreated with 0.5 mg/mL TNC blocking peptide and cultured with high-glucose (Anti-TNC, 30 mmol/L glucose), RMCs were treated with 2.5 ug/mL recombinant TNC (r-TNC), RMCs were transfected with siRNA-TLR4 to silence TLR4 expression for 6 h, and the media were then replaced with normal-glucose (NG, 5.5 mmol/L glucose) and treated with 2.5 ug/mL r-TNC(r-TNC+siTLR4). All RMCs were treated for 24 h and collected for subsequent experiments. aP < 0.05 compared with RMCs cultured with normal glucose concentrations; cP < 0.05 compared with RMCs cultured with high glucose concentrations; eP < 0.05 compared with RMCs treated with r-TNC. TLR4, p-NF-κB p65, NF-κB p65, connective tissue growth factor, and fibronectin levels were all detected using Western blot. The results are presented as the mean ± SD of six independent experiments after normalization to GAPDH levels. TLR4: Toll-like receptor-4; p-NF-κB p65: Phosphorylated nuclear factor-κB p65 (Ser536); NF-κB p65: Nuclear factor-κB p65; CTGF: Connective tissue growth factor; FN: Fibronectin; N: Normal control; H: High-glucose.
Metformin regulates the expression of inflammatory and fibrosis factors in DKD models by regulating TNC levels
Metformin inhibits the inflammatory response of endothelial cells by inhibiting NF-κB through an AMPK-dependent pathway[38]. However, metformin has not been reported to inhibit the inflammatory response through TNC. By reviewing the literature on the effect of metformin on NF-κB, we concluded that metformin may inhibit TNC expression through NF-κB p65. Therefore, we designed the experiment described below.
In previous studies, colon cancer cells were treated with metformin at concentrations of 0.5, 1, 5, 10, and 50 mmol/L[39]. In a study of the protective effect of metformin on nerve cells, the effect was most pronounced at 20 mmol/L[40]. Based on these studies, we selected the following concentration of metformin to treat cells.
RMCs were divided into the MET, 24H, H+1, H+5, H+10, H+20, and H+50 groups. Western blot showed that increasing concentrations of metformin decreased levels of phosphorylated NF-κB p65 and TNC (P < 0.05) (Figure 12). Then, we treated the cells with 20 mmol/L metformin, and observed the levels of downstream inflammatory and fibrosis proteins using Western blot. Metformin significantly reduced the levels of CTGF and FN proteins (P < 0.05) (Figure 12).
Figure 12.

Inhibitory effects of metformin on rat mesangial cells. A-C: Protein bands and protein expression of tenascin-C (TNC) and phosphorylated nuclear factor-κB p65 (Ser536) (p-NF-κB p65). The MET (5.5 mmol/L glucose + 10 μmol/L metformin), 24H (30 mmol/L glucose), H+1 (30 mmol/L glucose + 1 μmol/L metformin), H+5 (30 mmol/L glucose + 5 μmol/L metformin), H+10 (30 mmol/L glucose + 10 μmol/L metformin), H+20 (30 mmol/L glucose + 20 μmol/L metformin), and H+50 (30 mmol/L glucose + 50 μmol/L metformin) groups were cultured with the appropriate medium for 24 h. aP < 0.05 compared with rat mesangial cells (RMCs) cultured with normal glucose concentrations; cP < 0.05 compared with RMCs cultured with high glucose concentrations; D-F: Protein bands and protein expression of connective tissue growth factor (CTGF) and fibronectin (FN). RMCs were divided into normal-glucose (NG, 5.5 mmol/L glucose), high-glucose (HG, 30 mmol/L glucose), and H+20 (30 mmol/L glucose + 20 μmol/L metformin) groups and cultured with the appropriate medium for 24 h. aP < 0.05 compared with RMCs cultured with normal glucose concentrations; cP < 0.05 compared with RMCs cultured with high glucose concentrations. TNC, p-NF-κB p65, and NF-κB p65 levels were detected using Western blot. The results are presented as the mean ± SD of six independent experiments after normalization to GAPDH levels. TNC: Tenascin-C; p-NF-κB p65: Phosphorylated nuclear factor-κB p65 (Ser536); NF-κB p65: Nuclear factor-κB p65; CTGF: Connective tissue growth factor; FN: Fibronectin; N: Normal control; H: High-glucose.
DISCUSSION
The main pathological manifestations of DKD are thickened glomerular basement membrane and increased mesangial matrix synthesis. Degradation of the glomerular filtration barrier leads to the development of proteinuria, an important manifestation of DKD, and proteinuria develops in a dynamic rather than linear manner[15,41,42]. As an ECM protein, TNC activates downstream signalling pathways by binding structural proteins in the ECM and the cell surface receptors EGF and integrin to regulate cell adhesion, migration, and proliferation[37]. According to Mackie et al[43], TNKO mice showed increased mesangial cell apoptosis, reduced proliferation, and reduced ECM deposition, while the addition of exogenous TNC reversed the changes.
Serum TNC levels were increased in patients with T2DM patients with an increase in the UACR, and an increased blood pressure further increased the TNC levels in the present study. In addition, serum TNC levels in patients with T2DM were positively correlated with HbA1c levels and BMI. TNC levels were increased significantly in the group with hypertension compared to the control group. As shown in previous studies, TNC is a tensor protein, and thus tension traction and Ang II may lead to increased TNC expression[44]. In addition, Fujimoto et al[45] observed that TNC promoted cerebral vasoconstriction through TLR4 through a MAPK-dependent pathway. Because hypertension exacerbates proteinuria, we assayed the hypertensive group to observe the effect of hypertension on the expression of TNC in different groups stratified by urine albumin excretion rates, and hypertension increased serum TNC levels in patients with an increasing UACR. TNC may play a role in the development of proteinuria in individuals with DKD. However, the specific mechanism by which TNC promotes the development of DKD remains unclear. In addition, a significantly higher level of TNC protein was observed in the glomeruli of diabetic rats than in the normal control group. Therefore, we performed in vitro cell culture to elucidate this molecular mechanism at the cellular and molecular levels.
Exogenous TNC induces the expression of collagen genes and the transformation of myofibroblasts through TLR4. TLR4 signalling promotes the production and accumulation of TNC in damaged tissues. TNC increases the expression and activity of TLR4 in turn, producing an inflammatory response loop that continuously increases the expression of TNC[46]. TNC and TLR4 not only regulate each other in an inflammatory state but also change cell function through TLR4. For example, TNC stimulation significantly increases the number of osteoclasts, a change that is antagonized by inhibitory antibody against TLR4[47]. In TNKO mice, the degree of fibrosis in cardiomyocytes is significantly reduced compared to control mice. Moreover, TNC regulates the polarization of M1/M2 macrophages through the TLR4 pathway. Furthermore, TNC promotes reactive oxygen species (ROS) production in bone marrow macrophages through TLR4[48].
In this study, it was found that the expression of TNC in the kidney tissues of diabetic rats was significantly increased, and the expression of TNC in the serum of diabetic rats was increased and had a positive correlation with urea nitrogen and creatinine.
TNC expression was significantly increased in RMCs cultured under high-glucose conditions. TNC has been reported to function as an endogenous activator of TLR4-mediated inflammation, and the functional domain of TNC, the fibrinogen-like globe domain, is the major contributor to TLR4 production[6,23,24]. In our study, TLR4 expression was significantly decreased when TNC was silenced by an siRNA in high glucose-cultured RMCs. We then used an siRNA to silence TLR4 expression and observed a significant decrease in the expression of TNC in cells cultured under high-glucose conditions, confirming the interaction between TNC and TLR4 in an anti-inflammatory reaction loop in high glucose-cultured RMCs.
In the present study, the silencing of TLR4 downregulated the expression of the inflammatory factor NF-κB p65 in high glucose-cultured RMCs. The inhibition of TNC also decreased NF-κB p65 phosphorylation. In addition, the expression of TLR4, CTGF, and FN was reduced when TNC expression was inhibited by blocking antibody in the presence of high glucose, while the expression of TLR4, CTGF, and FN was increased when r-TNC protein was added, and the expression levels of CTGF and FN were restored when knocking down TLR4 under r-TNC stimulation. Based on these results, TNC induces the phosphorylation of NF-κB p65 by TLR4 in high-glucose-cultured RMCs.
LPS stimulation of macrophages increases the expression of miR-155 and decreases the expression of SHIP1, which is believed to negatively regulate TLRs. Activation of TLR4 increases the expression of miR-155 and decreases the expression of the negative regulator SHIP1, thus prolonging TLR4 signalling[27]. MiR-155 is located on human chromosome 21[49]. The specific mechanism of miR-155 in the inflammatory response in individuals with T2DM remains unknown. Some studies reported a significant decrease in the expression of miR-155 in the platelets from patients with T2DM[50]. In contrast to its expression in platelets from patients with T2DM, miR-155 expression is significantly upregulated in skin samples from patients with diabetes, and the inhibition of miR-155 significantly reduces the wound inflammatory response[51]. Similarly, increased expression of miR-155 was observed in the endothelial progenitor cells from another group of patients with diabetes. The overexpression of miR-155 significantly reduces cell activity, migration, tubule formation, and NO production, and promotes LDH release, ROS production, and apoptosis[52].
The level of miR-155 is significantly increased in the urine of patients with DKD. Researchers speculated that the excess miR-155 in the urine of patients with DKD is released into the urine through glomerular ultrafiltration. As shown in the analysis of renal biopsies, the expression of miR-155 is significantly increased in patients with DKD and significantly associated with serum creatinine levels. In addition, the absence of miR-155 significantly alleviates kidney injury and IL-17 expression in mice with DKD induced by streptomycin[18]. Further studies of DKD models showed a significant increase in miR-155-5p expression in HK-2 cells stimulated with high glucose concentrations[53]. Reduced podocyte cell damage was observed in high-glucose-stimulated miR-155 −/− mice[54]. Thus, miR-155 has an important role in DKD. However, with the exception of the studies described above, the specific mechanism of miR-155 in DKD has not been studied.
In our study, high glucose concentrations induced the expression of miR-155-5p in RMCs in a time-dependent manner. In RMCs cultured under high-glucose conditions, the silencing of TLR4 with an siRNA altered the phosphorylation of the inflammatory factor NF-κB p65, and miR-155-5p expression was significantly reduced after TLR4 silencing. Similarly, after the silencing of TNC, the levels of TLR4, phosphorylated NF-κB p65, and miR-155-5p were decreased, indicating that TNC induced the expression of miR-155-5p through the TLR4/NF-κB pathway. In RMCs cultured under high-glucose conditions, the inhibition of miR-155-5p decreased the expression of TNC and TLR4 and the phosphorylation of NF-κB p65, indicating that miR-155-5p also induces the expression of TNC and TLR4/NF-κB in turn, thereby forming a complete inflammatory response loop that functions together with downstream fibrosis factors and promotes the development of DKD. Although TNC primarily functions as an ECM protein, it produces a series of subsequent inflammatory responses by communicating with TLR4, a membrane receptor and a bridge between the intracellular and extracellular environments, closely linking extracellular TNC with intracellular NF-κB p65 and miR-155-5p.
Metformin has been widely studied as a first-line drug for treating diabetes. It functions by reducing the hepatic production of glycogen and improving the sensitivity to insulin. Based on accumulating evidence obtained recently, metformin not only reduces blood glucose levels but also protect endothelial cells through its anti-inflammatory, anti-apoptotic, and anti-oxidant activities. Metformin significantly inhibits the migration of NF-κB from the cytoplasm to the nucleus, thereby inhibiting the subsequent inflammatory cascade[19]. Sekino et al[55] performed immunostaining to determine the intracellular localization of NF-κB. In the control group, NF-κB was localized in the cytoplasm and nucleus, while in the metformin-treated group, nuclear NF-κB expression was significantly decreased. Thus, metformin affects the level of NF-κB in the nucleus, thereby altering the activation of the NF-κB signalling pathway and further modulating downstream inflammatory pathways[55]. Therefore, metformin relieves inflammation by inhibiting the expression of NF-κB.
In the present study, we treated RMCs with different concentrations of metformin and detected the levels of TNC and phosphorylated NF-κB p65 using Western blot. The levels of NF-κB p65 phosphorylation and TNC protein were significantly reduced by increasing metformin concentrations. In addition, metformin not only reduced the levels of TNC and phosphorylated NF-κB p65 proteins but also reduced the levels of CTGF and FN proteins, suggesting that metformin might delay the occurrence of inflammation and fibrosis in individuals with DKD by inhibiting TNC expression.
In conclusion, serum TNC levels were increased in patients with DKD with an increasing UACR, and hypertension further increased the TNC levels. In addition, significantly higher TNC expression was observed in the glomeruli of diabetic rats than in the normal control group. In cell-based experiments, TNC induced the expression of miR-155-5p through the TLR4/NF-κB pathway and miR-155-5p induced the expression of TNC and TLR4/NF-κB in turn, thus forming a complete inflammatory response loop. The components of this loop jointly affect the levels of downstream inflammatory and fibrosis factors. After treatment with metformin, the level of TNC protein was reduced, thereby inhibiting downstream inflammatory and fibrotic responses. We explored the mechanism by which increased serum TNC levels promote the development of DKD at the cellular and molecular levels. Our results are expected to further refine the pathological mechanism of DKD and provide new insights into the treatment of DKD.
CONCLUSION
TNC can promote the occurrence and development of DKD. Interfering with the TNC/TLR4/NF-κB p65/mi-155-5p pathway can become a new target for DKD treatment.
ARTICLE HIGHLIGHTS
Research background
With the increasing incidence of diabetes, the incidence of diabetic kidney disease (DKD) continues to rise, which has become the leading cause of end-stage renal disease. However, current treatments like angiotensin-converting enzyme inhibitors only partially inhibited the DKD progression. Thus, the mechanisms underlying DKD should be clarified and disclosed, and new strategies for delaying the progression are urgently needed.
Research motivation
By testing the serum tenascin-C (TNC) level in patients with type 2 diabetic mellitus (T2DM), we aim to provide insights into the pathogenesis of the DKD and suggest that TNC can serve as a therapeutic target for this disease.
Research objectives
In the present study, we evaluated the alterations of TNC expression levels in the serum or/and glomeruli of patients with T2DM and rats with streptozotocin-induced diabetes. We also evaluated the diagnose indexes of DKD, including glycosylated hemoglobin (HbA1c) level, body mass index (BMI), systolic blood pressure (SBP), and urinary albumin to creatinine ratio (UACR) in the serum of patients. In addition, we explored the specific molecular mechanism of TNC on DKD by culturing rat glomerular mesangial cells.
Research methods
Diabetes patient serum samples were collected to detect the expression level of TNC in serum and make analysis with other related factors in diabetes. Diabetic rat models were used to observe the expression of TNC in diabetic rat kidney, and enzyme-linked immunosorbent assay was used to detect the expression of TNC in diabetic rat serum, and analyze the associated renal function indexes. SiRNA transfection technique was used in cultured rat glomerular mesangial cells stimulated with high glucose to explore the molecular mechanisms of TNC in DKD.
Research results
Diabetic patients had significantly increased serum levels of TNC, and TNC was positively correlated with UACR, BMI, SBP, and DBP. TNC expression in diabetic rat kidney increased obviously, and diabetic rats had significantly higher serum TNC expression levels compared with normal rats. Urea nitrogen and creatinine were positively correlated with the increase of TNC in diabetic rats. Rat glomerular mesangial cells stimulated with high glucose had significantly increased protein expression of TNC, and TNC can promote inflammation and fibrosis through the Toll-like receptor 4/nuclear factor-κB pathway. Metformin can inhibit the expression of TNC and delay the progress of DKD.
Research conclusions
We demonstrated that increased TNC expression is involved in the cascade of DKD. Importantly, inhibition of TNC delays the development of DKD, indicating that TNC represents a potential therapeutic target in DKD.
Research perspectives
By targeting TNC expression, the occurrence and development of DKD can be delayed.
Footnotes
Institutional review board statement: This study was approved by The Ethics Committee of The First Affiliated Hospital of China Medical University. All procedures were performed in accordance with the ethical standards mentioned in the 1964 Declaration of Helsinki and its subsequent amendments or comparable ethical standards.
Institutional animal care and use committee statement: All animal experiments conformed to the internationally accepted principles for the care and use of laboratory animals. All experiments were approved by The Institutional Animal Care and Use Committee (IACUC) of The China Medical University Animal Experiment Department, Approval No. 2017112.
Conflict-of-interest statement: The author(s) have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article to declare.
ARRIVE guidelines statement: The authors have read the ARRIVE guidelines, and the manuscript was prepared and revised according to the ARRIVE guidelines.
Manuscript source: Unsolicited manuscript
Peer-review started: August 21, 2020
First decision: September 21, 2020
Article in press: October 26, 2020
Specialty type: Endocrinology and metabolism
Country/Territory of origin: China
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Bartakova V, Fiorina P S-Editor: Huang P L-Editor: Wang TQ P-Editor: Ma YJ
Contributor Information
Yang Zhou, Department of Endocrinology, The First Affiliated Hospital of China Medical University, Shenyang 110000, Liaoning Province, China.
Xiao-Yu Ma, Department of Gerontology, The First Affiliated Hospital of China Medical University, Shenyang 110000, Liaoning Province, China.
Jin-Yu Han, Department of Gerontology, The First Affiliated Hospital of China Medical University, Shenyang 110000, Liaoning Province, China.
Min Yang, Department of Clinical Laboratory, The First Affiliated Hospital of China Medical University, Shenyang 110000, Liaoning Province, China.
Chuan Lv, Department of Endocrinology, The People’s Hospital of China Medical University, Shenyang 110000, Liaoning Province, China.
Ying Shao, Department of Endocrinology, Shengjing Hospital of China Medical University, Shenyang 110000, Liaoning Province, China.
Yi-Li Wang, Department of Endocrinology, The First Affiliated Hospital of China Medical University, Shenyang 110000, Liaoning Province, China.
Jia-Yi Kang, Department of Endocrinology, The First Affiliated Hospital of China Medical University, Shenyang 110000, Liaoning Province, China.
Qiu-Yue Wang, Department of Endocrinology, The First Affiliated Hospital of China Medical University, Shenyang 110000, Liaoning Province, China. wqycmu123@163.com.
Data sharing statement
No additional data are available.
References
- 1.National Kidney Foundation. KDOQI Clinical Practice Guideline for Diabetes and CKD: 2012 Update. Am J Kidney Dis. 2012;60:850–886. doi: 10.1053/j.ajkd.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 2.Jia Y, Guan M, Zheng Z, Zhang Q, Tang C, Xu W, Xiao Z, Wang L, Xue Y. miRNAs in Urine Extracellular Vesicles as Predictors of Early-Stage Diabetic Nephropathy. J Diabetes Res. 2016;2016:7932765. doi: 10.1155/2016/7932765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhattacharjee N, Barma S, Konwar N, Dewanjee S, Manna P. Mechanistic insight of diabetic nephropathy and its pharmacotherapeutic targets: An update. Eur J Pharmacol. 2016;791:8–24. doi: 10.1016/j.ejphar.2016.08.022. [DOI] [PubMed] [Google Scholar]
- 4.Alicic RZ, Rooney MT, Tuttle KR. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin J Am Soc Nephrol. 2017;12:2032–2045. doi: 10.2215/CJN.11491116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiquet-Ehrismann R, Tucker RP. Tenascins and the importance of adhesion modulation. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zuliani-Alvarez L, Marzeda AM, Deligne C, Schwenzer A, McCann FE, Marsden BD, Piccinini AM, Midwood KS. Mapping tenascin-C interaction with toll-like receptor 4 reveals a new subset of endogenous inflammatory triggers. Nat Commun. 2017;8:1595. doi: 10.1038/s41467-017-01718-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiquet-Ehrismann R, Chiquet M. Tenascins: regulation and putative functions during pathological stress. J Pathol. 2003;200:488–499. doi: 10.1002/path.1415. [DOI] [PubMed] [Google Scholar]
- 8.Liabeuf S, Barreto DV, Kretschmer A, Barreto FC, Renard C, Andrejak M, Choukroun G, Massy Z. High circulating levels of large splice variants of tenascin-C is associated with mortality and cardiovascular disease in chronic kidney disease patients. Atherosclerosis. 2011;215:116–124. doi: 10.1016/j.atherosclerosis.2010.11.038. [DOI] [PubMed] [Google Scholar]
- 9.Ishizaki J, Takemori A, Suemori K, Matsumoto T, Akita Y, Sada KE, Yuzawa Y, Amano K, Takasaki Y, Harigai M, Arimura Y, Makino H, Yasukawa M, Takemori N, Hasegawa H Research Committee of Intractable Vasculitis Syndrome and the Research Committee of Intractable Renal Disease of the Ministry of Health; Labour and Welfare of Japan. Targeted proteomics reveals promising biomarkers of disease activity and organ involvement in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Res Ther. 2017;19:218. doi: 10.1186/s13075-017-1429-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hörstrup JH, Gehrmann M, Schneider B, Plöger A, Froese P, Schirop T, Kampf D, Frei U, Neumann R, Eckardt KU. Elevation of serum and urine levels of TIMP-1 and tenascin in patients with renal disease. Nephrol Dial Transplant. 2002;17:1005–1013. doi: 10.1093/ndt/17.6.1005. [DOI] [PubMed] [Google Scholar]
- 11.Truong LD, Pindur J, Barrios R, D'Agati V, Lechago J, Suki W, Majesky M. Tenascin is an important component of the glomerular extracellular matrix in normal and pathologic conditions. Kidney Int. 1994;45:201–210. doi: 10.1038/ki.1994.24. [DOI] [PubMed] [Google Scholar]
- 12.Godfroy JI 3rd, Roostan M, Moroz YS, Korendovych IV, Yin H. Isolated Toll-like receptor transmembrane domains are capable of oligomerization. PLoS One. 2012;7:e48875. doi: 10.1371/journal.pone.0048875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hosoi T, Yokoyama S, Matsuo S, Akira S, Ozawa K. Myeloid differentiation factor 88 (MyD88)-deficiency increases risk of diabetes in mice. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Everard A, Geurts L, Caesar R, Van Hul M, Matamoros S, Duparc T, Denis RG, Cochez P, Pierard F, Castel J, Bindels LB, Plovier H, Robine S, Muccioli GG, Renauld JC, Dumoutier L, Delzenne NM, Luquet S, Bäckhed F, Cani PD. Intestinal epithelial MyD88 is a sensor switching host metabolism towards obesity according to nutritional status. Nat Commun. 2014;5:5648. doi: 10.1038/ncomms6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wada J, Makino H. Innate immunity in diabetes and diabetic nephropathy. Nat Rev Nephrol. 2016;12:13–26. doi: 10.1038/nrneph.2015.175. [DOI] [PubMed] [Google Scholar]
- 16.Bi Y, Liu G, Yang R. MicroRNAs: novel regulators during the immune response. J Cell Physiol. 2009;218:467–472. doi: 10.1002/jcp.21639. [DOI] [PubMed] [Google Scholar]
- 17.Guay C, Kruit JK, Rome S, Menoud V, Mulder NL, Jurdzinski A, Mancarella F, Sebastiani G, Donda A, Gonzalez BJ, Jandus C, Bouzakri K, Pinget M, Boitard C, Romero P, Dotta F, Regazzi R. Lymphocyte-Derived Exosomal MicroRNAs Promote Pancreatic β Cell Death and May Contribute to Type 1 Diabetes Development. Cell Metab 2019; 29: 348-361. :e6. doi: 10.1016/j.cmet.2018.09.011. [DOI] [PubMed] [Google Scholar]
- 18.Beltrami C, Simpson K, Jesky M, Wonnacott A, Carrington C, Holmans P, Newbury L, Jenkins R, Ashdown T, Dayan C, Satchell S, Corish P, Cockwell P, Fraser D, Bowen T. Association of Elevated Urinary miR-126, miR-155, and miR-29b with Diabetic Kidney Disease. Am J Pathol. 2018;188:1982–1992. doi: 10.1016/j.ajpath.2018.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Tao L, Li D, Liu H, Jiang F, Xu Y, Cao Y, Gao R, Chen G. Neuroprotective effects of metformin on traumatic brain injury in rats associated with NF-κB and MAPK signaling pathway. Brain Res Bull. 2018;140:154–161. doi: 10.1016/j.brainresbull.2018.04.008. [DOI] [PubMed] [Google Scholar]
- 20.Dhanesha N, Doddapattar P, Chorawala MR, Nayak MK, Kokame K, Staber JM, Lentz SR, Chauhan AK. ADAMTS13 Retards Progression of Diabetic Nephropathy by Inhibiting Intrarenal Thrombosis in Mice. Arterioscler Thromb Vasc Biol. 2017;37:1332–1338. doi: 10.1161/ATVBAHA.117.309539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh S, Mohanty A. In silico identification of potential drug compound against Peroxisome proliferator-activated receptor-gamma by virtual screening and toxicity studies for the treatment of Diabetic Nephropathy. J Biomol Struct Dyn. 2018;36:1776–1787. doi: 10.1080/07391102.2017.1334596. [DOI] [PubMed] [Google Scholar]
- 22.Tahara A, Takasu T. Prevention of progression of diabetic nephropathy by the SGLT2 inhibitor ipragliflozin in uninephrectomized type 2 diabetic mice. Eur J Pharmacol. 2018;830:68–75. doi: 10.1016/j.ejphar.2018.04.024. [DOI] [PubMed] [Google Scholar]
- 23.Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, Drexler S, Sofat N, Kashiwagi M, Orend G, Brennan F, Foxwell B. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15:774–780. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- 24.Piccinini AM, Zuliani-Alvarez L, Lim JM, Midwood KS. Distinct microenvironmental cues stimulate divergent TLR4-mediated signaling pathways in macrophages. Sci Signal. 2016;9:ra86. doi: 10.1126/scisignal.aaf3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo H, Wang J, Qiao C, Zhang X, Zhang W, Ma N. ATF3 Inhibits Tenascin-C-induced Foam Cell Formation in LPS-Stimulated THP-1 Macrophages by Suppressing TLR-4. J Atheroscler Thromb. 2015;22:1214–1223. doi: 10.5551/jat.28415. [DOI] [PubMed] [Google Scholar]
- 26.Goh FG, Piccinini AM, Krausgruber T, Udalova IA, Midwood KS. Transcriptional regulation of the endogenous danger signal tenascin-C: a novel autocrine loop in inflammation. J Immunol. 2010;184:2655–2662. doi: 10.4049/jimmunol.0903359. [DOI] [PubMed] [Google Scholar]
- 27.Machino-Ohtsuka T, Tajiri K, Kimura T, Sakai S, Sato A, Yoshida T, Hiroe M, Yasutomi Y, Aonuma K, Imanaka-Yoshida K. Tenascin-C aggravates autoimmune myocarditis via dendritic cell activation and Th17 cell differentiation. J Am Heart Assoc. 2014;3:e001052. doi: 10.1161/JAHA.114.001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding H, Jin M, Liu D, Wang S, Zhang J, Song X, Huang R. Tenascin‑C promotes the migration of bone marrow stem cells via toll‑like receptor 4‑mediated signaling pathways: MAPK, AKT and Wnt. Mol Med Rep. 2018;17:7603–7610. doi: 10.3892/mmr.2018.8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Rotellar F, Valentí V, Silva C, Gil MJ, Salvador J, Frühbeck G. Increased tenascin C and Toll-like receptor 4 levels in visceral adipose tissue as a link between inflammation and extracellular matrix remodeling in obesity. J Clin Endocrinol Metab. 2012;97:E1880–E1889. doi: 10.1210/jc.2012-1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao W, Huang H, Xia T, Liu C, Muhammad S, Sun C. Homeobox a5 Promotes White Adipose Tissue Browning Through Inhibition of the Tenascin C/Toll-Like Receptor 4/Nuclear Factor Kappa B Inflammatory Signaling in Mice. Front Immunol. 2018;9:647. doi: 10.3389/fimmu.2018.00647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krasovska V, Doering LC. Regulation of IL-6 Secretion by Astrocytes via TLR4 in the Fragile X Mouse Model. Front Mol Neurosci. 2018;11:272. doi: 10.3389/fnmol.2018.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benbow JH, Thompson KJ, Cope HL, Brandon-Warner E, Culberson CR, Bossi KL, Li T, Russo MW, Gersin KS, McKillop IH, deLemos AS, Schrum LW. Diet-Induced Obesity Enhances Progression of Hepatocellular Carcinoma through Tenascin-C/Toll-Like Receptor 4 Signaling. Am J Pathol. 2016;186:145–158. doi: 10.1016/j.ajpath.2015.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Piccinini AM, Midwood KS. Endogenous control of immunity against infection: tenascin-C regulates TLR4-mediated inflammation via microRNA-155. Cell Rep. 2012;2:914–926. doi: 10.1016/j.celrep.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bala S, Csak T, Kodys K, Catalano D, Ambade A, Furi I, Lowe P, Cho Y, Iracheta-Vellve A, Szabo G. Alcohol-induced miR-155 and HDAC11 inhibit negative regulators of the TLR4 pathway and lead to increased LPS responsiveness of Kupffer cells in alcoholic liver disease. J Leukoc Biol. 2017;102:487–498. doi: 10.1189/jlb.3A0716-310R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bala S, Csak T, Saha B, Zatsiorsky J, Kodys K, Catalano D, Satishchandran A, Szabo G. The pro-inflammatory effects of miR-155 promote liver fibrosis and alcohol-induced steatohepatitis. J Hepatol. 2016;64:1378–1387. doi: 10.1016/j.jhep.2016.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bala S, Marcos M, Kodys K, Csak T, Catalano D, Mandrekar P, Szabo G. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor {alpha} (TNF{alpha}) production via increased mRNA half-life in alcoholic liver disease. J Biol Chem. 2011;286:1436–1444. doi: 10.1074/jbc.M110.145870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu H, Tian Y, Zhou L, Zhou D, Tan RJ, Stolz DB, Liu Y. Tenascin-C Is a Major Component of the Fibrogenic Niche in Kidney Fibrosis. J Am Soc Nephrol. 2017;28:785–801. doi: 10.1681/ASN.2016020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nafisa A, Gray SG, Cao Y, Wang T, Xu S, Wattoo FH, Barras M, Cohen N, Kamato D, Little PJ. Endothelial function and dysfunction: Impact of metformin. Pharmacol Ther. 2018;192:150–162. doi: 10.1016/j.pharmthera.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 39.Park JH, Kim YH, Park EH, Lee SJ, Kim H, Kim A, Lee SB, Shim S, Jang H, Myung JK, Park S, Lee SJ, Kim MJ. Effects of metformin and phenformin on apoptosis and epithelial-mesenchymal transition in chemoresistant rectal cancer. Cancer Sci. 2019;110:2834–2845. doi: 10.1111/cas.14124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bibi F, Ullah I, Kim MO, Naseer MI. Metformin attenuate PTZ-induced apoptotic neurodegeneration in human cortical neuronal cells. Pak J Med Sci. 2017;33:581–585. doi: 10.12669/pjms.333.11996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manda G, Checherita AI, Comanescu MV, Hinescu ME. Redox Signaling in Diabetic Nephropathy: Hypertrophy versus Death Choices in Mesangial Cells and Podocytes. Mediators Inflamm. 2015;2015:604208. doi: 10.1155/2015/604208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gallagher H, Suckling RJ. Diabetic nephropathy: where are we on the journey from pathophysiology to treatment? Diabetes Obes Metab. 2016;18:641–647. doi: 10.1111/dom.12630. [DOI] [PubMed] [Google Scholar]
- 43.Mackie EJ, Tucker RP. The tenascin-C knockout revisited. J Cell Sci. 1999;112 (Pt 22):3847–3853. doi: 10.1242/jcs.112.22.3847. [DOI] [PubMed] [Google Scholar]
- 44.Aguado A, Rodríguez C, Martínez-Revelles S, Avendaño MS, Zhenyukh O, Orriols M, Martínez-González J, Alonso MJ, Briones AM, Dixon DA, Salaices M. HuR mediates the synergistic effects of angiotensin II and IL-1β on vascular COX-2 expression and cell migration. Br J Pharmacol. 2015;172:3028–3042. doi: 10.1111/bph.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujimoto M, Suzuki H, Shiba M, Shimojo N, Imanaka-Yoshida K, Yoshida T, Kanamaru K, Matsushima S, Taki W. Tenascin-C induces prolonged constriction of cerebral arteries in rats. Neurobiol Dis. 2013;55:104–109. doi: 10.1016/j.nbd.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 46.Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X, Lafyatis R, Lee J, Hinchcliff M, Feghali-Bostwick C, Lakota K, Budinger GR, Raparia K, Tamaki Z, Varga J. Tenascin-C drives persistence of organ fibrosis. Nat Commun. 2016;7:11703. doi: 10.1038/ncomms11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kiyeko GW, Hatterer E, Herren S, Di Ceglie I, van Lent PL, Reith W, Kosco-Vilbois M, Ferlin W, Shang L. Spatiotemporal expression of endogenous TLR4 ligands leads to inflammation and bone erosion in mouse collagen-induced arthritis. Eur J Immunol. 2016;46:2629–2638. doi: 10.1002/eji.201646453. [DOI] [PubMed] [Google Scholar]
- 48.Kimura T, Tajiri K, Sato A, Sakai S, Wang Z, Yoshida T, Uede T, Hiroe M, Aonuma K, Ieda M, Imanaka-Yoshida K. Tenascin-C accelerates adverse ventricular remodelling after myocardial infarction by modulating macrophage polarization. Cardiovasc Res. 2019;115:614–624. doi: 10.1093/cvr/cvy244. [DOI] [PubMed] [Google Scholar]
- 49.Pordzik J, Pisarz K, De Rosa S, Jones AD, Eyileten C, Indolfi C, Malek L, Postula M. The Potential Role of Platelet-Related microRNAs in the Development of Cardiovascular Events in High-Risk Populations, Including Diabetic Patients: A Review. Front Endocrinol (Lausanne) 2018;9:74. doi: 10.3389/fendo.2018.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elgheznawy A, Shi L, Hu J, Wittig I, Laban H, Pircher J, Mann A, Provost P, Randriamboavonjy V, Fleming I. Dicer cleavage by calpain determines platelet microRNA levels and function in diabetes. Circ Res. 2015;117:157–165. doi: 10.1161/CIRCRESAHA.117.305784. [DOI] [PubMed] [Google Scholar]
- 51.Moura J, Sørensen A, Leal EC, Svendsen R, Carvalho L, Willemoes RJ, Jørgensen PT, Jenssen H, Wengel J, Dalgaard LT, Carvalho E. microRNA-155 inhibition restores Fibroblast Growth Factor 7 expression in diabetic skin and decreases wound inflammation. Sci Rep. 2019;9:5836. doi: 10.1038/s41598-019-42309-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao J, Zhao G, Li W, Zhang J, Che Y, Song M, Gao S, Zeng B, Wang Y. MiR-155 targets PTCH1 to mediate endothelial progenitor cell dysfunction caused by high glucose. Exp Cell Res. 2018;366:55–62. doi: 10.1016/j.yexcr.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 53.Wang Y, Zheng ZJ, Jia YJ, Yang YL, Xue YM. Role of p53/miR-155-5p/sirt1 loop in renal tubular injury of diabetic kidney disease. J Transl Med. 2018;16:146. doi: 10.1186/s12967-018-1486-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin X, You Y, Wang J, Qin Y, Huang P, Yang F. MicroRNA-155 deficiency promotes nephrin acetylation and attenuates renal damage in hyperglycemia-induced nephropathy. Inflammation. 2015;38:546–554. doi: 10.1007/s10753-014-9961-7. [DOI] [PubMed] [Google Scholar]
- 55.Sekino N, Kano M, Matsumoto Y, Sakata H, Akutsu Y, Hanari N, Murakami K, Toyozumi T, Takahashi M, Otsuka R, Yokoyama M, Shiraishi T, Okada K, Hoshino I, Iida K, Akimoto AK, Matsubara H. Antitumor effects of metformin are a result of inhibiting nuclear factor kappa B nuclear translocation in esophageal squamous cell carcinoma. Cancer Sci. 2018;109:1066–1074. doi: 10.1111/cas.13523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No additional data are available.