

Here we review recent developments in mass spectrometry that have enabled its growing contribution to the field of structural virology. Evidently, there is a strong renewed general interest in viruses and the interactions with their host. Recent years have been especially exciting for MS-based structural virology, largely due to breakthrough developments in high-resolution native MS, single-particle approaches, and new developments in glycoproteomics as well as a growing role for MS in integrative structural biology approaches. Consequently, we focus primarily on recent developments in native MS, glycoproteomics, and hydrogen–deuterium exchange MS in this review. We will describe recent analytical developments and several of the latest case studies, selecting prominent examples of the contribution of MS in structural virology.
First, we review developments in analytical methodology geared toward the improved mass analysis of intact viruses and virus-like particles, covering instrumentation, sample preparation, and data analysis. We will discuss developments in native mass spectrometry, charge detection mass spectrometry, ion mobility mass spectrometry, as well as nanoelectromechanical-based mass spectrometry and how these advances have expanded our ability to study macromolecular assemblies such as intact viruses, virus-like particles, bacterial encapsulins, as well as synthetic designed nanocontainers. We will highlight several exciting applications but also discuss remaining analytical challenges.
Second, we will review how mass spectrometry can be used to study conformational dynamics of viruses and viral proteins. The study of dynamic structural behavior in proteins is particularly challenging for most analytical techniques, whereby especially crystallography and cryo-EM are biased to well-ordered structural components and generally rely on interpolation of rigid structural snapshots to infer dynamics. It is well-known that structural dynamics are essential for viral infection and replication. For instance, some capsid shells can expand their diameters by as much as 25%,1 or dynamically flip internal capsid components to the outside to bind receptors or help lyse the host membrane to enter the cell.2 For enveloped viruses, the structural dynamics of the surface glycoproteins play a crucial role in membrane fusion and cell entry, and conformational changes of receptor binding domains play an important part in balancing immune evasion with host interactions.3,4 These “breathing” motions and the capsid maturation process happen through cooperative structural and conformational changes in the proteins of the capsid, matrix, and envelope. Also, self-assembly and disassembly of the capsid proteins is a major quaternary structural rearrangement, often guided by conformational changes in the assembling building block. Especially hydrogen–deuterium exchange mass spectrometry is sensitive to monitor such conformational changes and dynamics and we will describe here how this technique has advanced over the last years to tackle larger macromolecular machineries including viruses, and how that has expanded our knowledge about virus assembly, stability and conformational dynamics.
Third, we will review recent advances in mass spectrometry to discover how viral proteins, especially those in the viral envelope, are extensively decorated by protein glycosylation and how this influences the interactions with the host. The field of structural virology has generated beautiful high-resolution structures of viral glycoproteins through crystallography and electron microscopy, especially of the polypeptide chain, whereas the attached glycans have remained largely elusive or rather even ignored. A major analytical challenge to characterize the glycans on these viral proteins is that they are notoriously heterogeneous and dynamic, making it hard to either crystallize or assign densities in the reconstructed three-dimensional maps. Advances in cryo electron microscopy have made these heavily glycosylated viral proteins more feasible targets for structural studies, however, and the presence of these glycans has certainly also become more visible and is making its way to the forefront of the structural analyses. In parallel, recent advances in mass spectrometry have advanced the field of glycoproteomics, especially through new selective enrichment techniques, glycopeptide fragmentation techniques, and dedicated database search algorithms. Through these developments, in-depth qualitative and quantitative characterization of all glycoproteoforms of proteins has come within reach, including for very complex viral glycoproteins. The characterization and site-specific annotation of the glycans by mass spectrometry further helps to improve annotation of electron density in high-resolution cryoEM maps of viruses and viral glycoproteins. Moreover, as these glycans play a crucial role in virus-host interactions, through host receptor-binding and immune evasion, knowledge about their exact structure will advance our understanding of the viral replication cycle and ultimately lead to improved therapeutic routes to inhibit infection.
As we focus this review on the outlined structure-based topics, we certainly do not cover all contributions that mass spectrometry can make to the broader field of virology. Notable omissions are advances in mass spectrometry-based proteomics applied to virology, including studies on how host cells respond to viral infections, extensive interactome analyses of viral protein within host cells, or even cases where proteomics is used to detect viral proteins in serum and other bodily fluids as a diagnostic technique. To briefly highlight the broader utility of MS in virology research, in particular during this era of the COVID-19 pandemic, we finish off this review with a short series of case studies on how several mass spectrometry based analytical methods have been used to study this new virus.
Viruses and Virus-Like Particles
Viruses represent some of the most beautiful macromolecular structures present in nature. A general feature of viruses is that they use a proteinaceous capsid to encapsulate their genetic material, being either DNA or RNA. These capsids need to be very robust to survive the harsh conditions that are encountered when jumping between host cells. For instance, many nonenveloped viruses including coxsackieviruses, rotavirus, and poliovirus can remain infectious for extended periods outside the host, exposed on surfaces. While the capsids are sufficiently stable to survive these harsh conditions, they also need to be flexible and dynamic, allowing them to efficiently release their genomic content in a timely manner when infecting a new cell. Moreover, for nonenveloped viruses the capsid makes the first contact with the host, mediating host recognition and cell entry. Enveloped viruses contain an additional layer around the capsid composed of lipids and viral glycoproteins, which then take on the role of host recognition and cell entry. The bumps, knobs, and spikes, as seen in the well-known images of the enveloped virus SARS-CoV-2, depict such structures on the viral envelope.
Especially the capsids of nonenveloped viruses represent ideal model systems to study the assembly of protein complexes, since these protein shells have the astounding ability to self-organize their folding and assembly even in vitro without the help of chaperones. Moreover, their natural capacity of encapsulating material, i.e., the viral genome, renders virus capsids an interesting target for nanotechnological applications that exceed far beyond drug delivery. The detailed biophysical and biochemical characterization of the virus assembly and maturation processes is crucial, as this knowledge may potentially be used to interfere with viral infection. Moreover, viruses have important new medical applications as platforms for gene-delivery in vaccines and gene-therapies, with huge investments by pharmaceutical companies. Most of these medical applications are based on viral vectors that are relatively safe for humans, such as adeno-associated virus (AAV), which is used to package and deliver the gene specifically to the desired tissues and cells. With this new emerging class of future medicines, there is also a new demand for analytical technologies to characterize them and to be used for quality control.
Technically, studying virus assemblies is rather challenging as they are enormous molecular machineries, and their composition can be very heterogeneous. Another problem is posed by the transient nature of the intermediates formed during assembly and maturation, impeding their purification and analysis. A few decades ago, through pioneering work by the groups of Siuzdak,5 Robinson,6 and Heck,7 native mass spectrometry entered this area of research and tackled several important questions that were less accessible through other techniques. In particular, native MS represents a method that can unambiguously reveal the constituents of a virus, the structural integrity of the particles, and the stoichiometry of the viral structural proteins as well as monitor virus assembly and detect assembly intermediates.
Native Mass Spectrometry of Viruses and Virus-Like Particles
Over the last 2 decades native MS has matured into a valuable technique in structural biology of protein assemblies.8−11 In contrast to denatured top-down approaches, in native MS, samples are sprayed from solutions which preserve noncovalent interactions and quarternary structural arrangements of protein complexes throughout their transition into the gas phase. With the obtained protein complex masses, it is then possible to deduce their composition and stoichiometry and even to observe post-translational modifications or binding of small ligands. The capsids of especially icosahedral viruses and VLPs typically assemble from one or a handful of different capsid proteins at a well-defined stoichiometry. This strict capsid organization makes them tractable targets for native MS. In fact, most of the reported high mass native MS studies have been performed on viruses or virus-like particles (VLP), which were often used as a benchmark for the development of new native MS technologies. In this respect, native MS method development and studies into virus assembly have gone hand in hand over the last years.12 Native MS analysis of intact viruses and VLPs can give insights into capsid assembly, composition, and cargo load but also on the shape and stability of the virus through measurements of collisional cross section (CCS) or collision induced dissociation pathways. Although native MS has also been used to study other aspects of virus structure and replication,13−15 here we focus on recent advances in the mass analysis of intact viruses and VLPs.
Instrument Development for High Mass Analytes
Virus structures are often highly organized and symmetric, with the majority of virus capsids exhibiting either helical or icosahedral structures. In icosahedral viruses, the number and arrangement of capsid proteins can be classified using the “quasi-equivalence principle” proposed by Caspar and Klug.16 Consequently, icosahedral viruses and VLPs are typically composed of at least 60, or a multiple of 60, capsid protein subunits (with molecular weights of 10–100 kDa), such that the mass of the whole virus can extend well into the megadalton (MDa) range. The analysis of MDa assemblies poses many challenges, which were tackled through ongoing instrumental developments over the last decades.17
The first challenge lies in the high mass-to-charge ratio (m/z) of the virus capsid ions. The main difference between native and denatured proteins, when analyzed by electrospray ionization (ESI), is the number of charges the gas-phase ions acquire. For native protein assemblies, the average number of charges in ESI scales with the solvent accessible surface area of the complex and has been empirically shown to scale roughly with the square root of the mass.18,19 ESI under native conditions thereby produces ions with a substantially lower number of charges compared to denatured protein ions, resulting in the higher m/z. High m/z ions have more inertia when they enter into the vacuum of the mass spectrometer and are thus transmitted poorly by the ions optics that are conventionally developed for the analysis of smaller peptides and denatured proteins. Hence, instrument modifications are required to accommodate high mass samples for native mass spectrometry.
The efficiency with which radio frequency (RF)-only guides are capable to focus and transmit ions is highly m/z dependent and decreases with increasing m/z. Traditionally, a way to counteract this was by dropping the frequency of the RF ion guides and increasing the gas pressure in the front end of the instrument to allow collisional cooling as described for Q-ToF instruments in the early 2000s.20−22 Such modifications enabled transmission of virus capsid ions up to 20 MDa. Early proof-of-principle studies on the bacteriophage MS26 capsid and empty hepatitis B virus (HBV) capsids were followed by detailed studies on the assembly and stability of HBV and norovirus.23 Native MS was also used to study the structure and assembly of the Triatoma Virus and monitor the pH triggered genome release from the particles.24 This monitoring of the cargo load in a viral capsid is another powerful application of native MS, as was also demonstrated in studies monitoring genome loading within the plant viruses Cowpea Chlorotic Mottle Virus (CCMV) and Brome Mosaic Virus (BMV), small synthetic molecule loading in CCMV for nanotechnology applications, and fluorescent protein loading in the bacterial virus-like nanocompartment encapsulin.25−27
These studies extended to masses as high as 10 MDa. Later studies on the even larger bacteriophage HK97 capsid have been illustrative of current mass limitations in native MS.28 The HK97 capsid assembles with the aid of a scaffolding domain on the capsid protein, which is subsequently cleaved by the coassembled viral maturation protease. The empty procapsids, assembled without the protease, represent some of the largest reported macromolecular assemblies with resolved charge states measured on quadrupole time-of-flight (QToF) analyzers, weighing approximately 18 MDa.19 While masses for these enormous ions could be determined, the individual charge states were barely resolved. When the viral maturation protease was coassembled with the procapsids, the resulting 21 MDa assembly could also be detected, but charge states could no longer be resolved, hampering precise mass determination. It was argued that this lack of resolution originated partly due to poor desolvation and partly due to the co-occurrence of several different procapsid-protease stoichiometries. After cleavage of the scaffolding domain and self-cleavage of the protease, a mature HK97 capsid is formed, with a reduced molecular weight of about 13 MDa. Charge states on this assembly could be readily distinguished in the mass spectrum, and co-occurring assemblies with mass differences as small as 15 kDa could be resolved (representing a mere 0.1% of the total mass of the complex).
The limited resolving power in the high mass range on QToF platforms was recently partly overcome by the introduction of the Exactive plus EMR, which combines a high resolution Orbitrap mass analyzer with modified ion optics that enhance the transmission of high m/z ions.29 In addition to the improved resolving power of this mass analyzer, improved desolvation of the ions, resulting in narrower mass distributions, gave rise to better-resolved spectra, especially for high-mass ions (see Figure 1a). Although ion transmission efficiency was still low for ion species with an m/z above 20 000, it was possible to obtain baseline charge-state resolved mass spectra for the intact 4.6 MDa Cowpea Chlorotic Mottle Virus.25
Figure 1.
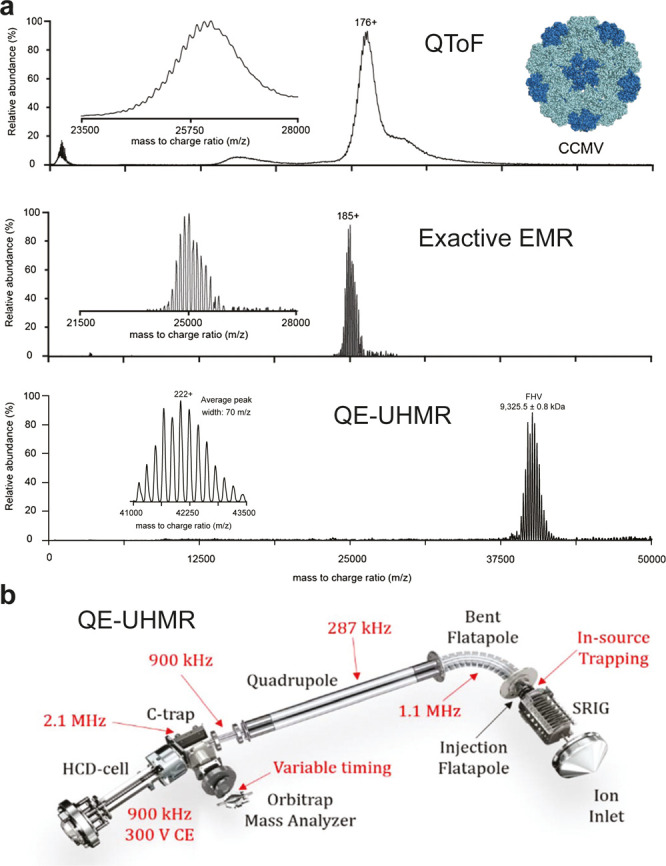
Advances in mass resolving power for the analysis of viruses by native mass spectrometry. (a) Native mass spectra of the 4.6-MDa intact Cowpea chlorotic mottle virus (CCMV) virions and 9.3 MDa Flock House virus (FHV) acquired with several platforms for native MS. (Top) Mass spectrum acquired using a quadrupole time-of-flight (QToF) instrument. A single series of partly resolved charge states can be observed. (Middle) Spectrum acquired for the sample on an Orbitrap EMR with extended mass range, displaying clearly baseline resolved ion signals. (Bottom) Intact FHV analyzed on an QE-UHMR instrument. The native mass spectrum of FHV, detected at 42 000 m/z shows baseline resolved charge states at a resolution of approximately 600. (b) Schematic of the Q-Exactive UHMR Orbitrap mass spectrometer indicating the various ion optic modifications made to enable the analysis of intact viruses in red. Adapted with permission from van de Waterbeemd, M.; Snijder, J.; Tsvetkova, I. B.; Dragnea, B. G.; Cornelissen, J. J.; Heck, A. J. R. 2016 Examining the Heterogeneous Genome Content of Multipartite Viruses BMV and CCMV by Native Mass Spectrometry. J. Am. Soc. Mass Spectrom.27(6): 1000–1009 (ref (25)). Copyright 2016 American Chemical Society. Adapted by permission from Macmillan Publishers Ltd.: Nat. Methods, van de Waterbeemd, M.; Fort, K. L.; Boll, D.; Reinhardt-Szyba, M.; Routh, A.; et al. 2017 High-fidelity mass analysis unveils heterogeneity in intact ribosomal particles. Nat. Methods, pp. 1–7 (ref (30)). Copyright 2017. Fort, K. L.; Van De Waterbeemd, M.; Boll, D.; Reinhardt-Szyba, M.; Belov, M. E. et al. 2018 Expanding the structural analysis capabilities on an Orbitrap-based mass spectrometer for large macromolecular complexes. Analyst. 143(1): 100–105 (ref (31)). Reproduced by permission of The Royal Society of Chemistry.
More recently, a further improved Orbitrap-based platform was launched as the QE-UHMR, with yet additional improvements to the ion optics for the transmission of high mass ions as illustrated in Figure 1b.30 The modifications included increased amplitude-to-amplitude voltages and reduced RF-frequencies for the ion guides, a quadrupole for the selection of ions with m/z up to 35 000, and a variable timing for ion injection into the Orbitrap. Furthermore, in-source trapping was developed to counteract the jet-expansion in the source region, the maximum activation energy in the HCD cell was increased to 300 V, and the possibility to switch between xenon and nitrogen as collision gas was implemented.31 These developments increased the transmission of higher mass ions substantially and allowed the mass analysis of 9.4 MDa intact Flock House Virus (FHV) particles with baseline resolved charge states.30 Notably, these FHV particles are authentic virions, packaging both genomic RNA segments of the virus.
Spectra Interpretation and Complexity for High Mass Analytes
The resolving power obtained in spectra of high mass ions is not just limited by the performance of the mass analyzer.32 Poor desolvation of the high-mass ions turns out to be another crucial limiting factor, as this results in many closely spaced solvent adducts that crowd the spectrum and give rise to very broad charge states, far beyond the resolving power of the mass analyzer itself. In many cases, this sample heterogeneity is further increased if the analyzed particles harbor variable modifications on their monomeric building blocks (e.g., small N- or C-terminal protein truncations) or if the capsid is composed of variable combinations of different protein subunits.27,33 Due to the stochastic nature by which these subunits are incorporated in the assembled capsid, the mass distribution can broaden to such an extent that charge states overlap and can no longer be resolved. While ion transmission is already a big bottleneck, the low intensity signals will also spread out over a wider m/z range, resulting in worse signal-to-noise (S/N).27
The poor desolvation and microheterogeneity of the megadalton assemblies makes it difficult to resolve individual charge states and determine a precise peak position, while both are imperative for unambiguous charge state assignment. The two most employed approaches for charge state assignments in native mass spectra were proposed initially by Mann and colleagues34 and Robinson and colleagues, respectively.6 Following Mann et al. the charge state of two neighboring peaks can be calculated with the formula: zi = (mzj-mproton)/(mzi – mzj) with mzj < mzi. The approach by Robinson follows the iterative assignment of a range of potential charges over the charge state distribution. By calculating the corresponding masses for all peaks and minimizing the standard deviation across the charge state distribution it is possible to infer the correct charge assignment. Charge state assignment through either one of these approaches is trivial for well-resolved spectra of properly desolvated ions but becomes increasingly challenging for larger and poorly desolvated ions. The problem originates from the fact that ions with a higher charge experience proportionally higher collision energies and are therefore typically better desolvated than their lower charged counterparts. In both charge state assignment strategies, this “activation bias” can lead to underestimated charges and masses. A similar case presents itself if the composition of the capsid particle is highly variable, which can be caused by a stochastic number of encapsulated cargo particles or variability in the encapsulated genome.25,27 The resulting charge state distributions of the co-occurring particle compositions can interfere in such a manner that they overlay over wide ranges, appearing as a wrongly interpreted single species in the final deconvoluted mass spectra.
Experimental Approaches for Resolving Complex Mass Spectra
The challenges described above can be partly overcome by the addition of charge reduction reagents like triethylammonium acetate (TEAA) to the electrospray solution35 or by subjecting the ions to (asymmetric) charge partitioning after collision induced dissociation. In both cases, either the charge reduced ions or product ions will populate a higher m/z range where the spacing between charge states is increased and previously convoluted charge state series are resolved. This increased spacing between charge states aids the charge assignment, as it becomes less sensitive to errors in determined peak positions or mass differences due to solvent adducts.25,28,33 Moreover, ion activation as employed in collision-induced dissociation (CID) may further assist in desolvating the formed fragment ions.
Another possible way to circumvent the need for charge state assignment is the use of alterative ionization methods, such as matrix-assisted laser desorption/ionization (MALDI), which produces mainly +1 and +2 ions.36 For ToF mass analyzers, equipped with a dedicated ion detector for high mass ions, the detection of N-terminal capping/methyltransferase domain (CAP) oligomers of the Brome Mosaic Virus (BMV) at 1.3 MDa has been demonstrated using MALDI as an ionization method.36
Ion-mobility spectrometry-mass spectrometry (IMS-MS) provides and an extra dimension of particle separation and provides as such the potential of resolving complex spectra by measuring the ions collisional cross section (CCS) alongside their m/z.38 In the context of intact viruses and VLPs, with the structural characterization of virus assembly intermediates by their CCS, ion mobility and native MS were combined to investigate the assembly pathway for the HBV and Norovirus capsids.23 Due to the unfavorable scaling of mass and CCS, it gets more challenging to resolve small conformational changes of intact viruses populating the same m/z region. However, the average CCS of complete viruses can give valuable information on, e.g., binding of antibodies39 and can be used as an alternative way of mass estimation by inference from CCS-mass scaling, as discussed below.
Other Means for Mass Approximation of Virus-Like Particles
Even when charge state series cannot be resolved in the spectrum, it is often still possible to estimate an average mass based on empirical scaling of mass with m/z. Several native MS studies have shown that the charging of globular protein complexes in ESI scales approximately with the square root of the mass and that this relationship can be used to infer masses from unresolved ion signals.18,19 Using such an approach, it was possible to estimate masses for otherwise unresolvable signals for the HK97 protease-filled Prohead-1 capsid (∼21 MDa) and for full and empty bacterial encapsulins (∼8 and ∼10 MDa).28,33
Another possible approach for mass approximation is done by correlating the shape of the particle with its mass, especially for globular proteins assemblies. This has been demonstrated for several viruses by measuring the electrophoretic mobility diameter (EMD) using a gas-phase electrophoretic mobility molecular analyzer (GEMMA). In GEMMA, multiply charged ions initially produced by native ESI are first charge reduced in a bipolar atmosphere utilizing a 210Po α-particle emitter. The resulting singly charged ions then pass through a differential mobility analyzer (DMA) where particles are sorted based on their electrophoretic mobility diameter using a laminar flow and an orthogonal electric field. An excellent correlation between EMDs and mass has been demonstrated for viruses and VLPs exhibiting masses of up to 27 MDa.40,41 Recent developments in the instrumentation replaced the radioactive 210Po emitter with a nonradioactive corona discharge-based module for charge equilibration, making the method likely more attractive for a broader field of users.42
Notably, both these approaches only provide reasonable estimates if the particles follow the charging and mobility of globular proteins. In the case of m/z-based mass estimation, nucleic acid containing particles typically acquire less charges and populate higher m/z regions than purely proteinaceous assemblies,30 and for GEMMA, particles with variable amounts of cargo, and thus mass, will provide indistinguishable EMDs.40,41
Charge Detection MS
One possibility to circumvent the convolution of signals from heterogeneous macromolecular ions is the measurement of individual particles, one at a time, instead of bulk particle ensembles. This is especially attractive in cases wherein the native mass spectra provide no resolved features for charge and mass assignment. If the measured individual ions m/z can be combined with an independent measurement of its charge, mass distributions can be directly calculated, thereby avoiding the need for revolving charge states like in conventional native MS. Thus, far the most widely used experimental setup for charge detection MS (CDMS) is based on a conductive tube, through which the individual ions pass. From the time it takes the ion to travel through the tube, the m/z can be calculated (when the ion energy is known, typically by applying an energy filter), and the charge can be directly determined from the amplitude of the current that is induced in the tube while the ion passes (see Figure 2a).43
Figure 2.
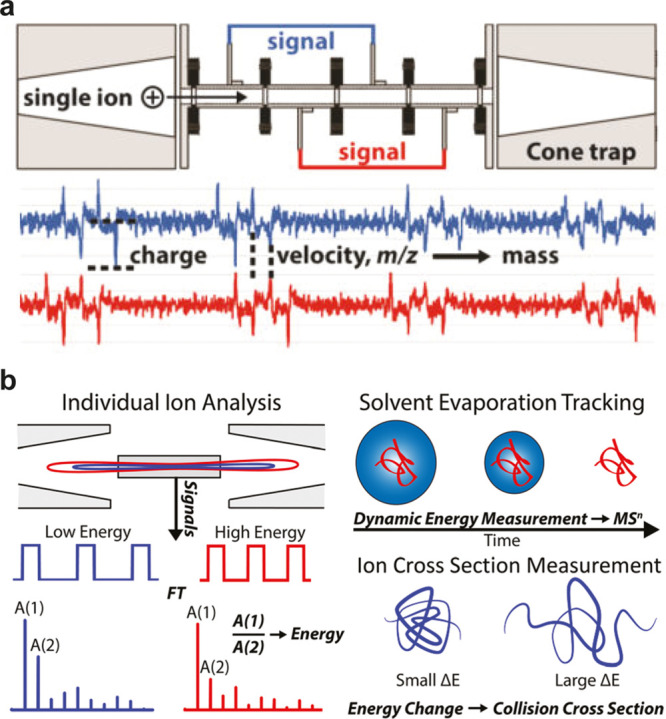
Ion trap charge detection mass analyzers and dynamic ion energy calibrations. (a) Schematic diagrams of CDMS mass analyzers using an ion trap and detector array with below the recorded transient for an oscillating single ion. The amplitude of the pulse signal indicates the charge and the measured velocity of the ion indicates its m/z. With both measured parameters it is possible to calculate each ions mass directly. (b) Alternative CDMS setup, using a conductive tube instead of a detector array. The m/z is derived here from the fundamental frequency of the pulse signal calculated by using a Fourier transform. The ratio of the fundamental frequency and second harmonic (HAR) depends on the ion energy, which is an essential parameter for measuring ion mass in CDMS. The HAR is determined dynamically over the entire trapping period, making it possible to observe the change in ion energy that takes place as solvent evaporates from the ion due to collisions with the background gas. The rate of change in ion energy correlates also with the collision cross section (CCS) of the analyzed particle. Reprinted from Int. J. Mass Spectrom.414, Elliott, A. G.; Merenbloom, S. I.; Chakrabarty, S.; Williams, E. R. Single Particle Analyzer of Mass: A Charge Detection Mass Spectrometer with a Multi-Detector Electrostatic Ion Trap, pp. 45–55 (ref (44)). Copyright 2017, with permission from Elsevier. Reprinted with permission from Harper, C. C.; Elliott, A. G.; Lin, H. W.; Williams, E. R. 2018 Determining Energies and Cross Sections of Individual Ions Using Higher-Order Harmonics in Fourier Transform Charge Detection Mass Spectrometry (FT-CDMS). J. Am. Soc. Mass Spectrom.29(9): 1861–1869 (ref (45)). Copyright 2018 American Chemical Society.
The accuracy of this approach has been greatly improved by putting this cylinder in between a linear ion trap, allowing the ions to oscillate several times through the device, as opposed to only a single pass.44 The time domain signal can then be analyzed by using a Fourier transform, whereby the frequency is used to calculate the m/z and the amplitude of the signal represents a measure of its charge. The performance of these devices has been further improved by cooling the cylinder, increasing the trapping time and using dynamic ion energy calculations,45 compensating for changes in the ion’s energy caused by collisions with background gas molecules (see Figure 2b). These modifications allowed more accurate charge determination for ions with up to 500 charges.46 The developed dynamic ion energy calculations also opened the door for the analysis of several ions at once, as the narrow energy filtering before the trapping event is no longer required.47
Mass analysis by CDMS can be applied to highly heterogeneous samples and can give unique insight into virus structure and assembly. Some examples of recent studies are the resolution of full, empty, and partially packed adeno-associated viruses (AAVs),48 the identification of disassembly intermediates of the Brome Mosaic Virus,49 and the determination of the population of virus-antibody conjugates for MS2 VLPs.50 By combining all the improvements in both m/z and charge accuracy, Jarrold et al. were able to increase the effective mass resolution by almost an order of magnitude to ∼300, being able to resolve intermediate structures present in the HBV capsid assembly pathway.51
In addition to these home-built charge detection mass spectrometers, there have also been developments in modifying commercial mass spectrometers for charge detection of individual ions. For instance, Bier and co-workers52 recently coupled a MALDI ionization source, producing mainly +1 and +2 ions to a charge detection mass analyzer to characterize large macromolecular assemblies. By using superconducting tunneling junction (STJ) cryo-detection, they were able to resolve +1, +2, and +3 ions for Holo and Apo-Ferritin, with masses of up to 1.63 MDa and derived estimates for the number of encapsulated iron atoms within these cages.
Most recently, the capacity for single ion CDMS with Orbitrap mass analyzers has been demonstrated.53,54 The Orbitraps’ capability to resolve individual multiply charged ions and the general scaling between the number of charges and peak intensities was demonstrated already early on by Makarov et al.55 While the detection of smaller and denatured proteins is difficult as single collisions with background gas can cause fragmentation and ion decay, large biomolecules seem to be more stable and can be trapped in the Orbitrap for up to several seconds. This can be rationalized, as the center-of-mass collision energies decrease for larger molecules and poorly desolvated megadalton particles have more degrees of freedom to distribute the transferred collision energies when compared to denatured proteins. The linear relationship between signal intensity and charge could be established over a wide mass range (150 kDa to 9.4 MDa, RMSD ∼3.5 charges, see Figure 3a) and enabled charge detection mass spectrometry on the QE-UHMR Orbitrap platform without any substantial instrument modifications.53 Along a similar line, Kafader et al. demonstrated that, while accessing transient information, it is possible to correlate the slope of the integrated ion signal over the transient length with the ions charge.54 The linear relationship was demonstrated for particles within a range of charges (10–80). Both Orbitrap-based CDMS approaches were used for the analysis of several viruses and VLPs, including the bacteriophage MS2 VLPs (1 and 3.1 MDa), the engineered AaLS-neg nanocontainer (3 MDa), genome-filled and empty adeno-associated viruses (AAVs; 3.7 and 4.9 MDa, see Figure 3b,c), and the intact Flock House Virus (FHV; 9.4 MDa).
Figure 3.
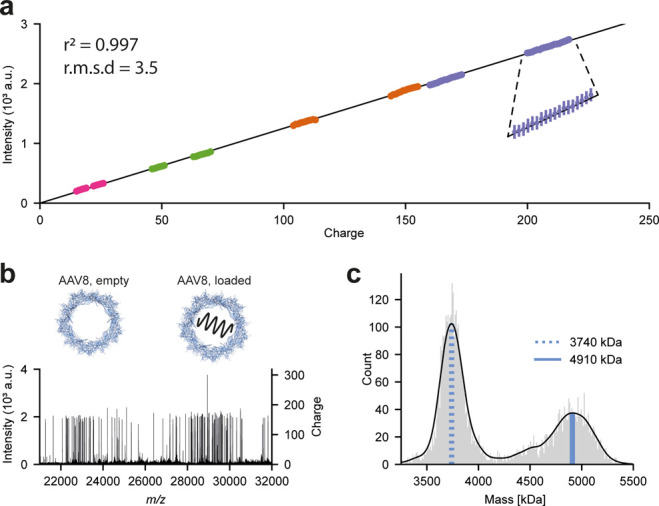
Orbitrap-based charge detection mass spectrometry applied to adeno-associated viruses. (a) Analyzing a large range of protein assemblies, a linear regression model was fitted to 200 sampled single-ion intensities per charge state (n = 15 600) in the range of 20–220 charges. The resulting fit, Intensity = 12.521 × Charge, with an r2 of 0.997, demonstrates the capability of charge detection mass spectrometry on Orbitrap platforms. (b) Individual scan of single particles for a mixture of empty and genome-loaded adeno-associated virus Serotype 8 (AAV8) capsids with the calculated charges indicated in the right y-axis. (c) Mass histogram for AAV8 particles directly calculated from the number of single-ions detected. Blue lines indicate top masses of the empty AAV8 (dotted) and the loaded AAV8 (solid) particle. Adapted by permission from Macmillan Publishers Ltd.: Nat. Methods, Wörner, T. P.; Snijder, J.; Bennett, A.; Agbandje-McKenn, M.; Makarov, A. A.; Heck, A. J. R.2020 Resolving heterogeneous macromolecular assemblies by Orbitrap-based single-particle charge detection mass spectrometry. Nat. Methods. 17(4): 395–98 (ref (53)). Copyright 2020.
NEMS
Another emerging attractive alternative for the mass analysis of large macromolecular assemblies is nanoelectromechanical system-mass spectrometry (NEMS-MS). In this approach, individual particles are deposited on a NEMS resonator, whose frequency is highly dependent on the deposited particle mass as depicted in Figure 4. Early implementation of the method only allowed the average mass determination over a larger set of deposited particles, as the frequency shift is also dependent on the deposition location.56 This was solved by recording two, instead of one, vibrational modes simultaneously, allowing one to correct the measured frequencies for the particle’s deposition position. By tracking the frequency shift upon each particle deposition, it became possible to measure the mass for each individual particle in real-time.57 In contrast to previously described methods, NEMS-MS offers a charge-independent measure of particle mass. This allows also the analysis of neutral particles like presented by Masselon et al. and depicted in Figure 4.58 They demonstrated the mass analysis of empty and filled T5 capsids (27 and 108 MDa), nebulized via surface acoustic wave nebulization (SAWN), at an instrument resolution of 100. NEMS therefore provides great potential especially for mass spectrometry of very high mass viruses.
Figure 4.

Nanoelectromechanical systems mass spectrometry (NEMS) applied to the bacteriophage T5 virus. (a, Top) Negatively stained electron microscopy image of the native bacteriophage T5. (Bottom) The table shows the molecular components of the capsid, with theoretical molecular mass calculations for both types of capsid. (b) High-transmission system architecture for nanomechanical resonator-based charge-independent single-particle mass sensing. The setup consists of three chambers with decreasing pressures. Analytes in solution are nebulized by surface acoustic wave nebulization (SAWN) or nanoelectrospray ionization (nano-ESI) and aspirated through a heated metal capillary inlet at atmospheric pressure. An aerodynamic lens focuses the particle stream (shaded blue area), which is then transferred onto an array of frequency-addressed nanomechanical resonators. (c) Accumulated mass histograms of 363 empty (left) and 648 filled (right) capsids nebulized using nano-ESI from which the molecular masses of the bacteriophage T5 capsid could be determined. From Dominguez-Medina, S.; Fostner, S.; Defoort, M.; Sansa, M.; Stark, A.-K. et al. 2018 Neutral mass spectrometry of virus capsids above 100 megadaltons with nanomechanical resonators. Science (Washington, DC, U. S.)362(6417): 918–922 (ref (58)). Reprinted with permission from AAAS.
Selected Highlights in Native Mass Spectrometry of Viruses and Virus-Like-Particles
Although we focused on recent developments in analytical technologies for the mass analysis of viruses, we also like to close this section by highlighting a few exciting applications to show how native mass spectrometry can be used to study virus structure, stability, assembly, and maturation.
Snijder et al.24 used a combination of atomic force microscopy (AFM) and native mass spectrometry to probe the biophysical interplay between a viral genome and its capsid in the picorna-like Triatoma virus (see Figure 5). Starting with the 8 MDa intact virus, they observed that at more basic pH the genome became uncoated and released, whereafter the capsid proteins reassembled into empty capsid particles with a Mw of about 5.4 MDa. From the data, they proposed an assembly model in which heterotrimeric pentons that consist of five copies of structural proteins VP1, VP2, and VP3 are the principal units of assembly. Both the AFM and MS data also showed that the genome is used to stabilize the very densely packed virion particles.
Figure 5.

Alkaline-triggered uncoating of Triatoma virus (TrV) monitored by native mass spectrometry. (a) Spectra of TrV virions, incubated at different pHs. Signal corresponding to virion is highlighted in red (m/z ∼40000; Mw ∼8.3 MDa), that of empty capsids in yellow (m/z ∼28000; Mw ∼5.4 MDa). (b) Model of the alkaline-triggered uncoating of TrV. Under neutral pH, TrV confines a very large genome. This comes at a high energetic cost, but the ssRNA stabilizes the capsid, thereby preventing premature uncoating. At higher pH, this stabilizing interaction is lost and electrostatic self-repulsion of the ssRNA increases due to loss of charge on counterions; the capsid bursts and falls apart into pentons. The genome and VP4 are released into solution and pentons reassemble into empty capsids. Adapted by permission from Macmillan Publishers Ltd.: Nat. Chem. Snijder, J.; Uetrecht, C.; Rose, R. J.; Sanchez-Eugenia, R.; Marti, G. A. et al. 2013 Probing the biophysical interplay between a viral genome and its capsid. Nat. Chem.5(6): 502–509 (ref (24)). Copyright 2013.
The interactions between viral capsids and antibodies play a crucial role in the immune response against viral infection. Moreover, such antibodies are also explored for biomedical applications as VLP conjugates. Native MS and CDMS offer unique capabilities for characterizing these interactions by monitoring the relative mass increase upon antibody binding. Bereszczak et al.39 first demonstrated this by combining native MS and GEMMA to investigate the binding characteristics of several antibodies to HBV capsids (see Figure 6a). Titration experiments with two different antibodies (Fab 3120 and Fab E1) on the T = 3 and T = 4 capsid showed that the binding capacity is dependent on the available binding sites on the capsid. Interestingly, some differences between saturation concentration for the capsids were observed for the two different Fabs. For Fab E1, binding to the spikes, a saturation for the T = 3 and T = 4 capsid could be reached with a mixing ratio of Fab E1/Cp149 dimer at 1.2:1 ± 0.1. For Fab 3120, with the epitope located on the subunit interfaces, different mixing ratios were required for saturation (Fab 3120/Cp149 dimer ratio of 2.4:1 ± 0.1 and 1.4:1 ± 0.1 for T = 3 and T = 4). The lower affinity of the Fab 3120 to the T = 3 capsid was attributed to a slight distortion of the 3120 epitope caused by the greater curvature of the T = 3 lattice. Bond et al.50 analyzed the binding of antibodies to MS2 capsids by CDMS (see Figure 6b). Remarkably, the achieved mass resolution allowed them to resolve individual bound antibodies to the capsid and utilizing the mass distribution of the empty capsid it was possible to determine the distributions of bound antibodies for different mixing ratios. They concluded that both distributions fitted very well to a stochastic binding mechanism of the antibodies to the virus.
Figure 6.

Assessment of antibody binding to viruses by native mass spectrometry. (a) Mass spectra corresponding to Fab 3120 binding to hepatitis B core antigen (HBcAg) showing the effect of different mixing ratios of Fab and HBcAg on the HBcAg-Fab assemblies formed. Mixing ratios were 0.65:1, 1:1, 2:1, and 4:1 (bottom to top) in 200 mM ammonium acetate, pH 6.8. (HBcAg concentration in terms of Cp149 dimer concentration). (b) CDMS spectra of MS2-antibody conjugates. (I) CDMS spectrum measured for hybrid-MS2 capsids (black line). (II and III) CDMS spectrum measured for MS2-antibody conjugates from a reaction with 3 and 10 equiv of antibody, respectively (black traces). The red line in I shows the fit obtained for a peak fitting function with five Gaussians. The peak fitting function is used to fit the CDMS spectra for the MS2-antibody conjugates in II and III (red traces for distinct number of antibodies bound and blue traces for summed antibody distribution). (IV) The antibody population distributions derived from the fits to the CDMS spectra in II and III. The orange and green lines are Poisson distributions calculated for 3 equiv of antibody (green) and 10 equiv (orange). Adapted by permission from Macmillan Publishers Ltd.: Anal. Bioanal. Chem. Bereszczak, J. Z.; Havlik, M.; Weiss, V. U.; Marchetti-Deschmann, M.; van Duijn, E. et al. Sizing up large protein complexes by electrospray ionization-based electrophoretic mobility and native mass spectrometry: morphology selective binding of Fabs to hepatitis B virus capsids., Anal. Bioanal. Chem. (ref (39)). Copyright 2013. Adapted with permission from Bond, K. M.; Aanei, I. L.; Francis, M. B.; Jarrold, M. F. 2020 Determination of Antibody Population Distributions for Virus-Antibody Conjugates by Charge Detection Mass Spectrometry. Anal. Chem.92(1): 1285–1291 (ref (50)). Copyright 2020 American Chemical Society.
Capsid assembly and uncoating are crucial phases in the viral replication cycle and have therefore been investigated extensively. Uetrecht et al.23 analyzed small oligomers of the HBV and norovirus in equilibrium combining IMS and native MS. The detected intermediate capsid-protein oligomers were, based on their sheetlike shape, identified as assembly intermediates that suggested defined pathways for the assembly of both capsids. Bond et al.49 investigated the disassembly intermediates of BMV by CDMS, suggesting two distinct pathways dependent on the different experimental conditions. Through a sudden pH jump into a basic environment, the capsid breaks into two fragments corresponding to an almost complete empty capsid and the released RNA in complex with a few capsid proteins. Through a slow buffer exchange and by scavenging the divalent cations present in the capsid, the interactions between the capsid proteins were disrupted and the capsid swelled, but no genome material was released. Over time, an increase in mass was observed as free capsid monomers seemingly did bind to the exposed genome. It was hypothesized that this behavior might be closely related to the in vivo process as it could shield the genome from degradation.
HDX-MS Analysis of Structural Dynamics in Viruses
Hydrogen–deuterium exchange (HDX) mass spectrometry is an isotope labeling technique that probes the interactions of a biomolecule with its solvent. It has been applied to many protein-based systems over the years.59,60 It has also been applied to study the structural dynamics of viruses at all steps of the replication cycle; from host–cell binding and membrane fusion, to genome replication, transcription, mRNA capping, nucleocapsid assembly, and budding of new virions.61 In an HDX experiment, the targeted viral protein is transferred into deuterated “heavy” water (D2O), and mass spectrometry is used to monitor the extent and kinetics of deuterons exchanging with hydrogens on the proteins’ various functional groups. HDX-MS can be performed on the intact proteins, but the precise molecular location of hydrogen–deuterium exchange can also be narrowed down using a bottom-up, peptide-centric LC–MS readout or even by top-down fragmentation in some advanced implementations.62,63
Analytical Workflow of an HDX-MS Experiment
The hydrogen–deuterium exchange reaction depends mainly on three local structural features of the protein: the intrinsic exchange rates of functional groups, hydrogen-bonding interactions, and solvent accessibility. The combination of these local structural features results in a characteristic hydrogen–deuterium exchange profile across the molecule and importantly, the profile responds to local structural changes when the biomolecule changes conformation, refolds, binds ligands, or forms higher order complexes with additional interaction partners. It is therefore very well suited to monitor structural dynamics such as “breathing” of virus capsids, the pre- to postfusion transitions of viral spike proteins, or to map interactions with host receptors and neutralizing antibodies.61 Besides local structural features of the viral biomolecule, the rate of the HDX reaction is highly dependent on temperature and catalyzed under both alkaline and extremely acidic conditions, with a minimum exchange rate at pH ∼2.5.
Most HDX experiments implement a bottom-up LC–MS/MS type readout of deuterium labeling. This way, the extent of deuterium uptake can be resolved for every individual region of the viral protein for which a corresponding peptide is identified. After HDX labeling of the protein under native conditions, the reaction is quenched by dropping the pH to 2.5 and the temperature to zero degrees Celsius. Whereas this quenching step reduces the rate of HDX to a minimum, so-called back-exchange with H2O from solvent in downstream steps, like proteolytic digestion and LC, remains an issue. HDX on hydrocarbon functional groups is so slow that virtually no deuterium is picked up in the labeling reaction to begin with, but HDX is so fast on most heteroatom functional groups (e.g., carboxyl, amine, etc.) that all deuterium is immediately lost to back-exchange. Only HDX on the amide groups of the peptide backbone occurs at an experimentally feasible rate, namely, within seconds to hours during the labeling reaction. Even after quenching the reaction at pH 2.5, the deuterium on backbone amides is still gradually exchanged back for hydrogens within minutes to hours. The characteristic HDX profile of the structured protein is luckily preserved, as the rate of back-exchange is largely uniform across the denatured polypeptide chain and digested peptides. Nevertheless, back-exchange puts strong time-pressure on the sample preparation and analytical steps of the workflow. A measurement needs to be completed within 10–30 min after quenching, before too much of the site-specific information on deuterium uptake is lost to back-exchange.
To cope with this time-pressure, quenching is typically combined with rapid denaturation and disulfide bond reduction in a single step, before proteolytic digestion with a protease that is sufficiently active at pH 2.5 to digest the sample within minutes at moderate temperatures, i.e., pepsin (or type VIII protease, EndoPro, and others).64−67 Moreover, the LC step is also performed on the order of 10 min at reduced temperatures close to zero degrees Celsius, with a well-established negative trade-off for peak separation. The need for a relatively short LC gradient puts similar pressure on the tandem MS selection and fragmentation strategy employed to identify peptides, favoring also Data Independent Acquisition (DIA) and All Ion Fragmentation (AIF) approaches, even though Data Dependent Acquisition on both linear-ion-trap-FT and quadrupole-time-of-flight instruments is still used in the majority of HDX studies on virus proteins.
The restrictions that back-exchange imposes on the experimental workflow currently limits HDX to relatively simple samples like single proteins (with ligands) or protein complexes of up to half a dozen components, depending on the length of the individual chains. Still, on such simple samples, a sequence coverage of 80–99% can be readily achieved, aided by the unspecific digestion of pepsin, which typically yields many overlapping short peptides. Moreover, the whole HDX workflow, from sample mixing and labeling to quenching, digestion, and LC–MS is amenable to automation, resulting in high sample throughput. This also puts HDX in a favorable position for use in drug screening efforts. HDX-MS is only limited to sample complexity on the level of the peptide mixture, so there is no effective upper size limit of protein complexes that can be monitored with the technique. This makes HDX-MS very well suited to characterize intact virus particles into the megadalton range, from nonenveloped icosahedral viruses to pleomorphic enveloped viruses.68−73
HDX-MS Perspective on the Viral Replication Cycle
HDX-MS has been used to study all steps of the viral replication cycle in recent years, including host cell recognition and entry. Whereas most available methods in structural biology provide a static structural picture of a viral protein, HDX-MS is capable of probing structural dynamics and detect multiple co-occurring conformational states. This is an especially useful perspective to study the typical pre- to postfusion conformation changes in viral envelope glycoproteins. For example, Gutmann, Lee and colleagues used HDX-MS to follow the pH triggered pre- to postfusion transition of the Influenza A Virus (IAV) hemeagglutinin (HA) spike protein.73 They uncovered a distinct set of early and late conformational changes in HA, monitored directly on whole virions (see Figure 7). Their HDX experiments describe the pathway of this dynamic structural transition in unprecedented detail. Multiple recent studies have investigated the mechanisms of fusion activation of IAV-HA by HDX-MS.73−75 In addition, a recent study investigated host receptor binding and detected allosteric changes in Nipah Virus Glycoprotein–ephrinB2 interactions,76 illustrating yet another aspect of host cell recognition and entry elucidated by HDX-MS.
Figure 7.
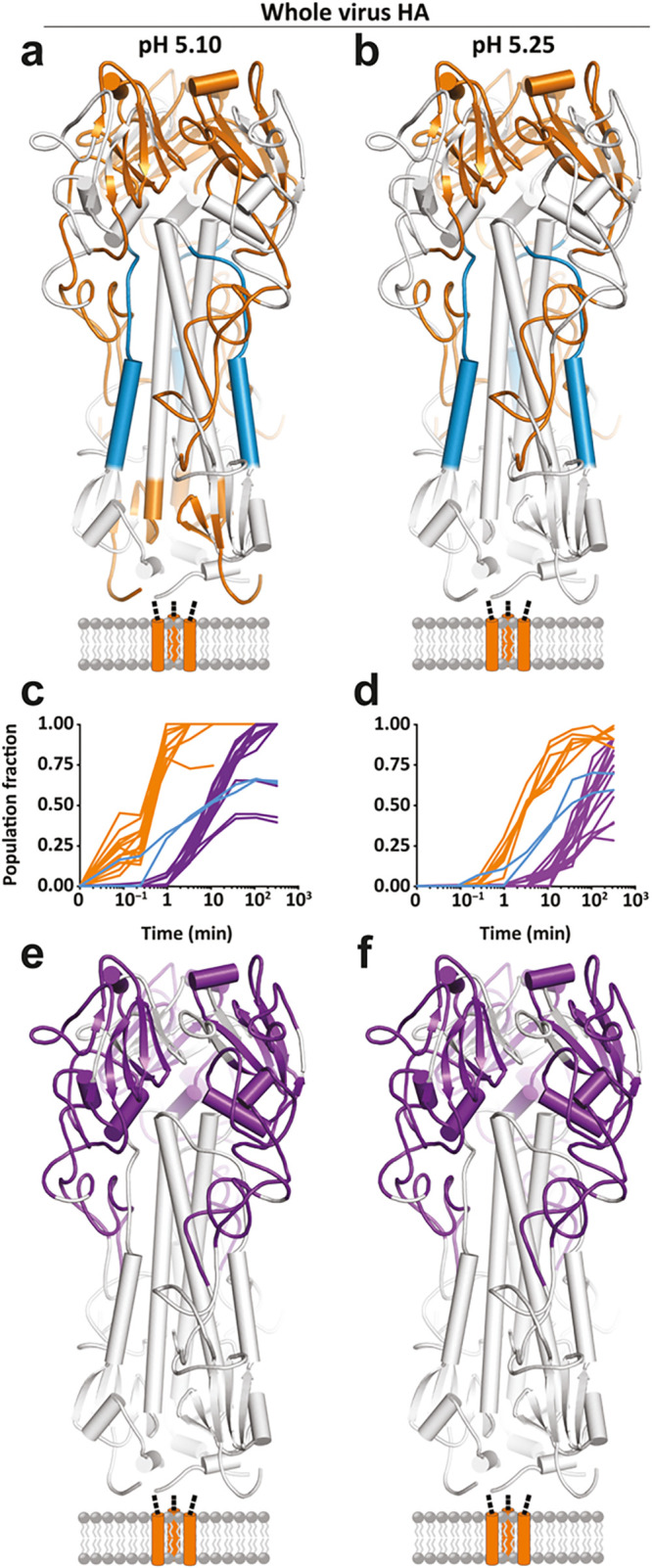
Global kinetic comparison and pH dependence of the fusion activation in influenza hemagglutinin (HA) investigated by HDX-MS. HDX-MS of full-length HA embedded in the whole virion is used to monitor the pH triggered refolding from the pre- to postfusion state. The specific pH of activation did not alter the nature (a and b) or sequence of any observed structural changes in full-length HA but simply accelerated the onset and rate of change of each conformational event (c and d). Conformational transitions that take place before 3.5 min are colored in orange. The last transition reported by the HA1 peptides is to a largely unfolded, highly flexible state, which begins at 3.5 min [purple; (c and d) and (e and f)]. Formation of the postfusion helical bundle in full-length HA is delayed because of the formation of the intermediate state [(a–d); blue peptides and traces]. Reprinted from Benhaim, M. A.; Mangala Prasad, V.; Garcia, N. K.; Guttman, M.; Lee, K. K. 2020 Structural monitoring of a transient intermediate in the hemagglutinin fusion machinery on influenza virions. Sci. Adv.6(18): eaaz8822 (ref (73)). Copyright The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. Distributed under a Creative Commons Attribution Non-Commercial License 4.0 (CC BY-NC) http://creativecommons.org/licenses/by-nc/4.0/.
Upon host cell entry, viral transcription and genome replication kick-starts host cell remodeling and the production of new virus particles. HDX-MS has also been used to understand this phase of the replication cycle at a structural level. For example, the interdomain interactions and flexibility of the RNA-dependent RNA polymerase (RdRP) of dengue virus, NS5,77 and primer extension and polyadenylation activity of the Chikungunya Virus RdRP nsP4 have been studied by HDX-MS.78 Another study by Griffin and colleagues shed light on RNA recognition by the Chikungunya helicase nsP2, which is essential for replication and transcription.79 Oligomerization of the Marburg Virus (MARV, a filovirus related to ebola virus) polymerase cofactor VP35 has also been investigated by HDX-MS as well as binding of a viral processivity factor to the DNA polymerase E9 of Vaccinia Virus.80,81 In addition to transcription, capping of the viral mRNA is an essential early step in the viral replication cycle that evades the host immune response, and some viruses do it by snatching the required structures from the hosts own mRNAs. This process of cap-snatching has also been investigated by HDX-MS for the bunyavirus SFTSV.82
Following transcription and genome replication, viral gene products are expressed as proteins, ultimately resulting in assembly and budding of new virions. Moreover, the host cell is remodeled, and the innate immune response suppressed by viral gene products through a wide variety of pathways. Recent HDX-MS studies have aided our understanding of capsid assembly for Hepatitis B Virus capsids, highlighting the role of dynamics in the dimeric building block through a network of allosteric interactions in the growing capsid.83 Likewise, two important alpha helices responsible for oligomerization of the Ebola virus nucleocapsid protein NP could be identified with HDX-MS.84 In relation to budding, interactions of the MARV matrix protein VP40 with phospholipid bilayers were studied as well as oligomerization of the Lassa virus matrix protein Z.85,86 In examples of host remodeling, HDX-MS has been used to uncover how MARV VP24 interacts with the Kelch domain of the host Kaep1 protein to alter the antioxidative stress response,87 study zinc binding in the HBx protein of HBV in complex with the host DNA-damage response protein DDB1,88 probe the structural dynamics of Human Immunodeficiency Virus (HIV) Nef variants and how it interacts with host Src kinase,89,90 and how enterovirus c10orf76 interacts with PI4KB.91,92
HDX-MS for Antiviral Drug and Vaccine Development
Beyond the viral replication cycle itself, HDX-MS has also been used to understand the host immune response to viral infections by mapping epitopes of neutralizing antibodies. The viral glycoproteins of enveloped viruses have been an important focus of these studies. For example, Gross and colleagues mapped out the epitopes of a panel of five antibodies directed against the Envelope (E) protein of Japanese Encephalitis Virus (JEV), revealing the structural basis for differential neutralization potency among the monoclonal antibodies.93 Similarly, HDX-MS was used to map the epitopes of monoclonal antibodies binding to the prM and E components of the related dengue virus (DENV)94,95 as well as the Hepatitis C Virus envelope protein E2,96 IAV HA,97,98 Respiratory Syncytial Virus Fusion (RSV-F) protein,99 and the MARV polymerase cofactor VP35.100
These epitope mapping experiments with HDX-MS do not only serve to understand the mechanisms of neutralizing immunity, they also offer valuable information for screening and optimization of these monoclonal antibodies for therapeutic applications. HDX-MS has been applied more broadly in antiviral drug and vaccine development studies. For example, HDX-MS was used to screen and assess stabilizing mutations to HIV Envelope (Env) protein and RSV-F to develop better immunogens101−103 and to engineer and optimize anti-HIV Env monoclonal antibodies.104 Epitopes of IAV HA inhibitors have also been mapped with the method.105−107 In addition, small molecule inhibitor binding to the Hepatitis C Virus RdRP, HIV integrase, and HIV reverse transcriptase enzymes has been investigated by HDX-MS.108−111 The technique has thus been widely applied to characterize and optimize novel antiviral drug leads.
Biophysical Characterization of Viruses by HDX-MS
HDX-MS can also be used for biophysical characterization of virus particles, using it as a structurally resolved readout to monitor the response of the virus particle to chemical denaturation or changes in temperature and pH. In one recent example HDX-MS was used to monitor pH-triggered expansion of the DENV virion, a transition that is relevant to fusion of the viral envelope with the host membrane and subsequent entry into the cell.69,70 It was also used to monitor how a small icosahedral plant virus responded to increasing concentrations of denaturing urea, revealing exactly where the capsid starts to unravel in response to this chemical stress.71 Similarly, HDX-MS was used to monitor the heat-response of the parvovirus Minute Virus of Mice (MVM) capsid.68 Heating of the capsid is known to trigger externalization of the capsid proteins N-termini, exposing a nuclear localization signal that is crucial for intracellular trafficking and infectivity. It thereby mimics an important structural transition during viral replication and by studying how HDX kinetics scaled with temperature, the precise regions that melt and enable externalization of the capsid protein N-termini could be identified.
Outlook on Integrated Structural Virology by HDX-MS
HDX-MS has thus proven its great value in structural virology in studies that essentially cover the entire replication cycle, neutralizing immunity, drug development, and biophysical characterization. A common theme in many of the studies we reviewed here is how HDX-MS is used as part of an integrative structural biology approach to these common and fundamental problem in virology. Whereas atomic structural models derived from X-ray crystallography and cryo electron microscopy experiments provide a more static structural view of the viral protein, they also build an essential framework to interpret the HDX patterns and dynamics that can be monitored by MS. More examples of this integrative structural virology approach with other MS-based techniques are reviewed in the following sections.
Whereas the technical basis of a typical HDX-MS experiment has remained virtually the same over the past decades, the accumulated improvements in mass spectrometry-based proteomics in sample preparation, LC, MS, data analysis, and automation have improved throughput tremendously and made the technique much more widely accessible throughout academia and the biopharmaceutical industry. However, the viral envelope glycoproteins remain very challenging targets due to their heterogeneous glycosylation and the associated challenges of identifying heterogeneous glycopeptides under the time-pressure restrictions of an HDX-MS experiment. Similarly, HDX-MS on a system- or virion-wide scale would get us closer to studying these viral targets in their native environments and tackle more complex problems with less prior knowledge on the system. With this in mind, we look forward to future improvements in RP-LC at subzero temperatures, more applications of modern DIA technologies in HDX-MS, as well as implementation of the trialed and tested new glycopeptide fragmentation schemes and we review in the following section. Such developments would push HDX-MS to a new level for an even broader and deeper understanding of the structural dynamics of viruses.
Mass Spectrometry-Based Analysis of Viral Glycoproteins and Their Functional Role
The viral glycoproteins of enveloped viruses are notoriously heavily glycosylated.112 These viral glycans constitute the outermost surface of the enveloped virion and thereby play an important role in molecular recognition of the host. In addition to the envelope glycoproteins, some viruses can also secrete glycoproteins (e.g., NS1 protein of flaviviruses, sGP of Ebola virus, sgG of herpes simplex virus, and HBsAg of HBV).112 Whether they are embedded in the envelope or secreted from infected cells, these glycoproteins are involved in many steps in the viral replication cycle, e.g., by binding to host glycans and lectin-type receptors (e.g., DC-SIGN, L-SIGN, mannose receptor, sialic acids, etc.),112−115 through evasion of the host immune system by glycan shielding,116−121 correct viral protein folding,112,122,123 structure maintenance,118,121 and receptor destruction for virion release (e.g., neuraminidase and hememagglutinin esterase).124 The viral glycoproteins are decorated with glycans through the host cell’s own glycosylation machinery.112 This way the virus ends up with a similar glycosylation pattern as its host, which is thought to hamper immune recognition of the virus.112 Some giant viruses like chlorovirus and mimiviruses form notable exceptions, as they carry their own glycosylation machinery that gives rise to unconventional and unique glycosylation patterns.112,125,126
This pattern of extensive glycosylation in the viral surface antigens is observed in many highly pathogenic viruses. Among the most extreme examples are the extraordinary glycan shields present on the surfaces of the HIV envelope protein Env (25–30 glycosites per protomer), coronavirus spike proteins (23–38 glycosites per protomer), the Ebola virus GP (∼17 glycosites per heterodimer and a mucin-like domain), and influenza A virus hememagglutinin HA (∼13 glycosites per protomer)112,116 (see Figure 8). In each case, glycosylation is thought to hide conserved immunodominant epitopes from the immune system of the host (e.g., receptor binding domains, RBDs). On the other hand, viral glycans play a dual role in immune recognition: being the outermost virion layer and a critical component on the main surface antigens, they are often also integral parts of neutralization epitopes.115,117,127−129 All things considered, viral glycans play a major role in replication, disease, and immunity and are therefore important targets in the design of next-generation structure-guided vaccines and therapeutics, especially against the viruses for which conventional vaccine design has proven to be ineffective.
Figure 8.

Oligomannose patches in viral glycan shields are quite distinct as revealed by glycoproteomics. From left to right, MERS-CoV S, SARS-CoV-1 S, SARS-CoV-2 S, LASV GPC, and HIV-1 Env. Under-processing of viral glycan shields is shown as site-specific N-linked glycan oligomannose quantifications and colored according to the key. All glycoproteins were expressed as soluble trimers in HEK 293F cells apart from LASV GPC, which was derived from virus-like particles from Madin-Darby canine kidney II cells. From Watanabe, Y.; Allen, J. D.; Wrapp, D.; McLellan, J. S.; Crispin, M. 2020 Site-specific glycan analysis of the SARS-CoV-2 spike. Science (Washington, DC, U. S.)369(6501): 330–333 (ref (148)). Reprinted with permission from AAAS.
To our knowledge, not a single glycosylation study has been performed on host or patient derived material, due to the scarce amounts of viral glycoproteins that can be extracted from blood, let alone tissue. The true glycosylation patterns of “wild” viruses therefore remain elusive. Notable exceptions are HIV and flaviviruses like dengue virus, which can replicate in peripheral blood monocytes130 and dendritic cells,131 such that the in vitro culturing methods emulate natural infection rather closely. For most other viruses, however, the difficulty of obtaining virion material from natural infection necessitates the usage of various mammalian, insect, and plant expression systems to produce viral glycoproteins for structural studies and vaccines.122,130,132,133 In some cases, like production of live attenuated influenza vaccine, chicken eggs are the system of choice.134,135 We do reiterate, viral glycosylation is strictly dependent on the glycosylation machinery of the host or expression system used in a study, which remains an important theme and restriction in the field.
Glycans are known for their heterogeneity and structural complexity that stems from the number of enzymes in the ER and Golgi involved in glycan processing (around 1% of the mammalian genome).112,136 Glycans consist of relatively rigid monosaccharide residues coupled by flexible glycosidic linkages, which are responsible for the countless numbers of branched and linear structures.136 N-Linked glycans decorate virtually all enveloped viruses and, therefore, represent also the bulk of viral glycosylation studies reported to date, although some examples of O-linked glycosylation are also well-documented, such as those investigating the mucin-like domain of EBOV GP.112,137 EBOV GP represents an interesting case as it is also found to carry additional C-linked mannose on the tryptophan within the canonical WXXW motif (X is any amino acid), normally found only on selected mammalian proteins carrying thrombospondin repeats.138
The specificity of the N-glycosylation initiation step that occurs on the asparagine within an NXT/S sequon (and rarely NXC sequon; X can be any amino acid except proline), makes N-glycosylation sites most predictable.126 Whereas the sites of N-linked glycans are predictable, N-linked site occupancy can vary substantially throughout the sequence of a viral glycoprotein. Moreover, many viruses contain a multitude of N-glycosites within one glycoprotein, each of which can be modified with up to 100 different glycoforms. Even within the same viral glycoprotein, N-glycan composition can vary substantially from site to site, recently defined as meta-heterogeneity,139 indicating that beyond the restrictions of the host expression system, local structural features of the viral antigen can further regulate the extent of glycan processing. The situation for O-linked viral glycosylation is even more complicated, as it can potentially occur on every serine or threonine within mucin-like domains (e.g. GP and sGP proteins of Ebola virus, gC of herpes simplex virus and the G protein of respiratory syncytial virus), characterized by S/T/P-rich stretches in the sequence, without strict sequence motifs that can be used to mark the sites of glycosylation.112,140 Each serine and threonine residue in a mucin-like domain can be decorated with at least eight different glycan cores, each of which can be extended further.112 O-Linked glycans are, therefore, similarly heterogeneous but often spaced more closely together in the sequence, complicating structural characterization by mass spectrometry. In other words, glycans of all forms as present on many viral proteins are challenging entities for analytical and structural studies.
In the past, most studies involving viral glycosylation were performed on bulk released glycans, using a wide range of glycomics techniques.126,133,141 Although valuable, these studies usually provide a list of all the glycoforms present on the viral glycoprotein, without any information to assign which glycan is attached to which glycosylation site.126 Site-specific information on glycosylation had been largely limited to site occupancy of N-glycans, inferred from the 1 Da difference between tryptic nonglycosylated and deglycosylated peptides upon the deamidation of asparagine to aspartic acid during enzymatic release of the N-glycan.133 Deamidation, in its turn, can also occur spontaneously, resulting in a potential overestimation of site-occupancy. As mentioned above, viral glycoproteins may carry up to 40 glycosylation sites per protomer and to fully understand the structural, virological, and immunological role that glycosylation plays, we need to consider the ensemble of glycoforms at each individual site in its unique local structural context. Higher order structural organization of specific sites into clusters, carrying specific glycans can impact the attachment to the host cells or recognition and neutralization by the host immune system, i.e., by neutralizing antibodies.115,118 To complicate things further, viral glycans can also carry site-specific modifications like phosphorylation and sulfation, as was shown for the receptor binding HA glycoprotein derived from seven different influenza vaccines.142 These modifications were thought to affect viral replication, receptor binding, and antigenicity.142 Interestingly, influenza virus has evaded the host immune response by acquiring new glycosylation sites on the HA glycoprotein.113,134,143 Detailed quantitative characterization of site-specific glycosylation patterns is essential to understand these processes. Recent developments in glycoproteomics technologies, encompassing sample preparation, powerful contemporary MS methodology, and bioinformatic tools for automated glycopeptide analysis have enabled this site-specific analysis of viral glycosylation. Hence, we would like to acknowledge the pivotal role that glycan-based technology still plays in the field, but focus our review on the glycoproteomic approaches for site-specific glycan characterization at the glycopeptide level as applied in structural virology.
Glycoproteomic Toolbox of Viral Glycosylation Studies
The basis of glycoproteomics is a glycopeptide-centric bottom-up approach, in which glycoproteins are digested by proteases into (glyco)peptides and subsequently analyzed by reverse phase LC–MS/MS. Considering the differences in N-linked and O-linked glycosylation machineries, different types of analyses are required for the corresponding site-specific glycan localization and composition determination for each of these glycosylation types.
N-Linked Glycoproteomic Sample Preparation
Most viral glycoproteomic analyses to date are still performed on purified recombinant glycoproteins, expressed in non-natural host cells,119,144 although sometimes whole virus like particles,120 spikes from pseudoviruses130 or even authentic virions are targeted.135,145 A major recent theme advancing viral N-glycoproteomics, enabling precise and confident mapping of glycosylation sites and glycoforms, is the adaptation of multiple proteases in parallel in the workflows, extending beyond the trypsin gold standard used in most proteomics studies. Trypsin, although still one of the best proteases for proteomics, yields limited sequence and site coverage and can be hampered by missed cleavages close to glycosylation sites.146 Instead, multiple proteases with different complementary specificities are widely used nowadays, especially in the glycoproteomics field, to obtain close to full sequence coverage and multiple peptides for each glycosylation site, improving confidence in the identifications. Often, the sequential digestion by multiple proteases is required to create a glycopeptide of suitable size for subsequent LC–MS/MS analysis containing just a single N-glycan sequon. This approach was especially valuable in the characterization of the HIV-1 Env trimers (around 90 N-glycosites per trimer),130,144,147 of various coronavirus spike glycoproteins,117,119,121,148 and when studying the Lassa virus glycan shield.120 The most popular proteases adopted in these workflows include trypsin, chymotrypsin, alpha lytic protease, and GluC in particular and to a lesser extent also AspN, ArgC, LysC, elastase, subtilisin, and combinations thereof.
The generated glycopeptides are then analyzed directly with LC–MS/MS or following further enrichment with various strategies like HILIC-SPE.149 The latter was used in the identification of a unique protein degradation pathway, based on the nonenzymatic site-specific glycan shedding from the stabilized form of the RSV prefusion F protein.150 Glycopeptides are generated from bulk sample by in-solution digestion or in the case of whole virion samples often by targeted in-gel digestion of the glycoprotein band,117,145,151 for instance in HIV gp120 glycoproteome mapping, whereby the protein was derived from virions propagated in T-lymphocytes.145 Glycopeptide enrichment can usually be omitted when targeting the highly glycosylated envelope proteins but may offer benefits in analyses of whole virion digests with a high background of nonglycosylated peptides. The acquired site-specific glycosylation data allows the detailed analysis of glycan processing for each individual site of the viral glycoproteins, focusing on the prevalence of the N-glycosylation type (oligomannosidic, hybrid, or complex).
Monitoring the extent of glycan processing can also be addressed with a complementary approach where the viral glycoproteins are first digested with multiple proteases, followed by parallel glycan release from the generated peptides using the enzymes PNGaseF and EndoH in “heavy” water, i.e., H2O18.152 In this case, glycopeptides harboring oligomannose glycans will retain the N-acetylglucosamine residue (+ 203 Da) on the asparagine and the glycopeptides decorated with complex glycans will go through the deamidation with the +3 Da difference from the original asparagine.152 The ratio between the glycopeptides carrying + 203, + 3 Da masses and unchanged nonglycosylated peptides is then used to determine site-specific glycan processing and occupancy.152 This approach was elegantly applied in HIV research116,147,152 and lately in the characterization of feline coronavirus spike and IAV HA glycosylation.121,153 Both approaches brought to light the so-called oligomannose patches present in viral glycoproteins, e.g., of HIV-1 Env,118,123,152 Lassa virus glycoprotein,120 and to lesser extent MERS116,117 (see also Figure 8). These oligomannose patches are thought to originate from local crowding of the glycans, resulting in an inability of the glycosyltransferases in the secretory pathway to extensively process (elongate, branch, and cap) the glycan.120,123 While these oligomannose patches shield conserved neutralization epitopes on the peptide backbone, they are themselves also targeted by neutralizing antibodies.115,129 Furthermore, neutralizing antibodies targeting glycans at various specific glycosites, within and outside these oligomannose patches, have also been described.115,127,129,148
O-Linked Glycoproteomic Sample Preparation
Compared to site-specific viral N-glycosylation profiling by MS, site-specific O-linked glycosylation characterization is even more challenging, in part due to the absence of a consensus sequence motif for O-glycosites, hampering prediction of sites.140 Although only a limited number of studies have been reported, O-glycosylation was shown to be involved in virus infectivity and immunomodulation.140 Recently, exciting new workflows for efficient O-glycopeptide generation have been developed for both recombinant proteins154 and virus-infected cells.140 The critical step is the removal of the highly abundant viral N-linked glycans with PNGaseF, prior to subsequent LC–MS/MS analysis.140,154 This step reduces the complexity of the sample, improves the digestion, and facilitates correct O-glycosite assignments. Furthermore, to enrich the O-linked glycopeptides from the cell lysates, lectin affinity chromatography based on PNA and VVA lectins and isoelectric focusing fractionation can be applied.140 Another approach that allows the effective digestion of O-glycoproteins and a reduction of the O-glycosite numbers per peptide is the use of novel glycoproteases that specifically cleave either N-terminally (OpeRATOR)154 or C-terminally (StcE)155 from O-glycosylated serines and threonines. These strategies were successfully implemented for the proteome-wide O-glycan characterization of herpes viruses against the background of infected cells and demonstrated the significance of the extended O-glycans for virus propagation.137,140 Moreover, it revealed that viral O-glycosylation is widely distributed beyond clearly identifiable mucin-like domains (e.g., in cases of Zika virus envelope protein and herpesviruses).137,154
MS Fragmentation Strategies in Viral Glycoproteomics
Once glycopeptides are generated with the appropriate proteases, they are subjected to LC–MS/MS analysis where fragmentation of the glycopeptides plays a crucial role in the site-specific glycan identification. To enable confident glycopeptide identification, fragments should be identified from the peptide backbone, the glycan attached to the peptide, and the complementary oxonium ions that appear upon the fragmentation of glycosidic linkages.133,156 To achieve the formation of all these fragment ions, various fragmentation strategies have been applied over the years. When applied to glycopeptides, the most common fragmentation methods in conventional proteomics, collision-induced dissociation (CID) and higher energy collision-induced dissociation (HCD), predominantly break the labile glycosidic linkages. This provides mostly glycan fragments (b- and y-ions) and thus limited peptide backbone fragmentation to enable confident peptide identification. On the other hand, electron transfer dissociation (ETD) and electron capture dissociation (ECD) preferentially fragment the peptide backbone of glycopeptides with the conjugated glycan left intact (c- and z-ions) and, therefore, can provide peptide sequence coverage.156 Furthermore, by alternating the collision energies of CID and HCD, differential sets of fragments can be acquired, with lower energy resulting primarily in glycan fragmentation and higher energy resulting in additional peptide backbone fragmentation.133 The latter approach with parallel acquisition of CID/HCD or CID/ETD spectra for the same glycopeptide has recently been more widely adopted in the viral glycoproteomic field, as it generates two very complementary and informative data sets. Such combined fragmentation methods to identify viral glycopeptides were applied in the bulk of pioneering studies on HIV Env glycosylation,116,130,144,145,147,152 for the characterization of the N-glycans decorating glycoprotein B of cytomegalovirus and E2 of Hepatitis C virus.122,132 In the case of glycosylation characterization of heamagglutinin-neuraminidase from Newcastle disease virus, a fragmentation strategy was implemented wherein MS/MS spectra were acquired with HCD fragmentation, which upon identification of oxonium ions triggered either ETD or CID fragmentation of the same precursor, a technique called HCD-pd-ETD(CID), which boosts specifically the numbers of identified glycopeptide fragmentation spectra and improves the glycopeptide assignments.135
In recent years, a hybrid fragmentation strategy known as EThcD (electron transfer/higher energy collision-induced dissociation), in which peptides are fragmented with ETD and subsequent supplemental HCD activation,157 was shown to be beneficial for the characterization of glycopeptides and had a positive effect on the field of viral glycoproteomics.157,158 This scheme takes advantage of two complementary dissociation techniques and, therefore, generates dual fragment ion series from the glycan, peptide, and glycopeptide conjugate in a single mass spectrum.157 Similar results can be obtained with the recently developed and related hybrid fragmentation technique activated ion-ETD (AI-ETD).159 Consequently, EThcD and AI-ETD fragmentation approaches provide richer and more informative mass spectra and facilitate reliable peptide identification, glycosite localization, and partial glycan identification (Figure 9). EThcD fragmentation is especially useful for the localization of O-glycans, since multiple sites can be present within one peptide160 and was successfully applied in the global O-glycosylation mapping of various clinically relevant herpesviruses that showed the presence of O-glycans in the envelope protein regions important for the virus entry.137 Furthermore, EThcD played a vital role in unraveling of the glycan shield compositions for a variety of coronaviruses and Lassa virus,117,119,120 and in determination of the site-specific glycan shielding on the HIV Env against a neutralizing antibody.128 Although highly advantageous, EThcD is still limited to a narrow set of instruments. Nowadays, many instruments can apply multiple collision energies during the CID/HCD fragmentation in one scan, known as stepped HCD, in which the glycopeptide is independently fragmented with three different collision energies and the fragments are recorded in a single mass spectrum.161 By alternating the collisional energies to induce both glycan dissociation and peptide backbone fragmentation, more comprehensive information for the characterization of the glycopeptide is collected.161 Following such an approach, the site-specific glycosylation pattern of the SARS-CoV-2 spike derived from authentic virions could be established.151
Figure 9.

Hybrid EThcD fragmentation yields rich structural data on viral glycopeptides. (a) EThcD MS/MS spectrum of a glycopeptide derived from the MERS-CoV S protein as described in ref (117). A multitude of b/c/y/z-ions originating from peptide backbone fragmentation are detected and enable accurate site localization. In addition fragments of the glycan, conjugated to the peptide, are observed as well as oxonium signature fragment ions, all in one MS/MS spectrum. (b) Overview of the assigned fragments in the EThcD spectrum of panel a, enabling structural reconstruction of the glycopeptide.
Glycoproteomic Data Analysis Tools
Due to their complex fragmentation pathways, very much dependent on the nature of the glycopeptide and fragmentation method used, analysis of glycopeptide MS/MS data is not trivial. Until recently, glycoproteomic data analysis relied primarily on the manual interpretation of acquired mass spectra, as nearly all widely available database search algorithms developed for standard proteomics experiments are not suited for the analysis of glycopeptides. Accordingly, the introduction of dedicated software tools facilitating automated glycopeptide identifications has boosted the field of (viral) glycoproteomics. Currently available tools for glycopeptide identification include the commercial software package Byonic162 and pGlyco,163 the open source programs GlycoPep,164 MS Fragger,165 the ProLuCID algorithm from the Integrated Proteomics Pipeline-IP2,166 and a manual glycan assignment facilitated by GlycoMod.167 Despite the introduction of automation into glycopeptide identification, manual validation is still indispensable due to common misassignments (additional unexpected peptide modifications, isobaric or near-isobaric glycan subcompositions, or noncovalent glycopeptide dimer formation).156
Relative quantification of the site-specific glycosylation patterns of the viral glycoproteins is often done based on the elution peak area for each assigned glycopeptide.124,139,152 Software solutions for peak integration, available from the broader proteomics field, can be adapted for this and include Skyline168 and Byologic from Protein Metrics. It allows one to dissect in a semiquantitative manner site-specific glycan type prevalence and glycosite occupancy that may influence the epitope accessibility for recognition by the host immune system,114,116−118,127,143 fucosylation patterns that may influence the antibody binding as in the case with the SARS-CoV-2 spike,148 sialylation patterns that may influence receptor binding and transmission,124,135 and comparison of the glycosylation patterns for several viral glycoproteins derived from different cell types,122,132,134 viral strains,169 and constructs,144,147 which are important to advance vaccine design and production, since they may influence vaccine efficacy and safety.134 Although such quantitative approaches do not account for differences in detection efficiency between the various glycoforms, various studies have provided evidence that the ionization efficiency of glycopeptides is predominantly driven by the biochemical nature of the peptide component,170 and several quantitative bottom-up glycoproteomics studies showed that the quantitative patterns are consistently observed with orthogonal methods like native MS.171,172
Remaining Challenges in Viral Glycoproteomics and Future Perspectives
Evidently, MS-based glycoproteomics provides essential information for structural virology. Nonetheless, the major disadvantage of current workflows lies in the difficulty of obtaining enough material from natural infections for analysis. Numerous studies have shown that site-specific glycosylation of the viral glycoprotein preparations may differ in detail when produced in different expression systems. However, site-specific glycosylation patterns such as the degree of processing appear to be governed in large part by the local structure of the glycoprotein, which can be monitored even if the expression and culturing systems may differ from the natural host.
Another remaining challenge in MS-based glycoproteomics is the inability to determine the exact nature of the linkages by which monosaccharides are connected within a glycan, as the linkage isomers have identical masses.114,126 The glycan linkages can be very important to viral replication, however. For instance, human influenza viruses bind α2,6-linked sialic acids while avian influenza viruses bind preferentially α2,3-linked sialic acids, information that determines the tropism and the transmission of the virus in different hosts.114 In addition, some of the more common monosaccharide residues are stereoisomers with identical and therefore indistinguishable masses (galactose/mannose/glucose or N-acetyl galactosamine/glucosamine). Unfortunately, most glycoproteomics methods to date are blind to linkage variations and unable to distinguish between stereoisomers, although many of these elusive structural features can also be inferred from the known biosynthetic pathways of glycans.
In the past few years, native MS has also entered the field of intact glycoprotein analysis, delivering stunning results on proteoform characterization of glycosylated proteins and allowing direct assessment of the combinatorial PTMs.172 Viral glycoproteins remain challenging targets for this technique, however, due to the excessive amounts of glycans present on most viral proteins. Despite these challenges, a limited number of studies have started to tackle such complex viral systems.13,173
Glycoproteomics has developed very rapidly in recent years with fruitful applications in structural virology. We look forward to further analytical advances in sample preparation that enable the extraction of also low-abundance viral glycoproteins from natural infection. In addition, a more widespread implementation of novel glycopeptide fragmentation strategies like EThCD, AI-ETD, stepped HCD, HCD-pd-EThcD, and improvements in automated glycopeptide assignments will help to bring the field of the viral glycoproteomics to a higher level and contribute to improved vaccine design and therapeutics as well as a deeper understanding of the structural role of glycans in host cell recognition and immune evasion.
Integrating Mass Spectrometry into Structural Studies of Viruses
Although mass spectrometry, in all its flavors, provides extremely valuable information for structural virology, ideally these approaches are combined and integrated with complementary approaches to study in detail all different processes important in virology. Several of the HDX-MS and viral glycoproteomics studies reviewed above were in fact part of such an integrative structural biology approach, in which mass spectrometry is combined with for instance X-ray crystallography or cryo-electron microscopy.
As mentioned earlier, glycans are abundant in many viral glycoproteins, but also highly heterogeneous and flexible, which makes glycans a difficult target for the more conventional high-resolution structural studies. Usually, high-resolution structures of the viral glycoproteins decorated with the mammalian glycans deal with a lack of electron density for the regions with glycosylation sites117,124,128,174,175 or low-resolution densities surrounding the peptide backbone carrying the glycans.176 For instance, glycan shields of Lassa virus and coronaviruses contribute 25% to the molecular weight of the glycoprotein.116 Despite representing such a substantial portion of the viral antigen, the glycans can typically only be described in the electron density maps as a blob surrounding the proteinaceous structure, missing essential information on the site-specific glycosylation patterns.
To tackle this issue, several groups implemented contemporary MS-based glycoproteomic techniques to chart dozens of site-specific glycan compositions,124,152 thereby providing valuable information to interpret the low-resolution glycan blobs in the EM maps.116,117,119,121,124,128,177−179 Such combined approaches were used, for instance, to reveal holes in the glycan shields of SARS- and MERS-CoVs that can be targeted by vaccines or therapeutics.117 Taken together, these integrative structural biology approaches provide much more comprehensive information about, among others, the density of the glycan shields,116 organization of weakly immunogenic oligo-mannose patches around the protein backbone,117,123 conservation of specific glycans and glycosylation sites,178 as well as important glycan epitopes and accessibility for broadly neutralizing antibodies.127,128
Beyond HDX-MS and glycoproteomics, native MS has also been used in several integrative structural virology studies, for example, in studying capsid self-assembly. The propensity of virus capsids for self-assembly is increasingly used in the design of virus-based polyvalent nanoparticles180 that are nonreplicating and combine all the features of the native virion surface with natural immunogens.181 The integrative structural biology approach including native MS was applied to monitor nanoparticle formation and the proportion of unassembled protomers in the case of 60-valent icosahedral nanoparticles displaying the fused pathogenic rotavirus spike protein.181 The same approach was used to describe the heterogeneity of the norovirus capsid symmetries at pH 7.5180 and in the integrative structural study of VSV (vesicular stomatitis virus) and CHAV (Chandipura virus) glycoprotein spikes that are responsible for host cell membrane fusion under acidic conditions.182
In structural virology, the whole is greater than the sum of its parts when it comes to our research methodologies. High-resolution structures obtained by X-ray crystallography and cryo-electron microscopy can, and should, be complemented by HDX-MS, glycoproteomics, and native MS to fill in blind spots on virus assembly, composition, and heterogeneity as well as structural dynamics.
Mass Spectrometry in the Era of COVID-19
In late 2019, the world was confronted by a new virus that led to the outbreak of a pandemic. The virus has been named SARS-CoV-2, due to its close relationship with the earlier SARS coronavirus from 2002. Since the outbreak of the pandemic, the scientific community at large has reached out to study this new virus and its effect on infected humans. By September 2020, the COVID-19 Open Research Data set resource (CORD-19) contained over 52 000 articles on COVID-19 and related coronaviruses.
Mass spectrometry in all its different implementations has also contributed to this humongous worldwide research effort. With the daily growth in papers published about COVID-19 in journals and public depositories, it is rather impossible to review the full body of work. Still, we did think that a review about mass spectrometry-based structural virology should not leave COVID-19 unmentioned. Therefore, we chose to highlight some of the work that has been published in the last year, in response to the pandemic outbreak. Here we chose not to limit ourselves to only structural or glycoproteomics studies on SARS-CoV-2 but also mention other flavors of mass spectrometry-based analysis. Indeed, leaders in all areas of mass spectrometry-based proteomics, e.g., interaction proteomics, quantitative phosphoproteomics, and clinical proteomics adapted their methodologies, efficiently applying them to studies targeting COVID-19. Additionally, the first studies on SARS-CoV-2 appeared using native MS, HDX-MS, and glycoproteomics, which we will briefly mention here as well.
We do likely miss many other recent studies in which mass spectrometry was used to study SARS-CoV-2 and its impact on the host. Still, these chosen highlights already make a clear case that mass spectrometry in all its forms can be used and applied in structural and functional virology, including that of SARS-CoV-2.
Mass Spectrometry-Based SARS-CoV-2 Interactome Analysis
A consortium led by Krogan et al. used their well-established affinity-proteomics-mass spectrometry (AP-MS) pipeline for identifying interactions between all predicted viral proteins from SARS-CoV-2 (27 proteins) and host cell proteins.183 Not much later, a similar AP-MS study on the interactome appeared by Stukalov et al.184 Related to that, Gingras, Raugth et al. targeted specifically interactions between SARS-CoV-2 at host cell membrane proteins using not classical AP-MS but proximity-dependent biotin labeling (BioID).185 These studies provide an extensive network of interactions, albeit mostly in a somewhat unnatural cellular environment, HEK-293T/17 cells or A549 lung carcinoma cells.
Mass Spectrometry-Based Analysis of SARS-CoV-2 Infection in Vitro
The same consortium led by Krogan reported a quantitative mass spectrometry-based proteomics and phosphoproteomics analysis of Vero E6 cells infected by SARS-CoV-2. Vero E6 cells are the most widely used cells to replicate and isolate SARS-CoV-2. As expected, this analysis revealed an extensive rewiring of phosphorylation on both host and viral proteins. Upon infection by the virus, casein kinase II (CK2) and p38 MAPK became activated, diverse cytokines were higher expressed, and mitotic kinases became inactivated, resulting in cell cycle arrest. Inhibition of several kinases was shown to hamper infection in vitro, thereby providing leads for putative COVID-19 therapies.
Mass Spectrometry-Based Detection of SARS-CoV-2 in Body Fluids
Mass spectrometry (MS) can deliver valuable diagnostic data that complements genomic information and allows us to increase our current knowledge of the COVID-19 disease caused by SARS-CoV-2. For instance, Sinz et al. developed a MS-based method to detect SARS-CoV-2 proteins from gargle solution samples of COVID-19 patients using a targeted MS analysis, focusing on unique peptides from the nucleoprotein of SARS-CoV-2.186 Similar studies appeared almost simultaneously with detection by mass spectrometry of SARS-CoV-2 virus peptide signatures in scrapings/swaps of the epithelium of the nasopharynx of infected people.187,188 Messner et al. used a targeted SWATH-MS based high-throughput proteomics approach to identify clinical classifiers in patients that had been affected by COVID-19.189 Using first a cohort of 31 COVID-19 patients to identify classifiers, these were validated on an additional cohort of 17 new patients and 15 healthy volunteers. Although they identified protein signatures to classify COVID-19 patients, most of these proteins are classical proteins involved in inflammation. It needs to be seen whether these classifiers are indeed COVID-19 selective, being able to distinguish them from patients with other forms of infection and disease that leads to inflammation.
Mass Spectrometry-Based Analysis of SARS-CoV-2 Glycoproteins
The envelope of SARS-CoV-2 is decorated with the trimeric spike glycoprotein (S) molecules consisting of S2 and S1 subunits. The latter is in possession of the RBD and promotes host cell attachment and entry. Being surface exposed, spike protein is a major target for the humoral immune response and a focus for the vaccine and therapeutic development. Importantly, SARS-CoV-2 S protein is decorated with 22 predicted N-glycosylation sites per monomer. Previously, such glycan shields have been described in detail for other coronaviruses as SARS, MERS, and HKU1. These glycans are known to mask the virus from the host immune surveillance and influence host cell attachment. As a result, the SARS-CoV-2 glycan shield has drawn the attention of the glycoproteomic community early in the pandemic. A first comprehensive quantitative site-specific characterization of the S protein glycosylation was reported by Watanabe et al.148 and appeared only a couple of months into the pandemic. The analysis was performed on the native like S-trimers locked in the prefusion conformation. The authors found all 22 predicted sites to be occupied. The RBD of the spike protein is known to be an important target for neutralizing antibodies and was found to be partly shielded by nearby glycans. The majority of the sites were occupied by processed complex glycans. No distinctive oligomannose patches were observed on the SARS-CoV-2 S protein. Furthermore, minimal traces of O-glycosylation were detected. Similar results were obtained later by Shajahan et al.190 and Zhou et al.191 In a complementary study, Zhao et al.192 also described the N-glycosylation of the ACE2 receptor and provided molecular dynamics simulations of both S and ACE2 glycoproteins decorated with the identified glycans interacting with each other. Molecular dynamics simulations predicted the interactions of ACE2 glycans with the RBD of the S glycoprotein as well as interactions between the glycans of both glycoproteins. All these studies were performed on virus proteins generated recombinantly in human HEK293 cells. Lately, Yao et al.151 provided a site-specific N-glycan characterization of the spikes derived from authentic virions propagated in Vero cells. Notably, the overall N-glycan processing state of the native spikes were similar to those of the earlier studied recombinant trimers. Taken together, these data provide important information on the epitopes available for the neutralizing antibodies and the accessibility thereof, which will impact immunogen design strategies.
Mass Spectrometry-Based Structural and Functional Analysis of SARS-CoV-2 Proteins
The SARS-CoV-2 RNA genome encodes for more than two dozen different proteins with their own specific functions. Although evidently the SARS-CoV-2 spike proteins receive a lot of attention, several studies have now appeared characterizing the structure and function of some of the other proteins. For instance, Chen et al. reported on the structure of the SARS-CoV-2 replication-transcription complex (RTC) alone and bound with the nsp13 helicase presenting high-resolution structures obtained by cryoEM. The RTC complex is essential for replication and transcription of the genome of the virus and represent therefore alternative putative targets for treating the disease. Mass spectrometry contributed to this study as the mass extracted from native mass spectrometry measurements for the holo-RdRp:RNA complex corroborated the 1:2:1:1 stoichiometry for nsp7–nsp8–nsp12–RNA. Addition of the 67.5-kDa nsp13 helicase to holo-RdRp sample revealed an efficient complex formation with a 1:1 stoichiometry.
Work of Robinson and Vakonakis193 focused on the protease M encoded by the SARS-COV-2 RNA genome. Its role is to process several of the virus structural proteins. This processing is essential for viral replication, and therefore the protease may provide another therapeutic target. Using native mass spectrometry, they did show that the protease has a high tendency to dimerize, Kd = 0.14 μM. Moreover, using mass spectrometry they set up an assay to monitor protease activity and how that could be diminished by adding small inhibiting molecules representing therefore again a possible alternative way for treating the disease.
Mass Spectrometry-Based Analysis of SARS-CoV-2 Presented HLA Class II Ligands
Parker et al.194 used mass spectrometry to monitor HLA class II peptide antigens from the SARS-CoV-2 spike presented on the cell surface of dendritic cells. Such peptides may be recognized by T-cells and elicit a protective immune response. They identified more than 200 unique HLA-II- peptides, many originating from nested sets. Several of these represented glycosylated peptides. This data may be relevant for vaccine design and could aid analysis of CD4+ T cell responses in infected patients as well as future vaccine recipients.
Concluding Remarks
Here we have reviewed how mass spectrometry has contributed to the field of structural virology, focusing primarily on studies that emerged in the last couple of years. We have seen how developments in, for instance, native mass spectrometry and glycoproteomics are readily picked up by the structural virology field, whereas HDX-MS has long since been part of the repertoire and is becoming ever more widespread. Other mass spectrometry techniques, not covered here, will likely also enter the field of structural virology. Here, we like to mention especially cross-linking mass spectrometry, as this provides structural information via distance constraints and can be used to look at interaction networks on a system-wide scale (e.g., whole virions or replication compartments) and at conformational changes occurring within a single protein (pre- to postfusion changes and receptor binding). All in all, it is clear that mass spectrometry is here to stay in structural virology, and we look forward to see its limits expand in the near future.
Acknowledgments
We thank all the members of the Heck and Snijder laboratories for general support. This research received funding through The Netherlands Organization for Scientific Research (NWO) through the Spinoza Award SPI.2017.028 to A.J.R.H. and the ICI 024.002.009 grant to J.S.
Biographies
Tobias P. Wörner (1989) is currently a Ph.D. candidate at Utrecht University, The Netherlands. He obtained his Master degree in Biophysics from the Humboldt University of Berlin, Germany. His research focuses on the analysis of high-mass biomolecular systems by native mass spectrometry. He has a special focus on developing new analytical techniques for the analysis of complex systems, like viruses and virus-like particles, primarily by using Orbitrap-based mass analyzers.
Tatiana M. Shamorkina (1984) is currently a Ph.D. candidate at Utrecht University, The Netherlands. She obtained her Master degree (cum laude) in Biomedical Sciences from Leiden University, The Netherlands. Her Ph.D. research focuses on the role of glycosylation in virus–host interactions with specific interest in the impact of glycosylation on vaccine design for the Dengue virus combining mass spectrometry with structural biology.
Joost Snijder (1988) is an Assistant Professor at Utrecht University, The Netherlands. His research focuses on the role of glycosylation in virus–host interactions, using an integrated structural biology approach, which combines mass spectrometry and cryo electron microscopy. He received his Ph.D. from Utrecht University in 2015, followed by a postdoctoral research position at the University of Washington in Seattle, WA, USA. He is the recipient of an EMBO long-term fellowship and an NWO Rubicon grant. In 2018, he was awarded the KNAW Heineken Young Scientist Award. With the support of the NWO Gravitation Program, The Institute for Chemical Immunology, he started his own research group at Utrecht University in 2019.
Albert J. R. Heck (1964) received his Ph.D. from the University of Amsterdam. Since 1998 he has been a Professor in Biomolecular Mass Spectrometry and Proteomics at Utrecht University (NL), becoming a Distinguished Faculty Professor in 2017. His research focuses on the development and applications of mass spectrometry-based proteomics and structural biology. Heck introduced proteomics technologies for phospho-enrichment and the use of alternative proteases and hybrid peptide fragmentation techniques. Heck is also known for his expertise in structural biology, being a pioneer in native mass spectrometry and cross-linking mass spectrometry. Heck is the recipient of several awards including the Field and Franklin Award (ACS), The Thomson Medal Award (IMSF), the Krebs Medal (FEBS), and the Spinoza Prize (NWO). He is a member of the Royal Netherlands Academy of Sciences and Arts (KNAW) and EMBO.
Author Contributions
# T.P.W. and T.M.S. contributed equally.
The authors declare no competing financial interest.
References
- Conway J. F.; Duda R. L.; Cheng N.; Hendrix R. W.; Steven A. C. Proteolytic and Conformational Control of Virus Capsid Maturation: The Bacteriophage HK97 System. J. Mol. Biol. 1995, 253 (1), 86–99. 10.1006/jmbi.1995.0538. [DOI] [PubMed] [Google Scholar]
- Mateu M. G. Assembly, stability and dynamics of virus capsids. Arch. Biochem. Biophys. 2013, 531 (1–2), 65–79. 10.1016/j.abb.2012.10.015. [DOI] [PubMed] [Google Scholar]
- Dowd K. A.; Pierson T. C. The Many Faces of a Dynamic Virion: Implications of Viral Breathing on Flavivirus Biology and Immunogenicity. Annu. Rev. Virol. 2018, 5 (1), 185–207. 10.1146/annurev-virology-092917-043300. [DOI] [PubMed] [Google Scholar]
- Harrison S. C. Viral membrane fusion. Virology 2015, 479–480 (7), 498–507. 10.1016/j.virol.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuerstenau S. D.; Benner W. H.; Thomas J. J.; Brugidou C.; Bothner B.; Siuzdak G. Mass Spectrometry of an Intact Virus. Angew. Chem., Int. Ed. 2001, 40 (3), 541–44. . [DOI] [PubMed] [Google Scholar]
- Tito M. A.; Tars K.; Valegard K.; Hajdu J.; Robinson C. V. Electrospray Time-of-Flight Mass Spectrometry of the Intact MS2 Virus Capsid. J. Am. Chem. Soc. 2000, 122 (14), 3550–51. 10.1021/ja993740k. [DOI] [Google Scholar]
- Uetrecht C.; Versluis C.; Watts N. R.; Roos W. H.; Wuite G. J. L.; et al. High-resolution mass spectrometry of viral assemblies: Molecular composition and stability of dimorphic hepatitis B virus capsids. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (27), 9216–20. 10.1073/pnas.0800406105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo J. A. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom. Rev. 1997, 16 (1), 1–23. . [DOI] [PubMed] [Google Scholar]
- Leney A. C.; Heck A. J. R. Native Mass Spectrometry: What is in the Name?. J. Am. Soc. Mass Spectrom. 2017, 28, 5–13. 10.1007/s13361-016-1545-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck A. J. R. Native mass spectrometry: A bridge between interactomics and structural biology. Nat. Methods 2008, 5 (11), 927–33. 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- Mehmood S.; Allison T. M.; Robinson C. V. Mass Spectrometry of Protein Complexes: From Origins to Applications. Annu. Rev. Phys. Chem. 2015, 66 (1), 453–74. 10.1146/annurev-physchem-040214-121732. [DOI] [PubMed] [Google Scholar]
- Uetrecht C.; Heck A. J. R. Modern Biomolecular Mass Spectrometry and its Role in Studying Virus Structure, Dynamics, and Assembly. Angew. Chem., Int. Ed. 2011, 50 (36), 8248–62. 10.1002/anie.201008120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Du Y.; Kaltashov I. A. The Utility of Native MS for Understanding the Mechanism of Action of Repurposed Therapeutics in COVID-19: Heparin as a Disruptor of the SARS-CoV-2 Interaction with Its Host Cell Receptor. Anal. Chem. 2020, 92 (16), 10930–34. 10.1021/acs.analchem.0c02449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehaffey M. R.; Lee J.; Jung J.; Lanzillotti M. B.; Escobar E. E.; Morgenstern K. R.; Georgiou G.; Brodbelt J. S. Mapping a Conformational Epitope of Hemagglutinin A Using Native Mass Spectrometry and Ultraviolet Photodissociation. Anal. Chem. 2020, 92, 11869. 10.1021/acs.analchem.0c02237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty R.; Lokareddy R. K.; Roy A.; Yang Y.; Lander G. C.; et al. Architecture of the Complex Formed by Large and Small Terminase Subunits from Bacteriophage P22. J. Mol. Biol. 2015, 427 (20), 3285–99. 10.1016/j.jmb.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspar D. L. D.; Klug A. Physical Principles in the Construction of Regular Viruses. Cold Spring Harbor Symp. Quant. Biol. 1962, 27, 1–24. 10.1101/SQB.1962.027.001.005. [DOI] [PubMed] [Google Scholar]
- Snijder J.; Heck A. J. R. Analytical Approaches for Size and Mass Analysis of Large Protein Assemblies. Annu. Rev. Anal. Chem. 2014, 7 (1), 43–64. 10.1146/annurev-anchem-071213-020015. [DOI] [PubMed] [Google Scholar]
- Heck A. J. R.; Van Den Heuvel R. H. H. Investigation of intact protein complexes by mass spectrometry. Mass Spectrom. Rev. 2004, 23 (5), 368–89. 10.1002/mas.10081. [DOI] [PubMed] [Google Scholar]
- Snijder J.; Rose R. J.; Veesler D.; Johnson J. E.; Heck A. J. R. Studying 18 MDa virus assemblies with native mass spectrometry. Angew. Chem., Int. Ed. 2013, 52 (14), 4020–23. 10.1002/anie.201210197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutchinsky A. N.; Chernushevich I. V.; Spicer V. L.; Ens W.; Standing K. G. Collisional damping interface for an electrospray ionization time-of-flight mass spectrometer. J. Am. Soc. Mass Spectrom. 1998, 9 (6), 569–79. 10.1016/S1044-0305(98)00027-0. [DOI] [Google Scholar]
- Van Den Heuvel R. H. H.; Van Duijn E.; Mazon H.; Synowsky S. A.; Lorenzen K.; et al. Improving the performance of a quadrupole time-of-flight instrument for macromolecular mass spectrometry. Anal. Chem. 2006, 78 (21), 7473–83. 10.1021/ac061039a. [DOI] [PubMed] [Google Scholar]
- Sobott F.; Hernández H.; McCammon M. G.; Tito M. A.; Robinson C. V. A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal. Chem. 2002, 74 (6), 1402–7. 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- Uetrecht C.; Barbu I. M.; Shoemaker G. K.; Van Duijn E.; Heck A. J. R. Interrogating viral capsid assembly with ion mobility-mass spectrometry. Nat. Chem. 2011, 3 (2), 126–32. 10.1038/nchem.947. [DOI] [PubMed] [Google Scholar]
- Snijder J.; Uetrecht C.; Rose R. J.; Sanchez-Eugenia R.; Marti G. A.; et al. Probing the biophysical interplay between a viral genome and its capsid. Nat. Chem. 2013, 5 (6), 502–9. 10.1038/nchem.1627. [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd M.; Snijder J.; Tsvetkova I. B.; Dragnea B. G.; Cornelissen J. J.; Heck A. J. R. Examining the Heterogeneous Genome Content of Multipartite Viruses BMV and CCMV by Native Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2016, 27 (6), 1000–1009. 10.1007/s13361-016-1348-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luque D.; Escosura A. d. l.; Snijder J.; Brasch M.; Burnley R. J.; Koay M. S. T.; Carrascosa J. L.; Wuite G. J. L.; Roos W. H.; Heck A. J. R.; Cornelissen J. J. L. M.; Torres T.; Caston J. R. Self-assembly and characterization of small and monodisperse dye nanospheres in a protein cage. Chem. Sci. 2014, 5, 575–581. 10.1039/C3SC52276H. [DOI] [Google Scholar]
- Snijder J.; Grinfeld D.; Bennett A.; Makarov A.; Agbandje-McKenna M.; et al. Defining the stoichiometry and cargo load of viral and bacterial nanoparticles by orbitrap mass spectrometry. J. Am. Chem. Soc. 2014, 136 (20), 7295–99. 10.1021/ja502616y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veesler D.; Khayat R.; Krishnamurthy S.; Snijder J.; Huang R. K. K.; et al. Architecture of a dsDNA viral capsid in complex with its maturation protease. Structure 2014, 22 (2), 230–37. 10.1016/j.str.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose R. J.; Damoc E.; Denisov E.; Makarov A.; Heck A. J. R. High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nat. Methods 2012, 9 (11), 1084–86. 10.1038/nmeth.2208. [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd M.; Fort K. L; Boll D.; Reinhardt-Szyba M.; Routh A.; Makarov A.; Heck A. J R High-fidelity mass analysis unveils heterogeneity in intact ribosomal particles. Nat. Methods 2017, 14, 283–286. 10.1038/nmeth.4147. [DOI] [PubMed] [Google Scholar]
- Fort K. L.; Van De Waterbeemd M.; Boll D.; Reinhardt-Szyba M.; Belov M. E.; et al. Expanding the structural analysis capabilities on an Orbitrap-based mass spectrometer for large macromolecular complexes. Analyst 2018, 143 (1), 100–105. 10.1039/C7AN01629H. [DOI] [PubMed] [Google Scholar]
- Lossl P.; Snijder J.; Heck A. J. R. Boundaries of mass resolution in native mass spectrometry. J. Am. Soc. Mass Spectrom. 2014, 25 (6), 906–917. 10.1007/s13361-014-0874-3. [DOI] [PubMed] [Google Scholar]
- Sigmund F.; Massner C.; Erdmann P.; Stelzl A.; Rolbieski H.; Desai M.; Bricault S.; Worner T. P.; Snijder J.; Geerlof A.; Fuchs H.; Hrabe de Angelis M.; Heck A. J. R.; Jasanoff A.; Ntziachristos V.; Plitzko J.; Westmeyer G. G. Bacterial encapsulins as orthogonal compartments for mammalian cell engineering. Nat. Commun. 2018, 9, 1990. 10.1038/s41467-018-04227-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M.; Meng C. K.; Fenn J. B. Interpreting Mass Spectra of Multiply Charged Ions. Anal. Chem. 1989, 61 (15), 1702–8. 10.1021/ac00190a023. [DOI] [Google Scholar]
- Pacholarz K. J.; Barran P. E. Use of a charge reducing agent to enable intact mass analysis of cysteine-linked antibody-drug-conjugates by native mass spectrometry. EuPa Open Proteomics 2016, 11, 23–27. 10.1016/j.euprot.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root K.; Frey R.; Hilvert D.; Zenobi R. High-Mass MALDI-MS Analysis for the Investigation of Protein Encapsulation within an Engineered Capsid Forming Protein. Helv. Chim. Acta 2017, 100 (10), e1700166. 10.1002/hlca.201700166. [DOI] [Google Scholar]
- Wang H.; Eschweiler J.; Cui W.; Zhang H.; Frieden C.; et al. Native Mass Spectrometry, Ion Mobility, Electron-Capture Dissociation, and Modeling Provide Structural Information for Gas-Phase Apolipoprotein E Oligomers. J. Am. Soc. Mass Spectrom. 2019, 30 (5), 876–85. 10.1007/s13361-019-02148-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereszczak J. Z.; Havlik M.; Weiss V. U.; Marchetti-Deschmann M.; van Duijn E.; et al. Sizing up large protein complexes by electrospray ionisation-based electrophoretic mobility and native mass spectrometry: morphology selective binding of Fabs to hepatitis B virus capsids. Anal. Bioanal. Chem. 2014, 406 (5), 1437–46. 10.1007/s00216-013-7548-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss V. U.; Bereszcazk J. Z.; Havlik M.; Kallinger P.; Gösler I.; et al. Analysis of a Common Cold Virus and Its Subviral Particles by Gas-Phase Electrophoretic Mobility Molecular Analysis and Native Mass Spectrometry. Anal. Chem. 2015, 87 (17), 8709–17. 10.1021/acs.analchem.5b01450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss V. U.; Pogan R.; Zoratto S.; Bond K. M.; Boulanger P.; et al. Virus-like particle size and molecular weight/mass determination applying gas-phase electrophoresis (native nES GEMMA). Anal. Bioanal. Chem. 2019, 411 (23), 5951–62. 10.1007/s00216-019-01998-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss V. U.; Frank J.; Piplits K.; Szymanski W. W.; Allmaier G. Bipolar Corona Discharge-Based Charge Equilibration for Nano Electrospray Gas-Phase Electrophoretic Mobility Molecular Analysis of Bio- And Polymer Nanoparticles. Anal. Chem. 2020, 92 (13), 8665–69. 10.1021/acs.analchem.0c01904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keifer D. Z.; Motwani T.; Teschke C. M.; Jarrold M. F. Measurement of the accurate mass of a 50 MDa infectious virus. Rapid Commun. Mass Spectrom. 2016, 30 (17), 1957–62. 10.1002/rcm.7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott A. G.; Merenbloom S. I.; Chakrabarty S.; Williams E. R. Single Particle Analyzer of Mass: A Charge Detection Mass Spectrometer with a Multi-Detector Electrostatic Ion Trap. Int. J. Mass Spectrom. 2017, 414, 45–55. 10.1016/j.ijms.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper C. C.; Elliott A. G.; Lin H. W.; Williams E. R. Determining Energies and Cross Sections of Individual Ions Using Higher-Order Harmonics in Fourier Transform Charge Detection Mass Spectrometry (FT-CDMS). J. Am. Soc. Mass Spectrom. 2018, 29 (9), 1861–69. 10.1007/s13361-018-1987-x. [DOI] [PubMed] [Google Scholar]
- Keifer D. Z.; Shinholt D. L.; Jarrold M. F. Charge Detection Mass Spectrometry with Almost Perfect Charge Accuracy. Anal. Chem. 2015, 87 (20), 10330–37. 10.1021/acs.analchem.5b02324. [DOI] [PubMed] [Google Scholar]
- Harper C. C.; Williams E. R. Enhanced Multiplexing in Fourier Transform Charge Detection Mass Spectrometry by Decoupling Ion Frequency from Mass to Charge Ratio. J. Am. Soc. Mass Spectrom. 2019, 30 (12), 2637–45. 10.1007/s13361-019-02330-3. [DOI] [PubMed] [Google Scholar]
- Pierson E. E.; Keifer D. Z.; Asokan A.; Jarrold M. F. Resolving Adeno-Associated Viral Particle Diversity With Charge Detection Mass Spectrometry. Anal. Chem. 2016, 88 (13), 6718–25. 10.1021/acs.analchem.6b00883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond K. M.; Lyktey N. A.; Tsvetkova I. B.; Dragnea B.; Jarrold M. F. Disassembly Intermediates of the Brome Mosaic Virus Identified by Charge Detection Mass Spectrometry. J. Phys. Chem. B 2020, 124 (11), 2124–2131. 10.1021/acs.jpcb.0c00008. [DOI] [PubMed] [Google Scholar]
- Bond K. M.; Aanei I. L.; Francis M. B.; Jarrold M. F. Determination of Antibody Population Distributions for Virus-Antibody Conjugates by Charge Detection Mass Spectrometry. Anal. Chem. 2020, 92 (1), 1285–91. 10.1021/acs.analchem.9b04457. [DOI] [PubMed] [Google Scholar]
- Todd A. R.; Barnes L. F.; Young K.; Zlotnick A.; Jarrold M. F. Higher Resolution Charge Detection Mass Spectrometry. Anal. Chem. 2020, 92, 11357–11364. 10.1021/acs.analchem.0c02133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe D. M.; Plath L. D.; Aksenov A. A.; Feldman J. S.; Bier M. E. Characterization of Mega-Dalton-Sized Nanoparticles by Superconducting Tunnel Junction Cryodetection Mass Spectrometry. ACS Nano 2018, 12 (3), 2591–2602. 10.1021/acsnano.7b08541. [DOI] [PubMed] [Google Scholar]
- Wörner T. P.; Snijder J.; Bennett A.; Agbandje-McKenna M.; Makarov A. A.; Heck A. J. R. Resolving heterogeneous macromolecular assemblies by Orbitrap-based single-particle charge detection mass spectrometry. Nat. Methods 2020, 17 (4), 395–98. 10.1038/s41592-020-0770-7. [DOI] [PubMed] [Google Scholar]
- Kafader J. O.; Melani R. D.; Durbin K. R.; Ikwuagwu B.; Early B. P.; et al. Multiplexed mass spectrometry of individual ions improves measurement of proteoforms and their complexes. Nat. Methods 2020, 17 (4), 391–94. 10.1038/s41592-020-0764-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov A. Electrostatic Axially Harmonic Orbital Trapping: A High-Performance Technique of Mass Analysis. Anal. Chem. 2000, 72 (6), 1156–62. 10.1021/ac991131p. [DOI] [PubMed] [Google Scholar]
- Naik A. K.; Hanay M. S.; Hiebert W. K.; Feng X. L.; Roukes M. L. Towards single-molecule nanomechanical mass spectrometry. Nat. Nanotechnol. 2009, 4 (7), 445–50. 10.1038/nnano.2009.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage E.; Brenac A.; Alava T.; Morel R.; Dupre C.; Hanay M. S.; Roukes M. L.; Duraffourg L.; Masselon C.; Hentz S. Neutral particle mass spectrometry with nanomechanical systems. Nat. Commun. 2015, 6, 6482. 10.1038/ncomms7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Medina S.; Fostner S.; Defoort M.; Sansa M.; Stark A.-K.; et al. Neutral mass spectrometry of virus capsids above 100 megadaltons with nanomechanical resonators. Science (Washington, DC, U. S.) 2018, 362 (6417), 918–22. 10.1126/science.aat6457. [DOI] [PubMed] [Google Scholar]
- Konermann L.; Pan J.; Liu Y.-H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 2011, 40 (3), 1224–34. 10.1039/C0CS00113A. [DOI] [PubMed] [Google Scholar]
- Lossl P.; Waterbeemd M.; Heck A. J. The diverse and expanding role of mass spectrometry in structural and molecular biology. EMBO J. 2016, 35, 2634. 10.15252/embj.201694818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Lee K. K. Isotope Labeling of Biomolecules: Structural Analysis of Viruses by HDX-MS. In Methods in Enzymology; Academic Press Inc., 2016; Vol. 566, pp 405–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.; Han J.; Borchers C. H.; Konermann L. Hydrogen/deuterium exchange mass spectrometry with top-down electron capture dissociation for characterizing structural transitions of a 17 kDa protein. J. Am. Chem. Soc. 2009, 131 (35), 12801–8. 10.1021/ja904379w. [DOI] [PubMed] [Google Scholar]
- Brodie N. I.; Huguet R.; Zhang T.; Viner R.; Zabrouskov V.; et al. Top-Down Hydrogen-Deuterium Exchange Analysis of Protein Structures Using Ultraviolet Photodissociation. Anal. Chem. 2018, 90 (5), 3079–82. 10.1021/acs.analchem.7b03655. [DOI] [PubMed] [Google Scholar]
- Ahn J.; Cao M. J.; Yu Y. Q.; Engen J. R. Accessing the reproducibility and specificity of pepsin and other aspartic proteases. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834 (6), 1222–29. 10.1016/j.bbapap.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiatsiani L.; Akeroyd M.; Olsthoorn M.; Heck A. J. R. Aspergillus Niger Prolyl Endoprotease for Hydrogen-Deuterium Exchange Mass Spectrometry and Protein Structural Studies. Anal. Chem. 2017, 89 (15), 7966–73. 10.1021/acs.analchem.7b01161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamuro Y.; Zhang T. High-Resolution HDX-MS of Cytochrome c Using Pepsin/Fungal Protease Type XIII Mixed Bed Column. J. Am. Soc. Mass Spectrom. 2019, 30 (2), 227–34. 10.1007/s13361-018-2087-7. [DOI] [PubMed] [Google Scholar]
- Nirudodhi S. N.; Sperry J. B.; Rouse J. C.; Carroll J. A. Application of Dual Protease Column for HDX-MS Analysis of Monoclonal Antibodies. J. Pharm. Sci. 2017, 106 (2), 530–36. 10.1016/j.xphs.2016.10.023. [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd M.; Llauró A.; Snijder J.; Valbuena A.; Rodríguez-Huete A.; et al. Structural Analysis of a Temperature-Induced Transition in a Viral Capsid Probed by HDX-MS. Biophys. J. 2017, 112 (6), 1157–65. 10.1016/j.bpj.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim X.-X.; Chandramohan A.; Lim X. Y. E.; Bag N.; Sharma K. K.; Wirawan M.; Wohland T.; Lok S.-M.; Anand G. S. Conformational changes in intact dengue virus reveal serotype-specific expansion. Nat. Commun. 2017, 8, 14339. 10.1038/ncomms14339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K. K.; Lim X.-X.; Tantirimudalige S. N.; Gupta A.; Marzinek J. K.; Holdbrook D.; Lim X. Y. E.; Bond P. J.; Anand G. S.; Wohland T. Infectivity of Dengue Virus Serotypes 1 and 2 is Correlated to E Protein Intrinsic Dynamics But Not to Envelope Conformations. Structure 2019, 27 (4), 618–630.e4. 10.1016/j.str.2018.12.006. [DOI] [PubMed] [Google Scholar]
- Ramesh R.; Lim X. X.; Raghuvamsi P. V.; Wu C.; Wong S. M.; Anand G. S. Uncovering metastability and disassembly hotspots in whole viral particles. Prog. Biophys. Mol. Biol. 2019, 143, 5–12. 10.1016/j.pbiomolbio.2018.12.006. [DOI] [PubMed] [Google Scholar]
- Bereszczak J. Z.; Watts N. R.; Wingfield P. T.; Steven A. C.; Heck A. J. R. Assessment of differences in the conformational flexibility of hepatitis B virus core-antigen and e-antigen by hydrogen deuterium exchange-mass spectrometry. Protein Sci. 2014, 23 (7), 884–96. 10.1002/pro.2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhaim M. A.; Mangala Prasad V.; Garcia N. K.; Guttman M.; Lee K. K. Structural monitoring of a transient intermediate in the hemagglutinin fusion machinery on influenza virions. Sci. Adv. 2020, 6 (18), eaaz8822 10.1126/sciadv.aaz8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranaweera A.; Ratnayake P. U.; Ekanayaka E. A. P.; Declercq R.; Weliky D. P. Hydrogen-Deuterium Exchange Supports Independent Membrane-Interfacial Fusion Peptide and Transmembrane Domains in Subunit 2 of Influenza Virus Hemagglutinin Protein, a Structured and Aqueous-Protected Connection between the Fusion Peptide and Soluble Ectodomain, and the Importance of Membrane Apposition by the Trimer-of-Hairpins Structure. Biochemistry 2019, 58 (19), 2432–46. 10.1021/acs.biochem.8b01272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia N. K.; Guttman M.; Ebner J. L.; Lee K. K. Dynamic changes during acid-induced activation of influenza hemagglutinin. Structure 2015, 23 (4), 665–76. 10.1016/j.str.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J. J. W.; Young T. A.; Zhang J.; Liu S.; Leser G. P.; Komives E. A.; Lamb R. A.; Zhou Z. H.; Salafsky J.; Jardetzky T. S. Monomeric ephrinB2 binding induces allosteric changes in Nipah virus G that precede its full activation. Nat. Commun. 2017, 8, 781. 10.1038/s41467-017-00863-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Soh T. S.; Zheng J.; Chan K. W. K.; Phoo W. W.; Lee C. C.; Tay M. Y. F.; Swaminathan K.; Cornvik T. C.; Lim S. P.; Shi P.-Y.; Lescar J.; Vasudevan S. G.; Luo D. A Crystal Structure of the Dengue Virus NS5 Protein Reveals a Novel Inter-domain Interface Essential for Protein Flexibility and Virus Replication. PLoS Pathog. 2015, 11, e1004682. 10.1371/journal.ppat.1004682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. W.; Tan Y. B.; Zheng J.; Zhao Y.; Lim B. T.; et al. Chikungunya virus nsP4 RNA-dependent RNA polymerase core domain displays detergent-sensitive primer extension and terminal adenylyltransferase activities. Antiviral Res. 2017, 143, 38–47. 10.1016/j.antiviral.2017.04.001. [DOI] [PubMed] [Google Scholar]
- Law Y. S.; Utt A.; Tan Y. B.; Zheng J.; Wang S.; et al. Structural insights into RNA recognition by the Chikungunya virus nsP2 helicase. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (19), 9558–67. 10.1073/pnas.1900656116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruhn J. F.; Kirchdoerfer R. N.; Urata S. M.; Li S.; Tickle I. J.; Bricogne G.; Saphire E. O. Crystal Structure of the Marburg Virus VP35 Oligomerization Domain. J. Virol. 2017, 91 (2), e01085-16. 10.1128/JVI.01085-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarbouriech N.; Ducournau C.; Hutin S.; Mas P. J.; Man P.; Forest E.; Hart D. J.; Peyrefitte C. N.; Burmeister W. P.; Iseni F. The vaccinia virus DNA polymerase structure provides insights into the mode of processivity factor binding. Nat. Commun. 2017, 8, 1455. 10.1038/s41467-017-01542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Shin W. J.; Zhang B.; Choi Y.; Yoo J. S.; et al. The Cap-Snatching SFTSV Endonuclease Domain Is an Antiviral Target. Cell Rep. 2020, 30 (1), 153–163. 10.1016/j.celrep.2019.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson A.; Zhao Z.; Waymire E.; Zlotnick A.; Bothner B. Dynamics of Hepatitis B Virus Capsid Protein Dimer Regulate Assembly through an Allosteric Network. ACS Chem. Biol. 2020, 15 (8), 2273–80. 10.1021/acschembio.0c00481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z.; Wu C.; Shi L.; Luthra P.; Pintilie G. D.; et al. Electron Cryo-microscopy Structure of Ebola Virus Nucleoprotein Reveals a Mechanism for Nucleocapsid-like Assembly. Cell 2018, 172 (5), 966–978. 10.1016/j.cell.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijesinghe K. J.; Urata S.; Bhattarai N.; Kooijman E. E.; Gerstman B. S.; et al. Detection of lipid-induced structural changes of the Marburg virus matrix protein VP40 using hydrogen/deuterium exchange-mass spectrometry. J. Biol. Chem. 2017, 292 (15), 6108–22. 10.1074/jbc.M116.758300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie K. M.; Zandonatti M.; Liu T.; Li S.; Woods V. L.; Saphire E. O. Crystal Structure of the Oligomeric Form of Lassa Virus Matrix Protein Z. J. Virol. 2016, 90 (9), 4556–62. 10.1128/JVI.02896-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B.; Li J.; Adhikari J.; Edwards M. R.; Zhang H.; et al. Dimerization Controls Marburg Virus VP24-dependent Modulation of Host Antioxidative Stress Responses. J. Mol. Biol. 2016, 428 (17), 3483–94. 10.1016/j.jmb.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan D.; Xing W.; Beran R. K.; Chemuru S.; Rohrs H.; Niedziela-Majka A.; Marchand B.; Mehra U.; Zabransky A.; Dolezal M.; Hubalek M.; Pichova I.; Gross M. L.; Kwon H. J.; Fletcher S. P. Hepatitis B Virus X Protein Function Requires Zinc Binding. J. Virol. 2019, 93 (16), e00250-19. 10.1128/JVI.00250-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroco J. A.; Alvarado J. J.; Staudt R. P.; Shi H.; Wales T. E.; et al. Remodeling of HIV-1 Nef Structure by Src-Family Kinase Binding. J. Mol. Biol. 2018, 430 (3), 310–21. 10.1016/j.jmb.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wales T. E.; Poe J. A.; Emert-Sedlak L.; Morgan C. R.; Smithgall T. E.; Engen J. R. Hydrogen Exchange Mass Spectrometry of Related Proteins with Divergent Sequences: A Comparative Study of HIV-1 Nef Allelic Variants. J. Am. Soc. Mass Spectrom. 2016, 27 (6), 1048–61. 10.1007/s13361-016-1365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhail J. A; Lyoo H.; Pemberton J. G; Hoffmann R. M; Elst W.; Strating J. R.; Jenkins M. L; Stariha J. T.; Powell C. J; Boulanger M. J; Balla T.; Kuppeveld F. J.; Burke J. E Characterization of the c10orf76-PI4KB complex and its necessity for Golgi PI4P levels and enterovirus replication. EMBO Rep. 2020, 21, e48441. 10.15252/embr.201948441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhail J. A.; Ottosen E. H.; Jenkins M. L.; Burke J. E. The Molecular Basis of Aichi Virus 3A Protein Activation of Phosphatidylinositol 4 Kinase IIIβ, PI4KB, through ACBD3. Structure 2017, 25 (1), 121–31. 10.1016/j.str.2016.11.016. [DOI] [PubMed] [Google Scholar]
- Fernandez E.; Kose N.; Edeling M. A.; Adhikari J.; Sapparapu G.; Lazarte S. M.; Nelson C. A.; Govero J.; Gross M. L.; Fremont D. H.; Crowe J. E.; Diamond M. S. Mouse and human monoclonal antibodies protect against infection by multiple genotypes of japanese encephalitis virus. mBio 2018, 9 (1), e00008-18. 10.1128/mBio.00008-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim X. X.; Chandramohan A.; Lim X. Y. E.; Crowe J. E.; Lok S. M.; Anand G. S. Epitope and Paratope Mapping Reveals Temperature-Dependent Alterations in the Dengue-Antibody Interface. Structure 2017, 25 (9), 1391–1402. 10.1016/j.str.2017.07.007. [DOI] [PubMed] [Google Scholar]
- Wirawan M.; Fibriansah G.; Marzinek J. K.; Lim X. X.; Ng T. S.; et al. Mechanism of Enhanced Immature Dengue Virus Attachment to Endosomal Membrane Induced by prM Antibody. Structure 2019, 27 (2), 253–267. 10.1016/j.str.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L.; Lee D. E.; Kadam R. U.; Liu T.; Giang E.; et al. Structural flexibility at a major conserved antibody target on Hepatitis C virus E2 antigen. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (45), 12768–73. 10.1073/pnas.1609780113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg N. J.; Zhang H.; Bangaru S.; Sapparapu G.; Kose N.; et al. H7N9 influenza virus neutralizing antibodies that possess few somatic mutations. J. Clin. Invest. 2016, 126 (4), 1482–94. 10.1172/JCI85317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangaru S.; Lang S.; Schotsaert M.; Vanderven H. A.; Zhu X.; et al. A Site of Vulnerability on the Influenza Virus Hemagglutinin Head Domain Trimer Interface. Cell 2019, 177 (5), 1136–1152. 10.1016/j.cell.2019.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousa J. J.; Sauer M. F.; Sevy A. M.; Finn J. A.; Bates J. T.; et al. Structural basis for nonneutralizing antibody competition at antigenic site II of the respiratory syncytial virus fusion protein. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (44), E6849–58. 10.1073/pnas.1609449113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatya P.; Wagner N.; Chen G.; Luthra P.; Shi L.; et al. Inhibition of Marburg Virus RNA Synthesis by a Synthetic Anti-VP35 Antibody. ACS Infect. Dis. 2019, 5 (8), 1385–96. 10.1021/acsinfecdis.9b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais N.; Gagne M.; Hamuro Y.; Rheault P.; Boyer M.; Steff A.-M.; Baudoux G.; Dewar V.; Demers J.; Ruelle J.-L.; Martin D. Characterization of Pre-F-GCN4t, a Modified Human Respiratory Syncytial Virus Fusion Protein Stabilized in a Noncleaved Prefusion Conformation. J. Virol. 2017, 91 (13), e02437-16. 10.1128/JVI.02437-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrents de la Peña A.; Julien J. P.; de Taeye S. W.; Garces F.; Guttman M.; et al. Improving the Immunogenicity of Native-like HIV-1 Envelope Trimers by Hyperstabilization. Cell Rep. 2017, 20 (8), 1805–17. 10.1016/j.celrep.2017.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Guttman M.; Williams J. A.; Verkerke H.; Alvarado D.; et al. Changes in Structure and Antigenicity of HIV-1 Env Trimers Resulting from Removal of a Conserved CD4 Binding Site-Proximal Glycan. J. Virol. 2016, 90 (20), 9224–36. 10.1128/JVI.01116-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman J.; Soto C.; Yang M. M.; Davenport T. M.; Guttman M.; et al. Structures of HIV-1 Env V1V2 with broadly neutralizing antibodies reveal commonalities that enable vaccine design. Nat. Struct. Mol. Biol. 2016, 23 (1), 81–90. 10.1038/nsmb.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchades C.; Kukrer B.; Diefenbach O.; Sneekes-Vriese E.; Juraszek J.; Koudstaal W.; Apetri A. Epitope mapping of diverse influenza Hemagglutinin drug candidates using HDX-MS. Sci. Rep. 2019, 9, 4735. 10.1038/s41598-019-41179-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauch E. M.; Bernard S. M.; La D.; Bohn A. J.; Lee P. S.; et al. Computational design of trimeric influenza-neutralizing proteins targeting the hemagglutinin receptor binding site. Nat. Biotechnol. 2017, 35 (7), 667–71. 10.1038/nbt.3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner H. L.; Pallesen J.; Lang S.; Bangaru S.; Urata S.; Li S.; Cottrell C. A.; Bowman C. A.; Crowe J. E.; Wilson I. A.; Ward A. B. Potent anti-influenza H7 human monoclonal antibody induces separation of hemagglutinin receptor-binding head domains. PLoS Biol. 2019, 17 (2), e3000139 10.1371/journal.pbio.3000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L.; Dharmarajan V.; Serrao E.; Hoyte A.; Larue R. C.; et al. The Competitive Interplay between Allosteric HIV-1 Integrase Inhibitor BI/D and LEDGF/p75 during the Early Stage of HIV-1 Replication Adversely Affects Inhibitor Potency. ACS Chem. Biol. 2016, 11 (5), 1313–21. 10.1021/acschembio.6b00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P. D. M.; Zheng J.; Gremminger T. J.; Qiu L.; Zhang D.; et al. Binding interface and impact on protease cleavage for an RNA aptamer to HIV-1 reverse transcriptase. Nucleic Acids Res. 2020, 48 (5), 2709–22. 10.1093/nar/gkz1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuske S.; Zheng J.; Olson E. D.; Ruiz F. X.; Pascal B. D.; et al. Integrative structural biology studies of HIV-1 reverse transcriptase binding to a high-affinity DNA aptamer. Curr. Res. Struct. Biol. 2020, 2, 116–29. 10.1016/j.crstbi.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deredge D.; Li J.; Johnson K. A.; Wintrode P. L. Hydrogen/Deuterium Exchange Kinetics Demonstrate Long Range 7 Effects of Thumb Site 2 Inhibitors of Hepatitis C Viral RNA-dependent RNA Polymerase. J. Biol. Chem. 2016, 291 (19), 10078–88. 10.1074/jbc.M115.708370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Bowden T. A.; Wilson I. A.; Crispin M. Exploitation of glycosylation in enveloped virus pathobiology. Biochim. Biophys. Acta, Gen. Subj. 2019, 1863 (10), 1480–97. 10.1016/j.bbagen.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri K.; Klein J. A.; White M. R.; Grant O. C.; Leymarie N.; et al. Integrated omics and computational glycobiology reveal structural basis for influenza a virus glycan microheterogeneity and host interactions. Mol. Cell. Proteomics 2016, 15 (6), 1895–1912. 10.1074/mcp.M116.058016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagdonaite I.; Wandall H. H. Global aspects of viral glycosylation. Glycobiology. 2018, 28 (7), 443–67. 10.1093/glycob/cwy021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss I. J. Antibody recognition of HIV and dengue glycoproteins. Glycobiology 2016, 26 (8), 813–19. 10.1093/glycob/cww031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Berndsen Z. T.; Raghwani J.; Seabright G. E.; Allen J. D.; Pybus O. G.; McLellan J. S.; Wilson I. A.; Bowden T. A.; Ward A. B.; Crispin M. Vulnerabilities in coronavirus glycan shields despite extensive glycosylation. Nat. Commun. 2020, 11, 2688. 10.1038/s41467-020-16567-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A. C.; Xiong X.; Park Y. J.; Tortorici M. A.; Snijder J.; et al. Unexpected Receptor Functional Mimicry Elucidates Activation of Coronavirus Fusion. Cell 2019, 176 (5), 1026–1039. 10.1016/j.cell.2018.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargett A. A.; Wei Q.; Knoppova B.; Hall S.; Huang Z.-Q.; Prakash A.; Green T. J.; Moldoveanu Z.; Raska M.; Novak J.; Renfrow M. B. Defining HIV-1 Envelope N-Glycan Microdomains through Site-Specific Heterogeneity Profiles. J. Virol. 2018, 93 (1), e01177-18. 10.1128/JVI.01177-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X.; Tortorici M. A.; Snijder J.; Yoshioka C.; Walls A. C.; Li W.; McGuire A. T.; Rey F. A.; Bosch B.-J.; Veesler D. Glycan shield and fusion activation of a deltacoronavirus spike glycoprotein fine-tuned for enteric infections. J. Virol. 2018, 92 (4), e01628-17. 10.1128/JVI.01628-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Raghwani J.; Allen J. D.; Seabright G. E.; Li S.; et al. Structure of the Lassa virus glycan shield provides a model for immunological resistance. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (28), 7320–25. 10.1073/pnas.1803990115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T. J.; Chang Y. C.; Ko T. P.; Draczkowski P.; Chien Y. C.; et al. Cryo-EM analysis of a feline coronavirus spike protein reveals a unique structure and camouflaging glycans. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (3), 1438–46. 10.1073/pnas.1908898117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smargiasso N.; Nader J.; Rioux S.; Mazzucchelli G.; Boutry M.; De Pauw E.; Chaumont F.; Navarre C. Exploring the N-glycosylation profile of glycoprotein b from human cytomegalovirus expressed in CHO and nicotiana tabacum BY-2 cells. Int. J. Mol. Sci. 2019, 20 (15), 3741. 10.3390/ijms20153741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens A.-J.; Harvey D. J.; Milne E.; Cupo A.; Kumar A.; Zitzmann N.; Struwe W. B.; Moore J. P.; Crispin M. Molecular Architecture of the Cleavage-Dependent Mannose Patch on a Soluble HIV-1 Envelope Glycoprotein Trimer. J. Virol. 2017, 91 (2), e01894-16. 10.1128/JVI.01894-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurdiss D. L.; Drulyte I.; Lang Y.; Shamorkina T. M.; Pronker M. F.; van Kuppeveld F. J. M.; Snijder J.; de Groot R. J. Cryo-EM structure of coronavirus-HKU1 haemagglutinin esterase reveals architectural changes arising from prolonged circulation in humans. Nat. Commun. 2020, 11 (1), 4646. 10.1038/s41467-020-18440-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speciale I.; Duncan G. A.; Unione L.; Agarkova I. V.; Garozzo D.; et al. The N-glycan structures of the antigenic variants of chlorovirus PBCV-1 major capsid protein help to identify the virus-encoded glycosyltransferases. J. Biol. Chem. 2019, 294 (14), 5688–99. 10.1074/jbc.RA118.007182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey D. J. Mass spectrometric analysis of glycosylated viral proteins. Expert Rev. Proteomics 2018, 15 (5), 391–412. 10.1080/14789450.2018.1468756. [DOI] [PubMed] [Google Scholar]
- Seabright G. E.; Cottrell C. A.; van Gils M. J.; D’addabbo A.; Harvey D. J.; et al. Networks of HIV-1 Envelope Glycans Maintain Antibody Epitopes in the Face of Glycan Additions and Deletions. Structure 2020, 28 (8), 897–909. 10.1016/j.str.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst A. J; Weidle C. E; Gray M. D; Frenz B.; Snijder J.; Joyce M G.; Georgiev I. S; Stewart-Jones G. B.; Kwong P. D; McGuire A. T; DiMaio F.; Stamatatos L.; Pancera M.; Veesler D. Germline VRC01 antibody recognition of a modified clade C HIV-1 envelope trimer and a glycosylated HIV-1 gp120 core. eLife 2018, 7, e37688. 10.7554/eLife.37688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart-Jones G. B. E.; Soto C.; Lemmin T.; Chuang G. Y.; Druz A.; et al. Trimeric HIV-1-Env Structures Define Glycan Shields from Clades AB, and G. Cell 2016, 165 (4), 813–26. 10.1016/j.cell.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard L. K.; Harvey D. J.; Bonomelli C.; Crispin M.; Doores K. J. Cell- and Protein-Directed Glycosylation of Native Cleaved HIV-1 Envelope. J. Virol. 2015, 89 (17), 8932–44. 10.1128/JVI.01190-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Nicolás O.; Lewandowska M.; Ricklin M. E.; Summerfield A. Monocyte-Derived Dendritic Cells as Model to Evaluate Species Tropism of Mosquito-Borne Flaviviruses. Front. Cell. Infect. Microbiol. 2019, 9, 5. 10.3389/fcimb.2019.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanowicz R. A.; Wang R.; Schiel J. E.; Keck Z.-y.; Kerzic M. C.; Lau P.; Rangarajan S.; Garagusi K. J.; Tan L.; Guest J. D.; Ball J. K.; Pierce B. G.; Mariuzza R. A.; Foung S. K. H.; Fuerst T. R. Antigenicity and Immunogenicity of Differentially Glycosylated Hepatitis C Virus E2 Envelope Proteins Expressed in Mammalian and Insect Cells. J. Virol. 2019, 93 (7), e01403-18. 10.1128/JVI.01403-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipollo J. F.; Parsons L. M. Glycomics and glycoproteomics of viruses: Mass spectrometry applications and insights toward structure-function relationships. Mass Spectrom. Rev. 2020, 39 (4), 371–409. 10.1002/mas.21629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Y; Parsons L. M.; Jankowska E; Melnyk D; Joshi M; Cipollo J. F. N-Glycosylation of Seasonal Influenza Vaccine Hemagglutinins: Implication for Potency Testing and Immune Processing. J. Virol. 2019, 93 (2), e01693-18. 10.1128/JVI.01693-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegg C. L.; Hoogland C.; Gorman J. J. Site-specific glycosylation of the Newcastle disease virus haemagglutinin-neuraminidase. Glycoconjugate J. 2017, 34 (2), 181–97. 10.1007/s10719-016-9750-7. [DOI] [PubMed] [Google Scholar]
- Seeberger P. H. Monosaccharide Diversity. In Essentials of Glycobiology;Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 2015; pp 19–30. [Google Scholar]
- Bagdonaite I.; Nordén R.; Joshi H. J.; King S. L.; Vakhrushev S. Y.; et al. Global mapping of o-glycosylation of varicella zoster virus, human cytomegalovirus, and Epstein-Barr virus. J. Biol. Chem. 2016, 291 (23), 12014–28. 10.1074/jbc.M116.721746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzarano D.; Krokhin O.; Van Domselaar G.; Wolf K.; Seebach J.; et al. Ebola sGP-The first viral glycoprotein shown to be C-mannosylated. Virology 2007, 368 (1), 83–90. 10.1016/j.virol.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caval T; Heck A. J. R.; Reiding K. R. Meta-heterogeneity: evaluating and describing the diversity in glycosylation between sites on the same glycoprotein. Mol. Cell. Proteomics 2020, mcp.R120.002093. 10.1074/mcp.R120.002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagdonaite I.; Nordén R.; Joshi H. J.; Dabelsteen S.; Nyström K.; et al. A Strategy for O-Glycoproteomics of Enveloped Viruses—the O-Glycoproteome of Herpes Simplex Virus Type 1. PLoS Pathog. 2015, 11 (4), e1004784 10.1371/journal.ppat.1004784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhaak L. R.; Xu G.; Li Q.; Goonatilleke E.; Lebrilla C. B. Mass Spectrometry Approaches to Glycomic and Glycoproteomic Analyses. Chem. Rev. 2018, 118 (17), 7886–7930. 10.1021/acs.chemrev.7b00732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She Y. M.; Li X.; Cyr T. D. Remarkable Structural Diversity of N-Glycan Sulfation on Influenza Vaccines. Anal. Chem. 2019, 91 (8), 5083–90. 10.1021/acs.analchem.8b05372. [DOI] [PubMed] [Google Scholar]
- Chang D.; Zaia J. Why glycosylation matters in building a better flu vaccine. Mol. Cell. Proteomics 2019, 18 (12), 2348–58. 10.1074/mcp.R119.001491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go E. P.; Ding H.; Zhang S.; Ringe R. P.; Nicely N.; Hua D.; Steinbock R. T.; Golabek M.; Alin J.; Alam S. M.; Cupo A.; Haynes B. F.; Kappes J. C.; Moore J. P.; Sodroski J. G.; Desaire H. Glycosylation Benchmark Profile for HIV-1 Envelope Glycoprotein Production Based on Eleven Env Trimers. J. Virol. 2017, 91 (9), e02428-16. 10.1128/JVI.02428-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panico M.; Bouche L.; Binet D.; O’Connor M.-J.; Rahman D.; Pang P.-C.; Canis K.; North S. J.; Desrosiers R. C.; Chertova E.; Keele B. F.; Bess J. W.; Lifson J. D.; Haslam S. M.; Dell A.; Morris H. R. Mapping the complete glycoproteome of virion-derived HIV-1 gp120 provides insights into broadly neutralizing antibody binding. Sci. Rep. 2016, 6, 32956. 10.1038/srep32956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsa D.; Baiwir D.; La Rocca R.; Zimmerman T. A.; Hanozin E.; et al. Multi-Enzymatic Limited Digestion: The Next-Generation Sequencing for Proteomics?. J. Proteome Res. 2019, 18 (6), 2501–13. 10.1021/acs.jproteome.9b00044. [DOI] [PubMed] [Google Scholar]
- Cao L.; Pauthner M.; Andrabi R.; Rantalainen K.; Berndsen Z.; Diedrich J. K.; Menis S.; Sok D.; Bastidas R.; Park S.-K. R.; Delahunty C. M.; He L.; Guenaga J.; Wyatt R. T.; Schief W. R.; Ward A. B.; Yates J. R.; Burton D. R.; Paulson J. C. Differential processing of HIV envelope glycans on the virus and soluble recombinant trimer. Nat. Commun. 2018, 9, 3693. 10.1038/s41467-018-06121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Allen J. D.; Wrapp D.; McLellan J. S.; Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science (Washington, DC, U. S.) 2020, 369 (6501), 330–333. 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Bertozzi C. R.; Pitteri S. J. A Pragmatic Guide to Enrichment Strategies for Mass Spectrometry-based Glycoproteomics. Mol. Cell. Proteomics 2020, mcp.R120.002277. 10.1074/mcp.R120.002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J.; Yearley E.; Tian S.; Jing L.; Balsaraf A.; et al. Non-Enzymatic and Site-Specific Glycan Shedding: A Novel Protein Degradation Pathway Observed in a Stabilized Form of RSV Prefusion F Protein. Anal. Chem. 2018, 90 (18), 10897–902. 10.1021/acs.analchem.8b02402. [DOI] [PubMed] [Google Scholar]
- Yao H.-P.; Song Y.; Chen Y.; Wu N.; Xu J.; Sun C.; Zhang J.; Weng T.; Zhang Z.; Wu Z.; Cheng L.; Shi D.; Lu X.; Lei J.; Crispin M.; Shi Y.; Li L.; Li S. Molecular Architecture of the SARS-CoV-2 Virus. Cell 2020, 10.2139/ssrn.3654623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L.; Diedrich J. K.; Kulp D. W.; Pauthner M.; He L.; Park S.-K. R.; Sok D.; Su C. Y.; Delahunty C. M.; Menis S.; Andrabi R.; Guenaga J.; Georgeson E.; Kubitz M.; Adachi Y.; Burton D. R.; Schief W. R.; Yates III J. R.; Paulson J. C. Global site-specific N-glycosylation analysis of HIV envelope glycoprotein. Nat. Commun. 2017, 8, 14954. 10.1038/ncomms14954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. J.; Cao L.; Ma Y.; Wang X.; Diedrich J. K.; et al. Human Influenza Virus Hemagglutinins Contain Conserved Oligomannose N-Linked Glycans Allowing Potent Neutralization by Lectins. Cell Host Microbe 2020, 27 (5), 725–735. 10.1016/j.chom.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.; Onigman P.; Wu W. W.; Sjogren J.; Nyhlen H.; et al. Deciphering Protein O-Glycosylation: Solid-Phase Chemoenzymatic Cleavage and Enrichment. Anal. Chem. 2018, 90 (13), 8261–69. 10.1021/acs.analchem.8b01834. [DOI] [PubMed] [Google Scholar]
- Malaker S. A.; Pedram K.; Ferracane M. J.; Bensing B. A.; Krishnan V.; et al. The mucin-selective protease StcE enables molecular and functional analysis of human cancer-associated mucins. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (15), 7278–87. 10.1073/pnas.1813020116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaysen-Andersen M.; Packer N. H.; Schulz B. L. Maturing Glycoproteomics Technologies Provide Unique Structural Insights into the N -glycoproteome and Its Regulation in Health and Disease. Mol. Cell. Proteomics 2016, 15 (6), 1773–90. 10.1074/mcp.O115.057638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Zhou H.; Taus T.; Altelaar A. F. M.; Mechtler K.; et al. Unambiguous phosphosite localization using electron-transfer/higher-energy collision dissociation (EThcD). J. Proteome Res. 2013, 12 (3), 1520–25. 10.1021/pr301130k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q.; Wang B.; Chen Z.; Urabe G.; Glover M. S.; et al. Electron-Transfer/Higher-Energy Collision Dissociation (EThcD)-Enabled Intact Glycopeptide/Glycoproteome Characterization. J. Am. Soc. Mass Spectrom. 2017, 28 (9), 1751–64. 10.1007/s13361-017-1701-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Hebert A. S.; Westphall M. S.; Coon J. J. Capturing site-specific heterogeneity with large-scale N-glycoproteome analysis. Nat. Commun. 2019, 10, 1311. 10.1038/s41467-019-09222-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Malaker S. A.; Driessen M. D.; Bertozzi C. R. Optimal Dissociation Methods Differ for N- A nd O-Glycopeptides. J. Proteome Res. 2020, 19 (8), 3286–3301. 10.1021/acs.jproteome.0c00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Yang C.; Sun T. Characterization of glycopeptides using a stepped higher-energy C-trap dissociation approach on a hybrid quadrupole orbitrap. Rapid Commun. Mass Spectrom. 2018, 32 (16), 1353–62. 10.1002/rcm.8191. [DOI] [PubMed] [Google Scholar]
- Roushan A.; Wilson G. M.; Kletter D.; Sen K. I.; Tang W. H.; Kil Y. J.; Carlson E.; Bern M. Peak Filtering, Peak Annotation, and Wildcard Search for Glycoproteomics. Mol. Cell. Proteomics 2020, mcp.RA120.002260. 10.1074/mcp.RA120.002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.-Q.; Zeng W.-F.; Fang P.; Cao W.-Q.; Liu C.; Yan G.-Q.; Zhang Y.; Peng C.; Wu J.-Q.; Zhang X.-J.; Tu H.-J.; Chi H.; Sun R.-X.; Cao Y.; Dong M.-Q.; Jiang B.-Y.; Huang J.-M.; Shen H.-L.; Wong C. C. L.; He S.-M.; Yang P.-Y. PGlyco 2.0 enables precision N-glycoproteomics with comprehensive quality control and one-step mass spectrometry for intact glycopeptide identification. Nat. Commun. 2017, 8, 438. 10.1038/s41467-017-00535-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W.; Su X.; Zhu Z.; Go E. P.; Desaire H. GlycoPep MassList: software to generate massive inclusion lists for glycopeptide analyses. Anal. Bioanal. Chem. 2017, 409 (2), 561–70. 10.1007/s00216-016-9896-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polasky D. A.; Yu F.; Teo G. C.; Nesvizhskii A. I. Fast and comprehensive N- and O-glycoproteomics analysis with MSFragger-Glyco. Nat. Methods 2020, 17, 1125–1132. 10.1038/s41592-020-0967-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T.; Park S. K.; Venable J. D.; Wohlschlegel J. A.; Diedrich J. K.; et al. ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J. Proteomics 2015, 129, 16–24. 10.1016/j.jprot.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper C. A.; Gasteiger E.; Packer N. H. GlycoMod - A software tool for determining glycosylation compositions from mass spectrometric data. Proteomics 2001, 1 (2), 340–49. . [DOI] [PubMed] [Google Scholar]
- MacLean B.; Tomazela D. M.; Shulman N.; Chambers M.; Finney G. L.; et al. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26 (7), 966–68. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Y.; McCullers J. A.; Alymova I.; Parsons L. M.; Cipollo J. F. Glycosylation analysis of engineered H3N2 influenza A virus hemagglutinins with sequentially added historically relevant glycosylation sites. J. Proteome Res. 2015, 14 (9), 3957–69. 10.1021/acs.jproteome.5b00416. [DOI] [PubMed] [Google Scholar]
- Hargett A. A.; Renfrow M. B. Glycosylation of viral surface proteins probed by mass spectrometry. Curr. Opin. Virol. 2019, 36, 56–66. 10.1016/j.coviro.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiding K. R.; Franc V.; Huitema M. G.; Brouwer E.; Heeringa P.; Heck A. J. R. Neutrophil myeloperoxidase harbors distinct site-specific peculiarities in its glycosylation. J. Biol. Chem. 2019, 294 (52), 20233–45. 10.1074/jbc.RA119.011098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Franc V.; Heck A. J. R. Glycoproteomics: A Balance between High-Throughput and In-Depth Analysis. Trends Biotechnol. 2017, 35 (7), 598–609. 10.1016/j.tibtech.2017.04.010. [DOI] [PubMed] [Google Scholar]
- Struwe W. B.; Stuckmann A.; Behrens A. J.; Pagel K.; Crispin M. Global N-Glycan Site Occupancy of HIV-1 gp120 by Metabolic Engineering and High-Resolution Intact Mass Spectrometry. ACS Chem. Biol. 2017, 12 (2), 357–61. 10.1021/acschembio.6b00854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H.; De Val N.; Lyumkis D.; Ward A. B. Model building and refinement of a natively glycosylated HIV-1 Env protein by high-resolution cryoelectron microscopy. Structure 2015, 23 (10), 1943–51. 10.1016/j.str.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R; Edwards R. J.; Mansouri K; Janowska K; Stalls V; Kopp M.; Haynes B. F.; Acharya P. Glycans on the SARS-CoV-2 Spike Control the Receptor Binding Domain Conformation. bioRxiv 2020, 2020.06.26.173765. 10.1101/2020.06.26.173765. [DOI] [Google Scholar]
- Frenz B.; Rämisch S.; Borst A. J.; Walls A. C.; Adolf-Bryfogle J.; et al. Automatically Fixing Errors in Glycoprotein Structures with Rosetta. Structure 2019, 27 (1), 134–139. 10.1016/j.str.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndsen Z. T.; Chakraborty S.; Wang X.; Cottrell C. A.; Torres J. L.; Diedrich J. K.; Lopez C. A.; Yates J. R.; van Gils M. J.; Paulson J. C.; Gnanakaran S.; Ward A. B. Visualization of the HIV-1 Env Glycan Shield Across Scales. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (45), 28014–28025. 10.1073/pnas.2000260117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi R.; Pallesen J.; Allen J. D.; Song G.; Zhang J.; et al. The Chimpanzee SIV Envelope Trimer: Structure and Deployment as an HIV Vaccine Template. Cell Rep. 2019, 27 (8), 2426–2441. 10.1016/j.celrep.2019.04.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder J.; Ortego M. S.; Weidle C.; Stuart A. B.; Gray M. D.; et al. An Antibody Targeting the Fusion Machinery Neutralizes Dual-Tropic Infection and Defines a Site of Vulnerability on Epstein-Barr Virus. Immunity 2018, 48 (4), 799–811. 10.1016/j.immuni.2018.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M.; Jiang X. Norovirus Capsid Protein-Derived Nanoparticles and Polymers as Versatile Platforms for Antigen Presentation and Vaccine Development. Pharmaceutics 2019, 11 (9), 472. 10.3390/pharmaceutics11090472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M.; Huang P.; Sun C.; Han L.; Vago F. S.; et al. Bioengineered Norovirus S 60 Nanoparticles as a Multifunctional Vaccine Platform. ACS Nano 2018, 12 (11), 10665–82. 10.1021/acsnano.8b02776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquero E.; Albertini A. A.; Raux H.; Abou-Hamdan A.; Boeri-Erba E.; et al. Structural intermediates in the fusion-associated transition of vesiculovirus glycoprotein. EMBO J. 2017, 36 (5), 679–92. 10.15252/embj.201694565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D. E.; Jang G. M.; Bouhaddou M.; Xu J.; Obernier K.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583 (7816), 459–68. 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukalov A.; Girault V.; Grass V.; Bergant V.; Karayel O.; et al. Multi-level proteomics reveals host-perturbation strategies of SARS-CoV-2 and SARS-CoV. bioRxiv 2020, 2020.06.17.156455. 10.1101/2020.06.17.156455. [DOI] [Google Scholar]
- St-Germain J. R.; Astori A.; Samavarchi-Tehrani P.; Abdouni H.; Macwan V.; Kim D.; Knapp J. J.; Roth F. P.; Gingras A.; Raught B. A SARS-CoV-2 BioID-based virus-host membrane protein interactome and virus peptide compendium: new proteomics resources for COVID-19 research. bioRxiv 2020, 2020.08.28.269175. 10.1101/2020.08.28.269175. [DOI] [Google Scholar]
- Ihling C.; Tanzler D.; Hagemann S.; Kehlen A.; Huttelmaier S.; Arlt C.; Sinz A. Mass Spectrometric Identification of SARS-CoV-2 Proteins from Gargle Solution Samples of COVID-19 Patients. J. Proteome Res. 2020, 19 (11), 4389–4392. 10.1021/acs.jproteome.0c00280. [DOI] [PubMed] [Google Scholar]
- Nikolaev E. N.; Indeykina M. I.; Brzhozovskiy A. G.; Bugrova A. E.; Kononikhin A. S. Mass-Spectrometric Detection of SARS-CoV-2 Virus in Scrapings of the Epithelium of the Nasopharynx of Infected Patients via Nucleocapsid N Protein. J. Proteome Res. 2020, 19 (11), 4393–4397. 10.1021/acs.jproteome.0c00412. [DOI] [PubMed] [Google Scholar]
- Gouveia D.; Miotello G.; Gallais F.; Gaillard J.-C.; Debroas S.; Bellanger L.; Lavigne J.-P.; Sotto A.; Grenga L.; Pible O.; Armengaud J. Proteotyping SARS-CoV-2 Virus from Nasopharyngeal Swabs: A Proof-of-Concept Focused on a 3 min Mass Spectrometry Window. J. Proteome Res. 2020, 19 (11), 4407–4416. 10.1021/acs.jproteome.0c00535. [DOI] [PubMed] [Google Scholar]
- Messner C. B.; Demichev V.; Wendisch D.; Michalick L.; White M.; et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst. 2020, 11 (1), 11–24. 10.1016/j.cels.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajahan A.; Supekar N. T.; Gleinich A. S.; Azadi P. Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020, cwaa042. 10.1093/glycob/cwaa042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D; Tian X; Qi R; Peng C; Zhang W Identification of 22 N-glycosites on spike glycoprotein of SARS-CoV-2 and accessible surface glycopeptide motifs: Implications for vaccination and antibody therapeutics. Glycobiology 2020, cwaa052. 10.1093/glycob/cwaa052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P.; Praissman J. L.; Grant O. C.; Cai Y.; Xiao T.; et al. Virus-Receptor Interactions of Glycosylated SARS-CoV-2 Spike and Human ACE2 Receptor. Cell Host Microbe 2020, 28 (4), 586–601. 10.1016/j.chom.2020.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Baba T. J.; Lutomski C. A.; Kantsadi A. L.; Malla T. R.; John T.; Mikhailov V.; Bolla J. R.; Schofield C. J.; Zitzmann N.; Vakonakis I.; Robinson C. V. Allosteric inhibition of the SARS-CoV-2 main protease - insights from mass spectrometry-based assays. Angew. Chem., Int. Ed. 2020, 10.1002/anie.202010316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R; Partridge T; Wormald C; Kawahara R; Stalls V Mapping the SARS-CoV-2 spike glycoprotein-derived peptidome presented by HLA class II on dendritic cells. bioRxiv 2020, 2020.08.19.255901. 10.1101/2020.08.19.255901. [DOI] [PMC free article] [PubMed] [Google Scholar]