

Abstract
Background and purpose: Lipopolysaccharide (LPS) is an important factor that induce severe inflammation, resulting in multiple types of diseases. It is reported that LPS-induced inflammation is related to the activation of the NF-κB signal pathway and reactive oxygen species (ROS)-induced oxidative stress. Azilsartan, an angiotensin II type 1 (AT1) receptor blocker, has been licensed as a new generation of Sartan antihypertensive drugs. However, the effects of azilsartan in LPS-induced inflammation have not been reported before. The present study aims to investigate the anti-inflammatory effects of azilsartan on LPS-stimulated macrophages and explore the underlying mechanism. Methods: The release of lactic dehydrogenase (LDH), secretion of HMGB-1, and concentrations of IL-6, IL-1β, MCP-1, MMP-2, MMP-9, and PGE2 were evaluated using the enzyme-linked immunosorbent assay (ELISA). The gene expression levels of IL-6, IL-1β, MCP-1, MMP-2, MMP-9, and COX-2 were determined by quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR). Western blot analysis was used to detect the protein expression level of COX-2, Nrf2, TLR2, MyD-88, and NF-κB. The level of ROS was determined using the dihydroethidium (DHE) staining assay. The activity of NF-κB was evaluated using the luciferase activity assay. Results: The release of LDH, HMGB-1, IL-6, IL-1β, MCP-1, MMP-2, MMP-9, and PGE2 was significantly promoted by LPS stimulation, whereas it was greatly suppressed by azilsartan. The upregulated COX-2, TLR2, MyD-88, and NF-κB in the LPS-treated macrophages were significantly downregulated by azilsartan. Interestingly, the expression level of Nrf2 was elevated by azilsartan. On the contrary, ROS levels were greatly increased by LPS but suppressed by azilsartan. Mechanistically, it was found that azilsartan suppressed LPS-induced activation of the TLR2/Myd-88/NF-κB signaling pathway. Conclusion: Azilsartan might suppress LPS-induced inflammation in U937 macrophages through suppressing oxidative stress and inhibiting the TLR/MyD88 signal pathway.
1. Introduction
Lipopolysaccharide (LPS) is an important factor that induces sepsis and multiple organ dysfunction syndrome.1 As the main component of the membrane on Gram-negative bacilli, LPS combines with CD14 expressed on the membrane of macrophages to induce significant inflammation by regulating the production of inflammatory factors and nitric oxide (NO) through mediating the NF-κB signal pathway.2 LPS is reported to be directly or indirectly involved in the pathogenesis of multiple types of pulmonary diseases, including chronic obstructive pulmonary disease (COPD), asthma, and allergic lung injury.3 Therefore, stimulating in vitro cells with LPS is an effective way to explore the possible mechanism and therapeutic routine for pulmonary diseases. It is reported that the expression levels of NF-κB and cAMP could be significantly promoted by stimulating the alveolar macrophages with LPS, through which the macrophages could be activated.4
Oxidative stress is one of the mechanisms underlying the inflammation-inducing effects of LPS, by which large amounts of reactive oxygen species (ROS) and reactive nitrogen species (RNS) are produced.5,6 ROS and RNS are involved in a variety of physiological functions, such as regulating the expression of specific genes and the apoptosis of cells. However, excessive production of ROS and RNS will directly induce tissue injury and an inflammatory cascade.7 Oxidative stress will be induced by the accumulation of cellular ROS, which cannot be effectively degraded by antioxidative materials.8 Cellular lipids, proteins, and organelles are damaged by excessive ROS and toxic materials are produced due to decreased biological function of these biomacromolecules, which eventually impact the activity of lysosomes and contribute to cell death.9 In addition, RNS will result from oxidative stress induced by excessive ROS, and the ion channels on the membrane of mitochondria are opened, which decreases the concentration of ATP and Ca2+ in the mitochondria. Subsequently, cytochrome C is released and mitochondrial swelling is induced through which the mitochondria are significantly injured. It is reported that autophagy, cell death, apoptosis, and cell necrosis are induced by the damaged mitochondria.10 Therefore, suppressing oxidative stress may be an effective way to prevent cell death and inflammation, which are crucial steps for the treatment of LPS-induced diseases.
Azilsartan is a new generation of sartan antihypertensive drugs developed by Takeda Pharmaceutical Co., the molecular structure of which is shown in Figure 1. The antihypertensive drug was introduced to the market in 2012 with the commercial name “Azilva”. As a new generation of angiotensin II antagonist, its antihypertensive mechanism is similar to other sartan drugs, which inhibit the vasoconstriction and excessive secretion of aldosterone induced by angiotensin II by combining with the angiotensin II receptor.11 Furthermore, the expression level of the peroxisome proliferator-activated receptor-γ (PPAR-γ) in adipose tissues could potentially improve the glucose tolerance and metabolism.12 Currently, it is reported that the secretion of adiponectin in adipose tissues is induced by azilsartan, which eventually inhibits the expression of TNF-α and suppresses insulin resistance.13 Aurigena also reported that azilsartan could reduce TNF-α levels, increase IL-10 levels, and upregulate VEGF, FGF, KGF, and TGF-α in an oral mucositis model.14 However, it is unknown whether azilsartan possesses a protective effect against LPS in macrophages. In the present study, the anti-inflammatory effects and antioxidative stress effects of azilsartan in macrophages will be investigated to explore the novel therapeutic purpose of azilsartan.
Figure 1.
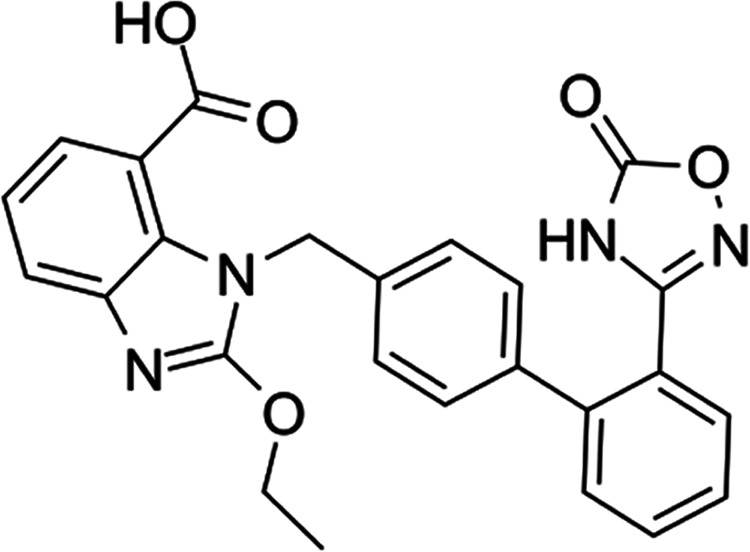
Molecular structure of azilsartan.
2. Results
2.1. Production of LDH and HMGB1 in U937 Macrophages Induced by LPS was Suppressed by Azilsartan
As shown in Figure 2A, the release of LDH by U937 macrophages was significantly promoted by the stimulation of LPS at a value from 6.6 to 43.1%, but greatly decreased to 39.8, 31.6, and 22.5% by the introduction of 1, 5, and 10 μM azilsartan, respectively. A significant difference was observed in the 5 and 10 μM groups. Figure 2B shows the concentrations of HMGB1 in different groups. The secretion of HMGB1 in the U937 macrophages was promoted from 133.6 to 698.6 pg/mL (increased by 423%) by the treatment with LPS but was reduced to 635.5 (decreased by 9%), 532.1 (decreased by 23.8%), and 355.7 (decreased by 49.1%) pg/mL by the introduction of 1, 5, and 10 μM azilsartan, respectively. A significant difference was observed in the 5 and 10 μM groups.
Figure 2.
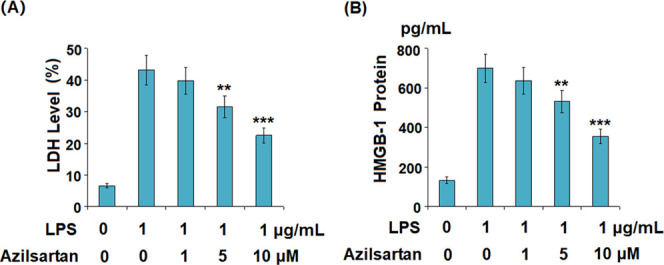
Azilsartan prevented LPS-induced release of LDH and HMGB1 in U937 macrophages. Cells were treated with 1 μg/mL LPS in the presence or absence of azilsartan (1, 5, or 10 μM) for 24 h. (A) Release of LDH and (B) secretion of HMGB-1 measured using the enzyme-linked immunosorbent assay (ELISA) (**P < 0.01, ***P < 0.005 vs LPS treatment group).
2.2. LPS-Induced Production of Inflammatory Factors in U937 Macrophages was Reduced by Azilsartan
As shown in Figure 3A, the elevated gene expression levels of MCP-1, IL-6, and IL-1β were significantly inhibited by azilsartan in a dose-dependent manner. Figure 3B shows the concentrations of inflammatory factors in the supernatant of macrophages. The concentration of secreted MCP-1 was promoted from 323.3 to 3025.7 pg/mL (increased by 835.8%) under the stimulation of LPS but was decreased to 2176.5 pg/mL (decreased by 28.1%) and 1349.9 pg/mL (decreased by 55.4%) by the introduction of 5 and 10 μM azilsartan, respectively. The concentration of secreted IL-1β was elevated from 166.5 to 798.5 pg/mL (increased by 379.6%) under the stimulation of LPS but was suppressed to 632.1 pg/mL (decreased by 20.8%) and 495.5 pg/mL (decreased by 37.9%) by the treatment with 5 and 10 μM azilsartan with a significant difference, respectively. The promoted concentration of secreted IL-6 (256.8–1833.6 pg/mL) induced by LPS was significantly inhibited by azilsartan to 1366.8 pg/mL (decreased by 25.5%) and 851.3 pg/mL (decreased by 53.6%) at dosages of 5 and 10 μM, respectively.
Figure 3.
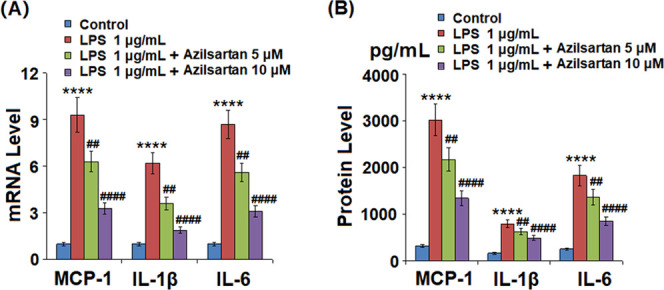
Azilsartan reduced LPS-induced expressions and secretions of pro-inflammatory cytokines in U937 macrophages. Cells were treated with 1 μg/mL LPS in the presence or absence of azilsartan (5, 10 μM) for 24 h. (A) mRNA of MCP-1, IL-1β, and IL-6 and (B) secretions of MCP-1, IL-1β, and IL-6 (****P < 0.0001 vs vehicle group; ##P < 0.01, ####P < 0.0001 vs LPS group).
2.3. Azilsartan Prevented LPS-Induced Expression of MMP-2, MMP-9, Cyclooxygenase 2 (COX-2), and Prostaglandin E2 (PGE2) in U937 Macrophages
As shown in Figure 4, the elevated expression levels of MMP-2 and MMP-9 in the U937 macrophages induced by LPS were significantly suppressed by the introduction of Azilsartan. As shown in Figure 5A,B, COX-2 in the U937 macrophages was significantly upregulated by LPS, whereas it was greatly downregulated by the treatment with azilsartan at both the gene and protein levels. Figure 5C shows the data of released PGE2. We found that the concentration of PGE2 in the supernatant of macrophages was increased from 367.9 to 1368.2 pg/mL (increased by 271.8%) by the stimulation of LPS but was decreased by 38.9% to 835.5 pg/mL and 42% to 787.8 pg/mL by the treatment with 5 and 10 μM azilsartan, respectively.
Figure 4.
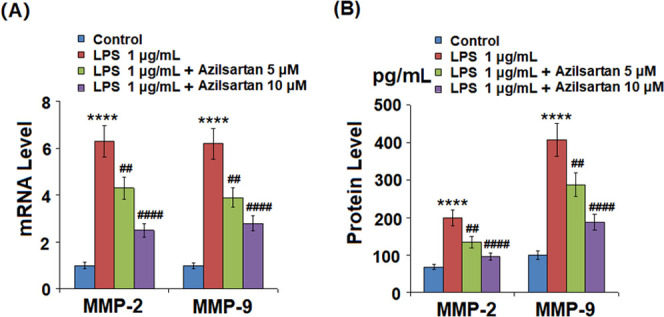
Azilsartan decreased LPS-induced expression of MMP-2 and MMP-9 in U937 macrophages. Cells were treated with 1 μg/mL LPS in the presence or absence of azilsartan (5, 10 μM) for 24 h. (A) mRNA of MMP-2 and MMP-9 and (B) protein levels of MMP-2 and MMP-9 (****P < 0.0001 vs vehicle group; ##P < 0.01, ####P < 0.0001 vs LPS group).
Figure 5.
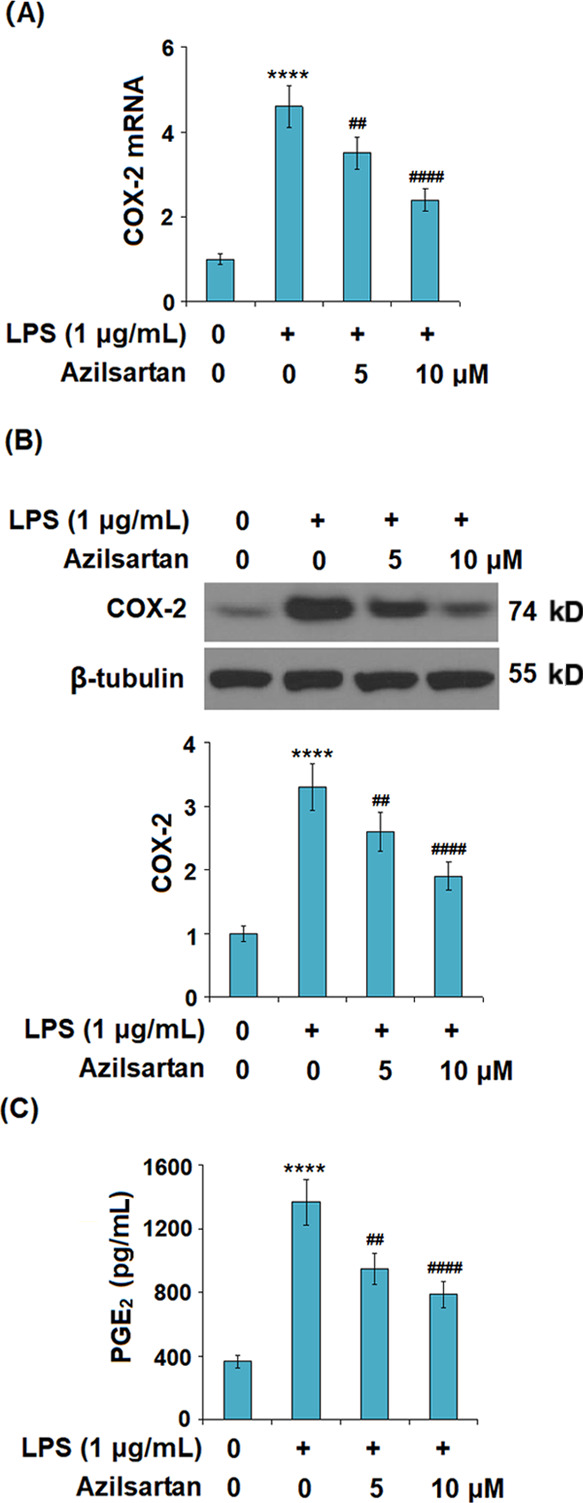
Azilsartan prevented LPS-induced expression of cyclooxygenase 2 (COX-2) and secretion of prostaglandin E2 (PGE2) in U937 macrophages. Cells were treated with 1 μg/mL LPS in the presence or absence of azilsartan (5, 10 μM) for 24 h. (A) mRNA of COX-2; (B) protein levels of COX-2; and (C) secretion of PGE2 (****P < 0.0001 vs vehicle group; ##P < 0.01, ####P < 0.0001 vs LPS group).
2.4. Oxidative Stress Induced by LPS was Alleviated by Azilsartan
As shown in Figure 6A, the production of ROS was significantly elevated by LPS but greatly inhibited by the introduction of azilsartan in a dose-dependent manner. Figure 6B shows the expression level of the antioxidant factor, nuclear factor erythroid 2-related factor 2 (Nrf2), in each group. We found that Nrf2 was significantly upregulated by the treatment with azilsartan.
Figure 6.
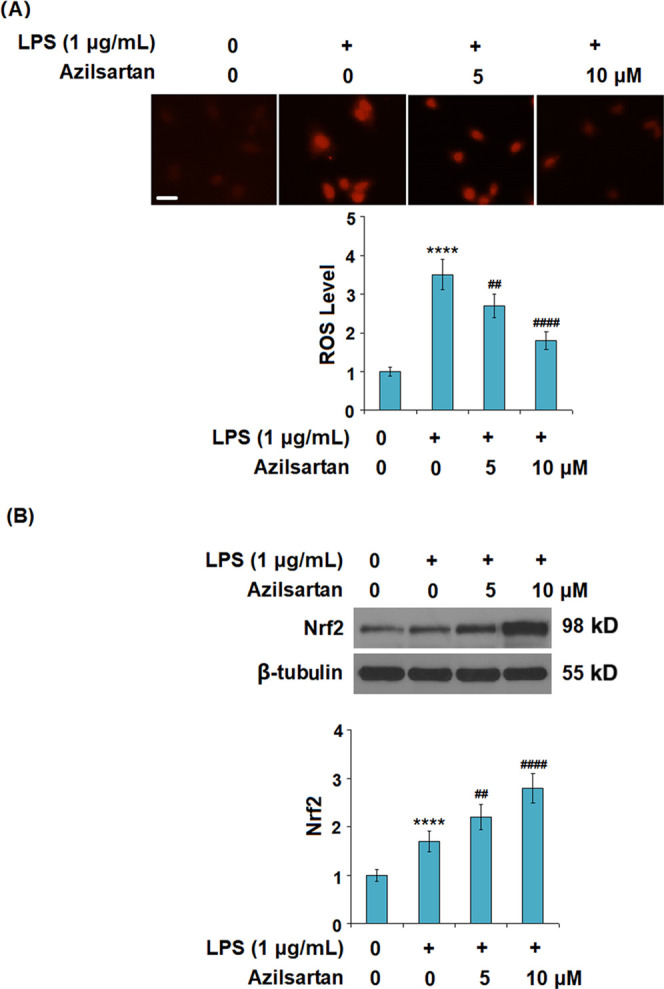
Azilsartan prevented LPS-induced oxidative stress in U937 macrophages. Cells were treated with 1 μg/mL LPS in the presence or absence of azilsartan (5, 10 μM) for 24 h. (A) Production of reactive oxygen species (ROS) (scale bar, 100 μm) and (B) expression of the antioxidant factor, nuclear factor erythroid 2-related factor 2 (Nrf2), as measured using western blot (****P < 0.0001 vs vehicle group; ##P < 0.01, ####P < 0.0001 vs LPS group).
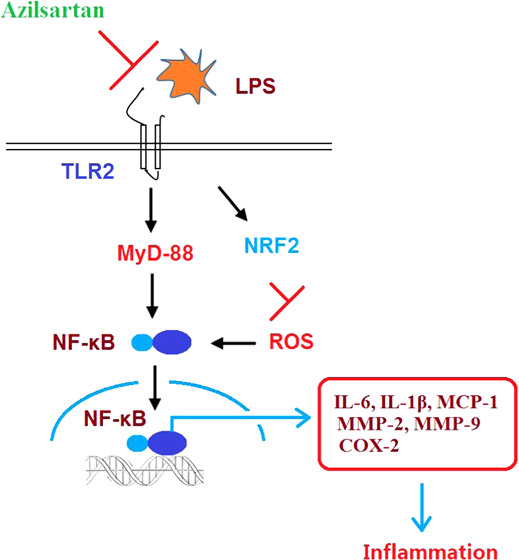
2.5. Azilsartan Might Exert an Anti-Inflammatory Effect by Inhibiting the TLR2/Myd-88/NF-κB Signaling Pathway
As shown in Figures 7 and 8A, we found that the elevated expression levels of TLR2, Myd-88, and NF-κB were significantly inhibited by the introduction of azilsartan in a dose-dependent manner. The transcriptional activity of NF-κB was measured using the luciferase activity. The results indicated that the activated function of the NF-κB promoter induced by LPS was extremely inhibited by azilsartan.
Figure 7.
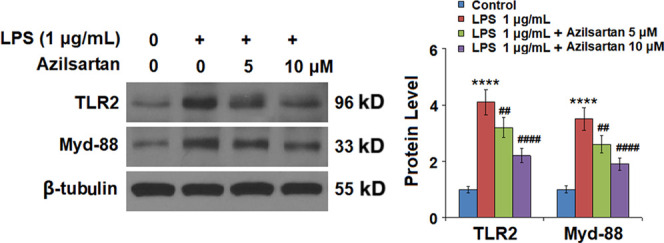
Azilsartan reduced LPS-induced expression of TLR2 and Myd-88 in U937 macrophages. Cells were treated with 1 μg/mL LPS in the presence or absence of azilsartan (5, 10 μM) for 24 h. Expressions of TLR2 and Myd-88 were measured using western blot analysis (****P < 0.0001 vs vehicle group; ##P < 0.01, ####P < 0.0001 vs LPS group).
Figure 8.
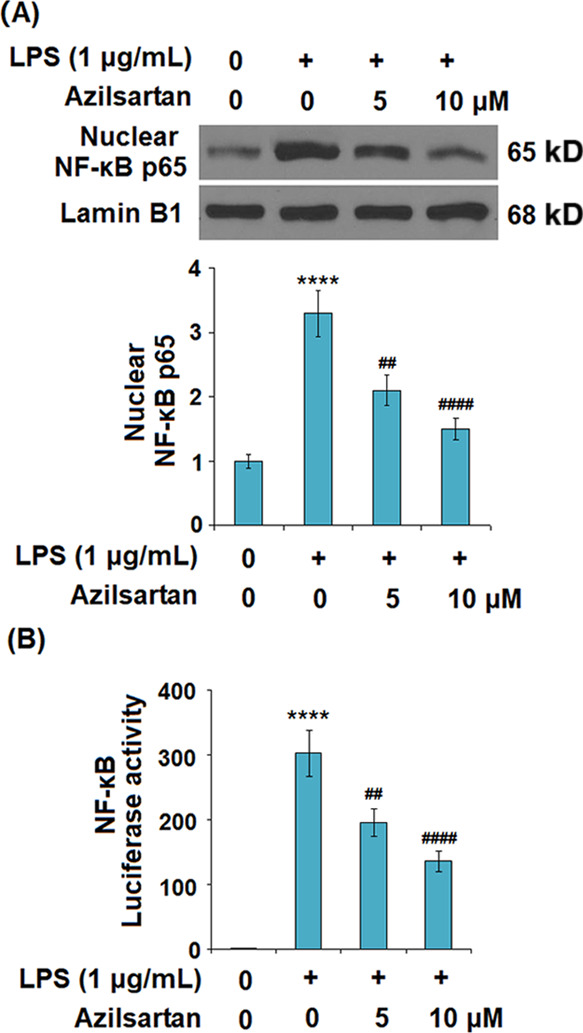
Azilsartan prevented LPS-induced activation of NF-κB in U937 macrophages. Cells were treated with 1 μg/mL LPS in the presence or absence of azilsartan (5, 10 μM) for 24 h. (A) Nuclear levels of NF-κB p65 as measured by western blot analysis and (B) luciferase activity of the NF-κB gene reporter (****P < 0.0001 vs vehicle group; ##P < 0.01, ####P < 0.0001 vs LPS group).
3. Discussion
Inflammation is a basic pathological state found in injured tissues. It is induced by multiple types of pro-inflammatory factors and is an important mechanism underlying many diseases. Appropriate inflammatory reactions are reported to be beneficial to self-protection. However, severe tissue or organ pathological injury is induced by excessive inflammation.15,16 In the present study, LPS was used to stimulate macrophages to release inflammatory factors, which was verified by elevated production of IL-6, IL-1β, and MCP-1 in LPS-incubated macrophages. The excessive secretion of inflammatory factors is also accompanied by upregulated LDH and HMGB1, which were reported to be markers for cell death17 and serve inflammation.18 Through treatment with different dosages of azilsartan, the secretion of inflammatory factors was significantly suppressed, along with the downregulated LDH and HMGB1 expression. These data indicated that the injury and pro-inflammatory effects of LPS on macrophages were significantly reversed by azilsartan, indicating the promising anti-inflammatory property of azilsartan.
It is reported that TLR2 can be activated by LPS stimulation, which contributes to a severe immunoreaction in the body. Excessive immunoreaction is a stumbling block to self-repair.19 The expression level of NF-κB can be upregulated by TLR2 through activating the MyD88-dependent signal pathway, which will eventually induce the production of inflammatory factors such as IL-6, IL-1β, and TNF-α.20 Recently, the TLR2/MyD88 signal pathway has been found to be an effective way to alleviate cellular inflammation. As the main downstream effector molecule of TLR2, MyD88 plays an important role in the development and processing of inflammatory reactions.21 In the present study, to further investigate the possible mechanism underlying the anti-inflammatory effects of azilsartan, the impact of azilsartan on the TLR2/MyD88 signal pathway was evaluated. We found that LPS stimulation significantly activated the TLR2/MyD88 signal pathway in macrophages, accompanied by the upregulation of NF-κB activity, indicating that LPS induced the excessive production of inflammatory factors by activating the TLR2/MyD88 signal pathway to enhance the bio-function of NF-κB. Through the introduction of azilsartan, the activated TLR2/MyD88 signal pathway in LPS-induced macrophages was greatly inhibited, accompanied by an obvious downregulation of NF-κB activity. These data claimed that azilsartan might exert anti-inflammatory effects in the LPS-stimulated macrophages by preventing the activation of NF-κB through suppressing the TLR2/MyD88 signal pathway. Consistent with our results, a previous study demonstrated that the administration of azilsartan exerted a robust anti-inflammatory effect in ligature-induced periodontitis in rats by reducing the expression of IL-1β, MMP-2, MMP-9, COX-2, RANK, and RANKL.22 Another study reported that treatment with azilsartan could restore endothelial function by ameliorating vascular inflammation and reducing the expression of MCP-1, NOX-2, NOX-4, and TNF-α.23 However, further investigation will be helpful to explore the molecular mechanism underlying the bio-function of azilsartan on TLR2 in our future work to better understand the anti-inflammatory property of azilsartan.
Excessive inflammation is reported to be closely related to the production of ROS, which are important inducers in the activation of NF-κB in macrophages to aggravate the inflammation.24 It is also reported that dissociation of the thioredoxin interacting protein (TXNIP) from thioredoxin-1 is induced by the accumulated ROS under oxidative stress, which will activate the NLRP3 inflammasome by binding with NLRP3.25 Nrf2 is one of the most important defense systems against oxidative stress. The Nrf2 signal pathway will be activated under oxidative stress state, and subsequently, the DNA sequences on the antioxidant response element (ARE) will be recognized and bound by Nrf2, which triggers the transcription of anti-oxidant genes to promote the expression level of antioxidants and related enzymes, including reduced nicotinamide adenine dinucleotide phosphate (NADPH), quinone oxidoreductase (NQO-1), hemeoxygenase 1 (HO-1), superoxide dismutase (SOD), glutathione peroxidase (GPx), and glutathione transferase (GST). The cellular injuries induced by oxidative stress can be defended against these antioxidants and related enzymes.26−29 Followed by the treatment of azilsartan, the ROS levels in the incubated macrophages were significantly suppressed, accompanied by an elevated expression level of Nrf2 and decreased expression levels of MMP-2, MMP-9, PGE2, and COX-2. These data indicated that the induced oxidative stress by LPS was greatly reversed by azilsartan, indicating another possible mechanism underlying the anti-inflammatory effects of azilsartan. However, further detailed investigations are needed to explore the impact of azilsartan on the expression level of Nrf2 to better understand the inhibitory effect of azilsartan on oxidative stress in our future work.
Taken together, our data indicate that azilsartan might suppress LPS-induced inflammation in U937 macrophages by suppressing oxidative stress and inhibiting the TLR2/MyD88 signal pathway.
4. Methods and Materials
4.1. Cell Culture and Treatments
The human monocyte U937 cell line was purchased from American Type Culture Collection (ATCC, Rockville, MD), which was cultured in RPMI 1640 medium (Cat#31800, Solarbio life sciences, Beijing) containing 10% fetal bovine serum (FBS) with penicillin (50 U/mL) and streptomycin (100 μg/mL) at 37 °C in an incubator with 5% humidified CO2 and 95% air. The U937 cells were cultured in a 6-well cell culture dish for treatment. Cells were differentiated into macrophage-like cells by being treated with 50 nM phorbol myristate acetate (PMA, P1585, Sigma-Aldrich) for 24 h. U937 macrophages were treated with LPS (Cat#abs47014848, absin, China) at the final concentration of 1 μg/mL (100 ng total LPS per well for 96-well cell culture plate) in the presence or absence of Azilsartan (Cat#CC3202, Chemcatch, China) (1, 5, or 10 μM) for 24 h.
4.2. Reverse Transcriptase-Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted using the TRIzol Reagent according to the manufacturer’s protocol. Briefly, cDNA synthesis was performed using a TaqMan MicroRNA Array Kit (Thermo Fisher Scientific, Waltham). Real-time quantitative PCR was performed using a SYBR Green PCR Master Mix (Cat#4309155, Thermo Fisher Scientific) mixed with primers at 95 °C for 3 min, then 40 cycles of 95 °C for 12 s, and 60 °C for 40 s. U6 was used as an endogenous control to normalize expression. The following primers were used in this study: MCP-1 (F: 5′-TTCTGTGCCTGCTGCTCAT-3′; R: 5′-GGGGCATTGATTTGCATCT-3′); IL-6 (F: 5′-TTGGGAAGGTTACATCAGATCAT-3′; R: 5′-GGGTTGGTCCATGTCAATTT-3′); IL-1β (F: 5′-TACCTGTCCTGCGTGTTGAA-3′, R: 5′-TCTTTGGGTAATTTTTGGGATCT-3′); MMP-2 (F: 5′-TAACCTGGATGCCGTCGT-3′; R: 5′-TTCAGGTAATAAGCACCCTTGAA-3′); and MMP-9 (F: 5′-GAACCAATCTCACCGACAGG-3′; R: 5′-GCCACCCGAGTGTAACCATA-3′). Each experiment was performed in triplicate, and all of the relative expression levels were measured using the 2–ΔΔCT method.
4.3. Western Blotting Assay
After being washed three times with cold phosphate-buffered saline (PBS), the treated cells were harvested using a Mammalian Cell Lysis Kit (Thermo Fisher Scientific, Waltham). The nuclear protein was isolated using an EpiQuik Nuclear Extraction Kit (Cat#OP-0002, EpiGentek). The expression of NF-κB p65 was measured using nuclear fragmentations. The expressions of other proteins were measured using total cell lysates. Equal amounts of the proteins were subjected to SDS-PAGE (12%) under reducing conditions; the separated proteins were transferred onto PVDF membranes and then blocked with 5% nonfat dry milk in Tris-buffered saline with Tween-20 (TBST) at room temperature for 1 h. The membranes were probed with the indicated antibodies against COX-2 (1:500, Cat#sc-19999, Santa Cruz Biotechnology), Nrf2 antibody (1:2000, Cat#4399, Cell Signaling Technology), TLR2 (1:1000, Cat#66645-1-Ig, Proteintech, China), Myd-88 (1:2000, Cat#3699, Cell Signaling Technology), NF-κB p65 (1:1000, Cat#3034, Cell Signaling Technology), β-tubulin (1:5000, #2146, Cell Signaling Technology), and lamin B1 (1:2000, Cat#9087, Cell Signaling Technology) overnight at 4 °C. After three washes, the membranes were incubated with the alkaline phosphatase-conjugated goat anti-mouse/rabbit IgG secondary antibody (1:5000 dilution). A chemiluminescence detection system was used to detect the signals. The intensity of the protein bands was quantified by densitometry using ImageJ software (NIH). Each western blot was repeated at least three times.
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
HMGB-1, IL-6, MCP-1, IL-1β, and PGE2 levels in the supernatant of macrophages were determined using ELISA kits, including a human HMGB-1 ELISA Kit (Cat#EH0084, FineTest, China), human IL-6 ELISA Kit (Cat#KAC1261, Invitrogen), human MCP-1 ELISA Kit (Cat#BMS281, Invitrogen), human IL-1β ELISA Kit (Cat#SEKH-0002, Sobarbio life sciences), and human PGE2 ELISA Kit (Cat#SEKH-0414, Solarbio life sciences, Beijing), according to the manufacturer’s protocol. The concentration range of the protein standards is from 0 to 10 000 pg/mL. Briefly, 50 μL of standard samples or target samples was added to the ELISA plate. After incubation for 2 h and washing three times, 50 μL of the test solution was added and incubated for another 30 min. After three washes, 50 μL of horseradish peroxidase (HRP)-conjugated secondary antibodies was added and incubated for 30 min. The reaction was developed with a substrate solution and terminated with a stop solution. After the procedure, the plates were read on a spectrometer at a wavelength of 450 nm. The results were converted to numeric values using standard curves.
4.5. Dihydroethidium (DHE) Staining
The intracellular ROS levels in macrophages were detected using dihydroethidium (DHE). Macrophages were cultured in 96-well plates at a density of 1 × 105cells/mL in 1640 medium containing 10% FBS. Subsequently, the cells were gently washed with Hanks’ balanced salt solution (HBSS), followed by incubation with 5 μM DHE at 37 °C for 30 min. The dye was then removed and replaced with fresh HBSS. Fluorescence of the cells was measured immediately on a microplate reader (Ex (λ) 535 nm; Em (λ) 610 nm).
4.6. Luciferase Activity of NF-κB
Macrophages (3×106 cells/mL) were planted in the plates and transfected with NF-κB Luc or a plasmid encoding β-galactosidase (0.25 μg/mL) by the polyethylenimine (PEI) method. The cells were collected after incubating for 24 h and lysed by freezing at −70 °C for at least 3 h. The luminescence was measured using a luminometer to evaluate the luciferase reporter activity, which was then normalized to the β-galactosidase activity.
4.7. Statistical Analysis
Mean ± standard deviation (SD) was used to show data. GraphPad was used to analyze data. Analysis of variance (ANOVA) was utilized for the contrast among different groups followed by Tukey’s post-hoc test. P < 0.05 was regarded as a statistically significant difference between the two groups.
Acknowledgments
This study was supported by the Dongying Health Scientific Project (No. DYH-201804012).
Author Contributions
∥ Q.D. and Y.L. contributed equally.
The authors declare no competing financial interest.
References
- Yuan H.; Perry C. N.; Huang C.; Iwai-Kanai E.; Carreira R. S.; Glembotski C. C.; Gottlieb R. A. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am. J. Physiol.: Heart Circ. Physiol. 2009, 296, H470–H479. 10.1152/ajpheart.01051.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters J. J.; Sommer J. A.; Pfeiffer Z. A.; Prabhu U.; Guerra A. N.; Bertics P. J. A differential role for the mitogen-activated protein kinases in lipopolysaccharide signaling: the MEK/ERK pathway is not essential for nitric oxide and interleukin 1beta production. J. Biol. Chem. 2002, 277, 9077–9087. 10.1074/jbc.M104385200. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Lou J.; Mao Y. Y.; Lai T. W.; Liu L. Y.; Zhu C.; Zhang C.; Liu J.; Li Y. Y.; Zhang F.; Li W.; Ying S. M.; Chen Z. H.; Shen H. H. Activation of MTOR in pulmonary epithelium promotes LPS-induced acute lung injury. Autophagy 2016, 12, 2286–2299. 10.1080/15548627.2016.1230584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon E. Y.; Pyo S. Lipopolysaccharide stimulates Epac1-mediated Rap1/NF-kappaB pathway in Raw 264.7 murine macrophages. Immunol. Lett. 2007, 110, 121–125. 10.1016/j.imlet.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Zhang W. B.; Yang F.; Wang Y.; Jiao F. Z.; Zhang H. Y.; Wang L. W.; Gong Z. J. Inhibition of HDAC6 attenuates LPS-induced inflammation in macrophages by regulating oxidative stress and suppressing the TLR4-MAPK/NF-kappaB pathways. Biomed. Pharmacother. 2019, 117, 109166 10.1016/j.biopha.2019.109166. [DOI] [PubMed] [Google Scholar]
- Tanaka M.; Kishimoto Y.; Sasaki M.; Sato A.; Kamiya T.; Iida K.; Kondo K. Terminalia bellirica (Gaertn.) Roxb. Extract and Gallic Acid Attenuate LPS-Induced Inflammation and Oxidative Stress via MAPK/NF-kappaB and Akt/AMPK/Nrf2 Pathways. Oxid. Med. Cell. Longevity 2018, 2018, 9364364 10.1155/2018/9364364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morcillo E. J.; Estrela J.; Cortijo J. Oxidative stress and pulmonary inflammation: pharmacological intervention with antioxidants. Pharmacol. Res. 1999, 40, 393–404. 10.1006/phrs.1999.0549. [DOI] [PubMed] [Google Scholar]
- Fleury C.; Mignotte B.; Vayssiere J. L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2002, 84, 131–141. 10.1016/S0300-9084(02)01369-X. [DOI] [PubMed] [Google Scholar]
- Yorimitsu T.; Klionsky D. J. Eating the endoplasmic reticulum: quality control by autophagy. Trends Cell Biol. 2007, 17, 279–285. 10.1016/j.tcb.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Lemasters J. J.; Nieminen A. L.; Qian T. L.; Trost C.; Elmore S. P.; Nishimura Y.; Crowe R. A.; Cascio W. E.; Bradham C. A.; Brenner D. A.; Herman B. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta 1998, 1366, 177–196. 10.1016/S0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- Angiotensin II Receptor Antagonists. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; 2012. [PubMed]
- Iwai M.; Chen R.; Imura Y.; Horiuchi M. TAK-536, a new AT1 receptor blocker, improves glucose intolerance and adipocyte differentiation. Am. J. Hypertens. 2007, 20, 579–586. 10.1016/j.amjhyper.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Kurtz T. W.; Klein U. Next generation multifunctional angiotensin receptor blockers. Hypertens. Res. 2009, 32, 826–834. 10.1038/hr.2009.135. [DOI] [PubMed] [Google Scholar]
- de Araújo A. A.; Varela H.; de Medeiros C. A.; de Castro Brito G. A.; de Lima K. C.; de Moura L. M.; de Araujo Júnior R. F. Azilsartan reduced TNF-alpha and IL-1beta levels, increased IL-10 levels and upregulated VEGF, FGF, KGF, and TGF-alpha in an oral mucositis model. PLoS One 2015, 10, e0116799 10.1371/journal.pone.0116799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras E. M. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood 2017, 130, 1693–1698. 10.1182/blood-2017-06-780882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuprash D. V.; Nedospasov S. A. Molecular and Cellular Mechanisms of Inflammation. Biochemistry 2016, 81, 1237–1239. 10.1134/S0006297916110018. [DOI] [PubMed] [Google Scholar]
- Wei R.; Cao J.; Yao S. Matrine promotes liver cancer cell apoptosis by inhibiting mitophagy and PINK1/Parkin pathways. Cell Stress Chaperones 2018, 23, 1295–1309. 10.1007/s12192-018-0937-7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Andersson U.; Tracey K. J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 2011, 29, 139–162. 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar M. A.; Basith S.; Choi S. Negative regulatory approaches to the attenuation of Toll-like receptor signaling. Exp. Mol. Med. 2013, 45, e11 10.1038/emm.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas K.; Maes M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: possible treatments targeting the TLR4 pathway. Mol. Neurobiol. 2013, 48, 190–204. 10.1007/s12035-013-8425-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong E.; Lee J. Y. Intrinsic and extrinsic regulation of innate immune receptors. Yonsei Med. J. 2011, 52, 379–392. 10.3349/ymj.2011.52.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Araújo A.; Varela H.; de Castro Brito G.; de Medeiros C.; de Souza Araújo L.; do Nascimento J.; de Araújo Júnior R. Azilsartan increases levels of IL-10, down-regulates MMP-2, MMP-9, RANKL/RANK, Cathepsin K and up-regulates OPG in an experimental periodontitis model. PLoS One 2014, 9, e96750 10.1371/journal.pone.0096750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto S.; Shimabukuro M.; Fukuda D.; Soeki T.; Yamakawa K.; Masuzaki H.; Sata M. Azilsartan, an angiotensin II type 1 receptor blocker, restores endothelial function by reducing vascular inflammation and by increasing the phosphorylation ratio Ser(1177)/Thr(497) of endothelial nitric oxide synthase in diabetic mice. Cardiovasc. Diabetol. 2014, 13, 30. 10.1186/1475-2840-13-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S. K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox?. Oxid. Med. Cell. Longevity 2016, 2016, 5698931 10.1155/2016/5698931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R.; Tardivel A.; Thorens B.; Choi I.; Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- Donovan E. L.; McCord J. M.; Reuland D. J.; Miller B. F.; Hamilton K. L. Phytochemical activation of Nrf2 protects human coronary artery endothelial cells against an oxidative challenge. Oxid. Med. Cell. Longevity 2012, 2012, 132931 10.1155/2012/132931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T. J.; Lan D. H.; He S. Z.; Ye Z.; Li P.; Zhai W.; Chen W. Q.; Huang Y.; Fu Y.; Sun A.; Wang Y. B.; Ye Z.; Li J. L.; Gao Y.; Yan X. L.; Li Z. H. Nrf2 protects human lens epithelial cells against H2O2-induced oxidative and ER stress: The ATF4 may be involved. Exp. Eye Res. 2018, 169, 28–37. 10.1016/j.exer.2018.01.018. [DOI] [PubMed] [Google Scholar]
- Gong W.; Ma Y.; Li A.; Shi H.; Nie S. Trimetazidine suppresses oxidative stress, inhibits MMP-2 and MMP-9 expression, and prevents cardiac rupture in mice with myocardial infarction. Cardiovasc. Ther. 2018, 36, e12460 10.1111/1755-5922.12460. [DOI] [PubMed] [Google Scholar]
- Song Q.; Feng Y. B.; Wang L.; Shen J.; Li Y.; Fan C.; Wang P.; Yu S. Y. COX-2 inhibition rescues depression-like behaviors via suppressing glial activation, oxidative stress and neuronal apoptosis in rats. Neuropharmacology 2019, 160, 107779 10.1016/j.neuropharm.2019.107779. [DOI] [PubMed] [Google Scholar]