

Abstract
Late recurrences of breast cancer are hypothesized to originate from disseminated tumor cells that re-activate after a long period of dormancy, ≥ 5 years for estrogen-receptor positive (ER+) tumors. An outstanding question remains as to what the key microenvironment interactions are that regulate this complex process, and well-defined human model systems are needed for probing this. Here, a robust, bioinspired 3D ER+ dormancy culture model is established and utilized to probe the effects of matrix properties for common sites of late recurrence on breast cancer cell dormancy. Formation of dormant micrometastases over several weeks is examined for ER+ cells (T47D, BT474), where the timing of entry into dormancy versus persistent growth depends on matrix composition and cell type. In contrast, triple negative cells (MDA-MB-231), associated with early recurrence, are not observed to undergo long-term dormancy. Bioinformatic analyses quantitatively support an increased ‘dormancy score’ gene signature for ER+ cells (T47D) and reveal differential expression of genes associated with different biological processes based on matrix composition. Further, these analyses support a link between dormancy and autophagy, a potential survival mechanism. This robust model system will allow systematic investigations of other cell-microenvironment interactions in dormancy and evaluation of therapeutics for preventing late recurrence.
Keywords: ER+ breast cancer dormancy, late recurrence, synthetic extracellular matrices, 3D culture models, autophagy
Graphical Abstract
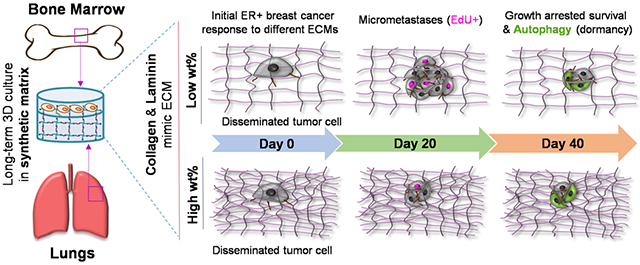
Bioinspired synthetic matrices for examining dormancy and reactivation of the ER+ breast cancer for insights into late recurrence
Introduction
Cancer metastasis remains the main cause of breast cancer-related deaths.[1] In the body, disseminated tumor cells (DTCs) escape from the primary tumor and circulate through the blood stream, entering a metastatic site where they can undergo proliferation, apoptosis, or dormancy (growth arrest) (Figure 1A).[2] Reactivation of dormant DTCs at metastatic sites is hypothesized to be one mechanism of both early and late recurrence, where the elapsed time of cancer recurrence after successful treatment of the primary tumor is dependent on breast cancer subtype. For example, triple negative (TN) breast cancers typically recur after 1-2 years, whereas estrogen receptor positive (ER+) breast cancers are associated with late recurrence 5 to 20+ years after remission and occur almost exclusively at metastatic sites.[3-5] The large time frame for these late recurrences of ER+ cancers makes them inherently difficult to study clinically, and few effective tools exist for the detection of dormant DTCs or prediction and treatment of associated late recurrences that affect approximately 20% of disease-free patients.[4,6] Innovative human model systems are needed for mechanistic studies and insights into key regulators of breast cancer dormancy and reactivation toward improved diagnosis and identification of potential therapeutic targets.
Figure 1. Approach for studying ER+ breast cancer dormancy and regulators of late recurrence.
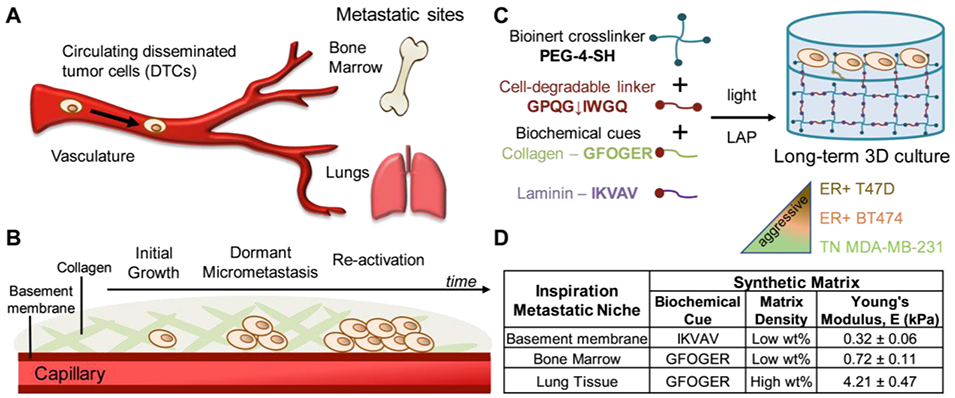
A) Disseminated tumor cells (DTCs) escape from the primary tumor and circulate in the blood stream until cells extravasate from capillary into a metastatic site. At these sites, DTCs have a number of different fates, including dormancy which is thought to be the origin of late recurrences which most frequently occur in bone marrow and lung tissues. B) Specifically, after dissemination, DTCs i) can initially exhibit growth followed by the ii) formation of dormant micrometastases that iii) re-activate or recommence proliferation at late times. Collagen is the most prevalent ECM protein in bone marrow and lung niches, and the basement membrane, enriched in laminin, comprises the endothelium surrounding the capillary. C) To study this, well-defined bioinspired synthetic matrices were created with light-triggered thiol-ene ‘click’ chemistry for long-term 3D culture of breast cancer cells that are ER+ (T47D, BT474), which clinically undergo late recurrence, and TN (MDA-MB-231), which clinically form secondary tumors or undergo early recurrence. A peptide linker derived from collagen (GPQG↓IWGQ) that degrades in response to a variety of proteinases was selected to allow both cell-driven and externally-triggered remodeling of matrix upon the exogenous application of collagenase. D) Based on key compositional differences between the ECM of these sites, pendant peptides derived from collagen (GFOGER) or laminin (IKVAV) and different matrix densities (low or high wt%) with relevant mechanical properties were selected, measured by rheometry (Figure S1).
The microenvironment surrounding the DTCs at metastatic sites, particularly the extracellular matrix (ECM), is thought to play a key role in regulating both breast cancer cell dormancy and reactivation, a cause of late recurrences which are most frequently observed in the bone marrow, lungs, and liver, respectively.[7] Specifically, in response to the metastatic site microenvironment, DTCs are hypothesized to exhibit low levels of initial proliferation, a long stage of growth arrest as dormant single cells or micrometastases, and recommence proliferation in response to stimuli, leading to cancer growth and relapse at late times (Figure 1B).[8] To study this complex process outside of the human body, in vitro culture models with different levels of complexity have been developed and have demonstrated the importance of a few specific cell-microenvironment interactions in cancer dormancy.[3,9-12]
Traditional tissue culture plates coated with different ECM proteins have been used for well-defined two-dimensional (2D) cultures that promote dormancy of ER+ breast cancer cells over short times (up to ~ 1 week), where basic fibroblast growth factor and ß1-integrin binding to fibronectin were observed to be important.[13,14] Moving into three dimensions, ER+ breast cancer cells have been cultured in basement membrane extract (BME harvested from Engelbreth-Holm-Swarm tumors rich in laminin I and type IV collagen)[15] or in cell-secreted ECMs generated in vitro with metastatic niche cells:[16,17] ß1-integrin signaling remained critical and perivascular niche cues were revealed to be important in the dormant-to-proliferative switch (e.g., TGFß1, POSTN for metastatic outgrowth; Laminin, Col IV, TSP-1 for dormancy). While these culture systems have provided great insights, examining how different biochemical content and matrix densities, like found in different sites of late recurrence, regulate dormancy can be challenging. Matrix density and related mechanical properties vary widely in vivo (e.g., bone marrow, Young’s modulus (E) ~ 0.5 kPa; lung, E ~ 1-5 kPa)[18,19] yet harvested or naturally-derived protein material systems have limited control of matrix density and mechanics. Further, compositional batch-to-batch variation is a significant concern for reproducibility and often only short to mid-term cultures (e.g., ≤ 2 weeks) can be performed.[20] Approaches that allow precise control of matrix properties are needed for hypothesis testing and for probing their individual and synergistic roles in dormancy and late recurrence over long times in 3D culture.
Synthetic materials for controlled 3D cell culture are of continued and growing interest owing to their reduced batch-to-batch variability for enhanced reproducibility and because they can be designed with a range of mechanical properties relevant for mimicking different tissues in the body.[21-25] Indeed, synthetic and hybrid matrices, such as those comprised of multifunctional polymers (e.g., poly(ethylene glycol) (PEG), poly(vinyl alcohol), poly(lactic acid)) and bioactive peptides or proteins, have been utilized for 3D culture of cancer cells with a focus on growth and metastasis,[26-28] including breast cancer cells.[29-31] Few studies have applied synthetic biomaterials to the study of breast cancer dormancy in 3D culture.[10] These include examination of breast cancer cell morphology and proliferation when seeded on non-bioactive stiff electrospun poly(capralactone) fibers[32] or entrapped within crosslinked poly(siloxanes).[30] Recently, synthetic PEG-based hydrogels formed by free radical chain growth polymerization have been used for the 3D culture of TN breast cancer cells associated with early recurrence, where the presence and concentration of the integrin-binding ligand RGDS was observed to influence dormancy vs. growth.[33] Cumulatively, these studies demonstrate the relevance of well-defined synthetic matrices for the study of breast cancer and the opportunity they present for probing cell-matrix interactions in breast cancer dormancy. The application of soft synthetic ECMs to the study of ER+ breast cancer dormancy has the potential to provide new insights into late recurrence and improved 3D model systems for a range of investigations into this intractable disease.
To address this need, in this work, we establish a well-defined 3D human culture model of ER+ breast cancer dormancy and utilize it to probe the effects of matrix composition, inspired by common metastatic sites, on ER+ breast cancer dormancy. We hypothesized that matrix composition, including biochemical content and matrix density, and cell type influence the propensity for dormancy of breast cancer cells. To test this, synthetic bioactive hydrogels were formed by photoinitiated thiol-ene ‘click’ chemistry as reductionist models of the ECM in common metastatic sites for late recurrence: specifically, the interstitial space of i) bone marrow and ii) lung tissues (collagen rich and E ~ 0.5 or 5 kPa, respectively) and iii) the basement membrane (laminin rich and E ~ 0.1 kPa) of the vascular endothelium, where DTCs reside.[18,19,34] Breast cancer cells with different levels of aggressiveness and propensity for late recurrence were selected: ER+ breast cancer cells (luminal A ER+ T47D, luminal B ER+ HER2+ BT474) and TN breast cancer cells (basal B MDA-MB-231) (in order of increasing aggressiveness and decreasing propensity for late recurrence). These cells were encapsulated as a single cell suspension in these well-defined, bioinspired matrices and cultured in three dimensions over ≥ 5 weeks to probe their relative responses to differences in the synthetic matrix. To assess dormancy, the metabolism, viability, and proliferation of these prototypical lines were evaluated, where luminal ER+ and basal TN breast cancer cells were expected to be more to less likely to undergo long-term dormancy, respectively.
The formation of ER+ dormant micrometastases was observed in these long-term bioinspired 3D cultures with cells that are: viable, not growing, and capable of recommencing growth upon stimulation (e.g., matrix ‘remodeling’ by enzymatic degradation of the synthetic matrix). Interestingly, the occurrence and timing of entry into dormancy versus persistent growth was observed to depend on both matrix density and cell type for ER+ cell lines, whereas TN MDA-MB-231 cells showed variable response to the biochemical cue within the bioinspired matrix composition but did not exhibit dormancy. To further study ER+ cell response within the 3D dormancy culture model, next generation sequencing and related bioinformatics analyses were used to probe ER+ T47D cells in these microenvironments. Significant differential expression of a variety of genes associated with cell cycle, cell motility, or cell-ECM interactions was observed in response to different matrix compositions. Further, gene signatures for dormancy and autophagy were observed over time that confirm phenotypic observations and suggest a correlation between dormancy and autophagy, a potential mechanism of survival. These studies provide a new tool for studying ER+ breast cancer dormancy for examining late recurrence and provide insights into the role of matrix composition on ER+ breast cancer dormancy.
Results
Well-defined hydrogel-based synthetic matrices, inspired by common metastatic sites, for probing breast cancer dormancy
To evaluate breast cancer cell response to metastatic-site-like microenvironments, breast cancer cells (luminal A ER+ T47D, luminal B ER+ HER2+ BT474, basal B TN MDA-MB-231) were each encapsulated and cultured in 3D synthetic hydrogel-based matrices inspired by common sites for late recurrence: the interstitial space of the bone marrow and lung tissues, which DTCs enter following extravasation, are composed primarily of collagen I amongst other proteins with differences in mechanical properties (E ~ 0.5 kPa and ~ 5 kPa, respectively) (Figure 1A, B).[18,19] Immediately upon extravasation, DTCs are in contact with the basement membrane of the endothelium, where basement membrane is composed primarily of laminin I with mechanical properties of E ~ 0.1 kPa.[34]
Inspired by such microenvironments, well-defined PEG-peptide hydrogels formed by photoinitiated thiol-ene ‘click’ chemistry were used as reductionist models of the biophysical and biochemical properties of the ECM in these metastatic sites (bone marrow, lung, and basement membrane) (Figure 1C). The initial density of these synthetic matrices, as measured by mechanical properties, were tuned by varying the concentration of the multi-arm bioinert macromer (PEG) and incorporating integrin-binding peptides GFOGER or IKVAV derived from collagen or laminin proteins, respectively, to control biochemical content. Specifically, collagen biochemical cue GFOGER is derived from collagen I and, when flanked with repeats of proline-hydroxyproline-glycine (POG) repeats, forms a triple-helical structure for binding of β1-integrin binding.[35,36] The laminin biochemical cue IKVAV is derived from the laminin α1 chain that forms protein-polysaccharide complexes for cell adhesion.[37] Hydrogels were formed by crosslinking a four-arm PEG-thiol (PEG-4-SH, Mn ~ 20 kDa) with a cell- degradable bis-alkene peptide crosslinker derived from collagen I (K(alloc)GGPQG↓IWGQGK(alloc)), which undergoes a rapid step growth polymerization reaction in the presence of cytocompatible doses of long wavelength UV light (10 mW/cm2 at 365 nm for 1 minute).[31] Pendant peptides (2 mM of collagen cue GFOGER or laminin cue IKVAV at preparation), were functionalized with a K(alloc) group for facile incorporation as pendant groups during hydrogel formation. This approach for matrix formation was selected based on our prior work that shows it allows i) consistent peptide incorporation, ii) independent control of hydrogel mechanical properties and biochemical cues for mimicking different soft tissues like those of interest here, and iii) a uniform encapsulation of single cancer cells within a thin hydrogel layer for quantitative analysis of cell responses in three dimensions using imaging-based techniques.[31,38,39] We previously have established the relevance of this materials system for short term studies of the growth and morphology of mesenchymal stem cells or breast cancer cells and now exploit the property control it affords for studying the effects of specific bioinspired matrix compositions on breast cancer dormancy in long term 3D cultures.
Bioinspired metastatic-site-like matrices were established as i) 6 wt% PEG (low wt%) + laminin cue IKVAV for basement membrane, ii) 6 wt% PEG (low wt%) + collagen cue GFOGER for bone marrow, and iii) 10 wt% (high wt%) + collagen cue GFOGER for lung. Mechanical properties of the equilibrium swollen hydrogels were consistent with reported mechanical properties for these tissues, with Young moduli for the synthetic matrices of E = 0.32 ± 0.06 kPa, E = 0.72 ± 0.11 kPa, and E = 4.21 ± 0.47 kPa similar to mechanical properties of basement membrane, bone marrow, and lung tissue microenvironments, respectively (Figure 1D). Mechanical properties of the equilibrium swollen hydrogels were measured with rheometry, where measured storage modulus (G′) was converted to E for ease of comparison of mechanical properties to the inspiration tissues (Figure S1). The resulting density of these synthetic matrices was estimated by calculation of respective equilibrium swollen crosslink densities with rubber elasticity theory, using measured values of storage modulus G′ and swelling (Q): crosslink density (ρx) of i) 0.21 ± 0.03, ii) 0.44 ± 0.04, and iii) 2.5 ± 0.2 mol*m−3, respectively.[40] This reductionist bioinspired matrix approach allowed the parsing of the relative effects of matrix composition on breast cancer cell dormancy: matrix density and biochemical content controlled with PEG-4-SH wt% and peptide selection, respectively.
ER+ breast cancer cell subtypes exhibit dormancy or continued growth in different matrix compositions
As previously mentioned, during the cascade of DTC dissemination to metastatic sites, ER+ DTCs can initially proliferate and over time become growth arrested (exit the cell-cycle into stages G0-G1) and form dormant micrometastases – non-proliferating cells that are still alive and have the potential to re-activate or recommence proliferation at a later time (Figure 1A, B). In this context, metabolic activity often is the ‘gold standard’ measure for initial evaluation of dormancy: limited increases in metabolic activity with confirmation of persistent cell viability often are used as indicators of dormancy (i.e., viable cells that are no longer growing).[9,15] To test our hypothesis that ER+ cells would form dormant micrometastases in a matrix-dependent manner, cell viability and metabolic activity first were measured for ER+ T47D breast cancer cells in all three metastatic-site-like compositions, and cell proliferation subsequently was examined to confirm any observations of dormancy.
A membrane integrity assay (live/dead cytotoxicity assay, live cells fluoresce green, dead cells red) was employed to determine viability. Confocal imaging was used to examine stained cells within the 3D volume (Figure 2A) and the percentage of live cells was quantified (Figure 2B). ER+ T47D cells were successfully encapsulated as single cells (Figure 2A, day 3) and exhibited some initial growth (formation of small clusters, Figure 2A, day 10-20) while maintaining high viability both initially and over long times in these 3D cultures (most > 90% viable cells).
Figure 2. Examination of viability and growth/dormancy of ER+ breast cancer cells in long-term 3D culture.
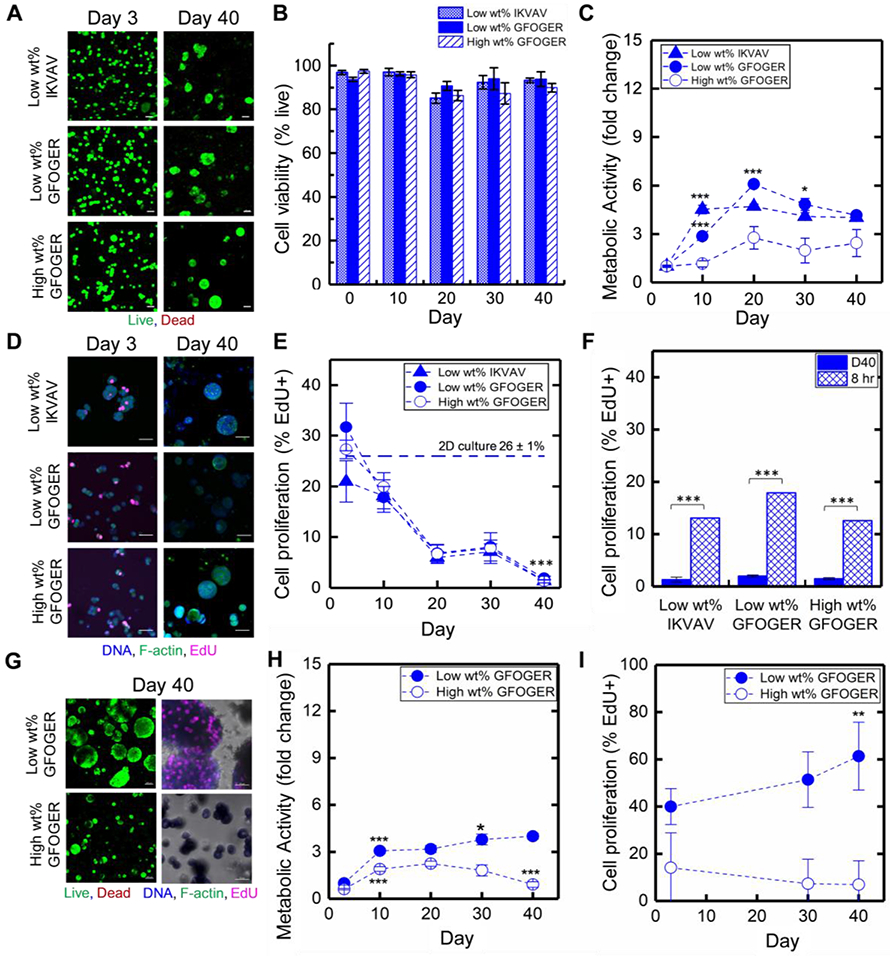
In each bioinspired matrix composition over long times in culture, A) Luminal A ER+ T47D cell viability was examined with live/dead cytotoxicity assay (representative confocal z-stack projections, live cells stained green, dead cells stained red, Scale bar = 50 μm) and B) quantitatively analyzed. C) Metabolic activity was assessed with AlamarBlue assay (fold change relative to day 3). Statistical differences determined with one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test, where differences shown are for each time point in comparison to the prior time point (*p < 0.05, **p < 0.01; ***p < 0.001; full set of p-values for other comparisons can be found in Table S1.) D) Proliferation was assessed with EdU proliferation assay (representative confocal z-stack projections, f-actin green, DNA blue, EdU pink, Scale bar = 50 μm) and E) quantitatively analyzed. Statistical differences determined with one-way ANOVA with Tukey’s test, where differences shown are for each time point in comparison to day 3 (*p < 0.05, **p < 0.01; ***p < 0.001; full set of p-values for other comparisons can be found in Table S2). F) Cells were released from hydrogels by matrix degradation with collagenase and briefly plated for assessing their ability to recommence proliferation, quantitatively analyzing the percentage of proliferating cells (%EdU+) before (D40) and after (8 hr) hydrogel degradation. Statistical differences determined with one-way ANOVA with Tukey’s test, where differences are shown for 8 hr after release from hydrogel in comparison to day 40 in 3D culture (*p < 0.05; ***p < 0.001; full set of p-values for other comparisons can be found in Table S2). G) Luminal B ER+ HER2+ BT474 breast cancer cell viability (left) and proliferation (right) similarly was examined (representative confocal z-stack projections, Scale bar = 100 μm and 50 μm, respectively); H) metabolic activity was assessed (fold change relative to day 3); and I) proliferation quantitatively analyzed (% EdU+ cells). Statistical differences determined with one-way ANOVA with Tukey’s test, where differences are shown for each time point in comparison to the prior time point (metabolic activity) and day 3 (EdU) (*p < 0.05, **p < 0.01; ***p < 0.001; full set of p-values for other comparisons can be found in Tables S1 and S2, respectively).
Importantly, while viability was consistently high in all compositions, significant differences of T47D metabolic activity were observed in the different matrix compositions, as measured with AlamarBlue (Figure 2C). In the low wt% matrix compositions, a statistically significant increase in metabolic activity was initially observed with both the collagen and laminin cues (day 3 to day 10, *** p < 0.001). Within the low wt% laminin cue IKVAV matrix composition, metabolic activity of T47D cells remained flat for the remainder of the culture, indicating limited growth and suggesting potential induction of dormancy (day 10 to day 40, no statistical differences). Within the low wt% collagen cue GFOGER matrix composition, metabolic activity increased until day 20 (day 10 to day 20, *** p < 0.001) followed by a slight decrease (day 20 to day 30, * p < 0.05) and plateau through day 40, indicating more persistent initial growth in the collagen rich matrix that ultimately slows. In contrast, in the high wt% matrix with the collagen cue GFOGER, a low metabolic activity was maintained throughout these cultures, indicating potential induction of dormancy even at early culture times and the importance of matrix density. These trends in metabolic activity generally were consistent with observations of cluster volume and cell number over time, with some initial growth (from day 3 to days 10 or 20, respectively) followed by limited to no growth (Figure S2A & B, Figure S3D & E, supporting information). Consequently, all studies were taken out to 40 days in culture, providing for all samples approximately two data points past which growth had slowed or ceased for observation of trends. These data suggested that ER+ breast cancer cells in 3D metastatic-site-inspired matrices could survive with minimal cell death and form small clusters reminiscent of dormant micrometastases where the time frame for entry into dormancy depended on matrix composition.
We next measured cell proliferation where we hypothesized that, owing to the lack of changes in cell growth (high viability, levelled metabolic activity, and arrested cluster size growth), cells would exhibit limited proliferation at long culture times. Measurement of proliferation was performed with an EdU proliferation assay (4 hr EdU pulse measuring the number of cells in the synthesis (S)-phase of the cell cycle), and samples subsequently were fixed, stained for EdU (AF647, magenta), nuclei (DAPI, blue) and cytoskeleton (F-actin, green), and imaged with confocal microscopy (Figure 2D, E). A substantial decrease in proliferation was observed over time in all three metastatic-site-like conditions. Initially, similar proliferation (percentage of EdU positive (EdU+) cells) was observed after 3 days in culture, in all three compositions, compared to the 2D plated culture control, T47D cells cultured on tissue culture polystyrene (TCPS). By 40 days in 3D culture, few to no EdU+ cells were observed (≤ 2%), indicating cells were no longer in proliferative S-phase. To confirm these observations, a subset of samples was stained for proliferation marker Ki-67, a nuclear protein expressed when cells are in the cell cycle. Trends observed with Ki-67 were similar to those with EdU, with a significant percentage of Ki-67+ cells at day 3 in 3D culture, indicating proliferation, and <1% Ki-67+ cells at day 40 (statistically the same as zero) indicative of dormancy (low wt% with GFOGER or IKVAV examined) (Figure S3A-C). Taken together, these observations (no to few EdU+ or Ki-67+ cells at day 40) suggested that ER+ T47D cell clusters were growth arrested and in a stage of dormancy reminiscent of dormant micrometastases. In dormancy, cells remain viable and not growing but capable of growing upon stimulation. Accordingly, as a final phenotypic assessment of dormancy, we evaluated the ability of dormant ER+ breast cancer cells in these bioinspired cultures to recommence proliferation. The synthetic matrix was ‘remodeled’ and completely degraded with the exogenous application of enzyme (collagenase) at day 40 in 3D culture. Dissociated cells were plated onto tissue culture treated plastic and their proliferation (EdU assay) was assessed. After sufficient time for the released cells to settle on and adhere to the plate (8 hours after matrix degradation and re-plating), ER+ T47D cells exhibited proliferation with significantly increased EdU expression as compared to observations at day 40 in 3D culture (from approximately 0% at day 40 to 15% EdU+ cells upon matrix degradation, *** p < 0.001) (Figure 2F). These released cells continued to proliferate and grow as expected in 2D culture (monitored up to 1 week) (Figure S6). With the increased number of cells in S-phase after release from the 3D matrix, these results support the ability of these ER+ T47D cells to recommence proliferation after 40 days of suppressed or limited growth in 3D culture. Overall, ER+ breast cancer cells were able to switch fate, from small non-proliferative clusters reminiscent of dormant micrometastases to proliferating cells upon complete matrix degradation, further serving to validate the model system for studying ER+ breast cancer cell dormancy.
Having established that ER+ T47D cells were exhibiting phenotypic markers of dormancy, we wanted to probe if the effects of increased matrix density on promoting limited growth and dormancy at early times in culture applied to other ER+ cell types. Luminal B ER+ HER2+ BT474 breast cancer cells similarly were encapsulated and cultured in synthetic matrices inspired by the bone marrow and lung microenvironments, low and high wt% matrices with the collagen cue GFOGER. These ER+ BT474 cells also exhibited good viability and some initial growth that then subsided in the high wt% matrix (Figure 2G, H). Slowed metabolic activity by day 20 correlated with a decrease in the percentage of EdU+ cells to approximately zero while good viability was maintained indicating dormancy (Figure 2H, I and Figure S4, Supporting Information). In contrast, ER+ BT474 cells continued growing in the low wt% matrix over 40 days, with increasing metabolic activity, cluster size, and percentage of EdU+ cells. These observations further support the significant influence that matrix density has on promoting the formation of dormant micrometastases by ER+ breast cancer cells.
TN breast cancer cells did not exhibit dormancy in these bioinspired synthetic matrices
Clinically, TN breast cancer is associated with ‘early’ recurrence (typically within 1-2 years after primary tumor treatment). Accordingly, in comparison to the ER+ breast cancer cells, we expected basal TN MDA-MB-231 breast cancer cells would be highly proliferative and less likely to undergo long-term dormancy. To examine this, TN MDA-MB-231 cells were encapsulated in the three bioinspired synthetic matrix compositions and their growth and viability were assessed, as was done with ER+ cells. In contrast to ER+ cells, TN MDA-MB-231 cells exhibited a stellate or spread morphology in the bone marrow inspired matrix (low wt% matrix with collagen cue GFOGER) (Figure 3A and Figure S2C, D, Supporting Information), a morphology commonly observed for this cell type and other cells exhibiting a mesenchymal phenotype in 3D culture.[41] Changing the composition, TN MDA-MB-231 cells exhibited a more rounded morphology in the low wt% matrix with the laminin cue IKVAV and a combination of rounded and spread morphologies in the high wt% GFOGER matrix. These observations at both short and long times in culture were consistent with previous reports that TN MDA-MB-231 are particularly responsive to the integrin-binding biochemical content of the matrix.[29,31,33] Quantifying viability over time, increased variance was observed for TN MDA-MB-231 cells, relative to what had been observed with ER+ cells, with differential responses to matrix composition (Figure 3B). In the low wt% matrix with collagen cue GFOGER, viability was initially high (> 90%) and then decreased slightly to approximately 80-90% over days 10-40. In the high wt% GFOGER matrix, viability remained high both initially and over time (~ 90%). In contrast, in the low wt% IKVAV matrix, viability was maintained but was lower both initially and over time, varying between approximately 70-85%.
Figure 3. Examination of viability and growth of TN breast cancer cells in long-term 3D culture.

In each bioinspired matrix composition over long times in culture, A) TN MDA-MB-231 cell viability was examined with live/dead cytotoxicity assay (representative confocal z-stack projections, live cells stained green, dead cells stained red, Scale bar = 50 μm) and B) quantitatively analyzed. C) Metabolic activity was assessed with AlamarBlue assay (fold change relative to day 3). Statistical differences determined with one-way ANOVA Tukey’s test, where differences shown are for each time point in comparison to the prior time point (*p < 0.05, **p < 0.01; ***p < 0.001; full set of p-values for other comparisons can be found in Table S1). D) Proliferation was assessed with 5-ethynyl-2′-deoxyuridine (EdU) proliferation assay (representative confocal z-stack projections, f-actin green, DNA blue, EdU pink, Scale bar = 50 μm) and E) quantitatively analyzed. Statistical differences determined with one-way ANOVA with Tukey’s test; differences shown are for cells in each composition at day 40 in comparison to day 3 (*p < 0.05, **p < 0.01; ***p < 0.001; full set of p-values for other comparisons can be found in Table S2), where the percentage of EdU+ cells significantly decreased between day 3 and day 10 and no significant differences were observed between day 10 and day 40 in 3D culture. F) Cells were released from hydrogels by matrix degradation with collagenase and briefly plated for assessing any changes in proliferation, quantitatively analyzing the percentage of proliferating cells (%EdU+) before (D40) and after (8 hr) hydrogel degradation. Statistical differences determined with one-way ANOVA Tukey’s test, where differences shown for 8 hr after release from hydrogel in comparison to day 40 in 3D culture (*p < 0.05; ***p < 0.001; full set of p-values for other comparisons can be found in Table S2).
Metabolic activity was measured to assess TN MDA-MB-231 cell growth and viability over time. In both the low and high wt% collagen cue GFOGER matrices, TN MDA-MB-231 cells exhibited significant growth and had the highest metabolic activity over time (Figure 3C). A peak in metabolic activity was reached around day 20 (day 10 to day 20, **p < 0.01 and ***p < 0.001, respectively; day 20 to day 30, no statistical difference) and at longer time periods a slight decrease in metabolic activity was observed (day 30 to 40, p < 0.01). In contrast, in the low wt% matrix with laminin cue IKVAV, minimal, non-statistical changes in metabolic activity were observed over time (day 3 through day 40). Additionally, these trends in MDA-MB-231 metabolic activity and viability were consistent with observations of cluster volume and cell number over time, with limited initial growth followed by a plateau or decrease at later times (Figure S2C & D, Figure S5D & E, supporting information). Overall, these data further support the importance of biochemical cues in TN MDA-MB-231 viability and growth.
Examination of cell proliferation provided a more complete viewpoint of TN MDA-MB-231 cell function in these different matrix compositions. Initially (day 3), TN MDA-MB-231 cells encapsulated in all compositions had a slight, but not statistically significant, decrease in the percentage of EdU+ cells relative to the plated 2D growth control (Figure 3D, E). After 10 days in culture, the percentage of proliferating EdU+ cells decreased to a level that was relatively constant over long culture times (through day 40), with ~ 10% EdU+. To confirm these observations, a subset of samples was stained for Ki-67. Trends observed with Ki-67 were similar to those with EdU with a significant percentage of Ki-67+ cells at day 3 in 3D culture, indicating proliferation, and decreased but statistically non-zero percentage of Ki-67+ at day 40, indicating continued proliferation (low wt% with GFOGER or IKVAV examined) (Figure S5A-C). Additionally, at day 40 in 3D culture, these cells were subjected to externally-triggered, complete degradation of the 3D matrices with exogenous application of collagenase followed by re-plating (Figure 3F, Figure S7 Supporting Information). Consistent with the continued MDA-MB-231 proliferation analyses in 3D culture, appreciable percentages of EdU+ cells were observed in 2D culture after release from the matrix. Cells released from low wt% IKVAV and high wt% GFOGER conditions had statistically similar proliferation (% EdU+ cells) to that observed in 3D culture at day 40. Overall, these data support that a significant percentage of TN MDA-MB-231 cells were proliferating consistently even at longer culture times. These observations, persistent proliferation in the context of plateaued metabolic activity and variable cell viability over time, suggest a balance between TN MDA-MB-231 cell proliferation and cell death in these 3D synthetic matrix compositions.
Quantitative examination of ‘dormancy score’ gene signature further supports the ER+ dormancy culture model
To further assess dormancy beyond phenotypic observations, a dormancy signature was examined at the gene level with next generation sequencing (RNA-seq). A variety of gene signatures are being established for breast cancer dormancy toward ultimately predicting the likelihood of late recurrence. The ‘dormancy score’ is an open-source method for calculating the likelihood for dormancy and late recurrence.[42] Specifically, the dormancy score consists of 49 up- and down-regulated dormancy-related genes that have been identified both in vitro and in vivo with human cell lines, patient data, and animal models, and summation of their normalized expression is utilized to calculate a numerical value or ‘score’, where higher positive values indicate dormancy and an increased propensity for late recurrence. Consequently, the dormancy score provides a quantitative metric for assessment of a dormancy gene signature and an opportunity to compare our model system to in vivo and clinical observations of dormancy at the gene level. Here, a dormancy score was calculated for ER+ T47D and TN MDA-MB-231 cells in each matrix composition at an early and late times in culture: i) day 3, where cells were observed to be actively proliferating serving effectively as a 3D growth control for each cell type in each matrix composition, and ii) day 40, where ER+ T47D were observed to exhibit dormancy and TN MDA-MB-231 remained proliferative based on phenotypic assays (Figure 4).
Figure 4. Examination of ‘dormancy score’ for assessing expression of dormancy gene signature in long-term 3D culture.
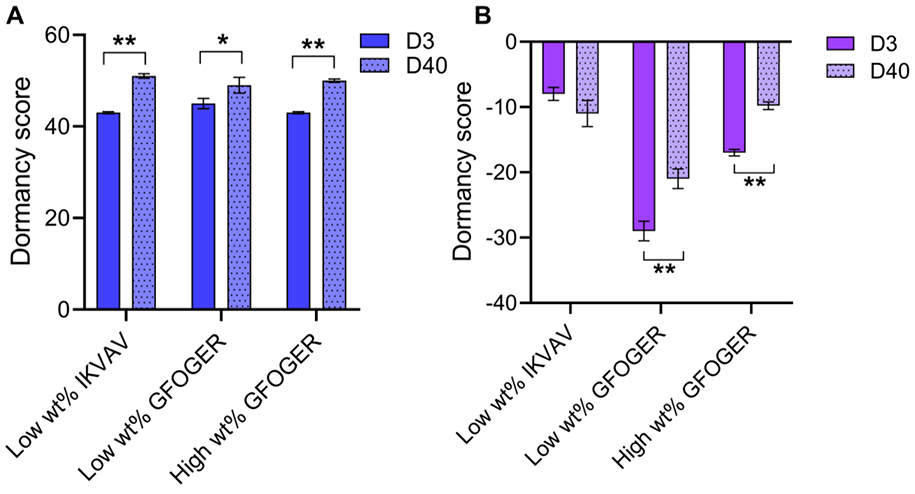
Next generation sequencing (RNA-seq) was performed on A) T47D (ER+) and B) MDA-MB-231 (TN) in each bioinspired matrix composition at early and late times (day 3 and day 40, respectively) in culture and a dormancy score was calculated with the RNA-seq data. Statistical differences in dormancy score determined with Student’s two-sided t-test, where differences shown are for day 40 time point in comparison to day 3 (*p < 0.05; full set of p-values for other comparisons can be found in Table S4).
ER+ T47D cells had positive dormancy scores at day 3, indicating a propensity of these cells for dormancy and late recurrence (Figure 4A). Importantly, ER+ T47D exhibited statistical increases in dormancy score magnitude from day 3 to day 40 in all three metastatic-site-like compositions, indicating an increased dormancy gene signature and further validating phenotypic observations of dormancy. Additionally, pair-wise comparisons of dormancy score genes between day 3 and day 40 showed correct directionality in gene expression, with appropriate up- or down-regulation at day 40 (Figure S8A, Supporting Information). These observations correspond to those made with patient data, where a high dormancy score suggests prolonged dormancy before undergoing recurrence.[42] With substantially decreased proliferation over long-term 3D culture and statistically increased positive dormancy scores, these data further supported the formation of small dormant cell clusters reminiscent of dormant micrometastases within the culture model.
For TN MDA-MB-231 cells, dormancy scores were negative in all cases, indicating these aggressive basal B cells do not have a propensity for late recurrence and were not dormant (Figure 4B).[42] While an increase in magnitude of the dormancy score (smaller negative number) was observed at long times for cells cultured in low wt% or high wt% matrices with collagen cue GFOGER, values remained negative, indicating down-regulated dormancy genes were still dominant and were upregulated for these TN cells in the 3D cultures (Figure S8B, Supporting Information). Interestingly, a decrease in the magnitude of the dormancy score (a larger negative number) was observed for cells cultured in the low wt% matrix with laminin cue IKVAV over time; this correlates with the minimal change in phenotypic response that also was observed in this matrix composition where TN MDA-MB-231 exhibited slow yet persistent growth over time. These observations further support that TN MDA-MB-231 breast cancer cells were not broadly exhibiting dormancy in these bioinspired matrices, although different responses to the biochemical content of the matrix were observed.
Bioinformatics analyses for probing cellular responses at the gene level
Differentially expressed genes, identified with RNA-seq, were analyzed over time and between conditions to better understand differences in cell responses to matrix composition and identify potential mechanisms for ER+ T47D cell survival in dormancy (Figure 5A). Differential gene expression was determined for cells in each matrix composition by comparing day 40, where dormancy was observed, to day 3, where cells were observed actively proliferating at levels similar to 2D culture. There were significant numbers of differentially expressed genes both in common (overlapping circles) and unique (non-overlapping circles) for ER+ T47D cells cultured in the different matrix compositions. Notably, more genes were differentially expressed in the high wt% GFOGER over time in 3D culture, indicating a significant transcriptomic effect of increased matrix density on cell response in addition to the significant suppression of growth this condition promoted in phenotypic assays (e.g., statistically flat metabolic activity). Amongst these genes, a substantial increase in gene expression related to glycolysis (EIF4B, PGK1, LDHA) was found for T47D cells cultured in the high wt% GFOGER matrix while these gene showed low expression at both time points in the low wt% matrices (Figure S9, Supporting Information), suggestive of changes in metabolism which were observed at the cellular level. Further, amongst the 50 most differentially expressed genes, there also were several in common with similar directionality in up- or down- regulation, respectively, including down-regulation of HSPA8 (gene encoding for Heat shock 70 kDa protein 8 whose down-regulation is correlated with induction of autophagy) and up-regulation of IGFBP5 (gene encoding for insulin-like growth factor-binding protein 5 whose up-regulation is correlated with suppression of tumor growth).[43,44] Gene ontology (GO) analysis of these differentially expressed genes was used to identify key enriched biological processes among the matrix compositions (Figure 5B and Figure S10, Supporting Information). Focusing on the top 20 most enriched GO terms, for low wt% matrix density compositions, cell-cycle related biological processes were most enriched, along with those related to response to external stimuli and cell motility. Interestingly, in the higher wt% GFOGER composition, biological processes associated with matrix interactions and cell cycle were most enriched over time, in addition to observation of enriched GO terms associated with cell motility and developmental processes. Overall, the observation of differential expression of genes associated with cell cycle correlated with our prior analyses and observation of dormancy for the ER+ T47D cells. More importantly, these bioinformatics analyses provided insights into some of the commonalities and differences in cell response to matrix density, where biological processes associated with cell-matrix interactions and developmental processes were enriched in the higher density matrix.
Figure 5. Differential expression and related analyses for mechanistic insights into dormancy.

RNA-seq was performed on ER+ T47D cells in each matrix composition at early and late times, where differentially expressed genes from day 3 (D3, active state) to day 40 (D40, dormant state) were examined at a significance cut of FDR < 0.05 and log2FC >± 1. A) Venn diagram of the number of differentially expressed genes for T47D ER+ cells in the three matrix compositions. B) Analysis of enriched gene ontology (GO) terms with top 20 significant processes enriched from D3 to D40 (full names of biological processes in Figure S10). C) Heat map of significantly differentially expressed associated with autophagy (autophagy signature genes): up-regulated (red) and down-regulated (blue) genes from D3 to D40 with a significance cut off of FDR < 0.05. D) Immunostaining for ATG9B autophagy-associated protein at D40 in 3D culture (representative confocal z-stack projections, DNA blue, F-actin red, and ATG9B green, Scale bar = 50 μm) and quantitatively analyzed the ATG9B protein (Figure S13). Similarly, expression of MAP1LC3B autophagy-associated protein was observed (Figure S13, which also contains immunostaining controls). E) RT-qPCR used to assess ATG9B and MAP1LC3B gene expression (presented as relative fold change from D3 to D40). Relative fold change of 1 corresponds to D3 expression. Significance of *p< 0.05 and **p<0.01 based on Mann-Whitney.
The same bioinformatics analyses were performed for TN MDA-MB-231 cells in three matrix compositions (Figure S11, Supporting Information). Few genes were differentially expressed for the low wt% IKVAV culture, mirroring the observations of few phenotypic differences for this condition. A substantial number of differentially expressed genes were measured for MDA-MB-231 cells cultured in the low and high wt% matrices with GFOGER (>4500 unique to high wt%, >500 unique to low wt%, >950 in common between low and high wt% GFOGER), including upregulation of FN1, ITGB1, VEGFA, and MMP1 that are characteristic key cell-matrix in metastasis.[9,15] Additional GO term enrichment analyses indicated biological processes related to cell cycle were most changed from day 3 to day 40 for MDA-MB-231 cells culture in the high wt% matrix with GFOGER, whereas developmental processes were most enriched in the low wt% matrix with GFOGER (Figure S12, Supporting Information). These observations further demonstrate the effect of matrix density of cell response. While TN MDA-MB-231 did not exhibit many changes at the cellular level (e.g., consistently proliferating with high metabolic activity in both low and high wt% matrices with GFOGER), they do manifest significant differences at the gene level.
Expression of markers of autophagy in the ER+ breast cancer dormancy culture model
A potential mechanism of ER+ breast cancer cell survival in dormancy is through autophagy, a self-degradative process to balance sources of energy for cell survival. To further examine the role of autophagy in the 3D ER+ dormancy culture model, we next looked at the individual genes that were differentially expressed and noticed that many were genes associated with autophagy, such as HSP8A noted above amongst the top 50 most differentially expressed genes. More broadly, we observed significant differential expression of an array of autophagy-related genes that are part of a known 234-gene autophagy signature (Human Autophagy Database, HADb) (Figure 5C), with greater than 100 of these genes expressed in each composition with the same 65 differentially expressed across all 3 compositions.[45-47] Autophagy is the process where cells degrade and recycle cellular components, by formation of an autophagosome, to maintain activity and viability when under stress, and the HADb is a public repository that provides a complete “list of human genes and proteins involved directly or indirectly in autophagy as described in literature” (http://autophagy.lu/index.html).[48,49] While autophagy has been implicated in cancer for decades, autophagy more recently has been reported as a survival mechanism for dormant cancer cells and breast cancer stem cells.[50-52]
As autophagy is a multi-step process, including induction, phagophore formation, elongation and autophagosome formation, lysosomal fusion, and degradation, we examined two specific proteins important in different steps in autophagy, ATG9B and MAP1LC3B (LC3B), for probing autophagy within the 3D ER+ dormancy model. ATG9B is a transmembrane protein that plays key roles in the early stages of autophagy, in phagophore and autophagosome formation. LC3B is a cytosolic protein that becomes integrated into the transmembrane of the autophagosome and promotes autophagosome formation. Given their importance for autophagosome formation, both often are used as indicators of autophagy.[48,49,53,54] The genes that encode these proteins, as well as others critical in autophagy (ULK1 and ULK2 indicative of induction, p62/SQSTM1 indicative of autophagosome formation), were amongst those observed by RNA-seq to have significantly increased expression in dormant ER+ T47D cells within our bioinspired 3D culture system.
Enrichment of ATG9B and LC3B was examined further at both the protein and gene level with immunocytochemistry and real-time quantitative reverse transcription polymerase chain reaction (RT-qPCR) to validate observations made with RNA-seq. Expression of both proteins, ATG9B (Figure 5D, Figure S13A, Supporting Information) and LC3B (Figure S13C, Supporting Information), was confirmed by immunostaining: cytosolic expression of proteins was present in dormant cell clusters at 40 days in 3D culture yet not in actively growing cells on day 3 in 3D culture (growth control in Figure S13A & C, Supporting Information). Differential expression of the ATG9B at the protein level was confirmed by semi-quantitative analysis of the resulting confocal z-stacks, with increased fluorescence per cell in all conditions at day 40 (dormant) relative to day 3 (active) (Figure S13B). Additionally, RT-qPCR analysis quantitatively confirmed increased expression of genes associated with these proteins (Figure 5E). Of note, at both the gene and protein level, differential expression of ATG9B was significantly increased with increased matrix density. Probing this effect further, immunostaining for expression of LC3B in 3D cultures of ER+ HER2+ BT474 cells qualitatively revealed expression in high wt% GFOGER condition in which dormancy was observed but not in low wt% GFOGER condition in which cells remained proliferative (Figure S13D). From bioinformatics analyses to experimental confirmation at the gene and protein level, a link between dormancy and autophagy within this well-defined 3D ER+ dormancy model was established, suggesting a potential mechanism whereby ER+ T47D dormant micrometastases survive for future investigations in further studying and ultimately targeting late recurrence.
Discussion
Clinical observations support the formation of dormant micrometastases as a mechanism for DTC survival over long times where their re-activation is hypothesized to be the origin of late recurrence.[3] In vivo models of breast cancer recurrence further support this mechanism. For example, Ghajar et al. demonstrated, in an immunocompromised mouse model, dissemination and arrival of breast cancer cells at a metastatic site leads to a short growth period followed by formation of dormant micrometastases that recommence proliferation after long periods of time.[16] In the work presented here, we sought to probe how key aspects of matrix density and biochemical content influenced the dormancy of ER+ breast cancer cells and establish a well-defined in vitro 3D culture system for parsing the role of such microenvironment cues in dormancy and late recurrence. Benchmarking the model system against clinical and in vivo observations of the disease, our in vitro model showed a similar trend for ER+ T47D cells in all 3 bioinspired matrix compositions where: 1) there was initial growth (volume and metabolic activity) of cells in 3D culture followed by a plateau; 2) the small cell clusters formed were highly viable over time; 3) importantly, decreased proliferation was observed over time while viability was maintained, and 4) these cells were capable of recommencing proliferation after a change in the microenvironment. Dormancy of ER+ T47D cells in these environments was further examined using RNA-seq bioinformatics tools, where increasingly positive dormancy scores were observed over time that indicate proper directionality in gene expression for the in vitro model in comparison to dormant cells in vitro and in vivo. Thus, from the cellular to gene level, the reductionist bioinspired culture model established here was observed to recapitulate key aspects of ER+ breast cancer dormancy.
With this validated dormancy model, we then went about probing both potential mechanisms and differences in cell response to matrix compositions. One mechanism for dormancy of disseminated tumor cells from other types of cancers has been elucidated by Aguirre-Ghiso and coworkers and demonstrates the importance of ECM interactions in the development and maintenance of dormancy. Specifically, they have demonstrated that the presence of fibronectin (FN) in the ECM surrounding the DTC and up-regulation of TGFβ2 and BMP4 or BMP7 is promoting of dormancy, which upregulates SMAD1 or SMAD5 intracellularly.[9] From our RNA-seq analyses, these genes (FN1, TGFB2, BMP4 or BMP7, SMAD 5 or SMAD1) had increased differential expression from day 3 to day 40 for T47D cells cultured in all three matrices (Figure S14, Supporting Information) and may suggest these same types of interactions also are important in the promotion of dormancy in these well-defined bioinspired 3D cultures.
More broadly, autophagy was examined as a potential mechanism for how dormant cells survive for longer periods of time, which could provide a future target for further investigations and prevention of late recurrence. While autophagy is known to play an important role in cancer and other disorders where cells survive in harsh environments, it has more recently been implicated in survival of dormant DTCs, including breast cancer. For example, Green and coworkers demonstrated that dormant murine mammary tumor cells (D2.0R) cultured in BME or disseminated in vivo to the lung microenvironment in a murine model exhibited dormancy and autophagy, where inhibition of autophagy modulated either a dormancy to apoptosis or dormancy to proliferation switch.[50] In this context, observation of autophagy in the new model system presented here further validates the relevance of this well-defined bioinspired 3D culture system for probing dormancy of human ER+ breast cancer cells. With this robust in vitro model system now established, the effects of matrix density and integrin binding on key processes in dormancy and autophagy can be independently probed, with opportunities for future mechanistic and therapeutic evaluation studies.
As noted in the introduction, a few synthetic biomaterials have been established for probing dormancy of breast cancer cells and other cell types.[10] For example, crosslinked poly(siloxanes) have been used to entrap breast cancer cells (ER+ MCF-7 and TN MDA-MB-231) in a non-degradable, non-bioactive matrix of different densities, inducing dormancy in short time studies over days.[30] In a more recent report in parallel with our investigations, TN MDA-MB-231 cells were encapsulated in synthetic PEG-based hydrogels formed by free radical chain growth polymerization and cultured over longer times. In a highly crosslinked hydrogel formulation absent integrin binding ligands, low cell viability was observed (< 50%) with the surviving cell population exhibiting dormancy, and inclusion of the integrin-binding ligand RGDS was observed to influence the switch between dormancy and growth.[33] Dormancy of TN MDA-MB-231 in response to high matrix density also has been observed previously in a harvested matrix of crosslinked gelatin.[55] Interestingly, in our studies, TN MDA-MB-231 were not observed to cease proliferation in well-defined bioinspired PEG-peptide synthetic matrices with physiologically relevant moduli and select integrin binding peptides derived from collagen or laminin, respectively. Cumulatively, these observations from the literature and our studies suggest the importance of both matrix density and composition in TN dormancy or proliferation, where the matrix densities we examined may have been insufficient for inducing dormancy in the presence of relevant bioactive ligands.
More importantly, our studies established the relevance of the well-defined bioinspired 3D culture system for the study of ER+ breast cancer cell dormancy over long times for investigations related to combatting late recurrence. Most studies of the dormancy of ER+ breast cancer cells to date have been within harvested or secreted matrices with limited control of matrix density and modulus. Here, with the independent property control afforded by a synthetic matrix, we uniquely observed the impact of matrix density on ER+ cells. For example, both luminal A T47D and luminal B BT474 breast cancer cells exhibited limited increases metabolic activity/growth in the higher density matrix inspired by lung tissue. However, over longer times, T47D cells were observed to exhibit dormancy even in the low matrix density, whereas BT474 cells exhibited continued proliferation and growth in the low density matrix. These observations of differential responses to ER+ cell types to differences in matrix composition highlight the opportunity that this new model system affords for future studies of ER+ breast cancer dormancy, including with other ER+ cell types, different matrix compositions, and the introduction of niche cells. Additionally, these studies point to future opportunities for understanding mechanistically how matrix density influences dormancy. For example, in seminal studies, Loebel, Burdick, and coworkers recently have demonstrated how the density and degradability of synthetic matrices influence the secretion and deposition of extracellular matrix proteins by mesenchymal stem cells and thereby regulate cell fate.[56] Applying such innovative approaches and analyses to examining the effects of matrix degradability and protein deposition within the well-defined 3D ER+ dormancy model established here could provide great insights into the underlying regulators of ER+ dormancy toward preventing late recurrence. Further, this dormancy model system provides future opportunities for studying dormant cells after re-activation, where dormant cells in 3D culture can be released by complete degradation of the synthetic matrix and subsequently integrated within other similar or more complex 3D microenvironments.
Conclusion
Late recurrences of breast cancer continue to be an outstanding challenge for patients with ER+ primary tumors, and their underlying causes remain difficult to study. Such late recurrences are hypothesized to originate from DTCs that re-activate after a long period of dormancy, owing in part to cell-matrix interactions. In the work presented here, we established a well-defined, bioinspired 3D culture model for probing the effects of key matrix properties, matrix density and biochemical content, for common sites of late recurrence on breast cancer cell dormancy. Based on matrix composition and cell type (luminal A ER+ T47D, luminal B ER+ HER2+ BT474), ER+ breast cancer cells were observed to persistently proliferate or form dormant micrometastases over several weeks, with leveling metabolic activity, decreased proliferation, up- and down-regulation of relevant dormancy genes, and recommenced proliferation upon a matrix remodeling event. The timing of entry into dormancy was dependent on matrix composition, where increased matrix density was identified to be particularly important for the induction of dormancy at early times. Under these same conditions, TN MDA-MB-231 cells associated with early recurrence were not observed to undergo long-term dormancy, although differential responses to matrix composition were observed. For dormant ER+ T47D cells within the 3D culture model, bioinformatic analyses revealed expression of a dormancy gene signature and significant differential expression of genes associated with different biological processes in different matrix compositions, as well as expression of autophagy markers suggesting a potential survival mechanism. In conclusion, we have developed a robust, well-defined, tunable model system for studying long-term ER+ breast cancer cell dormancy that can be further utilized in future studies to probe other breast cancer cell-ECM interactions, identify and study mechanisms of dormancy, and aid in the identification of therapeutics for prevention of late recurrence.
Materials and Methods
Synthesis and characterization of peptides
Peptides containing alloxycarbonyl (alloc)-protected lysines, K(alloc), providing a reactive alkene, were synthesized via solid phase peptide synthesis. The crosslinking enzymatically-degradable peptide Ac-KK(alloc)G[GPQG↓IWGQ]GK(alloc)K and pendant peptides K(alloc)G(POG)4-FOGERG-(POG)4-G (GFOGER) and K(alloc)(PEG2)2W(PEG2)IKVAV (IKVAV) were successfully synthesized on automated peptide synthesizers using standard FMOC-chemistry (Liberty Blue™ Automated Microwave Peptide Synthesizer; CEM, Matthews, NC, and PS3 Peptide Synthesizer; Protein Technologies, Inc, Tucson, AZ). The crosslinker and IKVAV peptides were synthesized on MBHA rink amide resin (Novabiochem) and a high-swelling Chemmatrix resin (Protein Technologies) was used for the GFOGER sequence. All amino acids were double coupled. Peptides were cleaved from resin for 4 hours in 95% triflouroacetic acid (Arcos Organics), 2.5% triisopropylsilane (Arcos Organics), and 2.5% water (all percentages v/v) supplemented with 25 mg/mL Phenol (Research Products International). After cleavage, all peptides were precipitated in cold diethyl ether (9x excess volume) overnight at 4 °C and purified by reverse-phase high performance liquid chromatography (HPLC; XBridge BEH C18 OBD 5 μm column; Waters, Milford, MA) with a linear water-acetonitrile (ACN) gradient (Water:ACN 95:5 to 45:5; 1.17% change in water per minute). Purified peptides were lyophilized, and their molecular weights were verified by mass spectrometry (Figure S15-17, Supporting Information).
Formation and rheological characterization of PEG hydrogels
Hydrogels were polymerized and mechanical and biochemical properties characterized using established methods.[31] 20 kDa 4-arm PEG-SH (JenKem Technology, Plano, TX) and crosslinking and pendant peptides (IKVAV or GFOGER) were dissolved in phosphate buffered saline (PBS, Invitrogen) supplemented with 1% penicillin streptomycin (PS, Invitrogen) and 0.5 μg/mL fungizone (FZ, Invitrogen). Hydrogel precursor solution, containing 6 or 10 percent PEG by weight (wt%), was prepared by mixing PEG with 2 mM pendant peptides and the balance in crosslinking peptide on stoichiometry (1:1 SH:alloc). Hydrogel compositions formed include 6 wt% PEG with 2 mM IKVAV (Low wt% IKVAV), 6 wt% PEG with 2 mM GFOGER (Low wt% GFOGER), and 10 wt% PEG with 2 w mM GFOGER (High wt% GFOGER). Hydrogel precursor solutions were pipetted into a sterile, cut syringe (1 mL with the tip removed). Collimated light at 365 nm and 10 mW/cm2 (Inpro Technologies collimating adaptor, Exfo Omnicure Series 2000) was applied for 1 minute (min). After polymerization, hydrogels were removed from syringe tips and placed in appropriate buffer (PBS or growth medium) for analysis.
Mechanical properties of the hydrogels were measured by rheometry (AR-G2; TA Instruments, New Castle, DE). Modulus measurements were performed on each hydrogel composition polymerized (10 mW/cm2 at 365 nm) for 1 min and equilibrium swollen in PBS overnight. Swollen hydrogels were placed between parallel plates (20 mm geometry and Peltier Plate) on the rheometer and compressed to 0.01 N normal force to prevent slip. Strain- and frequency-sweeps were performed to determine the linear viscoelastic (LVE) regime, and 2% strain and 2 rad/s frequency within the LVE regime were selected to perform measurements of modulus for equilibrium swollen hydrogels (n = 4). Young’s modulus (E) was estimated by Rubber Elasticity Theory, E = 2G(1 + ν), where ν is Poisson’s ratio and is taken to be 0.5 for these incompressible PEG hydrogels, and G is the shear modulus from the rheometry measurements. Additionally, swelling was measured and crosslink density (ρx) was calculated for equilibrium swollen hydrogels as previously described[40] using rubber elasticity theory, where G = ρxRTQ−1/3.
Breast cancer cell culture
T47D, MDA-MB-231, and BT474 breast cancer cells (passages 16-24) were cultured on tissue culture polystyrene in Dulbecco’s Modified Eagle Medium (DMEM, Corning Cellgro) supplemented with 10 % v/v fetal bovine serum (FBS, Invitrogen) and 1% PS and 0.5 μg/mL FZ. Growth medium was replaced every 48-72 hours during culture. At 80% confluency, cells were removed from plates (trypsin/EDTA, 5 min, Corning Cellgro), counted via hemocytometer, and aliquoted for the desired number of cells to be passaged or encapsulated for experiments.
Cell encapsulation
T47D, MDA-MB-231, or BT474 cells were encapsulated in different hydrogel compositions (T47D, MDA-MB-231: Low wt% IKVAV, Low wt% GFOGER, High wt% GFOGER; BT474: Low wt% GFOGER, High wt% GFOGER). Cells were encapsulated in 5 μL layer on top of a 15 μL cell-free hydrogel layer for ease of handling and allow the observation of homogenous cell responses as previously.[31] Briefly, a 15 μL gel-prep solution was pipetted into a 1 mL syringe and irradiated with light (365 nm, 10 mW/cm2) for 1 min. After trypsinization of cells, aliquots of cells were centrifuged (1200 rpm, 3 min) and re-suspended in hydrogel precursor solution to form the top cell layer. Specifically, a 5 μL gel prep solution with cells was pipetted on-top and irradiated with light (365 nm, 10 mW/cm2) for 1 min. Cells were encapsulated at a density of 5000 cells/μL in the top 5-μL layer (25000 cells/gel). Post-polymerization, hydrogels were placed in a sterile, untreated 48-well plate and rinsed with fresh culture medium. Culture medium was replaced every 48-72 hours, and cells were cultured in all three gel compositions for 40 days.
Viability assay
Viability of T47D, MDA-MB-231, or BT474 cells encapsulated in different hydrogel compositions was assessed by a LIVE/DEAD Viability Kit (ThermoFisher Scientific) on days (D) 3, 10, 20, 30, and 40. Hydrogels (n = 3) were rinsed of culture medium (2x 5 min, PBS), stained (45 min, 2 μM calcein AM and 4 μM ethidium homodimer-1, 37 °C and 5% CO2). After staining, hydrogels were washed (2x 5 min, PBS) before imaging. Hydrogels were transferred to a chamber slide (Nunc Lab-Tek™ II Chamber Slide, Glass, 1 well) and imaged with a confocal microscope (Zeiss LSM 800, 10x objective and image frame size of 1024 x 1024, 250 μm z-stack, 3 images per hydrogel sample). Orthogonal projections were made of each z-stack, and live (green) and dead (red) cells were counted using Volocity software. The percentage of viable cells was calculated by the number of green cells/total number of cells x 100%.
Metabolic activity
AlamarBlue cell viability reagent (Thermo Fisher) was used to measure metabolic activity for cells encapsulated in different hydrogel compositions at days 3, 10, 20, 30 and 40. Alamar Blue was diluted 1:10 to Phenol red-free growth medium, and hydrogels were incubated (37 °C and 5% CO2) with solution for 4 hours (500 μL per hydrogel). Conditioned culture media was collected from each well, and hydrogels were replenished with fresh standard culture medium. Conditioned media (100 μL) were transferred to a black 96-well plate, and fluorescence was measured (BioTek Synergy H4 Hybrid Reader, ex/em ~ 560 nm/590 nm).
Proliferation assay
Click-it 5-ethynyl-2’-deoxyuridine (EdU) (ThermoFisher), a nucleoside analog of thymidine, was used for measuring proliferation of cells in hydrogel cultures at time points D3, D10, D20, D30, and D40. The manufacturer’s protocol was followed. At each timepoint, 10 μM EdU was added to cell culture media, and samples were incubated (37 °C and 5% CO2) for 4 hours (500 μL per hydrogel). After the 4 hour EdU pulse, media was removed, and samples were fixed with 4% paraformaldehyde (PFA) for 15 min. Afterwards, hydrogels were rinsed with 1% BSA in PBS and permeabilized with Saponin-based permeabilization solution (300 μL per hydrogel) for 30 min at room temperature. A click-it reaction cocktail was prepared, following the manufacturer’s protocol, with an AF647 azide fluorophore for the copper-catalyzed click reaction. 200 μL of the reaction cocktail was incubated on samples for 30 min in the dark. Afterwards, hydrogels were rinsed 2x 1% BSA in PBS. F-actin and nuclei (phalloidin-TRITC and DAPI) were stained in all samples, and confocal z-stack images were taken, as described in the immunostaining section. For ease of viewing within the resulting confocal z-stack projections, the F-actin channel was represented as green, nuclei blue, and EdU pink. The percentage of EdU+ cells was determined by counting the number of EdU cells (AF647) / total number of cells (DAPI stained) x 100% and averaged across replicates.
RNA isolation and RNA-seq pipeline analysis
RNA was isolated for T47D or MDA-MB-231 breast cancer cells cultured for 3 and 40 days in the three matrix compositions: Low wt% IKVAV, Low wt% GFOGER, High wt% GFOGER. RNA was isolated in triplicate for each sample type (breast cancer cell line in specific matrix composition at desired time point). At each time point, hydrogel samples for each condition were transferred to microcentrifuge tubes and degraded with collagenase (250 U/mL) for 20 minutes at 37 °C and 5% CO2. Once hydrogels were completely degraded (e.g., liquid solution that could be pipetted), cells were centrifuged (1200 RPM, 3 min), rinsed to remove residual polymer and collagenase (500 μL, PBS), centrifuged, and then lysed with buffer for RNA isolation. Three degraded hydrogels were combined for each RNA isolation replicate on day 3, and two degraded hydrogels were combined for each RNA isolation replicate on day 40 to obtain sufficient concentrations of RNA for downstream analyses. Total RNA was extracted using the Nucleospin miRNA kit (Takara). RNA concentrations were quantified using a Qubit Fluorometer (Invitrogen), and RNA integrity was assessed using the AATI Fragment Analyzer (Advanced Analytical Technologies, Inc.). Samples with RQN (RNA quality number) ≥ 6 were used for this study.
Extracted RNA (3 replicates per condition) were sequenced by the University of Delaware Sequencing and Genotyping Center at the Delaware Biotechnology Institute. Briefly, indexed libraries were constructed from 100-500 ng of total RNA using the Universal Plus mRNA-seq Workflow Kit (Nugen) following the manufacturer’s instructions. The molar quantity and quality of the libraries also were assessed by Qubit and AATI Fragment Analyzer, respectively. The average library size was 400 bp. Sequencing was performed on an Illumina HiSeq 2500 platform with 51-bp single-end reads to obtain ca. 25 million reads/sample.
Raw sequence data was analyzed by the Center for Bioinformatics and Computational Biology Core Facility at the University of Delaware using an existing RNA-Seq analysis pipeline (adapted from Kalari et al., 2014).[57] Briefly, initial quality control was performed using fastqc; reads were aligned to the human reference genome (Hg19) using Tophat2;[58] mapping metrics were assessed using RseQC;[59] and gene/exon feature counts were calculated using HTseq.[60] Raw sequence data and gene feature counts have been submitted to the NCBI under Gene Expression Omnibus (GEO) studies GSE145696 and GSE121179.
Dormancy score calculations
A dormancy score for T47D or MDA-MB-231 breast cancer cells in 3D cultures were calculated at days 3 and 40 using CPM (count per million) values. The dormancy score equation was adapted from Kim et al. and is based on 22 up-regulated and 27 down-regulated dormancy-associated genes.[42] Here, CPM values were used as gene expression values within the published R code, which takes the ratio of each up-regulated or down-regulated dormancy gene, respectively, to the average of that gene across all samples in the dataset and subsequently uses these ratios in the calculation of a ‘dormancy score’.
A heatmap of the CPM for the different matrix compositions at day 3 and day 40 for each cell line (T47D and MDA-MB-231) was generated with the R function heatmap.2 from the gplots package version 3.0.1.[61] CPM were averaged among the three replicates for each composition. Genes that did not appear in the RNA-seq data are marked as black. Each row of the heatmap was scaled to a mean of 0 and standard deviation of 1 (z-score) (Figure S8, Supporting Information).
Differential expression analysis
Differential expression analysis was performed for each cell line comparing day 3 to day 40 in all three hydrogel matrix compositions using EdgeR.[62] Differential expression p-values were corrected using the false discovery rate (FDR) method of Benjamini and Hochberg.[63] Genes with FDR-adjusted p values < 0.05 and fold change > 2 or < 0.5 (log2FC > 1 or log2FC < - 1) were considered significantly differentially expressed. Venn diagrams were generated to illustrate the number of genes differentially expressed for the two cell lines and their overlap (Figure 5A and Figure S11A, Supporting Information). Heat maps of the top 50 differentially expressed genes were generated as above for T47D and MDA-MB-231 cells in the three culture conditions, and genes that did not appear in the RNA-seq data were removed (Figure S9, 11, Supporting Information). Additionally, a heatmap of the differentially expressed autophagy genes (Human Autophagy Database) was generated as above for T47D cells in all 3 matrix compositions at day 3 and day 40 in culture (Figure 5C).[61]
Enrichment analysis
DAVID (version 6.8 via the R package, RDAVIDWebService (version 1.16.0))[64] was used to perform GO enrichment analysis for differentially expressed genes. Enrichment was tested for the GO Biological Process terms in GOTERM_BP_FAT. Terms with Benjamini-Hochberg corrected p-values < 0.05 were considered significantly enriched. The top 20 enriched terms were categorized into biological processes (Figure 5B and Figure S10, 12, Supporting Information).
RT-qPCR
iTaq Universal One-Step RT-qPCR kit (Bio-Rad; Hercules, CA) was used for RT-qPCR analysis of ATG9B and LC3B autophagy genes in D3 and D40 RNA samples. GAPDH was the measured housekeeping gene to calculate ∆Ct’s at each timepoint and sample. A total reaction mix volume of 15 μL was run for each sample on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad; Hercules, CA) to measure Ct values. These reactions included 300 nM forward and reverse primers (primers listed in Table S7), 0.188 μL iScript reverse transcriptase, 7.5 μL iTaq universal SYBR Green reaction mix (2x), 1 μL of RNA (~50 ng), and nuclease-free H2O for the remaining volume. ∆Ct values were calculated by subtracting GAPDH from the gene of interest (ATG9B or LC3B) Ct values for each condition at the early and late time point, in triplicate. ∆∆Ct’s were calculated as the difference between ∆Ct at D40 and D3 for ATG9B and LC3B. Averages and standard errors were calculated for each composition (n = 3) and plotted (log2 scale).
Immunostaining
Immunostaining for autophagy-related proteins ATG9B and LC3B and proliferation marker Ki-67 was performed on 3D hydrogel compositions at D3 and D40. Hydrogels were fixed with cold 4% PFA for 15 min at room temperature. Hydrogels were rinsed (2x PBS) and then blocked and permeabilized solutions were prepared, BPSoln1 (3% w/v bovine serum albumin/BSA + 0.05% v/v Triton-X in PBS) and BPSoln2 (5% BSA w/v + 0.1% v/v Triton-X in PBS). After fixation, hydrogels were washed (1 x 5 min PBS, 2 x 5 min BPSoln1), and then permeabilized and blocked (1 x 60 min, BPSoln2). After blocking, cells were incubated overnight at 4 °C with primary antibody (1:500 dilution of rabbit anti-human LC3B, Novus Biologicals, 1:150 dilution of rabbit anti-human ATG9B, Novus Biologicals, or 1:100 dilution of mouse anti-human Ki-67, Abcam) in BPSoln2. The next day, hydrogels were rinsed (3 x 60 min, BPSoln1) and incubated with secondary antibody Alexa Fluor 488 goat-anti-mouse/rabbit (ThermoFisher, 1:300 dilution) and phalloidin-TRITC to stain F-actin (Sigma Aldrich, 1:200 dilution) in BPSoln2 overnight at 4 °C, protected from light. On the final day of immunostaining, hydrogels were rinsed (3 x 45 min, BPSoln1) and incubated with DAPI (700 nM in PBS) for 1 hour to stain nuclei. Hydrogels were rinsed (3 x 30 min, PBS) and stored at 4 °C in PBS, protected from light, until imaging. Samples were imaged on a confocal microscope (Zeiss LSM 800). Three z-stacks (100 images per stack, 2 μm spacing) were taken per hydrogel (n = 3), for a total of 9 images. The quantification analysis of Ki-67+ cells, cell numbers, and intensity of ATG9B protein per cells were performed using open source Fiji ImageJ.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8. Data are reported as means ± standard error (SE) with replication noted with each specific assay. Statistical significance was evaluated using one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test for multiple comparisons or Student’s two-sided t-test for pair-wise comparisons as noted. Significance is denoted with * p< 0.05, ** p≤ 0.01, and *** p≤ 0.001.
Supplementary Material
Acknowledgements
This work was supported by a grant from the Susan G. Komen Foundation (CCR16377327) made possible by funding from American Airlines and an Institutional Development Award (IDeA) for a Center of Biomedical Research Excellence (COBRE) from National Institute of General Medical Sciences within the National Institutes of Health (P20GM104316). The authors would like to thank Dr. Jaysheel Bhavsar at the University of Delaware Bioinformatics Core Facility for assistance with RNA-Seq analysis, Dr. Karol Miaskiewicz at the Delaware Biotechnology Institute (DBI) for computational assistance, and Dr. Brewster Kingham and Ms. Summer Thompson in the Sequencing and Genotyping Core at DBI for assistance with sequencing runs. Use of the BIOMIX computational cluster by this project was made possible through funding from Delaware INBRE (NIGMS P20 GM103446) and the Delaware Biotechnology Institute.
Footnotes
Dedicated to the late Kimberly Newman-McCown, Breast Cancer Advocate and early member of our breast cancer research team
References
- [1].Zhang XHF, Giuliano M, Trivedi MV, Schiff R, Kent Osborne C, Clin. Cancer Res 2013, 19, 6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chambers AF, Groom AC, MacDonald IC, Nat. Rev. Cancer 2002, 2, 563. [DOI] [PubMed] [Google Scholar]
- [3].Aguirre-Ghiso JA, Nat. Rev. Cancer 2007, 7, 834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Brewster AM, Hortobagyi GN, Broglio KR, Kau SW, Santa-Maria CA, Arun B, Buzdar AU, Booser DJ, Valero V, Bondy M, Esteva FJ, J. Natl. Cancer Inst 2008, 100, 1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pan H, Gray R, Braybrooke J, Davies C, Taylor C, McGale P, Peto R, Pritchard KI, Bergh J, Dowsett M, Hayes DF, N. Engl. J. Med 2017, 377, 1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Davies C, Pan H, Godwin J, Gray R, Arriagada R, Raina V, Abraham M, Medeiros Alencar VH, Badran A, Bonfill X, Bradbury J, Clarke M, Collins R, Davis SR, Delmestri A, Forbes JF, Haddad P, Hou MF, Inbar M, Khaled H, Kielanowska J, Kwan WH, Mathew BS, Mittra I, Müller B, Nicolucci A, Peralta O, Pernas F, Petruzelka L, Pienkowski T, Radhika R, Rajan B, Rubach MT, Tort S, Urrútia G, Valentini M, Wang Y, Peto R, Lancet 2013, 381, 805.23219286 [Google Scholar]
- [7].Bragado P, Sosa MS, Keely P, Condeelis J, Aguirre-Ghiso JA, Recent Results Cancer Res. 2012, 195, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ghajar CM, Nat. Publ. Gr 2015, 15, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sosa MS, Bragado P, Aguirre-Ghiso JA, Nat. Rev. Cancer 2014, 14, 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rao SS, Kondapaneni RV, Narkhede AA, J. Biol. Eng 2019, 13, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Endo H, Inoue M, Cancer Sci. 2019, 110, 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hen O, Barkan D, Semin. Cancer Biol 2019. [DOI] [PubMed] [Google Scholar]
- [13].Barrios J, Wieder R, Cancer Microenviron. 2009, 2, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L, Mol. Biol. Cell 2001, 12, 863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Barkan D, El Touny LH, Michalowski AM, Smith JA, Chu I, Davis AS, Webster JD, Hoover S, Simpson RM, Gauldie J, Green JE, Cancer Res. 2010, 70, 5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, Almeida D, Koller A, Hajjar KA, Stainier DYR, Chen EI, Lyden D, Bissell MJ, Nat. Cell Biol 2013, 15, 807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Carlson P, Dasgupta A, Grzelak CA, Kim J, Barrett A, Coleman IM, Shor RE, Goddard ET, Dai J, Schweitzer EM, Lim AR, Crist SB, Cheresh DA, Nelson PS, Hansen KC, Ghajar CM, Nat. Cell Biol 2019, 21, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Booth AJ, Hadley R, Cornett AM, Dreffs AA, Matthes SA, Tsui JL, Weiss K, Horowitz JC, Fiore VF, Barker TH, Moore BB, Martinez FJ, Niklason LE, White ES, Am. J. Respir. Crit. Care Med 2012, 186, 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jansen LE, Birch NP, Schiffman JD, Crosby AJ, Peyton SR, J. Mech. Behav. Biomed. Mater 2015, 50, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tibbitt MW, Anseth KS, Biotechnol. Bioeng 2009, 103, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lutolf MP, Raeber GP, Zisch AH, Tirelli N, Hubbell JA, Adv. Mater 2003, 15, 888. [Google Scholar]
- [22].Kundu S, Reis R, Eds. , Biomaterials for 3D Tumor Modeling, Elsevier, 2020. [Google Scholar]
- [23].Caliari SR, Burdick JA, Nat. Methods 2016, 13, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Murphy NP, Lampe KJ, J. Mater. Chem. B 2015, 3, 7867. [DOI] [PubMed] [Google Scholar]
- [25].Madl CM, Heilshorn SC, Adv. Funct. Mater 2018, 28, 1706046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Holt SE, Ward ES, Ober RJ, Alge DL, MRS Commun. 2017, 7, 427. [Google Scholar]
- [27].Bray LJ, Werner C, ACS Biomater. Sci. Eng 2018, 4, 337. [DOI] [PubMed] [Google Scholar]
- [28].Weigelt B, Ghajar CM, Bissell MJ, Adv. Drug Deliv. Rev 2014, 69, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Taubenberger AV, Bray LJ, Haller B, Shaposhnykov A, Binner M, Freudenberg U, Guck J, Werner C, Acta Biomater. 2016, 36, 73. [DOI] [PubMed] [Google Scholar]
- [30].Preciado JA, Reátegui E, Azarin SM, Lou E, Aksan A, Technology 2017, 05, 129. [Google Scholar]
- [31].Sawicki LA, Ovadia EM, Pradhan L, Cowart JE, Ross KE, Wu CH, Kloxin AM, APL Bioeng. 2019, 3, 016101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Guiro K, Patel SA, Greco SJ, Rameshwar P, Arinzeh TL, PLoS One 2015, 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pradhan S, Slater JH, Biomaterials 2019, 215, 2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Miller RT, Matrix Biol. 2017, 57, 366. [DOI] [PubMed] [Google Scholar]
- [35].Smithmyer ME, Sawicki L. a., Kloxin AM, Biomater. Sci 2014, 2, 634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yamazaki CM, Kadoya Y, Hozumi K, Okano-Kosugi H, Asada S, Kitagawa K, Nomizu M, Koide T, Biomaterials 2010, 31, 1925. [DOI] [PubMed] [Google Scholar]
- [37].Yamada Y, Hozumi K, Nomizu M, Chem. - A Eur. J 2011, 17, 10500. [DOI] [PubMed] [Google Scholar]
- [38].Sawicki LA, Kloxin AM, Biomater. Sci 2014, 2, 1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sawicki LA, Kloxin AM, J. Vis. Exp 2016, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Rehmann MS, Skeens KM, Kharkar PM, Ford EM, Maverakis E, Lee KH, Kloxin AM, Biomacromolecules 2017, 18, 3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kenny PA, Lee GY, Myers CA, Neve RM, Semeiks JR, Spellman PT, Lorenz K, Lee EH, Barcellos-Hoff MH, Petersen OW, Gray JW, Bissell MJ, Mol. Oncol 2007, 1, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kim RS, Avivar-Valderas A, Estrada Y, Bragado P, Sosa MS, Aguirre-Ghiso JA, Segall JE, PLoS One 2012, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Butturini E, Carcereri de Prati A, Boriero D, Mariotto S, Int. J. Mol. Sci 2019, 20, 4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Akkiprik M, Feng Y, Wang H, Chen K, Hu L, Sahin A, Krishnamurthy S, Ozer A, Hao X, Zhang W, Breast Cancer Res. 2008, 10, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mizushima N, Levine B, Cuervo AM, Klionsky DJ, Nature 2008, 451, 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Levine B, Kroemer G, Cell 2008, 132, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Moussay E, Kaoma T, Baginska J, Muller A, Van Moer K, Nicot N, Nazarov PV, Vallar L, Chouaib S, Berchem G, Janji B, Autophagy 2011, 7, 760. [DOI] [PubMed] [Google Scholar]
- [48].Yang ZJ, Chee CE, Huang S, Sinicrope FA, Mol. Cancer Ther 2011, 10, 1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tandon M, Othman AH, Ashok V, Stein GS, Pratap J, J. Cell. Physiol 2018, 233, 559. [DOI] [PubMed] [Google Scholar]
- [50].Vera-Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE, Nat. Commun 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chaterjee M, van Golen KL, Bone Marrow Res. 2011, 2011, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Patel LR, Camacho DF, Shiozawa Y, Pienta KJ, Taichman RS, Futur. Oncol 2011, 7, 1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, Ichikawa R, Kinjo M, Ohsumi Y, J. Cell Biol 2012, 198, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yu L, Chen Y, Tooze SA, Autophagy 2018, 14, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Fang JY, Tan SJ, Wu YC, Yang Z, Hoang BX, Han B, J. Transl. Med 2016, 14, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Loebel C, Mauck RL, Burdick JA, Nat. Mater 2019, 18, 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kalari KR, Nair AA, Bhavsar JD, O’Brien DR, Davila JI, Bockol MA, Nie J, Tang X, Baheti S, Doughty JB, BMC Bioinformatics 2014, 15, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL, Genome Biol. 2013, 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wang L, Wang S, Li W, Bioinformatics 2012, 28, 2184. [DOI] [PubMed] [Google Scholar]
- [60].Anders S, Pyl PT, Huber W, Bioinformatics 2015, 31, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Warnes MGR, Bolker B, Bonebakker L, Gentleman R, Var. R Program. Tools Plotting Data 2016, 3. [Google Scholar]
- [62].Robinson MD, McCarthy DJ, Smyth GK, Bioinformatics 2010, 26, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Url S, R. S. Society, R. S. Society, 2007, 57, 289. [Google Scholar]
- [64].Fresno C, Fernández EA, Bioinformatics 2013, 29, 2810. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.