

Abstract
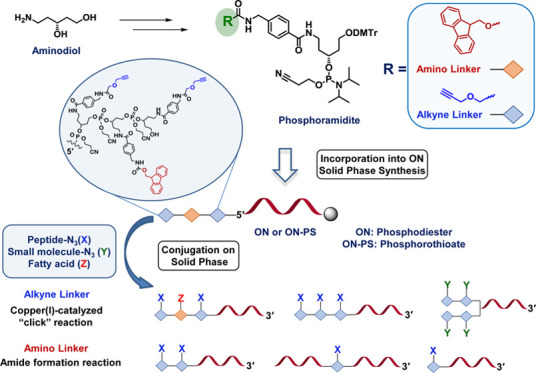
Oligonucleotide (ON) conjugates are increasingly important tools for various molecular diagnostics, nanotechnological applications, and for the development of nucleic acid-based therapies. Multiple labeling of ONs can further equip ON-conjugates and provide improved or additional tailored properties. Typically, the preparation of ON multiconjugates involves additional synthetic steps and/or manipulations in post-ON assembly. This report describes the simplified methodology allowing for multiple labeling of ONs on a solid support and is compatible with phosphodiester as well as phosphorothioate (PS) ONs. The current approach utilizes two novel alkyne- and amino-functionalized linker phosphoramidites that can be readily synthesized from a common aminodiol intermediate in three steps. The combination of new linkers provides orthogonal functionalities, which allow for multiple attachments of similar or varied moieties. The linkers are incorporated into ONs during automated solid-phase ON synthesis, and the conjugation with functional entities is achieved by either amide bond formation or by copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC). The versatility of the approach is demonstrated by the synthesis of 5′-site ON multiconjugates with small molecules, peptides, and fatty acids as well as in the preparation of an internal peptide–ON conjugate.
Introduction
Synthetic oligonucleotides (ONs) are widely used for various applications in life sciences, biotechnology, diagnostics, and represent a major part of currently used nucleic acid therapeutics.1,2 The conjugation of ONs with various ligands, reporter groups, peptides, or proteins can provide the ON conjugate with new valuable properties for various nanotechnological applications and pharmaceutical development.3 Consequently, ON conjugates are emerging as an important subgroup of this class of macromolecules and are considered promising candidates for clinical use.3−5 Despite many ongoing clinical trials and several recent FDA approvals,6 the ON delivery to the site of action remains one of the main challenges of ON-based therapeutics.7−9 However, ON cellular uptake and the ability to target specific tissues can be significantly enhanced by the conjugation of uptake promoting molecules, such as N-acetylgalactosamine (GalNAc) for targeting the liver hepatocytes,10−14 or glucagon-like peptide-1 (GLP-1) for the delivery to pancreatic β-cells.15 In addition, ON-fatty acid conjugates showed enhanced potency of antisense ONs (AONs) in muscles16 and high potential for the treatment of myelofibrosis.17−19 A number of methods for the conjugation of specific molecules to ONs have been developed, including in-line conjugation20 and post-conjugation in solution3 or on the solid phase.21,22 Among various chemistries developed for the preparation of ON conjugates,3,23,24 the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC), which is a variant of Huisgen azide–alkyne 1,3-dipolar cycloaddition25,26 and known as “click” reaction, has become an efficient method for conjugation of small molecules,27,28 fluorophores,29 peptides,21,30 and carbohydrates31,32 to ONs. Multiple labeling of ONs13,33 and incorporation of several different moieties into ONs, to combine the effect of various entities,22,34−36 can improve and provide additional tailored properties of ON conjugates. Several approaches have been established for the preparation of ON multiconjugates using copper(I)-catalyzed “click” chemistry, including preparation of DNA constructs with different fluorescent labels by sequential conjugation/deprotection reactions,37 in combination with thiol-Michael-type addition,38 using a “click/reverse-click” procedure,39 or by solid-phase CuAAC approach for the preparation of ON-GalNAc dendrimer conjugates.27 Recently, we have reported an efficient method for the synthesis of multiple functionalized ONs in a stepwise procedure that utilizes “click cycles” using CuAAC,22 and a modified method allowing for efficient conjugation of peptides to phosphorothioate (PS) ONs.40 Both approaches involve ON conjugation on the solid support utilizing a H-phosphonate-based alkyne linker. Although the above methodologies are efficient for the preparation of ON conjugates, they involve several additional derivatization steps of the ON prior conjugation reaction. Herein, we report the development of novel alkyne- and amino-functionalized linkers derived from the same scaffold and compatible with automated ON synthesis using phosphoramidite chemistry. New linkers are directly incorporated during automated solid-phase ON synthesis and allow for multiple attachments of moieties, such as small molecules and/or peptides, to phosphodiester as well as PS ONs by CuAAC on a solid phase (Figure 1). In addition, conjugation of two different entities to the ON is enabled by the orthogonal functionality of alkyne and amino linkers.
Figure 1.

Schematic representation of synthesized ON conjugates, structures of alkyne and amino linker phosphoramidites, and azido-modified moieties of benzylguanine (BG-N3), P4 peptide (P4-N3), MIF peptide (Ac-MIF-N3), and palmitic acid (PA).
Results and Discussion
In this study, we aimed to develop a methodology that enables direct incorporation of linker units into ONs during automated solid-phase ON synthesis and further mono- and multiple conjugation of the ON with moieties of interest on a solid support without postassembly derivatization of ON. The first novel phosphoramidite-based linker carries a sufficiently active triple bond for efficient copper(I)-catalyzed “click” reaction on a solid support. To allow conjugation of the ON with different entities, the second new amine-containing linker (Figure 1) is employed, thus enabling orthogonal chemistry based on amide bond formation. The alkyne linker phosphoramidite was developed with two variants of 5′-OH protecting group, the 4-methoxytrityl (MMTr) and 4,4′-dimethoxytrityl (DMTr) (Scheme 1). Initially, the MMTr derivative 7, having a more stable protecting group, was synthesized (Method A) for the postsynthetic attachment to solid-supported ON (that can also be purchased, if a synthesizer is not available) and further conjugation procedures. In Method B, the synthesis route was improved to fewer steps and the standard DMTr 5′-OH protecting group was introduced for the automated in-line ON synthesis to give alkyne linker 11.
Scheme 1. Synthesis of 4-Monomethoxytrityl- and 4,4′-Dimethoxytrityl-Protected Alkyne Linker Phosphoramidites 7 and 11.

The synthesis of MMTr-protected alkyne linker 7 (Scheme 1, Method A) began with the conversion of the low cost and commercially readily available ethyl (3R)-4-cyano-3-hydroxybutanoate 1 into aminodiol 2 by reduction with lithium aluminum hydride (Supporting Information). The aminodiol 2 was further converted into trifluoroacetyl (TFA) derivative 3 for purification purposes. Regioselective protection of the primary hydroxyl group in intermediate 3 using 1.1 equiv of 4-methoxytrityl chloride in pyridine afforded compound 4 in 54% yield. Deprotection of the TFA group was performed in aqueous ammonia–ethanol (2:1, v/v) mixture at room temperature for 36 h. The crude compound 5 was then subjected to the reaction with the preactivated triple bond donor 4-((2-(prop-2-yn-1-yloxy)acetamido)methyl) benzoic acid (8, PAMBA)22 to afford 6 in high yield, which was converted into phosphoramidite derivative 7 in 72% yield.
The synthesis methodology was further modified for the preparation of DMTr-protected alkyne linker 11 (Scheme 1, Method B). The synthesis of phosphoramidite 11 was started by direct condensation reaction between the preactivated PAMBA (8) and crude aminodiol 2 using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 1-hydroxybenzotriazole (HOBt) as condensing agents to obtain 9 in 49% yield. The primary hydroxyl group in diol derivative 9 was then protected with a DMTr group to obtain compound 10, which was then converted into phosphoramidite 11 in 81% yield. Even though the coupling of PAMBA (8) with crude aminodiol 2 was less efficient compared to the coupling with protected intermediate 5 described in Method A, the overall yields of the final phosphoramidites 7 and 11 were comparable. Method B also reduced the number of synthetic steps making the method more amenable to an increase in synthesis scale.
To enable orthogonal conjugation of entities to ONs, a fluorenylmethoxycarbonyl (Fmoc)-protected amino linker (Figure 1), which allows conjugation through amide bond formation, was developed based on the same aminodiol scaffold (2) as the alkyne-type linker. The synthesis was started with the protection of the amino group of p-(aminomethyl)benzoic acid 12 with the Fmoc-protecting group (Scheme 2, Supporting Information). Fmoc-protected derivative 13 was then condensed with aminodiol 2 using EDC/HOBt as a condensing agent in N-methylpyrrolidone (NMP). The primary hydroxyl group of crude 14 was protected with a DMTr group using DMTrCl in pyridine, and 4,4′-dimethoxytritylated compound 15 was then converted into phosphoramidite 16 in 62% yield.
Scheme 2. Synthesis of Fmoc-Protected Amino Linker Phosphoramidite 16.

As the first example of an ON construct utilizing the new alkyne linker, a branched DNA multiconjugate carrying four units of benzylguanine (BG) was synthesized (ON1-(BG)4 in Figure 1 and Table 1). BG-labeled DNA can be used as a tool in SNAP-display technology for the selection of proteins.41,42 The synthesis of the ON1-(BG)4 conjugate was started from postsynthetic incorporation of a commercial doubler modifier at the 5′-end of the solid-supported DNA followed by sequential attachment of four units of alkyne linker 7 by two cycles on the automated DNA synthesizer (Figure 2). The reversed-phase HPLC (RP-HPLC) analysis of alkyne-modified DNA intermediate (ON1 in Figure 2) showed a rather moderate coupling efficiency of the doubler modifier as evidenced from the presence of free DNA (Figure S25 in the Supporting Information). Nevertheless, addition of the doubler resulted in a sufficient conversion to demonstrate the use of the new linker. Apart from the major 5′-tetra-linker-modified ON1, only minor amounts of bis- and tris-alkyne-modified variants were detected. The solid-supported DNA conjugate with four BG units was then prepared by simultaneous regioselective conjugation using azido-functionalized BG (BG-N3, Supporting Information) and alkyne-modified DNA (ON1) utilizing the CuAAC reaction with copper(I) iodide. The “click” reaction was performed directly on the solid support at ambient temperature, minimizing purification steps and achieving efficient removal of the residual copper by an ethylenediaminetetraacetic acid (EDTA) wash procedure. The final ON1-(BG)4 construct obtained after deprotection and cleavage from the solid support was analyzed by RP-HPLC (Figures 2 and S26 in the Supporting Information) and its identity was confirmed by mass spectrometry.
Table 1. Oligonucleotides and ON Conjugates Used in the Study.
| ES-TOF
mass m/zc |
|||
|---|---|---|---|
| ONa | sequence 5′ → 3′b | calcd | found |
| ON1 | (Lalkyne)2-(Lalkyne)2-doubler-GCGTTGATGCAATTTCTATGC | 2104.23d | 2104.43d |
| ON2 | Lalkyne-G*G*C*C*A*A*A*C*C*U*C*G*G*C*U*U*A*C*C*U | 1827.49d | 1827.81d |
| ON3 | Lalkyne-Lalkyne-G*G*C*C*A*A*A*C*C*U*C*G*G*C*U*U*A*C*C*U | 1934.11d | 1933.96d |
| ON4 | Lalkyne-Lalkyne-Lalkyne-G*G*C*C*A*A*A*C*C*U*C*G*G*C*U*U*A*C*C*U | 1632.37e | 1632.21e |
| ON5 | TCAAGGAAG-Lalkyne-ATGGCATTTCT | 1640.59d | 1640.77d |
| ON6 | Lalkyne-LNHFmoc-Lalkyne-TCAAGGAAGATGGCATTTCT | 1821.75d,f | 1821.88d,f |
| ON1-(BG)4 | (BG-L)2-(BG-L)2-doubler-GCGTTGATGCAATTTCTATGC | 2036.34e | 2036.54e |
| ON2-P4g | (P4-L)-G*G*C*C*A*A*A*C*C*U*C*G*G*C*U*U*A*C*C*U | 2060.25d | 2060.40d |
| ON3-(P4)2g | (P4-L)-(P4-L)-G*G*C*C*A*A*A*C*C*U*C*G*G*C*U*U*A*C*C*U | 1919.49e | 1919.63e |
| ON4-(P4)3g | (P4-L)-(P4-L)-(P4-L)-G*G*C*C*A*A*A*C*C*U*C*G*G*C*U*U*A*C*C*U | 2190.90e | 2191.19e |
| ON5-P4 | TCAAGGAAG-(P4-L)-ATGGCATTTCT | 1873.34d | 1873.82d |
| ON6-(PA) | Lalkyne-(PA-L)-Lalkyne-TCAAGGAAGATGGCATTTCT | 1881.35d | 1881.31d |
| ON6-(MIF)-(PA)-(MIF)h | (MIF-L)-(PA-L)-(MIF-L)-TCAAGGAAGATGGCATTTCT | 2079.08d | 2079.50d |
Cleaved from the solid support and deprotected ONs. See Table S1 for the yield of ON conjugation and the total yield of ON conjugates.
In all sequences, A = 2′-deoxyadenosine, G = 2′-deoxyguanosine, C = 2′-deoxycytidine, T = thymidine, A = 2′-OMe-adenosine, G = 2′-OMe-guanosine, C = 2′-OMe-cytidine, U = 2′-OMe-uridine, * = PS linkage. L = linker, BG = benzylguanine, P4 = LGAQSNF, MIF = (Ac)PLG (N → C), PA = palmitic acid.
Negative mode.
[M]−4.
[M]−5.
ES-TOF mass after Fmoc removal.
Figure 2.

Synthesis of tetra-benzylguanine-labeled ON1-(BG)4.
Thereafter, we investigated the efficiency of the new alkyne linker for the preparation of PS-ON peptide multiconjugates. The model 2′-O-methyloligoribonucleotide phosphorothioate (2′-OMe-PS ON) targets exon 23 of the dystrophin pre-mRNA in mdx mice. Mono-conjugation of the muscle homing P4 peptide (LGAQSNF) derivative to the corresponding ON results in increased exon-skipping levels in the diaphragm and the cardiac muscle tissue of mdx mice.43 To explore the potency of the developed alkyne linker, we performed the synthesis of 2′-OMe-PS ON conjugates with one, two, and three P4 peptide units attached to the 5′-end of the PS ONs (ON2-P4, ON3-(P4)2, and ON4-(P4)3 in Figure 1). The ONs with PS backbone equipped with one, two, and three units of alkyne linker 11 (ONs2-4 in Figure 3, Table 1) were in-line assembled on an automated ON synthesizer. The copper(I)-catalyzed conjugation of the solid-supported ONs2-4 to the P4 peptide was performed using a P4 peptide equipped with azido-lysine at the N-terminus (P4-aza-peptide) and the recently developed protocol where the CuBr × Me2S complex is used as copper(I) source40 (Figure 3). The conjugation reaction was followed by subsequent washing of the solid-supported ON-peptide conjugates with DMSO, 50 mM EDTA solution, and acetonitrile to remove residual copper(I) salts and other reagents. Subsequent deprotection and cleavage from the solid support resulted in the final mono-, bis-, and tris-ON-P4 peptide conjugates, which were analyzed by RP-HPLC, and their identities were confirmed by mass spectrometry (ON2-P4, ON3-(P4)2, and ON4-(P4)3 in Figure 3, and Figures S28, S30, and S32 in the Supporting Information). The HPLC profiles of the resulting ON-P4 conjugates revealed the presence of the corresponding residual starting ON3 and ON4 as well as ON conjugates with only one unit of P4 peptide attached for ON3-(P4)2 (Figure S30 in the Supporting Information), and with one or two units of P4 peptides attached for ON4-(P4)3 (Figure S32 in Supporting Information). The reason for not complete attachment of two and three units of P4 peptide, in comparison to the rather efficient mono-conjugation of the P4 peptide, could possibly be due to steric hindrance brought about by the peptide. Also, hypothetical coordination of copper between two neighboring alkyne moieties, which may come in close proximity, could lead to a reduced rate of reaction. The final ON3-(P4)2 and ON4-(P4)3 bis- and tris-conjugates were, however, obtained as main products. Incomplete conjugation will result in difficulties during isolation and purification to obtain homogenous conjugates and will lead to a decrease in overall yields. Separation during chromatography will of course depend also on, apart from the HPLC gradient, the nature of the peptide attached.
Figure 3.

Synthesis of mono-, bis-, and tris-P4 peptide PS-ON conjugates ON2-P4, ON3-(P4)2, and ON4-(P4)3, via 1–3 cycles of linker 11 incorporation.
The new alkyne linker was also evaluated for internal labeling of ONs. Such constructs with an internally incorporated entity could be utilized as a tool in various diagnostic applications. Recent studies revealed that internal labeling of ONs with short peptides, which influence specificity and affinity to the target, improve their target recognition and stability toward nucleases.44 A solid-supported DNA (ON5 in Figure 4 and Table 1) with the internally incorporated alkyne linker was assembled on an automated ON synthesizer using phosphoramidite 11 and subjected to conjugation on a solid support with the P4-aza peptide using copper(I) iodide in a CuAAC reaction. The conjugation reaction of ON5 appeared to be somewhat slower, and 8 equiv of P4-aza peptide was used instead of 6 equiv of P4-aza peptide, which was required for the preparation of the 5′-monoconjugate ON2-P4. The need for a larger amount of P4-aza peptide could possibly be explained by the additional hindrance at the reaction site by the formation of the secondary structure in ON5 with an internally incorporated abasic site modification. A structural effect is also suggested by the HPLC profile of alkyne-modified ON5 (Figure S33 in the Supporting Information), which after deprotection and cleavage from the support, displays a split peak with the same mass data. The internally P4-conjugated DNA (ON5-P4) was cleaved from the solid support, deprotected, and analyzed by RP-HPLC (Figures 4 and S34 in the Supporting Information), and its identity was confirmed by mass spectrometry.
Figure 4.

Synthesis of ON internal conjugate with P4 peptide (ON5-P4) utilizing ON5 with internally incorporated linker 11 and P4-aza peptide.
The orthogonality of the developed alkyne and amine linkers was exploited for the synthesis of ON multiconjugates containing different entities at the 5′-terminal position of an ON. Multifunctionalized ONs bearing different functional moieties are of great interest for various biotechnological applications and for further development of ON therapeutics. The conjugation of different entities to ONs has been investigated, e.g., for the preparation of GalNAc–cholesterol-modified ONs,34 GalNAc–pentylamine-modified siRNA,35 cell-penetrating peptide (CPP)–myristic acid-modified ONs,36 and multivalent ON glycoconjugates.45,46 The methodology utilizing a combination of the alkyne and amine linkers is exemplified by the preparation of a 5′-site ON tris-conjugate ON6-(MIF)-(PA)-(MIF) by incorporating an acetylated form of the MIF-1 peptide47 (MIF), a palmitoyl unit, and a second MIF peptide moiety (Figure 1). The MIF-1 is an endogenous brain peptide with a variety of pharmacological effects on the central nervous system, whereas the attachment of long-chain fatty acyl groups can modulate lipophilicity of the ON and facilitate cellular uptake.48 In addition, palmitoylation of ONs can enhance the affinity to albumin and has been demonstrated to facilitate functional uptake of the ON into skeletal and cardiac muscles.16 The synthesis of the ON6-(MIF)-(PA)-(MIF) conjugate was started with in-line sequential addition of linkers 11, 16, and then again 11 at the 5′-end of the ON during automated solid-phase ON synthesis, followed by the removal of the Fmoc-protecting group from the amino linker on the solid support (Figure 5). The standard treatment for Fmoc removal (20% piperidine solution in DMF) resulted in the formation of significant amounts of side product corresponding to a dibenzofulvene adduct (Figure S35 in the Supporting Information). Therefore, several scavengers and conditions were evaluated to overcome this problem, e.g., 2% 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 10% piperidine solution in DMF, 20% diethylamine (DEA) in acetonitrile, 2% DBU and 10% morpholine in DMF. The treatment of ON6 attached to Glen UnySupport FC for 2 min with a mixture of 2% DBU and 5% piperazine solution in DMF49 gave the best results in the suppression of the side product formation (Figure S36 in the Supporting Information). Conjugation of palmitic acid was performed using HBTU and N-methylmorpholine (NMM), and the resulting ON-palmitic acid conjugate ON6-(PA) was then reacted with the MIF peptide functionalized with an azido linker (Ac-Pro-Leu-Gly-CH2CH2-N3, Ac-MIF-N3)21 using the CuAAC reaction. The solid-supported ON conjugate was deprotected and cleaved from the solid support resulting in the final tris-conjugate ON6-(MIF)-(PA)-(MIF) (Figures 5 and S38 in the Supporting Information), which was analyzed by RP-HPLC and its identity was confirmed by mass spectrometry.
Figure 5.

Synthesis of ON tris-conjugate ON6-(MIF)-(PA)-(MIF) with attached palmitoyl residue and two units of the MIF peptide. Chromatograms A–C demonstrate the RP-HPLC profile of (A) crude ON6 after Fmoc deprotection and cleavage from the solid support, (B) crude ON6-(PA) conjugate, and (C) final crude tris-conjugate ON6-(MIF)-(PA)-(MIF) (see enlarged Figures S36–S38 in the Supporting Information). RP-HPLC is performed on a C18 column (Supelco Discovery BIO Wide Pore C18-5) with a linear gradient from 0 to 50% (for A), and from 50 to 100% (for B and C) of buffer B in buffer A over 45 min at 25 °C.
In conclusion, we have developed new versatile phosphoramidite-based alkyne and amine linkers that allow for multiple conjugation of ONs. Both linkers are derived from the same chemical scaffold that can be readily obtained from a low-cost commercial material, and they are compatible with in-line addition during automated solid-phase ON synthesis as well as for orthogonal conjugation on a solid support. Assembly of ONs, containing the activated alkyne linker at the 5′-end of ONs or internally, allowed for direct conjugation of multiple units of peptides or low-molecular-weight compounds to ONs using the CuAAC reaction on a solid support. The described methodology is also compatible with phosphorothioate ONs as exemplified by the synthesis of a series of PS ON conjugates with one, two, and three P4 peptides. Sequential incorporation of alkyne- and amine-based linkers at the 5′-terminus of an ON enabled the synthesis of a tris-conjugate bearing two entities of the MIF peptide and one palmitoylated unit. The alkyne linker allows for the conjugation reaction at low concentrations and under mild conditions. Additionally, conjugation on a solid support has the advantage of simplifying the purification of ON conjugates since the excess of copper can be readily removed by washing the solid support with the EDTA solution. The multiconjugation of an ON with a low-molecular-weight molecule (benzylguanine, BG) as well as with a short MIF peptide proceeded efficiently with negligible amounts of the residual nonconjugated ON. The attachment of a longer P4-peptide, and especially its multiple conjugation, appeared to be more challenging and resulted in the presence of incomplete conjugation products, but with the final ON conjugate being the major product. The main advantage of the presented strategy is the flexibility in the design of ON conjugates, allowing for conjugation of various entities at different positions including multiple attachments. The methodology is applicable for both phosphodiester and PS ONs including preparation of ON multiconjugates with different entities of interest for numerous biotechnological applications and ON therapeutics.
Materials and Methods
General Information
All reagents and solvents used in chemical synthesis were of commercial grade. The (3R)-4-cyano-3-hydroxybutanoate (1) and 6-((4-(aminomethyl)benzyl)oxy)-9H-purin-2-amine (SNAP-Tag) were obtained from Fluorochem Ltd. (U.K.), 2-[2-(2-azidoethoxy)ethoxy]acetic acid hydroxysuccinimide ester was from MCAT GmbH (Germany), and palmitic acid was from Sigma-Aldrich Sweden. Azido-functionalized P4 peptide (P4-aza peptide),40 azido-functionalized MIF peptide (Ac-MIF-N3),21 and PAMBA22 were synthesized according to the respective published procedures. Azido-functionalized benzylguanine (BG-N3, SNAP-Tag azide) was synthesized as described in the Supporting Information. Methanol (MeOH, Analytical Grade) and acetonitrile (CH3CN, Analytical Grade) were additionally dried over 3 Å molecular sieves and pyridine (Analytical Grade) was dried over 4 Å molecular sieves. Tetrahydrofuran (THF, Puriss p.a.) was distilled at atmospheric pressure over LiAlH4 prior to use. Thin-layer chromatography (TLC) was performed on Merck precoated silica gel 60 F254 glass-backed or aluminum plates and visualized by UV and/or by charring with 8% (v/v) sulfuric acid in methanol, with 6% (m/v) ninhydrin in ethanol or standard potassium permanganate solution. Chromatographic separations were performed on Merck G60 silica gel. Automated chromatography was performed on CombiFlash Rf 200 by Teledyne ISCO system using RediSep normal-phase silica flash or RediSep Rf Gold normal-phase silica columns. Mass analysis was performed with a Micromass LCT ESI-TOF mass spectrometer. All NMR spectra were recorded on a Bruker AVANCE DRX-400 (400.1 MHz in 1H, 100.6 MHz in 13C) or Bruker AV 500 MHz (500.13 MHz in 1H, 125.76 MHz in 13C, and 202.47 MHz in 31P) spectrometers using either tetramethylsilane (TMS) or the deuterated solvent as an internal standard. Chemical shifts (δ scale) are reported in parts per million (ppm). Coupling constants (J values) are given in Hertz (Hz). RP-HPLC was carried out on a Jasco HPLC system with UV detection at 254 nm using a Supelco Discovery BIO Wide Pore C18-5 (250 × 4.6 mm2) column with 1 mL/min flow rate and a linear gradient from 0 to 50% of buffer B in buffer A over 45 min at ambient temperature or as specified. Buffers for RP-HPLC were as follows: (A) 50 mM triethylammonium acetate (TEAA), pH 6.5; (B) 50 mM TEAA, pH 6.5, in 50% CH3CN. The 21-mer DNA (5′-d(GCGTTGATGCAATTTCTATGC)-3′) on a solid support used for the synthesis of ON1-(BG)4 was purchased from Eurogentec S.A., Belgium. ONs were analyzed in the negative mode as solutions in acetonitrile/water, 1:1 (v/v), and their molecular weights were obtained from the m/z values of the multiple charged ions.
(R)-5-(N-Trifluoroacetyl)-aminopentane-1,3-diol (3)
A solution of compound 1 (0.66 g, 4.2 mmol) in anhydrous tetrahydrofuran (THF, 8 mL) was added dropwise to a cooled (0 °C) suspension of LiAlH4 (0.87 g, 22.9 mmol) in anhydrous THF (4 mL). The resulting mixture was left to attain ambient temperature and then stirred overnight. Next, the mixture was cooled to 0 °C and treated sequentially with Milli-Q water (0.38 mL), 10 N NaOH (1.04 mL), and Milli-Q water (0.38 mL). The mixture was then kept at ambient temperature with stirring for 1 h. The solids were removed by filtration, washed with 2-propanol, and the filtrate was concentrated in vacuo. The residue was re-dissolved in Milli-Q water (10 mL), and the pH of the solution was adjusted to 7.0 using 0.5 M HCl. The mixture was concentrated in vacuo, and traces of water were removed by evaporation of the added methanol. Crude aminodiol (compound 2) was dissolved in anhydrous methanol (42 mL), and triethylamine (5.8 mL, 42 mmol) was added followed by the addition of ethyl trifluoroacetate (5.0 mL, 42 mmol). The reaction mixture was stirred at room temperature for 20 h. Volatiles were evaporated in vacuo, and the residue was purified by flash column chromatography using 0–6% methanol in dichloromethane as eluent to afford compound 3 (0.69 g, 3.21 mmol, 76% for two steps). 1H NMR (400.1 MHz, CDCl3): δ = 7.90 (br s, 1H), 4.06–3.98 (m, 1H), 3.96–3.89 (m, 1H), 3.87–3.79 (m, 1H), 3.72–3.63 (m, 1H), 3.40–3.31 (m, 1H), 1.81–1.62 (m, 4H) ppm. 13C NMR (100.6 MHz, CDCl3): δ = 157.3 (q, J = 36.3 Hz), 115.9 (q, J = 287.4 Hz), 71.3, 61.5, 38.0, 37.8, 35.0 ppm. ES–MS calcd for C7H11F3NO3 [M – H]− 214.07, found 214.05.
(R)-1-O-(4-Methoxytrityl)-5-(N-trifluoroacetyl)-aminopentane-1,3-diol (4)
Compound 3 (0.635 g, 2.95 mmol) was dried by evaporation of the added anhydrous pyridine and was dissolved in the same solvent (29 mL). The resulting solution was cooled to 0 °C, and 4-methoxytritylchloride (1.0 g, 3.24 mmol) was added under a nitrogen atmosphere. The reaction mixture was stirred overnight allowing the temperature to rise to the ambient. The reaction was quenched with 10% aqueous NaHCO3, and the mixture was extracted with dichloromethane. The organic phase was washed with water, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was subjected to flash column chromatography using 0–2.5% methanol in dichloromethane as eluent to give compound 4 (0.78 g, 1.6 mmol, 54%). 1H NMR (400.1 MHz, CDCl3): δ = 7.59 (br s, 1H), 7.35–7.31 (m, 4H), 7.27–7.14 (m, 8H), 6.78 (d, J = 8.7 Hz, 2H), 3.92–3.83 (m, 1H), 3.73 (s, 3 H), 3.67–3.57 (m, 1H), 3.41–3.34 (m, 1H), 3.28–3.16 (m, 2H), 1.83–1.71 (m, 1H), 1.68–1.47 (m, 3H) ppm. 13C NMR (100.6 MHz, CDCl3): δ = 158.7, 157.0 (q, J = 36.5 Hz), 144.0, 143.8, 135.1, 130.2, 128.18, 128.15, 128.0, 127.1, 115.9 (q, J = 287.7 Hz), 113.3, 87.4, 72.2, 62.6, 55.2, 38.2, 36.4, 34.5 ppm; ES–MS calcd for C27H27F3NO4 [M – H]− 486.19, found 486.17.
(R)-1-O-(4-Methoxytrityl)-5-N-[4-((2-(prop-2-yn-1-yloxy)acetamido)methyl)benzoyl]-aminopentane-1,3-diol (6)
Compound 4 (0.45 g, 0.92 mmol) was dissolved in a mixture of aqueous ammonia (28%) and ethanol (1:1 v/v) (10 mL) containing triethylamine (0.17 mL, 1.2 mmol) and stirred for 24 h at the ambient temperature. After that time, an additional 6 mL of aqueous ammonia (28%) was added to the reaction mixture and the reaction was kept overnight. Volatiles were partially removed in vacuo, and the residue was partitioned between ethyl acetate and saturated aqueous solution of NaHCO3. The aqueous phase was re-extracted with ethyl acetate, and the combined organic phases were dried over Na2SO4, filtered and concentrated under reduced pressure to afford 5. The crude compound 5 was dissolved in anhydrous N,N-dimethylformamide (DMF, 4.5 mL) containing N,N-diisopropylethylamine (DIPEA, 0.16 mL, 0.93 mmol) and was added to a solution of 4-((2-(prop-2-yn-1-yloxy)acetamido)methyl) benzoic acid (PAMBA, 0.21 g, 0.84 mmol) in anhydrous DMF (4.5 mL), which was preactivated with 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU, 0.32 g, 0.84 mmol) and DIPEA (0.29 mL, 1.69 mmol) for 30 min. The reaction mixture was stirred for 3 h at the ambient temperature and then partitioned between ethyl acetate and water. The aqueous phase was washed with ethyl acetate, and the combined organic phases were dried over Na2SO4, filtered and concentrated in vacuo. The residue was subjected to flash column chromatography using 0–4% methanol in dichloromethane as eluent to give compound 6 (0.51 g, 0.82 mmol, 97%). 1H NMR (400.1 MHz, CDCl3): δ = 7.66 (d, J = 8.3 Hz, 2H), 7.36–7.31 (m, 4H), 7.28–7.12 (m, 10H), 7.05 (t, J = 5.1 Hz, 1H), 6.82–6.72 (m, 3H), 4.45 (d, J = 6.1 Hz, 2H), 4.15 (d, J = 2.4 Hz, 2H), 4.05 (s, 2H), 3.92–3.84 (m, 1H), 3.79–3.69 (m, 4H), 3.37–3.25 (m, 2H), 3.22–3.14 (m, 1H), 2.40 (t, J = 2.4 Hz, 1H), 1.84–1.73 (m, 1H), 1.70–1.51 (m, 3H) ppm. 13C NMR (100.6 MHz, CDCl3): δ = 168.9, 167.1, 158.6, 144.3, 144.1, 141.3, 135.3, 133.9, 130.2, 128.2, 127.9, 127.7, 127.3, 127.0, 113.2, 87.1, 78.1, 75.9, 70.6, 69.0, 62.3, 58.6, 55.2, 42.4, 37.8, 36.6, 36.0 ppm; ES–MS calcd for C38H39N2O6 [M – H]− 619.28, found 619.29.
(R)-3-O-(N,N-Diisopropylamino-(2-cyanoethoxy)phosphinyl)-1-O-(4-methoxytrityl)-5-N-[4-((2-(prop-2-yn-1-yloxy)acetamido)methyl)benzoyl]-aminopentane-1,3-diol (7)
To a cooled (0 °C) solution of compound 6 (0.21 g, 0.34 mmol) in THF (3.5 mL), DIPEA (0.29 mL, 1.7 mmol) was added under a nitrogen atmosphere followed by 2-cyanoethyl N,N-diisopropylphosphoramidochloridite (0.15 mL, 0.68 mmol). The reaction mixture was stirred for 30 min, then allowed to warm to ambient temperature, and stirred for an additional 2 h. The mixture was partitioned between ethyl acetate and a 10% aqueous solution of NaHCO3, and the aqueous phase was re-extracted with ethyl acetate. The combined organic phases were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography using 0–2% acetonitrile in ethyl acetate containing 0.1% of triethylamine as eluent to give compound 7 (0.2 g, 72%). 31P NMR (202.47 MHz, DMSO-d6): δ = 146.7, 146.3 ppm; ES–MS calcd for C47H58N4O7P [M + H]+ 821.40, found 821.36.
(R)-5-N-[4-((2-(Prop-2-yn-1-yloxy)acetamido)methyl)benzoyl]-aminopentane-1,3-diol (9)
PAMBA (8, 12.5 g, 50.56 mmol) and aminodiol 2 (8.434 g, 70.78 mmol) were dissolved in water/THF mixture (3:2). HOBt (1.37 g, 10.11 mmol) was added to the resulting solution followed by the addition of EDC (9.42 g, 60.67 mmol), and the reaction mixture was stirred overnight. Brine solution (62 mL) was added and the mixture was extracted with THF. The organic phase was concentrated under reduced pressure and purified by flash column chromatography using 0–12% THF in acetonitrile as eluent to afford compound 9 (8.68 g, 24.91 mmol, 49%). 1H NMR (400.1 MHz, CD3OD): δ = 7.77 (d, J = 8.3 Hz, 2H), 7.38 (d, J = 8.4 Hz, 2H), 4.47 (s, 2H), 4.28 (d, J = 2.4 Hz, 2H), 4.10 (s, 2H), 3.87–3.79 (m, 1H), 3.74–3.67 (m, 2H), 3.59–3.42 (m, 2H), 2.94 (t, J = 2.4 Hz, 1H), 1.86–1.63 (m, 4H) ppm. 13C NMR (100.6 MHz, CD3OD): δ = 172.2, 170.1, 143.8, 134.6, 128.55, 128.49, 79.7, 77.1, 69.5, 67.9, 60.1, 59.4, 43.2, 40.8, 38.1 ppm; ES–MS calcd for C18H23N2O5 [M – H]− 347.16, found 347.16.
(R)-1-O-(4,4′-Dimethoxytrityl)-5-N-[4-((2-(prop-2-yn-1-yloxy)acetamido)methyl)-benzoyl]-aminopentane-1,3-diol (10)
Method A
Compound 9 (5.43 g, 15.59 mmol) was dissolved in DMF (27 mL) and 2,6-lutidine (3.34 g, 31.17 mmol) was added to the reaction mixture followed by 4,4′-dimethoxytrityl chloride (5.02 g, 14.81 mmol). The reaction mixture was stirred for 18 h and then water (11 mL) and saturated aqueous NaHCO3 solution (5.5 mL) were added to the reaction mixture, and the crude product was extracted with toluene. The combined organic phases were washed with 20% citric acid (14 mL), concentrated in vacuo, then methanol and dichloromethane were added and the solution concentrated under reduced pressure. The crude material was purified by flash column chromatography using 0–10% methanol in dichloromethane as eluent to afford compound 10 (4.98 g, 7.65 mmol, 49%).
Method B
Compound 9 (2.61 g, 7.5 mmol) was dried by evaporation of the added anhydrous pyridine and was dissolved in the same solvent (75 mL). The resulting solution was cooled to 0 °C and 4,4′-dimethoxytrityl chloride (1.78 g, 5.25 mmol) was added under nitrogen atmosphere. The reaction mixture was kept at 0 °C, and after 2 h an additional amount of 4,4′-dimethoxytrityl chloride (1.02 g, 3.0 mmol) was added under a nitrogen atmosphere. The mixture was then allowed to warm to ambient temperature and stirred for 40 h. After that time, saturated aqueous NaHCO3 was added and the product was extracted with dichloromethane. The combined organic phases were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was subjected to flash column chromatography using acetonitrile in ethyl acetate containing 0.1% of triethylamine as eluent to give compound 10 (4.34 g, 6.67 mmol, 89%). 1H NMR (500.1 MHz, CD3OD): δ = 7.77–7.74 (m, 2H), 7.41–7.35 (m, 4H), 7.29–7.25 (m, 4H), 7.25–7.20 (m, 2H), 7.19–7.14 (m, 1H), 6.82–6.78 (m, 4H), 4.29 (d, J = 2.40 Hz, 2H), 4.11 (s, 1H), 3.58–3.49 (m, 1H), 3.49–3.41 (m, 1H), 3.29–3.23 (m, 1H), 3.20–3.13 (m, 1H), 2.93 (m, 1 H), 1.82–1.70 (m, 3H), 1.65–1.56 (m, 1H) ppm. 13C NMR (125.76 MHz, CD3OD): δ = 172.3, 170.3, 160.1, 146.9, 143.9, 137.9, 137.8, 134.7, 131.3, 129.4, 128.8, 128.7, 128.6, 127.8, 114.2, 87.5, 79.8, 77.2, 69.7, 68.0, 61.7, 59.5, 55.9, 43.3, 38.7, 38.1 ppm; ES–MS calcd for C39H41N2O7 [M – H]− 649.29, found 649.27.
(R)-3-O-(N,N-Diisopropylamino-(2-cyanoethoxy)phosphinyl)-1-O-(4,4′-dimethoxytrityl)-5-N-[4-((2-(prop-2-yn-1-yloxy)acetamido)methyl)benzoyl]-aminopentane-1,3-diol (11)
To a cooled (0 °C) solution of compound 10 (1.63 g, 2.5 mmol) in THF (25 mL), DIPEA (2.18 mL, 12.5 mmol) was added under a nitrogen atmosphere followed by the addition of 2-cyanoethyl N,N-diisopropylphosphoramidochloridite (1.39 mL, 6.25 mmol). The reaction mixture was stirred for 30 min, then allowed to warm to ambient temperature, and was additionally stirred for 2 h. The mixture was partitioned between ethyl acetate and 10% aqueous solution of NaHCO3, and the aqueous phase was re-extracted with ethyl acetate. The combined organic phases were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography using 0–2% acetonitrile in ethyl acetate containing 0.1% of triethylamine as eluent to give compound 11 (1.73 g, 81%). 31P NMR (202.47 MHz, DMSO-d6): δ = 146.7, 146.3 ppm; ES–MS calcd for C48H60N4O8P [M + H]+ 851.41, found 851.45.
(R)-1-O-(4,4′-Dimethoxytrityl)-5-N-[((9H-fluoren-9-yl-methoxycarbonylamino)methyl)-benzoyl]-aminopentane-1,3-diol (15)
To a solution of compound 13 (2.24 g, 8.2 mmol) in NMP (20 mL), HOBt (0, 22 g, 1.64 mmol) and EDC (1.526 g, 9.83 mmol) were added, and the reaction mixture was stirred for 20 min. A solution of aminodiol 2 (1.1 g, 9.23 mmol) in NMP (5 mL) was added dropwise, and the reaction mixture was stirred for 4 h at ambient temperature. After the completion of the reaction (monitored by RP-HPLC), 0.05 M HCl (30 mL) was added to the reaction mixture and water/ethyl acetate work-up was performed. The crude residue was subjected to flash column chromatography (ISCO system) using 0–25% methanol in dichloromethane to give intermediate 14 (1.52 g 3.2 mmol), which was co-evaporated with pyridine and re-dissolved in the same solvent (15 mL). DMTrCl (1.30 g, 3.84 mmol) was added under a nitrogen atmosphere and the reaction mixture was stirred overnight. After that time, the reaction was quenched with methanol (1 mL) followed by the addition of saturated NaHCO3 (5 mL). Volatiles were evaporated under reduced pressure and a water/dichloromethane work-up was performed. The organic phase was collected and washed with water and a 10% citric acid solution, dried over Na2SO4, and filtered. The crude product was concentrated on celite (for solid loading) and chromatographed (ISCO chromatography system) using 0–100% gradient of ethyl acetate in heptane to yield compound 15 (1.2 g, 48%) as a white foam. 1H NMR (500.1 MHz, CD3OD): δ = 7.78–7.68 (m, 4H), 7.62 (d, J = 7.5 Hz, 2H), 7.39–7.32 (m, 4H), 7.32–7.02 (m, 11H), 6.77 (dd, J = 8.8, 1.5 Hz, 4H), 4.46–4.38 (m, 2H), 4.33–4.29 (s, 2H), 4.20–4.15 (t, 1H), 3.95–3.87 (m, 1H), 3.73–3.67 (s, 6H), 3.59–3.50 (m, 1H), 3.50–3.40 (m, 1H), 3.28–3.21 (m, 1H), 3.18–3.11 (m, 1H), 1.82–1.69 (m, 3H), 1.65–1.54 (m, 1 H) ppm. 13C NMR (125.76 MHz, CD3OD): δ = 169.5, 159.3, 158.3, 146.0, 144.6, 142.0, 137.0, 130.5, 128.6, 128.1, 127.8, 127.6, 127.5, 127.0, 125.5, 120.3, 113.3, 86.6, 67.2, 66.9, 60.9, 55.0, 44.4, 37.9, 37.3, ppm. ES–MS calcd for C49H48N2O7 [M + Na]+ 799.34, found 799.08.
(R)-3-O-(N,N-Diisopropylamino-(2-cyanoethoxy)phosphinyl)-1-O-(4,4′-dimethoxytrityl)-5-N-[((9H-fluoren-9-yl-methoxycarbonylamino)methyl)benzoyl]-aminopentane-1,3-diol (16)
Compound 15 (1.15 g, 1.48 mmol) was dissolved in THF (16 mL) and cooled in an ice-bath. DIPEA (516 μL, 2.96 mmol) was added to the resulting solution under a nitrogen atmosphere followed by 2-cyanoethyl N,N-diisopropylphosphoramidochloridite (495 μL, 2.22 mmol). The reaction mixture was stirred for 2 h, quenched with methanol (300 μL), and concentrated in vacuo. Then water/EtOAc work-up was performed, the organic phases were combined, dried over Na2SO4, and filtered. Triethylamine (100 μL) was added to the filtrate, which was then concentrated on celite (for solid loading) and chromatographed (ISCO chromatography system) using 0–100% ethyl acetate in heptane to give compound 16 (0.89 g, 62%) as a white foam. 31P NMR (202.47 MHz, DMSO-d6): δ = 146.7, 146.3 ppm; ES–MS calcd for C58H66N4O8P [M + H]+ 977.46, found 977.46.
Synthesis of Linker-Modified ONs
The tetra-alkyne linker modified ON1 (Table 1) was assembled starting from commercial CPG-supported DNA (Eurogentec S.A.) by postsynthetic coupling with the symmetric doubler modifier amidite (Glen Research) followed by the sequential coupling of activated alkyne linker phosphoramidite 7. The ONs2-6 (Table 1) were assembled in-line in a TWIST synthesis column (Glen Research) on an Applied Biosystems 394 DNA/RNA synthesizer using 0.1 M anhydrous acetonitrile solution of commercial dATac, dGTac, T, dCAc (Sigma-Aldrich), or 2′-OMe-APac, 2′-OMe-CAc, 2′-OMe-U, 2′-OMe-GiPr-Pac phosphoramidites (Glen Research), and the synthesized alkyne linker phosphoramidite 11, and amino linker phosphoramidite 16. PADS (0.2 M phenylacetyl disulfide in CH3CN/3-methylpyridine, 120 s treatment) was used as a sulfurizing reagent for the synthesis of phosphorothioate ONs2-4. The ON syntheses were performed on a 1 μmol scale using Glen UnySupport FC, 0.3 M 5-benzylthio-1-H-tetrazole (BTT) as an activator and 60 s coupling time for DNA phosphoramidites, 600 s coupling time for 2′-OMe phosphoramidites, and 900 s coupling time for the symmetric doubler modifier amidite, and linker phosphoramidites 7, 11, and 16.
Synthesis of Benzylguanine (BG)-Labeled ON (ON1-(BG)4)
The CPG-supported tetra-linker-modified ON1 (0.25 μmol) containing the tetra-5′-alkyne functionality was placed in 2 mL Eppendorf tube with a screw cap, and the azido-functionalized BG (BG-N3, 7.1 mg, 4 μmol, 16 equiv) dissolved in dimethyl sulfoxide (DMSO, 560 μL) was added. To the resulting mixture, DIPEA solution (5 μmol, 20 μL from a stock solution of 1 mL of DMSO containing 43 μL of DIPEA) was added followed by the addition of CuI (2 μmol, 200 μL from a stock solution of 1.9 mg of CuI in 1 mL of DMSO). The reaction mixture was agitated on a vortex for 24 h at ambient temperature. After that time, the mixture was centrifuged and the solution above the support was carefully removed using a syringe with a needle. The CPG-supported ON-(BG)4 conjugate was then washed sequentially with DMSO (3 × 0.6 mL), tert-butanol-H2O (1:1 v/v, 1 × 0.6 mL), 1 mM solution of EDTA in tert-butanol-H2O (1:1 v/v, 3 × 0.6 mL), tert-butanol-H2O (1:1 v/v, 1 × 0.6 mL), and CH3CN (2 × 0.6 mL), and dried by a 1 min stream with nitrogen gas. The resulting solid-supported BG-labeled ON was treated with aqueous ammonia (28%) for 48 h at ambient temperature. The CPG was removed by filtration using a Millex syringe-driven filter (0.22 μm) into a 50 mL round-bottomed flask, and the residue in the Eppendorf tube and filter were washed with milli-Q water (3 × 1 mL). The combined filtrate was concentrated under reduced pressure at 30 °C, and the residue was re-dissolved in water, transferred to an Eppendorf tube, and lyophilized. The aqueous EDTA solution (0.25 μmol) was added to the deprotected ON-(BG)4, and the crude conjugate was subjected to RP-HPLC using C18 column with a linear gradient from 0 to 100% of buffer B in buffer A over 45 min at 25 °C; tR = 26.5 min. ES–TOF mass m/z calcd (M–5) 2036.34, found 2036.54.
Synthesis of Oligonucleotide-mono-P4 Conjugate (ON2-P4)
To the solid-supported ON2, containing one unit of 5′-alkyne linker (15 mg, 0.5 μmol) in an Eppendorf tube, the P4-aza peptide (2.8 mg, 3 μmol, 6 equiv, in 50.6 μL of DMSO-H2O, 4:1 v/v) was added, followed by addition of a DIPEA solution (1.74 μL, 10 μmol, 20 equiv in 5 μL of H2O), CuBr × Me2S (0.41 mg, 2 μmol, 4 equiv, in 15 μL of DMSO), and 50 μL of DMSO. The reaction was agitated on a vortex for 24 h at ambient temperature. The resulting solid-supported ON-peptide conjugate was washed with DMSO (3 times), 50 mM EDTA (3 times), and CH3CN (3 times), and then air-dried. The solid-supported ON2-P4 conjugate was then placed in an Eppendorf tube with a screw cap, and 1 mL of aqueous ammonia (28%) was added to deprotect and cleave the ON from the solid support at room temperature overnight. The resulting solution was then removed with a syringe, evaporated and subjected to RP-HPLC on a C18 column with a linear gradient from 10 to 35% of buffer B in buffer A over 30 min at 25 °C, tR = 19.2 min. ES–TOF mass m/z calcd (M–4) 2060.25, found 2060.40.
Synthesis of Oligonucleotide-bis-P4 Conjugate (ON3-(P4)2)
To the solid-supported ON3, containing two units of the 5′-alkyne linker (15 mg, 0.5 μmol) in an Eppendorf tube, the P4-aza peptide (4.65 mg, 5 μmol, 10 equiv, in 133 μL of DMSO-H2O, 4:1 v/v) was added, followed by the addition of a DIPEA solution (1.74 μL, 10 μmol, 20 equiv in 5 μL of H2O), CuBr × Me2S (0.82 mg, 4 μmol, 8 equiv, in 15 μL of DMSO), and 50 μL of DMSO. The reaction was agitated on a vortex for 24 h at ambient temperature. The resulting solid-supported ON-peptide conjugate was washed with DMSO (3 times), 50 mM EDTA (3 times), and CH3CN (3 times), and then air-dried. The solid-supported ON3-(P4)2 conjugate was then placed in an Eppendorf tube with a screw cap, and 1 mL of aqueous ammonia (28%) was added to deprotect and cleave the ON from the solid support at room temperature overnight. The resulting solution was then removed with a syringe, evaporated, and subjected to RP-HPLC on a C18 column with a linear gradient from 0 to 100% of buffer B in buffer A over 30 min at 25 °C; tR = 13.3 min. ES–TOF mass m/z calcd (M–5) 1919.49, found 1919.63.
Synthesis of Oligonucleotide-tris-P4 Conjugate (ON4-(P4)3)
To the solid-supported ON4, containing three units of the 5′-alkyne linker (15 mg, 0.5 μmol) in an Eppendorf tube, the P4-aza peptide (7 mg, 7.5 μmol, 15 equiv, in 133 μL of DMSO-H2O, 4:1 v/v) was added, followed by the addition of a DIPEA solution (2.4 μL, 30 μmol, 60 equiv in 5 μL of H2O), CuBr × Me2S (5.2 mg, 6 μmol, 12 equiv, in 15 μL of DMSO), and 50 μL of DMSO. The reaction was agitated on a vortex for 24 h at ambient temperature. The resulting solid-supported ON-peptide conjugate was washed with DMSO (3 times), 50 mM EDTA (3 times), and CH3CN (3 times), and then air-dried. The solid-supported ON4-(P4)3 conjugate was then placed in an Eppendorf tube with a screw cap, and 1 mL of aqueous ammonia (28%) was added to deprotect and cleave the ON from the solid support at room temperature overnight. The resulting solution was then removed with a syringe, evaporated, and subjected to RP-HPLC on a C18 column with a linear gradient from 0 to 50% of buffer B in buffer A over 30 min at 25 °C; tR = 21.3 min. ES–TOF mass m/z calcd (M–5) 2190.90, found 2191.19.
Synthesis of Oligonucleotide-P4 Internal Conjugate (ON5-P4)
To the solid-supported ON5, containing an internally incorporated alkyne unit (0.1 μmol) in an Eppendorf tube, the P4-aza peptide (0.74 mg, 0.8 μmol, 8 equiv, in 68 μL of DMSO-H2O, 9:1 v/v) was added, followed by the addition of a DIPEA solution (2.0 μL, 0.5 μmol, from a stock solution of 957 μL of H2O and 43 μL of DIPEA) and CuI (0.2 μmol, 20 μL from a stock solution of 1.9 mg of CuI in 1 mL of DMSO). The reaction was agitated on a vortex for 24 h at ambient temperature. The resulting solid-supported ON-peptide conjugate was washed with DMSO (3 times), 50 mM EDTA (3 times), and CH3CN (3 times), and then air-dried. The solid-supported ON5-P4 conjugate was then placed in an Eppendorf tube with a screw cap, and 1 mL of aqueous ammonia (32%) was added to deprotect and cleave the ON from the solid support at ambient temperature for 24 h. The resulting solution was then removed with a syringe, evaporated, and subjected to RP-HPLC on a C18 column with a linear gradient from 0 to 50% of buffer B in buffer A over 45 min at 25 °C; tR = 20.1 min. ES–TOF mass m/z calcd (M–4) 1873.34, found 1873.82.
Synthesis of Oligonucleotide–Palmitic Acid Conjugate ON6-(PA)
CPG-supported ON6 (0.25 μmol) containing the 5′-alkyne–(Fmoc)amino–alkyne sequence of linkers was placed in a 2 mL Eppendorf tube with a screw cap, and the solution containing 2% DBU and 5% piperazine in DMF (400 μL) was added. The resulting mixture was gently agitated on a vortex for 2 min at ambient temperature. After that time, the mixture was centrifuged and the solution above the support was carefully removed using a syringe with a needle. The CPG-supported oligonucleotide was then washed sequentially with DMF (5 × 0.5 mL), CH3CN (3 × 0.5 mL), and dichloromethane (3 × 0.5 mL), and then air-dried for 20 min. A small part of the resulting Fmoc-deprotected ON6 (1 mg) was cleaved from the solid support and deprotected for analytical purposes using aqueous ammonia (32%) at 55 °C for 24 h. The crude Fmoc-deprotected ON6 was analyzed using RP-HPLC on a C18 column with a linear gradient from 0 to 50% of buffer B in buffer A over 45 min at 25 °C; tR = 19.7 min. ES–TOF mass m/z calcd (M–4) 1821.75, found 1821.88. The main portion of the Fmoc-deprotected CPG-supported ON (0.22 μmol) was placed in a 2 mL Eppendorf tube with a screw cap. Palmitic acid (6.4 mg, 0.025 mmol) was preactivated with HBTU (9.48 mg, 0.025 mmol) and NMM (27.5 μL 0.025 mol) in 250 μL DMF-dichloromethane (1:1, v/v) by agitation for 25 min on a vortex, and was added to the Eppendorf tube with the solid-supported ON. The resulting mixture was agitated on a vortex for 2 h at ambient temperature. After that time, the mixture was centrifuged and the solution above the solid support was removed using a syringe with a needle. The CPG-supported oligonucleotide conjugate was washed sequentially with DMF (3 × 0.5 mL), CH3CN (3 × 0.5 mL), and dichloromethane (3 × 0.5 mL), and the air-dried. The resulting CPG-supported ON–palmitic acid conjugate (1 mg) was treated with aqueous ammonia (32%) for 24 h at 55 °C. The CPG was removed by filtration using a Millex syringe-driven filter (0.22 μm) into a 50 mL round-bottomed flask. The residue in the Eppendorf tube and filter were washed with Milli-Q water (3 × 1 mL). The combined filtrate was concentrated under reduced pressure at 30 °C, and the residue was dissolved in water, transferred to an Eppendorf tube, and lyophilized. The crude deprotected ON conjugate was subjected to RP-HPLC on a C18 column with a linear gradient from 50 to 100% of buffer B in buffer A over 45 min at 25 °C to obtain ON6-(PA); tR = 42.9 min. ES–TOF mass m/z calcd (M–4) 1881.35, found 1881.31.
Synthesis of Oligonucleotide tris-Conjugate ON6-(MIF)-(PA)-(MIF)
The CPG-supported ON6-(PA) (0.1 μmol) containing two alkyne units interspaced by the palmitoylated amino-modifier at the 5′-terminus was placed in a 2 mL Eppendorf tube with a screw cap. The azido-functionalized MIF peptide (0.6 mg, 1.5 μmol, 15 equiv), dissolved in 76 μL of H2O, was added. To the resulting mixture, a DIPEA solution (4 μL from a stock solution of 1 mL of H2O containing 43 μL of DIPEA) was added followed by the addition of CuI (0.2 μmol, 20 μL from a stock solution of 19 mg of CuI in 5 mL of DMSO). The reaction mixture was agitated on a vortex for 24 h at ambient temperature. After that time, the mixture was centrifuged and the solution above the solid support was removed using a syringe with a needle. The CPG-supported ON conjugate was then washed sequentially with DMSO (3 × 0.5 mL), EDTA in tert-butanol-H2O (1:1 v/v, 4 × 0.5 mL), tert-butanol-H2O (1:1 v/v, 1 × 0.5 mL), and CH3CN (2 × 0.5 mL), and air-dried. The resulting solid-supported ON conjugate was treated with aqueous ammonia (32%) for 48 h at ambient temperature. The CPG was removed by filtration using Millex syringe-driven filter (0.22 μm) into a 50 mL round-bottomed flask, and the residue in the Eppendorf tube and filter were washed with Milli-Q water (3 × 1 mL). The combined filtrate was concentrated under reduced pressure at 30 °C, the residue was transferred to Eppendorf tube, and lyophilized. The crude deprotected ON conjugate was analyzed (an aqueous solution containing EDTA was added to the solution of crude ON-conjugate) using RP-HPLC on C18 column with a linear gradient from 50 to 100% of buffer B in buffer A over 45 min at 25 °C; tR = 38.6 min. ES–TOF mass m/z calcd (M–4) 2079.08, found 2079.50.
Acknowledgments
The authors gratefully acknowledge funding from the Swedish Research Council (Grant No. 2016-03283), Duchenne Parent Project NL (Grant No. 17.013), Muscular Dystrophy Association (Grant No. MDA602835), and the European Commission: H2020 Marie Skłodowska-Curie Actions, “MMBIO” (Grant No. 721613).
Glossary
Abbreviations
- ON
oligonucleotide
- PS
phosphorothioate
- AON
antisense oligonucleotide
- PAMBA
4-((2-(prop-2-yn-1-yloxy)acetamido)methyl) benzoic acid
- TFA
trifluoroacetyl
- MMTrCl
4-methoxytrityl chloride
- HOBt
1-hydroxybenzotriazole
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- DMF
N,N-dimethylformamide
- DMTrCl
4,4′-dimethoxytrityl chloride
- THF
tetrahydrofuran
- MeOH
methanol
- Et3N
triethylamine
- EtOH
ethanol
- HBTU
N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate
- DIPEA
N,N-diisopropylethylamine
- Fmoc
fluorenylmethoxycarbonyl
- Su
succinimide
- NMP
N-methylpyrrolidone
- BG
benzylguanine
- EDTA
ethylenediaminetetraacetic acid
- P4-N3
P4-aza peptide
- Ac-MIF-N3
azido-functionalized acetylated MIF-1 peptide
- PA
palmitic acid
- BG-N3 or SNAP-Tag azide
azido-functionalized benzylguanine
- L
linker
- DMSO
dimethyl sulfoxide
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- NMM
N-methylmorpholine
- DEA
diethylamine
- Tac
4-tert-butylphenoxyacetyl
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c05075.
Author Contributions
§ D.H. and K.D. contributed equally to this paper. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Astakhova K.; Ray R.; Taskova M.; Uhd J.; Carstens A.; Morris K. “Clicking” gene therapeutics: A successful union of chemistry and biomedicine for new solutions. Mol. Pharmaceutics 2018, 15, 2892–2899. 10.1021/acs.molpharmaceut.7b00765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taskova M.; Mantsiou A.; Astakhova K. Synthetic nucleic acid analogues in gene therapy: An update for peptide–oligonucleotide conjugates. ChemBioChem 2017, 18, 1671–1682. 10.1002/cbic.201700229. [DOI] [PubMed] [Google Scholar]
- Benizri S.; Gissot A.; Martin A.; Vialet B.; Grinstaff M. W.; Barthélémy P. Bioconjugated oligonucleotides: Recent developments and therapeutic applications. Bioconjugate Chem. 2019, 30, 366–383. 10.1021/acs.bioconjchem.8b00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer A. D.; Dowdy S. F. GalNAc-siRNA Conjugates: Leading the way for delivery of RNAi therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. 10.1089/nat.2018.0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooding M.; Malhotra M.; Evans J. C.; Darcy R.; O’Driscoll C. M. Oligonucleotide conjugates – Candidates for gene silencing therapeutics. Eur. J. Pharm. Biopharm. 2016, 107, 321–340. 10.1016/j.ejpb.2016.07.024. [DOI] [PubMed] [Google Scholar]
- Aartsma-Rus A.; Corey D. R. The 10th Oligonucleotide Therapy Approved: Golodirsen for Duchenne Muscular Dystrophy. Nucleic Acid Ther. 2020, 30, 67–70. 10.1089/nat.2020.0845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W.; Rogge M. Targeting RNA: A transformative therapeutic strategy. Clin. Transl. Sci. 2019, 12, 98–112. 10.1111/cts.12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus A. New momentum for the field of oligonucleotide therapeutics. Mol. Ther. 2016, 24, 193–194. 10.1038/mt.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano R. L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. 10.1093/nar/gkw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentijn A. R. P. M.; van der Marel G. A.; Sliedregt L. A. J. M.; van Berkel T. J. C.; Biessen E. A. L.; van Boom J. H. Solid-phase synthesis of lysine-based cluster galactosides with high affinity for the asialoglycoprotein receptor. Tetrahedron 1997, 53, 759–770. 10.1016/S0040-4020(96)01018-6. [DOI] [Google Scholar]
- Rensen P. C. N.; van Leeuwen S. H.; Sliedregt L. A. J. M.; van Berkel T. J. C.; Biessen E. A. L. Design and synthesis of novel N-acetylgalactosamine-terminated glycolipids for targeting of lipoproteins to the hepatic asialoglycoprotein receptor. J. Med. Chem. 2004, 47, 5798–5808. 10.1021/jm049481d. [DOI] [PubMed] [Google Scholar]
- Khorev O.; Stokmaier D.; Schwardt O.; Cutting B.; Ernst B. Trivalent, Gal/GalNAc-containing ligands designed for the asialoglycoprotein receptor. Bioorg. Med. Chem. 2008, 16, 5216–5231. 10.1016/j.bmc.2008.03.017. [DOI] [PubMed] [Google Scholar]
- Nair J. K.; Willoughby J. L. S.; Chan A.; Charisse K.; Alam M. R.; Wang Q.; Hoekstra M.; Kandasamy P.; Kel’in A. V.; Milstein S.; Taneja N.; O’Shea J.; Shaikh S.; Zhang L.; van der Sluis R. J.; Jung M. E.; Akinc A.; Hutabarat R.; Kuchimanchi S.; Fitzgerald K.; Zimmermann T.; van Berkel T. J. C.; Maier M. A.; Rajeev K. G.; Manoharan M. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- Miller C. M.; Tanowitz M.; Donner A. J.; Prakash T. P.; Swayze E. E.; Harris E. N.; Seth P. P. Receptor-mediated uptake of phosphorothioate antisense oligonucleotides in different cell types of the liver. Nucleic Acid Ther. 2018, 28, 119–127. 10.1089/nat.2017.0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ämmälä C.; Drury W. J. III; Knerr L.; Ahlstedt I.; Stillemark-Billton P.; Wennberg-Huldt C.; Andersson E.-M.; Valeur E.; Jansson-Löfmark R.; Janzén D.; Sundström L.; Meuller J.; Claesson J.; Andersson P.; Johansson C.; Lee R. G.; Prakash T. P.; Seth P. P.; Monia B. P.; Andersson S. Targeted delivery of antisense oligonucleotides to pancreatic β-cells. Sci. Adv. 2018, 4, eaat3386 10.1126/sciadv.aat3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash T. P.; Mullick A. E.; Lee R. G.; Yu J.; Yeh S. T.; Low A.; Chappell A. E.; Østergaard M. E.; Murray S.; Gaus H. J.; Swayze E. E.; Seth P. P. Fatty acid conjugation enhances potency of antisense oligonucleotides in muscle. Nucleic Acids Res. 2019, 47, 6029–6044. 10.1093/nar/gkz354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert B.-S.; Gellert G. C.; Hochreiter A.; Pongracz K.; Wright W. E.; Zielinska D.; Chin A. C.; Harley C. B.; Shay J. W.; Gryaznov S. M. Lipid modification of GRN163, an N3′ → P5′ thio-phosphoramidate oligonucleotide, enhances the potency of telomerase inhibition. Oncogene 2005, 24, 5262–5268. 10.1038/sj.onc.1208760. [DOI] [PubMed] [Google Scholar]
- Burchett K. M.; Yan Y.; Ouellette M. M. Telomerase inhibitor Imetelstat (GRN163L) limits the lifespan of human pancreatic cancer cells. PLoS One 2014, 9, e85155 10.1371/journal.pone.0085155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldblatt E. M.; Erickson P. A.; Gentry E. R.; Gryaznov S. M.; Herbert B.-S. Lipid-conjugated telomerase template antagonists sensitize resistant HER2-positive breast cancer cells to trastuzumab. Breast Cancer Res. Treat. 2009, 118, 21–32. 10.1007/s10549-008-0201-4. [DOI] [PubMed] [Google Scholar]
- Zaramella S.; Yeheskiely E.; Strömberg R. A method for solid-phase synthesis of oligonucleotide 5′-peptide-conjugates using acid-labile α-amino protections. J. Am. Chem. Soc. 2004, 126, 14029–14035. 10.1021/ja046945o. [DOI] [PubMed] [Google Scholar]
- Wenska M.; Alvira M.; Steunenberg P.; Stenberg A.; Murtola M.; Stroemberg R. An activated triple bond linker enables “click” attachment of peptides to oligonucleotides on solid support. Nucleic Acids Res. 2011, 39, 9047–9059. 10.1093/nar/gkr603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jezowska M.; Honcharenko D.; Ghidini A.; Stroemberg R.; Honcharenko M. Enabling multiple conjugation to oligonucleotides using “click cycles”. Bioconjugate Chem. 2016, 27, 2620–2628. 10.1021/acs.bioconjchem.6b00380. [DOI] [PubMed] [Google Scholar]
- Lönnberg H. Solid-phase synthesis of oligonucleotide conjugates useful for delivery and targeting of potential nucleic acid therapeutics. Bioconjugate Chem. 2009, 20, 1065–1094. 10.1021/bc800406a. [DOI] [PubMed] [Google Scholar]
- Weisbrod S. H.; Marx A. Novel strategies for the site-specific covalent labelling of nucleic acids. Chem. Commun. 2008, 5675–5685. 10.1039/b809528k. [DOI] [PubMed] [Google Scholar]
- Verma S. A novel loom of click chemistry in drug discovery. Int. J. Drug Dev. Res. 2015, 7, 18–26. [Google Scholar]
- Meldal M.; Tornøe C. W. Cu-catalyzed azide–alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- Farzan V. M.; Ulashchik E. A.; Martynenko-Makaev Y. V.; Kvach M. V.; Aparin I. O.; Brylev V. A.; Prikazchikova T. A.; Maklakova S. Y.; Majouga A. G.; Ustinov A. V.; Shipulin G. A.; Shmanai V. V.; Korshun V. A.; Zatsepin T. S. Automated solid-phase click synthesis of oligonucleotide conjugates: From small molecules to diverse N-acetylgalactosamine clusters. Bioconjugate Chem. 2017, 28, 2599–2607. 10.1021/acs.bioconjchem.7b00462. [DOI] [PubMed] [Google Scholar]
- Jezowska M.; Romanowska J.; Bestas B.; Tedebark U.; Honcharenko M. Synthesis of biotin linkers with the activated triple bond donor [p-(N-propynoylamino) toluic acid] (PATA) for efficient biotinylation of peptides and oligonucleotides. Molecules 2012, 17, 14174–14185. 10.3390/molecules171214174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav S.; Gulumkar V.; Deshpande P.; Coffey E. T.; Lönnberg H.; Virta P. Synthesis of azide-modified chondroitin sulfate precursors: Substrates for “click”- conjugation with fluorescent labels and oligonucleotides. Bioconjugate Chem. 2018, 29, 2382–2393. 10.1021/acs.bioconjchem.8b00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCulloch T.; Buchberger A.; Stephanopoulos N. Emerging applications of peptide–oligonucleotide conjugates: Bioactive scaffolds, self-assembling systems, and hybrid nanomaterials. Org. Biomol. Chem. 2019, 17, 1668–1682. 10.1039/C8OB02436G. [DOI] [PubMed] [Google Scholar]
- Ligeour C.; Meyer A.; Vasseur J.-J.; Morvan F. Bis- and tris-alkyne phosphoramidites for multiple 5′-labeling of oligonucleotides by click chemistry. Eur. J. Org. Chem. 2012, 2012, 1851–1856. 10.1002/ejoc.201101763. [DOI] [Google Scholar]
- Karskela M.; Helkearo M.; Virta P.; Lönnberg H. Synthesis of oligonucleotide glycoconjugates using sequential click and oximation ligations. Bioconjugate Chem. 2010, 21, 748–755. 10.1021/bc900529g. [DOI] [PubMed] [Google Scholar]
- Singh Y.; Renaudet O.; Defrancq E.; Dumy P. Preparation of a multitopic glycopeptide–oligonucleotide conjugate. Org. Lett. 2005, 7, 1359–1362. 10.1021/ol050134n. [DOI] [PubMed] [Google Scholar]
- Wada F.; Yamamoto T.; Ueda T.; Sawamura M.; Wada S.; Harada-Shiba M.; Obika S. Cholesterol–GalNAc dual conjugation strategy for reducing renal distribution of antisense oligonucleotides. Nucleic Acid Ther. 2018, 28, 50–57. 10.1089/nat.2017.0698. [DOI] [PubMed] [Google Scholar]
- Zewge D.; Butora G.; Sherer E. C.; Tellers D. M.; Sidler D. R.; Gouker J.; Copeland G.; Jadhav V.; Li Z.; Armstrong J.; Davies I. W. Post-synthetic modification of oligonucleotides via orthogonal amidation and copper catalyzed cycloaddition reactions. Bioconjugate Chem. 2018, 29, 1859–1865. 10.1021/acs.bioconjchem.8b00298. [DOI] [PubMed] [Google Scholar]
- Tajik-Ahmadabad B.; Polyzos A.; Separovic F.; Shabanpoor F. Amphiphilic lipopeptide significantly enhances uptake of charge-neutral splice switching morpholino oligonucleotide in spinal muscular atrophy patient-derived fibroblasts. Int. J. Pharm. 2017, 532, 21–28. 10.1016/j.ijpharm.2017.08.116. [DOI] [PubMed] [Google Scholar]
- Gramlich P. M. E.; Warncke S.; Gierlich J.; Carell T. Click–click–click: Single to triple modification of DNA. Angew. Chem., Int. Ed. 2008, 47, 3442–3444. 10.1002/anie.200705664. [DOI] [PubMed] [Google Scholar]
- Meyer A.; Vasseur J.-J.; Morvan F. Synthesis of monoconjugated and multiply conjugated oligonucleotides by “click thiol” thiol-Michael-type additions and by combination with CuAAC “click Huisgen”. Eur. J. Org. Chem. 2013, 2013, 465–473. 10.1002/ejoc.201201311. [DOI] [Google Scholar]
- Pradère U.; Hall J. Site-specific difunctionalization of structured RNAs yields probes for microRNA maturation. Bioconjugate Chem. 2016, 27, 681–687. 10.1021/acs.bioconjchem.5b00661. [DOI] [PubMed] [Google Scholar]
- Honcharenko M.; Honcharenko D.; Strömberg R. Efficient conjugation to phosphorothioate oligonucleotides by Cu-catalyzed Huisgen 1,3-dipolar cycloaddition. Bioconjugate Chem. 2019, 30, 1622–1628. 10.1021/acs.bioconjchem.9b00217. [DOI] [PubMed] [Google Scholar]
- Houlihan G.; Gatti-Lafranconi P.; Kaltenbach M.; Lowe D.; Hollfelder F. An experimental framework for improved selection of binding proteins using SNAP display. J. Immunol. Methods 2014, 405, 47–56. 10.1016/j.jim.2014.01.006. [DOI] [PubMed] [Google Scholar]
- Houlihan G.; Lowe D.; Hollfelder F. SNAP Display - an in vitro method for the selection of protein binders. Curr. Pharm. Des. 2013, 19, 5421–5428. 10.2174/1381612811319300012. [DOI] [PubMed] [Google Scholar]
- Jirka S. M. G.; Heemskerk H.; Tanganyika-de Winter C. L.; Muilwijk D.; Pang K. H.; de Visser P. C.; Janson A.; Karnaoukh T. G.; Vermue R.; ‘t Hoen P. A. C.; van Deutekom J. C. T.; Aguilera B.; Aartsma-Rus A. Peptide conjugation of 2′-O-methyl phosphorothioate antisense oligonucleotides enhances cardiac uptake and exon skipping in mdx mice. Nucleic Acid Ther. 2014, 24, 25–36. 10.1089/nat.2013.0448. [DOI] [PubMed] [Google Scholar]
- Taskova M.; Madsen C. S.; Jensen K. J.; Hansen L. H.; Vester B.; Astakhova K. Antisense oligonucleotides internally labeled with peptides show improved target recognition and stability to enzymatic degradation. Bioconjugate Chem. 2017, 28, 768–774. 10.1021/acs.bioconjchem.6b00567. [DOI] [PubMed] [Google Scholar]
- Meyer A.; Noël M.; Vasseur J.-J.; Morvan F. Hetero-click conjugation of oligonucleotides with glycosides using bifunctional phosphoramidites. Eur. J. Org. Chem. 2015, 2015, 2921–2927. 10.1002/ejoc.201500165. [DOI] [Google Scholar]
- Kiviniemi A.; Virta P.; Drenichev M. S.; Mikhailov S. N.; Lönnberg H. Solid-supported 2′-O-glycoconjugation of oligonucleotides by azidation and click reactions. Bioconjugate Chem. 2011, 22, 1249–1255. 10.1021/bc200097g. [DOI] [PubMed] [Google Scholar]
- Khan R. S.; Yu C.; Kastin A. J.; He Y.; Ehrensing R. H.; Hsuchou H.; Stone K. P.; Pan W. Brain activation by peptide Pro-Leu-Gly-NH(2) (MIF-1). Int. J. Pept. 2010, 2010, 537639 10.1155/2010/537639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfrum C.; Shi S.; Jayaprakash K. N.; Jayaraman M.; Wang G.; Pandey R. K.; Rajeev K. G.; Nakayama T.; Charrise K.; Ndungo E. M.; Zimmermann T.; Koteliansky V.; Manoharan M.; Stoffel M. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 2007, 25, 1149–1157. 10.1038/nbt1339. [DOI] [PubMed] [Google Scholar]
- Ralhan K.; KrishnaKumar V. G.; Gupta S. Piperazine and DBU: a safer alternative for rapid and efficient Fmoc deprotection in solid phase peptide synthesis. RSC Adv. 2015, 5, 104417–104425. 10.1039/C5RA23441G. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.