

Abstract
New conjugates of substituted 1,2,3-triazoles linked to 1,2,4-triazoles were synthesized starting from the appropriate S-propargylated 1,2,4-triazoles 7 and 8. Ligation of 1,2,4-triazoles to the 1,2,3-triazole core was performed through Cu(I)-catalyzed cycloaddition of 1,2,4-triazole-based alkyne side chain 7 and/or 8 with several un/functionalized alkyl- and/or aryl-substituted azides 9–15 to afford the desired 1,4-disubstituted 1,2,3-triazoles 16–27, using both classical and microwave methods. After their spectroscopic characterization (infrared, 1H, 13C nuclear magnetic resonance, and elemental analyses), an anticancer screening was carried out against some cancer cell lines including human colon carcinoma (Caco-2 and HCT116), human cervical carcinoma (HeLa), and human breast adenocarcinoma (MCF-7). The outcomes of this exploration revealed that compounds 17, 22, and 25 had a significant anticancer activity against MCF-7 and Caco-2 cancer cell lines with IC50 values of 0.31 and 4.98 μM, respectively, in relation to the standard reference drug, doxorubicin. Enzyme-docking examination was executed onto cyclin-dependent kinase 2; a promising aim for cancer medication. Synthesized compounds acquiring highest potency showcased superior interactions with the active site residue of the target protein and exhibited minimum binding energy. Finally, the density functional theory (DFT) calculations were carried out to confirm the outcomes of the molecular docking and the experimental findings. The chemical reactivity descriptors such as softness (δ), global hardness (η), electronegativity (χ), and electrophilicity were calculated from the levels of the predicted frontier molecular orbitals and their energy gap. The DFT results and the molecular docking calculation results explained the activity of the most expectedly active compounds 17, 22, and 25.
1. Introduction
Recently, the development of new chemotherapeutic (anticancer) candidates has been attracting the attention of researchers in life science-oriented research.1 Among these, 1,2,3-triazole motifs were widely explored in medicinal chemistry and still their use is unavoidable because of their unique features such as strong dipole moments, hydrogen bond formation, dipole–dipole and π stacking interactions, and solubility.2 As per the literature, 1,4-disubstituted 1,2,3-triazoles were well known as crucial linkers and displayed bioisosteric effects.2 These effects are probably because of their similarity to amide bonds in terms of planarity and distance.3−6 Thus, 1,2,3-triazole cores were found in several drug structures and bioactive scaffolds showing anti-HIV,7 antimicrobial,8 antiviral,9 and anticancer10−15 activities.
Along with this, 1,2,4-triazole cores among the various heterocycles displayed a wide spectrum of biological properties, and are well documented in the literature.16 This class also serves as antiviral,17 antibacterial,18 antifungal,19 anti-inflammatory,20 and anticancer21 agents. The triazole skeletal moiety is an elite building block in the discovery of potent anticancer agents, and some of its analogues have already been made up to healthcare and clinics or are under clinical trials for fighting against various cancers. Hybrid compounds have captured an important position in cancer therapy, and hybridization of triazole scaffolds with other anticancer pharmacophores may furnish valuable therapeutic intervention for the cancer treatment (Figure 1), specifically drug-resistant cancer.22−25
Figure 1.

Biologically active anticancer drug-based 1,2,3-triazole and/or 1,2,4-triazole.
On the other hand, physicochemical, pharmacokinetic, and pharmacodynamic properties have emerged as some of the crucial properties to appraise the effects or risks of drug candidates on human body. High-rate drug withdrawals escalate the pressure on the pharmaceutical industry and regulators to improve preclinical safety trials. Because in vitro and in vivo screenings are costly and laborious, in silico approaches have been widely implemented to estimate these traits. Computational tools play a pivotal role in drug-discovery programs, right starting from identification of hits to the optimization of the lead compounds and so on. Diverse computational methods and techniques are approachable, which aid in sieving the candidate drug from other molecules on the basis of various properties viz. drug likeness, physicochemical, pharmacodynamic, and pharmacokinetic traits. Molecular docking is a computational method, which is extremely useful and reasonably attested for forecasting putative affinities and binding modes of ligands with macromolecules. Furthermore, the speed and accuracy of the molecular docking approaches have been enhanced and these methods now play a noteworthy role in structure-based drug design. The newly synthesized hybrids were gauged using in silico docking studies for acquiring an additional apprehension of the binding pattern of the target ligands with the cyclin-dependent kinase 2 (CDK2), whose overexpression causes dysfunction of the cell cycle, which is thoroughly allied with hyperproliferation of cancer cells.26−29 Numerous evidences have been put forward in the literature sustaining the concept of restraining cancer progression by aiming CDK2.30−33 In various categories of human tumors, the dysregulation of CDK2 is noticed, thereby for anticancer therapy, making CDK2 a promising drug target.26,34−41 In breast cancer progression, CDK2 is considered accountable for activating and phosphorylating the receptor hormones. CDK2 structure with Protein Data Bank (PDB) ID: 2R3I was recruited for carrying out molecular docking studies owing to its higher resolution (1.28 Å) and nonmutated monomeric form. The target triazole skeletons have been reported to be CDK2 inhibitors10,42−44 (Pathoor and Bahulayan 2018, Kassem, Abbas et al. 2019, Xu, Zhao et al. 2019, Elgogary, Khidre et al. 2020).10,42−44 Considering our interest in the design and synthesis of 1,2,3-triazole-based heterocyclic motifs45−54 as bioactive scaffolds and exploring their fascinating binding mode, we have attempted to synthesize a newer library of 1,2,3-triazoles encompassing 1,2,4-triazole cores using both classical and microwave irradiation (MWI) methods, and subsequently investigated their anticancer activity. The anticancer activity results were supported by molecular docking and density functional theory (DFT) study in terms of the levels of the frontier molecular orbitals (FMOs) as well as their energy gap. The predicted levels of the molecular orbitals were used in simulation of parameters that are important for explaining the activity of the prepared compounds. The tested compounds exhibiting highest anticancer activities also displayed minimum binding energy on the target receptor of CDK2 along with showing a better binding affinity and fitting inside the active site of the protein molecular surface.
2. Results and Discussion
2.1. Chemistry
The synthetic strategies adopted in the present work are depicted in Schemes 1 and 2. The targeted 1,2,4-triazole-1,2,3-triazole molecular conjugates 16–27 were synthesized from two 1,2,4-triazole units 5 and 6 as starting materials (Scheme 2), which in turn were obtained through mulistep synthesis from the appropriate acid hydrazides, 1 and 2, according to the reported procedures55,56 (Scheme 1). Thermal condensation of benzohydrazide (1) and acetohydrazide (2) with phenylisothiocyanate for 4 h gave the respective acid thiosemicarbazides 3 and 4 in 86 and 85% yields; MWI required 2 min to give 96 and 95% yield of 3 and 4, respectively.
Scheme 1. Microwave Versus Conventional Synthesis of 1,2,4-Triazoles 5 and 6.

Scheme 2. Microwave Versus Conventional Synthesis of 1,2,4-Triazole-1,2,3-triazole Molecular Conjugates 16–27.

MWI of a solution of acid thiosemicarbazide 3 and/or 4 in aqueous sodium hydroxide for 3 min resulted in the formation of the targeted precursors namely 4-methyl-5-phenyl-4H-1,2,4-triazole-3-thione (5) and 4,5-diphenyl-4H-1,2,4-triazole-3-thione (6) in 93 and 92% yields, respectively (Table 1). 1,2,4-Triazoles 5 and 6 were conventionally obtained in 84–85% yields, by thermal dehydrative intramolecular cyclization of the corresponding thiosemicarbazide derivatives 3 and/or 4 in aqueous sodium hydroxide for 4 h, as reported in the literature.
Table 1. Microwave Assisted Organic Synthesis Versus Conventional Synthesis of 1,2,3-Triazoles 16–27: Times and Yields.
| conventional
procedure (CP) |
microwave
procedure (MWI) |
|||
|---|---|---|---|---|
| compound no | time (h) | yield (%) | time (min) | yield (%) |
| 3 | 4 | 86 | 2 | 96 |
| 4 | 4 | 85 | 2 | 95 |
| 5 | 4 | 85 | 3 | 93 |
| 6 | 4 | 84 | 3 | 92 |
| 7 | 1 | 91 | 2 | 97 |
| 8 | 1 | 92 | 2 | 98 |
| 16 | 6 | 86 | 4 | 94 |
| 17 | 6 | 87 | 4 | 95 |
| 18 | 6 | 86 | 4 | 94 |
| 19 | 6 | 87 | 4 | 95 |
| 20 | 6 | 88 | 4 | 96 |
| 21 | 6 | 88 | 4 | 96 |
| 22 | 10 | 81 | 6 | 91 |
| 23 | 10 | 82 | 6 | 91 |
| 24 | 10 | 81 | 6 | 90 |
| 25 | 10 | 82 | 6 | 91 |
| 26 | 10 | 83 | 6 | 92 |
| 27 | 10 | 81 | 6 | 91 |
The success of the intramolecular cyclodehydration was also supported by the spectral data of the resulted 1,2,4-triazoles 5 and 6. Their infrared (IR) spectra exhibited common characteristic absorption bands at 3276–3389 cm–1 assigned to (NH), 1288–1295 cm–1 (C=S), and 1620–1625 cm–1 (C=N) confirming the formation of the triazole ring. In their 1H nuclear magnetic resonance (NMR) spectra, the appearance of the diagnostic triazole-NH singlet at δH 13.70–14.16 ppm was a clear proof for the formation of the targeted triazoles 5 and 6 in their thione forms. In addition, the signal belonging to the C=S group was recorded at δC 167.34–168.57 ppm confirming the predominance of the thione isomer. All these spectral data supported that compounds 3 and 4 underwent ring closure to give the corresponding 1,2,4-triazoles 5 and 6, respectively.
The 1,3-dipolar cycloaddition reaction of organoazides with terminal alkynes was reported as an efficient and convenient method for the regioselective synthesis of 1,4-disubstituted 1,2,3-triazoles.57 This strategy is right way a center of attention because of its high regioselectivity that runs into the demand of click chemistry in organic synthesis.57 Accordingly, the synthesis of the targeted 1,2,3-triazoles with thio-1,2,4-triazole connected via a flexible methylene spacer involved Huisgen 1,3-dipolar cycloaddition of the thiopropargylated 1,2,4-triazoles 7 and/or 8 as terminal alkynes with different un/functionalized alkyl- and/or aryl-substituted azides 9–15 (Scheme 2).
Therefore, the alkyne precursors 7 and 8 were successfully synthesized in 91–92% yields through the propargylation of the 1,2,4-triazole-3-thiones 5 and 6 in the presence of triethylamine as basic catalyst for 1 h. Under MWI, only 2 min was required to give the same thiopropargylated-1,2,4-triazoles 7 and 8 in excellent yield (97–98%) (Table 1). The structures of the thiopropargylated derivatives 7 and 8 were elucidated based on their spectral data. Their IR spectra exhibited their principal functional groups. The acetylenic hydrogen (≡C–H) and carbons (−C≡C−) were recorded at 3305–3328 and 2145–2155 cm–1, respectively.
The investigation of their 1H NMR spectra revealed the presence of a diagnostic triplet and a doublet in the aliphatic region at δH 2.24–3.28 and 3.94–4.00 ppm relative to sp-CH and SCH2 protons, respectively, confirming the incorporation of the propargyl side chain. In addition, their 13C NMR spectra confirmed the propargyl carbon signals (SCH2 and C≡C) resonating around δC 20.89–21.27 and 72.34–79.48 ppm, respectively. The remaining carbons were also recorded on their respected chemical shifts (see the Experimental Section).
The targeted 1,2,4-triazole-1,2,3-triazole molecular conjugates 16–27 were well synthesized via the Cu(I)-assisted 1,3-dipolar cycloaddition of the latest thiopropargylated 1,2,4-triazoles 7 and 8 with some selected azides 9–15 at room temperature for 8–10 h in the presence of copper sulfate and sodium ascorbate as catalysts. It was noticeable that the 1,2,3-triazoles carrying aliphatic side chain (benzyl, alkyl long chain and/or ester side chain) at N-1 atom 16–21 were obtained in good yields (86–88%) compared to their analogues bearing aromatic substituents 22–27 (81–83%). On the other hand, it was observed that the click ligation has been successfully accomplished under MWI and afforded the desired click products 16–27 in excellent yields (90–96%) with a great reduction in reaction times (4–6 min) (Table 1). It should be noted that the used azides 9–15 were synthesized from their respective alkyl halides and/or substituted anilines as reported in the literature.58−60
The structures of the 1,2,3-triazoles 16–27 have been established on the basis of their spectral data (IR, 1H NMR, 13C NMR, and elemental analysis). All the IR spectra showed clearly the disappearance of the alkyne groups (C≡C) and sp-H of their starting materials, confirming their involvement in the cycloaddition reaction.
Almost all the synthesized click adducts 16–27 displayed similar spectral patterns (1H NMR and 13C NMR). Their 1HNMR spectra confirmed the absence of the signals related to the lateral hydrogen of the ≡C–H group of their corresponding alkynes 7 and 8 around δH 2.24–2.28 ppm, and showed the presence of one distinct signal at δH 8.01–8.85 ppm attributed to the 1,2,3-triazole-CH proton supporting the formation of such a ring. The 1,2,3-triazole derivatives 16–21 exhibited the common NCH2 protons at 4.30–5.57 ppm. In addition, the 1H and 13C NMR spectra of compounds 16–19 were characterized by the appearance of extra aromatic protons and carbons. Moreover, a triplet and a quartet were observed at 1.21–1.22 and 4.15–4.18 ppm in the 1H NMR spectra attributed to the ethyl group of the ester functionality appended by the triazoles 20 and 21. Additionally, in their 13C NMR spectra, their structure was also evidenced by the appearance of the carbonyl ester carbon around 166.99–167.81 ppm. In contrast, the triazoles 22–27 appended with aromatic rings revealed the presence of three or four extra protons and carbons in their 1H and 13C NMR spectra (see the Experimental Section).
2.2. DFT Molecular Structure Estimation
The theoretical calculations by the DFT were done in the gas phase at the B3LYP 6-311G(d,p) basis set implemented into the Gaussian 9. This included the simulation of the geometrical optimization on each prepared compound to determine the minimum energy molecular structure, followed by the calculation of the frequency at the optimized structural geometry during which many thermochemical quantities are also calculated. All optimum structures of the prepared compounds (7, 8, and 16–27) are considered stable because of the absence of the imaginary frequency. The findings of the DFT of the optimized molecular structures for all investigated compounds show the nonplanarity of all compounds as shown in Figure 2.
Figure 2.

Optimized geometrical structures of the prepared compounds 7, 8, and 16–27.
2.3. In Vitro Anticancer Screening
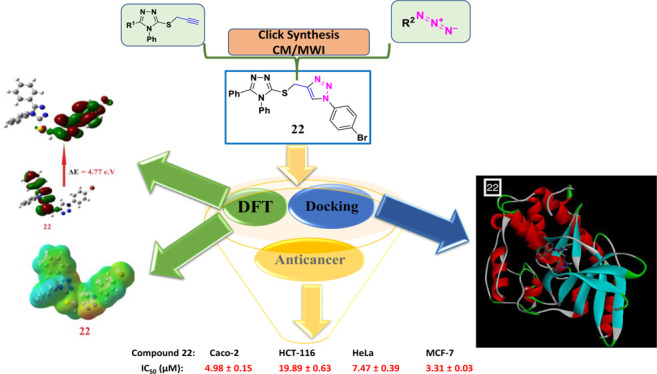
The synthesized compounds, 7, 8, and 16–27 were in vitro screened for anticancer activity using four different cancer cell lines namely human colon carcinoma (Caco-2 and HCT116), human cervical carcinoma (HeLa), and human breast adenocarcinoma (MCF-7). The standard reference drug considered was doxorubicin. The data are summarized in Table 2. The half maximal inhibitory concentration (IC50) of compound 17 exhibited increased selectivity against MCF-7 with an IC50 of 0.31 μM, while compound 22 showed more selectivity against MCF-7 and Caco-2 cell lines with IC50 of 3.31 and 4.98 μM, respectively, however, compound 25 showed moderate activity against MCF-7 and Caco-2 cell lines with IC50 of 4.46 and 7.22 μM, respectively. While compounds 7, 8, 16, 19, 20, and 21 revealed substandard activity against all the four cell lines tested in this study. The compounds 17, 18, and 23–27 displayed good anticancer activity against the Caco-2 cell line (IC50 = 7.22–19.09 μM). By gauging the inhibitory effect of the tested compounds in comparison to the standard drug doxorubicin against the HCT-116, HeLa, and MCF-7 cell lines, it was ascertained that the majority of the screened compounds possess good to moderate anticancer activity. Overall, among the synthesized compounds, compounds 17, 22, and 25 have demonstrated the most potent anticancer activity against all cancer cell lines explored in the present study.
Table 2. Anticancer Screenings of the Synthesized Compounds against Caco-2, HCT116, HeLa, and MCF7 Human Cancer Cell Linesa.
| compound no | Caco-2 | HCT-116 | HeLa | MCF-7 |
|---|---|---|---|---|
| 7 | 165.33 ± 2.45 | >200 | 124.67 ± 4.67 | 93.43 ± 3.56 |
| 8 | 168.71 ± 1.98 | >200 | 120.98 ± 4.84 | 98.26 ± 4.41 |
| 16 | 79.67 ± 1.67 | 91.19 ± 4.23 | 39.75 ± 2.14 | 34.79 ± 1.73 |
| 17 | 16.36 ± 1.12 | 43.45 ± 2.34 | 4.41 ± 0.23 | 0.31 ± 0.01 |
| 18 | 19.09 ± 1.02 | 55.60 ± 2.78 | 24.65 ± 1.87 | 17.29 ± 1.13 |
| 19 | 85.72 ± 2.12 | 97.23 ± 3.45 | 42.17 ± 2.46 | 36.06 ± 2.05 |
| 20 | 96.79 ± 3.18 | 124.32 ± 4.45 | 56.35 ± 3.78 | 52.75 ± 3.46 |
| 21 | 99.47 ± 3.15 | 126.87 ± 4.67 | 61.10 ± 3.90 | 54.68 ± 3.57 |
| 22 | 4.98 ± 0.15 | 19.89 ± 0.63 | 7.47 ± 0.39 | 3.31 ± 0.03 |
| 23 | 12.34 ± 1.41 | 35.78 ± 1.98 | 29.25 ± 1.23 | 23.09 ± 1.84 |
| 24 | 10.21 ± 0.38 | 30.09 ± 1.65 | 19.05 ± 0.45 | 14.78 ± 1.23 |
| 25 | 7.22 ± 0.26 | 22.34 ± 0.87 | 11.05 ± 0.56 | 4.46 ± 0.02 |
| 26 | 14.18 ± 1.51 | 39.55 ± 2.11 | 31.79 ± 2.04 | 28.46 ± 1.93 |
| 27 | 11.67 ± 1.34 | 32.41 ± 1.92 | 22.08 ± 1.29 | 18.14 ± 1.14 |
| doxorubicin | 5.17 ± 0.25 | 5.64 ± 0.17 | 1.25 ± 0.02 | 0.65 ± 0.01 |
IC50 values are expressed as mean ± SD of three independent experiments.
2.4. In Silico Analysis
In order to qualify to be a drug candidate, some prerequisites such as physicochemical, pharmacodynamic, and pharmacokinetic parameters should be attained by any chemical entity. Thus, the potential of a drug aspirant can be evaluated by forecasting in silico physicochemical attributes, drug likeness, or adsorption, distribution, metabolism, excretion, and toxicity (ADMET) traits. In silico analysis was performed to corroborate the reliability of in vitro anticancer screenings. Calculations were made for different physicochemical characteristics such as the enumeration of particular atom class, the count of rotatable bonds, lipophilicity, molar refractivity, and water solubility. Topological polar surface area (TPSA), an attribute that is explored to forecast the passive molecular transport trait of drugs, was also appraised. These physicochemical properties are shown in Table 3. The physicochemical attributes are in good agreement with the applied criteria and are expected to have good bioavailability score as all the compounds have TPSA ≤ 140 Å.
Table 3. Physicochemical Properties of the Selected Compounds.
| comp. no. | fraction C sp3a | no. of rotatable bonds | HBAb | HBDc | i log Pd | molar refractivity | log Se | TPSAf |
|---|---|---|---|---|---|---|---|---|
| 7 | 0.06 | 4 | 2 | 0 | 3.17 | 86.29 | MS | 56.01 |
| 8 | 0.17 | 3 | 2 | 0 | 2.55 | 65.82 | S | 56.01 |
| 16 | 0.08 | 7 | 4 | 0 | 3.67 | 122.33 | PS | 86.72 |
| 17 | 0.52 | 20 | 4 | 0 | 6.57 | 169.95 | I | 86.72 |
| 18 | 0.30 | 10 | 4 | 0 | 4.27 | 121.88 | PS | 86.72 |
| 19 | 0.16 | 6 | 4 | 0 | 3.05 | 101.86 | PS | 86.72 |
| 20 | 0.19 | 9 | 6 | 0 | 3.45 | 113.55 | PS | 113.02 |
| 21 | 0.31 | 8 | 6 | 0 | 2.38 | 93.08 | MS | 113.02 |
| 22 | 0.04 | 6 | 4 | 0 | 3.86 | 125.62 | PS | 86.72 |
| 23 | 0.04 | 6 | 5 | 0 | 3.92 | 117.88 | PS | 86.72 |
| 24 | 0.04 | 6 | 4 | 0 | 4.19 | 127.94 | PS | 86.72 |
| 25 | 0.11 | 5 | 4 | 0 | 3.63 | 105.15 | PS | 86.72 |
| 26 | 0.11 | 5 | 5 | 0 | 3.37 | 97.41 | PS | 86.72 |
| 27 | 0.11 | 5 | 4 | 0 | 3.64 | 107.47 | PS | 86.72 |
The ratio of sp3 hybridized carbons over the total carbon count of the molecule.
Number of hydrogen bond acceptors.
Number of hydrogen bond donors.
Lipophilicity.
Water solubility (SILICOS-IT [I = insoluble, PS = poorly soluble, MS = moderately soluble, and S = soluble]).
TPSA (Å2).
In silico pharmacokinetic/ADME evaluations were executed, and the forecasted data are presented in Table 4, which unveil that all the screened compounds showed high gastrointestinal absorptions (GI) except compound 17. All of them have no BBB (blood brain barrier) permeability except compounds 7 and 8. The majority of the compounds are P-gp (p-glycoprotein) noninhibitors. The prediction for BBB permeations, passive human GI absorption, and P-gp substrates is pooled in a built-in graphical classification model, that is, the BOILED-Egg diagram, as displayed in Figure 3. All the synthesized compounds were tested for inhibition of the Cytochrome P450 isomers, that is, CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4. Most of the compounds are CYP1A2 inhibitors except 16 and 21–24. All the tested compounds are inhibitors of CYP2C19 with the exception of compound 17 for CYP2C9 and compounds 7 and 8 for CYP3A4. The tested compounds emerged to be noninhibitors of CYP2D6. The tested compounds possess low skin permeability.
Table 4. Pharmacokinetic/ADME Properties of the Selected Compounds.
| pharmacokinetic/ADME properties |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| comp. no | GI absa | BBB permeantb | P-gpsubstratec | CYP1A2 inhibitord | CYP2C19 inhibitore | CYP2C9 inhibitorf | CYP2D6 inhibitorg | CYP3A4 inhibitorh | log Kpi |
| 7 | high | yes | no | yes | yes | yes | no | no | –5.44 |
| 8 | high | yes | no | yes | yes | yes | no | no | –5.97 |
| 16 | high | no | yes | no | yes | yes | no | yes | –5.71 |
| 17 | low | no | yes | yes | yes | no | no | yes | –2.20 |
| 18 | high | no | no | yes | yes | yes | no | yes | –5.19 |
| 19 | high | no | no | yes | yes | yes | no | yes | –6.22 |
| 20 | high | no | no | yes | yes | yes | no | yes | –6.54 |
| 21 | high | no | no | no | yes | yes | no | yes | –7.06 |
| 22 | high | no | yes | no | yes | yes | no | yes | –5.56 |
| 23 | high | no | yes | no | yes | yes | no | yes | –5.61 |
| 24 | high | no | yes | no | yes | yes | no | yes | –5.11 |
| 25 | high | no | no | yes | yes | yes | no | yes | –6.08 |
| 26 | high | no | no | yes | yes | yes | no | yes | –6.13 |
| 27 | high | no | no | yes | yes | yes | no | yes | –5.62 |
Gastrointestinal absorption.
Blood brain barrier permeant.
P-gp substrate.
CYP1A2: cytochrome P450 family 1 subfamily A member 2 (PDB: 2HI4).
CYP2C19: cytochrome P450 family 2 subfamily C member 19 (PDB: 4GQS).
CYP2C9: cytochrome P450 family 2 subfamily C member 9 (PDB: 1OG2).
CYP2D6: cytochrome P450 family 2 subfamily D member 6 (PDB: 5TFT).
CYP3A4: cytochrome P450 family 3 subfamily A member 4 (PDB: 4K9T).
Skin permeation in cm.
Figure 3.

BOILED-Egg diagram of the selected compounds.
In silico AMES toxicity and carcinogenicity are assessed and are given in Table 5. All the synthesized compounds were noncarcinogenic and AMES nontoxic in nature except compounds 7, 8, 16, and 19, which emerged to be AMES toxic in nature. Furthermore, the computed rat acute toxicity, that is, LD50 in mol/kg seems to be sufficiently safe in the range 2.238–2.74 mol/kg.
Table 5. In Silico-Predicted LD50, Toxicity, and Carcinogenicity Profiles of the Selected Compounds.
| <!—Col Count:5F0E0comp. no. | AMES toxicity | carcinogenicity | rat acute toxicity LD50, (mol/kg) |
|---|---|---|---|
| 7 | Toxic | noncarcinogenic | 2.238 |
| 8 | Toxic | noncarcinogenic | 2.329 |
| 16 | Toxic | noncarcinogenic | 2.329 |
| 17 | nontoxic | noncarcinogenic | 2.680 |
| 18 | nontoxic | noncarcinogenic | 2.680 |
| 19 | Toxic | noncarcinogenic | 2.354 |
| 20 | nontoxic | noncarcinogenic | 2.685 |
| 21 | nontoxic | noncarcinogenic | 2.744 |
| 22 | nontoxic | noncarcinogenic | 2.350 |
| 23 | nontoxic | noncarcinogenic | 2.396 |
| 24 | nontoxic | noncarcinogenic | 2.290 |
| 25 | nontoxic | noncarcinogenic | 2.371 |
| 26 | nontoxic | noncarcinogenic | 2.416 |
| 27 | nontoxic | noncarcinogenic | 2.321 |
The FMO and the highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals have been in focus for clarifying several forms in chemical reactivity. Recently, the impact of the FMOs of the materials on their biological reactivity have been taken into consideration. Many reports61,62 show the correlation between FMOs and several biological properties such as antimicrobial,63−65 anticancer,66−68 antifungal,69,70 and cytotoxic71−73 activities, which is a new drug-design field.74
The energy level as well as the energy gap of the FMOs (EHOMO, ELUMO and ΔE) are good tools to explore several important parameters such as the potential required for ionization from the HOMO level (I = −EHOMO) and the electron affinity from the energy of the LUMO (A = −ELUMO). In addition, the FMOs are excellent in appraising various chemical reactivity descriptors such as softness (δ), global hardness (η), electronegativity (χ), and electrophilicity (ω). These parameters are calculated according to the following equations75
| 1 |
| 2 |
| 3 |
| 4 |
The χ value is an appraisal for the behavior of the compound to accept electrons, that is, Lewis acidity (Table 6). The magnitude of the molecules for charge transfer prevention is calculated from the global hardness (η); however, the global softness (δ) describes the capability of a molecule for electronic transportation.75 Soft compounds have a small FMO energy gap and are more reactive to transfer their electrons to the acceptors than the harder one.
Table 6. Chemical Reactivity Descriptors and Dipole Moment (μ, Debye) of Investigated Compounds 7, 8, and 16–27.
| comp. no. | ELUMO | EHOMO | ΔE | χ | Η | δ | ω | I | A | μ |
|---|---|---|---|---|---|---|---|---|---|---|
| 7 | –1.28 | –6.28 | 5.00 | 3.78 | 2.50 | 0.40 | 2.86 | 6.28 | 1.28 | 4.87 |
| 8 | –1.12 | –6.45 | 5.33 | 3.79 | 2.67 | 0.38 | 2.69 | 6.45 | 1.12 | 4.69 |
| 16 | –1.31 | –6.26 | 4.95 | 3.79 | 2.48 | 0.40 | 2.89 | 6.26 | 1.31 | 9.95 |
| 17 | –1.33 | –6.27 | 4.94 | 3.80 | 2.47 | 0.40 | 2.92 | 6.27 | 1.33 | 8.92 |
| 18 | –1.31 | –6.25 | 4.94 | 3.78 | 2.47 | 0.40 | 2.89 | 6.25 | 1.31 | 8.82 |
| 19 | –1.12 | –6.41 | 5.29 | 3.77 | 2.65 | 0.38 | 2.68 | 6.41 | 1.12 | 5.43 |
| 20 | –1.24 | –6.20 | 4.96 | 3.72 | 2.48 | 0.40 | 2.79 | 6.20 | 1.24 | 8.86 |
| 21 | –1.11 | –6.39 | 5.28 | 3.75 | 2.64 | 0.38 | 2.66 | 6.39 | 1.11 | 8.66 |
| 22 | –1.63 | –6.40 | 4.77 | 4.02 | 2.39 | 0.42 | 3.38 | 6.40 | 1.63 | 8.60 |
| 23 | –1.50 | –6.41 | 4.91 | 3.96 | 2.46 | 0.41 | 3.19 | 6.41 | 1.50 | 8.02 |
| 24 | –1.89 | –6.52 | 4.63 | 4.21 | 2.32 | 0.43 | 3.82 | 6.52 | 1.89 | 9.13 |
| 25 | –1.58 | –6.56 | 4.98 | 4.07 | 2.49 | 0.40 | 3.33 | 6.56 | 1.58 | 8.57 |
| 26 | –1.47 | –6.56 | 5.09 | 4.02 | 2.55 | 0.39 | 3.17 | 6.56 | 1.47 | 8.10 |
| 27 | –1.88 | –6.68 | 4.80 | 4.28 | 2.40 | 0.42 | 3.82 | 6.68 | 1.88 | 9.34 |
The electrophilicity (ω) could be calculated from the predicted electronegativity and chemical hardness is an indicator for the lower energy because of the highest electronic gap between the acceptor LUMO and the donor HOMO.
The binding affinity in kcal/mol of all the docked ligands is given in Table 7. The ligands unveiled good to excellent binding affinities toward the target receptor CDK2 ranging from −9.9 to −6.9 kcal/mol. The molecular docking studies disclosed that the compounds 17, 22, and 25 were the most promising candidates, which is justified by lowest binding energy, that is, −9.4, −9.9. and −9.1 kcal/mol, respectively, with the CDK2 active site residues. Docking and 2D diagrams illustrating protein–ligand interactions of these compounds, that is, 17, 22, and 25, are shown in Figure 4a,b, respectively. These compounds entrenched a network of interactions at the molecular level, that is, H-bonds, atomic charge, and π–σ interactions with the CDK2 active site residues when examined in 2D plots. Distinct interactions were depicted by different colors: for example, purple indicates the covalent bond; pink denotes electrostatic interaction; and green indicates van der Waals molecular interaction. Light blue shading surrounding the atom or residue reveals the solvent accessibility of the ligand atom and the amino acid residues while high shading signifies more exposure to solvent. Compound 17 displayed various binding interactions including conventional H bonds with residue Asp86 and Gln132. Compound 22 established various interactions embracing a conventional hydrogen bond with Leu83. Likewise, 25 was involved in several binding interactions comprising H bonds with Ile10 and Leu83. Thus, the minimum binding energies and H bond interactions of these compounds 17, 22, and 25 with the active site residues of CDK2 might be one of the justifications for the good activities displayed by these molecules in in vitro screenings.
Table 7. Drug Likeness Predictions and Docking Scores of the Selected Compounds.
| comp. no. | Lipinski violations | Ghose violations | Veber violations | Egan violations | Muegge violations | bioavailability score | binding affinity (kcal/mol) |
|---|---|---|---|---|---|---|---|
| 7 | 0 | 0 | 0 | 0 | 0 | 0.55 | –8.0 |
| 8 | 0 | 0 | 0 | 0 | 0 | 0.55 | –6.9 |
| 16 | 1 | 0 | 0 | 0 | 0 | 0.55 | –7.8 |
| 17 | 2 | 4 | 1 | 1 | 2 | 0.17 | –9.4 |
| 18 | 1 | 0 | 0 | 0 | 1 | 0.55 | –8.5 |
| 19 | 0 | 0 | 0 | 0 | 0 | 0.55 | –7.5 |
| 20 | 0 | 0 | 0 | 0 | 0 | 0.55 | –8.4 |
| 21 | 0 | 0 | 0 | 0 | 0 | 0.55 | –7.9 |
| 22 | 1 | 1 | 0 | 0 | 1 | 0.55 | –9.9 |
| 23 | 1 | 0 | 0 | 0 | 0 | 0.55 | –8.1 |
| 24 | 1 | 1 | 0 | 1 | 1 | 0.55 | –8.8 |
| 25 | 0 | 0 | 0 | 0 | 0 | 0.55 | –9.1 |
| 26 | 0 | 0 | 0 | 0 | 0 | 0.55 | –8.9 |
| 27 | 1 | 0 | 0 | 0 | 0 | 0.55 | –8.9 |
Figure 4.

(a) Docking poses of selected compounds 17, 22, and 25 onto the active site of CDK2 (PDB ID: 2R3I).
To appraise the drug likeness, various rules like Lipinski,76 Ghose,77 Veber,78 Egan,79 and Muegge80 rules were implemented. Moreover, the bioavailability scores along with the number of violations to these rules are given in Table 7. Our results regarding the drug-like attributes showed that most of our compounds obeyed these rules with few exceptions. The entire tested compounds were in good agreement in terms of the bioavailability score being 0.17–0.55. In sequence of analyzing the binding affinity of the ligands with the target protein, molecular docking studies of newly synthesized target compounds on CDK2 (PDB ID: 2R3I) as the target receptor were carried out. Computational docking analysis was executed utilizing PyRx and AutoDockVina option based on the scoring functions. The minimum binding energy manifests that the CDK2 (target receptor) was successfully docked with our compounds.
The DFT calculations of FMOs to compare the level of energy of the HOMO and LUMO as well as with their energy gap of the most expectedly active compounds 17, 22, and 25 from the molecular docking calculations were investigated. Such an alternative factor, the FMOs (HOMO and LUMO energy values), that influences the binding affinity of compounds determines the way the compound interacts with receptor proteins.73 The energy levels of HOMOs of the compounds under investigation are in between −6.20 and −6.68 eV, however, the LUMOs are in between −1.11 and −1.89 eV depending on the degree of conjugation as well as the nature of the attached substituents around the triazole nucleus. Based on the energy level of the HOMOs of the most reactive compound from the molecular docking of 17, 22, and 25, compound 25 was predicted to have the least lying HOMO (−6.56 eV) as compared to the others. Furthermore, the order of the FMO energy gap was 4.49, 4.77, and 4.98 eV for 17, 22, and 25, respectively. It is clear that the compound 17 with the high lying HOMO, EHOMO = −6.27 eV, is the most susceptible to be an electron donor, however, compound 22 is considered the best electron acceptor ELUMO = −1.63 eV and consequently these data could illustrate the higher affinity of interaction of 22 in comparison to compound 17 of the same derivative and 25 of its methyl analogue. Moreover, the highest binding affinity of 22 could also be explained in terms of its lower energy gap between the FMOs, ΔE = 4.77 eV. Moreover, compound 22 with the higher basicity (χ = 4.02) rather than the other derivative 17 (χ = 3.80) could be another explanation for its binding affinity. Another influence that could impact the degree of molecular interaction of these compounds with the protein is the dipole moment.81−83 The calculated dipole moments of the prepared compounds 7, 8, and 17–27 are in the range of 4.69–9.95 debye, however, compound 17 showed the highest and 25 showed the least among the compounds with higher binding affinity (Table 7). All of these investigated factors show a range of significant influences on the affinity for their binding with active protein sites as shown in Figure 5.
Figure 5.

Calculated ground state isodensity surface plots for FMOs for the most reactive compounds expected from the molecular docking 17, 22, and 25.
2.5. Molecular Electrostatic Potential
To confirm the evidence for the interaction of these compounds as inhibitors, the molecular electrostatic potential (MEP) could be a significant trait to be calculated. Although the MEP shows information about the shape and the size of the electrostatic potential, it can also be a tool for predicting physiochemical property relationships with the molecular structure. Moreover, MEP is a useful tool for predicting the effectiveness of the studied compounds toward electrophilic and nucleophilic attacks.
The MEP of the best binding affinity compounds 17, 22, and 25 is estimated using the same method and basis sets and is given in Figure 6. It is evident that the size and shape as well as the orientation of the negative (red, electrophilic attack sites), positive (blue, nucleophilic attack sites), and neutral potentials differ according to the electronic nature of the attached groups. The difference in mapping this electrostatic potential around the drug-like compounds is mainly responsible for the degree to which the binding to the receptors occurs at the active sites of the targeted receptor.
Figure 6.

Molecular electrostatic potentials (MEP) of the most binding affinity compounds 17, 22, and 25.
3. Conclusions
A focused library of 1,2,3-triazoles encompassing 1,2,4-triazole cores was designed and successfully synthesized through a click reaction of a proper S-propargylated 1,2,4-triazoles 7 and 8 using the Cu(I)-catalyst. The reaction proceeded effectively either by classical or MWI and yielded the desired click adduct 16–27 in good to excellent yields. The anticancer screening was also investigated against some cancer cell lines including Caco-2 and HCT116, HeLa, and MCF-7. The results revealed that the compound 17 emerged as the most potent tested compounds against the MCF-7 cancer cell line with an IC50 value of 0.31 μM while compounds 22 and 25 exhibited good anticancer activity against the MCF-7 cancer cell line with IC50 values of 4.98 and 7.22 μM, respectively. The experimental results were proved using the enzyme docking examination onto CDK2 as well as DFT calculations. The inhibitory activity of compounds 17, 22, and 25 has been explained by two major factors: minimum binding energy and H-bond interactions of the ligand with the target protein CDK2. Using the DFT calculations and the molecular docking calculations, the activity of the most active compounds 17, 22, and 25 was explained in terms of the predicted chemical reactivity descriptors as well as molecular electrostatic potential.
4. Experimental Section
All reagents and solvents used were of the highest quality of analytical reagent grade and were used without further purification. Fine chemicals and solvents were purchased from BDH Chemicals Ltd. and Sigma-Aldrich. Melting points are uncorrected and were measured on a Stuart Scientific SMP. Thin layer chromatography (TLC) was performed on UV fluorescent Silica gel Merck 60 F254 plates, and the spots were visualized using a UV lamp (254 nm). FT-IR spectrometer type PerkinElmer 1430 series was used for the investigation of the functional groups in the range of 400–4000 cm–1. The NMR spectra were obtained using a Bruker spectrometer (400 MHz) with tetramethylsilane as an internal reference. Elemental analyses were performed using a GmbH-Vario EL III Elementar Analyzer.
4.1. General Procedure for the Synthesis of Acid Thiosemicarbazides 3–4
4.1.1. Conventional Procedure
Benzohydrazide (1) and/or acetohydrazide (2) (10 mmol) was dissolved in ethanol (30 mL) containing phenylisothiocyanate (10 mmol). The mixture was heated under reflux for 4 h, and the reaction was monitored by TLC. After cooling, the crude product was collected by filtration and recrystallized from ethanol to afford the targeted acid thiosemicarbazides 3–4.
4.1.2. Microwave Procedure
In a closed borosilicate glass vessel fitted with a silicone cap, a solution of 3-benzohydrazide (1) and/or acetohydrazide (2) (1 mmol) dissolved in ethanol (5 mL) containing phenylisothiocyanate (1 mmol) was exposed to irradiation for 2 min using a microwave reactor. The reaction was processed as described in the conventional procedure (CP) outlined earlier to afford the same acid thiosemicarbazides 3–4.
4.1.3. Characterization of 2-Benzoyl-N-phenylhydrazinecarbothioamide (3)
mp 165–166 °C (Lit. mp 163 °C);55 IR (KBr, ν): 3288–3346 (NH), 3076 (CH-Ar), 1685 (C=O), 1548 (C=C), 1283 (C=S). 1H NMR (400 MHz, DMSO-d6): δH 7.17 (1H, t, J = 8.0 Hz, HAr), 7.35 (2H, t, J = 8.0 Hz, HAr), 7.50–7.61 (5H, m, HAr), 8.01 (2H, d, J = 8.0 Hz, HAr), 9.72 (1H, br s, NH), 9.84 (H, br s, NH), 10.56 (1H, br s, NH). 13C NMR (100 MHz, DMSO-d6): δC 124.99, 125.81, 127.99, 128.21, 131.83, 132.52, 139.25 (CAr); 165.99 (C=O); 181.33 (C=S). Calcd for C14H13N3OS: C, 61.97; H, 4.83; N, 15.49. Found: C, 61.69; H, 4.66; N, 15.37.
4.1.4. Characterization of 2-Acetyl-N-phenylhydrazinecarbothioamide (4)
mp 175–176 °C (Lit. mp 178 °C);56 IR (KBr, ν): 3274–3339 (NH), 3045 (CH-Ar), 2986 (CH-Al), 1675 (C=O), 1555 (C=C), 1289 (C=S). 1H NMR (400 MHz, DMSO-d6): δH 1.90 (3H, s, CH3), 7.16 (1H, t, J = 8.0 Hz, HAr), 7.33 (2H, t, J = 8.0 Hz, HAr), 7.43–7.49 (2H, m, HAr), 9.53 (1H, br s, NH), 9.62 (H, br s, NH), 9.88 (1H, br s, NH). 13C NMR (100 MHz, DMSO-d6): δC 21.00 (CH3); 125.05, 125.91, 128.01, 139.11 (CAr); 169.17 (C=O); 180.94 (C=S). Calcd for C9H11N3OS: C, 51.65; H, 5.30; N, 20.08. Found: C, 51.89; H, 5.40; N, 20.25.
4.2. General Procedure for the Synthesis of 1,2,4-Triazole-3-thiones 5–6
4.2.1. Conventional Procedure
The appropriate acid thiosemicarbazide 3–4 (10 mmol) was dissolved in 10% aqueous solution of sodium hydroxide (100 mL). The mixture was allowed to react at reflux for 4 h, cooled, filtered, and acidified with hydrochloric acid. The crude product was collected by filtration and recrystallized from ethanol to afford the targeted 1,2,4-triazoles 5–6.
4.2.2. Microwave Procedure
In a closed borosilicate glass vessel fitted with a silicone cap, a solution of the appropriate acid thiosemicarbazide 3–4 (1 mmol) dissolved in 10% aqueous solution of sodium hydroxide (10 mL) was exposed to irradiation for 3 min using a microwave reactor. The reaction was processed as described in the CP outlined earlier to afford the same 1,2,4-triazoles 5–6.
4.2.3. Characterization of 4,5-Diphenyl-4H-1,2,4-triazole-3-thione (5)
mp 283–284 °C (Lit. mp 281–282 °C);55 IR (KBr, ν): 3276–3389 (NH), 3087 (CH-Ar), 1625 (C=N), 1556 (C=C), 1288 cm–1 (C=S). 1H NMR (400 MHz, DMSO-d6): δH 7.29–7.43 (7H, m, HAr), 7.47–7.50 (3H, m, HAr), 14.16 (1H, br s, NH). 13C NMR (100 MHz, DMSO-d6): δC 125.80, 128.24, 128.55, 128.74, 129.33, 129.42, 130.34, 134.54 (CAr); 150.56 (C=N); 168.57 (C=S). Calcd for C14H11N3S: C, 66.38; H,4.38; N, 16.59. Found: C, 66.65; H, 4.52; N, 16.78.
4.2.4. Characterization of 5-Methyl-4-phenyl-4H-1,2,4-triazole-3-thione (6)
mp 215–216 °C (Lit. mp 214–215 °C);56 IR (KBr, ν): 3284–3348 (NH), 3032 (CH-Ar), 2899 (CH-Al), 1620 (C=N), 1559 (C=C), 1295 cm–1 (C=S). 1H NMR (400 MHz, DMSO-d6): δH 2.14 (3H, s, CH3), 7.45–7.48 (2H, m, HAr), 7.55–7.60 (2H, m, HAr), 13.70 (1H, br s, NH). 13C NMR (100 MHz, DMSO-d6): δC 11.61 (CH3), 128.04, 129.26, 133.73 (CAr), 149.04 (C=N), 167.34 (C=S). Calcd for C9H9N3S: C, 56.52; H, 4.74; N, 21.97. Found: C, 56.38; H, 4.80; N, 21.89.
4.3. General Procedure for the Synthesis of Thiopropargylated 1,2,4-Triazoles 7–8
4.3.1. Conventional Procedure
Appropriate triazoles 5–6 (10 mmol) were dissolved in ethanol (30 mL) and triethylamine (10 mmol) was added followed by propargyl bromide (12 mmol). The mixture was allowed to react at reflux for 1 h, cooled, and excess ethanol was removed in vacuo. The crude product was recrystallized from ethanol to afford the targeted thiopropargylated-1,2,4-triazoles 7–8.
4.3.2. Microwave Procedure
In a closed borosilicate glass vessel fitted with a silicone cap, a solution of the appropriate triazoles 5–6 (1 mmol) dissolved in ethanol (5 mL) containing triethylamine (1 mmol) and propargyl bromide (1.2 mmol) was exposed to irradiation for 2 min using a microwave reactor. The reaction was processed as described in the CP outlined earlier to afford the same thiopropargylated-1,2,4-triazoles 7–8.
4.3.3. Characterization of 4,5-Diphenyl-3-(prop-2-yn-1-ylthio)-4H-1,2,4-triazole (7)
mp: 102–103 °C; IR (KBr, ν): 3305 (≡CH), 3029 (CH-Ar), 2945 (CH-Al), 2155 (C≡C), 1615 cm–1 (C=N). 1H NMR (400 MHz, DMSO-d6): δH3.28 (1H, t, J = 4.0 Hz, ≡CH), 4.00 (2H, d, J = 4.0 Hz, SCH2), 7.35–7.43 (7H, m, HAr), 7.55–7.57 (3H, m, HAr). 13C NMR (100 MHz, DMSO-d6): δC 20.89 (SCH2); 74.72, 79.48 (C≡C); 126.56, 127.77, 128.00, 128.64, 129.89, 130.02, 130.18, 133.77, 150.45, 154.68 (CAr, C=N). Calcd for C17H13N3S: C, 70.08; H, 4.50; N, 14.42. Found: C, 70.26; H, 4.39; N, 14.28.
4.3.4. Characterization of 5-Methyl-4-phenyl-3-(prop-2-yn-1-ylthio)-4H-1,2,4-triazole (8)
mp 105–106 °C; IR (KBr, ν): 3328 (≡CH), 3063 (CH-Ar), 2945 (CH-Al), 2145 (C≡C), 1623 cm–1 (C=N). 1H NMR (400 MHz, CDCl3): δH 2.24 (1H, t, J = 4.0 Hz, ≡CH), 2.32 (3H, s, CH3), 3.94 (2H, d, J = 4.0 Hz, SCH2), 7.25–7.28 (2H, m, HAr), 7.55–7.58 (3H, m, HAr). 13C NMR (100 MHz, DMSO-d6): δC 11.35 (CH3); 21.27 (SCH2); 72.34, 78.26 (C≡C); 126.94, 130.03, 130.10, 133.21, 149.57, 153.13 (CAr, C=N). Calcd for C12H11N3S: C, 62.86; H, 4.84; N, 18.33. Found: C, 62.67; H, 4.93; N, 18.45.
4.4. General Procedure for the Synthesis of 1,2,4-Triazole-1,2,3-triazole Conjugates 16–27
4.4.1. Conventional Procedure
To a stirred solution of the appropriate propargylated 1,2,4-triazole 7–8 (1 mmol) in DMSO: H2O (1:1, v/v) (20 mL), CuSO4·5H2O (0.10 g), sodium ascorbate (0.15 g), and the appropriate azide 9–15 (1 mmol) were added with stirring at room temperature for 8–10 h. The progress of the reaction was monitored using TLC. After completion of the reaction, ice cold water (100 mL) was added to the reaction mixture. The precipitate thus formed was collected by filtration, washed with saturated solution of ammonium chloride, and recrystallized from ethanol to give the targeted 1,2,3-triazoles 16–27.
4.4.2. Microwave Procedure
In a closed borosilicate glass vessel fitted with a silicone cap, a solution of the appropriate propargylated 1,2,4-triazole 7–8 (1 mmol) dissolved in DMSO: H2O (1:1, v/v) (20 mL) containing CuSO4·5H2O (0.10 g), sodium ascorbate (0.15 g), and the appropriate azide 9–15 (1 mmol) was exposed to irradiation for 4–6 min using a microwave reactor. The reaction was processed as described in the CP outlined earlier to afford the same 1,2,3-triazoles 16–27.
4.4.3. Characterization of 1-Benzyl-4-(((4,5-diphenyl-4H-1,2,4-triazol-3-yl)thio)methyl)-1H-1,2,3-triazole (16)
mp 214–215 °C; IR (KBr, ν): 3086 (CH-Ar), 2963 (CH-Al), 1623 (C=N), 1559 cm–1 (C=C). 1H NMR (600 MHz, DMSO-d6): δH 4.45 (2H, s, SCH2), 5.57 (2H, s, NCH2), 7.30–7.50 (15H, m, HAr), 8.07 (1H, s, CH-1,2,3-triazole). 13C NMR (100 MHz, DMSO-d6): δC 27.79 (SCH2); 53.45 (NCH2); 124.47, 127.27, 128.30, 128.57, 128.62, 128.81, 129.25, 129.43, 130.47, 130.58, 130.71, 131.95, 134.48, 136.67, 143.53, 151.89 (CAr, C=N). Calcd for C24H20N6S: C, 67.90; H, 4.75; N, 19.80. Found: C, 67.65; H, 4.84; N, 19.66.
4.4.4. Characterization of 4-(((4,5-Diphenyl-4H-1,2,4-triazol-3-yl)thio)methyl)-1-hexadecyl-1H-1,2,3-triazole (17)
mp 80–81 °C; IR (KBr, ν): 3087 (CH-Ar), 2898–2963 (CH-Al), 1625 (C=N), 1559 cm–1 (C=C). 1H NMR (600 MHz, DMSO-d6): δH 0.86 (3H, t, J = 6.0 Hz, CH3), 1.21–1.26 (28H, m, 14× CH2), 4.30 (2H, t, J = 6.0 Hz, NCH2), 4.45 (2H, s, SCH2), 7.34–7.43 (7H, m, HAr), 7.52–7.55 (3H, m, HAr), 8.02 (1H, s, CH-1,2,3-triazole). 13C NMR (150 MHz, DMSO-d6): δC 14.63 (CH3); 22.77, 26.45, 27.74, 29.02, 29.37, 29.50, 29.59, 29.65, 29.70, 30.97 (14× CH2); 31.97 (SCH2); 49.99 (NCH2); 124.24, 127.29, 128.32, 128.54, 129.25, 130.45, 130.60, 130.74, 134.50, 142.95, 151.81, 155.08 (CAr, C=N). Calcd for C33H46N6S: C, 70.93; H, 8.30; N, 15.04. Found C, 70.78; H, 8.39; N, 15.18.
4.4.5. Characterization of 4-(((4,5-Diphenyl-4H-1,2,4-triazol-3-yl)thio)methyl)-1-hexyl-1H-1,2,3-triazole (18)
mp 129–130 °C; IR (KBr, ν): 3091 (CH-Ar), 2891–2988 (CH-Al), 1630 cm–1 (C=N). 1H NMR (400 MHz, DMSO-d6): δH 0.83 (3H, t, J = 4.0 Hz, CH3), 1.11–1.22 (6H, m, 3× CH2), 1.75–1.78 (2H, m, NCH2CH2), 4.32 (2H, t, J = 4.0 Hz, NCH2), 4.55 (2H, s, SCH2), 7.34–7.40 (7H, m, HAr), 7.52–7.55 (3H, m, HAr), 8.09 (1H, s, CH-1,2,3-triazole). 13C NMR (100 MHz, DMSO-d6): δC 13.72 (CH3); 21.79, 25.34, 26.68, 29.51 (4× CH2); 30.44 (SCH2); 49.42 (NCH2); 123.69, 126.21, 127.57, 127.85, 128.45, 129.79, 129.84, 130.07, 133.52, 142.76, 151.54, 154.22 (CAr, C=N). Calcd for C23H26N6S: C, 66.00; H, 6.26; N, 20.08. Found C, 66.23; H, 6.33; N, 20.21.
4.4.6. Characterization of 1-Benzyl-4-(((5-methyl-4-phenyl-4H-1,2,4-triazol-3-yl)thio)methyl)-1H-1,2,3-triazole (19)
mp 115–116 °C; IR (KBr, ν): 3089 (CH-Ar), 2986 (CH-Al), 1635 cm–1 (C=N). 1H NMR (400 MHz, DMSO-d6): δH 2.18 (3H, s, CH3), 4.35 (2H, s, SCH2), 5.56 (2H, s, NCH2), 7.28–7.38 (7H, m, HAr), 7.53–7.55 (3H, m, HAr), 8.01 (1H, s, CH-1,2,3-triazole). 13C NMR (100 MHz, DMSO-d6): δC 10.88 (CH3); 27.14 (SCH2); 52.73 (NCH2); 123.64, 126.99, 127.90, 128.11, 128.72, 129.77, 129.80, 133.07, 135.94, 142.88, 148.89, 152.47 (CAr, C=N). Calcd for C19H18N6S: C, 62.96; H, 5.01; N, 23.19. Found: C, 62.78; H, 5.10; N, 23.38.
4.4.7. Characterization of Ethyl 2-(4-(((4,5-Diphenyl-4H-1,2,4-triazol-3-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)acetate (20)
mp 80–81 °C; IR (KBr, ν): 3087 (CH-Ar), 2899 (CH-Al), 1740 (C=O), 1625 cm–1 (C=N). 1H NMR (600 MHz, DMSO-d6): δH 1.21 (3H, t, J = 6.0 Hz, CH3), 4.15–4.18 (2H, q, J = 6.0 Hz, OCH2), 4.50 (2H, s, SCH2), 5.38 (2H, s, NCH2), 7.35–7.40 (7H, m, HAr), 7.52–7.54 (3H, m, HAr), 8.13 (1H, s, CH-1,2,3-triazole). 13C NMR (150 MHz, DMSO-d6): δC 14.64 (CH3); 27.64 (SCH2); 51.17 (NCH2); 62.15 (OCH2); 127.35, 128.35, 128.57, 129.25, 130.47, 130.61, 130.76, 134.58, 152.63, 156.46 (CAr, C=N); 167.81 (C=O). Calcd for C21H20N6O2S: C, 59.98; H, 4.79; N, 19.99. Found: C, 59.69; H, 4.72; N, 19.85.
4.4.8. Characterization of Ethyl 2-(4-(((5-Methyl-4-phenyl-4H-1,2,4-triazol-3-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)acetate (21)
mp 107–108 °C; IR (KBr, ν): 3075 (CH-Ar), 2945 (CH-Al), 1742 (C=O), 1628 cm–1 (C=N). 1HNMR (600 MHz, DMSO-d6): δH 1.22 (3H, t, J = 6.0 Hz, CH3CH2O), 2.16 (3H, s, CH3), 4.17 (2H, q, J = 6.0 Hz, CH2O), 4.44 (2H, s, SCH2), 5.39 (2H, s, NCH2CO), 7.26–7.29 (2H, m, HAr), 7.55–7.59 (3H, m, HAr), 8.06 (1H, s, CH-1,2,3-triazole). 13C NMR (150 MHz, DMSO-d6): δC 11.83 (CH3); 14.42 (CH3CH2O); 27.23 (SCH2); 50.95 (NCH2); 61.94 (OCH2); 120.34, 120.59, 126.03, 129.81, 133.08, 135.63, 150.65, 153.54 (CAr, C=N); 166.99 (C=O). Calcd for C16H18N6O2S: C, 53.62; H, 5.06; N, 23.45. Found: C, 53.81; H, 5.11; N, 23.62.
4.4.9. Characterization of 1-(4-Bromophenyl)-4-(((4,5-diphenyl-4H-1,2,4-triazol-3-yl)thio)methyl)-1H-1,2,3-triazole (22)
mp 210–211 °C; IR (KBr, ν): 3092 (CH-Ar), 2895 (CH-Al), 1630 cm–1 (C=N). 1H NMR (400 MHz, DMSO-d6): δH 4.55 (2H, s, SCH2), 7.35–7.40 (7H, m, HAr), 7.53–7.55 (3H, m, HAr), 7.80–7.87 (4H, m, HAr), 8.76 (1H, s, CH-1,2,3-triazole). 13C NMR (100 MHz, DMSO-d6): δC 27.26 (SCH2); 121.82, 122.45, 122.48, 127.03, 128.12, 128.36, 129.03, 130.26, 130.37, 130.54, 133.27, 134.23, 136.15, 144.50, 152.46, 155.26 (CAr, C=N). Calcd for C23H17BrN6S: C, 56.45; H, 3.50, N, 17.17. Found: C, 56.69; H, 3.60, N, 17.30.
4.4.10. Characterization of 4-(((4,5-Diphenyl-4H-1,2,4-triazol-3-yl)thio)methyl)-1-(4-fluorophenyl)-1H-1,2,3-triazole (23)
mp 144–145 °C; IR (KBr, ν): 3078 (CH-Ar), 2988 (CH-Al), 1625 cm–1 (C=N). 1H NMR (400 MHz, DMSO-d6): δH 4.60 (2H, s, SCH2), 7.35–7.53 (12H, m, HAr), 7.92 (2H, br s, HAr), 8.74 (1H, s, CH-1,2,3-triazole). 13C NMR (100 MHz, DMSO-d6): δC 26.72 (SCH2); 116.87, 122.35, 122.53, 126.27, 127.66, 127.99, 128.51, 129.99, 130.14, 132.96, 133.98, 144.10, 160.46, 162.90 (CAr, C=N). Calcd for C23H17FN6S: C, 64.47; H, 4.00; N, 19.61. Found: C, 64.67; H, 4.10; N, 19.75.
4.4.11. Characterization of 1-(3,4-Dichlorophenyl)-4-(((4,5-diphenyl-4H-1,2,4-triazol-3-yl)thio)methyl)-1H-1,2,3-triazole (24)
mp 185–186 °C; IR (KBr, ν): 3087 (CH-Ar), 2890 (CH-Al), 1632 cm–1 (C=N). 1H NMR (600 MHz, DMSO-d6): δH 4.55 (2H, s, SCH2), 7.35–7.40 (7H, m, HAr), 7.53–7.55 (3H, m, HAr), 7.89 (1H, d, J = 6.0 Hz, HAr), 7.97 (1H, dd, J = 6.0 Hz, 12.0 Hz, HAr), 8.26 (1H, d, J = 6.0 Hz, HAr), 8.85 (1H, s, CH-1,2,3-triazole). 13C NMR (150 MHz, DMSO-d6): δC 27.20 (SCH2); 120.55, 122.24, 122.73, 126.91, 127.03, 128.12, 128.35, 129.04, 130.38, 130.54, 132.30, 132.83, 134.19, 136.46, 144.61, 161.42 (CAr, C=N). Calcd for C23H16Cl2N6S: C, 57.63; H, 3.36; N, 17.53. Found: C, 57.80; H, 3.31; N, 17.39.
4.4.12. Characterization of 1-(4-Bromophenyl)-4-(((5-methyl-4-phenyl-4H-1,2,4-triazol-3-yl)thio)methyl)-1H-1,2,3-triazole (25)
mp 109–110 °C; IR (KBr, ν): 3095 (CH-Ar), 2933 (CH-Al), 1630 cm–1 (C=N). 1H NMR (400 MHz, DMSO-d6): δH 2.20 (3H, s, CH3), 4.43 (2H, s, SCH2), 7.40–7.43 (2H, m, HAr), 7.55–7.58 (3H, m, HAr), 7.78–7.85 (4H, m, HAr), 8.69 (1H, s, CH-1,2,3-triazole). 13C NMR (100 MHz, DMSO-d6): δC 10.91 (CH3); 26.87 (SCH2); 121.30, 121.84, 121.89, 127.03, 129.81, 132.76, 133.08, 135.63, 144.05, 148.65, 152.64 (CAr, C=N). Calcd for C18H15BrN6S: C, 50.59; H, 3.54; N, 19.67. Found: C, 50.81; H, 3.64; N, 19.81.
4.4.13. Characterization of 1-(4-Fluorophenyl)-4-(((5-methyl-4-phenyl-4H-1,2,4-triazol-3-yl)thio)methyl)-1H-1,2,3-triazole (26)
mp 89–90 °C; IR (KBr, ν): 3088 (CH-Ar), 2967 (CH-Al), 1628 cm–1 (C=N). 1H NMR (400 MHz, DMSO-d6): δH 2.20 (3H, s, CH3), 4.45 (2H, s, SCH2), 7.40–7.43 (2H, m, HAr), 7.55–7.58 (3H, m, HAr), 7.80–7.86 (4H, m, HAr), 8.70 (1H, s, CH-1,2,3-triazole). 13C NMR (100 MHz, DMSO-d6): δC 15.85 (CH3); 27.61 (SCH2); 116.59, 116.83, 122.15, 122.30, 122.41, 127.05, 127.78, 129.81, 129.86, 132.98, 133.01, 134.04, 144.00, 144.93, 160.38, 162.80 (CAr, C=N). Calcd for C18H15FN6S: C, 59.00; H, 4.13; N, 22.94. Found: C, 59.16; H, 4.19; N, 22.86.
4.4.14. Characterization of 1-(3,4-Dichlorophenyl)-4-(((5-methyl-4-phenyl-4H-1,2,4-triazol-3-yl)thio)methyl)-1H-1,2,3-triazole (27)
mp 111–112 °C; IR (KBr, ν): 3069 (CH-Ar), 2966 (CH-Al), 1622 cm–1 (C=N). 1H NMR (600 MHz, DMSO-d6): δH 2.11 (3H, s, CH3), 4.44 (2H, s, SCH2), 7.39–7.42 (2H, m, HAr), 7.54–7.58 (3H, m, HAr), 7.85–7.92 (2H, m, HAr), 8.21 (1H, br s, HAr), 8.75 (1H, s, CH-1,2,3-triazole). 13C NMR (150 MHz, DMSO-d6): δC 11.70 (CH3); 27.28 (SCH2); 120.57, 122.23, 127.50, 130.33, 130.41, 131.52, 132.30, 132.85, 136.44 (CAr); 160.38 (C=N); 162.80 (C=N). Calcd for C18H14Cl2N6S: C, 51.81; H, 3.38; N, 20.14. Found: C, 51.68; H, 3.33; N, 20.21.
4.5. Anticancer Activity
4.5.1. Cell Lines
The human colorectal adenocarcinoma cell line (Caco-2), the human colorectal carcinoma cell line (HCT-116), the human uterine cervical cancer cell line (HeLa), and the human breast adenocarcinoma cell line (MCF-7) used in this study were purchased from ATCC. Cell lines were cultured in high-glucose Dulbecco’s modified eagle medium containing 10% heat-inactivated fetal bovine serum, 2 mmol L–1 of l-glutamine, 50 U mL–1 of penicillin, and 50 μg mL–1 of streptomycin. Cells were plated at a density of 1 × 104 cell/well into 96-well culture plates and incubated at 37 °C in a humidified atmosphere containing 5% CO2 for 24 h.
4.5.2. MTT Assay
The cytotoxic activity was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) colorimetric assay as reported previously.84−86 In brief, the tumor cell lines were suspended in a medium at a concentration of 1 × 104 cell/well in Corning 96-well tissue culture plates and then incubated for 24 h. The tested compounds with concentrations were then added into 96-well plates to achieve different conc. for each compound. After incubating for 24 h, the numbers of viable cells were determined using the MTT test. Briefly, the medium was removed and the MTT solution (50 μL, 0.5 mg/mL in RPMI 1640 without phenol red) was added. After 3 h of incubation, the MTT solution was removed and the acquired formazan was dissolved in the isopropanol/HCl system. The 96-well plates were then incubated at 37 °C and 5% CO2 for 4 h. Finally, the optical density was measured at 570 nm with the microplate reader to determine the number of viable cells and the percentage of viability was calculated as [1 – (ODt/ODc)] × 100% where ODt is the mean optical density of wells treated with the tested sample and ODc is the mean optical density of untreated cells. The experiment was conducted in three independent iterations with four technical repetitions. Tests were conducted at concentrations ranging from 0.01 to 0.8 mM. The 50% inhibitory concentration (IC50), the concentration required to cause toxic effects in 50% of intact cells, was estimated from graphic plots of the dose–response curve for each concentration using CalcuSyn.
4.6. In Silico Analysis
Various physicochemical, pharmacokinetic, and pharmacodynamic traits (i.e., fragment-based drug likeness and violations, clog P, TPSA, ADMET, and so forth) of the synthesized compounds were forecasted employing various computational tools.
4.6.1. Molecular Docking Studies
The 3D atomic coordinates of the tested compounds were modeled using the Marvin sketch software (https://chemaxon.com/products/marvin). These target ligands were further optimized and energy minimized utilizing the Dundee PRODRG2 server.87 These energy optimized compounds were availed to execute the molecular docking studies. The 3D crystallographic structure of the molecular aspirant was acquired from Protein Data Bank (PDB) (www.rcsb.org): CDK-2 (PDB: 2R3I). The receptor was prepared by the removal of heteroatoms (water and ions). The active site of the target receptor was predicted by utilizing the BIOVIA Discovery studio visualizer.88 The ligands were docked into the predicted active site of the CDK2, in order to interpret their binding interaction in silico and to analyze their anticancer activity. The docking study was carried out by PyRx software using AutoDock vina as the engine for docking, which is based on the scoring function.89 Grid resolutions were utilized to perform docking simulations. The grid was defined as a spherical region that encloses all the active site residue within 15.0 Å of the confined crystallographic ligand atom. At every step of the docking simulation, the binding energy of the interaction between the ligand and protein was gauged by exercising atomic affinity potentials computed on a grid. All the calculations were conducted exploiting the default settings. For 2D and 3D visualizations, the BIOVIA Discovery studio visualizer was employed.
Author Contributions
All authors discussed the results and commented on the manuscript.
The authors declare no competing financial interest.
References
- Lombardino J. G.; Lowe J. A. The role of the medicinal chemist in drug discovery - then and now. Nat. Rev. Drug Discovery 2004, 3, 853–862. 10.1038/nrd1523. [DOI] [PubMed] [Google Scholar]
- Bonandi E.; Christodoulou M. S.; Fumagalli G.; Perdicchia D.; Rastelli G.; Passarella D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discovery Today 2017, 22, 1572–1581. 10.1016/j.drudis.2017.05.014. [DOI] [PubMed] [Google Scholar]
- Li H.; Aneja R.; Chaiken I. Click chemistry in peptide-based drug design. Molecules 2013, 18, 9797–9817. 10.3390/molecules18089797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angell Y. L.; Burgess K. Peptidomimetics via copper-catalyzed azide-alkyne cycloadditions. Chem. Soc. Rev. 2007, 36, 1674–1689. 10.1039/B701444A. [DOI] [PubMed] [Google Scholar]
- Agalave S. G.; Maujan S. R.; Pore V. S. Click Chemistry: 1,2,3-Triazoles as Pharmacophores. Chem.—Asian J. 2011, 6, 2696–2718. 10.1002/asia.201100432. [DOI] [PubMed] [Google Scholar]
- Muller T.; Bräse S. Click chemistry finds its way into covalent porous organic materials. Angew. Chem., Int. Ed. 2011, 50, 11844–11845. 10.1002/anie.201105707. [DOI] [PubMed] [Google Scholar]
- Gonzaga D. T. G.; Souza T. M. L.; Andrade V. M. M.; Ferreira V. F.; da Silva F. d. C. Identification of 1-aryl-1H-1, 2, 3-triazoles as potential new antiretroviral agents. Med. Chem. 2018, 14, 242–248. 10.2174/1573406413666170906121318. [DOI] [PubMed] [Google Scholar]
- El Malah T.; Nour H. F.; Satti A. A. E.; Hemdan B. A.; El-Sayed W. A. Design, Synthesis, and Antimicrobial Activities of 1,2,3-Triazole Glycoside Clickamers. Molecules 2020, 25, 790. 10.3390/molecules25040790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed W. A.; Khalaf H. S.; Mohamed S. F.; Hussien H. A.; Kutkat O. M.; Amr A. E. Synthesis and antiviral activity of 1,2,3-triazole glycosides based substituted pyridine via click cycloaddition. Russ. J. Gen. Chem. 2017, 87, 2444–2453. 10.1134/S1070363217100279. [DOI] [Google Scholar]
- Xu Z.; Zhao S.-J.; Liu Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700. 10.1016/j.ejmech.2019.111700. [DOI] [PubMed] [Google Scholar]
- Ríos-Malváez Z. G.; Cano-Herrera M.-A.; Dávila-Becerril J. C.; Mondragón-Solórzano G.; Ramírez-Apan M. T.; Morales-Morales D.; Barroso-Flores J.; Santillán-Benítez J. G.; Unnamatla M. V. B.; García-Eleno M. A.; González-Rivas N.; Cuevas-Yañez E. Synthesis, characterization and cytotoxic activity evaluation of 4-(1,2,3-triazol-1-yl) salicylic acid derivatives. J. Mol. Struct. 2021, 1225, 129149. 10.1016/j.molstruc.2020.129149. [DOI] [Google Scholar]
- Andrade P. d.; Fraga Dias A. d.; Figueiró F.; Torres F. C.; Kawano D. F.; Oliveira Battastini A. M.; Carvalho I.; Tomich de Paula da Silva C. H.; Campos J. M. 1,2,3-Triazole tethered 2-mercaptobenzimidazole derivatives: design, synthesis and molecular assessment toward C6 glioma cell line. Future Med. Chem. 2020, 12, 689–708. 10.4155/fmc-2019-0227. [DOI] [PubMed] [Google Scholar]
- Jia Y.; Li M.; Cao Y.; Feng W.; Li X.; Xue W.; Shi H. Discovery of a Novel Benzenesulfonamide Analogue That Inhibits Proliferation and Metastasis Against Ovarian Cancer OVCAR-8 Cells. Drug Des., Dev. Ther. 2020, 14, 207–216. 10.2147/dddt.s225201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipoline I. C.; da Fonseca A. C. C.; da Costa G. R. M.; de Souza M. P.; Rabelo V. W.-H.; de Queiroz L. N.; de Souza T. L. F.; de Almeida E. C. P.; Abreu P. A.; Pontes B. Molecular mechanism of action of new 1, 4-naphthoquinones tethered to 1, 2, 3-1H-triazoles with cytotoxic and selective effect against oral squamous cell carcinoma. Bioorg. Chem. 2020, 101, 103984. 10.1016/j.bioorg.2020.103984. [DOI] [PubMed] [Google Scholar]
- Sonego M. S.; Segatto N. V.; Damé L.; Fronza M.; Gomes C. B.; Oliveira T. L.; Seixas F. K.; Savegnago L.; Schachtschneider K. M.; Alves D.; Collares T. 7-Chloroquinoline-1,2,3-triazoyl carboxamides induce cell cycle arrest and apoptosis in human bladder carcinoma cells. Invest. New Drugs 2019, 38, 1020–1030. 10.1007/s10637-019-00861-w. [DOI] [PubMed] [Google Scholar]
- Maddila S.; Pagadala R.; Jonnalagadda S. 1,2,4-Triazoles: A Review of Synthetic Approaches and the Biological Activity. Lett. Org. Chem. 2013, 10, 693–714. 10.2174/157017861010131126115448. [DOI] [Google Scholar]
- Cui L.-J.; Xie Z.-F.; Piao H.-R.; Li G.; Chai K.-Y.; Quan Z.-S. Synthesis and Anticonvulsant Activity of 1-Substituted-7-Benzyloxy-4,5-dihydro-[1,2,4]triazolo[4,3-a]quinoline. Biol. Pharm. Bull. 2005, 28, 1216–1220. 10.1248/bpb.28.1216. [DOI] [PubMed] [Google Scholar]
- Stingaci E.; Zveaghinteva M.; Pogrebnoi S.; Lupascu L.; Valica V.; Uncu L.; Smetanscaia A.; Drumea M.; Petrou A.; Ciric A.; Glamoclija J.; Sokovic M.; Kravtsov V.; Geronikaki A.; Macaev F. New vinyl-1,2,4-triazole derivatives as antimicrobial agents: Synthesis, biological evaluation and molecular docking studies. Bioorg. Med. Chem. Lett. 2020, 30, 127368. 10.1016/j.bmcl.2020.127368. [DOI] [PubMed] [Google Scholar]
- Gupta D.; Jain D. Synthesis, antifungal and antibacterial activity of novel 1,2,4-triazole derivatives. J. Adv. Pharm. Technol. Res. 2015, 6, 141. 10.4103/2231-4040.161515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarigol D.; Uzgoren-Baran A.; Tel B. C.; Somuncuoglu E. I.; Kazkayasi I.; Ozadali-Sari K.; Unsal-Tan O.; Okay G.; Ertan M.; Tozkoparan B. Novel thiazolo[3,2-b]-1,2,4-triazoles derived from naproxen with analgesic/anti-inflammatory properties: Synthesis, biological evaluation and molecular modeling studies. Bioorg. Med. Chem. 2015, 23, 2518–2528. 10.1016/j.bmc.2015.03.049. [DOI] [PubMed] [Google Scholar]
- Mioc M.; Avram S.; Bercean V.; Kurunczi L.; Ghiulai R. M.; Oprean C.; Coricovac D. E.; Dehelean C.; Mioc A.; Balan-Porcarasu M. Design, synthesis and biological activity evaluation of S-substituted 1H-5-mercapto-1,2,4-triazole derivatives as antiproliferative agents in colorectal cancer. Front. Chem. 2018, 6, 373–392. 10.3389/fchem.2018.00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morak-Młodawska B.; Pluta K.; Latocha M.; Jeleń M.; Kuśmierz D. Design, Synthesis, and Structural Characterization of Novel Diazaphenothiazines with 1, 2, 3-Triazole Substituents as Promising Antiproliferative Agents. Molecules 2019, 24, 4388–4399. 10.3390/molecules24234388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur R.; Ranjan Dwivedi A.; Kumar B.; Kumar V. Recent developments on 1, 2, 4-triazole nucleus in anticancer compounds: a review. Anti-Cancer Agents Med. Chem. 2016, 16, 465–489. 10.2174/1871520615666150819121106. [DOI] [PubMed] [Google Scholar]
- Filho R. I.; Gonzaga D. T. G.; Demaria T. M.; Leandro J. G. B.; Costa D. C. S.; Ferreira V. F.; Sola-Penna M.; da Silva F. d. C.; Zancan P. A novel triazole derivative drug presenting in vitro and in vivo anticancer properties. Curr. Top. Med. Chem. 2018, 18, 1483–1493. 10.2174/1568026618666180917100640. [DOI] [PubMed] [Google Scholar]
- da Silva F. d. C.; do Carmo Cardoso M. F.; Ferreira P. G.; Ferreira V. F., Biological Properties of 1H-1, 2, 3-and 2H-1, 2, 3-Triazoles. Chemistry of 1,2,3- (Triazoles), ; Springer, 2014; pp 117–165. [Google Scholar]
- Tadesse S.; Caldon E. C.; Tilley W.; Wang S. Cyclin-dependent kinase 2 inhibitors in cancer therapy: An update. J. Med. Chem. 2018, 62, 4233–4251. 10.1021/acs.jmedchem.8b01469. [DOI] [PubMed] [Google Scholar]
- Gu Y.; Rosenblatt J.; Morgan D. O. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 1992, 11, 3995–4005. 10.1002/j.1460-2075.1992.tb05493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S.; Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. 10.1242/dev.091744. [DOI] [PubMed] [Google Scholar]
- Malumbres M.; Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- Harbour J. W.; Luo R. X.; Santi A. D.; Postigo A. A.; Dean D. C. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999, 98, 859–869. 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- Chohan T.; Qian H.; Pan Y.; Chen J.-Z. Cyclin-dependent kinase-2 as a target for cancer therapy: progress in the development of CDK2 inhibitors as anti-cancer agents. Curr. Med. Chem. 2014, 22, 237–263. 10.2174/0929867321666141106113633. [DOI] [PubMed] [Google Scholar]
- Brown N. R.; Lowe E. D.; Petri E.; Skamnaki V.; Antrobus R.; Johnson L. Cyclin B and cyclin A confer different substrate recognition properties on CDK2. Cell Cycle 2007, 6, 1350–1359. 10.4161/cc.6.11.4278. [DOI] [PubMed] [Google Scholar]
- Bramson H. N.; Corona J.; Davis S. T.; Dickerson S. H.; Edelstein M.; Frye S. V.; Gampe R. T.; Harris P. A.; Hassell A.; Holmes W. D.; Hunter R. N.; Lackey K. E.; Lovejoy B.; Luzzio M. J.; Montana V.; Rocque W. J.; Rusnak D.; Shewchuk L.; Veal J. M.; Walker D. H.; Kuyper L. F. Oxindole-based inhibitors of cyclin-dependent kinase 2 (CDK2): design, synthesis, enzymatic activities, and X-ray crystallographic analysis. J. Med. Chem. 2001, 44, 4339–4358. 10.1021/jm010117d. [DOI] [PubMed] [Google Scholar]
- Beale G.; Haagensen E. J.; Thomas H. D.; Wang L.-Z.; Revill C. H.; Payne S. L.; Golding B. T.; Hardcastle I. R.; Newell D. R.; Griffin R. J.; Cano C. Combined PI3K and CDK2 inhibition induces cell death and enhances in vivo antitumour activity in colorectal cancer. Br. J. Cancer 2016, 115, 682–690. 10.1038/bjc.2016.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B.; Lane M. E.; Wadler S. SU9516, a cyclin-dependent kinase 2 inhibitor, promotes accumulation of high molecular weight E2F complexes in human colon carcinoma cells. Biochem. Pharmacol. 2002, 64, 1091–1100. 10.1016/s0006-2952(02)01264-9. [DOI] [PubMed] [Google Scholar]
- Qu X.; Zhu L.; Song L.; Liu S., Circ_0084927 promotes cervical carcinogenesis by sponging miR-1179 that suppresses CDK2, a cell cycle-related gene. 2020, 20, 333DOI: doi.org/ 10.1186/s12935-020-01417-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abd El-Karim S. S.; Syam Y. M.; El Kerdawy A. M.; Abdelghany T. M. New thiazol-hydrazono-coumarin hybrids targeting human cervical cancer cells: Synthesis, CDK2 inhibition, QSAR and molecular docking studies. Bioorg. Chem. 2019, 86, 80–96. 10.1016/j.bioorg.2019.01.026. [DOI] [PubMed] [Google Scholar]
- Assirey E.; Alsaggaf A.; Naqvi A.; Moussa Z.; Okasha R. M.; Afifi T. H.; Abd-El-Aziz A. S. Synthesis, biological assessment, and structure activity relationship studies of new flavanones embodying chromene moieties. Molecules 2020, 25, 544. 10.3390/molecules25030544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weroha S. J.; Lingle W. L.; Hong Y.; Li S. A.; Li J. J. Specific Overexpression of Cyclin E·CDK2 in Early Preinvasive and Primary Breast Tumors in Female ACI Rats Induced by Estrogen. Horm. Cancer 2010, 1, 34–43. 10.1007/s12672-009-0004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queiroz T. M.; Orozco E. V. M.; Silva V. R.; Santos L. S.; Soares M. B. P.; Bezerra D. P.; Porto A. L. M. Semi-synthesis of β-keto-1,2,3-triazole derivatives from ethinylestradiol and evaluation of the cytotoxic activity. Heliyon 2019, 5, e02408 10.1016/j.heliyon.2019.e02408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara H.; Ezhevsky S. A.; Vocero-Akbani A. M.; Kaldis P.; Solomon M. J.; Dowdy S. F. Transforming growth factor beta targeted inactivation of cyclin E:cyclin-dependent kinase 2 (Cdk2) complexes by inhibition of Cdk2 activating kinase activity. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 14961–14966. 10.1073/pnas.96.26.14961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgogary S. R.; Khidre R. E.; El-Telbani E. M. Regioselective synthesis and evaluation of novel sulfonamide 1,2,3-triazole derivatives as antitumor agents. J. Iran. Chem. Soc. 2020, 17, 765–776. 10.1007/s13738-019-01796-y. [DOI] [Google Scholar]
- Kassem A. F.; Abbas E. M. H.; El-Kady D. S.; Awad H. M.; El-Sayed W. A. Design, Synthesis and Anticancer Activity of New Thiazole-Tetrazole or Triazole Hybrid Glycosides Targeting CDK-2 via Structure-Based Virtual Screening. Mini-Rev. Med. Chem. 2019, 19, 933–948. 10.2174/1389557519666181231121217. [DOI] [PubMed] [Google Scholar]
- Pathoor R.; Bahulayan D. MCR-click synthesis, molecular docking and cytotoxicity evaluation of a new series of indole-triazole-coumarin hybrid peptidomimetics. New J. Chem. 2018, 42, 6810–6816. 10.1039/C8NJ00032H. [DOI] [Google Scholar]
- Aouad M. R.; Mayaba M. M.; Naqvi A.; Bardaweel S. K.; Al-blewi F. F.; Messali M.; Rezki N. Design, synthesis, in silico and in vitro antimicrobial screenings of novel 1,2,4-triazoles carrying 1,2,3-triazole scaffold with lipophilic side chain tether. Chem. Cent. J. 2017, 11, 117–129. 10.1186/s13065-017-0347-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezki N.; Messali M.; Al-Sodies S. A.; Naqvi A.; Bardaweel S. K.; Al-blewi F. F.; Aouad M. R.; El Ashry E. S. H. Design, synthesis, in-silico and in-vitro evaluation of di-cationic pyridinium ionic liquids as potential anticancer scaffolds. J. Mol. Liq. 2018, 265, 428–441. 10.1016/j.molliq.2018.06.045. [DOI] [Google Scholar]
- Rezki N.; Al-Sodies S. A.; Messali M.; Bardaweel S. K.; Sahu P. K.; Al-blewi F. F.; Sahu P. K.; Aouad M. R. Identification of new pyridinium ionic liquids tagged with Schiff bases: Design, synthesis, in silico ADMET predictions and biological evaluations. J. Mol. Liq. 2018, 264, 367–374. 10.1016/j.molliq.2018.05.071. [DOI] [Google Scholar]
- Al-Blewi F. F.; Rezki N.; Al-Sodies S. A.; Bardaweel S. K.; Sabbah D. A.; Messali M.; Aouad M. R. Novel amphiphilic pyridinium ionic liquids-supported Schiff bases: ultrasound assisted synthesis, molecular docking and anticancer evaluation. Chem. Cent. J. 2018, 12, 118–135. 10.1186/s13065-018-0489-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouad M. R.; Messali M.; Rezki N.; Said M. A.; Lentz D.; Zubaydi L.; Warad I. Hydrophobic pocket docking, double-proton prototropic tautomerism in contradiction to single-proton transfer in thione ⇔thiol Schiff base with triazole-thione moiety: Green synthesis, XRD and DFT-analysis. J. Mol. Struct. 2019, 1180, 455–461. 10.1016/j.molstruc.2018.12.010. [DOI] [Google Scholar]
- Aouad M. R.; Almehmadi M. A.; Rezki N.; Al-blewi F. F.; Messali M.; Ali I. Design, click synthesis, anticancer screening and docking studies of novel benzothiazole-1,2,3-triazoles appended with some bioactive benzofused heterocycles. J. Mol. Struct. 2019, 1188, 153–164. 10.1016/j.molstruc.2019.04.005. [DOI] [Google Scholar]
- Al-blewi F.; Rezki N.; Naqvi A.; Qutb Uddin H.; Al-Sodies S.; Messali M.; Aouad M. R.; Bardaweel S. A Profile of the In Vitro Anti-Tumor Activity and In Silico ADME Predictions of Novel Benzothiazole Amide-Functionalized Imidazolium Ionic Liquids. Int. J. Mol. Sci. 2019, 20, 2865–2885. 10.3390/ijms20122865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezki N.; Al-Blewi F. F.; Al-Sodies S. A.; Alnuzha A. K.; Messali M.; Ali I.; Aouad M. R. Synthesis, Characterization, DNA Binding, Anticancer, and Molecular Docking Studies of Novel Imidazolium-Based Ionic Liquids with Fluorinated Phenylacetamide Tethers. ACS Omega 2020, 5, 4807–4815. 10.1021/acsomega.9b03468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albalawi A. H.; Almutairi S. M.; Aljuhani A.; Sahu P. K.; Sahu P. K.; Rezki N.; Aouad M. R.; Messali M. Novel Fluorinated Imidazolium Ionic Liquids: Eco-friendly, Facile and Efficient Construction, Characterization, in vitro Anticancer Activity, Toxicity and in silico Analysis. Asian J. Chem. 2020, 32, 690–696. 10.14233/ajchem.2020.22491. [DOI] [Google Scholar]
- Al-Sodies S. A.; Aouad M. R.; Ihmaid S.; Aljuhani A.; Messali M.; Ali I.; Rezki N. Microwave and conventional synthesis of ester based dicationic pyridinium ionic liquids carrying hydrazone linkage: DNA binding, anticancer and docking studies. J. Mol. Struct. 2020, 1207, 127756. 10.1016/j.molstruc.2020.127756. [DOI] [Google Scholar]
- El-Ashry E. S. H.; Rashed N.; Awad L. F.; Ramadan E. S.; Abdel-Maggeed S. M.; Rezki N. Synthesis of 5-Aryl-3-Glycosylthio-4-Pheny-4H-1, 2,4-Triazoles and Their Acyclic Analogs Under Conventional and Microwave Conditions. J. Carbohydr. Chem. 2008, 27, 70–85. 10.1080/07328300802030795. [DOI] [Google Scholar]
- Rostamizadeh S.; Mollahoseini K.; Moghadasi S. A One-Pot Synthesis of 4,5-Disubstituted-1,2,4-triazole-3-thiones on Solid Support under Microwave Irradiation. Phosphorus, Sulfur Silicon Relat. Elem. 2006, 181, 1839–1845. 10.1080/10426500500542992. [DOI] [Google Scholar]
- Khau V. V.; Martinelli M. J. 1,3-Dipolar cycloreversion of a 1,3,4-oxadiazolidine as a controlled azomethine imine surrogate for pyrazolidine synthesis. Tetrahedron Lett. 1996, 37, 4323–4326. 10.1016/0040-4039(96)00836-2. [DOI] [Google Scholar]
- Minvielle M. J.; Bunders C. A.; Melander C. Indole-triazole conjugates are selective inhibitors and inducers of bacterial biofilms. MedChemComm 2013, 4, 916–919. 10.1039/c3md00064h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devender N.; Gunjan S.; Tripathi R.; Tripathi R. P. Synthesis and antiplasmodial activity of novel indoleamide derivatives bearing sulfonamide and triazole pharmacophores. Eur. J. Med. Chem. 2017, 131, 171–184. 10.1016/j.ejmech.2017.03.010. [DOI] [PubMed] [Google Scholar]
- Wang G.; Peng Z.; Wang J.; Li X.; Li J. Synthesis, in vitro evaluation and molecular docking studies of novel triazine-triazole derivatives as potential α-glucosidase inhibitors. Eur. J. Med. Chem. 2017, 125, 423–429. 10.1016/j.ejmech.2016.09.067. [DOI] [PubMed] [Google Scholar]
- Novak J.; Zitterl-Eglseer K.; Deans S. G.; Franz C. M. Essential oils of different cultivars ofCannabis sativa L. and their antimicrobial activity. Flavour Fragrance J. 2001, 16, 259–262. 10.1002/ffj.993. [DOI] [Google Scholar]
- Vemuri V. K.; Makriyannis A. Medicinal chemistry of cannabinoids. Clin. Pharmacol. Ther. 2015, 97, 553–558. 10.1002/cpt.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover M.; Singh B.; Bakshi M.; Singh S. Quantitative structure-property relationships in pharmaceutical research - Part 1. Pharm. Sci. Technol. Today 2000, 3, 28–35. 10.1016/s1461-5347(99)00214-x. [DOI] [PubMed] [Google Scholar]
- Malhotra R.; Ravesh A.; Singh V. Synthesis, characterization, antimicrobial activities, and QSAR studies of organotin (IV) complexes. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 1–31. 10.1080/10426507.2016.1225054. [DOI] [Google Scholar]
- Kumer A.; Sarker M. N.; Paul S. The Simulating Study of HOMO, LUMO, thermo physical and Quantitative Structure of Activity Relationship (QSAR) of Some Anticancer Active Ionic Liquids. Eurasian J. Environ. Sci. 2019, 3, 1–10. [Google Scholar]
- Khodair A. I.; Awad M. K.; Gesson J.-P.; Elshaier Y. A. M. M. New N-ribosides and N-mannosides of rhodanine derivatives with anticancer activity on leukemia cell line: Design, synthesis, DFT and molecular modelling studies. Carbohydr. Res. 2020, 487, 107894. 10.1016/j.carres.2019.107894. [DOI] [PubMed] [Google Scholar]
- Kumar S. S.; Athimoolam S.; Sridhar B. Structural, spectral, theoretical and anticancer studies on new co-crystal of the drug 5-fluorouracil. J. Mol. Struct. 2018, 1173, 951–958. 10.1016/j.molstruc.2018.07.079. [DOI] [Google Scholar]
- Hagar M.; Ahmed H. A.; El-Sayed T. H.; Alnoman R. Mesophase behavior and DFT conformational analysis of new symmetrical diester chalcone liquid crystals. J. Mol. Liq. 2019, 285, 96–105. 10.1016/j.molliq.2019.04.083. [DOI] [Google Scholar]
- Joshi R.; Pandey N.; Yadav S. K.; Tilak R.; Mishra H.; Pokharia S. Synthesis, spectroscopic characterization, DFT studies and antifungal activity of (E)-4-amino-5-[N′-(2-nitro-benzylidene)-hydrazino]-2,4-dihydro-[1,2,4]triazole-3-thione. J. Mol. Struct. 2018, 1164, 386–403. 10.1016/j.molstruc.2018.03.081. [DOI] [Google Scholar]
- Joshi R.; Kumari A.; Singh K.; Mishra H.; Pokharia S. Triorganotin(IV) complexes of Schiff base derived from 1,2,4-triazole moiety: Synthesis, spectroscopic investigation, DFT studies, antifungal activity and molecular docking studies. J. Mol. Struct. 2020, 1206, 127639. 10.1016/j.molstruc.2019.127639. [DOI] [Google Scholar]
- Ali M. S.; Farah M. A.; Al-Lohedan H. A.; Al-Anazi K. M. Comprehensive exploration of the anticancer activities of procaine and its binding with calf thymus DNA: a multi spectroscopic and molecular modelling study. RSC Adv. 2018, 8, 9083–9093. 10.1039/c7ra13647a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachedi K. O.; Ouk T.-S.; Bahadi R.; Bouzina A.; Djouad S.-E.; Bechlem K.; Zerrouki R.; Ben Hadda T.; Almalki F.; Berredjem M. Synthesis, DFT and POM analyses of cytotoxicity activity of α-amidophosphonates derivatives: Identification of potential antiviral O,O-pharmacophore site. J. Mol. Struct. 2019, 1197, 196–203. 10.1016/j.molstruc.2019.07.053. [DOI] [Google Scholar]
- da Costa R. M.; Bastos J. K.; Costa M. C. A.; Ferreira M. M. C.; Mizuno C. S.; Caramori G. F.; Nagurniak G. R.; Simão M. R.; Dos Santos R. A.; Veneziani R. C. S.; Ambrósio S. R.; Parreira R. L. T. In vitro cytotoxicity and structure-activity relationship approaches of ent-kaurenoic acid derivatives against human breast carcinoma cell line. Phytochemistry 2018, 156, 214–223. 10.1016/j.phytochem.2018.10.005. [DOI] [PubMed] [Google Scholar]
- Lewis D. F. V. Quantitative structure-activity relationships (QSARs) within the cytochrome P450 system: QSARs describing substrate binding, inhibition and induction of P450s. Inflammopharmacology 2003, 11, 43–73. 10.1163/156856003321547112. [DOI] [PubMed] [Google Scholar]
- Rangel H. R.; Ortega J. T.; Serrano M. L.; Pujol F. H. Unrevealing sequence and structural features of novel coronavirus using in silico approaches: The main protease as molecular target. EXCLI J. 2020, 19, 400. 10.17179/excli2020-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Ghose A. K.; Viswanadhan V. N.; Wendoloski J. J. Prediction of hydrophobic (lipophilic) properties of small organic molecules using fragmental methods: an analysis of ALOGP and CLOGP methods. J. Phys. Chem. A 1998, 102, 3762–3772. 10.1021/jp980230o. [DOI] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H.-Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Egan W. J.; Lauri G. Prediction of intestinal permeability. Adv. Drug Delivery Rev. 2002, 54, 273–289. 10.1016/s0169-409x(02)00004-2. [DOI] [PubMed] [Google Scholar]
- Muegge I.; Heald S. L.; Brittelli D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. 10.1021/jm015507e. [DOI] [PubMed] [Google Scholar]
- Kouza M.; Banerji A.; Kolinski A.; Buhimschi I.; Kloczkowski A. Role of Resultant Dipole Moment in Mechanical Dissociation of Biological Complexes. Molecules 2018, 23, 1995–2004. 10.3390/molecules23081995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shawon J.; Khan A. M.; Rahman A.; Hoque M. M.; Khan M. A. K.; Sarwar M. G.; Halim M. A. Molecular Recognition of Azelaic Acid and Related Molecules with DNA Polymerase I Investigated by Molecular Modeling Calculations. Interdiscip. Sci.: Comput. Life Sci. 2018, 10, 525–537. 10.1007/s12539-016-0186-3. [DOI] [PubMed] [Google Scholar]
- Uzzaman M.; Hoque M. J. Physiochemical, molecular docking, and pharmacokinetic studies of Naproxen and its modified derivatives based on DFT. Int. J. Sci. Res. Manag. 2018, 6, 2018–2025. 10.18535/ijsrm/v6i9.c01. [DOI] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Demirci F.; Başer K. H. C. Bioassay Techniques for Drug Development By Atta-ur-Rahman, M. Iqbal Choudhary (HEJRIC, University of Karachi, Pakistan), William J. Thomsen (Areana Pharmaceuticals, San Diego, CA). Harwood Academic Publishers, Amsterdam, The Netherlands. 2001. xii + 223 pp. 15.5 × 23.5 cm. $79.00. ISBN 90-5823-051-1. J. Nat. Prod. 2002, 65, 1086–1087. 10.1021/np020725b. [DOI] [Google Scholar]
- Omar A. M.; Bajorath J.; Ihmaid S.; Mohamed H. M.; El-Agrody A. M.; Mora A.; El-Araby M. E.; Ahmed H. E. A. Novel molecular discovery of promising amidine-based thiazole analogues as potent dual Matrix Metalloproteinase-2 and 9 inhibitors: Anticancer activity data with prominent cell cycle arrest and DNA fragmentation analysis effects. Bioorg. Chem. 2020, 101, 103992. 10.1016/j.bioorg.2020.103992. [DOI] [PubMed] [Google Scholar]
- Schüttelkopf A. W.; Van Aalten D. M. F. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. 10.1107/s0907444904011679. [DOI] [PubMed] [Google Scholar]
- Dassault Systèmes . Pharmacophore And Ligand-Based Design With Biovia Discovery Studio, 2014. [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]