

Abstract
Vancomycin is a glycopeptide antibiotic produced by Amycolaptopsis orientalis used to treat serious infections by Gram-positive pathogens including methicillin-resistant Staphylococcus aureus. Vancomycin inhibits cell wall biosynthesis by targeting lipid II, which is the membrane-bound peptidoglycan precursor. The heptapeptide aglycon structure of vancomycin binds to the d-Ala-d-Ala of the pentapeptide stem structure in lipid II. The third residue of vancomycin aglycon is asparagine, which is not directly involved in the dipeptide binding. Nonetheless, asparagine plays a crucial role in substrate recognition, as the vancomycin analogue with asparagine substituted by aspartic acid (VD) shows a reduction in antibacterial activities. To characterize the function of asparagine, binding of vancomycin and its aspartic-acid-substituted analogue VD to l-Lys-d-Ala-d-Ala and l-Lys-d-Ala-d-Lac was investigated using molecular dynamic simulations. Binding interactions were analyzed using root-mean-square deviation (RMSD), two-dimensional (2D) contour plots, hydrogen bond analysis, and free energy calculations of the complexes. The analysis shows that the aspartate substitution introduced a negative charge to the binding cleft of VD, which altered the aglycon conformation that minimized the repulsive lone pair interaction in the binding of a depsipeptide. Our findings provide new insight for the development of novel glycopeptide antibiotics against the emerging vancomycin-resistant pathogens by chemical modification at the third residue in vancomycin to improve its binding affinity to the d-Ala-d-Lac-terminated peptidoglycan in lipid II found in vancomycin-resistant enterococci and vancomycin-resistant S. aureus.
1. Introduction
Vancomycin is a glycopeptide antibiotic discovered in the 1950s1 by Eli Lilly from the fermentation broth of a soil microbe Amycolaptopsis orientalis. The newly discovered antibiotic was named vancomycin, derived from the word “vanquish,” because of its potent antibacterial activities. Vancomycin is highly active against a broad spectrum of Gram-positive pathogens including bacilli and all aerobic cocci including staphylococci, streptococci, and enterococci. Despite its early Food and Drug Administration (FDA) approval in 1958, vancomycin was not widely used due to the high cost and adverse side effects associated with purification. Instead, methicillin, a powerful semisynthetic β-lactam antibiotic that was developed in the 1950s became the antibiotic of choice against penicillin-resistant pathogens. Then in the 1980s, vancomycin became one of the most important antibiotics due to the emergence of methicillin-resistant Staphylococcus aureus (MRSA) responsible for an alarming increase in mortality associated with serious nosocomial infections. MRSA is highly virulent, easily transmittable through contact, and it is typically resistant to multiple classes of antibiotics. Against MRSA, vancomycin was one of few remaining antibiotics that were highly effective, and thus, for a period vancomycin was considered as “the drug of last resort.” Currently, there are more than a hundred different types of glycopeptide antibiotics produced by different organisms,2 and even a greater number of chemically modified semisynthetic glycopeptides have been synthesized.3
The mode of action of vancomycin and related glycopeptide antibiotics is that it inhibits peptidoglycan (PG) biosynthesis by binding to the membrane-bound PG precursor, lipid II. PG is the primary component of the cell wall in Gram-positive bacteria with a repeat unit consisting of a disaccharide (GlcNAc-MurNAc), a pentapeptide-stem structure consisting of l-Ala-d-iso-Glu-l-Lys-d-Ala-d-Ala, and a pentaglycine. Pentapeptide-stem is attached to MurNAc, and the pentaglycine to the ε-nitrogen side of the Lys (Figure 1a). PG biosynthesis is catalyzed by two important enzymatic steps, transglycosylation and transpeptidation, which occur outside of the bacterial membrane.4 Transglycosylation is the polymerization of the PG-repeat unit by forming a long glycan chain of repeating GlcNAc-MurNAc. Transpeptidation is a cross-linking between the two adjacent neighboring glycan chains. Unlike β-lactam antibiotics, which also target the cell wall by inhibiting the transpeptidation step of PG biosynthesis through binding to the penicillin-binding proteins, vancomycin binds to lipid II and thereby inhibits the transglycosylation step of PG biosynthesis.5 The lipid II sequestration prevents the regeneration of the lipid transporter (C55) from lipid II, which requires transglycosylase activity. Since the number of C55 copies in the bacterium is found in small numbers, vancomycin sequestration of lipid II is a potent means of inhibiting bacterial cell wall biosynthesis.6
Figure 1.

Chemical structures of S. aureus peptidoglycan (PG) and vancomycin. (a) The PG-repeat unit in S. aureus consists of a GlcNAc-MurNAc disaccharide, a pentapeptide stem with a sequence l-Ala-d-iso-Glu-l-Lys-d-Ala-d-Ala, and a pentaglycine bridge structure that is attached to the ε-nitrogen of the side chain L-Lys. Vancomycin binds to the d-Ala-d-Ala dipeptide of the PG-stem structure (green circle). In vancomycin-resistant pathogens, including vancomycin-resistant enterococci (VRE) and vancomycin-resistant S. aureus (VRSA), the dipeptide is replaced by a depsipeptide d-Ala-d-Lac. (b) Chemical structure of vancomycin and its interactions with the bound PG-stem structure. The vancomycin binding affinity to depsipeptide (X = O) is 1000-fold less than that to dipeptide (X = NH). The minimal inhibitory concentration of vancomycin against VRE and VRSA increases by 1000-fold. The lost efficacy is attributed to the depsipeptide substitution, which replaces a hydrogen bond with an electrostatic repulsion (red dotted line). (c) A model structure of vancomycin bound to an acyl-d-Ala-d-Ala. Vancomycin is shown as a space-filling model with an electrostatic surface and the dipeptide as a stick-and-ball model.
The structures of vancomycin bound to a tripeptide PG analogue, Acyl-l-Lys-d-Ala-d-Ala, have been well characterized since the early 1980s by the solution-state nuclear magnetic resonance (NMR)7 spectroscopy and X-ray crystallography.8 The tripeptide-bound structures show that the d-Ala-d-Ala of PG stem is bound to a cleft formed by the vancomycin aglycon and it is stabilized by five hydrogen bonds: three hydrogen bonds between the amide protons of residues 1, 2, and 3 of vancomycin and the C-terminus of the dipeptide d-Ala-d-Ala, a fourth bond between the carbonyl of the 4th residue and an amide proton of the terminal d-Ala, and a fifth hydrogen bond between the amide proton of the 7th residue of the aglycon and the carbonyl of the penultimate d-Ala from the C-terminal end (Figure 1b). The drug sugars, d-glucose and l-vancosamine, are attached to the phenolic moiety on the 4-hydroxyphenylglycine side chain of the fourth amino acid and they are known to increase the vancomycin activity even though they do not participate in the dipeptide binding (Figure 1c).
Initially, the emergence of vancomycin resistance was thought to be highly unlikely. Then, in 1986, the first clinical case of infection by high-level vancomycin-resistant enterococci (VRE) was reported.9 Since then, there has been a rapid increase in VRE-associated nosocomial infections worldwide. The mechanism of high-level vancomycin resistance in VRE involves the modification of the d-Ala-d-Ala terminus of PG stems from a dipeptide d-Ala-d-Ala to a depsipeptide d-Ala-d-Lac.10−13 The depsipeptide-terminated lipid II in VRE replaces an amide group of the terminal alanine with the oxygen of an ester bond. This modification replaces one of the hydrogen bonds by electrostatic repulsion (Figure 1b, red line) and reduces vancomycin binding affinity by a 1000-fold with KD from 1 μM to 1 mM. The minimal inhibition concentration (MIC) of vancomycin increases from 1 μg/mL against vancomycin-susceptible enterococci to approximately 1000 μg/mL for the VRE. Subsequently, in 2002, the first case of infection by vancomycin-resistant S. aureus (VRSA) with a MIC greater than 256 μg/mL was reported.14 The genetic analysis confirmed that the mechanism of vancomycin resistance in VRSA was identical to that of VRE, possessing a transposal plasmid in VRSA containing the genetic determinants for vancomycin resistance in VRE.15
There have been several important developments in glycopeptide antibiotics to overcome resistance through chemical modifications. Some of the developments include (1) C-terminus modifications,16 (2) homo- and heterodimerization of glycopeptides,3 (3) drug sugar modifications,17 and (4) aglycon modifications.18,19 So far, the alkylation of hydrophobic adducts to the drug sugar has been the most effective, leading to the development of oritavancin (LY333328) by Eli Lilly,20,21 which was FDA approved in 2014. Oritavancin is a lipoglycopeptide antibiotic that has potent bactericidal activities against a broad spectrum of multidrug-resistant pathogens including MRSA,22 and it is effective against vancomycin-resistant pathogens including VRE23 and VRSA.24 The mode of action of oritavancin differs from vancomycin by exhibiting dual inhibition of transglycosylase and transpeptidase activities.25−29
Another exciting recent development is the chemical modification of the vancomycin aglycon structure to overcome the vancomycin resistance. Boger et al. determined that the reduced vancomycin binding affinity to the depsipeptide-terminated PG-stem is less affected by the loss of a hydrogen bond (Figure 1b) but more due to the electrostatic repulsion between the lone pair of a carbonyl oxygen atom at the 4th residue of vancomycin and the ester oxygen atom on the d-Ala-d-Lac.18 A rational approach was taken to re-design the aglycon structure of vancomycin through total synthesis by replacing the carbonyl oxygen atom at the 4th residue of vancomycin with a protonated amidine nitrogen.30 The incorporation of amidine nitrogen in the aglycon removes the destabilizing electron lone pair interactions and replaces it with the stabilizing interactions between the proton of the amidine nitrogen and the oxygen on the ester of d-Lac when bound to d-Ala-d-Lac.31 The modification on the glycopeptide resulted in a 600-fold increase in the binding affinity to the depsipeptide ligand and restored the activity against the VRE of VanA type with a MIC below 1 μg/mL.32 This demonstrated a proof of principle that the rational approach to design glycopeptide aglycon will play an important role in the future development of novel antibiotics.
To facilitate the development through rational drug design, the function of each amino acid in the aglycon structure needs to be investigated. All amino acids in the vancomycin core are involved in the formation of a highly crosslinked core structure, except for the amino acids at positions 1 and 3, which are N-methylleucine and asparagine, respectively. These amino acids are not involved in the ridged cleft formation and do not participate in the dipeptide binding; nonetheless, they are essential for the activity of vancomycin. In the case of N-methylleucine, the removal by Edman degradation results in desleucyl-vancomycin, which is devoid of any antimicrobial activity.33 For the asparagine, the third amino acid of vancomycin substitution by aspartic acid or glutamine has been shown to reduce the antimicrobial activity against S. aureus by approximately 8- and 4-fold, respectively.34 The introduction of the negative charge in the binding cleft by aspartic acid substitution or the lengthening of the side chain by glutamine substitution is presumed to interfere with the dipeptide binding by electrostatic or steric interactions; however, the exact role of these core amino acids is not well understood.
In this study, we investigate the role and function of asparagine at the 3rd position of vancomycin using classical molecular dynamics (MD) simulations. MD simulations were carried out for vancomycin (VN), and its aspartate-substituted analogue (VD) bound to the PG tripeptide mimics representing vancomycin-susceptible and vancomycin-resistant bacteria. The four glycopeptide–PG complexes analyzed are as follows: (1) vancomycin bound to the acyl-d-Ala-d-Ala (VN-Ala), (2) vancomycin bound to the acyl-d-Ala-d-Lac (VN-Lac), (3) aspartate-substituted vancomycin bound to the acyl-d-Ala-d-Ala (VD-Ala), and (4) aspartate-substituted vancomycin bound to the acyl-d-Ala-d-Lac (VD-Lac). From these simulations, we find that the aspartate-substituted analogue of vancomycin (VD) shows improved binding to the acyl-d-Ala-d-Lac, the PG-stem structure found in VRE and VRSA. This indicates that the amino acids in the aglycon structure that do not directly participate in the PG-stem binding play a crucial role in stabilizing the glycopeptide–PG complex. Furthermore, the study provides a new insight for the chemical modifications of the aglycon structure for the development of new glycopeptide antibiotics to mediate the improvement in the binding to the d-Ala-d-Lac-terminated PG precursors found in vancomycin-resistant pathogens.
2. Results and Discussion
2.1. Total Hydrogen Bonds and Binding Free Energies
The total number of hydrogen bonds formed between the glycopeptide and ligand as well as the total free energy of binding (ΔGbind) during MD simulations are shown in Table 1. The contributions from van der Waals (ΔEvdw) and electrostatic energy (ΔEele) to the ΔGbind are also shown in the table. The average number of hydrogen bonds formed during MD simulations ranged from 2 to 5. The most stable complex was VN-Ala, which has the maximum number of 5 stable hydrogen bonds,35 and the least stable complex was VD-Ala with only 2 hydrogen bonds. The order of stability, ranked based on the overall free energy of binding from the most to least stable is as follows: VN-Ala > VD-Lac > VN-Lac > VD-Ala.
Table 1. Number of Hydrogen Bonds and Binding Energies of VN-Ala, VN-Lac, VD-Lac, and VD-Ala.
| glycopeptide–PG complex | average number of H-bonds | ΔEvdw (kJ/mol) | ΔEele (kJ/mol) | ΔGbind (kJ/mol) |
|---|---|---|---|---|
| VN-Ala (R = NH2, X = NH) | 5.08 ± 0.19 | –41.48 ± 1.36 | –80.53 ± 5.33 | –3.81 ± 0.33 |
| VN-Lac (R = NH2, X = O) | 4.33 ± 0.24 | –32.32 ± 1.82 | –65.45 ± 1.26 | 14.31 ± 0.68 |
| VD-Ala (R = O–, X = NH) | 2.10 ± 0.04 | –35.38 ± 1.22 | 31.55 ± 0.59 | 15.82 ± 0.61 |
| VD-Lac (R = O–, X = O) | 3.39 ± 0.12 | –45.34 ± 1.02 | 23.27 ± 2.11 | 9.60 ± 0.02 |
The strongest electrostatic attraction between the glycopeptide and its ligand was observed in the VN-Ala complex with a ΔEele value of −80.53 kJ/mol (Table 1). This strong electrostatic attraction is attributed to the interactions between the C-terminus of the acyl-d-Ala-d-Ala and the aglycon structure.35 In contrast, the weakest attraction was observed in the VD-Ala complex with a ΔEele value of 31.55 kJ/mol, due to the loss of interaction by the displacement of the C-terminus of acyl-d-Ala-d-Ala out of the binding pocket (Figure 3b). It is interesting to note that the VD-Lac complex is more stable (ΔGbind of 9.60 kJ/mol) than the VN-Lac complex (ΔGbind of 14.31 kJ/mol) by −4.71 kJ/mol. This was surprising since the ΔEele of the VN-Lac complex (−65.45 kJ/mol) is much stronger than the VD-Lac (23.27 kJ/mol) complex with a difference ΔΔEele value of 88.42 kJ/mol. The van der Waals’s contribution to the ΔGbind, though some variations are observed between the complexes, nonetheless, all ranged between −32 and −45 kJ/mol. Thus, the difference in the van der Waals contributions between these two complexes (ΔΔEvdw) is only −13.02 kJ/mol, which is not enough to offset a large ΔΔEele value of 88.42 kJ/mol to attain a ΔΔGbind of −4.71 kJ/mol. This led to the possibility that the increased binding stability of the VD-Lac complex, compared to that of VN-Lac, is not significantly dependent on the number of H-bonds formed or the interactions between the C-terminus of ligand and the aglycon of glycopeptide. Instead, VD-Lac is more stable than VN-Lac because of the conformation change associated with the aspartate substitution, which led to the minimization of the electrostatic repulsion between the electron lone pair on the carbonyl oxygen at the 4th position of glycopeptide and the ester oxygen atom of the d-Ala-d-Lac.18 It is worth noting that the molecular mechanics Poisson–Boltzmann surface area (MM/PBSA) binding energy method is not a reliable technique for calculating the absolute binding free energies.36 However, the approach has been shown to be good for ranking the binding energies of similar ligands.37
Figure 3.

Initial structure of the VD-Ala complex at t = 0 ns and the final structure at 30 ns MD simulation. (a) The initial structure of the VD-Ala complex showing the acyl-d-Ala-d-Ala bound to the aglycon structure of the glycopeptide. (b) The final structure of the VD-Ala complex after 30 ns of MD simulation. The C-terminus of the d-Ala is displaced from the binding cleft, resulting in the loss of three hydrogen bonds between the C-terminus of d-Ala and the amide protons of residues 1, 2, and 3 of the aglycon.
2.2. Root-Mean-Square Deviation
The time evolution of RMSD values for the four glycopeptide–PG complexes are shown in Figure 2. The RMSD for each glycopeptide–PG complex was calculated during MD simulations by comparing the structures along the trajectories to the reference structure. The low fluctuation pattern in the RMSD plot of a complex indicates minimal changes in conformation, which is indicative of a stable complex. In contrast, high fluctuation indicates the low stability of a complex during MD simulation. As expected, the unmodified vancomycin bound to acyl-d-Ala-d-Ala (VN-Ala), which is the most stable complex of the four systems, had the lowest average RMSD value of 1.70 Å, followed by the VD-Lac with 2.07 Å, VN-Lac with 2.24 Å, and VD-Ala with 2.90 Å. The stability of the complex, inferred from the RMSD plots, when ordered from the most to the least stable is VN-Ala > VD-Lac > VN-Lac > VD-Ala. An identical order was also observed when the overall binding energy (ΔGbind) was calculated for the four complexes (Table 1). This indicated a direct correlation between the stability and binding energy of the complex. Unexpectedly, the VD-Lac complex shows increased stability (Figure 2c) compared to the VN-Lac complex (Figure 2b), based on the smaller average RMSD value and the reduced fluctuating amplitudes of the RMSD plot. As stated previously, VD-Lac had more stable binding energy than the VN-Lac (Table 1). This suggests that the aspartate substitution, which introduces a negative change in the aglycon structure of VD, diminished the effects of the electrostatic repulsion between the oxygen atom in the ester bond of the ligand d-Ala-d-Lac and the oxygen carbonyl carbon at the 4th residue of the glycopeptide that has been attributed as the primary cause of the loss in binding affinity to the depsipeptide.18
Figure 2.

RMSD plots of three independent 30 ns MD simulations for the complexes (a) VN-Ala, (b) VN-Lac, (c) VD-Lac, and (d) VD-Ala. The RMSD values of three independent simulations are plotted using red, blue, and black colors. The average RMSD values for each complex is shown as a figure inset.
The least-stable complex was VD-Ala, which shows an unusual RMSD fluctuating pattern (Figure 2d) that is not observed in other complexes. The initial structure of the VD-Ala complex (Figure 3a) undergoes a large RMSD fluctuation during the first 3 ns of the MD simulation. Then, the RMSD amplitude rapidly stabilizes as the structure reaches the final conformation (Figure 3b) with an average RMSD value of 2.90 Å. Figure 3 shows the initial and final structures of the VD-Ala complex, where VD is represented as a space-filling model with an electrostatic surface and the bound-dipeptide ligand as a stick-and-ball model. The observed instability during the first 3 ns is associated with the rearrangement of the aglycon structure and its bound ligand, resulting in the partial displacement of the terminal d-Ala from the binding cleft (Figure 3b). The final structure of the VD-Ala complex reveals the displacement of the d-Ala-d-Ala C-terminus from the binding cleft, which results in the loss of three hydrogen bonds between the C-terminus of the dipeptide and the amide protons of residues 1, 2, and 3 of the vancomycin peptide backbone (Figure 1b). This loss of three hydrogen bonds is consistent with the calculated 2.1 hydrogen bonds found in the VD-Ala complex (Table 1).
The 2D RMSD contour plots shown in Figure 4 was used to further characterize the stability and conformational distribution of the different glycopeptide–ligand complexes during MD simulation. The VN-Ala complex, which is the most stable complex, is used as a reference to compare the similarities of the binding distribution to that of other complexes (Figure 4). The numbers on the contour lines represent the density of the joint distribution between the conformational space of two different complexes along the RMSD trajectories. The closer the attraction basin (area of highest density) for a distribution to be centered on the same points, the more similarity between the two different complexes under comparison. From Figure 4a, it can be seen that the binding distribution of the VD-Lac complex is closest to that of VN-Ala due to their attraction basin centered at approximately (0.17, 0.20) Å and the presence of a single attraction basin. This is followed by the VN-Lac complex (Figure 4b), whose joint distribution with VN-Ala is centered at approximately (0.17, 0.23) Å, including a minor contribution at (0.17, 0.35) Å. The presence of multiple attraction basins in Figure 4b is indicative of greater differences between VN-Lac and VN-Ala complexes when compared to those of VD-Lac and VN-Ala complexes. The binding distribution of the VD-Ala complex is least similar to that of VN-Ala. This is shown by the high-density attraction basin centered at approximately (0.17, 0.30) Å, including an extension to about (0.18, 0.37) Å.
Figure 4.

Comparative 2D RMSD (Å) contour plots of the VN-Ala complex relative to VD-Lac, VN-Lac, and VD-Ala complexes. Comparative 2D RMSD contour plots of (a) VN-Ala vs VD-Lac, (b) VN-Ala vs VN-Lac, and (c) VN-Ala vs VD-Ala complexes. The contour lines represent the density of trajectories that are located within the area. The red color denotes the region of highest density and the blue color denotes the region of lowest density.
2.3. Glycopeptide Interactions with the C-Terminus of Peptidoglycan Precursor
The interactions between the glycopeptide and the C-terminus of the bound ligand are summarized in Table 2. The average number of H-bonds formed with the C-terminus and the interaction energy is determined to gain insight into the C-terminus contribution to the ligand binding. The interaction energy (ΔEtot) when ordered from the most stable to the least is as follows: VN-Ala > VN-Lac > VD-Lac > VD-Ala. This stability trend is identical to the trend observed for the average number of H-bonds shown in the table.
Table 2. Interactions between C-Terminus of the Peptidoglycan and Glycopeptide.
| glycopeptide–PG complex | average number of H-bonds | ΔEvdw (kJ/mol) | ΔEele (kJ/mol) | ΔEtot (kJ/mol) |
|---|---|---|---|---|
| VN-Ala (R = NH2, X = NH) | 3.28 ± 0.29 | 0.97 ± 3.32 | –306.87 ± 26.31 | –307.84 ± 26.52 |
| VN-Lac (R = NH2, X = O) | 2.73 ± 0.19 | –3.79 ± 1.43 | –254.93 ± 5.03 | –258.72 ± 5.23 |
| VD-Ala (R = O–, X = NH) | 0.11 ± 0.05 | –1.49 ± 0.74 | –49.05 ± 4.11 | –50.54 ± 4.18 |
| VD-Lac (R = O–, X = O) | 2.22 ± 0.03 | –6.63 ± 0.29 | –199.19 ± 6.41 | –205.82 ± 6.42 |
In the VN-Ala complex, the C-terminus of d-Ala-d-Ala forms approximately 3H-bonds per time frame (Figure 5a) with the aglycon during the simulation. From Table 2, the VN-Ala complex has the most stable interaction energy followed by the VN-Lac, VD-Lac, and VD-Ala complexes. For vancomycin, the number of H-bonds formed between the aglycon structure to the C-terminus of d-Ala-d-Ala is 3.28 but reduces to 2.73 when bound to d-Ala-d-Lac. In the VN-Lac complex, the replacement of an amide by ester oxygen in d-Ala-d-Lac replaces an H-bond (Figure 1b) with an electrostatic repulsion. This results in large RMSD fluctuation of the VN-Lac complex (Figure 2b) and it negatively impacted the H-bond interactions between the C-terminus of the bound ligand and the vancomycin aglycon. The VD-Ala complex is the least stable without any H-bond interaction between the C-terminus of d-Ala-d-Ala and the glycopeptide (Table 2). An analysis of the number of H-bond formed per time frame between the C-terminus of d-Ala-d-Ala and the modified vancomycin in the VD-Ala complex (Figure 5d) shows that the number of H-bonds reduces from 3 to 0 within 3 ns of the simulation.
Figure 5.

Number of intermolecular H-bonds formed per time frame between the C-terminus of the ligand and the glycopeptide. The number of stable H-bonds formed between the (a) C-terminus of d-Ala-d-Ala and vancomycin in the VN-Ala complex, (b) C-terminus of d-Ala-d-Lac and vancomycin in the VN-Lac complex, (c) C-terminus of d-Ala-d-Lac and the modified vancomycin in the VD-Lac complex, and (d) C-terminus of d-Ala-d-Ala and the modified vancomycin in the VD-Ala complex.
The H-bonds between the C-terminus of d-Ala-d-Ala and the amide protons of aglycon residues 1,2, and 3 are disrupted by the competing H-bond formed between the aspartate side chain of the glycopeptide and the hydrogen donor, the amide proton, of the C-terminus d-Ala. This preferential interaction between the aspartate side chain of residue 3 in the glycopeptide with d-Ala-d-Ala in the VD-Ala complex was monitored by measuring the intermolecular distance between the amide nitrogen atom at the C-terminus of the dipeptide and the carbonyl oxygen atom of the aspartate side chain (Figure 6d). The intermolecular distance starting at 0.5 nm was rapidly reduced to an overall average of 0.29 nm within 3 ns and remained stable for the remaining duration of the simulation (Figure 6d). The replacement of asparagine at the 3rd amino acid position in vancomycin with an aspartate introduces a negative charge within the binding cleft. This destabilizes the d-Ala-d-Ala binding through the electrostatic repulsion between the negative charges of aspartic acid and the C-terminus of the dipeptide. The formation of a new H-bond between the aspartate side chain and the amide proton of the terminal d-Ala of the dipeptide (Figure 6d) indicates a conformational change associated with the displacement of the bound C-terminal d-Ala out from the binding pocket (Figure 3b). The reduced binding stability in the VD-Ala complex is consistent with the eightfold reduction in the antimicrobial activity of VD against S. aureus with PG-stems terminating in d-Ala-d-Ala.34
Figure 6.

Monitoring intermolecular distance between the bound ligand and the side chain at the 3rd amino acid position in vancomycin per time frame during the 30 ns MD simulation. Intermolecular distances between the amide nitrogen of the asparagine side chain of residue 3 in vancomycin and (a) the C-terminus amide nitrogen of d-Ala-d-Ala for the VN-Ala complex and (b) the ester oxygen of d-Ala-d-Lac for VN-Lac complex. The fluctuating intermolecular distances in the VN-Lac complex, compared to the VN-Ala complex, indicate conformational changes associated with the depsipeptide binding. Intermolecular distances between the carbonyl oxygen atom of the aspartate side chain of residue 3 in the glycopeptide and (c) the C-terminus ester oxygen of d-Ala-d-Lac for the VD-Lac complex and (d) the C-terminus amide nitrogen of d-Ala-d-Ala for the VD-Ala complex. In the VD-Ala complex, the aspartate substitution in vancomycin introduces a negative charge that destabilizes d-Ala-d-Ala binding through electrostatic repulsion with the C-terminus of the bound ligand. After 3 ns, the formation of a new H-bond between the aspartate side chain and the amide proton of the terminal d-Ala is associated with the removal of the terminal d-Ala out of the binding pocket (Figures 5d and 3b).
In the case of the VD-Lac complex, the aspartate side chain of the glycopeptide does not form a destabilizing H-bond with the amide proton of the ligand due to ester oxygen in d-Lac (Figure 6c). The depsipeptide binding in the VD-Lac complex with the overall ΔGbind of 9.60 ± 0.02 kJ/mol is more stable than VN-Lac with ΔGbind of 14.31 ± 0.68 kJ/mol (Table 1). However, Table 2 shows that the localized C-terminus interaction of the bound ligand with the amide protons of residues 1, 2, and 3 of the aglycon is less stable in the VD-Lac complex than that in the VN-Lac by approximately 53 kJ/mol. This indicates that the unaccounted interactions that involve non-C-terminus of ligand play an essential role in stabilizing the VD-Lac complex.
In the VD-Lac complex, the negatively charged carboxyl side chain of aspartate interacts constantly with the amide protons of the amino acids at positions 1, 2, and 3 of the aglycon with an interaction energy of −22.23 ± 5.65 kJ/mol. The measured interaction energy between the amide side chain of asparagine with the amide protons at positions 1, 2, and 3 of the aglycon in the VN-Lac complex is 32.11 ± 4.19 kJ/mol. The attraction of the aspartate side chain to the neighboring amide protons in the aglycon of the VD-Lac complex makes the aspartate side chain less available for interaction with the non-C-terminus of the bound ligand, especially the ester oxygen. This is evident in the stable fluctuation pattern shown in Figure 6c, where the intermolecular distance is measured between the carbonyl oxygen atom of the aspartate side chain and the C-terminal ester oxygen of d-Ala-d-Lac. On the other hand, the intermolecular distance between the amide nitrogen of the asparagine sidechain and the ester oxygen of d-Ala-d-Lac in the VN-Lac complex shows an unstable fluctuation pattern implying more interaction (Figure 6b). Thus, the aspartate side chain in the VD-Lac complex competes for the amide protons of the amino acids at positions 1, 2, and 3 of the aglycon. The attraction of this negatively charged carboxyl side chain of aspartate for the neighboring amide protons in the VD-Lac complex weakens the H-bond network between the aglycon and the C-terminus of the ligand d-Lac (Table 2). This results in conformational changes in both the aglycon and the bound depsipeptide, which stabilizes the depsipeptide binding cleft but weakens the aglycon interaction with the C-terminus of the ligand d-Lac. Both the bound ligand and the glycopeptide aglycon in the VD-Lac complex undergo conformational rearrangements that stabilize the complex by minimization of the strong repulsive lone pair interaction, which has been attributed to the loss of binding affinity in the VN-Lac complex. This conformational rearrangement that minimizes the strong repulsive lone pair interaction in the VD-Lac complex is evident in Figure 7, which is a time-dependent plot of the intermolecular distance between the ester oxygen of d-Ala-d-Lac and the carbonyl oxygen of residue 4 in the glycopeptide during 30 ns MD simulation. The VN-Lac complex shows greater atomic distance fluctuation than the VD-Lac complex, which is relatively more stable with a lower average distance value. The average distance value for the VD-Lac complex is 3.9 Å and the VN-Lac complex is 4.9 Å (Figure 7). We hypothesize that the conformational change caused by the substitution of aspartate for asparagine in the VD-Lac complex leads to a reorientation of the amide carbonyl in the glycopeptide’s residue 4. This reorientation minimizes the repulsive lone pair interaction between the carbonyl oxygen of the glycopeptide and the d-Ala-d-Lac ester oxygen in the VD-Lac complex relative to the VN-Lac complex.
Figure 7.

Average atomic distance per time frame during the 30 ns MD simulation of oxygen–oxygen atoms involved in repulsive interactions between the peptidoglycan precursor and the glycopeptide in the different complexes. Average atomic distance between (a) d-Ala-d-Lac ester oxygen of the ligand and the carbonyl oxygen of residue 4 in vancomycin of the VN-Lac complex and (b) d-Ala-d-Lac ester oxygen of the ligand and the carbonyl oxygen of residue 4 in the modified vancomycin of the VD-Lac complex.
2.4. Intramolecular Interaction Between the Side Chain of Residue 3 of the Glycopeptide and Other Parts of the Glycopeptide
An analysis of the intramolecular interaction energy between the side chain of residue 3 to the rest of the glycopeptide in various complexes was carried out during the MD simulation. Intramolecular interactions within the glycopeptide can be useful to yield conformational changes in the glycopeptide that can favor the binding of the glycopeptide to the d-Ala-d-Lac of peptidoglycan. In general, the glycopeptide with aspartate at residue 3 showed stronger intramolecular interaction energy (ΔEtot) than the glycopeptide with asparagine (Table 3). The strongest intramolecular interaction was observed for the VD-Ala complex followed by VD-Lac, VN-Ala, and VN-Lac complexes. In the case of VD-Lac and VN-Lac complexes, the intramolecular interaction energy of VD-Lac is greater than the VN-Lac by approximately 60 kJ/mol (Table 3). This strong intramolecular interaction in the VD-Lac complex stabilizes the binding cleft but weakens the intermolecular interaction with the C-terminus of the ligand (Table 2) allowing for the conformational rearrangement of the bound complex. The conformational changes minimize the lone pair/lone pair electrostatic repulsion, making the VD-Lac complex more stable than the VN-Lac. In the VN-Lac complex, the opposite is observed where the intramolecular interaction is weaker but the intermolecular interaction with the C-terminus of the ligand is much stronger than the VD-Lac complex (Table 2).
Table 3. Intramolecular Interactions between the Side Chain of Residue 3 in the Glycopeptide of Different Complexes with Other Parts of the Glycopeptide.
| glycopeptide–PG complex | ΔEvdw (kJ/mol) | ΔEele (kJ/mol) | ΔEtot (kJ/mol) |
|---|---|---|---|
| VN-Ala (R = NH2, X = NH) | –17.49 ± 1.02 | –79.90 ± 2.82 | –97.39 ± 3.00 |
| VN-Lac (R = NH2, X = O) | –15.85 ± 0.65 | –74.91 ± 4.82 | –90.76 ± 4.86 |
| VD-Ala (R = O–, X = NH) | –10.70 ± 0.20 | –183.96 ± 1.26 | –194.66 ± 1.28 |
| VD-Lac (R = O–, X = O) | –14.03 ± 0.36 | –136.03 ± 4.86 | –150.06 ± 4.87 |
3. Conclusions
Vancomycin represents an important class of antibiotics reserved for the treatment of serious infection by multidrug-resistant Gram-positive pathogens. All side chains of amino acids in vancomycin, except for the residues at positions 1 and 3, are involved in cross-linking to form a binding cleft. The amino acids at 1 and 3 are N-methylleucine and asparagine, respectively. Although these residues are not directly involved in the PG-stem binding, the removal or chemical modifications dramatically affects both the efficacy of the compound and its binding affinity to the PG-stem. For example, a natural product of vancomycin analogue that contains aspartate in place of asparagine at position 3 is known to have an eight-fold reduction in the antimicrobial activity against S. aureus;(34) however, the effect of aspartate substitution on the d-Ala-d-Ala binding is not well understood.
To investigate the role of the asparagine side chain of residue 3 in vancomycin, the following four glycopeptide–PG complexes were analyzed using molecular dynamics simulation: VN-Ala, VN-Lac, VD-Ala, and VD-Lac. While VN-Ala was found to be the most stable complex, VD-Lac showed greater stability over VN-Lac and VD-Ala. The VD-Lac complex is more stable than the VN-Lac complex by −4.71 kJ/mol (ΔΔGbind) and by −6.22 kJ/mol for the VD-Ala complex. The reduced binding stability of the VN-Lac and VD-Ala complexes determined by MD simulations (Table 1) are consistent with the observed antimicrobial activities of the compounds. In the case of vancomycin (VN), its activity against VRE is devoid of antimicrobial activity with MIC exceeding 1000 μg/mL. For VD, its activity against S. aureus, compared to vancomycin, is reduced by eight-fold. Hence, the increased stability of the VD-Lac complex strongly suggests that the aspartate-substituted analogue of vancomycin will exhibit antimicrobial activities against VRE and VRSA. Our analysis shows that the aspartate side chain in VD is not directly involved in the depsipeptide binding. Instead, the aspartate substitution changed the conformations of both aglycon and its bound ligand such that it minimized the destabilizing lone pair interaction between the ester oxygen of the d-Lac and carbonyl oxygen at the 4th residue in VD. The mechanism by which the aspartate side change affects the conformations of both aglycon and ligand is by forming transient intramolecular attraction with the amide protons at positions 1, 2, and 3 of the aglycon. These intramolecular attractions compete with the intermolecular H-bonds between aglycon and the C-terminus of the d-Lac to allow rearrangement of the bound depsipeptide, which minimizes the electrostatic repulsion. The MD simulations of glycopeptide–PG complexes provided insight into the function of residue 3 in vancomycin aglycon and its effects on the conformational dynamics associated with the PG-stem binding necessary for the future development of novel glycopeptides to counter the continual emergence of glycopeptide resistance in pathogens.
4. Methods
4.1. Molecular Dynamics Simulations
The initial crystal structure of vancomycin complexed with the cell wall precursor analogue was obtained from the Protein Data Bank (PDB code: 1FVM).38 The structure was modified with GaussView 6.1.1 of the Gaussian16 software package39 to generate the other complexes used in the simulations. The VN-Ala complex represents the unmodified vancomycin and acyl-d-Ala-d-Ala, while VN-Lac is a complex with acyl-d-Ala-d-Ala modified to acyl-d-Ala-d-Lac. In the VD-Ala complex, the residue 3 of vancomycin is modified from asparagine to aspartate, and VD-Lac is the complex with residue 3 of vancomycin modified to aspartate and acyl-d-Ala-d-Ala modified to acyl-d-Ala-d-Lac. A CHARMM general force field (CGenFF) was generated for the simulation system using the online paramchem/CGenFF-4.0 server.40,41 Partial charges for the atoms were obtained from a series of Natural Population Analyses using the density functional theory (DFT) at the B3LYP/6-311G(d,p) level with the Gaussian16 software package.39 All simulations were carried out under periodic boundary conditions using the GROMACS version 2018.3.42 A cubic box size of approximately 46.1 Å was used. The number of atoms in the complexes ranges from 199 to 202. The system was solvated with transferable intermolecular potential with 3 points (TIP3P) explicit water molecules in a cubic periodic box and neutralized using sodium and chlorine ions under physiological conditions. Following energy minimization via steepest descent, constant volume and temperature (NVT) equilibration was performed for 200 ps at 1 fs time step using the leap-frog integrator and modified Berendsen thermostat to equilibrate the system to a temperature of 300 K. Thereafter, constant pressure and temperature (NPT) equilibration was performed to stabilize the pressure of the system at 1 bar for 10 ns with 2 fs time step using Parrinello–Rahman barostat with a compressibility of 4.5 × 10–5 bar–1. The pressure was controlled by the isotropic position scaling protocol applied in GROMACS. Initial velocities were assigned from a Maxwellian distribution. The particle mesh Ewald (PME) method was used for the electrostatic interactions, and the length of all covalent bonds was constrained using the linear constraint solver (LINCS) algorithm.43 A 30 ns molecular dynamics simulation was performed at a time step of 2 fs for each protein–ligand complex and the output saved after every 10 ps. The cutoff distance for the nonbonded interaction is 12 Å. Three simple harmonic distance restraints were applied to all of the systems during the MD simulation based on experimental solid-state NMR data.44 The distances were defined and discussed in a previous publication.33
4.2. Root-Mean-Square Deviation
The root-mean-square deviation (RMSD) was used to quantitatively measure the conformational difference between the structures in the trajectories of the complex during MD simulations and a stable reference structure to estimate structural similarity. This was done by least-squares fitting the dynamic structure to the reference structure. The calculation was done using GROMACS built-in functions, illustrated by the equation for a molecular structure represented by a cartesian coordinate vector ri (i = 1 – N) of N atoms
| 1 |
where 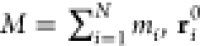 is the position of atom i in the reference structure, rit is
the position of atom i at time t, and mi is the mass
of atom i. The RMSD values for different complexes
were obtained by comparing their structure at time t = 0 (i.e., following the complete equilibration process) with the
various structures along the trajectories during MD simulations.
is the position of atom i in the reference structure, rit is
the position of atom i at time t, and mi is the mass
of atom i. The RMSD values for different complexes
were obtained by comparing their structure at time t = 0 (i.e., following the complete equilibration process) with the
various structures along the trajectories during MD simulations.
4.3. Hydrogen Bond Analysis
The hydrogen bond analysis monitored the stability of the hydrogen bonds formed between the glycopeptide and peptidoglycan analogues along the trajectory of the MD simulation. Hydrogen bond profiles between the selected glycopeptide and the peptidoglycan precursor were calculated with the g_hbond utility in GROMACS. All possible donors and acceptors are considered in the hydrogen bond analysis. The donor–acceptor distance is defined within 3.5 Å and the angle cutoff as 30° within a linear configuration. The average number of hydrogen bonds per time frame for different complexes can be used to compare their stability during the MD simulation.
4.4. Interaction Energy
The interaction energy was computed for each complex to characterize the nonbonded ligand–macromolecular interactions. The short-range nonbonded energies are decomposed using GROMACS g_energy analysis tool by defining the energy group of interest to recalculate energies from the existing simulation trajectory. The short-range van der Waals (Evdw) and electrostatic (Eelec) interactions were modeled using the Lennard–Jones short-range (LJ-SR) and Coulombic short-range interaction energy (Coul-SR), respectively. The total interaction energy (Etot) is the sum of the short-range Lennard–Jones energy (ELJ-SR) and the short-range Coulombic energy (ECoul-SR).
| 2 |
The measured intermolecular interaction energy is the interactions between the C-terminus of the bound ligand and the glycopeptide, while the intramolecular interaction energy was measured for the interactions within the glycopeptide, specifically between the side chain of residue 3 and other parts of the aglycon.
4.5. Binding Free Energy Calculations
The binding free energy between the glycopeptide and peptidoglycan analogues was computed using the molecular mechanics Poisson–Boltzmann surface area (MM/PBSA) method36 to quantify the strength of the interaction in a complex between the glycopeptide and its bound ligand. This was implemented in GROMACS using an external tool, g_mmpbsa, which integrates high-throughput MD simulations with binding energy calculations.45 The free energies of binding are described with the following set of equations
| 3 |
| 4 |
| 5 |
where ΔGbind is the total free energy of binding in the solution, Gcomplex, Greceptor, and Gligand are the free energies of the complex, glycopeptide, and ligand in the solution, respectively. ΔEMM is the molecular mechanic’s potential energy in a vacuum, which includes ΔEinternal (bond, angle, and dihedral energies), ΔEelec (electrostatic), and ΔEvdw (van der Waals) energies. ΔGsol is the free energy of solvation, and T and S denote the temperature and entropy, respectively. ΔEMM is evaluated directly from the force field terms and ΔGsol can be decomposed into polar and nonpolar contribution states. The polar term is estimated by solving the Poisson–Boltzmann equation, whereas the nonpolar term is estimated using the solvent-accessible volume model. The implementation of MM/PBSA within g-mmpbsa does not include entropic terms and thus is unable to provide the absolute binding free energy directly,45 although the tool is suited for calculating relative binding energies of similar systems.36,37,45,46
Acknowledgments
O.P.O. and K.L.S. were supported by the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy under Award Number DE-SC0019327, and S.J.K. was supported by the National Institutes of Health under grant number GM116130. The authors thank Baylor University High Performance Research Computing services for computing resources on the Kodiak HPC cluster. The authors also thank Dr. Emvia Calixte for meaningful discussions.
Glossary
Abbreviations
- lipid II
N-acetylglucosamine-N-acetyl-muramyl-pentapeptide-pyrophosphoryl-undecaprenol
- MD
molecular dynamics
- MRSA
methicillin-resistant Staphylococcus aureus
- PG
peptidoglycan
- VRE
vancomycin-resistant enterococci
- VRSA
vancomycin-resistant Staphylococcus aureus
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c05353.
Cartesian coordinates (Tables S1–S4) for the VN-Ala, VN-Lac, VD-Ala, and VD-Lac complexes are available (PDF)
Author Contributions
O.P.O, S.J.K., and K.L.S. designed the experiment. O.P.O carried out MD simulations. All authors contributed to the data analysis and writing of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- McCormick M. H.; McGuire J. M.; Pittenger G. E.; Pittenger R. C.; Stark W. M. Vancomycin, a new antibiotic. I. Chemical and biologic properties. Antibiot. Annu. 1955, 3, 606–611. [PubMed] [Google Scholar]
- Nagarajan R.Glycopeptide Antibiotics; Marcel Dekker, New York, 1994; Vol. 63. [Google Scholar]
- Nicolaou K. C.; Hughes R.; Cho S. Y.; Winssinger N.; Labischinski H.; Endermann R. Synthesis and biological evaluation of vancomycin dimers with potent activity against vancomycin-resistant bacteria: target-accelerated combinatorial synthesis. Chemistry 2001, 7, 3824–3843. . [DOI] [PubMed] [Google Scholar]
- Heijenoort J. v. Formation of the glycan chains in the synthesis of bacterial peptidoglycan. Glycobiology 2001, 11, 25R–36R. 10.1093/glycob/11.3.25R. [DOI] [PubMed] [Google Scholar]
- Cegelski L.; Kim S. J.; Hing A. W.; Studelska D. R.; O’Connor R. D.; Mehta A. K.; Schaefer J. Rotational-echo double resonance characterization of the effects of vancomycin on cell wall synthesis in Staphylococcus aureus. Biochemistry 2002, 41, 13053–13058. 10.1021/bi0202326. [DOI] [PubMed] [Google Scholar]
- Müller A.; Klöckner A.; Schneider T. Targeting a cell wall biosynthesis hot spot. Nat. Prod. Rep. 2017, 34, 909–932. 10.1039/C7NP00012J. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M.; Jones P. G.; Kennard O.; Williams D. H.; Smith G. A. Structure of vancomycin and its complex with acetyl-D-alanyl-D-alanine. Nature 1978, 271, 223–225. 10.1038/271223a0. [DOI] [PubMed] [Google Scholar]
- Schäfer M.; Schneider T. R.; Sheldrick G. M. Crystal structure of vancomycin. Structure 1996, 4, 1509–1515. 10.1016/S0969-2126(96)00156-6. [DOI] [PubMed] [Google Scholar]
- Leclercq R.; Derlot E.; Duval J.; Courvalin P. Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N. Engl. J. Med. 1988, 319, 157–161. 10.1056/NEJM198807213190307. [DOI] [PubMed] [Google Scholar]
- Bugg T. D.; Dutka-Malen S.; Arthur M.; Courvalin P.; Walsh C. T. Identification of vancomycin resistance protein VanA as a D-alanine:D-alanine ligase of altered substrate specificity. Biochemistry 1991, 30, 2017–2021. 10.1021/bi00222a002. [DOI] [PubMed] [Google Scholar]
- Bugg T. D.; Wright G. D.; Dutka-Malen S.; Arthur M.; Courvalin P.; Walsh C. T. Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry 1991, 30, 10408–10415. 10.1021/bi00107a007. [DOI] [PubMed] [Google Scholar]
- Derlot E.; Courvalin P. Mechanisms and implications of glycopeptide resistance in enterococci. Am. J. Med. 1991, 91, S82–S85. 10.1016/0002-9343(91)90348-2. [DOI] [PubMed] [Google Scholar]
- Arthur M.; Molinas C.; Bugg T. D.; Wright G. D.; Walsh C. T.; Courvalin P. Evidence for in vivo incorporation of D-lactate into peptidoglycan precursors of vancomycin-resistant enterococci. Antimicrob. Agents Chemother. 1992, 36, 867–869. 10.1128/AAC.36.4.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S.; Sievert D. M.; Hageman J. C.; Boulton M. L.; Tenover F. C.; Downes F. P.; Shah S.; Rudrik J. T.; Pupp G. R.; Brown W. J.; Cardo D.; Fridkin S. K. Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N. Engl. J. Med. 2003, 348, 1342–1347. 10.1056/NEJMoa025025. [DOI] [PubMed] [Google Scholar]
- Périchon B.; Courvalin P. Synergism between beta-lactams and glycopeptides against VanA-type methicillin-resistant Staphylococcus aureus and heterologous expression of the vanA operon. Antimicrob. Agents Chemother. 2006, 50, 3622–3630. 10.1128/AAC.00410-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miroshnikova O. V.; Printsevskaya S. S.; Olsufyeva E. N.; Pavlov A. Y.; Nilius A.; Hensey-Rudloff D.; Preobrazhenskaya M. N. Structure-activity relationships in the series of eremomycin carboxamides. J. Antibiot. 2000, 53, 286–293. 10.7164/antibiotics.53.286. [DOI] [PubMed] [Google Scholar]
- Allen N. E.; LeTourneau D. L.; Hobbs J. N. Jr. The role of hydrophobic side chains as determinants of antibacterial activity of semisynthetic glycopeptide antibiotics. J. Antibiot. 1997, 50, 677–684. 10.7164/antibiotics.50.677. [DOI] [PubMed] [Google Scholar]
- McComas C. C.; Crowley B. M.; Boger D. L. Partitioning the loss in vancomycin binding affinity for D-Ala-D-Lac into lost H-bond and repulsive lone pair contributions. J. Am. Chem. Soc. 2003, 125, 9314–9315. 10.1021/ja035901x. [DOI] [PubMed] [Google Scholar]
- Olsufyeya E. N.; Berdnikova T. F.; Miroshnikova O. V.; Reznikova M. I.; Preobrazhenskaya M. N. Chemical modification of antibiotic eremomycin at the asparagine side chain. J. Antibiot. 1999, 52, 319–324. 10.7164/antibiotics.52.319. [DOI] [PubMed] [Google Scholar]
- Butler M. S.; Hansford K. A.; Blaskovich M. A. T.; Halai R.; Cooper M. A. Glycopeptide antibiotics: back to the future. J. Antibiot. 2014, 67, 631–644. 10.1038/ja.2014.111. [DOI] [PubMed] [Google Scholar]
- Cooper R. D.; Snyder N. J.; Zweifel M. J.; Staszak M. A.; Wilkie S. C.; Nicas T. I.; Mullen D. L.; Butler T. F.; Rodriguez M. J.; Huff B. E.; Thompson R. C. Reductive alkylation of glycopeptide antibiotics: synthesis and antibacterial activity. J. Antibiot. 1996, 49, 575–581. 10.7164/antibiotics.49.575. [DOI] [PubMed] [Google Scholar]
- Vaudaux P.; Huggler E.; Arhin F. F.; Moeck G.; Renzoni A.; Lew D. P. Comparative activity of oritavancin against meticillin-resistant Staphylococcus aureus (MRSA) bloodstream isolates from Geneva University Hospital. Int. J. Antimicrob. Agents 2009, 34, 540–543. 10.1016/j.ijantimicag.2009.07.016. [DOI] [PubMed] [Google Scholar]
- Sweeney D.; Stoneburner A.; Shinabarger D. L.; Arhin F. F.; Belley A.; Moeck G.; Pillar C. M. Comparative in vitro activity of oritavancin and other agents against vancomycin-susceptible and -resistant enterococci. J. Antimicrob. Chemother. 2017, 72, 622–624. 10.1093/jac/dkw451. [DOI] [PubMed] [Google Scholar]
- McKay G. A.; Beaulieu S.; Arhin F. F.; Belley A.; Sarmiento I.; Parr T. Jr.; Moeck G. Time-kill kinetics of oritavancin and comparator agents against Staphylococcus aureus, Enterococcus faecalis and Enterococcus faecium. J. Antimicrob. Chemother. 2009, 63, 1191–1199. 10.1093/jac/dkp126. [DOI] [PubMed] [Google Scholar]
- Kim S. J.; Singh M.; Sharif S.; Schaefer J. Desleucyl-Oritavancin with a Damaged d-Ala-d-Ala Binding Site Inhibits the Transpeptidation Step of Cell-Wall Biosynthesis in Whole Cells of Staphylococcus aureus. Biochemistry 2017, 56, 1529–1535. 10.1021/acs.biochem.6b01125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M.; Chang J.; Coffman L.; Kim S. J. Hidden mode of action of glycopeptide antibiotics: inhibition of wall teichoic acid biosynthesis. J. Phys. Chem. B 2017, 121, 3925–3932. 10.1021/acs.jpcb.7b00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J. D.; Foster E. E.; Thadani A. N.; Ramirez A. J.; Kim S. J. Inhibition of Staphylococcus aureus cell wall biosynthesis by desleucyl-oritavancin: a quantitative peptidoglycan composition analysis by mass spectrometry. J. Bacteriol. 2017, 199, e00278-17 10.1128/JB.00278-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. J.; Cegelski L.; Stueber D.; Singh M.; Dietrich E.; Tanaka K. S. E.; Parr T. R.; Far A. R.; Schaefer J. Oritavancin exhibits dual mode of action to inhibit cell-wall biosynthesis in Staphylococcus aureus. J. Mol. Biol. 2008, 377, 281–293. 10.1016/j.jmb.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti G. J.; Kim S. J.; Yu T.-Y.; Dietrich E.; Tanaka K. S. E.; Parr T. R. Jr.; Far A. R.; Schaefer J. Vancomycin and oritavancin have different modes of action in Enterococcus faecium. J. Mol. Biol. 2009, 392, 1178–1191. 10.1016/j.jmb.2009.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James R. C.; Pierce J. G.; Okano A.; Xie J.; Boger D. L. Redesign of glycopeptide antibiotics: back to the future. ACS Chem. Biol. 2012, 7, 797–804. 10.1021/cb300007j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J.; Okano A.; Pierce J. G.; James R. C.; Stamm S.; Crane C. M.; Boger D. L. Total synthesis of [Ψ[C(=S)NH]Tpg4]vancomycin aglycon, [Ψ [C(=NH)NH]Tpg4]vancomycin aglycon, and related key compounds: reengineering vancomycin for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J. Am. Chem. Soc. 2012, 134, 1284–1297. 10.1021/ja209937s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J.; Pierce J. G.; James R. C.; Okano A.; Boger D. L. A redesigned vancomycin engineered for dual D-Ala-D-ala And D-Ala-D-Lac binding exhibits potent antimicrobial activity against vancomycin-resistant bacteria. J. Am. Chem. Soc. 2011, 133, 13946–13949. 10.1021/ja207142h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Zhou H.; Olademehin O. P.; Kim S. J.; Tao P. Insights into Key Interactions between Vancomycin and Bacterial Cell Wall Structures. ACS Omega 2018, 3, 37–45. 10.1021/acsomega.7b01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan R. Structure-activity relationships of vancomycin-type glycopeptide antibiotics. J. Antibiot. 1993, 46, 1181–1195. 10.7164/antibiotics.46.1181. [DOI] [PubMed] [Google Scholar]
- Williams D. H.; Bardsley B. The Vancomycin Group of Antibiotics and the Fight against Resistant Bacteria. Angew. Chem., Int. Ed. 1999, 38, 1172–1193. . [DOI] [PubMed] [Google Scholar]
- Hou T.; Wang J.; Li Y.; Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Kollman P. A. Free energy calculations on dimer stability of the HIV protease using molecular dynamics and a continuum solvent model. J. Mol. Biol. 2000, 303, 567–582. 10.1006/jmbi.2000.4057. [DOI] [PubMed] [Google Scholar]
- Nitanai Y.; Kikuchi T.; Kakoi K.; Hanamaki S.; Fujisawa I.; Aoki K. Crystal structures of the complexes between vancomycin and cell-wall precursor analogs. J. Mol. Biol. 2009, 385, 1422–1432. 10.1016/j.jmb.2008.10.026. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, revision C.01; Gaussian, Inc.: Wallingford CT, 2016.
- Guvench O.; Mallajosyula S. S.; Raman E. P.; Hatcher E.; Vanommeslaeghe K.; Foster T. J.; Jamison F. W. 2nd; Mackerell A. D. Jr. CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate-protein modeling. J. Chem. Theory Comput. 2011, 7, 3162–3180. 10.1021/ct200328p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Raman E. P.; MacKerell A. D. Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Hess B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- Singh M.; Kim S. J.; Sharif S.; Preobrazhenskaya M.; Schaefer J. REDOR constraints on the peptidoglycan lattice architecture of Staphylococcus aureus and its FemA mutant. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 363–368. 10.1016/j.bbamem.2014.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari R.; Kumar R.; Lynn A. g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D. A.; Cheatham T. E. 3rd Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.