

Abstract
Hepatocellular carcinoma (HCC) is the most common form of hepatic malignancies. The diagnosis of HCC remains challenging due to the low sensitivity and specificity of the diagnostic method. Exosomes, which are abundant in various proteins from parent cells, play pivotal roles in intercellular communication and have been confirmed as promising sources of disease biomarkers. Herein, we performed a simple but robust proteomic profiling on exosomes derived from 1 μL of serum using a data-independent acquisition (DIA) method for the first time, to screen potential biomarkers for the diagnosis of HCC. Ten pivotal differentially expressed proteins (DEPs) (von Willebrand factor (VWF), LGALS3BP, TGFB1, SERPINC1, HPX, HP, HBA1, FGA, FGG, and FGB) were screened as a potential candidate biomarker panel, which could completely discriminate patients with HCC from normal control (NC). Interestingly, Gene Expression Profiling Interactive Analysis (GEPIA) revealed that the expression levels of four genes increased and those of six genes decreased in HCC tissues compared with normal tissues, which were in concordance with protein expression levels. In conclusion, we screened 10 exosomal proteins holding promise for acting as a potential candidate biomarker panel for detection of HCC through a simple but robust proteomic profiling.
Introduction
Hepatocellular carcinoma (HCC) is the most common type of liver cancer. Liver cancer is the fourth on incidence and sixth on mortality among cancers worldwide,1 with the 1-year to 5-year survival rate of 47 to 12%. Although early detection of HCC is critical to reduce high HCC mortality rates, it remains challenging due to the low sensitivity and specificity of the diagnostic method.2,3 The serum biomarker α-fetoprotein (AFP) is commonly used to screen and diagnose HCC in the clinic.4 However, the sensitivity and specificity of AFP are limited to 40 and 62%.5 Clearly, a sensitive, simple, and rapid biomarker panel for the detection of HCC is an imperative need.
Exosomes are phospholipid bilayer-enclosed extracellular vesicles, which carry active substances such as proteins and nucleic acids derived from their parent cells.6 Exosomes have been found in various bodily fluids, including serum, plasma, urine, and saliva.7 These exosomes greatly facilitate the exchange of substances and information between cells and have been confirmed as promising sources of disease biomarkers.8 Besides, exosome is one of the three great liquid biopsies (circulating tumor cell (CTC) and circulating tumor DNA (ctDNA)),9 carrying various stable information for early diagnosis and treatment. Exosomes make up the drawbacks that the CTC contains comprehensive non-tumor cell information and ctDNA is easily degraded.10 Recently, multiple retrospective clinical diagnostic analyses reported exosomes as potential biomarkers in cancer, such as breast cancer,11 lung cancer,12 liver cancer,13 prostate cancer,14 pancreatic cancer,15 etc. An analysis indicated that exosome surfaceome could be used as noninvasive diagnosis of pancreatic cancer.16 A blood test of tumor exosomal RNA was developed for detecting lung cancer.17 Another study revealed that exosomal proteins were eventually selected as potential diagnostic and prognostic biomarkers of cancer.18 Therefore, exosomes in bodily fluids, specifically in serum, could be utilized as early diagnostic biomarkers in a minimally invasive manner.19
Data-independent acquisition (DIA) is a robust and reproducible proteomic technology for large-scale digital qualitative and quantitative research. The DIA mode divides the entire mass range into several sequential windows. All of the precursor ions in each window were fragmented sequentially. Therefore, DIA not only overcomes the loss of low abundance peptide information for accurate quantification in the data-dependent acquisition (DDA) mode but also avoids the drawback of only targeted interesting peptides in the selected reaction monitoring (SRM).20,21 A throughput optimized DIA workflow was allowed for the efficient simultaneous discovery and verification of candidate biomarkers.22 An analysis revealed carbonic anhydrase 2 as a potential new diagnostic biomarker for nasopharyngeal carcinoma in SWATH-MS analysis.23 Another analysis established a rapid and robust DIA-based quantitative proteomic profiling, describing 10 proteins as potential biomarkers.24,25 Overall, the DIA method acquires all high throughput peptide information without discrimination and is successfully used to detect candidate biomarkers. Unfortunately, there were few studies about the exosomal candidate biomarker panel of HCC using the DIA approach.
In this study, we investigated the proteomic profiling of exosomes from HCC patient serum samples by the DIA method to screen the potential candidate biomarker panel. Cluster analysis revealed 10 significant differentially expressed proteins (DEPs) that could completely distinguish HCC from normal control (NC). Gene Expression Profiling Interactive Analysis (GEPIA) revealed that the expression levels of four genes increased and those of six genes decreased in HCC tissues compared with normal tissues, which were in concordance with protein expression levels, suggesting exosomal proteins’ clinical values as a potential biomarker panel for HCC detection.
Results
Procedure of Exosomal Proteomics from Serum Samples
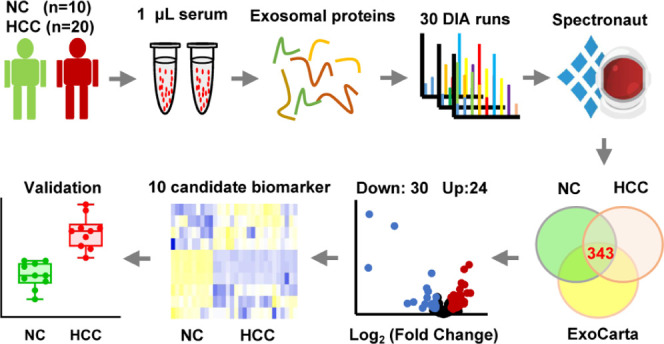
The procedure of exosomal proteomics from serum samples is shown in Figure 1A. In brief, the spectral library was generated with a mixture of 10 NC and 10 HCC samples in the DDA method. Then, the exosomal proteins from 10 independent NC and 20 independent HCC samples in DIA mode were analyzed by bioinformatic tools to screen potential candidate biomarkers of HCC. Finally, the potential vital exosomal candidate biomarker expression levels were validated by the Cancer Genome Atlas (TCGA) data set. On the evaluation of the minimal size of samples for proteomic analysis, the accumulation curve of the identified proteins from 10 samples in our previous study revealed that the accumulated protein number reached saturation when the sample size was 10 (Figure 1B). It was consistent with the results of the pooling strategy for quantitative proteomics.26,27
Figure 1.
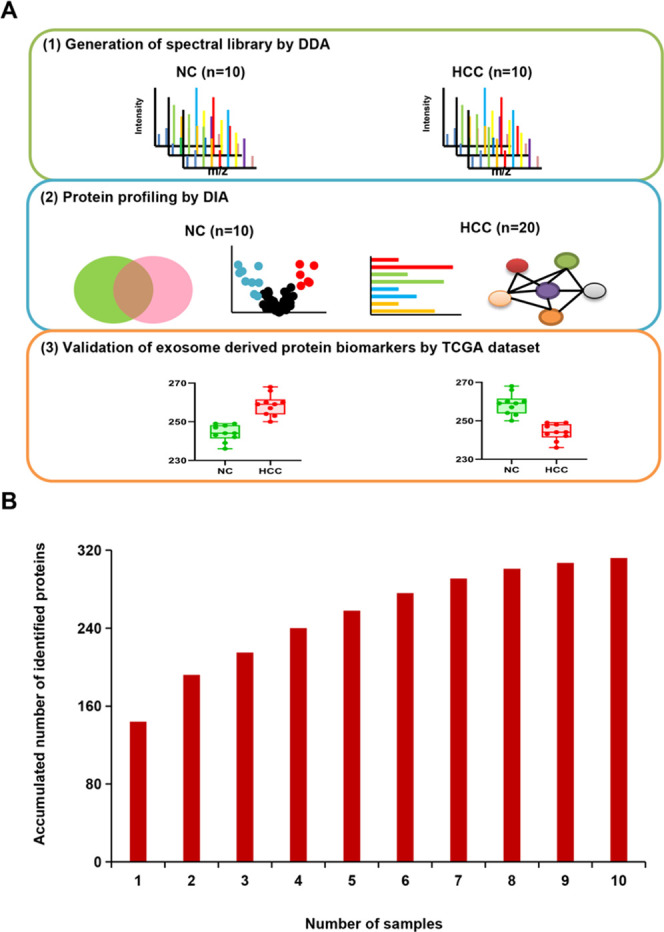
Procedure of exosomal proteomics from serum samples. (A) The procedure of the study. (B) The accumulation curve of the identified proteins from 10 samples. The accumulated number of proteins (y-axis) increased as the number of the samples (x-axis) increased. When the sample size reached 10, the accumulated protein number reached saturation.
Generation of Spectral Library
The spectral library is the basis of qualitative and quantitative analyses of the DIA data, which was generated by a mixture of NC and HCC samples in the DDA method. Clinical information of NC samples and HCC patients was listed, including cases, gender, and age (Table S1). The spectral library contained 627 protein groups, 1251 proteins, and 5529 peptides from NC and HCC samples (Tables S2 and S3). The spectral library data were evaluated with unique peptides and peptide length distribution. The results showed that the percentage of proteins with two or more unique peptides reached 83% (Figure S1A). The ratio of amino acid residues between 7 and 24 was over 95% (Figure S1B), suggesting that the spectral library data is available.
Evaluation of Experimental Variation
Since the reproducibility of the experimental methods and the LC system is a prerequisite for the reliable comparison of different runs, we tested run-to-run variation by repeated LC/MS/MS runs. Base peak profiles for the three consequent raw data were almost identical (data are not shown). The protein intensities of each run range from 102 to 107 (Figure S1C), suggesting that the stability of experimental methods. In addition, the synthetic unique peptide (LSASIAR) retention times were 28.94, 28.85, and 28.75 min, respectively. The intensities were 90.29, 100, and 90.03%, respectively (Figure S1D). The data strongly supported the high reproducibility of the automated LC/MS/MS system used in this study.
DEPs as an Indicator for the Classification of Samples
In total, 30 DIA raw data of LC-MS/MS were processed using Spectronaut software. The number of proteins identified in each individual serum exosome sample of NC and HCC was analyzed in a box plot, in which the numbers from HCC were significantly higher than those in NC (p-value < 0.05) (Figure 2A). Among the 478 proteins from NC and 490 proteins from HCC, 473 proteins (95.56%) were shared by both samples. Among them, 343 exosomal proteins could be found in 6515 human exosomal proteins from the ExoCarta database (Figure 2B). The fold change (FC) and p-value (student’s t-test) were calculated based on the protein information following crude screening. Upregulated and downregulated proteins were selected by the criteria of FC > 2, FC < 1/2, and p-value < 0.05. Eventually, 54 exosomal proteins were screened as DEPs, which were performed in a volcano plot (Figure 2C and Table S4). Twenty-four red dots represented upregulated exosomal proteins, while 30 green dots represented downregulated exosomal proteins. Furthermore, the clustered heat map of 54 DEPs was performed after normalization with z-score, and the exosomal proteins were grouped into four clusters according to the Euclidian distance. Upregulated and downregulated exosomal proteins completely separated the samples into HCC and NC, respectively (Figure 2D), suggesting that DEPs could be used as an indicator for the classification of the samples.
Figure 2.

DEPs as an indicator for the classification of the samples. (A) The box plot showed a significant difference in the number of proteins between NC (n = 10) and HCC (n = 20). (B) Venn diagrams of 343 coidentified exosomal proteins of NC and HCC, and 6515 human exosomal proteins from the ExoCarta database. (C) The volcano plot for DEPs. Blue dots represent the downregulated exosomal proteins, grey dots represent exosomal proteins that are not significantly expressed, and the red dots represent the upregulated exosomal proteins. (D) The clustered heat map of 54 DEPs. The exosomal proteins were grouped into four clusters according to the Euclidian distance. Upregulated and downregulated exosomal proteins completely separated the samples into HCC and NC, respectively.
DEPs are Associated with HCC
Gene ontology (GO) analysis of DEPs was performed to understand versatile biological and functional meanings. In Figure 3A, the 10 most statistically significant items (p-value < 0.05) of GO analysis are listed. For a biological process (BP), the exosomal proteins were mainly involved in platelet degranulation, blood coagulation, platelet aggregation and activation, cellular protein metabolic process, fibrinolysis, and innate immune response, which could indicate that serum exosome proteins contribute to the platelet aggregation and activation. The aberrant regulation of body immunity could partially involve the tumorigenesis and metastasis of HCC. A similar study revealed that exosomes facilitate intercellular communication in the tumor microenvironment, thereby remodeling normal macrophages to a tumor-activated phenotype with the assistance of hypoxia-inducible factors.28 As for the cellular components (CC), these DEPs are mainly associated with the extracellular region, blood microparticle, extracellular exosomes, extracellular space, etc., suggesting that these proteins were originated from exosomes. Molecular functions (MF) were also conducted with heparin binding, serine-type endopeptidase activity, glycoprotein binding, and receptor binding.
Figure 3.

Bioinformatic analysis of the DEPs of HCC. (A) GO and KEGG analyses for DEPs. The p-value of each enrichment entry was represented by the x-axis. (B) Visualization of the protein–protein interaction (PPI) network with DEPs. The red central nodes represented 10 candidate biomarkers. (C) Visualization of the PPI network with the 10 potential candidate biomarkers of HCC. (D) The clustered heat map for the 10 potential candidate biomarkers of HCC.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was used to analyze significantly affected metabolic and signal transduction pathways of DEPs. The KEGG pathway contained complement and coagulation cascades, platelet activation, and focal adhesion (Figure 3A), which were closely related to platelet degranulation, platelet aggregation, and activation of BP. The protein–protein interaction (PPI) network of DEPs were established (Figure 3B) based on the Search Tool for the Retrieval of Interacting Genes (STRING) database with a combined score > 0.4. It was worth noting that 10 exosomal proteins were screened as potential candidate biomarkers at central nodes, namely, von Willebrand factor (VWF), galectin-3-binding protein (LGALS3BP), transforming growth factor-β (TGFB1), antithrombin-III (SERPINC1), hemopexin (HPX), haptoglobin (HP), hemoglobin subunit α (HBA1), fibrinogen α chain (FGA), fibrinogen γ chain (FGG), and fibrinogen β chain (FGB) (Table S5). KEGG pathway enrichment analysis revealed that VWF, FGG, FGA, and FGB were relevant to both complement and coagulation cascades and platelet activation, implying that exosomal proteins could be key proteins regulating the coagulation cascade in patients with HCC. Moreover, TGFB1, VWF, LGALS3BP, FGG, FGA, and FGB were linked to platelet degranulation, which suggested that DEPs played complex roles in HCC tumorigenesis and development.
Ten Exosomal Proteins as a Potential Candidate Biomarker Panel of HCC
Correlation among the 10 potential candidate biomarker panel with STRING software showed that the 10 proteins were closely associated (Figure 3C). Furthermore, the clustered heat map revealed that the 10 potential candidate biomarkers also were grouped into four clusters, and upregulated and downregulated candidate biomarkers completely distinguish HCC from NC (Figure 3D), insinuating exosomal proteins as a potential candidate biomarker panel of HCC.
Validation of the Exosomal Potential Candidate Biomarker Panel
Gene expression levels of the 10 exosomal candidate biomarkers were estimated by GEPIA. The box plots revealed that the expression levels of VWF and LGALS3BP genes significantly increased in HCC tissues compared with normal tissues (p-value < 0.05), and the levels of TGFB1 and SERPINC1 genes also increased. However, the expression levels of HPX, HP, HBA1, and FGB genes significantly decreased (p-value < 0.05), and the levels of FGA and FGG genes also decreased (Figure 4), which were in concordance with protein expression levels.
Figure 4.

Gene expression levels of the 10 potential candidate biomarkers of HCC. The red and gray boxes represented cancer and normal tissues, respectively. (A, B) Gene expression levels of the four upregulated and six downregulated potential candidate biomarkers of HCC, respectively.
Discussion
Although various HCC biomarkers derived from omics data have been described,29,30 simple but robust proteomic analysis based on the DIA method with serum-derived exosomes from HCC patients has not been reported. In this study, we discovered that the serum-derived exosomes completely distinguish HCC from NC, suggesting that the exosomal proteins could be used as a potential complementary tool in diagnosing HCC.
Our study demonstrated that exosomes derived from HCC patients contained more exosomal proteins than those derived from NC. Similarly, the exosomes from HCC cell lines carried higher amounts of protein content than exosomes from immortalized hepatocyte lines.31 BP of HCC was enriched in our study, such as platelet degranulation, blood coagulation, platelet aggregation and activation, fibrinolysis, and innate immune response. An analysis revealed that platelet degranulation and human immune response played important roles in tumor growth and metastasis.32 An analysis indicated that platelet cargo, platelet adhesion, and activation were pivotal for nonalcoholic steatohepatitis and subsequent hepatocarcinogenesis.33 Another analysis indicated that tumor cell-induced platelet activation and blood coagulation system resulted in increased proliferation, migration, and metastatic potential of HCC.34 The similar finding also reported that coagulation and complement hematogenous metastasis pathway is implicated in HCC malignant ascites formation, and the coagulation products (such as fibrinogen and plasminogen) might predict prognosis of the patients with HCC.35 In addition, KEGG pathway analysis revealed that differentially expressed genes from HCC-associated data set GSE14323 significantly enriched complement and coagulation cascades and focal adhesion, which is consistent with our study.36
Among the DEPs, 10 potential candidate biomarkers (VWF, TGFB1, LGALS3BP, SERPINC1, HPX, HP, HBA1, FGA, FGG, and FGB) were identified in previous studies of HCC. VWF is a blood glycoprotein involved in the process of hemostasis, increased plasma levels in many cardiovascular and neoplastic, and could predict an increased risk of thrombosis.37 The VWF antigen level was higher in patients with severe liver fibrosis stage and/or HCC, suggesting that VWF is a potential biomarker for liver fibrosis and HCC development.38,39 TGFB1 is a secreted protein controlling cell growth, proliferation, differentiation, and apoptosis. An analysis revealed that TGFB1 participated in tumor invasion and migration through remodeling the tumor microenvironment and inducing inflammation.40 SERPINC1 was screened as a potentially novel biomarker from the liver-specific proteins through initial enrichment analysis with statistical significance, which was more amenable to downstream clinical validation experiments.41 The marker of HPX glycan differentiated patients with HCC and cirrhosis from healthy volunteers and patients with cirrhosis or fibrosis with a sensitivity and specificity of 79 and 93%, respectively. The elevated bifucosylation degree of HP could discriminate early-stage HCC patients from cirrhosis in each etiologic category.42 Arbelaiz et al. revealed LGALS3BP with higher diagnostic capacity than AFP by serum exosomal proteomic profiling of HCC.43 Fibrinogen, a blood-borne glycoprotein, is composed of three pairs of nonidentical polypeptide chains (FGA, FGB, and FGG). Significant downregulation of FGG genes compared to their nonmalignant counterparts highlighted FGG as a potential candidate of frequent allelic losses on chromosomes of HCC.44 The above data strongly support that exosomal proteins are closely related to the tumorigenesis and development of HCC. In addition, VWF, TGFB1, LGALS3BP, SERPINC1, HP, FGA, FGG, and FGB are membrane proteins. Membrane proteins play very important roles in cell proliferation and differentiation, energy conversion, signal transduction, and material transportation.
Furthermore, gene expressed levels of the 10 exosomal candidate biomarkers between HCC and NC by GEPIA were consistent with protein expression levels. The result provided a clue that exosomal proteins could be potential candidate biomarkers of HCC. However, it should be noted that the expressed levels were derived from the results of the gene expression but not the protein itself. Further studies are required for the validation and verification of these protein expressed levels in HCC samples.
Conclusions
In this study, we performed a simple but robust DIA-based quantitative exosomal proteomic profiling for distinguishing HCC from NC. The workflow is promising in clinical applications because of small serum amounts, various parent proteins from exosomes, and reproducible and rapid DIA method. The findings suggested that exosomal proteins from serum could be potential, promising, and efficient candidate biomarkers for HCC diagnosis. Although exosomal candidate biomarkers were required to validate our study, the procedure is expected to be highly instructive for other cancers.
Materials and Methods
Reagents
Titansphere TiO2 beads (5 μm) were purchased from GL Sciences (Tokyo, Japan). DMEM was provided by Gibco Brl (Grand Island, NY). EDTA-free protease inhibitor cocktail was from Roche (Basel, Switzerland). Ac-trypsin was provided by Enzyme & Spectrum (Beijing, China). Serum samples of 20 HCC patients were collected from Zhongshan Hospital, Fudan University.
Exosome Isolation and Protein Extraction
Exosomes were isolated from serum using TiO2 enrichment technology as described before with slight modification.45 Briefly, the cells and dead cells of serum were removed by centrifuging at 2000g for 30 min, and then the cells debris were removed consistently by centrifuging at 12 000g for 45 min. Next, 1 μL of serum was mixed with 2 mg of TiO2 microspheres in 49 μL of DMEM, followed by a 5 min incubation in a shaker MS-100 (Thermo Fisher Scientific, Palo Alto, CA). Sequentially, the exosomes on the TiO2 microsphere surface were lysed with 20 μL of lysis buffer (4% SDS, EDTA-free protease inhibitor cocktail, 0.1 M Tris-HCl, pH 7.5), followed by ultrasonication on ice for 20 min. The exosomal proteins were collected by centrifuging at 12 000g for 3 min.
Protein Digestion
Exosomal proteins were digested with the filter aided sample preparation (FASP) method after reduced with 5 mM dithiothreitol (DTT) and alkylated with 10 mM iodoacetamide (IAA).46 The exosomal proteins were sequentially digested with trypsin at a ratio of 1:50 (enzyme to substrate) at 37 °C overnight. The digested peptides for spectral library generation were divided into six fractions with a homemade C18 StageTip. A synthetic unique peptide (LSASIAR, 0.1 ng) was added into each sample as an internal standard before DIA analysis to evaluate the stability of the MS instrument. All peptides were desalted before LC-MS analysis.
DDA Mass Data Acquisition
Fractionated peptide samples were acquired with DDA mode on a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific, Palo Alto, CA) equipped with UltiMate 3000 high-pressure liquid chromatography (UHPLC) system (Thermo Fisher Scientific, Palo Alto, CA), with a homemade capillary column (75 μm i.d. × 12 cm; ReproSil-Pur C18-AQ, 3 μm; Dr. Maisch). In brief, the fractionated peptide samples were dissolved in 10 μL of loading buffer (0.1% formic acid in ddH2O), and then 5 μL of peptide solution was loaded onto an analytical column by an autosampler and eluted with an 88 min nonlinear gradient: 0–6% solvent B (0.1% formic acid in ACN) for 10 min, 6–10% for 5 min, 10–30% for 55 min, 30–40% for 10 min, 40–95% for 0.1 min, and finally holding at 95% for the last 4.9 min. The chromatographed peptides were ionized under a high voltage (2 kV). The full scan was acquired from m/z 350 to 1550 with a resolution of 120 000 at m/z 200. The automatic gain control (AGC) was set to 3 ×106, and the maximum ion injection time (MIT) was set to 20 ms. Data-dependent MS/MS scans were performed with a resolution of 15 000 at m/z 200. The top 25 most intense peptide ions with charge states in the range of 2–7 were subjected to further fragmentation in DDA mode via higher-energy collision dissociation (HCD) with 27% normalized collision energy and a target value of 2 × 104 for AGC. Dynamic exclusion was set to 15 s to avoid the redundancy detection.
Spectral Library Generation
For the generation of the spectral library, DDA raw data were analyzed with Spectronaut software and a mass spectrometer vendor-independent software from Biognosys.47 The DDA files were searched against the Swiss-Prot human database (20 353 entries released on Aug 10, 2020) and the decoy proteins were produced from pseudo-reversed sequences of the target proteins. The digestion of trypsin was allowed with a maximum of two missed cleavages. A minimal peptide length was set to seven amino acids. Cysteine carbamidomethylation was set as fixed modifications, while methionine oxidation and N-terminal acetylation were set as variable modifications. The mass tolerances were 20 ppm for precursor ions and fragment ions. The false discovery rates (FDR) of peptide and protein identifications were set to 0.01.
DIA Mass Data Acquisition and Analysis
DIA mass acquisition was performed with the same mass spectrometer, LC system, and nonlinear gradient as DDA, as described above. Full scans were analyzed over a mass range of 300–1400 m/z with a resolution of 120 000 at m/z 200. The AGC was set to 3 × 106, the MIT was set to 80 ms, and NCE was set to 27%. The isolation window was set to 35 DIA scans of 21 Da variable windows (containing 1 Da for the window overlap) as follows: 300–321, 320–341, 340–361, up to 980–1001 Da. Finally, a total of 30 DIA raw data of NC and HCC were analyzed against a spectral library as described previously with Spectronaut software. Default settings were performed, and the peptide and protein identifications were limited to a 1% FDR.
Statistical and Functional Analysis
The difference of identified proteins between NC and HCC was evaluated by a one-way ANOVA test. An overlap between the identified proteins and exosomal proteins on the ExoCarta database (http://www.exocarta.org/, release date: Aug 15, 2020) was exhibited on the online website (http://jvenn.toulouse.inra.fr/app/example.html).48 Statistical analysis of quantitative mass spectrometry-based proteomic experiments was performed for all samples with Perseus (version 1.6.6.0).49 A log2 transformation was applied to all intensity values. A protein was included if it was identified in at least 50% of samples in NC and HCC groups. Missing values were imputed using normal distribution. The cluster analysis of DEPs was performed by the online website (http://www.heatmapper.ca/expression/).50 The GO and KEGG analyses were performed with Database for Annotation, Visualization, and Integrated Discovery (DAVID) (http://david.abcc.ncifcrf.gov/).51 The protein–protein interaction (PPI) network was analyzed with the STRING (http://string.embl.de/)52 and constructed with Cytoscape software (version 3.8.0).53 The gene expressed levels were simulated by Gene Expression Profiling Interactive Analysis (GEPIA) (http://gepia.cancer-pku.cn/).54 The box plot and scatter plots were based on GraphPad Prism 8.3.0 (GraphPad Software, La Jolla, CA).55
Acknowledgments
This work was supported by the MOST (2017YFA0505700 and 2017YFA0505100), the National Natural Science Foundation of China (31670834, 31870824, 91839302, and 31901037), Innovation Foundation of Medicine (16CXZ027, BWS17J032, 19SWAQ17, and AWS17J008), National Megaprojects for Key Infectious Diseases (2018ZX10302302), the Foundation of State Key Lab of Proteomics (SKLP-K201704 and SKLP-K201901), Guangzhou Science and Technology Innovation & Development Project (201802020016), the Unilever 21th Century Toxicity Program (MA-2018–02170N), and the grant for Research Unit of Proteomics & Research and Development of New Drug of Chinese Academy of Medical Sciences (2019RU006).
Glossary
Abbreviations
- HCC
hepatocellular carcinoma
- NC
normal control
- DIA
data-independent acquisition
- FASP
filter aided sample preparation
- DTT
dithiothreitol
- IAA
iodoacetamide
- UHPLC
UltiMate 3000 high-pressure liquid chromatography
- HCD
higher-energy collision dissociation
- FDR
false discovery rates
- FC
fold change
- DEPs
differentially expressed proteins
- DAVID
Database for Annotation, Visualization, and Integrated Discovery
- PPI
protein–protein interaction
- STRING
Search Tool for the Retrieval of Interacting Genes
- GEPIA
Gene Expression Profiling Interactive Analysis
- TCGA
the Cancer Genome Atlas
- VWF
von Willebrand factor
- LGALS3BP
galectin-3-binding protein
- TGFB1
transforming growth factor β
- SERPINC1
antithrombin-III
- HPX
hemopexin
- HP
haptoglobin
- HBA1
hemoglobin subunit α
- FGA
fibrinogen α chain
- FGB
fibrinogen β chain
- FGG
fibrinogen γ chain
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c05408.
Evaluation of the spectral library and DIA data (Figure S1); patient information (Table S1); library information (Table S2); detailed information of library (Table S3); differentially expressed exosomal proteins between HCC and NC (Table S4); and information of 10 exosomal candidate biomarkers of HCC (Table S5) (PDF)
Serum-derived exosomal proteins as potential candidate biomarkers for hepatocellular carcinoma (XLSX)
The authors declare no competing financial interest.
Supplementary Material
References
- Villanueva A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. 10.1056/NEJMra1713263. [DOI] [PubMed] [Google Scholar]
- Bray F.; Ferlay J.; Soerjomataram I.; Siegel R. L.; Torre L. A.; Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2018, 68, 394–424. 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Craig A. J.; von Felden J.; Garcia-Lezana T.; Sarcognato S.; Villanueva A. Tumour evolution in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 139–152. 10.1038/s41575-019-0229-4. [DOI] [PubMed] [Google Scholar]
- Huang Z.; Zhou J. K.; Peng Y.; He W. F.; Huang C. H. The role of long noncoding RNAs in hepatocellular carcinoma. Mol. Cancer 2020, 19, 77 10.1186/s12943-020-01188-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrero J. A.; Lok A. S. Newer markers for hepatocellular carcinoma. Gastroenterology 2004, 127, S113–119. 10.1053/j.gastro.2004.09.024. [DOI] [PubMed] [Google Scholar]
- Théry C.; Zitvogel L.; Amigorena S. Exosomes: composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- Raposo G.; Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M.; Raposo G.; Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. 10.1146/annurev-cellbio-101512-122326. [DOI] [PubMed] [Google Scholar]
- Wang J. Y.; Chang S.; Li G. C.; Sun Y. L. Application of liquid biopsy in precision medicine: opportunities and challenges. Front. Med. 2017, 11, 522–527. 10.1007/s11684-017-0526-7. [DOI] [PubMed] [Google Scholar]
- Mader S.; Pantel K. Liquid Biopsy: Current Status and Future Perspectives. Oncol. Res. Treat. 2017, 40, 404–408. 10.1159/000478018. [DOI] [PubMed] [Google Scholar]
- Hannafon B. N.; Trigoso Y. D.; Calloway C. L.; Zhao Y. D.; Lum D. H.; Welm A. L.; Zhao Z. J.; Blick K. E.; Dooley W. C.; Ding W. Q. Plasma exosome microRNAs are indicative of breast cancer. Breast Cancer Res. 2016, 18, 90 10.1186/s13058-016-0753-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen K. R.; Paulsen B. S.; Bæk R.; Varming K.; Sorensen B. S.; Jørgensen M. M. Exosomal proteins as potential diagnostic markers in advanced non-small cell lung carcinoma. J. Extracell. Vesicles 2015, 4, 26659. 10.3402/jev.v4.26659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A. X.; Dong J.; Li S. H.; Wang C. N.; Ding H. M.; Li H.; Su X. T.; Ge X. F.; Sun L. Q.; Bai C. J.; Shen X. L.; Fang T.; Li J.; Shao N. S. Exosomal Transfer of Vasorin Expressed in Hepatocellular Carcinoma Cells Promotes Migration of Human Umbilical Vein Endothelial Cells. Int. J. Biol. Sci. 2015, 11, 961–969. 10.7150/ijbs.11943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez M.; Bajo-Santos C.; Hessvik N. P.; Lorenz S.; Fromm B.; Berge V.; Sandvig K.; Line̅ A.; Llorente A. Identification of non-invasive miRNAs biomarkers for prostate cancer by deep sequencing analysis of urinary exosomes. Mol. Cancer 2017, 16, 156. 10.1186/s12943-017-0726-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y. J.; Jin D. D.; Jiang F.; Liu J. X.; Qu L. S.; Ni W. K.; Liu Z. X.; Lu C. H.; Ni R. Z.; Zhu J.; Xiao M. B. Characterization and proteomic profiling of pancreatic cancer-derived serum exosomes. J. Cell. Biochem. 2019, 120, 988–999. 10.1002/jcb.27465. [DOI] [PubMed] [Google Scholar]
- Castillo J.; Bernard V.; San Lucas F. A.; Allenson K.; Capello M.; Kim D. U.; Gascoyne P.; Mulu F. C.; Stephens B. M.; Huang J.; Wang H.; Momin A. A.; Jacamo R. O.; Katz M.; Wolff R.; Javle M.; Varadhachary G.; Wistuba I. I.; Hanash S.; Maitra A.; Alvarez H. Surfaceome profiling enables isolation of cancer-specific exosomal cargo in liquid biopsies from pancreatic cancer patients. Ann. Oncol. 2018, 29, 223–229. 10.1093/annonc/mdx542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi K. R. The tumour trail left in blood. Nature 2016, 532, 269–271. 10.1038/532269a. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Ou X. X.; Wu X. H. Proteomics profiling of plasma exosomes in epithelial ovarian cancer: A potential role in the coagulation cascade, diagnosis and prognosis. Int. J. Oncol. 2019, 54, 1719–1733. 10.3892/ijo.2019.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R.; LeBleu V. S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977 10.1126/science.aau6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig C.; Gillet L.; Rosenberger G.; Amon S.; Collins B. C.; Aebersold R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol. Syst. Biol. 2018, 14, e8126 10.15252/msb.20178126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebersold R.; Mann M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- Muntel J.; Xuan Y.; Berger S. T.; Reiter L.; Bachur R.; Kentsis A.; Steen H. Advancing Urinary Protein Biomarker Discovery by Data-Independent Acquisition on a Quadrupole-Orbitrap Mass Spectrometer. J. Proteome Res. 2015, 14, 4752–4762. 10.1021/acs.jproteome.5b00826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y. Z.; Mok T. S.; Lin X. X.; Zhang W. L.; Cui Y. Z.; Guo J. H.; Chen X.; Zhang T.; Wang T. SWATH-based proteomics identified carbonic anhydrase 2 as a potential diagnosis biomarker for nasopharyngeal carcinoma. Sci. Rep. 2017, 7, 41191 10.1038/srep41191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L.; Zheng J. X.; Yu Q.; Chen W. D.; Xing J. C.; Chen C. X.; Tian R. J. High throughput and accurate serum proteome profiling by integrated sample preparation technology and single-run data independent mass spectrometry analysis. J. Proteomics 2018, 174, 9–16. 10.1016/j.jprot.2017.12.014. [DOI] [PubMed] [Google Scholar]
- Menon R.; Dixon C. L.; Sheller-Miller S.; Fortunato S. J.; Saade G. R.; Palma C.; Lai A.; Guanzon D.; Salomon C. Quantitative Proteomics by SWATH-MS of Maternal Plasma Exosomes Determine Pathways Associated With Term and Preterm Birth. Endocrinology 2019, 160, 639–650. 10.1210/en.2018-00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. Q.; Guo Y. B.; Song Y. P.; Sun W.; Yu C. H.; Zhao X. H.; Wang H. Y.; Jiang H. C.; Li Y. M.; Qian X. H.; Jiang Y.; He F. C. Proteomic analysis of individual variation in normal livers of human beings using difference gel electrophoresis. Proteomics 2006, 6, 5260–5268. 10.1002/pmic.200600006. [DOI] [PubMed] [Google Scholar]
- Xu P.; Tan H. P.; Duong D. M.; Yang Y. M.; Kupsco J.; Moberg K. H.; Li H.; Jin P.; Peng J. M. Stable isotope labeling with amino acids in Drosophila for quantifying proteins and modifications. J. Proteome Res. 2012, 11, 4403–4412. 10.1021/pr300613c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Zhou J. R.; Li X. D.; Wang X. J.; Lin Y. Y.; Wang X. P. Exosomes derived from hypoxic epithelial ovarian cancer cells deliver microRNAs to macrophages and elicit a tumor-promoted phenotype. Cancer Lett. 2018, 435, 80–91. 10.1016/j.canlet.2018.08.001. [DOI] [PubMed] [Google Scholar]
- Liu X. N.; Cui D. N.; Li Y. F.; Liu Y. H.; Liu G.; Liu L. Multiple ″Omics″ data-based biomarker screening for hepatocellular carcinoma diagnosis. World J. Gastroenterol. 2019, 25, 4199–4212. 10.3748/wjg.v25.i30.4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan G. S.; Lim K. H.; H T.; Khoo M. L.; Tan S. H.; Toh H. C.; Ching Ming Chung M. Novel proteomic biomarker panel for prediction of aggressive metastatic hepatocellular carcinoma relapse in surgically resectable patients. J. Proteome Res. 2014, 13, 4833–4846. 10.1021/pr500229n. [DOI] [PubMed] [Google Scholar]
- He M.; Hao Q.; Poon T. C. W.; Sze S. C.; Ding X. F.; Co N. N.; Ngai S. M.; Chan T. F.; Wong N. Hepatocellular carcinoma-derived exosomes promote motility of immortalized hepatocyte through transfer of oncogenic proteins and RNAs. Carcinogenesis 2015, 36, 1008–1018. 10.1093/carcin/bgv081. [DOI] [PubMed] [Google Scholar]
- Ehsani Ardakani M. J.; Safaei A.; Arefi Oskouie A.; Haghparast H.; Haghazali M.; Mohaghegh Shalmani H.; Peyvandi H.; Naderi N.; Zali M. R. Evaluation of liver cirrhosis and hepatocellular carcinoma using Protein-Protein Interaction Networks. Gastroenterol. Hepatol. Bed Bench 2016, 9, S14–S22. [PMC free article] [PubMed] [Google Scholar]
- Malehmir M.; Pfister D.; Gallage S.; Szydlowska M.; Inverso D.; Kotsiliti E.; Leone V.; Peiseler M.; Surewaard B. G. J.; Rath D.; Ali A.; Wolf M. J.; Drescher H.; Healy M. E.; Dauch D.; Kroy D.; Krenkel O.; Kohlhepp M.; Engleitner T.; Olkus A.; Sijmonsma T.; Volz J.; Deppermann C.; Stegner D.; Helbling P.; Nombela-Arrieta C.; Rafiei A.; Hinterleitner M.; Rall M.; Baku F.; Borst O.; Wilson C. L.; Leslie J.; O’Connor T.; Weston C. J.; Adams D. H.; Sheriff L.; Teijeiro A.; Prinz M.; Bogeska R.; Anstee N.; Bongers M. N.; Notohamiprodjo M.; Geisler T.; Withers D. J.; Ware J.; Mann D. A.; Augustin H. G.; Vegiopoulos A.; Milsom M. D.; Rose A. J.; Lalor P. F.; Llovet J. M.; Pinyol R.; Tacke F.; Rad R.; Matter M.; Djouder N.; Kubes P.; Knolle P. A.; Unger K.; Zender L.; Nieswandt B.; Gawaz M.; Weber A.; Heikenwalder M. Platelet GPIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat. Med. 2019, 25, 641–655. 10.1038/s41591-019-0379-5. [DOI] [PubMed] [Google Scholar]
- Zhuang M. J.; Xin G.; Wei Z. L.; Li S. Y.; Xing Z. H.; Ji C. J.; Du J. R.; Niu H.; Huang W. Dihydrodiosgenin inhibits endothelial cell-derived factor VIII and platelet-mediated hepatocellular carcinoma metastasis. Cancer Manage. Res. 2019, 11, 4871–4882. 10.2147/CMAR.S202225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Y.; Liang R.; Wei J. Z.; Ye J. X.; He Q.; Yuan C. L.; Ye J. Z.; Li Y. Q.; Liu Z. H.; Lin Y. Identification of Candidate Biomarkers in Malignant Ascites from Patients with Hepatocellular Carcinoma by iTRAQ-Based Quantitative Proteomic Analysis. BioMed Res. Int. 2018, 2018, 1–11. 10.1155/2018/5484976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C. Y.; Peng L.; Zhang Y. Q.; Liu Z. Y.; Li W. L.; Chen S. L.; Li G. C. The identification of key genes and pathways in hepatocellular carcinoma by bioinformatics analysis of high-throughput data. Med. Oncol. 2017, 34, 101 10.1007/s12032-017-0963-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidi M. Thrombosis and von Willebrand Factor. Adv. Exp. Med. Biol. 2017, 906, 285–306. 10.1007/5584_2016_122. [DOI] [PubMed] [Google Scholar]
- Takaya H.; Kawaratani H.; Tsuji Y.; Nakanishi K.; Saikawa S.; Sato S.; Sawada Y.; Kaji K.; Okura Y.; Shimozato N.; Kitade M.; Akahane T.; Moriya K.; Namisaki T.; Mitoro A.; Matsumoto M.; Fukui H.; Yoshiji H. von Willebrand factor is a useful biomarker for liver fibrosis and prediction of hepatocellular carcinoma development in patients with hepatitis B and C. United Eur. Gastroenterol J. 2018, 6, 1401–1409. 10.1177/2050640618779660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaya H.; Namisaki T.; Kitade M.; Kaji K.; Nakanishi K.; Tsuji Y.; Shimozato N.; Moriya K.; Seki K.; Sawada Y.; Saikawa S.; Sato S.; Kawaratani H.; Akahane T.; Noguchi R.; Matsumoto M.; Yoshiji H. VWF/ADAMTS13 ratio as a potential biomarker for early detection of hepatocellular carcinoma. BMC Gastroenterol. 2019, 19, 167 10.1186/s12876-019-1082-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fezza M.; Moussa M.; Aoun R.; Haber R.; Hilal G. DKK1 promotes hepatocellular carcinoma inflammation, migration and invasion: Implication of TGF-β1. PLoS One 2019, 14, e0223252 10.1371/journal.pone.0223252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awan F. M.; Nazm A.; Obaid A.; Ali A.; Ahmad J.; Anjum S.; Janjua H. A. Identification of Circulating Biomarker Candidates for Hepatocellular Carcinoma (HCC): An Integrated Prioritization Approach. PLoS One 2015, 10, e0138913 10.1371/journal.pone.0138913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J. H.; Warner E.; Parikh N. D.; Lubman D. M. Glycoproteomic markers of hepatocellular carcinoma-mass spectrometry based approaches. Mass Spectrom. Rev. 2019, 38, 265–290. 10.1002/mas.21583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbelaiz A.; Azkargorta M.; Krawczyk M.; Santos-Laso A.; Lapitz A.; Perugorria M. J.; Erice O.; Gonzalez E.; Jimenez-Agüero R.; Lacasta A.; Ibarra C.; Sanchez-Campos A.; Jimeno J. P.; Lammert F.; Milkiewicz P.; Marzioni M.; Macias R. I. R.; Marin J. J. G.; Patel T.; Gores G. J.; Martinez I.; Elortza F.; Falcon-Perez J. M.; Bujanda L.; Banales J. M. Serum extracellular vesicles contain protein biomarkers for primary sclerosing cholangitis and cholangiocarcinoma. Hepatology 2017, 66, 1125–1143. 10.1002/hep.29291. [DOI] [PubMed] [Google Scholar]
- Chan K. Y. Y.; Lai P. B. S.; Squire J. A.; Beheshti B.; Wong N. L. Y.; Sy S. M. H.; Wong N. Positional expression profiling indicates candidate genes in deletion hotspots of hepatocellular carcinoma. Mod. Pathol. 2006, 19, 1546–1554. 10.1038/modpathol.3800674. [DOI] [PubMed] [Google Scholar]
- Gao F. Y.; Jiao F. L.; Xia C. S.; Zhao Y.; Ying W. T.; Xie Y. P.; Guan X. Y.; Tao M.; Zhang Y. J.; Qin W. J.; Qian X. H. A novel strategy for facile serum exosome isolation based on specific interactions between phospholipid bilayers and TiO2. Chem. Sci. 2019, 10, 1579–1588. 10.1039/C8SC04197K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski J. R.; Zougman A.; Nagaraj N.; Mann M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Müller F.; Rappsilber J. A protocol for studying structural dynamics of proteins by quantitative crosslinking mass spectrometry and data-independent acquisition. J. Proteomics 2020, 218, 103721 10.1016/j.jprot.2020.103721. [DOI] [PubMed] [Google Scholar]
- Bardou P.; Mariette J. M.; Escudié F.; Djemiel C.; Klopp C. jvenn: an interactive Venn diagram viewer. BMC Bioinf. 2014, 15, 293 10.1186/1471-2105-15-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S.; Temu T.; Sinitcyn P.; Carlson A.; Hein M. Y.; Geiger T.; Mann M.; Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Babicki S.; Arndt D.; Marcu A.; Liang Y. J.; Grant J. R.; Maciejewski A.; Wishart D. S. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–153. 10.1093/nar/gkw419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Wei H.; Sherman B. T.; Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D.; Gable A. L.; Lyon D.; Junge A.; Wyder S.; Huerta-Cepas J.; Simonovic M.; Doncheva N. T.; Morris J. H.; Bork P.; Jensen L. J.; Mering C. V. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P.; Markiel A.; Ozier O.; Baliga N.; Wang J.; Ramage D.; Amin N.; Schwikowski B.; Ideker T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z. F.; Li C. X.; Kang B. X.; Gao G.; Li C.; Zhang Z. M. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujadas E.; Ibeh N.; Hernandez M. M.; Waluszko A.; Sidorenko T.; Flores V.; Shiffrin B.; Chiu N.; Young-Francois A.; Nowak M. D.; Paniz-Mondolfi A. E.; Sordillo E. M.; Cordon-Cardo C.; Houldsworth J.; Gitman M. R. Comparison of SARS-CoV-2 detection from nasopharyngeal swab samples by the Roche cobas 6800 SARS-CoV-2 test and a laboratory-developed real-time RT-PCR test. J. Med. Virol. 2020, 10, 1695–1698. 10.1002/jmv.25988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.