

Abstract

Deoxyartemisinin, a compound separated from Artemisinin annua L., shows anti-inflammatory and antiulcer activities. 10-Deoxoartemisinin is a novel compound with a strong antimalarial effect derivatized from artemisinin. Compared to the famous antimalarial natural compound artemisinin, deoxyartemisinin lacks the peroxide bridge structure, while 10-deoxoartemisinin remains this special peroxide bridge group but loses the 10-position keto group. To clarify their pharmacological differences, the absorption, distribution, metabolism, excretion (ADME) properties of artemisinin, deoxyartemisinin, and 10-deoxoartemisinin were first predicted using QikProp software. Also, their pharmacokinetic behaviors in rats were further evaluated by a rapid, sensitive, and specific liquid chromatography–tandem mass spectrometry (LC–MS/MS) method after oral and intravenous administration of each compound, in which deoxyartemisinin and 10-deoxoartemisinin were first evaluated for their pharmacokinetics. All parameters about ADME properties calculated by software met the criteria and the ADME performance order was 10-deoxoartemisinin > deoxyartemisinin > artemisinin. The oral bioavailability of artemisinin was calculated to be 12.2 ± 0.832%, which was about 7 times higher than that of deoxyartemisinin (1.60 ± 0.317%). For 10-deoxoartemisinin, its bioavailability (26.1 ± 7.04%) was superior to artemisinin at a degree of more than twice. Considering their chemical structures, losing the peroxide bridge might decrease the absorption rate of deoxyartemisinin in the gastrointestinal tract, while retaining the peroxide bridge but losing the 10-position ketone might improve the bioavailability of 10-deoxoartemisinin.
1. Introduction
Drug-likeness, a property that a candidate drug molecule should have, includes biological activities, good absorption, distribution, metabolism, excretion (ADME) properties, and safety.1 In the past few decades, the unpredictable nature of ADME/T (absorption, distribution, metabolism, elimination, toxicology) in the early stage of drug development has increased the failure rate in the late stage, resulting in huge investment waste.2 Therefore, taking rational control of chemical ADME properties of compounds is essential to improve the success rate of drug development.3
Artemisinin (Figure 1, CAS no. 63968-64-9, CC1CCC2C(C(=O)OC3C24C1CCC(O3)(OO4)C)C) and its derivatives are currently recommended by the World Health Organization (WHO) for treating malaria due to their rapid effects, low toxicity properties, and less drug resistance.4 The unique peroxide bridge structure and sesquiterpene lactone skeleton may be the main reason for the high antimalarial effect of artemisinin.5 However, artemisinin still shows the disadvantages of low bioavailability and large dosage in clinical applications when compared with its derivatives including dihydroartemisinin, artesunate, and artemether. The bioavailability of artemisinin could be greatly improved by different derivatization, which indicates that the ADME properties of these compounds should be further understood based on the basic maternal structure of artemisinin. In recent years, our team has been committed to those active ingredients in Artemisia annua L. (A. annua) that may not possess strong antimalarial activities but show anti-inflammatory, antipyretic, and antiasthmatic activities.6−12
Figure 1.
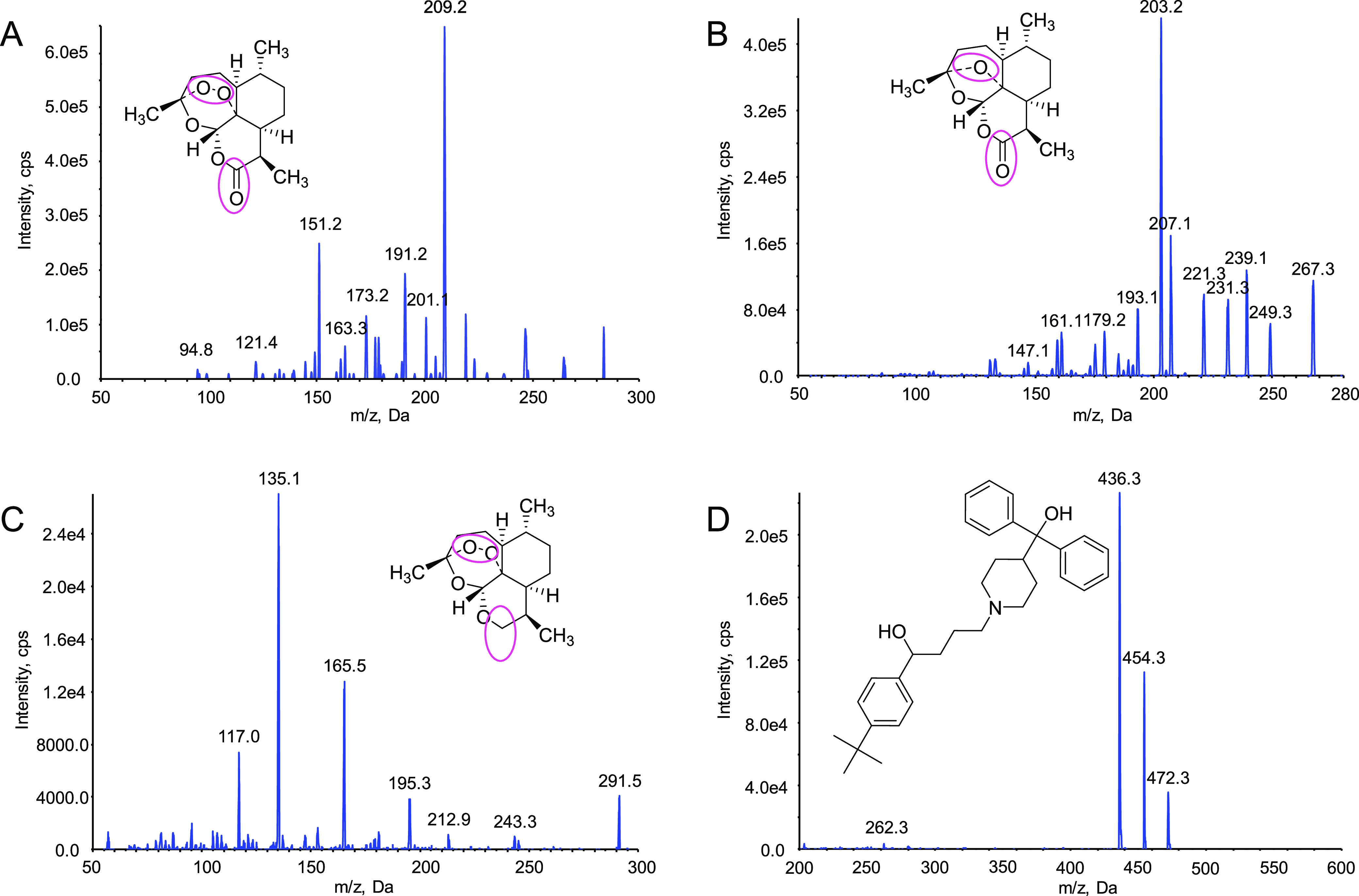
Chemical structures and product ion mass spectra of target compounds. The positions marked with colored circles indicated differences in the structure of the three compounds. (A) Artemisinin, (B) deoxyartemisinin, (C) 10-deoxoartemisinin, and (D) terfenadine.
Deoxyartemisinin (Figure 1, CAS no. 72826-63-2, CC1CCC2C(C(=O)OC3C24C1CCC(O3)(O4)C)C), a compound without peroxide bridge structure, was separated from A. annua by our experimental group at an early age.9 It is also one of the I-phase metabolites of artemisinin in vivo.13,14 Compared to artemisinin, deoxyartemisinin may not have significant antimalarial activity due to the absence of internal peroxide bridge but shows anti-inflammatory, antiulcer, and other pharmacological activities.15 10-Deoxoartemisinin (Figure 1, CAS no. 126189-95-5, CC1CC2CC(OCC23C(=CC(COO3)C)C1)(C)O), a novel bioactive component derivatized from artemisinin, was first prepared by a simple transformation in 1990.16 10-Deoxoartemisinin is made from artemisinin by reducing the 10-position keto group. It retains the peroxide bridge structure and thus leads to its highly efficient antimalarial effect, especially against multidrug-resistant malaria, which is more than 8 times that of artemisinin.16,17 Moreover, 10-deoxoartemisinin also shows obvious antitumor and antiangiogenesis activities.18−20 The structural changes of deoxyartemisinin and 10-deoxoartemisinin obviously affect their physical and chemical properties, resulting in different pharmacokinetic behaviors and pharmacological activities in vivo.
Since it has been reported that the therapeutic target was within the red blood cells,21,22 it is essential to study the in vivo processes of these compounds to clarify the differences in clinical efficacy. Therefore, it is necessary to characterize in vivo pharmacodynamic activity of artemisinin, deoxyartemisinin, and 10-deoxoartemisinin through their pharmacokinetic behaviors. The pharmacokinetic behavior of artemisinin in blood has already attracted widespread attention.13,23−26 It has low bioavailability and is rapidly and extensively metabolized in the body.13,25 Deoxyartemisinin, a metabolite of artemisinin, has also been reported to change over time in the blood.13 However, the comprehensive in vivo pharmacokinetics and bioavailability of deoxyartemisinin and 10-deoxoartemisinin have not been reported till now.
To explore the relationship between the structures and pharmacological activities of artemisinin derivatives, it is inevitable to characterize and compare the differences in the pharmacokinetic behaviors between artemisinin, deoxyartemisinin, and 10-deoxoartemisinin. Prior to investing in huge experimental costs, it is necessary to calculate their ADME properties to ensure their druggability. Thus, in the present study, ADME prediction was first performed on these three compounds, and then the pharmacokinetic study in rats was carried out using the high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry (HPLC-ESI-MS/MS) technique. Finally, the pharmacokinetic behaviors and oral bioavailability of the compounds were evaluated and compared. All of these provide theoretical support for subsequent drug development.
2. Results
2.1. ADME Properties
The physical properties and drug-related characteristics of these three compounds were calculated using the QikProp tool. All of the properties were identified based on Lipinski’s rule of 51,27 and other criteria.28,29Table 1 shows the calculated results of three compounds and all parameters are within the scope of the guidelines.
Table 1. Prediction of ADME Properties of Three Analytes Using QikProp.
| items | mol_MW | QP log Po/w | QP log S | QPPCaco | QP log BB | human oral absorption (%) | rule of five |
|---|---|---|---|---|---|---|---|
| artemisinin | 282.336 | 1.719 | –2.124 | 2040.288 | 0.021 | 96.25 | 0 |
| deoxyartemisinin | 266.336 | 2.059 | –2.236 | 3401.067 | 0.203 | 100 | 0 |
| 10-deoxoartemisinin | 268.352 | 2.725 | –2.796 | 7081.972 | 0.448 | 100 | 0 |
2.2. Method Optimization and Validation
Detailed validation results are exhibited in Tables 2–5. Appropriate linearity and good sensitivity, precision, accuracy, recovery, matrix effect, and stability values for each compound were obtained. Representative chromatograms of the blank rat plasma sample, lower limit of quantification (LLOQ), oral artemisinin, oral deoxyartemisinin, and oral 10-deoxoartemisinin are shown in Figure 2. No significant interference was observed. Linear ranges of the calibration curve for three analytes were all over 1.00–1000 ng/mL, in which the correlation coefficients (R) were more than 0.995. The LLOQs were all 1.00 ng/mL for each analyte. The precisions were less than 12%, and the accuracies ranged from 97.0 to 106.4%.
Table 2. Linearity, Range, and LLOQ of Artemisinin, Deoxyartemisinin, and 10-Deoxoartemisinin in Rat Plasma Determined by the LC–MS/MS Technique.
| compound | calibration equation | correlation coefficient (R) | range (ng/mL) | LLOQ (ng/mL) |
|---|---|---|---|---|
| artemisinin | y = 2.65 × 10–5x – 7.19 × 10–6 | 0.9961 | 1.00–1000 | 1.00 |
| deoxyartemisinin | y = 6.80 × 10–3x – 6.45 × 10–4 | 0.9968 | 1.00–1000 | 1.00 |
| 10-deoxoartemisinin | y = 2.32 × 10–4x + 7.79 × 10–5 | 0.9951 | 1.00–1000 | 1.00 |
Table 5. Stability Data for Artemisinin, Deoxyartemisinin, and 10-Deoxoartemisinin in Rat Plasma under Different Storage Conditions Determined by the LC–MS/MS Technique (n = 5).
| concentration (ng/mL) |
|||||
|---|---|---|---|---|---|
| compound | storage condition | spiked (ng/mL) | measured (ng/mL) | RSD (%) | accuracy (%, RE) |
| artemisinin | autosampler for 24 h (4 °C) | 2.00 | 1.91 ± 0.02 | 1.05 | 95.7 |
| 50.0 | 45.5 ± 0.66 | 1.45 | 91.0 | ||
| 800 | 777 ± 6.68 | 0.86 | 97.1 | ||
| three freeze/thaw cycles | 2.00 | 1.90 ± 0.02 | 1.06 | 95.2 | |
| 50.0 | 47.6 ± 0.52 | 1.09 | 95.2 | ||
| 800 | 770 ± 9.11 | 1.18 | 96.3 | ||
| long-term (30 days at –80 °C) | 2.00 | 1.95 ± 0.02 | 0.93 | 97.3 | |
| 50.0 | 45.7 ± 1.46 | 3.21 | 91.3 | ||
| 800 | 762 ± 9.29 | 1.22 | 95.2 | ||
| deoxyartemisinin | autosampler for 24 h (4 °C) | 2.00 | 2.18 ± 0.10 | 4.04 | 99.0 |
| 50.0 | 53.9 ± 1.66 | 4.04 | 99.0 | ||
| 800 | 782 ± 26.3 | 4.04 | 99.0 | ||
| three freeze/thaw cycles | 2.00 | 1.98 ± 0.08 | 4.49 | 109.1 | |
| 50.0 | 52.3 ± 0.99 | 4.49 | 109.1 | ||
| 800 | 774 ± 16.0 | 4.49 | 109.1 | ||
| long-term (30 days at –80 °C) | 2.00 | 2.03 ± 0.07 | 3.26 | 101.5 | |
| 50.0 | 54.2 ± 1.91 | 3.53 | 108.4 | ||
| 800 | 768 ± 50.2 | 6.53 | 96.0 | ||
| 10-deoxoartemisinin | autosampler for 24 h (4 °C) | 2.00 | 1.95 ± 0.11 | 9.41 | 97.5 |
| 50.0 | 46.7 ± 2.04 | 9.20 | 93.4 | ||
| 800 | 816 ± 43.2 | 5.51 | 102 | ||
| three freeze/thaw cycles | 2.00 | 2.05 ± 0.08 | 4.39 | 102.5 | |
| 50.0 | 56.1 ± 2.05 | 8.47 | 112.2 | ||
| 800 | 775 ± 42.8 | 1.67 | 96.9 | ||
| long-term (30 days at –80 °C) | 2.00 | 2.04 ± 0.05 | 6.63 | 102 | |
| 50.0 | 48.7 ± 1.22 | 5.09 | 97.4 | ||
| 800 | 827 ± 35.2 | 4.16 | 103.4 | ||
Figure 2.
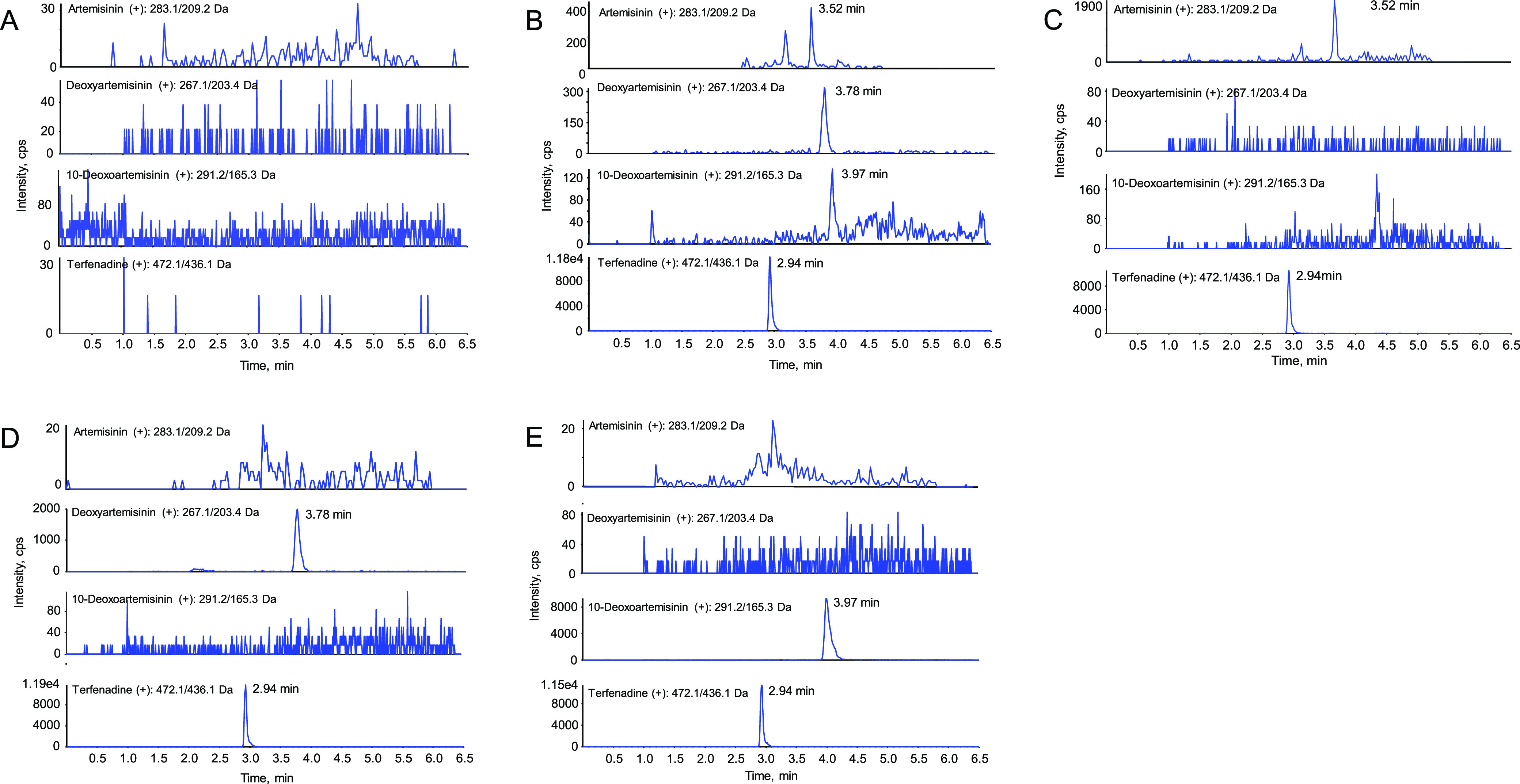
Representative MRM chromatograms of blank rat plasma sample (A), LLOQ (B), oral artemisinin after 5 min (C), oral deoxyartemisinin after 5 min (D), and oral 10-deoxoartemisinin after 5 min (E).
Table 3. Intra- and Interbatch Precision and Accuracy of Artemisinin, Deoxyartemisinin, and 10-Deoxoartemisinin in Rat Plasma Determined by the LC–MS/MS Technique (n = 5).
| concentration (ng/mL) |
precision (%, RSD) |
||||
|---|---|---|---|---|---|
| compound | spiked (ng/mL) | measured (ng/mL) | intra-batch | interbatch | accuracy (%, RE) |
| artemisinin | 1.00 | 1.03 ± 0.04 | 12.5 | 14.7 | 97.5 |
| 2.00 | 2.05 ± 0.12 | 6.14 | 6.02 | 102.4 | |
| 50.0 | 50.1 ± 2.87 | 5.77 | 5.73 | 100.1 | |
| 800 | 801 ± 47.0 | 6.08 | 5.87 | 100.2 | |
| deoxyartemisinin | 1.00 | 1.08 ± 0.02 | 8.23 | 10.3 | 99.3 |
| 2.00 | 1.94 ± 0.07 | 3.33 | 3.83 | 97.2 | |
| 50.0 | 49.4 ± 2.60 | 4.43 | 5.26 | 98.9 | |
| 800 | 787 ± 35.6 | 3.07 | 4.52 | 98.4 | |
| 10-deoxoartemisinin | 1.00 | 1.02 ± 0.05 | 11.5 | 7.42 | 96.2 |
| 2.00 | 1.96 ± 0.15 | 9.25 | 13.6 | 98.0 | |
| 50.0 | 53.2 ± 2.35 | 7.88 | 9.42 | 106.4 | |
| 800 | 776 ± 59.4 | 12.1 | 8.07 | 97.0 | |
Table 4. Matrix Effects and Recoveries of Artemisinin, Deoxyartemisinin, and 10-Deoxoartemisinin in Rat Plasma Determined by the LC–MS/MS Technique (n = 5).
| compound | spiked concentration (ng/mL) | recovery (%) | RSD (%) | matrix effect (%) | RSD (%) |
|---|---|---|---|---|---|
| artemisinin | 2.00 | 89.8 | 1.28 | 93.5 | 4.11 |
| 50.0 | 88.2 | 2.15 | 94.4 | 3.58 | |
| 800 | 85.8 | 2.71 | 92.1 | 4.61 | |
| deoxyartemisinin | 2.00 | 88.3 | 2.96 | 91.9 | 5.34 |
| 50.0 | 92.7 | 3.87 | 87.3 | 7.92 | |
| 800 | 89.7 | 3.67 | 90.3 | 4.34 | |
| 10-deoxoartemisinin | 2.00 | 86.8 | 8.41 | 91.3 | 4.59 |
| 50.0 | 89.4 | 6.45 | 92.5 | 8.55 | |
| 800 | 92.6 | 9.42 | 89.4 | 5.70 |
The recoveries of artemisinin, deoxyartemisinin, and 10-deoxoartemisinin varied from 85.8 to 92.7%, and the matrix effects ranged from 87.3 to 94.4%. The stabilities for room temperature (2 h), autosampler for 24 h (4 °C), three freeze/thaw cycles, and long-term (30 days at −80 °C) met all criteria. The relative deviations of stabilities were less than 9.41%.
2.3. Pharmacokinetics
The method validation in the present study was successfully applied to the pharmacokinetic study of artemisinin, deoxyartemisinin, and 10-deoxoartemisinin in rats after a single oral and intravenous administration. The pharmacokinetic profiles of intravenous and oral administration of artemisinin, deoxyartemisinin, and 10-deoxoartemisinin are shown in Figure 3. The main pharmacokinetic parameters are listed in Table 6.
Figure 3.
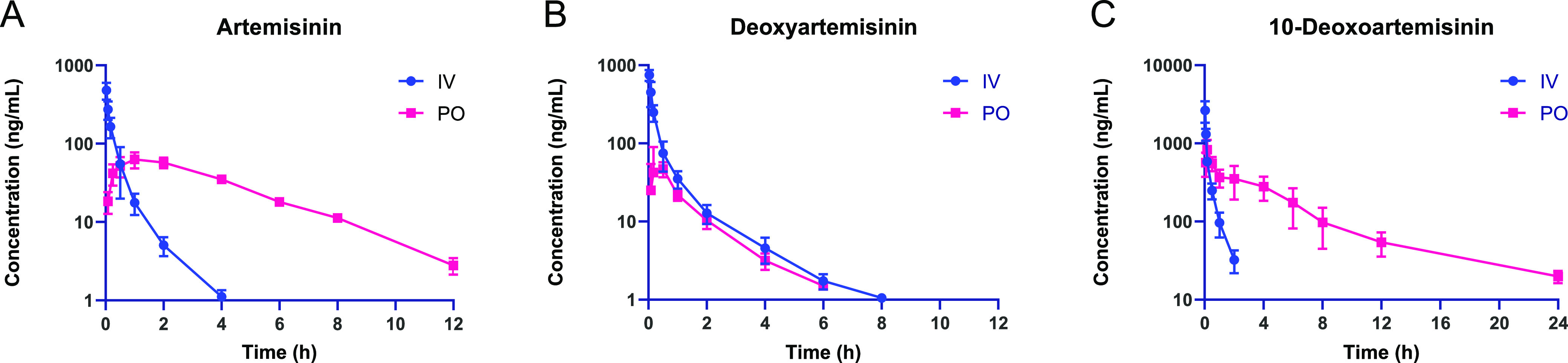
Mean plasma concentration profile of artemisinin (A), deoxyartemisinin (B), and 10-deoxoartemisinin (C) after intravenous administration (5 mg/kg) and oral administration (100 mg/kg) of artemisinin, deoxyartemisinin, and 10-deoxoartemisinin, respectively. Results are presented as mean ± standard deviation (SD) (n = 3).
Table 6. Pharmacokinetic Parameters of Three Compounds in Rat (n = 3) After Intravenous Administration of Artemisinin (5 mg/kg) or Oral Administration of Artemisinin (100 mg/kg), Respectively.
| dose | compound | t1/2 (h) | Tmax (h) | Cmax (ng/mL) | C0 (ng/mL) | AUClast (h·ng/mL) | AUCinf (h·ng/mL) | AUCextr (%) | Vz (L/kg) | CL (mL/min/kg) | MRTlast (h) | Fabs (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| i.v. 5 mg/kg | artemisinin | 0.773 ± 0.0598 | ND | ND | 700 ± 166 | 129 ± 38.9 | 130 ± 39.1 | 0.967 ± 0.110 | 45.1 ± 12.3 | 674 ± 174 | 0.408 ± 0.0660 | ND |
| deoxyartemisinin | 1.45 ± 0.059 | ND | ND | 1069 ± 173 | 219 ± 46.1 | 222 ± 47.1* | 1.33 ± 0.214 | 48.3 ± 8.00 | 385 ± 74.5* | 0.671 ± 0.183 | ND | |
| 10-deoxoartemisinin | 0.451 ± 0.0528 | ND | ND | 4367 ± 1956* | 583 ± 17.9** | 605 ± 14.7** | 3.57 ± 1.47* | 5.38 ± 0.642** | 138 ± 3.38** | 0.323 ± 0.0847 | ND | |
| p.o. 100 mg/kg | artemisinin | 2.22 ± 0.25 | 1.33 ± 0.58 | 65.1 ± 10.4 | ND | 309 ± 22.3 | 318 ± 21.6 | 2.89 ± 0.999 | ND | ND | 3.46 ± 0.249 | 12.2 ± 0.832 |
| deoxyartemisinin | 1.12 ± 0.156 | 0.389 ± 0.192 | 62.4 ± 31.3 | ND | 67.3 ± 14.2** | 71.0 ± 14.1** | 5.33 ± 1.92 | ND | ND | 1.21 ± 0.388** | 1.60 ± 0.317 | |
| 10-deoxoartemisinin | 5.22 ± 0.475 | 0.139 ± 0.0485 | 913 ± 138** | ND | 3008 ± 844** | 3156 ± 852** | 4.92 ± 1.35 | ND | ND | 5.36 ± 0.490** | 26.1 ± 7.04 |
p < 0.05 statistically significant difference against ART.
p < 0.01 statistically significant difference against ART.
2.3.1. Study of Intravenous and Oral Administration of Artemisinin
As shown in Table 6 and Figure 3, after intravenous injection of 5 mg/kg artemisinin in rats, the maximum concentration C0 was 700 ± 166 ng/mL and the elimination half-life t1/2 was 0.77 ± 0.06 h. Artemisinin was eliminated quickly in the blood because it could not be detected after 4 h. Also, it might be distributed to organs and metabolized in the body very fast. In addition, the Vz and CL of artemisinin in vivo are 45.1 ± 12.3 L/kg and 674 ± 174 mL/min/kg, respectively, and AUClast and AUCinf are 129 ± 38.9 and 130 ± 39.1 h·ng/mL, respectively.
After oral administration of 100 mg/kg artemisinin, the drug reached a peak concentration of 65.1 ± 10.4 ng/mL at a Tmax of 1.33 ± 0.58 h. Even though it reached a peak time fast, it was above the LLOQ in the plasma until 12 h. Compared with intravenous administration, the maximum concentration of artemisinin in blood after oral administration is lower, and the compound stays in the blood for a longer time. It could be speculated that the absorption time might be very long. In addition, the AUClast and AUCinf of artemisinin were 309 ± 22.3 and 318 ± 21.6 h·ng/mL, respectively. Finally, the absolute bioavailability was calculated to be 12.2 ± 0.83% according to the following formula, indicating that the oral bioavailability of artemisinin in SD rats by intragastric administration was relatively good. In the formula, Xiv represents the dose for intravenous administration and Xt represents the dose for extravascular administration.
2.3.2. Study of Intravenous and Oral Administration of Deoxyartemisinin
The pharmacokinetic profiles of intravenous and oral administration of deoxyartemisinin are illustrated in Figure 3B, and the main pharmacokinetic parameters are listed in Table 6.
After intravenous injection of 5 mg/kg deoxyartemisinin, the maximum concentration C0 in plasma was 1069 ± 173 ng/mL, and the elimination half-life t1/2 was 1.12 ± 0.16 h. However, the compound was not detected after 8 h, indicating that deoxyartemisinin was eliminated slowly from the blood. The Vz and CL were 48.3 ± 8.00 L/kg and 385 ± 74.5 mL/min/kg, respectively, and AUClast and AUCinf were 219 ± 46.1 and 222 ± 47.1 h·ng/mL, respectively.
After intragastric administration, deoxyartemisinin reached a peak concentration of 62.4 ± 31.3 ng/mL at about 0.390 ± 0.190 h, which was lower than that after intravenous administration. The elimination half-life t1/2 of deoxyartemisinin was 1.12 ± 0.160 h, and it disappeared from blood after 6 h, demonstrating that the amount of deoxyartemisinin absorbed in blood was very little. AUClast and AUCinf were 67.3 ± 14.2 and 71.0 ± 14.1 h·ng/mL, respectively. Finally, the absolute bioavailability was calculated to be 1.60 ± 0.32%, which indicated that deoxyartemisinin had a very low oral bioavailability.
2.3.3. Study of Intravenous and Oral Administration of 10-Deoxeartemisinin
The pharmacokinetic profiles of intravenous and oral administration of 10-deoxoartemisinin are illustrated in Figure 3C, and the main pharmacokinetic parameters are listed in Table 6.
After intravenous injection of 10-deoxyartemisinin in rats, the maximum concentration C0 in the blood was 4367 ± 1956 ng/mL. Vz and CL were 5.38 ± 0.64 L/kg and 138 ± 3.38 mL/min/kg, respectively. AUClast and AUCinf were 583 ± 17.9 and 605 ± 14.7 h·ng/mL, respectively. The elimination half-life t1/2 of 10-deoxyartemisinin was 0.45 ± 0.05 h, and the concentration cannot be detected after 2 h, indicating that it had a very short residence time in the blood as well as rapidly distributed or metabolized.
After intragastric administration of 100 mg/kg 10-deoxyartemisinin, the compound rapidly reached a peak concentration Cmax of 913 ± 138 at a time of about 0.140 ± 0.050 h, which was lower than intravenous administration. However, its elimination half-life t1/2 was 5.22 ± 0.47 h, and the concentration could still be detected after 24 h, indicating that it took a long time to absorb 10-deoxoartemisinin in the blood. The AUClast and AUCinf of the compound in rat blood were 3008 ± 844 and 3156 ± 852 h·ng/mL, respectively. Finally, the absolute bioavailability was calculated to be 26.1 ± 7.04%, indicating a high oral bioavailability of 10-deoxoartemisinin in rats.
3. Discussion
3.1. ADME Property Prediction
The molecular weights (mol_MW) were all less than 650. The partition coefficients between octanol and water (QP log Po/w) ranged from 1.719 to 2.725, and the solubility (QP log S) ranged from −2.796 to −2.124, which showed proper solubility in water and organic solvents. For the brain/blood partition coefficient (QP log BB) parameter, all of these compounds were between −3.0 and 1.2. Cerebral malaria is a common complication that occurred to malaria patients as well as an important cause of death,30 indicating that it is of great significance to evaluate QP log BB of the three compounds. The acceptable results demonstrated that they may play a therapeutic effect by penetrating the blood–brain barrier into the brain. Caco-2 is a human intestinal epithelial cell line that can imitate the intestinal–blood barrier to evaluate drug penetration cells. The QPPCaco results within the standard range indicated that the three compounds have good cell permeability in vivo. Human oral absorption of every compound was greater than 80%, showing an excellent absorption ability.
Taking the partition coefficients between octanol and water (QP log Po/w), brain/blood partition coefficient (QP log BB), and intestinal–blood barrier permeability (QPPCaco) together, the order from high to low was 10-deoxoartemisinin > deoxyartemisinin > artemisinin, which demonstrated that the reduction of 10-position keto greatly improved ADME property of 10-deoxoartemisinin and the loss of peroxide bridge also increased the ADME performance t of deoxyartemisinin.
3.2. Comparison among Artemisinin, Deoxyartemisinin, and 10-Deoxoartemisinin
Deoxyartemisinin is one of the I-phase metabolites of artemisinin in vivo, and 10-deoxoartemisinin is a derivative synthesized based on artemisinin. Therefore, it is necessary to compare their pharmacokinetic characteristics to find a better drug candidate.
For artemisinin and deoxyartemisinin, it could be simply calculated that the bioavailability of artemisinin was almost 7 times that of deoxyartemisinin, which is a large difference. As shown in Table 6 and Figure 4, only two parameters (AUClast and MRTlast) of oral administration showed significant differences between artemisinin and deoxyartemisinin (p < 0.01). Both parameters of deoxyartemisinin were obviously lower than artemisinin. Meanwhile, though there were no obvious differences in statistics for AUClast and MRTlast of intravenous administration, we could still see that these two parameters of deoxyartemisinin were higher than those of artemisinin. As a result, considering their chemical structures shown in Figure 1, it could be speculated that losing a peroxide bridge may lead to bad absorption of deoxyartemisinin in blood.
Figure 4.

Comparison among artemisinin, deoxyartemisinin, and 10-deoxoartemisinin based on different pharmacokinetic parameters. (A) i.v. 5 mg/kg and (B) p.o. 100 mg/kg.
For artemisinin and 10-deoxoartemisinin, the bioavailability of 10-deoxoartemisinin was more than twice that of artemisinin, which demonstrated an optimistic future as a candidate drug. As shown in Figure 4 and Table 6, almost every parameter exhibited a significant difference (p < 0.05) except MRTlast (i.v.) and AUCextr (%) (p.o.). The Cmax, C0, AUClast, and AUCinf of 10-deoxoartemisinin by either oral or intravenous administration were all much higher than that of artemisinin, which showed good absorption ability for 10-deoxoartemisinin in the blood. 10-Deoxoartemisinin remained the peroxide bridge structure and showed an excellent antimalaria effect. There have been many novel 10-deoxoartemisinin structures designed for bioactive experiments in recent years,31,32 implicating that it might become a hopeful candidate drug.
3.3. Oral Bioavailability of Three Compounds
The oral absorption process of drugs includes dissolution, gastric emptying, intestinal transport, drug transmembrane transport, and first-pass elimination caused by the intestinal wall and liver metabolism.33 These compounds were apparently fat-soluble and easily precipitated when they entered the gastrointestinal tract, resulting in a reduced amount in absorption. The oral bioavailability (Fabs), not equal to oral absorption, is not only related to absorption but also closely related to the amount and rate of metabolism and elimination. Therefore, even though the percent of human oral absorption (Table 1) calculated by software was more than 80%, the actual oral bioavailability (Fabs, Table 6) obtained from animal experiments for every compound was greatly reduced because of the several steps mentioned above. Fortunately, better ADME property for 10-deoxoartemisinin summarized in Section 3.1 led to better Fabs (26.1 ± 7.04%), which demonstrated that reduction of 10-position keto did improve the drug-likeness of 10-deoxoartemisinin and increase the antimalarial activity compared to artemisinin (12.2 ± 0.832%). For deoxyartemisinin, however, its Fabs (1.60 ± 0.317%) was not higher than artemisinin as speculated by software, demonstrating that it may suffer more severe influence than artemisinin and 10-deoxoartemisinin within the gastrointestinal tract due to its loss of peroxide bridge structure.
3.4. Difference between Human Oral Absorption and Rat Bioavailability
Drug absorption is a complex process that depends on the property of the drug, such as solubility and permeability, formulation factors, regional permeability differences, pH, luminal and mucosal enzymes, intestinal motility, etc.34 In the present research, the human oral absorption of three compounds calculated by the QikProp tool was close to 100%, which was theoretically good for their absorption. However, the practical experiment concluded that the bioavailability was just 1.6–26.1%, which does not correspond to the result of computer simulation. Of note, human oral absorption refers to the absorption degree of drugs by the human stomach and intestine, while oral bioavailability refers to the degree that drugs were absorbed in the blood. Therefore, one reason for the difference might be first-pass elimination, i.e., the liver probably metabolized most of them, resulting in a decrease in the concentration of the drugs in the plasma.
In addition, there have been literature studies on the oral bioavailability and intestinal permeability of various drugs with different absorption mechanisms in humans and rats. The results demonstrated that although rats and humans showed similar drug absorption curves and similar transporter expression patterns in the small intestine, the two species showed different expression levels and metabolic enzyme patterns in the intestine. Drug metabolizing enzymes, such as cytochrome P450, express in the intestine and liver to regulate the pharmacokinetics and oral bioavailability of drugs. Therefore, the rat model can be used to infer the oral drug absorption of the human small intestine, but it cannot fully predict the human oral bioavailability.35,36 In summary, the oral bioavailability results of this experiment in rats can provide evidence for the bioavailability of the three compounds in humans but has limitations to a certain extent.
4. Conclusions
Artemisinin, deoxyartemisinin, and 10-deoxoartemisinin were calculated using QikProp software and predicted to possess good ADME properties. A rapid, sensitive, and specific LC–MS/MS technique was developed and validated for the pharmacokinetic study of the three compounds in rats. After oral administration at a dose of 100 mg/kg and intravenous administration at a dose of 5 mg/kg of the three compounds, respectively, the oral bioavailability of artemisinin was 12.2 ± 0.832%, which was about sevenfold that of deoxyartemisinin (1.60 ± 0.317%). For 10-deoxoartemisinin, its bioavailability was highest (26.1 ± 7.04%), which is more than twice that of artemisinin.
5. Materials and Methods
5.1. Chemicals and Reagents
The reference standards of artemisinin, deoxyartemisinin, and 10-deoxoartemisinin were isolated from A. annua and then purified or semisynthesized and further standardized in our lab.37,38 Their chemical structures were further identified by UV, IR, 1H nuclear magnetic resonance (NMR), 13C NMR, and HR-ESI–MS techniques. The purities were detected to be all higher than 99.0% through the HPLC–diode-array detector (DAD) technique at a wavelength of 210 nm under an area normalization procedure. Terfenadine was purchased from Toronto Research Chemicals (TRC, Lot: 6-EOD-111-1, purity >99.0%, Toronto, Canada) and was used as the internal standard (IS). Methanol and acetonitrile of HPLC grade were purchased from Thermo Fisher Scientific. All other reagents belonged to analytical grade. The distilled water was obtained by a Milli Q water purification system from Millipore Corporation (MA).
5.2. Instruments and the LC–MS/MS Technique
The LC–MS/MS analysis procedure was operated on an Agilent 1200 HPLC system (CA) bridged with an AB Sciex 4000 Q Trap (ON, Canada). Data acquisition and quantification were implemented on Analyst 1.6 software (Applied Biosystems, MA). The elution procedure was performed on an Agilent Zorbax XDB C18 column (50 mm × 2.1 mm, 3.5 μm) at room temperature with a flow rate of 0.50 mL/min. The mobile phase A was water with 0.1% formic acid and B was methanol with 0.1% formic acid. In the LC gradient profile, the mobile phase B was 30% (v/v) for 0.50 min and linearly increased to 60% from 0.50 to 2.50 min. Then, it increased to 75% during 1.00 min and kept this state from 3.50 to 5.50 min. Finally, it went back to 30% at 5.51 min and maintained until 6.50 min.
The optimization of MS/MS conditions included source temperature (600 °C), ion spray voltage (5500 V), curtain gas (20 psi), nebulizing gas (60 psi), and turbo ion spray gas (60 psi). The quantitative ion pairs for multiple reaction monitoring (MRM) were m/z 283.1→209.2 for artemisinin, m/z 267.3→207.1 for deoxyartemisinin, m/z 291.2→165.3 for 10-deoxoartemisinin, and m/z 472.1→436.3 for terfenadine (IS). The product ion spectra of these compounds are shown in Figure 1. The declustering potentials (DP) were 86, 105, 92, and 130 V for artemisinin, deoxyartemisinin, 10-deoxoartemisinin, and terfenadine, respectively. The collision energies (CEs) were set at 11, 20, 22, and 50 eV artemisinin, deoxyartemisinin, 10-deoxoartemisinin, and terfenadine, respectively.
5.3. ADME Prediction
ADME properties of artemisinin, deoxyartemisinin, and 10-deoxoartemisinin were calculated using QikProp 5.8 tool (Schrodinger 2018, New York).39,40 QikProp can predict physicochemically significant descriptors as well as pharmacokinetically relevant properties. Not only does it provide a range of pharmacokinetic properties given by comparing a particular molecule with 95% known drugs but also it labels 30 kinds of reactive functional groups that may lead to false positives in high-throughput screening (HTS) analysis. In addition, it evaluates the acceptability of analogs based on Lipinski’s rule of five,1,27 which is necessary to obtain drugs with good drug-likeness.
In the current study, the parameters included are as follows: (a) molecular weight (mol_MW) (150–650), (b) octanol/water partition coefficient (QP log Po/w) (−2 to 6.5), (c) aqueous solubility (QP log S) (−6.5 to 0.5), (d) apparent Caco-2 cell permeability (QPPCaco) (nm/s; <25 poor, >500 great), (e) brain/blood partition coefficient (QP log BB) (−3.0 to 1.2), and (f) percent human oral absorption (≥80% is high, ≤25% is poor).
5.4. Animals
Male pathogen-free Sprague–Dawley rats (220–250 g) were purchased from the Charles River (Beijing, China, SYXK 2016-0006). The protocol was implemented according to the Animal Ethics Committee of Capital Medical University (Beijing, China) and followed the Guide for Care and Use of Laboratory Animals.41 The rats were fed in a room of special pathogen free (SPF) with an appropriate temperature of 25 ± 2 °C and a relative humidity of 40–60%. After 1-week adaptive breeding, all animals were fasted for 12 h until the experiment was started.
5.5. Preparation of Calibration Solutions and Quality Control (QC) Solutions
Stock solutions (1.00 mg/mL) for artemisinin, deoxyartemisinin, 10-deoxoartemisinin, and terfenadine were prepared in dimethyl sulfoxide (DMSO), respectively. The working solution of each compound was obtained by serially diluting the stock solutions with methanol to concentrations ranging from 10.0 to 10 000 ng/mL. To prepare calibration standards of the target compounds, 5 μL of working solutions were diluted in 50 μL of blank plasma to final concentrations of 1.00–1000 ng/mL for each compound. QC samples (2.00, 50.0, and 800 ng/mL) were independently prepared with the same method as calibration standards. All samples above were immediately stored at 4 °C.
5.6. Sample Preparation
All plasma samples were placed at room temperature for about 30 min until completely melted and then vortexed for 30 s. For calibration standards and QC samples, 100 μL of IS solution (100 ng/mL terfenadine in methanol/acetonitrile (50:50, v/v)) was added to corresponding plasma solutions. For plasma samples after administration, aliquots of 50 μL of rat plasma samples were collected and mixed with 5 μL of methanol and 100 μL of IS solution. All of the mixed suspensions contained IS were vortexed for 10 min and then centrifugated at 14 000g for 10 min, until aliquots of 100 μL of supernatants were transferred to HPLC vials for analysis.
5.7. Method Validation
According to the international guidelines and the established procedures in our group,42−46 the reliability of the current method for simultaneous quantification of artemisinin, deoxyartemisinin, and 10-deoxoartemisinin in plasma samples was proved by monitoring selectivity, linearity, lower limit of quantification (LLOQ), precision, accuracy, matrix effect, recovery, and stability.
The selectivity was evaluated by comparing the chromatogram of blank plasma with that of spiked matrix samples with artemisinin, deoxyartemisinin, 10-deoxoartemisinin, and terfenadine. Every calibration curve was plotted using the standard solution concentration as the abscissa and the ratio of the peak area of the analyte to the IS as the ordinate. The lower limit of quantification (LLOQ) referred to the lowest concentration whose precision and accuracy should be within −20 to 20%. The accuracy and precision were detected using QC samples (low, medium, and high concentrations) in six replicates and were characterized with relative standard deviation (RSD) and relative error (RE), respectively. The extraction recoveries were evaluated through the ratio of mean peak areas between regularly prepared QC samples (low, medium, and high concentrations) and spike-after-extraction plasma samples. Similarly, the matrix effect was assessed through the ratio of peak areas between postextraction samples spiked with analytes and mobile phase spiked with analytes at the same concentration. The stability was evaluated by analyzing QC samples set under different temperature conditions, including room temperature for 2 h (25 °C), autosampler for 24 h (4 °C), three freeze/thaw cycles, and long-term for 30 days (−80 °C).
5.8. Pharmacokinetics in Rats
Two days before starting the pharmacokinetic experiment, a polyethylene cannula was implanted in the jugular vein of each rat after receiving pentobarbital anesthesia (50 mg/kg, intravenous). The cannulas were exposed on the back of the neck and filled with heparin saline (20 units/mL). The formal experiment was operated on rats after 12 h fasting.
The intravenous solution (2.0 mg/mL) of each compound was prepared in DMSO and 30% HP-β-CD aqueous solution (5:95, v/v). Due to low polarity and poor solubility in water, each compound was dissolved to 80 mg/mL first using a small amount of cosolvent (DMSO). HP-β-CD aqueous solution (30%) was then added to make a final concentration of 2 mg/mL. As a kind of cyclodextrin inclusion agent, HP-β-CD can increase the water solubility of the compound by inclusion. Three groups of male SD rats (n = 3) were given solutions at a dose of 5 mg/kg through intravenous administration. The oral administration suspension solutions of these three compounds (10 mg/mL) were prepared using 0.5% CMC-Na. Three groups of male SD rats (n = 3) were treated with oral administration at a dose of 100 mg/kg. The choice of dosage is based on previous references reported by Birgersson and Dai.25,47 We calculated the equivalent dose for humans and rats and selected a compromised dose for the current experiment.
Blood samples with a volume of 200 μL were collected into heparinized tubes on ice at 0, 0.033, 0.083, 0.167, 0.50, 1.0, 2.0, 4.0, 6.0, 8.0, 12, and 24 h for intravenous, 0, 0.083, 0.25, 0.50, 1.0, 2.0, 4.0, 6.0, 8.0, 12, and 24 h for oral administration. The plasma samples were obtained via centrifugation of the blood samples at 14 000g for 10 min and were finally stored at −80 °C prior to analysis.
The pharmacokinetic parameters were calculated using WinNonlin software (version 6.4, Certara USA, Inc., Princeton, NJ), including half-life (t1/2), maximum plasma concentration (Cmax), area under the plasma concentration–time curve (AUClast), clearance (Cl), and the mean residence time (MRT). All data were shown with arithmetic mean ± standard deviation (SD).
Acknowledgments
This study was supported by the National Natural Science Foundation of China (Nos. 81573682 and 81841001) and the Major National Science and Technology Program of China for Innovative Drug (No. 2017ZX09101002-001-002).
Glossary
Abbreviations
- A. annua
Artemisia annua L.
- ESI
electrospray ionization
- MRM
multiple reaction monitoring
- DMSO
dimethyl sulfoxide
- QC
quality control
- CE
collision energy
- DP
declustering potential
- LLOQ
lower limit of quantification
- RSD
relative standard deviation
- RE
relative error
- QP log Po/w
octanol/water partition coefficient
- QP log S
aqueous solubility
- QPPCaco
apparent Caco-2 cell permeability
- QPlogBB
brain/blood partition coefficient
- i.v.
intravenous injection
- p.o.
oral administration
Author Contributions
The manuscript was written through the contribution of all authors. All authors have given approval to the final version of the manuscript
The authors declare no competing financial interest.
References
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Wang J. L. Comprehensive assessment of ADMET risks in drug discovery. Curr. Pharm. Des. 2009, 15, 2195–2219. 10.2174/138161209788682514. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Empfield J. R. Reducing the risk of drug attrition associated with physicochemical properties. Annu. Rep. Med. Chem. 2010, 45, 393–407. 10.1016/S0065-7743(10)45024-1. [DOI] [Google Scholar]
- Davis T. M.; Karunajeewa H. A.; Ilett K. Artemisinin-based combination therapies for uncomplicated malaria. Med. J. Aust. 2005, 182, 181–185. 10.5694/j.1326-5377.2005.tb06650.x. [DOI] [PubMed] [Google Scholar]
- Kamchonwongpaisan S.; Meshnick S. R. The mode of action of the antimalarial artemisinin and its derivatives. Gen. Pharmacol. Vasc. Syst. 1996, 27, 587–592. 10.1016/0306-3623(95)02047-0. [DOI] [PubMed] [Google Scholar]
- Fu C.; Yu P.; Wang M.; Qiu F. Phytochemical analysis and geographic assessment of flavonoids, coumarins and sesquiterpenes in Artemisia annua L. based on HPLC-DAD quantification and LC-ESI-QTOF-MS/MS confirmation. Food Chem. 2020, 312, 126070 10.1016/j.foodchem.2019.126070. [DOI] [PubMed] [Google Scholar]
- Qiu F.; Wu S.; Lu X. R.; Zhang C.; Li J.; Gong M. X.; Wang M. Y. Quality evaluation of the artemisinin-producing plant Artemisia annua L. based on simultaneous quantification of artemisinin and six synergistic components and hierarchical cluster analysis. Ind. Crops Prod. 2018, 118, 131–141. 10.1016/j.indcrop.2018.03.043. [DOI] [Google Scholar]
- Zhang D.; Yang L.; Yang L. X.; Wang M. Y.; Tu Y. Y. Determination of artemisinin,arteannuin B and artemisinic acid in Herba Artemisiae Annuae by HPLC-UV-ELSD. Acta Pharm. Sin. 2007, 42, 978–981. [PubMed] [Google Scholar]
- Tu Y. Y.; Ni M. Y.; Zhong Y. R.; Li L. N.; Cui S. L.; Zhang M. Q.; Wang X. Z.; Liang X. T. Studies on the Constituents of Artemisia annua Part I. Acta Pharm. Sin. 1981, 16, 366–370. [PubMed] [Google Scholar]
- Li Y. J.; Guo Y.; Yang Q.; Weng X. G.; Yang L.; Wang Y. J.; Chen Y.; Zhang D.; Li Q.; Liu X. C.; Kan X. X.; Chen X.; Zhu X. X.; Kmoniekova E.; Zidek Z. Flavonoids casticin and chrysosplenol D from Artemisia annua L. inhibit inflammation in vitro and in vivo. Toxicol. Appl. Pharmacol. 2015, 286, 151–158. 10.1016/j.taap.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Wang J. S. Casticin alleviates lipopolysaccharide-induced inflammatory responses and expression of mucus and extracellular matrix in human airway epithelial cells through Nrf2/Keap1 and NF-kappaB pathways. Phytother. Res. 2018, 32, 1346–1353. 10.1002/ptr.6067. [DOI] [PubMed] [Google Scholar]
- Yan H.; Li A. Y.; Zhao Y.; Lin Q. Y.; Li C. H. Studies on anti-heat-stress activity and its mechanisms of total coumarins from Artemisia annua. Chin. J. Exp. Tradit. Med. Formulae 2009, 15, 98–100. [Google Scholar]
- Du F.; Liu T.; Shen T.; Zhu F.; Xing J. Qualitative–(semi) quantitative data acquisition of artemisinin and its metabolites in rat plasma using an LTQ/Orbitrap mass spectrometer. J. Mass Spectrom. 2012, 47, 246–252. 10.1002/jms.2958. [DOI] [PubMed] [Google Scholar]
- Liu T.; Du F. Y.; Wan Y. K.; Zhu F. P.; Xing J. Rapid identification of phase I and II metabolites of artemisinin antimalarials using LTQ-Orbitrap hybrid mass spectrometer in combination with online hydrogen/deuterium exchange technique. J. Mass Spectrom. 2011, 46, 725–733. 10.1002/jms.1943. [DOI] [PubMed] [Google Scholar]
- de Faveri Favero F.; Grando R.; Nonato F. R.; Sousa I. M.; Queiroz N. C.; Longato G. B.; Zafred R. R.; Carvalho J. E.; Spindola H. M.; Foglio M. A. Artemisia annua L.: evidence of sesquiterpene lactones’ fraction antinociceptive activity. BMC Complementary Altern. Med. 2014, 14, 266 10.1186/1472-6882-14-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M.; Li X.; Bustos D. A.; ElSohly H. N.; McChesney J. D.; Milhous W. K. Synthesis and antimalarial activity of (+)-deoxoartemisinin. J. Med. Chem. 1990, 33, 1516–1518. 10.1021/jm00167a036. [DOI] [PubMed] [Google Scholar]
- Jung M.; Li X.; Bustos D. A.; ElSohly H. N.; McChesney J. D. A short and stereospecific synthesis of (+)-deoxoartemisinin and (−)-deoxodesoxyartemisinin. Tetrahedron Lett. 1989, 30, 5973–5976. 10.1016/S0040-4039(01)93831-6. [DOI] [Google Scholar]
- Lee C.-H.; Hong H.; Shin J.; Jung M.; Shin I.; Yoon J.; Lee W. NMR studies on novel antitumor drug candidates, deoxoartemisinin and carboxypropyldeoxoartemisinin. Biochem. Biophys. Res. Commun. 2000, 274, 359–369. 10.1006/bbrc.2000.3086. [DOI] [PubMed] [Google Scholar]
- Jung M.; Tak J.; Chung W.-Y.; Park K.-K. Antiangiogenic activity of deoxoartemisinin derivatives on chorioallantoic membrane. Bioorg. Med. Chem. Lett. 2006, 16, 1227–1230. 10.1016/j.bmcl.2005.11.074. [DOI] [PubMed] [Google Scholar]
- Jung M.; Lee S.; Ham J.; Lee K.; Kim H.; Kim S. K. Antitumor activity of novel deoxoartemisinin monomers, dimers, and trimer. J. Med. Chem. 2003, 46, 987–994. 10.1021/jm020119d. [DOI] [PubMed] [Google Scholar]
- Karunajeewa H. A.Artemisinins: Artemisinin, Dihydroartemisinin, Artemether and Artesunate. In Treatment and Prevention of Malaria; Springer: 2011; pp 157–190. [Google Scholar]
- Classen W.; Altmann B.; Gretener P.; Souppart C.; Skelton-Stroud P.; Krinke G. Differential effects of orally versus parenterally administered qinghaosu derivative artemether in dogs. Exp. Toxicol. Pathol. 1999, 51, 507–516. 10.1016/S0940-2993(99)80128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah F.; Zhang S.-Q.; Kandhari S. P.; Mukherjee P.; Chittiboyina A.; Avery M. A.; Avery B. A. In vitro erythrocytic uptake studies of artemisinin and selected derivatives using lc–ms and 2d-qsar analysis of uptake in parasitized erythrocytes. Bioorg. Med. Chem. 2009, 17, 5325–5331. 10.1016/j.bmc.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Lindegardh N.; Hanpithakpong W.; Kamanikom B.; Pattayaso J.; Singhasivanon P.; White N. J.; Day N. P. J. Quantification of dihydroartemisinin, artesunate and artemisinin in human blood: overcoming the technical challenge of protecting the peroxide bridge. Bioanalysis 2011, 3, 1613–1624. 10.4155/bio.11.158. [DOI] [PubMed] [Google Scholar]
- Dai T. M.; Jiang W. F.; Guo Z. Z.; Xie Y. X.; Dai R. K. Comparison of in vitro/in vivo blood distribution and pharmacokinetics of artemisinin,artemether and dihydroartemisinin in rats. J. Pharm. Biomed. Anal. 2019, 162, 140–148. 10.1016/j.jpba.2018.09.024. [DOI] [PubMed] [Google Scholar]
- Liu T.; Du F. Y.; Wan Y. K.; Zhu F. P.; Xing J. Rapid identification of phase I and II metabolites of artemisinin antimalarials using LTQ-Orbitrap hybrid mass spectrometer in combination with online hydrogen/deuterium exchange technique. J. Mass Spectrom. 2011, 46, 725–733. 10.1002/jms.1943. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; J F. P. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Kier L.Molecular Connectivity Indices in Chemistry and Drug Research; Academic Press: New York, NY, 2012; Vol. 14. [Google Scholar]
- Krämer S. D. Absorption prediction from physicochemical parameters. Pharm. Sci. Technol. Today 1999, 2, 373–380. 10.1016/S1461-5347(99)00188-1. [DOI] [PubMed] [Google Scholar]
- Seydel K. B.; Kampondeni S. D.; Valim C.; Potchen M. J.; Milner D. A.; Muwalo F. W.; Birbeck G. L.; Bradley W. G.; Fox L. L.; Glover S. J.; Hammond C. A.; Heyderman R. S.; Chilingulo C. A.; Molyneux M. E.; Taylor T. E. Brain Swelling and Death in Children with Cerebral Malaria. N. Engl. J. Med. 2015, 372, 1126–1137. 10.1056/NEJMoa1400116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khac V. T.; Van T. N.; Van S. T. Synthesis of novel 10-deoxoartemisinins. Bioorg. Med. Chem. Lett. 2005, 15, 2629–2631. 10.1016/j.bmcl.2005.03.050. [DOI] [PubMed] [Google Scholar]
- Bai Y.; Zhang D.; Sun P.; Zhao Y.; Chang X.; Ma Y.; Yang L. Evaluation of Microbial Transformation of 10-deoxoartemisinin by UPLC-ESI-Q-TOF-MS(E). Molecules 2019, 24, 3874 10.3390/molecules24213874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Lee S. L.; Yu L. X. Mechanistic approaches to predicting oral drug absorption. AAPS J. 2009, 11, 217–224. 10.1208/s12248-009-9098-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokoumetzidis A.; Valsami G.; Macheras P. Modelling and simulation in drug absorption processes. Xenobiotica 2007, 37, 1052–1065. 10.1080/00498250701502114. [DOI] [PubMed] [Google Scholar]
- Cao X.; Gibbs S. T.; Fang L.; Miller H. A.; Landowski C. P.; Shin H. C.; Lennernas H.; Zhong Y.; Amidon G. L.; Yu L. X.; Sun D. Why is it challenging to predict intestinal drug absorption and oral bioavailability in human using rat model. Pharm. Res. 2006, 23, 1675–1686. 10.1007/s11095-006-9041-2. [DOI] [PubMed] [Google Scholar]
- Hurst S.; Loi C. M.; Brodfuehrer J.; El-Kattan A. Impact of physiological, physicochemical and biopharmaceutical factors in absorption and metabolism mechanisms on the drug oral bioavailability of rats and humans. Expert Opin. Drug Metab. Toxicol. 2007, 3, 469–489. 10.1517/17425255.3.4.469. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Yang L.; Yang L. X.; Wang M. Y.; Tu Y. Y. Determination of artemisinin, arteannuin B and artemisinic acid in Herba Artemisiae Annuae by HPLC-UV-ELSD. Acta Pharm. Sin. 2007, 42, 978–981. [PubMed] [Google Scholar]
- Zhang C.; Gong M. X.; Qiu F.; Li J.; Wang M. Y. Effects of arteannuin B, arteannuic acid and scopoletin on pharmacokinetics of artemisinin in mice. Asian Pac. J. Trop. Med. 2016, 9, 677–681. 10.1016/j.apjtm.2016.05.004. [DOI] [PubMed] [Google Scholar]
- Pawar V. S.; Lokwani D. K.; Bhandari S. V.; Bothara K. G.; Chitre T. S.; Devale T. L.; Modhave N. S.; Parikh J. K. Design, docking study and ADME prediction of Isatin derivatives as anti-HIV agents. Med. Chem. Res. 2011, 20, 370–380. 10.1007/s00044-010-9329-y. [DOI] [Google Scholar]
- Mohd Amin S. N.; Md Idris M. H.; Selvaraj M.; Mohd Amin S. N.; Jamari H.; Kek T. L.; Salleh M. Z. Virtual screening, ADME study, and molecular dynamic simulation of chalcone and flavone derivatives as 5-Lipoxygenase (5-LO) inhibitor. Mol. Simul. 2020, 46, 487–496. 10.1080/08927022.2020.1732961. [DOI] [Google Scholar]
- National Research Council . Guide for the Care and Use of Laboratory Animals; National Academies Press, 2010. [Google Scholar]
- US Food and Drug Administration . Guidance for Industry: Bioanalytical Method Validation; US Deparment of Health and Human Services, 2018.
- European Medicines Agency . Guideline on Bioanalytical Method Validation, EMEA/CHMP/EWP/192217/2009; Committee for Medicinal Products for Human Use (CHMP), 2011.
- Wang M. Y.; Fu S. J.; Zhang X. S.; Li J.; Gong M. X.; Qiu F. LC-ESI-MS/MS analysis and pharmacokinetics of plantainoside D isolated from Chirita longgangensis var. hongyao, a potential anti-hypertensive active component in rats. Molecules 2014, 19, 15103 10.3390/molecules190915103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu F.; Fu S. J.; Zhang X. S.; Gong M. X.; Wang M. Y. Application of a sensitive and specific LC-MS/MS method for determination of eriodictyol-8-C-β-d-glucopyranoside in rat plasma for a bioavailability study. Biomed. Chromatogr. 2015, 29, 220–225. 10.1002/bmc.3263. [DOI] [PubMed] [Google Scholar]
- Qiu F.; Gu Y. N.; Wang T. T.; Gao Y. Y.; Li X.; Gao X. Y.; Cheng S. Quantification and pharmacokinetics of crizotinib in rats by liquid chromatography–tandem mass spectrometry. Biomed. Chromatogr. 2016, 30, 962–968. 10.1002/bmc.3636. [DOI] [PubMed] [Google Scholar]
- Birgersson S.; Van Toi P.; Truong N. T.; Dung N. T.; Ashton M.; Hien T. T.; Abelö A.; Tarning J. Population pharmacokinetic properties of artemisinin in healthy male Vietnamese volunteers. Malar. J. 2016, 15, 90 10.1186/s12936-016-1134-8. [DOI] [PMC free article] [PubMed] [Google Scholar]