

IL‐17 family was conducted with 6 members, most of them are widely involved in a variety of acute and chronic inflammatory responses, especially IL‐17A. After stroke, cerebral ischaemia and hypoxia lead nerve cell necrosis and release a large amount of damage‐associated molecular patterns and inflammation factors to activate IL‐17A. Then, IL‐17A promotes the development of stroke by inducing the secretion of inflammatory factors (such as TNF‐α, IL‐6, CXCL1), recruiting neutrophils to infiltrate into central nervous system and impairing the integrity of blood–brain barrier.
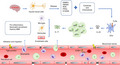
Keywords: IL‐17A, inflammation, interleukin‐17 (IL‐17), stroke, therapy
Summary
Interleukin‐17 (IL‐17) is a cytokine family that includes 6 members, IL‐17A through IL‐17F, most of them are reported to have pro‐inflammatory role. Through binding to their receptors (IL‐17Rs), IL‐17 activates the intracellular signalling pathways to play an important role in autoimmune diseases, including rheumatoid arthritis (RA) and multiple sclerosis (MS). Ischaemic stroke is a complex pathophysiological process mainly caused by regional cerebral ischaemia. Inflammatory factors contribute to the physiological process of stroke that leads to poor prognosis. IL‐17 plays a crucial role in promoting inflammatory response and inducing secondary injury in post‐stroke. Though immune cells and inflammatory factors have been reported to be involved in the damage of stroke, the functions of IL‐17 in this process need to be elucidated. This review focuses on the pathological modulation and the mechanism of IL‐17 family in ischaemic stroke and seeking to provide new insights for future therapies.
Abbreviations
- AP‐1
activator protein 1
- BBB
blood–brain barrier
- Bcl‐2
B‐cell lymphoma‐2
- BDNF
brain‐derived neurotrophic factor
- Blk
B‐lymphoid tyrosine kinase
- BMEC
brain microvascular endothelial cell
- CNS
central nervous system
- CREB
cAMP‐response element‐binding protein
- CTLA8
T‐lymphocyte‐associated antigen 8
- DAMP
damage‐associated molecular pattern
- DC
dendritic cell
- EAE
experimental autoimmune encephalitis
- ERK
extracellular signal‐related kinase
- Foxp3
forkhead box P3
- GSK‐3β
glycogen synthase kinase‐3β
- HIF‐1α
hypoxia‐inducible factor‐1α
- HuR
human antigen R
- IKK
inhibitor of kappa‐B kinase
- INF‐γ
interferon‐gamma
- JAK1
Janus kinase 1
- JNK
Janus kinase
- Lef1
lymphoid enhancer factor 1
- MKK
mitogen‐activated protein kinase kinases
- MLC
myosin light chain
- MS
multiple sclerosis
- NDR1
nuclear Dbf2‐related kinase 1
- NET
neutrophil extracellular trap
- NF‐κB
nuclear factor kappa‐B
- PI3K/AKT
phosphatidylinositol 3 kinase/protein kinase B
- pMCAO
permanent middle cerebral artery occlusion
- RA
rheumatoid arthritis
- RORγt
retinoid‐related orphan receptor gamma‐t
- SEFIR
SEFIR: SEF/IL‐17 receptor
- SF2
splicing factor 2
- STAT3
signal transducer and activator of transcription 3
- SYK
spleen tyrosine kinase
- TAB2/3
TGF‐β‐activated kinase 1/MAP 3K7 binding protein 2/3
- TAK1
TGF‐β activated kinase 1
- TJ
tight junction
- tMCAO
transient middle cerebral artery occlusion
- TRAF
tumour necrosis factor receptor‐associated factor
- TRPC6
transient receptor potential cation channel 6
- TYK2
tyrosine kinase 2
- MAPK
mitogen‐activated protein kinase
INTRODUCTION
The incidence, relapse and mortality rates of stroke are high around the world. From 2006 to 2016, stroke happens more frequently with advancing age in both males and females, and the number of strokes is predicted to increase by over 20% compared with 2012 in the United States by 2030. 1 A recent report from American Heart Association/American Stroke Association demonstrated the age‐adjusted stroke mortality decreased in comparison with previous years, regardless of gender, whereas the actual number of stroke deaths increased by almost 4%. 1 In 2016, stroke was accounted for 42.2% of deaths of all neurological disorders. The substantial high fatality rate makes it a serious threat to public health all over the world. 2 It is noteworthy that ischaemic stroke caused by regional cerebral blood supply disorders makes up 87% of all stroke patients. 1
The pathophysiological mechanism of ischaemic stroke is complex; various cellular, molecular and signalling pathways are involved in the process of stroke. The intricate process of stroke includes bioenergy failure, imbalance of ion homeostasis, apoptosis, excitotoxicity, oxidative stress, immune reaction, inflammation, etc. 3 , 4 The physical factors and chemical factors of anastomosis cause massive neurodegeneration. After cerebral infarction occlusion, the anoxic and malnourished nerve cells involving neurons, glial cells were necrotic. Necrotic nerve cells release excessive damage‐associated molecular patterns (DAMPs), as endogenous alarms, including high mobility group protein B1 (HMGB1), reactive oxygen species (ROS), peroxiredoxins (PRXs) and heat‐shock proteins (HSPs). Many DAMPs are sensed by Toll‐like receptors (TLRs) and receptor of advanced glycation end product (RAGE), both of which are the sensor of the innate immune system. 5 After combining with the TLRs and RAGE, DAMPs stimulate several inflammatory signalling pathways in the innate immune cells such as microglia and brain microvascular endothelial cells (BMECs), then release inflammatory mediators and thus cause central inflammatory events. 6 Notably, the up‐regulation of matrix metalloproteinases (MMPs), most of which are expressed at the undetected level in the normal model, shows the harmful effect of impairing the integrity of blood–brain barrier (BBB). Damaged BBB facilitates the transfer of DAMPs into peripheral blood and activates immune cells (including neutrophils, macrophages, and T and B lymphocytes) to migrate and infiltrate into cerebral parenchyma, finally leading to a robust activation of the immune system. 5 , 6 Studies have shown that 70–80% of nerve damage is related to post‐stroke inflammatory response. 7 Therefore, tempering the inflammatory response is essential for improving the prognosis of ischaemic stroke.
It has been widely accepted that the inhibition of lymphocyte activation and infiltration significantly improves the pathological outcome of a stroke. 8 , 9 In T‐ and B‐cell‐deficient mice, inflammatory factors, including TNF‐α and IL‐6, were significantly reduced with ameliorating infarct area after ischaemia. 10 However, reconstitution of B lymphocytes could not reverse this destructive change. 11 These results suggested that T cells might be more critical in modulating stroke damage.
T lymphocytes can be divided into αβ T cells and γδ T cells basing on the types of T‐cell receptor (TCR) on the cell membrane. Furthermore, depending on the surface molecules, αβ T cells can be divided into CD4+ T cells and CD8+ T cells; the former can be further divided into T helpers (Th) and regulatory T cells (Treg). 12 Notably, all types of T cells are closely involved in triggering inflammatory events in stroke. 13 In the acute phase of ischaemic stroke, Th1 and Th17 were destructive. 14 , 15 Since the 20th century, great progress has been made in the study of γδ T cells in stroke. CD4− IL‐17‐secreting γδ T cells (IL‐17+ γδ T cells), Th1 and Th17 were considered as harmful responders in sites of ischaemic hemisphere injury. 14
As previously reported, IL‐17+ γδ T cells, the principal recruited immune cells from the intestine, have been documented as a major source of neuroinflammation cascades in acute ischaemic brain damage. 16 , 17 According to Corinne et al., imbalance of intestinal flora homeostasis promoted the migration of IL‐17+ γδ T cells to meninges by altering the activity of dendritic cell, which therefore aggravated ischaemic brain injury by producing IL‐17 and its following by‐products, including CXCL1 and CXCL2. 16 IL‐17 is expressed by both Th17 and γδ T cells and is detrimental to stroke. 18 , 19
This review will focus on discussing the mechanism of IL‐17 in ischaemic stroke to provide a theoretical framework for the possibilities of targeting IL‐17 for the treatment of ischaemic stroke.
THE IL‐17 AND IL‐17 RECEPTOR FAMILY
In 1993, a mouse T‐cell hybridoma, cytotoxic T‐lymphocyte‐associated antigen 8 (CTLA8), was first cloned and then found to play an unusual role in the immune response. 20 In 1995, a nucleotide sequence related to mRNA instability was found in the 3ʹ untranslated region (3ʹ‐UTR) of CTLA8. Although the strand shares great similarities with most interleukins, CTLA8 was renamed as IL‐17. 21 In the same year, the human IL‐17 cDNA sequence was cloned from the CD4+ T cells, and it was found to exhibit 63% identity to the gene open reading frame of mouse CTLA8. This finding further confirmed the previous conclusion. 22 Since then, the five other cytokines of IL‐17 family, including IL‐17B, C, D, E and IL‐17F, were cloned (Fig. 1). Among them, IL‐17F has the most sequence identity with IL‐17A (50%), IL‐17E (also known as IL‐25) shares 17%, and IL‐17B, IL‐17D and IL‐17C share 29%, 25% and 23% identities with IL‐17A protein, respectively. 23 , 24 , 25 , 26 The above six cytokines form the IL‐17 family due to their similarities in structure.
Figure 1.
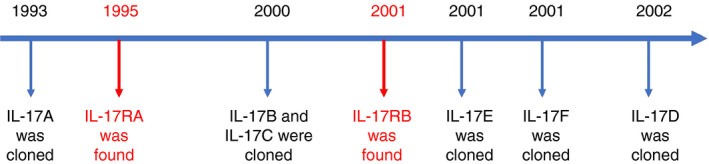
IL‐17 family of cytokines was found. A timeline showing the discovery of IL‐17 and its receptor family. References in 18, 19, 21, 22, 23, 24.
IL‐17 family activates downstream molecules by binding to the specific receptor complexes on the cellular membrane. The IL‐17 receptor (IL‐17R) family consists of five subunits: IL‐17RA to IL‐17RE. 27 Although the molecular weight of IL‐17R is large (about 860 amino acids), there is no evidence to show IL‐17R family is similar to other cytokine receptors. 27 The subunits of the IL‐17R family contain conserved functional domains, including the extracellular fibronectin III‐like domain and the intracellular signal transduction via motif “SEFIR” domain. 28 By binding to IL‐17R, IL‐17 mediates inflammatory responses via NF‐κB and MAPK/AP‐1 pathways. 29 (see below)
In the past 20 years, studies have confirmed that IL‐17A plays a vital role in infectious and non‐infectious diseases such as chronic hepatitis B virus, autoimmune diseases, cancer and ischaemic stroke. 4 , 12 , 30 In addition, CD4+ T‐cell subsets that secrete IL‐17A were named as Th17 cells. 31 Since then, other innate or adaptive immune cells including γδ T cells, natural killer T (NKT) cells, natural killer (NK) cells, group 3 innate lymphoid cells (ILC3s) and neutrophils have been found to also secrete IL‐17A. 32 The IL‐17A‐secreting cells were found to express CCL20 receptor (CCR6) and IL‐23 receptor (IL‐23R). 32 This finding suggested that chemokine CCL20 and IL‐23 may be important in the function of IL‐17A‐expressing cells.
In 2001, IL‐17F was successfully cloned. 26 Due to its high amino acid sequence identity with IL‐17A, 33 they were found to make up a heterodimer (IL‐17A/F), and share the same receptor (IL‐17RA/IL‐17RC heterodimer). 34 In addition, they show similar biological effects in many diseases, including psoriasis 33 and asthma. 35 IL‐17A has high affinity towards IL‐17RA expressed in haematopoietic cells. However, IL‐17F, which is secreted by Th17 cells, γδ T cells, monocytes, mast cells and basophils, has a high affinity towards IL‐17RC expressed on non‐haematopoietic cells. 36 This phenomenon determines the functional differences between IL‐17A and IL‐17F. 37 Studies have confirmed that IL‐17F plays an important role in inflammatory bowel disease (Crohn's disease and ulcerative colitis), 37 but whether IL‐17F plays a role in ischaemic stroke remains unknown.
IL‐17A/IL‐17R SIGNALLING PATHWAYS
IL‐17R, a constitutive heterodimer composed of two subunits IL‐17RA and IL‐17RC, is the receptor of IL‐17A/A, IL‐17A/F and IL‐17F/F. 38 IL‐17RC shares 23% amino acid sequence identity with IL‐17RA, both of which have a self‐conservation cytoplasmic SEFIR domain. 38 The key transcriptional factor in IL‐17A signalling is nuclear factor kappa‐B activator 1 (Act1). 39 Act1 is a protein with a SEFIR domain at the C‐terminal, and a tumour necrosis factor receptor‐associated factor (TRAF) family domain at the N‐terminal. When binding to IL‐17R, IL‐17A recruits and combines with Act1’s SEFIR domain, then recruits and ubiquitinates members of the TRAF family for through U‐box domain. 29 , 40 Ubiquitinated TRAF activate, TGF‐β‐activated kinase 1 (TAK1) that activates the complex of IKK (IKKα, IKKβ, IKKγ) 41 to ubiquitinate the two specific serine sequences of IκBα. Consequently, NF‐κB dimers dissociate from the complex. NF‐κB dimers translocate into the nucleus and bind to the corresponding target genes, finally promote the gene transcription of pro‐inflammatory cytokines. 41 , 42 In addition, TRAF6/TAK1 also promotes the transcription of IL‐17A target genes by activating MAPK/AP‐1 pathway 43 and C/EBP pathway 44 . However, extracellular signal‐related kinase (ERK) and glycogen synthase kinase‐3β (GSK‐3β) could phosphorylate the Thr188 and Thr179 of C/EBPβ, which in turn negatively regulate the expression of IL‐17A target genes 44 (Fig. 2).
Figure 2.
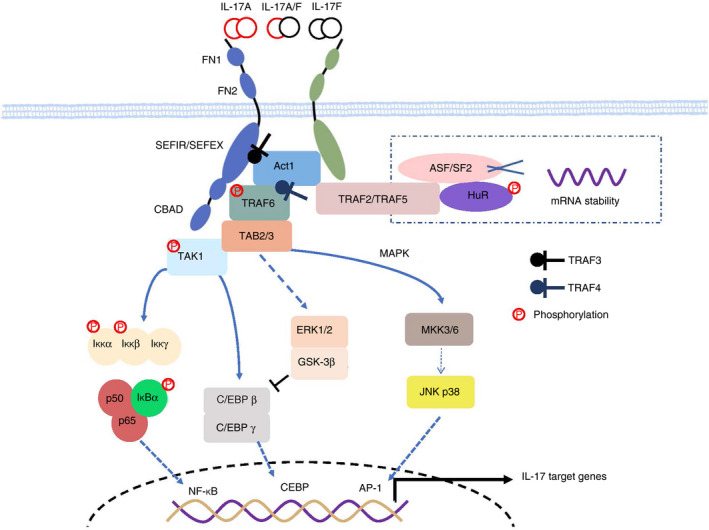
The IL‐17A signalling pathway. IL‐17 binds to its receptor, which is composed of IL17RA and IL‐17RC. Upon ligand binding, activated Act1 phosphorylated TRAF6 and triggers the transcription of TRAF6‐dependent target genes such as NF‐κB, CEBP and MAPK/AP‐1. The IL17A signalling pathway is regulated by many molecules, including TRAF family, HuR and so on. TRAF2‐TRAF5 complex and HuR stabilize the mRNA of IL‐17A target genes and contribute to the pathogenesis of stroke, while TRAF3 and TRAF4 seem to play a negative role. TRAF3 competes with Act1 to IL‐17R, while TRAF4 competes with TRAF6 for Act1. Besides, ERK1/2 and GSK‐3β phosphorylate the Thr188 and Thr179 of C/EBPβ, in turn, inhibits of CEBP pathway. References in 28, 41, 42, 44.
IL‐17A MODULATION IN STROKE
According to Tables 1 and 2, IL‐17A was elevated in brain tissue and peripheral blood after acute ischaemic brain injury in both experimental models and patients, and the level of IL‐17A was much higher than the level in recovery phases. Almost all the experimental data confirmed that IL‐17A was elevated within one day and peaked on the third day and then dropped a little bit on the following days after ischaemia injury in the mouse model. However, the actual situation of patients is different from that of experimental models. First of all, the patients’ data were usually taken from interval values, which is hard to track to a specific day. Secondly, it is not always clear whether there is ischaemia–reperfusion in the patients. Thirdly, the diagnostic criteria of stroke patients in different hospitals or different years are slightly different. Lastly, most of the patients are the elderly, who may have basic diseases such as hypertension or atherosclerosis, so individual differences are also relatively large. As a result, the experimental model in vivo is not always appropriate for meeting human conditions. The differences between experimental models and patients might be related to the following reasons: (1) species and genetic differences; (2) variable inclusion criteria; (3) individual differences among the elderly patients; and (4) data availability differences. Nevertheless, there is no doubt that the increased IL‐17A in brain tissue, serum and peripheral immune organs during acute stage of ischaemic stroke are related to brain injury in both experimental models and patients. (Tables 1 and 2)
Table 1.
IL‐17 was elevated in peripheral blood in ischaemic stroke patients and elevated in ischaemic hemisphere in patients died of ischaemic stroke
| Objects | Inclusion criteria | Detected organs or tissue | Analysed time‐points | Clinical characteristic | Releasing cells | Pub. year | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Age range | Type of stroke | Numerosity | Diagnosis criterion | |||||||
| Patients | 18 females, 11 males | 49–87 | Ischaemic stroke | 29 patients, 18 healthy individuals | Cerebral disturbance of clinical symptoms over 24 hours or recovery within 24 hours. | PBMCs | IL‐17 mRNA elevated more from day 1 to day 3 than from day 20 to day 31 after onset of symptoms. | – | – | 1999 | 45 |
| – | – | Ischaemic stroke | 26 | Patients died of ischaemic stroke. | Ischaemic hemisphere | IL‐17 was elevated on day 2 and peaked on day 3 to 5. | – | – | 2000 | 46 | |
| 28 females, 22 males | 45–76 | Ischaemic stroke | 50 patients, 30 healthy individuals | World Health Organization criteria. | Plasma | IL‐17A was elevated on day 7 significantly and remained at a low level on day 28. | Hypertension, diabetes, hyperlipidaemia | Th17 cells and γδ T cells | 2014 | 47 | |
| 19 females, 11 males | 66–74 | Ischaemic stroke | 30 patients, 30 healthy individuals | World Health Organization criteria. | PBMCs | IL‐17A was elevated on day 1, 5, 10 and peaked on day 1 after stroke onset. | – | Th17 cells | 2018 | 48 | |
| 300 | – | Ischaemic stroke | 300 patients, 300 healthy individuals | Ischaemic stroke diagnosed, regardless of gender and age. | Serum | IL‐17 was elevated after stroke onset. | – | – | 2019 | 49 | |
| 26 females, 78 males | 18–80 | Ischaemic stroke | 104 patients, 90 healthy individuals | Ischaemic stroke diagnosed within 7 days along with large‐artery atherosclerosis | – | – | Hypertension, diabetes mellitus, glucose, white blood cell count, high‐density lipoprotein | – | 2019 | 50 | |
PBMCs, peripheral blood mononuclear cells; Pub. year, published year.
Table 2.
IL‐17 was elevated in experimental models
| Objects | Sex and numerosity | Age/weight | Type of mouse | Modelling | Detected parts | Analysed time‐points and molecular results | Releasing cells | Pub. year | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Animals | 48 males | 250–300 g | SD rats | pMCAO | Brain tissue, blood, spleen, lymph node | IL‐17 mRNA was elevated at 1 h in the brain, blood, spleen and lymph node and then peaked on the sixth day, 6, 12 and 6 h, respectively. Moreover, INF‐γ, IL‐8, IP‐10, MIP‐2 and IL‐4 were up‐regulated and IL‐10 was down‐regulated in brain after pMCAO. | – | 2001 | 51 |
| 72 males | 250–300 g | SD rats | pMCAO | Ischaemic hemisphere | IL‐17 mRNA was elevated at 1 h and remained at a higher and then peaked on day 6. CD3+ was also elevated at 1 h and then peaked on day 6 in the brain. | T cells | 2005 | 46 | |
| Male | 7–17 weeks ole | C57BL/6 mice | MCAO/R | Ischaemic brain tissue | IL‐17 was elevated on day 3 followed by IL‐23, which was elevated on day 1 after MCAO‐R. Moreover, most of IL‐17‐producing cells were γδ T cells. | γδ T cells | 2009 | 17 | |
| ‐ | – | C57BL/6 mice | tMCAO | Ischaemic hemisphere | T cells were detectable at 1 h after tMCAO and then peaked on day 3, while IL‐17+ γδ T cells were found at 6 h and peaked at day 3. Moreover, CXCL‐1 and MMP3 were activated by IL‐17A. | γδ T cells | 2012 | 52 | |
| 48 males | 25–30 g | C57BL/6 mice | pMCAO | Ischaemic brain tissue, serum | Both IL‐23 and IL‐17 were elevated on day 6 in brain tissue and serum. | – | 2013 | 53 | |
| Male | 8–10 weeks old | C57BL/6 mice | tMCAO | Penumbra tissue | HMGB1 and IL‐17A were elevated on day 3 after tMCAO. IL‐17A knockout reduced the damage of brain, while rIL‐17A increased, but rHMGB1 failed to aggravate the injury. | γδ T cells | 2014 | 54 | |
| Male | 8–10 weeks old | C57BL/6 mice | tMCAO | Penumbra tissue | IL‐17A was elevated on day 1, peaked on day 3 and remained slightly decreased on day 6 in the hemisphere penumbra. IL‐17A activated TRPC6 channels on day 3 to aggravate the brain damage. | ‐ | 2014 | 55 | |
| 33 Males | 25 g | C57BL/6 mice | pMCAO | Ischaemic brain tissue | IL‐17 was elevated on day 5 after ischaemic injury, while IL‐23p19 knockout alleviated the damage. Moreover, IL‐23p19 knockout increased the expression of INF‐γ and Foxp3. | Th17 cells and γδ T cells | 2015 | 56 | |
| Male | 250–300 g | SD rats | tMCAO | Ischaemic hemisphere, peripheral blood | IL‐17 was elevated on days 1 and 3 after modelling. IL‐10 and TGF‐β were decreased on day 3, while IL‐1β and TNF‐α were increased on day 1 in brain. Treg cells were detected on day 3, while γδ T cells were increased on day 1 after cerebral ischaemic in peripheral blood. | γδ T cells | 2016 | 57 | |
| Male | – | C57BL/6 mice | tMCAO | Ischaemic hemisphere | γδ T cells were infiltrated at 12 h and kept rising on day 3. Most of them were Vγ6+/CCR6+ γδ T cells that produced IL‐17, not INF‐γ to aggravate the brain injury. | Vγ6+ γδ T cells | 2017 | 58 | |
| Male | 8–10 weeks old | C57BL/6J mice | MCAO/R | CSF, serum, peri‐infarct homogenates | IL‐17A was elevated at 12 h in peri‐infarct homogenates and CSF, while elevated on day 1 and peaked on day 3 in serum after MCAO/R. | Astrocytes | 2017 | 59 | |
| Male | – | C57BL/6 mice | tMCAO | Peripheral blood, cerebral hemisphere | IL‐17 was elevated on day 2 and peaked on day 3, then slightly decreased on day 5 in peripheral blood after tMCAO. IL‐17RA was slightly increased though 12 h to 5 days with no differences in cerebral. | – | 2018 | 60 | |
| 20 males | 6 weeks old | C57BL/6 mice | MCAO | Intestinal intraepithelial lymphocyte, spleen | IL‐17+ γδ T cells were up‐regulated on the third day in both small intestine and spleen, while CD4+ CD25+ T cells were down‐regulated in spleen and CD4+ Foxp3+ Treg cells were down‐regulated in small intestine. | γδ T cells | 2019 | 50 |
C57BL/6 mice, C57BL/6 background mice; Foxp3, forkhead box P3; INF‐γ, interferon‐gamma; MCAO, middle cerebral artery occlusion; MCAO/R, MCAO/reperfusion; pMCAO, permanent MCAO; rHMGB1, repletion of exogenous HMGB1; rIL‐17A, repletion of exogenous IL‐17A; SD rats, Sprague–Dawley rats; tMCAO, transient MCAO.
Previous studies mainly focused on the acute phase of stroke; however, more studies confirmed that IL‐17A might dampen specific stroke pathophysiological consequences in sub‐acute and convalescence stage. According to Lin et al., 61 IL‐17A played a biphasic role on different time‐points after ischaemic stroke in mice. IL‐17A‐releasing γδ T cells peaked on day 3 and aggravated the ischaemic injury. However, IL‐17A‐releasing astrocytes peaked on day 28, which seems to alleviate the damage by promoting neurogenesis and synaptogenesis, maintaining the neuronal differentiation and the survival of subventricular zone neural precursor cells via activated P38 MAPK/calpain 1 signalling. In this review, we still focused on the acute phase of stroke to study the possible mechanism of IL‐17A injury.
IL‐17A PROMOTES NEUTROPHIL INFILTRATION IN STROKE
IL‐17R is expressed on both glial cells and BMECs and up‐regulated after stroke in experimental models. 52 , 62 By binding with IL‐17R, IL‐17A promotes glia and BMECs to secrete large amounts of CXCL1 52 , 60 , 62 to induce neutrophils to infiltrate the cerebral parenchyma and inflict damage in stroke. 63 , 64 The main manifestations of neutrophils effects include the following: (1) activate MMPs (especially MMP‐2, ‐9) and hydrolyse endothelial cell tight junction proteins (TJs) to destroy the structural integrity of the BBB 65 , 66 ; and (2) secrete inflammatory cytokines, chemokines and adhesion molecules, and induce the infiltration of CD8+ T cells via CXCL12. 65 Infiltration of CD8+ T cells could induce the lysis and apoptosis of nerve cells through perforin pathway, granular enzyme pathway and Fas/FasL pathway; 12 (3) promote the formation of thrombosis. 67 Neutrophils not only adhere to endothelial cells through the interaction between leucocyte function‐associated antigen (LFA) and ICAM‐1 (thereby activating platelets and promoting thrombosis), 67 but also promote the formation of neutrophil extracellular trap (NET) through the release of DNA, histone and specific granule proteins. NET can capture fragments and dead cells to activate the exogenous coagulation pathway to form thrombus. 67 All the damaging effects lead to "no reflow" in the ischaemic area and aggravate the disturbance of downstream perfusion, leading to brain oedema, post‐embolization haemorrhage, larger infarction and worse outcome. 63 , 68 Notably, the expression of CXCL1, the main cytokine of neutrophils chemotaxis, depends on the induction of IL‐17A. 52 The blockage of IL‐17A signalling (in Il17ra −/−mouse model or with anti‐IL‐17A treatment) significantly inhibits neutrophil infiltration and reduces infarct size 52 (Fig. 3).
Figure 3.
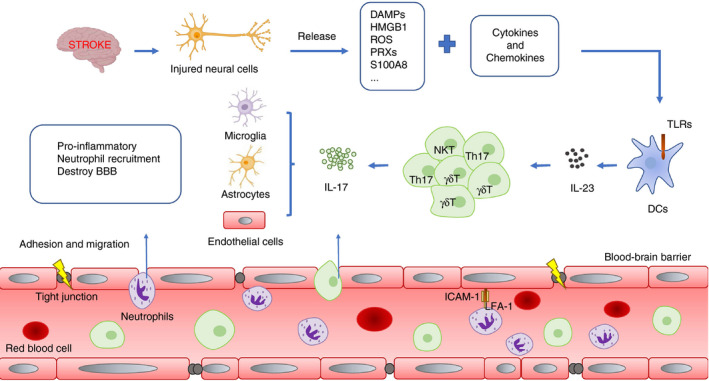
Production and function of IL‐17 after stroke. After stroke, cerebral ischaemia and hypoxia lead nerve cells necrosis and release a large amount of DAMPs and inflammation factors. When DAMPs are combined with TLRs on DCs, it promotes the DC‐derived IL‐23 releasing from DCs and then IL‐23 promotes the release of IL‐17 by γδ T cells, Th17 cells NKT cells and so on. Finally, IL‐17 leads to poor prognosis of stroke in three ways: (1) inducing the secretion of pro‐inflammatory factors; (2) recruiting neutrophils to infiltrate into CNS; (3) impairing the integrity of BBB.
IL‐17A IMPAIRS BBB INTEGRITY
BBB is an important component of the neurovascular unit (NVU) and is the first barrier to maintain the homeostasis of the microenvironment in the brain. 69 Destroyed BBB cohesiveness will cause a devastating effect on nerve cells’ survival and function. The mechanism of how IL‐17A impairs the cohesion of BBB is complex. Firstly, it down‐regulates the expression of tight junction (TJ) molecules such as occludin and claudin‐5 70 and promotes the elevation of MMP‐2 and MMP‐9 in BMECs to hydrolyse TJs. 71 As previously described, IL‐17A plays a powerful role in activating chemokine CXCL1 to induce neutrophils to infiltrate the cerebral parenchyma. Moreover, neutrophils produced and activated a large number of MMPs, such as MMP2, MMP3 and MMP9. MMPs destroy the integrity of BBB by hydrolysing TJs and improving BBB permeability. 52 , 66 Secondly, it activates BMECs to release reactive oxygen species (ROS) to induce oxidative stress and activate myosin light chain (MLC). 70 Phosphorylated MLC interacts with cytoskeletal actin and induces the contraction of BMECs and eventually leads to the enlarged gaps between endothelial cells. 70 , 72 IL‐17A induces inflammatory response by up‐regulating ICAM‐1 and IL‐6 in BMECs. 70 It consequently induces endothelial apoptosis through mitochondrial apoptosis pathway 73 and then results in the destruction of BBB. Experiments in vitro showed that IL‐17A up‐regulates the expression of IL‐17R in brain endothelial cells, while anti‐IL‐17A effectively protects the cohesion of BBB 71 (Fig. 3).
IL‐17A INDUCES NEURONAL APOPTOSIS
With current knowledge, transient receptor potential cation channel 6 (TRPC6) can phosphorylate cAMP‐response element‐binding protein (CREB), which in turn activates brain‐derived neurotrophic factor (BDNF) and anti‐apoptotic protein B‐cell lymphoma‐2 (Bcl‐2). 74 As a pro‐survival factor for neuron, CREB plays an important role in promoting both neuron survival and nutritional support. 75 TRPC6/CREB pathway is important to maintain nerve cell's survival and function after stroke by improving hypoxia tolerance of nerve cells. 76 , 77 Du et al. showed that inhibition of TRPC6/CREB pathway leads to reduce the expression of BDNF and Bcl‐2, as well as significantly reduced the hypoxia tolerance of neurons. 76 However, inhibiting TRPC6 degrades the defensive effect of ischaemic brain damage. After stroke, IL‐17A increases rapidly and activates calpain to mediate the TRPC6 hydrolysis. This process reduces the expression of BDNF and Bcl‐2 and results in neuron death. 55 It is noteworthy that IL‐17A promotes the expression of apoptosis‐related proteins including caspase‐3, caspase‐9 and Bax and increases the ratio of Bax/Bcl‐2 after brain injury, thus initiates the process of neuronal apoptosis 78 (Fig. 3).
SYNERGISTIC EFFECTS OF IL‐17A WITH IL‐6 AND IL‐22
Although IL‐17A is a mild stimulator of inflammatory response on its own, it activates the inflammatory signalling pathways of C/EBP, NF‐κB and MAPK that can promote expression of pro‐inflammatory cytokines and amplifying the inflammatory response. 32 It has been reported that the expression of C/EBP δ and NF‐κB is up‐regulated in glial cells, BMECs and macrophages after ischaemia and hypoxia, which is closely related to the activation of IL‐17A pathway. 44 , 79 After the activation of IL‐17R/Act1, TRAF6 is recruited and activates downstream C/EBP β, C/EBP δ and NF‐κB, 44 , 79 all of which are key proteins of IL‐6 transcription. When C/EBP β or C/EBP δ binds to the IL‐6 promoter, they induce macrophages, BMECs and fibroblasts to up‐regulate the expression of IL‐6, thereby amplify the inflammatory response.
IL‐22, a member of IL‐10 family, like IL‐17A, is secreted by CD4+ αβ T cells, γδ T cells, NKT cells and ILC cells. 80 However, the pro‐inflammatory effects of IL‐22 are weaker than IL‐17A. IL‐22 binds to the receptor (IL‐10Rβ/IL‐22R) and transmits downstream signals through Janus kinase 1 (JAK1) and TYK2. After phosphorylation of STAT3, it directly transmits into the nucleus to regulate the transcription of target genes, either AKT/mTOR or MAPK signalling pathways can be activated as well. 81 Ultimately, the expression of cytokines (IL‐6, G‐CSF) and chemokines (CXCL1, CXCL5, CXCL9) can be activated. 82 IL‐17A and IL‐22 improve their pro‐inflammatory effects by a synergistic effect. This synergistic effect has been well proved in asthma, airway inflammation, cancer and other autoimmune diseases. 80 , 83 Although there is no direct evidence showing IL‐17 and IL‐22 are synergistic in promoting stroke, the expression of both IL‐17 and IL‐22 are increased in post‐stroke might provide some counter‐evidence. 84 , 85 Therefore, the possible synergies between IL‐17 and IL‐22 in stroke need further investigation.
IL‐23/IL‐17A AXIS AND ISCHAEMIC STROKE
Both innate and adaptive immunity are involved and interacted with each other in ischaemic stroke. 86 After ischaemia, innate‐like lymphocytes are activated rapidly. Innate immunity triggers the first step of inflammatory cascade and then activates the adaptive immunity by presenting antigens. 86 Remarkably, the IL‐23/IL‐17 axis is mostly a bridging mechanism between innate immunity and adaptive immunity after stroke. 18 , 87
Dendritic cells (DCs) induce innate immune response in infarct area and rapidly spread to the whole body. This immune response occurs before the adaptive immune response and does not mediate antigen presentation. 88 Of note, the combination of DAMPs released by hypoxic neurons and TLRs can induce this immune response. 18 Then, DCs rapidly release a large number of cytokines and chemokines, especially DC‐derived IL‐23, 87 a heterodimer composed of p19 and p40 subunits. IL‐23R is highly expressed in mature Th17 cells and γδ T cells, but not in naïve T cell. 32 As a result, it is generally believed that IL‐23 can amplify the secretion of IL‐17A, while it is not enough to induce the T‐cell differentiation 89 (Fig. 3). The key combination that can effectively induce Th17 cell differentiation is TGF‐β+IL‐6, 90 , 91 TGF‐β+IL‐21 91 , 92 or IL‐23+IL‐6+IL‐1β. 93
When IL‐23 binds to IL‐23R on Th17 cells and γδ T cells, it activates the classic tyrosine‐dependent pathway and phosphorylates STAT3 through JAK2/STAT3 and PI3K/AKT signalling pathways. 94 What's more, it activates STAT3 by non‐tyrosine‐dependent pathway. 95 pSTAT3 then activates the downstream proteins of transcription factor c‐Maf, a member of the activator protein 1 (AP‐1) family, and then produces a large number of IL‐17 through four mechanisms. Firstly, STAT3 directly activates the nuclear transcription factor retinoid‐related orphan receptor gamma‐t (RORγt) of Th17 cells. 96 Secondly, STAT3 directly transmits into the nucleus and binds to the promoters of Il17a, Il17f, Il21 to promote the expression of IL‐17A. 96 Thirdly, STAT3 activates RORγt in a RORγt‐dependent manner through c‐Maf. 97 , 98 Lastly, STAT3 induces IL‐17A secretion in a RORγt‐independent manner through c‐Maf. 95
As IL‐23 is a powerful stimulant of IL‐17A, the concept of IL‐23/IL‐17 axis has been proposed and applied to a variety of autoimmune and inflammatory diseases, including ischaemic stroke. 87 , 99 After stroke, the expression of IL‐23 and IL‐17 in both serum and ischaemic brain tissue is significantly increased, 53 , 56 which eventually leads to the destruction of BBB, neuroinflammation and neuron death. Inhibition of IL‐23 (knockdown IL‐23p19) 56 or anti‐IL‐17 71 can significantly reduce the cerebral ischaemia as well as improve neurological behaviour. IL‐23/IL‐17 axis as an important pathway for post‐stroke inflammation is also plausible. Various targets are involved in the IL‐23/IL‐17 axis. Yet, few studies are on the roles for IL‐23/IL‐17 in stroke, which warrants further investigation as they could be important targets for stroke treatment.
REGULATION OF IL‐17A EXPRESSION AFTER STROKE
Th17 cells and γδ T cells infiltrate into the ischaemic area and serve as the main sources of IL‐17A during post‐stroke. 17 RORγt is a key transcription factor for IL‐17 expression in Th17 cells and γδ T cells. 100 The transcription and translation of RORγt are regulated by many molecules.
After stroke, the expression of hypoxia‐inducible factor‐1α (HIF‐1α) is up‐regulated. Although HIF‐1α usually acts as a neuroprotective factor for its role in angiogenesis, 101 glycolysis and glucose transport, 101 and neurogenesis, 102 the role of HIF‐1α in stroke is complex. Overexpression of HIF‐1α promotes neuronal inflammation and neuronal apoptosis. 103 HIF‐1α directly activates RORγt expression by binding to RORC promoter, in order for IL‐17A expression. 104 It is worth noting that histone acetyltransferase p300 recruited by HIF‐1α can greatly promote Th17 cells to secrete IL‐17A. 104 Recently, c‐Maf has been identified as a key transcription factor in γδ T 98 and Th17 cells. 105 As mentioned above, c‐Maf plays a key role in promoting the transcription of RORγt. Therefore, it promotes the expression of IL‐17A by up‐regulating RORγt, B‐lymphoid tyrosine kinase (Blk) and spleen tyrosine kinase (SYK), as well as inhibiting lymphoid enhancer factor 1 (Lef1). 98 In this manner, RORγt is important to the IL‐17A expression. Additional mechanisms likely exist to contribute to the complex regulation of RORγt; further research is needed to reveal new targets for the treatment of stroke.
REGULATION OF IL‐17R SIGNALLING AFTER STROKE
TRAF family, TRAF2 to TRAF6, is widely involved in multiple pathways, including IL‐17A signalling. 106 TRAF6 is involved in the inflammatory response of the central nervous system (CNS). Liu et al. 107 demonstrated that TRAF6 was significantly increased in rats with pMCAO model. As a key modulator in IL‐17A signalling pathway, TRAF6 positively regulates the expression of IL‐17A target genes. 42 TRAF2/5 maintains the stability of mRNA. 106 Act1 recruits and binds TRAF2/5 at Ser311 site and forms Act1‐TRAF2/5‐ASF complex. This complex blocks the binding of splicing factor alternative splicing factor (ASF) to mRNA 3'UTR region to prolong the half‐life period of chemokine mRNA. 106 , 108 TRAF2/5 recruits and phosphorylates human antigen R (HuR), and mRNA splicing factor 2 (SF2) competitively binds to RNA‐binding protein (RBP) in the 3'UTR region. Thereby, the binding stabilizes TNF‐α mRNA, IL‐6 mRNA, CXCL‐1 mRNA and ICAM‐1 mRNA to amplify the inflammatory effects. 41 , 42 , 109 HuR was reported to increase significantly in brain tissue of mice with ischaemia–reperfusion injury after tMCAO. 110
While TRAF6/2/5 promotes the expression of IL‐17A, TRAF3/4 inhibits IL‐17A signal transduction. TRAF3 competes with Act1 to IL‐17R, 111 while TRAF4 and TRAF6 competitively bind TB binding sites on Act1. 112 Thus, TRAF3/4 blocks the formation of IL‐17R‐Act1‐TRAF6 complex and negatively regulating the transcription. In HeLa cells, nuclear Dbf2‐related kinase 1 (NDR1) forms a complex with TRAF3 and blocks the interaction between TRAF3 and Act1, allowing the interaction between IL‐17R and Act1 to enhance inflammation. 113
Therefore, the activation of IL‐17A signalling pathway can prolong the half‐life of mRNA of pro‐inflammatory cytokines and chemokine, through TRAF and HuR to contribute to the strong pro‐inflammatory effects of IL‐17A. The research on TRAF family and HuR is thus of significance in ischaemic stroke.
IS IL‐17B INVOLVED IN STROKE?
In 2000, the expression of IL‐17B has for the first time been detected in adult digestive tract by Northern blot but not in activated T cells. 23 Soon after, high expression of IL‐17RB was detected in the intestines, accompanied by a large number of neutrophils infiltration in rats injected with indomethacin. 114 Thus, IL‐17B was proposed to be a potential pro‐inflammatory cytokine. Nowadays, IL‐17B has been proved to be prominent in inflammation and autoimmune diseases. 115 (Table 3) IL‐17B is highly expressed in experimental pneumonia and inducing the production of chemokine IL‐8 by activating inflammatory pathways such as PI3K/AKT, p38MAPK, ERK and NF‐κB in bronchial epithelial cells. Then, IL‐8 recruits neutrophils and promotes inflammation. 116 However, the expression of IL‐17RB is limited in digestive tract, lung and kidney. Whether IL‐17RB may specifically participate in the post‐stroke inflammatory response remains unknown. 114 At present, there is no evidence showing IL‐17B’s role in CNS, and it is still unclear whether IL‐17B is involved in the inflammatory cascade after stroke. Nonetheless, Moore et al. 117 detected high expression of IL‐17B in neurons, especially in grey matter projection neurons, although it is not clear whether IL‐17B is involved in the formation of neuronal axons, or the transmission of neuronal information. In a nutshell, whether IL‐17B is involved in injuring or repairing process after stroke needs elucidation.
Table 3.
IL‐17 family of cytokine and stroke
| IL‐17 subtype | Length (aa) | Homology with IL‐17A (%) | Expression cells | Receptor(s) | Reception cells | Functions | Stroke | Ref. |
|---|---|---|---|---|---|---|---|---|
| IL‐17A | 155 | 100 | γδ T cells, NKT cells, NK cells, ILC3s, CD8+T cells and neutrophils | IL17RA/IL‐17RC | Lymphocytes, endothelial cells, astrocytes, microglia | Pro‐inflammatory, neutrophil recruitment, destroy BBB | Pro‐inflammatory, neutrophil recruitment, destroy BBB | 29, 30 |
| IL‐17B | 180 | 29 | Pancreas, small intestine, stomach, neurons, epithelial cells | IL‐17RB | Fibroblasts, THP‐1 cells, stromal cells | Pro‐inflammatory, tissue regeneration, | – | 21, 112, 114 |
| IL‐17C | 197 | 23 | Prostate, epithelial cells | IL‐17RE/IL‐17RA | Monocyte, Th17 cells | Pro‐inflammatory, neutrophil recruitment | – | 21, 116, 117 |
| IL‐17D | 202 | 25 | Muscle, brain, heart, lung, pancreas, adipose tissue | Remain unknown | Endothelial cell and myeloid progenitor | Recruitment chemokines, anti‐tumour, antiviral | – | 22, 120 |
| IL‐17E | 161 | 17 | Epithelial cells, Th2 cells, eosinophils, mast cells, brain endothelial cells | IL‐17RB/IL‐17RA | NKT cells, basophils, eosinophils, mast cells, endothelial cells, macrophages, dendritic cells, ILC2s | Induce the immune response of Th2, suppress the immune response of Th1/Th17 | – | 23, 121 |
| IL‐17F | 153 | 50 | Monocytes, mast cells, basophils, mucosal epithelial cells, Th17, CD8 cells, γδ TCR+ T cells, NK, NKT, LTi | IL17RA/IL‐17RC | Non‐hematopoietic cell | Neutrophil recruitment, immunity to extracellular pathogens in inflammatory bowel disease and asthma | Pro‐inflammatory, neutrophil recruitment, destroy BBB | 35 |
Il‐17c: “AMPLIFIER” OF Th17 SIGNAL
IL‐17C was first cloned in 2001, the same time as IL‐17B. 23 Similar to IL‐17A and IL‐17F, IL‐17C acts as a pro‐inflammatory cytokine in the mucosal immune response. 118 , 119 After infection with Citrobacter in mice intestinal mucosa, IL‐17C was significantly increased. By binding to IL‐17RA/IL‐17RE receptor complex, IL‐17C activated NF‐κB signalling and drove the antibacterial reaction in the intestinal tract. 120 In CNS, IL‐17C seems to play a damaging role. IL‐17RE, a specific receptor for IL‐17C, is mainly expressed in epithelial cells. 118 In pathogenesis of experimental autoimmune encephalitis (EAE), IL‐17RE was expressed on Th17 cells. IL‐17C/IL‐17RE complex induces the expression of IκBζ, STAT3 and RORγt through Act1, thereby inducing high expression of IL‐17A, IL‐17F and IL‐22. The process finally leads to more severe immune damage. 119 (Table 3) As IL‐17C amplifies the inflammatory response and promotes the progression of disease in EAE, is it applicable to all the CNS inflammatory diseases? After stroke, Th17 cells are of great importance in causing CNS inflammatory damage. Although there is no evidence that the expression of IL‐17C increases after stroke, IL‐17C could trigger the inflammatory events in CNS. This suggests that the potential pro‐inflammatory role of IL‐17C and IL‐17RE in post‐stroke.
IL‐17D: A POTENTIAL PRO‐INFLAMMATORY CYTOKINE
IL‐17D is the least studied member of the IL‐17 cytokine family, its receptor remains unknown. 121 Early studies found that IL‐17D was mainly expressed in skeletal muscle, brain and heart. IL‐17D promotes the expression of IL‐6, IL‐8 and granulocyte macrophage colony‐stimulating factor (GM‐CSF) in a dose‐dependent manner. IL‐17D may be involved in local structural damage, such as ischaemic stroke. 24 IL‐17D has been found involved in cancers. Like most of the other IL‐17s, IL‐17D can induce tumour endothelial cells to produce chemokines CCL2 and MCP‐1 through the Nrf2‐IL‐17D pathway, which leads to the infiltration of NK cells and plays an anti‐tumour effect. 122 , 123 (Table 3) As a new immune axis, Nrf2‐IL‐17D might also participate in stroke.
IL‐17E REGULATES THE BALANCE OF TH1/TH2/TH17 CELLS
IL‐25, also known as IL‐17E, was first identified from the DNA sequence of the human genome. IL‐17E is mainly expressed in epithelial cells, Th2 cells, eosinophils and mast cells 124 and has the least homology with IL‐17A. 25 During asthma, IL‐17E binds to IL‐17RB/IL‐17RA receptor complexes on Th2 cells and type 2 lymphoid cells (ILC2s) to promote the expression of IL‐4, IL‐5, IL‐13, eotaxin and type 2 immunity for airway inflammation in asthma. 125 In contrast, IL‐17E inhibits the function of Th1 and Th17 cells. Low‐dose IL‐17E induced Th0 cells to differentiate into Th2 cells, the latter inhibited the differentiation of Th1 cells and Th17 cells by secreting IL‐4 and IL‐13 and thus plays an immune‐protective role in EAE. 126 Moreover, exogenous administration of IL‐17E significantly inhibits CNS inflammation of EAE. 126 (Table 3) Ischaemic stroke usually causes Th1/Th2/ Th17 cells subset imbalance and results in serious CNS inflammation and poor prognosis. 127 As IL‐17E uniquely induces type 2 immunity to counterbalance Th17/Th1‐mediated immune pathology, it is worthy of investigating whether IL‐17E may be used to benefit stroke.
CONCLUDING REMARKS
Ischaemic stroke is an increasing threat to endanger the health and safety in modern society. Its mechanisms need to be understood. There are clear correlations between nerve damage after stroke and inflammatory response. Decoding the immune inflammatory response will improve the understanding and treatment of stroke. The IL‐17 family proteins, especially IL‐17A, are widely involved in a variety of acute and chronic inflammatory responses. After stroke, IL‐17A and IL‐17F promote the development of stroke by inducing the secretion of inflammatory factors (such as TNF‐α, IL‐6 and CXCL1), recruiting neutrophils to infiltrate into CNS and impairing the integrity of BBB. While IL‐17B/IL‐17C/IL‐17D displays pro‐inflammatory effects in plethora of diseases, their roles in stroke need further investigation as evidences are available to show IL‐17B/IL‐17C/IL‐17D may be damaging in stroke. On the contrary, IL‐17E plays an immune‐protective role by balancing Th2 vs Th1/Th17 cell functions. In conclusion, the IL‐17 family proteins are prominent and pleiotropic to modulate both pro‐ and anti‐inflammatory responses and may be exceptional therapeutic targets for treating stroke.
AUTHOR CONTRIBUTIONS
Qiaohui Zhang and Yibo Tang conceived and wrote the manuscript; Yan Liao, Zhenquan Liu polished the manuscript; Yajie Dai, Yunxin Li and Yue Li provided many comments for the manuscript.
DISCLOSURES
The authors declare no conflict of interest.
ACKNOWLEDGEMENT
This work was supported by the National Natural Science Foundation of China (no. 81603453).
DATA AVAILABILITY STATEMENT
The data used to support this review are included within the article.
REFERENCES
- 1. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al Heart Disease and Stroke Statistics‐2019 Update: a report from the American Heart Association. Circulation 2019; 139:e56–e58. [DOI] [PubMed] [Google Scholar]
- 2. Neurology JTL. Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 2019; 18:459–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arumugam TV, Baik SH, Balaganapathy P, Sobey CG, Mattson MP, Jo DG. Notch signaling and neuronal death in stroke. Prog Neurogibol 2018; 165–167:103–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 2010; 87:779–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shichita T, Ito M, Morita R, Komai K, Noguchi Y, Ooboshi H, et al MAFB prevents excess inflammation after ischemic stroke by accelerating clearance of damage signals through MSR1. Nat Med 2017; 23:723–32. [DOI] [PubMed] [Google Scholar]
- 6. Richard SA, Sackey M, Su Z, Xu H. Pivotal neuroinflammatory and therapeutic role of high mobility group box 1 in ischemic stroke. Biosci Rep 2017; 37:BSR20171104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Macrez R, Ali C, Toutirais O, Le Mauff B, Defer G, Dirnagl U, et al Stroke and the immune system: from pathophysiology to new therapeutic strategies. Lancet Neurol 2011; 10:471–80. [DOI] [PubMed] [Google Scholar]
- 8. Brait VH, Arumugam TV, Drummond GR, Sobey CG. Importance of T lymphocytes in brain injury, immunodeficiency, and recovery after cerebral ischemia. J Cereb Blood Flow Metab 2012; 32:598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon‐gamma in ischemic stroke. Circulation 2006; 113:2105–12. [DOI] [PubMed] [Google Scholar]
- 10. Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, et al T‐ and B‐cell‐deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab 2007; 27:1798–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Schwarz T, et al Early detrimental T‐cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood 2010; 115:3835–42. [DOI] [PubMed] [Google Scholar]
- 12. Gu L, Jian Z, Stary C, Xiong X. T cells and cerebral ischemic stroke. Neurochem Res 2015; 40:1786–91. [DOI] [PubMed] [Google Scholar]
- 13. Bravo‐Alegria J, McCullough LD, Liu F. Sex differences in stroke across the lifespan: the role of T lymphocytes. Neurochem Int 2017; 107:127–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Swardfager W, Winer DA, Herrmann N, Winer S, Lanctôt KL. Interleukin‐17 in post‐stroke neurodegeneration. Neurosci Biobehav Rev 2013; 37:436–47. [DOI] [PubMed] [Google Scholar]
- 15. Gill D, Veltkamp R. Dynamics of T cell responses after stroke. Curr Opin Pharmacol 2016; 26:26–32. [DOI] [PubMed] [Google Scholar]
- 16. Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, et al Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat Med 2016; 22:516–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, et al Pivotal role of cerebral interleukin‐17‐producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med 2009; 15:946–50. [DOI] [PubMed] [Google Scholar]
- 18. Gelderblom M, Gallizioli M, Ludewig P, Thom V, Arunachalam P, Rissiek B, et al IL‐23 (Interleukin‐23)‐producing conventional dendritic cells control the detrimental IL‐17 (Interleukin‐17) response in stroke. Stroke 2018; 49:155–64. [DOI] [PubMed] [Google Scholar]
- 19. Wang DD, Zhao YF, Wang GY, Sun B, Kong QF, Zhao K, et al IL‐17 potentiates neuronal injury induced by oxygen‐glucose deprivation and affects neuronal IL‐17 receptor expression. J Neuroimmunol 2009; 212:17–25. [DOI] [PubMed] [Google Scholar]
- 20. Rouvier E, Luciani MF, Mattéi MG, Denizot F, Golstein P. CTLA‐8, cloned from an activated T cell, bearing AU‐rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J Immunol 1993; 150:5445–56. [PubMed] [Google Scholar]
- 21. Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, et al Herpesvirus Saimiri encodes a new cytokine, IL‐17, which binds to a novel cytokine receptor. Immunity 1995; 3:811–21. [DOI] [PubMed] [Google Scholar]
- 22. Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, et al Human IL‐17: a novel cytokine derived from T cells. J Immunol 1995; 155:5483–6. [PubMed] [Google Scholar]
- 23. Li H, Chen J, Huang A, Stinson J, Heldens S, Foster J, et al Cloning and characterization of IL‐17B and IL‐17C, two new members of the IL‐17 cytokine family. Proc Natl Acad Sci USA 2000; 97:773–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Starnes T, Broxmeyer HE, Robertson MJ, Hromas R. Cutting edge: IL‐17D, a novel member of the IL‐17 family, stimulates cytokine production and inhibits hemopoiesis. J Immunol 2002; 169:642–6. [DOI] [PubMed] [Google Scholar]
- 25. Lee J, Ho WH, Maruoka M, Corpuz RT, Baldwin DT, Foster JS, et al IL‐17E, a novel proinflammatory ligand for the IL‐17 receptor homolog IL‐17Rh1. J Biol Chem 2001; 27:1660–4. [DOI] [PubMed] [Google Scholar]
- 26. Starnes T, Robertson MJ, Sledge G, Kelich S, Nakshatri H, Broxmeyer HE, et al Cutting edge: IL‐17F, a novel cytokine selectively expressed in activated T cells and monocytes, regulates angiogenesis and endothelial cell cytokine production. J Immunol 2001; 167:4137–40. [DOI] [PubMed] [Google Scholar]
- 27. Aggarwal S, Gurney AL. IL‐17: prototype member of an emerging cytokine family. J Leukoc Biol 2002; 71:1–8. [PubMed] [Google Scholar]
- 28. Gaffen SL. Structure and signalling in the IL‐17 receptor family. Nat Rev Immunol 2009; 9:556–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Herjan T, Hong L, Bubenik J, Bulek K, Qian W, Liu C, et al IL‐17‐receptor‐associated adaptor Act1 directly stabilizes mRNAs to mediate IL‐17 inflammatory signaling. Nat Immunol 2018; 19:354–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tian CH, Dai J, Zhang W, Liu Y, Yang Y. Expression of IL‐17 and its gene promoter methylation status are associated with the progression of chronic hepatitis B virus infection. Medicine 2019; 98:e15924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 2005; 6:1133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Veldhoen M. Interleukin 17 is a chief orchestrator of immunity. Nat Immunol 2017; 18:612–21. [DOI] [PubMed] [Google Scholar]
- 33. Glatt S, Baeten D, Baker T, Griffiths M, Ionescu L, Lawson ADG, et al Dual IL‐17A and IL‐17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo‐controlled clinical trial that IL‐17F contributes to human chronic tissue inflammation. Ann Rheum Dis 2018; 77:523–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Patel DD, Kuchroo VK. Th17 cell pathway in human immunity: lessons from genetics and therapeutic interventions. Immunity 2015; 43:1040–51. [DOI] [PubMed] [Google Scholar]
- 35. Chenuet P, Fauconnier L, Madouri F, Marchiol T, Rouxel N, Ledru A, et al Neutralization of either IL‐17A or IL‐17F is sufficient to inhibit house dust mite induced allergic asthma in mice. Clin Sci (Lond) 2017; 131:2533–48. [DOI] [PubMed] [Google Scholar]
- 36. Taams LS, Steel KJA, Srenathan U, Burns LA, Kirkham BW. IL‐17 in the immuno‐ pathogenesis of spondyloarthritis. Nat Rev Rheumatol 2018; 14:453–66. [DOI] [PubMed] [Google Scholar]
- 37. Tang C, Kakuta S, Shimizu K, Kadoki M, Kamiya T, Shimazu T, et al Suppression of IL‐17F, but not of IL‐17A, provides protection against colitis by inducing Treg cells through modification of the intestinal microbiota. Nat Immunol 2018; 19:755–65. [DOI] [PubMed] [Google Scholar]
- 38. Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, et al Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol 2006; 177:36–9. [DOI] [PubMed] [Google Scholar]
- 39. Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin‐17 receptor. J Biol Chem 2006; 281:35603–7. [DOI] [PubMed] [Google Scholar]
- 40. Schwandner R, Yamaguchi K, Cao Z. Requirement of tumor necrosis factor receptor‐associated factor (TRAF)6 in interleukin 17 signal transduction. J Exp Med 2000; 191:1233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gu C, Wu L, Li X. IL‐17 family: cytokines, receptors and signaling. Cytokine 2013; 64:477–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Amatya N, Garg AV, Gaffen SL. IL‐17 Signaling: the Yin and the Yang. Trends Immunol 2017; 38:310–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Landström M. The TAK1‐TRAF6 signalling pathway. Int J Biochem Cell Biol 2010; 42:585–9. [DOI] [PubMed] [Google Scholar]
- 44. Shen F, Li N, Gade P, Kalvakolanu DV, Weibley T, Doble B, et al IL‐17 receptor signaling inhibits C/EBPbeta by sequential phosphorylation of the regulatory 2 domain. Sci Signal 2009; 2:ra8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kostulas N, Pelidou SH, Kivisäkk P, Kostulas V, Link H. Increased IL‐1beta, IL‐8, and IL‐17 mRNA expression in blood mononuclear cells observed in a prospective ischemic stroke study. Stroke 1999; 30:2174–9. [DOI] [PubMed] [Google Scholar]
- 46. Li GZ, Zhong D, Yang LM, Sun B, Zhong ZH, Yin YH, et al Expression of interleukin‐17 in ischemic brain tissue. Scand J Immunol 2005; 62:481–6. [DOI] [PubMed] [Google Scholar]
- 47. Hu Y, Zheng Y, Wu Y, Ni B, Shi S. Imbalance between IL‐17A‐producing cells and regulatory T cells during ischemic stroke. Mediators Inflamm 2014; 2014:813045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dolati S, Ahmadi M, Khalili M, Taheraghdam AA, Siahmansouri H, Babaloo Z, et al Peripheral Th17/Treg imbalance in elderly patients with ischemic stroke. Neurol Sci 2018; 39:647–54. [DOI] [PubMed] [Google Scholar]
- 49. Tian J, Bai Y, You A, Shen R, Yan J, Deng W, et al Interleukin‐17 receptor C gene polymorphism reduces treatment effect and promotes poor prognosis of ischemic stroke. Biosci Rep 2019; 39:BSR20190435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xia GH, You C, Gao XX, Zeng XL, Zhu JJ, Xu KY, et al Stroke Dysbiosis Index (SDI) in gut microbiome are associated with brain injury and prognosis of stroke. Front Neurol 2019; 10:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li HL, Kostulas N, Huang YM, Xiao BG, van der Meide P, Kostulas V, et al IL‐17 and IFN‐gamma mRNA expression is increased in the brain and systemically after permanent middle cerebral artery occlusion in the rat. J Neuroimmunol 2001; 116:5–14. [DOI] [PubMed] [Google Scholar]
- 52. Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, et al Neutralization of the IL‐17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood 2012; 120:3793–802. [DOI] [PubMed] [Google Scholar]
- 53. Ma S, Zhong D, Chen H, Zheng Y, Sun Y, Luo J, et al The immunomodulatory effect of bone marrow stromal cells (BMSCs) on interleukin (IL)‐23/IL‐17‐mediated ischemic stroke in mice. J Neuroimmunol 2013; 257:28–35. [DOI] [PubMed] [Google Scholar]
- 54. Zhang J, Wu Y, Weng Z, Zhou T, Feng T, Lin Y. Glycyrrhizin protects brain against ischemia‐reperfusion injury in mice through HMGB1‐TLR4‐IL‐17A signaling pathway. Brain Res 2014; 1582:176–86. [DOI] [PubMed] [Google Scholar]
- 55. Zhang J, Mao X, Zhou T, Cheng X, Lin Y. IL‐17A contributes to brain ischemia reperfusion injury through calpain‐TRPC6 pathway in mice. Neuroscience 2014; 274:419–28. [DOI] [PubMed] [Google Scholar]
- 56. Zheng Y, Zhong D, Chen H, Ma S, Sun Y, Wang M, et al Pivotal role of cerebral interleukin‐23 during immunologic injury in delayed cerebral ischemia in mice. Neuroscience 2015; 290:321–31. [DOI] [PubMed] [Google Scholar]
- 57. Zhang L, Huang Y, Lin Y, Shan Y, Tan S, Cai W, et al Anti‐inflammatory effect of cholera toxin B subunit in experimental stroke. J Neuroinflammation 2016; 13:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Arunachalam P, Ludewig P, Melich P, Arumugam TV, Gerloff C, Prinz I, et al CCR6 (CC Chemokine Receptor 6) is essential for the migration of detrimental natural interleukin‐17‐producing γδ T cells in stroke. Stroke 2017; 48:1957–65. [DOI] [PubMed] [Google Scholar]
- 59. Li S, Dai Q, Yu J, Liu T, Liu S, Ma L, et al Identification of IL‐17A‐derived neural cell type and dynamic changes of IL‐17A in serum/CSF of mice with ischemic stroke. Neurol Res 2017; 39:552–8. [DOI] [PubMed] [Google Scholar]
- 60. Sun H, Zhong D, Jin J, Liu Q, Wang H, Li G. Upregulation of miR‐215 exerts neuroprotection effects against ischemic injury via negative regulation of Act1/IL‐17RA signaling. Neurosci Lett 2018; 662:233–41. [DOI] [PubMed] [Google Scholar]
- 61. Lin Y, Zhang JC, Yao CY, Wu Y, Abdelgawad AF, Yao SL, et al Critical role of astrocytic interleukin‐17 A in post‐stroke survival and neuronal differentiation of neural precursor cells in adult mice. Cell Death Dis 2016; 7:e2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wojkowska DW, Szpakowski P, Glabinski A. Interleukin 17A promotes lymphocytes adhesion and induces CCL2 and CXCL1 release from brain endothelial cells. Int J Mol Sci 2017; 18:1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hermann DM, Kleinschnitz C, Gunzer M. Implications of polymorphonuclear neutrophils for ischemic stroke and intracerebral hemorrhage: predictive value, pathophysiological consequences and utility as therapeutic target. J Neuroimmunol 2018; 321:138–43. [DOI] [PubMed] [Google Scholar]
- 64. Hermann DM, Kleinschnitz C, Gunzer M. Role of polymorphonuclear neutrophils in the reperfused ischemic brain: insights from cell‐type‐specific immunodepletion and fluorescence microscopy studies. Ther Adv Neurol Disord 2018; 11:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lim K, Hyun YM, Lambert‐Emo K, Capece T, Bae S, Miller R, et al Neutrophil trails guide influenza‐specific CD8⁺ T cells in the airways. Science 2015; 349:aaa4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, et al Leukocyte‐derived matrix metalloproteinase‐9 mediates blood‐brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol 2005; 289:H558–H568. [DOI] [PubMed] [Google Scholar]
- 67. Ruhnau J, Schulze J, Dressel A, Vogelgesang A. Thrombosis, neuroinflammation, and poststroke infection: the multifaceted role of neutrophils in stroke. J Immunol Res 2017; 2017:5140679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Perez‐de‐Puig I, Miró‐Mur F, Ferrer‐Ferrer M, Gelpi E, Pedragosa J, Justicia C, et al Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol 2015; 129:239–57. [DOI] [PubMed] [Google Scholar]
- 69. Jiang X, Andjelkovic AV, Zhu L, Yang T, Bennett MVL, Chen J, et al Blood‐brain barrier dysfunction and recovery after ischemic stroke. Prog Neurogibol 2018; 163–164:144–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, et al Cellular mechanisms of IL‐17‐induced blood‐brain barrier disruption. FASEB J 2010; 24:1023–34. [DOI] [PubMed] [Google Scholar]
- 71. Ni P, Dong H, Wang Y, Zhou Q, Xu M, Qian Y, et al IL‐17A contributes to perioperative neurocognitive disorders through blood‐brain barrier disruption in aged mice. J Neuroinflammation 2018; 15:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kuhlmann CR, Tamaki R, Gamerdinger M, Lessmann V, Behl C, Kempski OS, et al Inhibition of the myosin light chain kinase prevents hypoxia‐induced blood‐brain barrier disruption. J Neurochem 2007; 102:501–7. [DOI] [PubMed] [Google Scholar]
- 73. Zhu F, Wang Q, Guo C, Wang X, Cao X, Shi Y, et al IL‐17 induces apoptosis of vascular endothelial cells: a potential mechanism for human acute coronary syndrome. Clin Immunol 2011; 141:152–60. [DOI] [PubMed] [Google Scholar]
- 74. Ravichandran VA, Kim M, Han SK, Cha YS. Stachys sieboldii extract supplementation attenuates memory deficits by modulating BDNF‐CREB and its downstream molecules, in animal models of memory impairment. Nutrients 2018; 10:917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kitagawa K. CREB and cAMP response element‐mediated gene expression in the ischemic brain. FEBS J 2007; 274:3210–7. [DOI] [PubMed] [Google Scholar]
- 76. Du W, Huang J, Yao H, Zhou K, Duan B, Wang Y. Inhibition of TRPC6 degradation suppresses ischemic brain damage in rats. J Clin Invest 2010; 120:3480–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Guo C, Ma Y, Ma S, Mu F, Deng J, Duan J, et al The Role of TRPC6 in the neuroprotection of calycosin against cerebral ischemic injury. Sci Rep 2017; 7:3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li T, Zhang YM, Han D, Hua R, Guo BN, Hu SQ, et al Involvement of IL‐17 in secondary brain injury after a traumatic brain injury in rats. Neuromolecular Med 2017; 19:541–54. [DOI] [PubMed] [Google Scholar]
- 79. Shen F, Hu Z, Goswami J, Gaffen SL. Identification of common transcriptional regulatory elements in interleukin‐17 target genes. J Biol Chem 2006; 281:24138–48. [DOI] [PubMed] [Google Scholar]
- 80. McAleer JP, Kolls JK. Directing traffic: IL‐17 and IL‐22 coordinate pulmonary immune defense. Immunol Rev 2014; 260:129–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL‐22‐IL‐22R1 system. Nat Rev Drug Discov 2014; 13:21–38. [DOI] [PubMed] [Google Scholar]
- 82. Yang X, Zheng SG. Interleukin‐22: a likely target for treatment of autoimmune diseases. Autoimmun Rev 2014; 13:615–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nardinocchi L, Sonego G, Passarelli F, Avitabile S, Scarponi C, Failla CM, et al Interleukin‐17 and interleukin‐22 promote tumor progression in human nonmelanoma skin cancer. Eur J Immunol 2015; 45:922–31. [DOI] [PubMed] [Google Scholar]
- 84. Wang J, Lin M, Ren H, Yu Z, Guo T, Gu B. Expression and clinical significance of serum miR‐497 in patients with acute cerebral infarction. Clin Lab 2019; 65:181001. [DOI] [PubMed] [Google Scholar]
- 85. Zhang Y, Niu C. The correlation of long non‐coding RNA intersectin 1–2 with disease risk, disease severity, inflammation, and prognosis of acute ischemic stroke. J Clin Lab Anal 2020; 34:e23053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Xu X, Jiang Y. The Yin and Yang of innate immunity in stroke. Biomed Res Int 2014; 2014:807978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL‐23‐IL‐17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 2014; 14:585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Satpathy AT, Briseño CG, Lee JS, Ng D, Manieri NA, Kc W, et al Notch2‐dependent classical dendritic cells orchestrate intestinal immunity to attaching‐and‐effacing bacterial pathogens. Nat Immunol 2013; 14:937–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. McGeachy MJ, Bak‐Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al TGF‐beta and IL‐6 drive the production of IL‐17 and IL‐10 by T cells and restrain T(H)‐17 cell‐mediated pathology. Nat Immunol 2007; 8:1390–7. [DOI] [PubMed] [Google Scholar]
- 90. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL‐17‐producing T cells. Immunity 2006; 24:179–89. [DOI] [PubMed] [Google Scholar]
- 91. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al IL‐6 programs T(H)‐17 cell differentiation by promoting sequential engagement of the IL‐21 and IL‐23 pathways. Nat Immunol 2007; 8:967–74. [DOI] [PubMed] [Google Scholar]
- 92. Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, et al IL‐21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 2007; 448:484–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al Generation of pathogenic T(H)17 cells in the absence of TGF‐β signalling. Nature 2010; 467:967–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cho ML, Kang JW, Moon YM, Nam HJ, Jhun JY, Heo SB, et al STAT3 and NF‐kappaB signal pathway is required for IL‐23‐mediated IL‐17 production in spontaneous arthritis animal model IL‐1 receptor antagonist‐deficient mice. J Immunol 2006; 176:5652–61. [DOI] [PubMed] [Google Scholar]
- 95. Floss DM, Mrotzek S, Klöcker T, Schröder J, Grötzinger J, Rose‐John S, et al Identification of canonical tyrosine‐dependent and non‐canonical tyrosine‐independent STAT3 activation sites in the intracellular domain of the interleukin 23 receptor. J Biol Chem 2013; 288:19386–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL‐17 and Th17 cells. Annu Rev Immunol 2009; 27:485–517. [DOI] [PubMed] [Google Scholar]
- 97. Sato K, Miyoshi F, Yokota K, Araki Y, Asanuma Y, Akiyama Y, et al Marked induction of c‐Maf protein during Th17 cell differentiation and its implication in memory Th cell development. J Biol Chem 2011; 286:14963–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zuberbuehler MK, Parker ME, Wheaton JD, Espinosa JR, Salzler HR, Park E, et al The transcription factor c‐Maf is essential for the commitment of IL‐17‐producing γδ T cells. Nat Immunol 2019; 20:73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wang H, Zhong D, Chen H, Jin J, Liu Q, Li G. NLRP3 inflammasome activates interleukin‐23/interleukin‐17 axis during ischaemia‐reperfusion injury in cerebral ischaemia in mice. Life Sci 2019; 227:101–13. [DOI] [PubMed] [Google Scholar]
- 100. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL‐17+ T helper cells. Cell 2006; 126:1121–33. [DOI] [PubMed] [Google Scholar]
- 101. Xue L, Chen H, Lu K, Huang J, Duan H, Zhao Y. Clinical significance of changes in serum neuroglobin and HIF‐1α concentrations during the early‐phase of acute ischemic stroke. J Neurol Sci 2017; 375:52–7. [DOI] [PubMed] [Google Scholar]
- 102. Wu X, Liu S, Hu Z, Zhu G, Zheng G, Wang G. Enriched housing promotes post‐stroke neurogenesis through calpain 1‐STAT3/HIF‐1α/VEGF signaling. Brain Res Bull 2018; 139:133–43. [DOI] [PubMed] [Google Scholar]
- 103. Singh N, Sharma G, Mishra V. Hypoxia inducible factor‐1: its potential role in cerebral ischemia. Cell Mol Neurobiol 2012; 32:491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al Control of T(H)17/T(reg) balance by hypoxia‐inducible factor 1. Cell 2011; 146:772–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tanaka S, Suto A, Iwamoto T, Kashiwakuma D, Kagami S, Suzuki K, et al Sox5 and c‐Maf cooperatively induce Th17 cell differentiation via RORγt induction as downstream targets of Stat3. J Exp Med 2014; 211:1857–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Swaidani S, Liu C, Zhao J, Bulek K, Li X. TRAF regulation of IL‐17 cytokine signaling. Front Immunol 2019; 10:1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Liu Z, He D, Zhang X, Li Y, Zhu C, Dong L, et al Neuroprotective effect of early and short‐time applying sophoridine in pMCAO rat brain: down‐regulated TRAF6 and up‐regulated p‐ERK1/2 expression, ameliorated brain infaction and edema. Brain Res Bull 2012; 88:379–84. [DOI] [PubMed] [Google Scholar]
- 108. Herjan T, Yao P, Qian W, Li X, Liu C, Bulek K, et al HuR is required for IL‐17‐induced Act1‐mediated CXCL1 and CXCL5 mRNA stabilization. J Immunol 2013; 191:640–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hartupee J, Liu C, Novotny M, Li X, Hamilton T. IL‐17 enhances chemokine gene expression through mRNA stabilization. J Immunol 2007; 179:4135–41. [DOI] [PubMed] [Google Scholar]
- 110. Ardelt AA, Carpenter RS, Iwuchukwu I, Zhang A, Lin W, Kosciuczuk E, et al Transgenic expression of HuR increases vasogenic edema and impedes functional recovery in rodent ischemic stroke. Neurosci Lett 2017; 661:126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhu S, Pan W, Shi P, Gao H, Zhao F, Song X, et al Modulation of experimental autoimmune encephalomyelitis through TRAF3‐mediated suppression of interleukin 17 receptor signaling. J Exp Med 2010; 207:2647–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Zepp JA, Liu C, Qian W, Wu L, Gulen MF, Kang Z, et al Cutting edge: TNF receptor‐associated factor 4 restricts IL‐17‐mediated pathology and signaling processes. J Immunol 2012; 189:33–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ma C, Lin W, Liu Z, Tang W, Gautam R, Li H, et al NDR1 protein kinase promotes IL‐17‐ and TNF‐α‐mediated inflammation by competitively binding TRAF3. EMBO Rep 2017; 18:586–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Shi Y, Ullrich SJ, Zhang J, Connolly K, Grzegorzewski KJ, Barber MC, et al A novel cytokine receptor‐ligand pair. Identification, molecular characterization, and in vivo immunomodulatory activity. Biol Chem 2000; 275:19167–76. [DOI] [PubMed] [Google Scholar]
- 115. Bie Q, Jin C, Zhang B, Dong H. IL‐17B: a new area of study in the IL‐17 family. Mol Immunol 2017; 90:50–6. [DOI] [PubMed] [Google Scholar]
- 116. Zhou J, Ren L, Chen D, Lin X, Huang S, Yin Y, et al IL‐17B is elevated in patients with pneumonia and mediates IL‐8 production in bronchial epithelial cells. Clin Immunol 2017; 175:91–8. [DOI] [PubMed] [Google Scholar]
- 117. Moore EE, Presnell S, Garrigues U, Guilbot A, LeGuern E, Smith D, et al Expression of IL‐17B in neurons and evaluation of its possible role in the chromosome 5q‐linked form of Charcot‐Marie‐Tooth disease. Neuromuscul Disord 2002; 12:141–50. [DOI] [PubMed] [Google Scholar]
- 118. Bordon Y. Cytokines: IL‐17C joins the family firm. Nat Rev Immunol 2011; 11:805. [DOI] [PubMed] [Google Scholar]
- 119. Chang SH, Reynolds JM, Pappu BP, Chen G, Martinez GJ, Dong C. Interleukin‐17C promotes Th17 cell responses and autoimmune disease via interleukin‐17 receptor E. Immunity 2011; 35:611–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Song X, Zhu S, Shi P, Liu Y, Shi Y, Levin SD, et al IL‐17RE is the functional receptor for IL‐17C and mediates mucosal immunity to infection with intestinal pathogens. Nat Immunol 2011; 12:1151–8. [DOI] [PubMed] [Google Scholar]
- 121. McGeachy MJ, Cua DJ, Gaffen SL. The IL‐17 family of cytokines in health and disease. Immunity 2019; 50:892–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Saddawi‐Konefka R, Seelige R, Gross ET, Levy E, Searles SC, Washington A, et al Nrf2 induces IL‐17D to mediate tumor and virus surveillance. Cell Rep 2016; 16:2348–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Seelige R, Washington A, Bui JD. The ancient cytokine IL‐17D is regulated by Nrf2 and mediates tumor and virus surveillance. Cytokine 2017; 91:10–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Xu M, Dong C. IL‐25 in allergic inflammation. Immunol Rev 2017; 278:185–91. [DOI] [PubMed] [Google Scholar]
- 125. Liu Y, Shao Z, Shangguan G, Bie Q, Zhang B. Biological properties and the role of IL‐25 in disease pathogenesis. J Immunol Res 2018; 2018:6519465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kleinschek MA, Owyang AM, Joyce‐Shaikh B, Langrish CL, Chen Y, Gorman DM, et al IL‐25 regulates Th17 function in autoimmune inflammation. J Exp Med 2007; 204:161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Luo Y, Zhou Y, Xiao W, Liang Z, Dai J, Weng X, et al Interleukin‐33 ameliorates ischemic brain injury in experimental stroke through promoting Th2 response and suppressing Th17 response. Brain Res 2015; 1597:86–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support this review are included within the article.