

Abstract

An expedient total synthesis of the title marine sponge alkaloids has been developed. The salient features of the synthesis are as follows: (i) preparation of the required 13- and 14-membered cyclic lactams with n + 4 ring-expansion strategy of cyclic β-keto esters and (ii) functional group manipulation of the resulted keto ester lactams. This approach used easily accessible and inexpensive materials/reagents, thus providing a promising alternative to the existing preparations.
Introduction
The prevalence of marine alkaloids containing azamacrocycle-based architectures have attracted tremendous chemical and biological research interests in recent years.1,2 In this regard, motuporamines A and B (1 and 2) (Figure 1), which were first isolated as an inseparable mixture from the tropical sponge Xestospongia exigua, represent a new class of this group.3 The crude mixtures were initially obtained by conversion to their diacetylated derivatives using the reversed-phase high-performance liquid chromatography (HPLC) technique. This subclass of marine alkaloid motuporamines features a 13- to 15-membered azamacrocyclic structure with the nitrogen atom linked to a spermidine-like side chain.
Figure 1.

Structures of motuporamines A (1) and B (2).
Later on, three new motuporamines D–F with one or two C=C double bonds embedded in the ring and a mixture of the methylated derivative motuporamines G–I were isolated from the same marine source along with motuporamines A and B.4 These compounds were found with various biological activities, including in vitro cytotoxicity against a panel of human cancer cell lines, inhibition against the invasion of MDA-MB-231 cells with IC50 values less than 15 μM, angiogenesis in the chick choriallantoic membrane assay to antimicrobial agents, and enhancement in antibiotic effect against resistant Gram-negative bacteria.5−7
The therapeutic potential and the unusual structure of these alkaloids have aroused much interest of many scholars for their syntheses. Motuporamines A (1) and B (2) are two minor components in the natural isolates. Due to their scarcity in natural resources, the total synthesis of 1 and 2 was reported shortly after their isolation by the groups of Baldwin and Weiler, which thus furnished samples for a preliminary study on pharmaceutical properties. Both pioneering works employed 13- and 14-membered lactams as precursors to the corresponding macrocyclic amine, with Baldwin’s group8 using a reductive amidation method to append the spermidine-like side chain, while Weiler’s group employed the Michael addition and amidation strategy to introduce the side unit.9 However, the lactams had to be prepared by ring-closing metathesis, and the respective bis(alkene)-amide in turn was synthesized by a multiple-step process.10 In 2008, Back and co-workers reported another synthesis of 1 and 2, relying on iterative 3-aza-Cope ring-expansion rearrangement of cyclic α-vinylamines to achieve the construction of the macrocyclic amine.11
In pursuit of our previous work in the field of cyclic polyamine alkaloids,12,13 we were prompted to search for a complementary and potentially scalable pathway to these two natural products by avoiding the use of inaccessible and expensive reagents. With this aim, the ring-expansion strategy of cyclic β-keto esters with the insertion of a β-amino acid ester segment, an elegant protocol established recently by Unsworth and co-workers,14−17 provided us with a promising solution. Herein, we reported a concept of implementing the [n + 4] ring expansion to the total synthesis of the two marine alkaloids (1 and 2).
Results and Discussion
Motuporamine A (1) was selected as the first synthesis target. The synthesis commenced with the preparation of 13-membered keto-lactam 10 as outlined in Scheme 1. Using the reported method,17 cyclo-octanone 3 was reacted with ethyl azoacetate 4 in the presence of BF3·Et2O to give the ring expanded cyclic β-keto ester 5 in 69% yield. Compound 5 occurs as a mixture of enol and ketone tautomers, and the ratio was estimated to be 1.1:1.0 by analysis of the diagnostic 1H nuclear magnetic resonances (NMR). Subsequently, C-acylation of 5 with 3 equiv of N-Fmoc-protected 3-amino acid chloride 6 in the presence of 2 equiv of MgCl2 led to the diketo ester compound 7 in 53% yield. Using the combination of pyridine and magnesium chloride was proved to be optimal for the soft enolization and acylation procedure. As reported by Rathke, other metal chlorides such as AlCl3, ZnCl2, or FeCl3 were ineffective in promoting the reaction.18 In addition, the use of low amounts of promoter MgCl2 or acid chloride 6 gave considerably decreased yields of 7.
Scheme 1. Synthetic Route to 13-Membered Keto-Lactam 10.

As compound 7 is a 9-fluorenylmethyl carbamate, its Fmoc group should be easily cleaved under nonhydrolytic condition, catalyzed by simple amines such as piperidine, morpholine, and dicyclohexylamine, whereby liberating the protected amine as its free base.19,20 Following Unsworth’s procedure, the N-Fmoc group was removed with piperidine, acting as the deblocking agent, giving rise to intermediate 8. To our delight, compound 8 concurrently underwent intramolecular nucleophilic addition of the free amino functionality to the ring’s carbonyl group, generating fused bicyclic aza-semiacetal specie 8′, which was spontaneously transformed to 13-membered ring product 9 under the reaction conditions. Acidic hydrolysis on 9 resulted in the formation of keto-lactam 10 in near-quantitative yield. Condition screening for the ester hydrolysis and decarboxylation suggested that the concentration of hydrochloric acid should not be lower than 6 M and the temperature should be higher than 60 °C. With the progression of the reaction, white solid 9 was slowly dissolved in the hydrochloric acid solution over 4 h, indicating that the reaction was completed.
At this juncture, the functional group manipulation on lactam 10 was carried out in three steps (Scheme 2) to synthesize macrocyclic amine 14. In the first step, 1,3-dithiane derivative 12 was prepared by the reaction of 10 with trimethylene mercaptan under the catalysis of BF3·Et2O, which was subjected to hydrogenolysis with the Raney-Ni catalyst to afford lactam 13. Our experimentations exhibited that this reduction step had to be carried out in two batches to ensure a high yield conversion (see Experimental Section). Thus, the conversion of the carbonyl group to methylene proceeded fast and cleanly, giving a 92% yield of lactam 13. Further reduction of 13 with LiAlH4 furnished the required macrocyclic amine 14 in 67% yield.
Scheme 2. Synthesis of TFA Salt of Motuporamine A (1·TFA).

Having the key cyclic amine fragment 14 in hand, the synthetic stage was set for the target motuporamine A (1). Reductive amination of 14 with bis(Boc)-protected diamino aldehyde 15(21) using sodium triacetoxyborohydride in methanol gave bis(Boc)-protected motuporamine A 16 in near-quantitative yield. Finally, quantitative removal of the Boc protecting group with trifluoroacetic acid successfully produced the trifluoroacetate of motuporamine A (1·TFA). By employing this protocol, we synthesized motuporamine A in its TFA salt form in a total yield of 14.8% through nine steps using cheap and readily available chemical reagents.
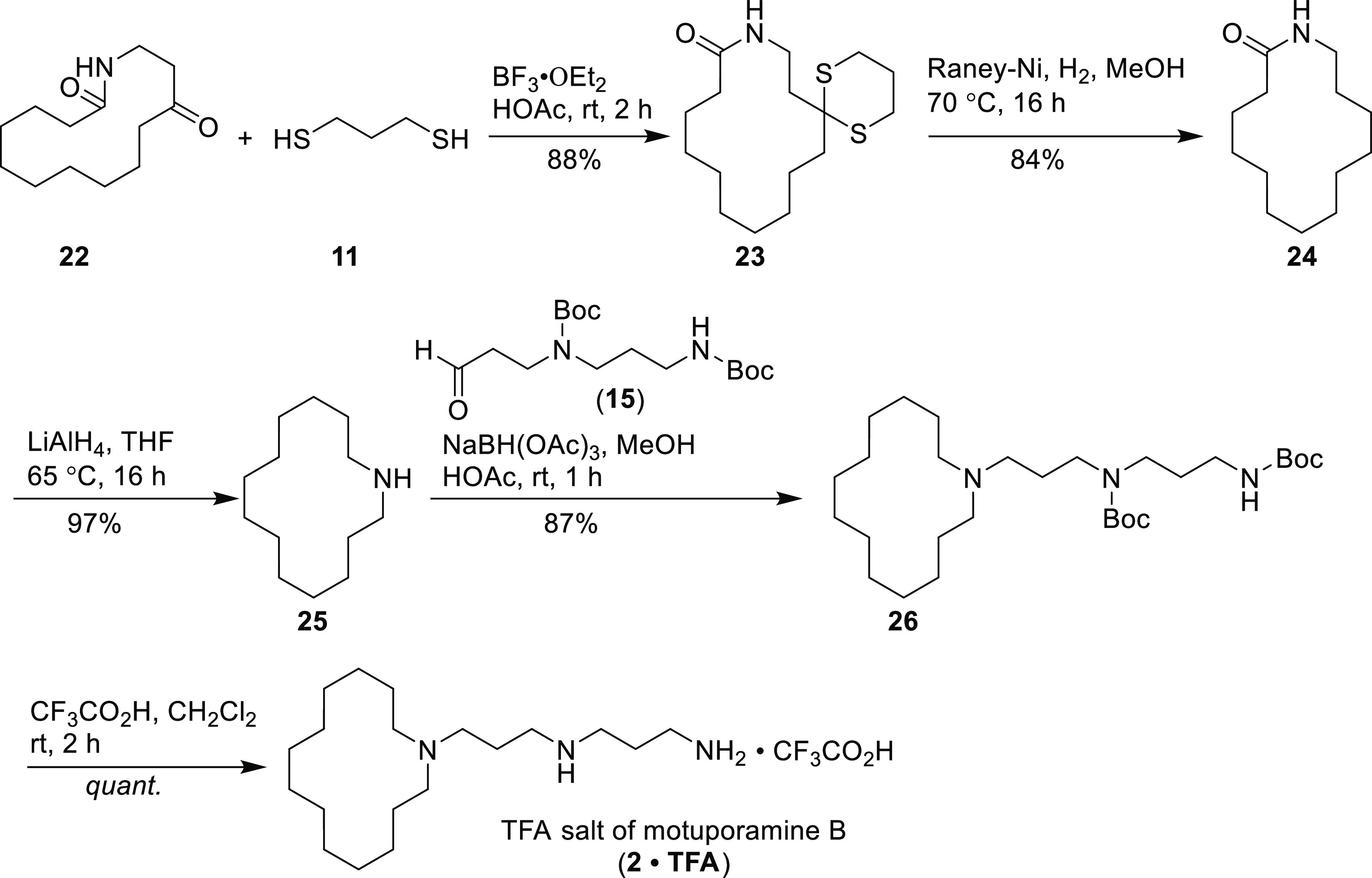
For the synthesis of motuporamine B (2), 10-membered cyclic β-keto ester 18 employed for the [10 + 4] ring expansion was first prepared by reacting cyclodecanone 17 with diethyl carbonate in the presence of 2 equiv of sodium hydride at room temperature for 16 h.22 Cyclic ketone 17 was, in turn, prepared from cyclo-octanone and methyl propiolate following the literature method.23 The remaining steps were similar to those employed in the synthesis of motuporamine A (1) as described above, and the detailed procedures are illustrated in Schemes 3 and 4. The total synthesis of motuporamine B, also in its TFA salt form (2·TFA), was accomplished with a total yield of 17.5% in 13 steps.
Scheme 3. Synthetic Route to 14-Membered Keto-Lactam 22.

Scheme 4. Synthesis of TFA Salt of Motuporamine B (2·TFA).
Conclusions
In conclusion, we have established the total syntheses of motuporamines A and B, both in the form of TFA salt, by conventional and readily available reagents. The key steps involved the application of the newly emerged ring-expansion strategy of the cyclic β-keto ester with the insertion of a linear β-amino acid fragment into the ring.
Experimental Section
General Information
NMR spectra were recorded at 298 K with a Bruker AW-400 spectrometer. Chemical shifts are given as values and are referenced to the residual solvent signal for CDCl3, d6-dimethyl sulfoxide (DMSO), and CD3OD. Internal references of hydrogen 7.26 were used for CDCl3, 2.50 for d6-DMSO, and 3.31 for CD3OD. 13C NMR spectra were recorded at 100 MHz. Internal references of carbon 77.16 were used for CDCl3, 39.52 for d6-DMSO, and 49.00 for CD3OD. High-resolution mass spectrometry-electrospray ionization (HRMS-ESI) spectra were recorded on a Bruker Micro-Tof 11 spectrometer with samples dissolved in CH3OH. IR spectra were recorded on a Nicolet 360 instrument in the 400–4000 cm–1 region. Thin-layer chromatography (TLC) was carried out on glass plates coated with silica 60 gel F254. The zones were detected with UV light when possible or by iodine fumigation. Chromatography was performed with silica gel (200–300 mesh). All of the commercial solvents were purified according to ref (24).
Ethyl 2-Oxocyclononanecarboxylate (5)
To a solution of cyclo-octanone (3) (5.00 g, 39.6 mmol) in anhydrous diethyl ether (100 mL) was added BF3·Et2O (8.44 g, 59.5 mmol) in anhydrous diethyl ether (30 mL) dropwise at 0 °C. Next, to the mixture was added ethyl 2-diazoacetate (4) (7.14 g, 95% pure, 59.5 mmol) in anhydrous diethyl ether (30 mL) dropwise at the same temperature for 15 min. The resulting mixture was stirred at 0 °C to room temperature for 16 h. Then, the mixture was cooled to 0 °C and quenched by a saturated aqueous NaHCO3 solution until pH 8. The mixture was extracted with diethyl ether (30 mL × 3), and the combined organic phases were washed with brine (30 mL), dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography (EtOAc/PE = 1:70) to afford 5 as a mixture of enol–keto tautomers in approximate ratio 1.1:1.0 (5.83 g, 69%) as a faint yellow oil: Rf = 0.60 (EtOAc/PE, 1:20); IR (film): 2928, 2856, 1747, 1641, 1470, 1239, 1196 cm–1; 1H NMR (400 MHz, CDCl3): δ = 12.71 (s, 1H, OH, enol), 4.16 (q, 2H, J = 7.2 Hz, OCH2Me), 4.09 (2H, q, J = 7.0 Hz, OCH2Me) (enol and keto), 3.59–3.55 (m, 1H, COCHCO2Et, keto), 2.60–2.48 (m, 2H, CH2COCH), 2.36–2.33 (m, 2H, CH2COH), 2.30–2.28 (m, 2H, CH2), 2.05–2.00 (m, 2H, CH2), 1.88–1.76 (m, 2H, CH2), 1.71–1.61 (m, 2H, CH2), 1.52–1.31 (m, 16H, 8 × CH2), 1.24 (t, 3H, J = 7.2 Hz), 1.18 (t, 3H, J = 6.8 Hz) (CH3, enol and keto); 13C NMR (100 MHz, CDCl3): δ = 211.8 (COCH2, keto), 175.8 (CO2Et, keto), 173.4 (COH, enol), 169.9 (CO2Et, enol), 100.1(CCO2Et, enol), 61.1 (OCH2), 60.2 (OCH2) (enol & keto), 58.7 (CHCO2Et, keto), 42.4 (CH2COCH, keto), 31.4, 27.0, 26.94, 26.14, 25.87, 25.18, 24.98, 24.64, 24.59, 24.49, 24.35, 23.93 (CH2, enol & keto), 14.3 (CH3, enol), 14.1 (CH3, keto).
Ethyl 1-(3-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)propanoyl)-2-oxocyclononanecarboxylate (7)
To a solution of ester 5 (1.74 g, 8.24 mmol) in dichloromethane (DCM) (20 mL) was added pyridine (3.90 g, 49.4 mmol) and MgCl2 (1.56 g, 16.5 mmol), stirred at room temperature for 30 min. Next, to the mixture was added (9H-fluoren-9-yl)methyl (3-chloro-3-oxopropyl) carbamate (6) (7.60 g, 23.0 mmol) in DCM (10 mL) and stirred at room temperature for additional 2 h. The mixture was diluted by DCM (40 mL), added HCl solution (30 mL, 1 M in water), and extracted with a mixture (EtOAc/DCM, 2:1, 30 mL × 3). The combined organic phases were dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography (EtOAc/PE = 1:10→1:4) to yield intermediate (7) (2.23 g, 53%) as a colorless amorphous solid: Rf = 0.65 (EtOAc/PE, 1:3); IR (KBr): 3408, 2933, 1713, 1519, 1449, 1243, 1136 cm–1; 1H NMR (400 MHz, CDCl3): δ = 7.75 (d, 2H, J = 7.6 Hz), 7.60 (d, 2H, J = 7.2 Hz), 7.39 (t, 2H, J = 7.2 Hz), 7.30 (t, 2H, J = 7.2 Hz), 5.32 (bs, 1H), 4.40–4.28 (m, 2H), 4.25–4.20 (m, 3H), 3.49–3.48 (m, 2H), 2.90–2.82 (m, 2H), 2.66–2.54 (m, 2H), 2.22–2.12 (m, 2H), 1.89 (m, 1H), 1.72–1.65 (m, 2H), 1.48–1.37 (m, 7H), 1.27 (t, 3H, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3): δ = 209.1, 202.9, 168.1, 156.4, 144.1, 144.0, 141.3, 127.7, 127.1, 125.2, 125.2, 120.0, 78.4, 66.8, 62.3, 47.2, 40.5, 38.2, 36.1, 28.2, 27.6, 24.4, 22.4, 20.6, 19.7, 14.0; HRMS (ESI): m/z calcd for C30H35NO6 [M + H]+: 506.2537, found: 506.2529.
Ethyl 4,13-dioxoazacyclotridecane-5-carboxylate (9)
To a solution of compound 7 (1.89 g, 3.74 mmol) in DCM (40 mL) was added piperidine (3.18 g, 37.4 mmol) and stirred at room temperature for 2 h. The mixture was evaporated under reduced pressure. The residue was dissolved in EtOAc (150 mL); washed with saturated aqueous NH4Cl solution (30 mL × 2), saturated aqueous NaHCO3 solution (30 mL), brine (30 mL); dried over Na2SO4; filtered; and concentrated. The residue was purified by flash column chromatography (EtOAc/PE = 1:10→pure EtOAc) to yield 9 (0.705 g, 66%) as a white solid: m.p. 128.0–129.3 °C; Rf = 0.50 (EtOAc/PE, 1:1); IR (KBr): 3331, 2933, 2869, 1731, 1640, 1541, 1253 cm–1; 1H NMR (400 MHz, CDCl3): δ = 6.11 (s, 1H), 4.17 (q, 2H, J = 6.8 Hz), 3.62–3.57 (m, 1H), 3.45–3.40 (m, 2H), 2.88 (m, 2H), 2.19–2.06 (m, 2H), 1.85–1.83 (m, 2H), 1.58 (m, 1H), 1.48 (m, 1H), 1.33–1.17 (m, 11H); 13C NMR (100 MHz, CDCl3): δ = 207.2, 173.5, 169.4, 61.4 58.3, 42.2, 36.7, 33.9, 27.7, 26.2, 25.7, 25.6, 24.4, 24.1, 14.1; HRMS (ESI): m/z calcd for C15H25NO4 [M + H]+: 284.1856, found: 284.1850.
Azacyclotridecane-2,11-dione (10)
Compound 9 (0.667 g, 2.35 mmol) was suspended in HCl (40 mL, 6 M in water); the mixture was heated at 66 °C for 4 h until all white solid dissolved. The mixture was cooled to room temperature, and cold NaOH solution (2.5 M in water) was added dropwise to adjust the basicity until pH 13–14. During the period, a lot of white solid separated out. The mixture was extracted with EtOAc (30 mL × 3), and the EtOAc extracts were washed with saturated aqueous NH4Cl solution (30 mL), saturated aqueous NaHCO3 solution (30 mL), and brine (30 mL); then dried over Na2SO4; filtered; and concentrated to yield ketone 10 (0.497 g, 100%) as a white solid: m.p. 133.1–134.0 °C; Rf = 0.40 (EtOAc/PE, 1:1); IR (KBr): 3238, 3072, 2932, 2866, 1702, 1629, 1560, 1462 cm–1; 1H NMR (400 MHz, CDCl3): δ = 6.12 (s, 1H), 3.49–3.45 (m, 2H), 2.78–2.75 (m, 2H), 2.33 (2H, t, J = 4.8 Hz), 2.11–2.09 (m, 2H), 1.59–1.55 (m, 4H), 1.23 (d, 8H, J = 12.0 Hz); 13C NMR (100 MHz, CDCl3): δ = 212.4, 173.6, 42.6, 41.9, 37.0, 34.1, 26.4, 26.3, 26.2, 25.8, 24.8, 23.8; HRMS (ESI): m/z calcd for C12H21NO2 [M + H]+: 212.1645, found: 212.1642.
1,5-Dithia-9-azaspiro[5.12]octadecan-10-one (12)
To a solution of ketone 10 (0.385 g, 1.82 mmol) and propane-1,3-dithiol (11) (0.394 g, 3.65 mmol) in AcOH (15 mL) was added BF3·Et2O (1.55 g, 10.9 mmol) at room temperature and stirred for 1 h. To the mixture was added NaOH solution (2.5 M in water) dropwise until the pH reached 14. The mixture was extracted with EtOAc (30 mL × 2). The organic phases were combined; washed with the saturated aqueous NH4Cl solution (30 mL), saturated aqueous NaHCO3 solution (30 mL), and brine (30 mL); then dried over Na2SO4; filtered; and concentrated. The residue was pulverized with a mixture (EtOAc/PE = 1:5, 15 mL) and filtered to afford thioketal 12 (0.550 g, 100%) as a white solid: m.p. 149.7–152.0 °C; Rf = 0.60 (EtOAc/PE, 1:1); IR (KBr): 3329, 2931, 2860, 1639, 1542, 1459, 1245 cm–1; 1H NMR (400 MHz, CDCl3): δ = 5.93 (s, 1H), 3.51 (s, 2H), 2.83–2.68 (m, 4H), 2.25–2.18 (m, 4H), 1.99 (s, 1H), 1.90–1.83 (m, 3H), 1.71 (s, 2H), 1.48 (s, 2H), 1.38 (s, 4H), 1.31 (s, 4H); 13C NMR (100 MHz, CDCl3): δ = 173.7, 53.2, 38.4, 37.0, 36.6, 36.3, 35.4, 27.7, 27.0, 26.2, 25.6, 25.3, 25.2, 23.4, 20.8; HRMS (ESI): m/z calcd for C15H27NOS2 [M + H]+: 302.1607, found: 302.1607.
Azacyclotridecan-2-one (13)
To a solution of thioketal 12 (0.400 g, 1.33 mmol) in MeOH (30 mL) was added Raney Ni (1.50 g with H2O). The mixture was heated at 70 °C under a H2 atmosphere for 10 h, to which was added additional Raney Ni (1.00 g with H2O) and stirred for another 3 h. The mixture was filtered through celite carefully, and the cake was washed with excess MeOH, keeping the filtrate cake wet. The filtrate liquid was concentrated, and the residue was purified by flash column chromatography (EtOAc/PE = 1:3→1:1) to yield 13 (0.241 g, 92%) as a white solid: 151.5–153.4 °C; Rf = 0.65 (EtOAc/PE, 1:1); IR (KBr): 3309, 2935, 2859, 1641, 1549, 1462, 744 cm–1; 1H NMR (400 MHz, CDCl3): δ = 5.71 (s, 1H), 3.28–3.27 (m, 2H), 2.18–2.16 (m, 2H), 1.65 (s, 2H), 1.48 (s, 2H), 1.30 (s, 14H); 13C NMR (100 MHz, CDCl3): δ = 173.6, 39.1, 37.0, 28.4, 26.8, 26.4, 26.2, 25.8, 25.3, 25.0, 24.7, 24.0; HRMS (ESI): m/z calcd for C12H23NO [M + H]+: 198.1852, found: 198.1851.
Azacyclotridecane (14)
To a mixture of LiAlH4 (0.127 g, 3.35 mmol) in anhydrous tetrahydrofuran (THF) (5 mL) was added lactam 13 (0.220 g, 1.12 mmol) in anhydrous THF (4 mL). The mixture was stirred at 65 °C under an inert atmosphere of N2 for 16 h. The mixture was diluted with THF (10 mL), quenched with Na2SO4·5H2O, and filtered through celite. The cake was washed with a mixture (MeOH/DCM, 1:20, 30 mL). The filtrate liquid was concentrated and purified by flash column chromatography (MeOH/DCM/NH3 = 10:100:1) to yield cyclic amine 14 (0.138 g, 67%) as a colorless oil (easy to evaporate): Rf = 0.60 (MeOH/DCM, 1:10); IR (film): 3365, 2924, 2853, 2808, 1446, 1339, 1114 cm–1; 1H NMR (400 MHz, CDCl3): δ = 2.64 (s, 4H), 1.49 (s, 4H), 1.38–1.24 (m, 16H); 13C NMR (100 MHz, CDCl3): δ = 47.9, 27.9, 26.6, 26.0, 25.5, 24.6; HRMS (ESI): m/z calcd for C12H25N [M + H]+: 184.2060, found: 184.2051.
Tert-butyl-(3-(azacyclotridecan-1-yl)propyl)(3-((tert-butoxy-carbonyl)amino)propyl)carbamate (16)
To a solution of amine 14 (0.980 g, 0.535 mmol) and aldehyde 15 (0.353 g, 1.07 mmol) in MeOH (5 mL) was added AcOH (one drop) and NaBH(OAc)3 (0.593 g, 2.67 mmol) at room temperature. The mixture was stirred for 1.5 h, poured into water (100 mL), and extracted with CHCl3 (15 mL × 6). The collected organic phases were dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography (MeOH/DCM = 1:100→1:20) to yield diprotected amine 16 (0.258 g, 100%) as a faint yellow amorphous solid: Rf = 0.50 (MeOH/DCM, 1:20); IR (KBr): 3412, 2932, 2863, 2575, 1694, 1479, 1250, 1173 cm–1; 1H NMR (400 MHz, CDCl3): δ = 4.96 (m, 1H), 3.22 (s, 4H), 3.05–2.87 (m, 8H), 2.09 (bs, 2H), 1.73–1.54 (m, 6H), 1.40 (d, 21H, J = 10.4 Hz), 1.31 (s, 13H); 13C NMR (100 MHz, CDCl3): δ = 156.1, 80.2, 78.9, 52.6, 51.0, 44.2, 37.6, 28.4, 26.0, 24.9, 24.8, 24.7, 23.7, 20.3, 20.2; HRMS (ESI): m/z calcd for C28H55N3O4 [M + H]+: 498.4265, found: 498.4266.
N1-(3-(azacyclotridecan-1-yl)propyl)propane-1,3-diamine 2,2,2-trifluoroacetate (1)
To a solution of Boc-protected amine 16 (70 mg, 0.141 mmol) in DCM (5 mL) was added CF3COOH (1 mL) and stirred at room temperature for 2 h. The mixture was concentrated under reduced pressure, azeotroped with MeOH three times, and treated with vacuum to afford TFA salt of motuporamine A (1) (58 mg, 100%) as a yellow solid: m.p. 145.7–148.4 °C; Rf = 0.10 (MeOH/DCM, 1:20); IR (KBr): 3426, 3029, 2936, 2868, 1676, 1479, 1201 cm–1; 1H NMR (400 MHz, CD3OD): δ = 3.32–3.04 (m, 12H), 2.23–2.06 (m, 4H), 1.79–1.77 (m, 4H), 1.48–1.28 (m, 16H); 13C NMR (100 MHz, CD3OD): δ = 162.5, 162.1, 119.2, 116.3, 53.3, 53.0, 45.9, 37.7, 26.7, 25.8, 25.6, 25.3, 25.2, 22.4, 22.2; HRMS (ESI): m/z calcd for C18H39N3 [M + H]+: 298.3217, found: 298.3236.
Ethyl 2-oxocyclodecanecarboxylate (18)
A 100 mL flask was charged with diethyl carbonate (40 mL), to which were added NaH (60% dispersion in mineral oil, 1.37 g, 34.4 mmol) and cyclodecanone 17 (2.65 g, 17.2 mmol) in diethyl carbonate (20 mL) dropwise at room temperature. The resulting mixture was stirred under an inert atmosphere of N2 for 16 h, which was quenched by HCl solution (30 mL, 1 M in water) and extracted with EtOAc (30 mL × 3). The organic extracts were washed with brine (30 mL × 2), dried over Na2SO4, filtered, and concentrated. After removal of almost all solvents and diethyl carbonate, the residue was purified by flash column chromatography (EtOAc/PE = 1:100) to afford β-ketoester 18 as a mixture of enol–keto tautomers in approximate ratio 2.7:1.0 (3.31 g, 85%) as a colorless oil: Rf = 0.70 (EtOAc/PE, 1:20); IR (film): 2929, 2857, 1746, 1640, 1474, 1228 cm–1; 1H NMR (400 MHz, CDCl3): δ = 12.94 (s, 1H, OH, enol), 4.19 (q, 2H, J = 7.2 Hz, OCH2Me), 4.11 (2H, q, J = 7.0 Hz, OCH2Me) (enol and keto), 3.80 (m, 1H, COCHCO2Et, keto), 2.67–2.66 (m, 2H, CH2COCH, keto), 2.49 (m, 2H), 2.38–2.35 (m, 2H), 2.22–2.13 (m, 2H), 1.95–1.90 (m, 2H), 1.75 (m, 4H), 1.54–1.41 (m, 20H) [7 × CH2 (keto and enol) and CH2CO (enol)], 1.28 (t, 3H, J = 7.2 Hz, OCH2CH3), 1.21 (t, 3H, J = 7.2 Hz, OCH2CH3) (enol and keto); 13C NMR (100 MHz, CDCl3): δ = 208.8 (COCH2, keto), 175.1 (CO2Et, keto), 173.6 (COH, enol), 169.9 (CO2Et, enol), 99.8 (CCO2Et, enol), 61.3 (OCH2), 60.2 (OCH2) (enol and keto), 57.9 (CHCO2Et, keto), 42.1(CH2COCH, keto), 30.22, 30.13, 30.11, 29.80, 27.53, 27.34, 26.1, 25.74, 25.50, 25.31, 25.25, 24.5, 23.66, 23.37, 22.8, 21.1, 20.6 (CH2, enol and keto), 14.3 (CH3, enol), 14.1 (CH3, keto); gas chromatography–mass spectrometry (GC-MS) (EI): m/z (%) = 226 (20) [M+], 197 (5), 181 (25), 162 (5), 152 (30), 124 (40), 98 (100), 84 (45).
Ethyl 1-(3-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)propanoyl)-2-oxocyclodecanecarboxylate (19)
To a solution of ester 18 (1.82 g, 8.04 mmol) in DCM (20 mL) was added pyridine (3.81 g, 48.2 mmol) and MgCl2 (1.53 g, 16.1 mmol) and stirred at room temperature for 30 min. Next, to the mixture was added (9H-fluoren-9-yl)methyl (3-chloro-3-oxopropyl) carbamate (6) (7.41 g, 22.5 mmol) in DCM (10 mL), and the solution was stirred for additional 2 h. The mixture was diluted by DCM (40 mL), added HCl solution (30 mL, 1 M in water), and extracted with a mixture (EtOAc/DCM, 2:1, 30 mL × 3). The organic combined phases were dried over Na2SO4 and filtered. Then, the solvent was evaporated under vacuum, and the crude product was further purified by flash column chromatography (EtOAc/PE = 1:10→1:4) to give 19 (3.28 g, 78%) as a colorless amorphous solid: Rf = 0.65 (EtOAc/PE, 1:3); IR (KBr): 3410, 2931, 1713, 1520, 1449, 1242 cm–1; 1H NMR (400 MHz, CDCl3): δ = 7.76 (d, 2H, J = 7.6 Hz), 7.59 (d, 2H, J = 7.2 Hz), 7.39 (t, 2H, J = 7.6 Hz), 7.30 (t, 2H, J = 7.2 Hz), 5.29 (bs, 1H), 4.38–4.36 (m, 1H), 4.33–4.31 (m, 1H), 4.25–4.20 (m, 3H), 3.49–3.48 (m, 2H), 2.97–2.64 (m, 3H), 2.31–2.27 (m, 1H), 2.14–2.10 (m, 1H), 2.04–1.98 (m, 1H), 1.74 (m, 2H), 1.58–1.33 (m, 10H), 1.26 (t, 3H, J = 7.2 Hz); 13C NMR (100 MHz, CDCl3): δ = 207.5, 203.2, 168.4, 156.4, 144.2, 144.1, 141.4, 127.8, 127.1, 125.3, 125.2, 120.3, 120.0, 77.8, 66.9, 62.3, 47.3, 40.8, 38.7, 36.3, 29.4, 27.1, 26.1, 25.0, 24.7, 23.3, 21.2, 14.0; HRMS (ESI): m/z calcd for C31H37NO6 [M + H]+: 520.2694, found: 520.2692.
Ethyl 4,14-dioxoazacyclotetradecane-5-carboxylate (21)
To a solution of compound 19 (3.28 g, 6.32 mmol) in DCM (40 mL) was added piperidine (6.77 g, 79.7 mmol); the mixture was stirred at room temperature for 2 h. Next, it was evaporated under reduced pressure, and the residue was dissolved in EtOAc (150 mL); washed with saturated aqueous NH4Cl solution (30 mL × 2), saturated aqueous NaHCO3 solution (30 mL), and brine (30 mL); then dried over Na2SO4; filtered; and concentrated. The residue was purified by flash column chromatography (EtOAc/PE = 1:10→pure EtOAc) to yield 21 (1.41 g, 75%) as a white solid: m.p. 112.0–112.7 °C; Rf = 0.50 (EtOAc/PE, 1:1); IR (KBr): 3338, 3293, 2934, 2859, 1707, 1640, 1257 cm–1; 1H NMR (400 MHz, CDCl3): δ = 6.13 (s, 1H), 4.17 (q, 2H, J = 6.8 Hz), 3.54–3.50 (m, 2H), 3.47–3.43 (m, 1H), 2.82 (m, 2H), 2.4 (t, 2H, J = 6.0 Hz), 2.01–1.95 (m, 1H), 1.79–1.77 (m, 1H), 1.64–1.63 (m, 2H), 1.34 (m, 2H), 1.22 (t, 6H, J = 6.8 Hz), 1.15 (s, 5H); 13C NMR (100 MHz, CDCl3): δ = 206.5, 172.8, 169.5, 61.4, 58.3, 41.5, 34.7, 33.2, 28.6, 26.7, 26.2, 25.4, 25.1, 24.7, 24.6, 14.1; HRMS (ESI): m/z calcd for C16H27NO4 [M + H]+: 298.2013, found: 298.2030.
Azacyclotetradecane-2,12-dione (22)
The above keto ester-lactam 21 (1.41 g, 4.76 mmol) was suspended in HCl solution (100 mL, 6 M in water), which was heated at 66 °C for 4 h until all white solid dissolved. The mixture was cooled to room temperature and added cold NaOH solution (2.5 M in water) dropwise to adjust pH 13–14. During the period, a lot of white solid separated out. The mixture was extracted with EtOAc (30 mL × 3), and the combined organic phases were washed with saturated aqueous NH4Cl solution (30 mL), saturated aqueous NaHCO3 solution (30 mL), and brine (30 mL); then dried over Na2SO4; filtered; and concentrated to afford keto-lactam 22 (1.07 g, 100%) as a white solid: m.p. 124.5–126.8 °C; Rf = 0.45 (EtOAc/PE, 1:1); IR (KBr): 3363, 3304, 2931, 2854, 1705, 1648, 1537, 1428 cm–1; 1H NMR (400 MHz, CDCl3): δ = 6.28–6.13 (m, 1H), 3.50 (s, 2H), 2.69 (s, 2H), 2.39 (s, 2H), 2.13 (s, 2H), 1.63 (s, 4H), 1.30–1.17 (m, 10H); 13C NMR (100 MHz, CDCl3): δ = 211.8, 172.9, 41.8, 35.0, 34.9, 33.5, 26.8, 25.9, 25.6, 25.5, 25.0, 24.1; HRMS (ESI): m/z calcd for C13H23NO2 [M + H]+: 226.1802, found: 226.1987.
1,5-Dithia-9-azaspiro[5.13]nonadecan-10-one (23)
To a solution of compound 22 (0.400 g, 1.78 mmol) and propane-1,3-dithiol (11) (0.480 g, 4.44 mmol) in AcOH (20 mL) was added BF3·Et2O (1.26 g, 8.88 mmol) at room temperature, and the mixture was stirred for 2 h. To the mixture, NaOH solution (2.5 M in water) was then added dropwise until pH 14. The mixture was extracted with EtOAc (30 mL × 2). The combined organic phases were washed with saturated aqueous NH4Cl solution (30 mL), saturated aqueous NaHCO3 solution (30 mL), and brine (30 mL); then dried over Na2SO4; filtered; and concentrated. The residue was pulverized with a mixture (EtOAc/PE, 1:6, 10 mL) and filtered to give thioketal 23 (0.497 g, 88%) as a white solid: m.p. 143.4–145.0 °C; Rf = 0.60 (EtOAc/PE, 1:1); IR (KBr): 3322, 2930, 2849, 1643, 1536, 1424, 1372, 1256 cm–1; 1H NMR (400 MHz, CDCl3): δ = 6.01 (1Hs,), 3.47 (s, 2H), 2.88–2.82 (m, 2H), 2.72–2.68 (m, 2H), 2.29 (m, 2H), 2.20–2.17 (m, 2H), 2.02–2.00 (m, 1H), 1.91–1.80 (m, 3H), 1.69 (m, 2H), 1.46–1.44 (m, 2H), 1.34–1.25 (m, 10H); 13C NMR (100 MHz, CDCl3): δ = 173.6, 53.2, 38.9, 36.7, 36.0, 35.3, 26.8, 26.6, 26.1, 25.6, 25.4, 24.7, 24.2, 22.1; HRMS (ESI): m/z calcd for C16H29NOS2 [M + H]+: 316.1763, found: 316.1761.
Azacyclotetradecan-2-one (24)
To a solution of thioketal 23 (0.400 g, 1.27 mmol) in MeOH (30 mL) was added Raney Ni (1.50 g with H2O), the resulting mixture was heated at 70 °C under a H2 atmosphere for 10 h, to which was added additional Raney Ni (1.00 g with H2O) and stirred for additional 3 h. The mixture was filtered through celite carefully and the cake was washed with excess MeOH, keeping the filtrate cake wet. The filtrate liquid was concentrated, and the residue was purified by flash column chromatography (EtOAc/PE = 1:3→2:3) to yield lactam (24) (0.227 g, 84%) as a white solid: m.p. 155.7–157.0 °C; Rf = 0.65 (EtOAc/PE, 1:1); IR (KBr): 3323, 2927, 2858, 1640, 1544, 1459, 1253 cm–1; 1H NMR (400 MHz, CDCl3): δ = 5.66 (bs, 1H), 3.30 (s, 2H), 2.20 (s, 2H), 1.65 (s, 2H), 1.47 (s, 2H), 1.34 (s, 12H), 1.24 (s, 4H); 13C NMR (100 MHz, CDCl3): δ = 173.2, 38.5, 36.3, 28.4, 26.6, 25.9, 25.8, 25.7, 25.5, 25.4, 24.1, 23.8, 23.2; HRMS (ESI): m/z calcd for C13H25NO [M + H]+: 212.2009, found: 212.1983.
Azacyclotetradecane (25)
To a mixture of LiAlH4 (0.114 g, 3.00 mmol) in anhydrous THF (5 mL) was added compound 24 (0.210 g, 1.00 mmol) in anhydrous THF (4 mL), which was stirred at 65 °C under an atmosphere of N2 for 16 h. The mixture was diluted with THF (10 mL), quenched with Na2SO4·5H2O, and filtered through celite. The cake was washed with a mixture (MeOH/DCM, 1:20, 30 mL). The filtrate liquid was concentrated and purified by flash column chromatography (MeOH/DCM/NH3 = 10:100:1) to yield cyclic amine (25) (0.192 g, 97%) as a faint yellow waxlike solid: Rf = 0.60 (MeOH/DCM, 1:10); IR (KBr): 3429, 2926, 2857, 2808, 1460, 1133, 714 cm–1; 1H NMR (400 MHz, CDCl3): δ = 2.63–2.62 (m, 4H), 1.53 (m, 4H), 1.36–1.25 (m, 18H); 13C NMR (100 MHz, CDCl3): δ = 46.1, 27.2, 26.1, 25.2, 24.3, 23.8, 23.3; HRMS (ESI): m/z calcd for C13H27N [M + H]+: 198.2216, found: 198.2235.
Tert-Butyl (3-(azacyclotetradecan-1-yl)propyl)(3-((tert-butoxycarbonyl)amino)propyl) carbamate (26)
To a solution of amine 25 (0.168 g, 0.853 mmol) and aldehyde (15) (0.563 g, 1.70 mmol) in MeOH (5 mL) were added AcOH (1 drop) and NaBH(OAc)3 (0.947 g, 4.26 mmol) at room temperature and stirred for 1.5 h. Next, the mixture was poured into water (100 mL) and extracted with CHCl3 (15 mL × 6). The organic phases were dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography (MeOH/DCM = 1:100→1:20) to afford 26 (0.380 g, 87%) as a colorless amorphous solid: Rf = 0.70 (MeOH/DCM, 1:10); IR (KBr): 3424, 2931, 2862, 2568, 1694, 1479, 1389, 1250, 1173 cm–1; 1H NMR (400 MHz, CDCl3): δ = 5.19–4.96 (m, 1H), 3.26–3.25 (m, 4H), 3.08 (m, 4H), 2.91 (m, 4H), 2.15 (bs, 2H), 1.68–1.57 (m, 6H), 1.44–1.41 (m, 26H), 1.27 (s, 10H); 13C NMR (100 MHz, CDCl3): δ = 156.2, 80.4, 77.4, 52.6, 49.7, 44.2, 37.7, 28.5, 26.0, 25.2, 23.9, 23.7, 23.5, 23.3, 18.6; HRMS (ESI): m/z calcd for C29H57N3O4 [M + H]+: 512.4422, found: 512.4416.
N1-(3-(Azacyclotetradecan-1-yl)propyl)propane-1,3-diamine 2,2,2-trifluoroacetate (2)
To a solution of compound 26 (60 mg, 0.117 mmol) in DCM (5 mL) was added CF3COOH (1 mL) and stirred at room temperature for 2 h. The mixture was concentrated under reduced pressure, azeotroped with MeOH three times, and treated with vacuum to afford TFA salt of motuporamine B (2) (50 mg, 100%) as a white solid: m.p. 163.0–164.9 °C; Rf = 0.10 (MeOH/DCM, 1:20); IR (KBr): 3439, 3045, 2934, 2867, 1678, 1466, 1211, 1127 cm–1; 1H NMR (400 MHz, CD3OD): δ = 3.30–3.04 (m, 12H), 2.20–2.08 (m, 4H), 1.71–1.67 (m, 4H), 1.48–1.28 (m, 18H); 13C NMR (100 MHz, CD3OD): δ = 52.5, 51.4, 45.9, 37.7, 27.0, 26.3, 25.3, 25.1, 24.7, 24.1, 22.6, 20.3; HRMS (ESI): m/z calcd for C19H41N3 [M + H]+: 312.3373, found: 312.3396.
Acknowledgments
This work has been supported by the National Natural Science Foundation of China (21971042). The authors are grateful to G. Tang for acquiring high-resolution mass spectrometry data.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c05484.
Compilation of copies of the NMR spectra of all synthetic intermediates and products (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Mudit M.; El Sayed K. A. Cancer control potential of marine natural product scaffolds through inhibition of tumor cell migration and invasion. Drug Discovery Today 2016, 21, 1745–1760. 10.1016/j.drudis.2016.06.032. [DOI] [PubMed] [Google Scholar]
- Althagbi H. I.; Alarif W. M.; Al-Footy K. O.; Abdel-Lateff A. Marine-Derived Macrocyclic Alkaloids (MDMAs): Chemical and Biological Diversity. Mar. Drugs 2020, 18, 368 10.3390/md18070368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D. E.; Lassota P.; Andersen R. J. Motuporamines A–C, Cytotoxic Alkaloids Isolated from the Marine Sponge Xestospongia exigua (Kirkpatrick). J. Org. Chem. 1998, 63, 4838–4841. 10.1021/jo980355p. [DOI] [PubMed] [Google Scholar]
- Williams D. E.; Craig K. S.; Patrick B.; McHardy L. M.; van Soest R.; Roberge M.; Andersen R. J. Motuporamines, Anti-Invasion and Anti-Angiogenic Alkaloids from the Marine Sponge Xestospongia exigua (Kirkpatrick): Isolation, Structure Elucidation, Analogue Synthesis, and Conformational Analysis. J. Org. Chem. 2002, 67, 245–258. 10.1021/jo016101c. [DOI] [PubMed] [Google Scholar]
- Borselli D.; Blanchet M.; Bolla J.-M.; Muth A.; Skruber K.; Phanstiel O.; Brunel J. M. Motuporamine Derivatives as Antimicrobial Agents and Antibiotic Enhancers against Resistant Gram-Negative Bacteria. ChemBioChem 2017, 18, 276–283. 10.1002/cbic.201600532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHardy L. M.; Sinotte R.; Troussard A.; Sheldon C.; Church J.; Williams D. E.; Andersen R. J.; Dedhar S.; Roberge M.; Roskelley C. D. The Tumor Invasion Inhibitor Dihydromotuporamine C Activates RHO, Remodels Stress Fibers and Focal Adhesions, and Stimulates Sodium-Proton Exchange. Cancer Res. 2004, 64, 1468–1474. 10.1158/0008-5472.CAN-03-2733. [DOI] [PubMed] [Google Scholar]
- Roskelley C. D.; Williams D. E.; McHardy L. M.; Leong K. G.; Troussard A.; Karsan A.; Andersen R. J.; Dedhar S.; Roberge M. Inhibition of Tumor Cell Invasion and Angiogenesis by Motuporamines. Cancer Res. 2001, 61, 6788–6794. [PubMed] [Google Scholar]
- Baldwin J. E.; Vollmer H. R.; Lee V. Total synthesis of cytotoxic sponge alkaloids motuporamines A and B. Tetrahedron Lett. 1999, 40, 5401–5404. 10.1016/S0040-4039(99)01016-3. [DOI] [Google Scholar]
- Goldring W. P. D.; Weiler L. Cytotoxic Alkaloids Motuporamines A–C: Synthesis and Structural Verification. Org. Lett. 1999, 1, 1471–1473. 10.1021/ol991029e. [DOI] [PubMed] [Google Scholar]
- Deiters A.; Mrtin S. F. Synthesis of Oxygen- and Nitrogen-Containing Heterocycles by Ring-Closing Metathesis. Chem. Rev. 2004, 104, 2199–2238. 10.1021/cr0200872. [DOI] [PubMed] [Google Scholar]
- Weston M. H.; Nakajima K.; Back T. G. Tandem Conjugate Additions and 3-Aza-Cope Rearrangements of Tertiary Allyl Amines and Cyclic α-Vinylamines with Acetylenic Sulfones. Applications to Simple and Iterative Ring Expansions Leading to Medium and Large-Ring Nitrogen Heterocycles. J. Org. Chem. 2008, 73, 4630–4637. 10.1021/jo800600a. [DOI] [PubMed] [Google Scholar]
- Zhou L.-J.; Li Z.-M.; Zou Y.; Wang Q.-R.; Sanhueza I. A.; Schoenebeck F.; Goeke A. Tandem Nucleophilic Addition/Oxy-2-azonia-Cope Rearrangement for the Formation of Homoallylic Amides and Lactams: Total Synthesis and Structural Verification of Motuporamine G. J. Am. Chem. Soc. 2012, 134, 20009–20012. 10.1021/ja310002m. [DOI] [PubMed] [Google Scholar]
- Mu W.-B.; Zhou L.-J.; Zou Y.; Wang Q.-R.; Goeke A. Irreversible Oxy-2-azonia-Cope Rearrangements for the Synthesis of Functionalized Allyl α-Amino Acid Derivatives. Eur. J. Org. Chem. 2014, 2014, 2379–2385. 10.1002/ejoc.201301818. [DOI] [Google Scholar]
- Kitsiou C.; Hindes J. J.; I’Anson P.; Jackson P.; Wilson T. C.; Daly E. K.; Felstead H. R.; Hearnshaw P.; Unsworth W. P. The Synthesis of Structurally Diverse Macrocycles By Successive Ring Expansion. Angew. Chem., Int. Ed. 2015, 54, 15794–15798. 10.1002/anie.201509153. [DOI] [PubMed] [Google Scholar]
- Baud L. G.; Manning M. A.; Arkless H. L.; Stephens T. C.; Unsworth W. P. Ring-Expansion Approach to Medium-Sized Lactams and Analysis of Their Medicinal Lead-Like Properties. Chem. – Eur. J. 2017, 23, 2225–2230. 10.1002/chem.201605615. [DOI] [PubMed] [Google Scholar]
- Stephens T. C.; Unsworth W. P. Consecutive Ring-Expansion Reactions for the Iterative Assembly of Medium-Sized Rings and Macrocycles. Synlett 2020, 31, 133–146. 10.1055/s-0037-1611500. [DOI] [Google Scholar]
- Stephens T. C.; Lodi M.; Steer A. M.; Lin Y.; Gill M. T.; Unsworth W. P. Synthesis of Cyclic Peptide Mimetics by the Successive Ring Expansion of Lactams. Chem. – Eur. J. 2017, 23, 13314–13318. 10.1002/chem.201703316. [DOI] [PubMed] [Google Scholar]
- Rathke M. W.; Cowan P. J. Procedures for the acylation of diethyl malonate and ethyl acetoacetate with acid chlorides using tertiary amine bases and magnesium chloride. J. Org. Chem. 1985, 50, 2622–2624. 10.1021/jo00215a003. [DOI] [Google Scholar]
- Carpino L. A. The 9-fluorenylmethyloxycarbonyl family of base-sensitive amino-protecting groups. Acc. Chem. Res. 1987, 20, 401–407. 10.1021/ar00143a003. [DOI] [Google Scholar]
- Carpino L. A.; Sadat-Aalaee D.; Beyermann M. Tris(2-aminoethyl)amine as substitute for 4-(aminomethyl)piperidine in the FMOC/polyamine approach to rapid peptide synthesis. J. Org. Chem. 1990, 55, 1673–1675. 10.1021/jo00292a050. [DOI] [Google Scholar]
- Fürstner A.; Rumbo A. Ring-Closing Alkyne Metathesis. Stereoselective Synthesis of the Cytotoxic Marine Alkaloid Motuporamine C. J. Org. Chem. 2000, 65, 2608–2611. 10.1021/jo991944d. [DOI] [PubMed] [Google Scholar]
- Silveira A. Jr.; Angelastro M.; Israel R.; Totino F.; Williamsen P. A new synthesis of cyclic allenic esters. J. Org. Chem. 1980, 45, 3522–3523. 10.1021/jo01305a034. [DOI] [Google Scholar]
- Brannock K. C.; Burpitt R. D.; Goodlett V. W.; Thweatt J. G. Enamine Chemistry. VI. Reactions with Propiolates. J. Org. Chem. 1963, 29, 818–823. 10.1021/jo01027a011. [DOI] [Google Scholar]
- Armarego W. L. F.; Chai C. L. L.. Purificaion of Laboratory Chemicals, 5th ed.; Elsevier Science: USA, 2003. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.