

Summary
Neurodegeneration is characterized by gradual onset and limited availability of specific biomarkers. Apart from various aetiologies such as infection, trauma, genetic mutation, the interaction between the immune system and CNS is widely associated with neuronal damage in neurodegenerative diseases. The immune system plays a distinct role in disease progression and cellular homeostasis. It induces cellular and humoral responses, and enables tissue repair, cellular healing and clearance of cellular detritus. Aberrant and chronic activation of the immune system can damage healthy neurons. The pro‐inflammatory mediators secreted by chief innate immune components, the complement system, microglia and inflammasome can augment cytotoxicity. Furthermore, these inflammatory mediators accelerate microglial activation resulting in progressive neuronal loss. Various animal studies have been carried out to unravel the complex pathology and ascertain biomarkers for these harmful diseases, but have had limited success. The present review will provide a thorough understanding of microglial activation, complement system and inflammasome generation, which lead the healthy brain towards neurodegeneration. In addition to this, possible targets of immune components to confer a strategic treatment regime for the alleviation of neuronal damage are also summarized.
Keywords: complement system, immune system, inflammasome, microglia, neurodegenerative disease
The present review aimed to provide a thorough information to the readers about microglial activation, complement system and inflammasome generation. Further, we discuss how the processes involved in these system lead the healthy brain towards neurodegeneration. Also discussed are the possible targets of immune components to confer a strategic treatment regime for the alleviation of neuronal damage.

Abbreviations
- AD
Alzheimer's disease
- PD
Parkinson's disease
- ALS
amyotrophic lateral sclerosis
- HD
huntington disease
- MS
multiple sclerosis
- SOD
superoxide dismutase
- mTT
mutant huntingtin
- CP
classical pathway
- LP
lectin pathway
- AP
alternative pathway
- CNS
central nervous system
- Fc
fragment crystallizable region
- MBL
mannan‐binding lectin
- MASPs
MBL‐associated serine proteases
- MAC
membrane attack complex
- TNF‐α
tumour necrosis factor
- IL‐1β
interleukin‐1β
- ROS
reactive oxygen species
- H2O2
hydrogen peroxide
- TLR
Toll‐like receptor
- IRAK
IL‐1R‐associated kinase
- NLR
NOD‐like receptor
- ASC
apoptosis‐associated spec‐like protein
- PYD
N‐terminal pyrin domain
- CARD
caspase recruitment domain
- NOD
nucleotide‐binding oligomerization domain
- LRR
leucine‐rich repeats
- GSDMD
gasdermin‐D
- DAMPs
damage‐associated molecular pattern
- PAMPs
pathogen‐associated molecular pattern
- PKR
protein kinase R
- AIM2
absent in melanoma 2
- Aβ
amyloid‐beta
- SNPs
single nucleotide polymorphisms
- CR1
complement receptor 1
- CLU
clusterin
- IHC
immunohistochemistry
- TCC
terminal C5b‐9 complement complex
- DAP12
DNAX‐activating protein of 12 kD
- TREM‐2
triggering receptor expressed on myeloid cells 2
- SN
substantia nigra
- Nox2
NADPH oxidase
- CSF
cerebrospinal fluid
- α‐syn
α‐synuclein
- MMP
metalloproteinase
- SIGLEC
sialic acid‐binding Ig‐like lectin
- PET
positron emission tomography
- LRRK2
leucine‐rich repeat kinase
- EGF
epidermal growth factor
- IDE
insulin‐degrading enzyme
- ATG7
autophagy‐related protein 7
- mAbs
monoclonal antibodies
- BBB
blood–brain barrier
- PPAR‐γ
peroxisome‐proliferated receptor gamma
- MAPK
mitogen‐activated protein kinase
- LFA‐1
lymphocyte function‐associated molecule‐1
- LCA
leucocyte common antigen
- CSF
colony‐stimulating factor
- MAIT
mucosal‐associated invariant T
- TRIF
TIR‐domain‐containing adaptor‐inducing interferon‐β
- FOXP3
forkhead box transcription factor
INTRODUCTION
Neuroinflammation is a hallmark of the vicious cycle of neurodegeneration. 1 It is responsible for the development and gradual progression of neurodegenerative conditions such as Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), huntington disease (HD) and multiple sclerosis (MS). It is also an obvious consequence of cerebral ischaemia. 2 These proteinopathies show ramification of organelles and synaptic impairment of nervous system. 2 An epidemiological study carried out in 2016 revealed that about 276 million people were suffering from neurological disorders worldwide, and among them, every year, the extent of fatality was about 9 million people. 3 , 4 As dementia is one of the outcomes of neurological disorders, the estimated count of a global dementia patient is around 35 million, and if compelling measures are not identified, it will rise threefold by 2050. 5 Neurodegenerative diseases affect both the immune system and the central nervous system. 6 The immune system can also be constructive as it triggers repair of the damaged tissues, neurotrophic factor production and remyelination in response to toxins, trauma and injury, thus maintaining the functional integrity of the brain. During neurodegeneration, T cells, microglia, astrocytes, oligodendrocytes, inflammasomes and the complement system can induce neuroinflammation. 7 , 8 Microglia and inflammasomes act as a host defence mechanism against infections and dysfunctional neurons. 9 , 10 However, aggregation of amyloid‐β, α‐synuclein, superoxide dismutase (SOD), mutant huntingtin (mHtt), etc. can cause aberrant activation of microglia, complement system and inflammasomes, which promotes the release of inflammatory cytokines leading to neuroinflammation. 11 , 12 The interplay between these two systems is of interest, albeit the mechanism is yet to be explored. 13 The present review focuses on the involvement of the immune system in regulating neurodegeneration and the available therapeutic approaches. Further, the detailed mechanism of immune component activation and how they contribute towards developing the pathology has been dealt with an elaborative summary of various therapeutic approaches of targeting immune components in neurodegenerative diseases.
COMPLEMENT SYSTEM
The complement system is an element of the body's innate immunity whose components are primarily synthesized in the liver and then enter into systemic circulation for activation. 14 , 15 These components cannot cross the blood–brain barrier; hence, neurons and glial cells synthesize them locally. 16 The complement system consists of more than 30 fluid phases to destroy pathogens and foreign cells. 17 The three pathways of complement system activation, that is classical (CP), lectin (LP) and alternative (AP), are mainly involved in protection against microbial infections, the connection of innate and adaptive immunity, and clearance of the debris of immune products. 18 , 19
Complement system in CNS
The complement system helps in maintaining homeostasis in the brain by acting against infections and by removal of waste products. 20 The classical pathway of the complement system starts with component C1q, which binds with the fragment crystallizable region (Fc region) of antibody. 21 Microglia is the primary source of C1q, which stimulates C1r and cleaves C1s to activate C1. 21 , 22 Activated C1 binds with C4 and cleaves it into C4a and C4b. C4b attaches itself to the surface of the pathogen and activates C2a and C2b. C2a and C4b together form C3 convertase (C4b2a and C3bBb). 23 The mannan‐binding lectin (MBL) pathway attaches to the carbohydrates of the pathogen and activates MBL‐associated serine proteases (MASPs). MASP‐2 splits C4 and C2 to form C3 convertase. 24 The other complement system pathway, that is the alternative pathway, is an independent antibody pathway responsible for distinguishing between self and non‐self. Hydrolysis of native C3 leads to activation of C3(H2O), which associates with factor B (serine protease, which stimulates B cell to instigate inflammatory responses) and gives rise to C3 convertase. 25 Properdin can directly activate and amplify AP and is also accepted as a recognition factor for AP. 26 C3 convertase in all three pathways then splits into C3a and C3b. C3b attaches to C3 convertase to give rise to C5 convertase, which then activates membrane attack complex (MAC), resulting in phagocytosis. 27 However, this activation of the complement system is highly specific. 28
ROLE OF MICROGLIA IN NEURODEGENERATION
Microglia are the resident macrophages of the CNS, which play an important role in maintaining host defence and at times can be detrimental to neurons. 29 These have self‐renewal properties and are responsible for eliciting immunological responses in the CNS. 30 As per different neurological disorders, it undergoes proliferation and configurational changes to become reactive, which is known as microgliosis. 31 These reactive microglia, on the one hand, stimulate the release of neurotrophic factors and guide stem cells towards injury. 32 On the other hand, chronic activation of microglia secrets pro‐inflammatory mediators such as tumour necrosis factor (TNF‐α), interleukin‐1β (IL‐1β) and reactive oxygen species (ROS), hydrogen peroxide (H2O2), hydroxyl radicals (OH−) and superoxide anions (O2−), which are detrimental for neurons. 33 , 34 , 35 Once microglia are activated by Toll‐like receptor (TLR), they stimulate the adaptor protein MyD88, which promotes autophosphorylation of IL‐1R‐associated kinase (IRAK) and releases pro‐inflammatory cytokines. 36 Furthermore, it promotes the generation of ROS, leading to neuroinflammation. 37 In addition to this, microgliosis also involved in the synaptic loss in diseases such as AD and PD, which correlates with the gradual progression of dementia. 38 It has been observed that M1 microglia releases pro‐inflammatory proteins such as IL‐6, TNF‐α and IL‐1β at 24 hours post‐stroke, which cause secondary injuries a few days afterwards. Unlike this, M2 microglia is anti‐inflammatory and reported to be increased and proliferated in the core region at the same time. Modulation of TLRs or IFN‐γ causes activation of pro‐inflammatory M1 phenotype, whereas anti‐inflammatory M2 phenotype is activated by regulatory factors such as IL‐4, IL‐10, IL‐13 and TGF‐β 39 (Figure 1).
FIGURE 1.
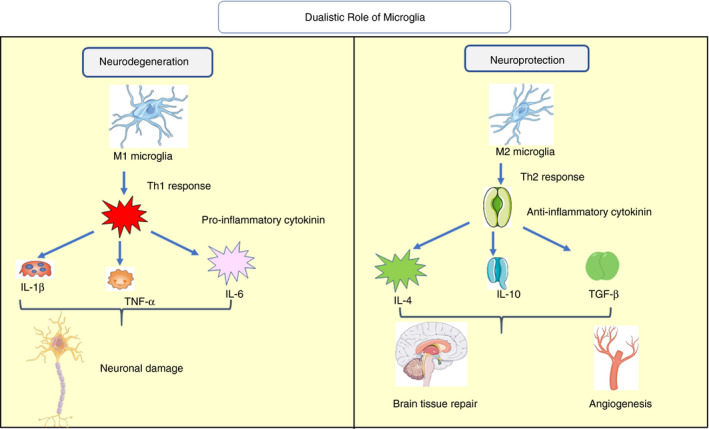
Dualistic role of microglia on the nervous system. M1 microglia produces pro‐inflammatory cytokines such as IL‐1β, TNF‐α and IL‐6 through Th‐1 response and leads to neurodegeneration. M2 microglia produces anti‐inflammatory cytokines such as IL‐4, IL‐10 and TGF‐β via Th2 response. These anti‐inflammatory cytokines help in brain tissue repair, angiogenesis and, thus, providing neuroprotection (IL – interleukin, TNF‐α – tumour necrotic factor‐alpha, TGF‐β – transforming growth factor‐beta, Th1 – T helper cell 1) (Adapted from Servier Medical Art by Servier, licensed under a Creative Commons Attribution 3.0 Unported Licence (http://smart.servier.com/))
INFLAMMASOME‐MEDIATED INFLAMMATION
The inflammasome is a complex protein that is responsible for the inflammation‐mediated response. The three chief components of inflammasomes are NOD‐like receptors (NLRs), apoptosis‐associated spec‐like protein (ASC) and pro‐caspase‐1. 40 NLRs, a tripartite structure, include N‐terminal pyrin domain (PYD) or caspase recruitment domain (CARD), NACHT or NOD (nucleotide‐binding oligomerization domain), and C‐terminal leucine‐rich repeats (LRRs). 40 Neurodegenerative diseases activate the NOD‐like receptor (NLR) pyrin domain containing 1 and 3 (NLRP1 and NLRP3) inflammasome, ASC and pro‐caspase‐1 via either MAPK or NF‐κB pathway. NLRP3 contributes to inflammation and injury in CNS and in neurodegenerative diseases such as AD, PD, MS and ALS, whereas both NLRP1 and NLRP3 play a role in the pathogenesis of ischaemic stroke. 41 Upon activation, the interaction of NLRP3 protein with ASC followed by the interaction of ASC with the CARD domain via PYD recruits pro‐caspase‐1 and forms the NLRP3–ASC–pro‐caspase‐1 complex (NLRP3 inflammasome). 42 It leads to the conversion of inactive pro‐inflammatory cytokines (pro‐IL‐1β, pro‐IL‐18) into active inflammatory cytokines (IL‐1β, IL‐18) and activation of caspase‐1 that ultimately cause neuroinflammation and pyroptosis. 43 Pyroptosis is mediated by producing gasdermin‐D (GSDMD), which is responsible for producing pores on the plasma membrane. 44 Damage‐associated molecular patterns (DAMPs) and pathogen‐associated molecular patterns (PAMPs) such as energy depletion, ion flux (Ca2+ influx, Na+ influx, K+ efflux, and Cl− efflux), cathepsin release, oxidized mitochondrial DNA release, PKR (protein kinase R) activation and ROS could activate inflammasome signalling 45 , 46 (Figure 2).
FIGURE 2.
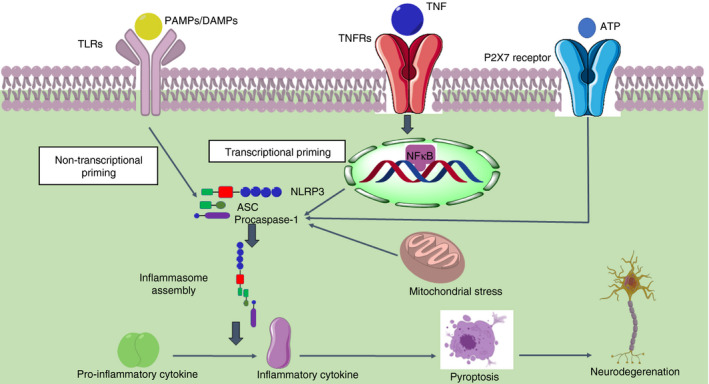
Inflammasome‐mediated neurodegeneration. PAMPs or DAMPs activate TLR and mediate non‐transcriptional priming of the inflammasome. TNF activates the TNF receptor and increases the expression of NLRP3, ASC and pro‐caspase‐1 via NF‐κB signalling. It also leads to the priming of inflammasome, which ultimately contributes to inflammasome assembly. It will further activate pro‐caspase‐1 into caspase‐1, which converts the pro‐inflammatory cytokines (pro‐IL‐1β, pro‐IL‐18) into inflammatory cytokines (IL‐1β, IL‐18). P2X7 receptor and mitochondrial stress also activate the inflammasome assembly. It will ultimately lead to pyroptosis and, thus, neurodegeneration (PAMPs – pathogen‐associated molecular pattern, DAMPs – damage‐associated molecular pattern, TNF – tumour necrosis factor, NLRP3 – nucleotide‐binding domain, leucine‐rich repeat‐containing receptor (NLR) family pyrin domain containing 3, ASC – apoptosis‐associated speck‐like protein containing CARD, NF‐κB – nuclear factor kappa‐light‐chain‐enhancer of activated B cells, IL – interleukin, P2X7‐P2 – purinergic receptor 2) (Adapted from Servier Medical Art by Servier, licensed under a Creative Commons Attribution 3.0 Unported Licence (http://smart.servier.com/))
The immune response is a harmonized interplay of potential threat recognition by the innate immune system initially and response by the adaptive immune system afterwards. 13 , 47 Studies support that pattern recognition receptor (PRR) not only is involved in recognition of DAMPs and PAMPs, but it also informs and influences the adaptive immune response via TLRs. 48 PRRs include (nucleotide‐binding oligomerization domain, leucine‐rich repeat and pyrin containing domain) NLRP‐1, NLRP‐3 NLRC‐4 and absent in melanoma 2(AIM2). 49 , 50 Microglia express these PRRs whose increased expression causes inflammasome signalling activation. 51
DESTRUCTION OF IMMUNE MACHINERY IN ALZHEIMER'S DISEASE AND PARKINSON'S DISEASE
Complement system: Associated pathways of activation
Alzheimer's disease (AD) is a neurodegenerative tauopathy that manifests as dementia and affects more than 6% of people older than 65 years. 18 , 52 AD is characterized by the formation of neurofibrillary tangles consisting of hyperphosphorylated tau and deposition of insoluble amyloid‐beta (Aβ) plaque. In genomic studies, it has been identified that single nucleotide polymorphisms (SNPs) of genes encoding complement protein complement receptor 1 (CR1) and clusterin (CLU) are associated with the risk of late‐onset AD. 53 , 54 CR1, a transmembrane protein that increases the phagocytosis of C3b, C4b and C1q, opsonized particles. 55 Interaction between CLU and Aβ prevents proteolytic degradation of oligomers leading to plaque formation. 56 The co‐localization of the classical pathway proteins (C1q, C3 and C4) with neurofibrillary tangles, amyloid fibrils and Aβ deposits, particularly in the hippocampus, temporal cortex and amygdala, has been observed in Immunohistochemistry (IHC) study of post‐mortem AD brains. 55 The terminal pathway marker of the complement system, terminal C5b‐9 complement complex (TCC), is predominantly present in the cortex of the AD brain, which is associated with the formation of neurofibrillary tangles and aggregated Aβ. 18 , 57 Dystrophic neurites in Aβ plaque and neurofibrillary tangles in neurons of the frontal cortex and hippocampus of AD brain express C5aR1 and C5L2 receptors predominantly. C5a fragment, generated by complement activation, is a chemotactic factor for glia. 58 C5a–CD88 (C5aR1) interaction has a detrimental effect in AD either by direct impact on a neuron or by indirectly activating the microglia, whereas the anti‐inflammatory effect of activated C5L2 receptor is reported. 58
Accumulation of toxic Aβ leads to activation of the complement system by secretion of C1q from neurons and the production of C3 by astrocytes. 14 C1q binds to Aβ plaque and C1q receptor (C1qR) on microglia, thus leads to synaptic pruning and initiates phagocytosis. 59 CD14, CD36, CD47, α6β1 integrins, TLRs and RAGE receptors of microglia get activated by soluble Aβ oligomer and Aβ fibrils, which in turn instigate phagocytosis. 59 Binding of Aβ oligomers to astrocytic TLR4 or CD36 receptors leads to the production of inflammatory cytokines or chemokines by activating NF‐κB signalling. 60 Furthermore, this activated signalling cascade along with microglial post‐phagocytic processes (lysosomal injury, acidification of cytosol, etc.) contributes to the activation of NLRP‐3 inflammasome. 61 C3 is further cleaved into C3a and C3b that ultimately produce MAC. C3b along with C3a contributes to the activation of DNAX‐activating protein of 12 kD (DAP12), which stabilizes the triggering receptor expressed on myeloid cells 2 (TREM‐2) to modulate microglial function leading to neuroinflammation. 61
In the case of Parkinson's disease, the presence of Lewy bodies and melanized neurons in the substantia nigra (SN) is responsible for the activation of the classical complement system. 62 These depositions show positive staining in IHC for early complement activation C3b, C4d and C7 and late activation of C9. 63 Later on, positive staining in IHC of microglia‐derived C1q and C5a levels was also found in SN. 64 Earlier studies have been reported that C1q also helps in removal of aggregated neuromelanin in the SN. 65 Lewy bodies and oligodendrocytes show MAC activation, which leads to phagocytosis in the SN in PD. 66 Microglial NADPH oxidase (Nox2) is stimulated by CR3, which suggests the role of the complement system in neuroinflammation‐mediated loss of dopaminergic neurons. 67 Increased neuroinflammation in PD is marked by elevated C3 and factor H (FH) along with reduced Aβ42 in cerebrospinal fluid (CSF), which helps to correlate with cognitive and motor dysfunction. 68 In contrast to this study, Rozemuller et al 69 found negative double staining for α‐synuclein (α‐syn) and components of complement, suggesting that reactive microglia and the complement system are not involved in the formation of Lewy body in the cortex of PD patients.
Implication of microglia
In AD, the structure of Aβ oligomer is sensed by the microglia as DAMP and they are morphologically altered to the ‘reactive’ or ‘primed’ microglia state. 13 Activation of microglia has both degenerative and protective effects in AD. 13 On a negative aspect, activation of microglia can produce pro‐inflammatory cytokines such as IL‐1β, IL‐6 and TNF‐α that act on the cholinergic neurons and induce apoptosis. 70 Conversely, it can also produce a proteolytic enzymes such as insulin‐degrading enzyme (IDE), matrix metalloproteinases (MMPs) and neprilysin to degrade Aβ plaque to provide neuroprotection. Phagocytic function of microglia is inhibited by a mutation in the TREM‐2 and CD33 protein 71 . TREM is a protein of immunoglobulin (Ig) superfamily, which is expressed in macrophages, microglia and dendritic cells of the brain, whereas CD33 is a cell‐surface protein of the sialic acid‐binding Ig‐like lectin (SIGLEC) family, which majorly expressed in myeloid and lymphoid cells. 72 TREM‐2 is involved in cell survival, phagocytosis, proliferation and production of inflammatory cytokines. 73 It has been observed in various studies that the risk of AD gets increased up to threefold to fourfold due to arginine to histidine mutation (R47H mutation) of TREM‐2, which results in functional loss of the protein. 74 Other mutant variants of TREM‐2 such as D87N, T96K and R62H have also been associated with AD generation. 75 , 76
Progression of PD is the result of the infiltration of lymphocytes from the periphery to CNS, excessive activation of microglial cells and astrogliosis. 77 Researchers have observed early and prolonged activation of microglia within the territory of damaged neuronal death using positron emission tomography (PET) scanning. 78 Reactive microglia phagocyte aggregated α‐syn and led to stimulation of Nox2 and generation of ROS to elevate dopaminergic neurodegeneration in SN. 79 Aggregation of α‐syn enables the upregulation of leucine‐rich repeat kinase (LRRK2) along with activation of iNOS and CD68 through macroglia and the invading peripheral cells. 80 Activated microglia facilitate the release of pro‐inflammatory cytokines such as interleukin‐1β (IL‐1β), IL‐23, IL‐12, epidermal growth factor (EGF) and TNF‐α and chemokines such as CCL5, CCL2 and CXCL10 in the striatum, CSF and peripheral blood 81 , 82 , 83 (Figure 3). Interferon‐gamma and TNF‐α are also stimulated in the SN. 84 Increased levels of TNF‐α in serum lead to four times of exacerbation in cognitive decline, which gives an idea about the role of inflammatory markers in cognitive function. 85 The release of IL‐1β impairs the spatial navigational learning and cognitive functions in female C57BL/6 mice. 86 These cytokines then trigger iNOS and COX‐2, which results in the generation of ROS. 87 , 88 A massive release of ROS leads to oxidative stress, which is responsible for aggravating neurodegeneration. 89
FIGURE 3.
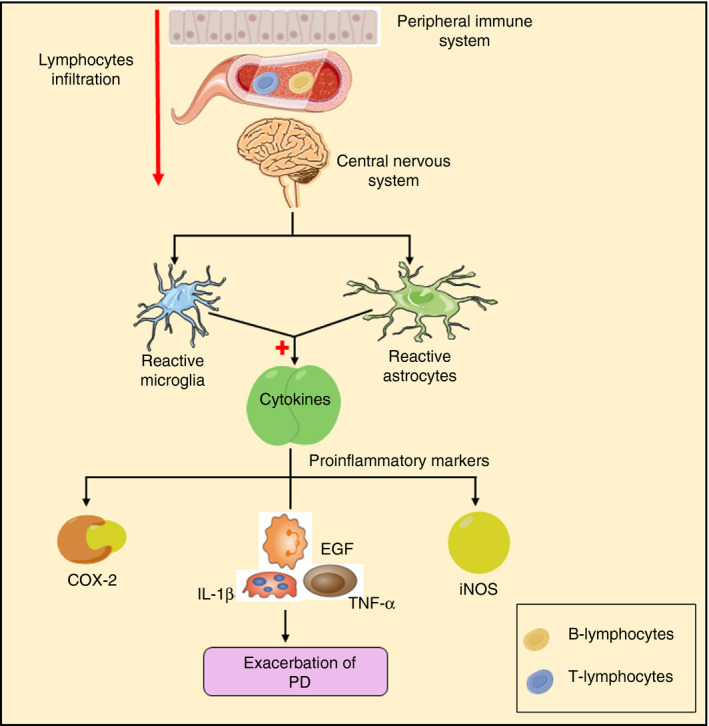
Microglial mediated neuroinflammation in PD. Infiltration of lymphocytes from the peripheral immune system to the central nervous system and aggregation of α‐syn stimulate the activation of the microglial cell, which is accompanied by the release of ROS and NO along with inflammatory markers such as IL‐1β, IL‐18, TNF‐α, EGF and COX‐2. All these inflammatory markers result in neuronal cell death (IL – interleukin, TNF‐α – tumour necrosis factor‐α, EGF – epidermal growth factor, COX‐2 – cyclooxygenase‐2, ROS – reactive oxygen species, NO – nitric oxide) (Adapted from Servier Medical Art by Servier, licensed under a Creative Commons Attribution 3.0 Unported Licence (http://smart.servier.com/))
Inflammasome activation
AD is the most studied disease for understanding inflammasome signalling. 90 , 91 Excessive generation of Aβ leads to its oligomerization and subsequently senile plaque formation. These Aβ oligomeric or fibrillar form can act as DAMPs to activate inflammasome signalling. 90 In experiments with AD patients, it has been observed that activation of NLRP3 inflammasome is an early‐stage event rather than a late‐stage event. 92 Another clinical study on AD has shown the release of IL‐18 and IL‐1β and coexpression of ASC, NLRP3 and caspase‐1 in the monocyte. 93 Fibrillar Aβ stimulates microglia to release IL‐1β based on the interaction of NLRP3 and ASC, whereas soluble Aβ peptides activate NLRP3 inflammasome majorly through CD36. 93 NLRP3 inflammasome activation in response to Aβ can also be regulated by autophagy‐related protein 7 (ATG7). Deficiency of cellular ATG7 causes increased cleavage of caspase 1, the formation of speck by ASC, and the release of IL‐1β in microglia and BV2 cell (alternative model for primary microglial culture). Moreover, microglia, which are deficient in ATG7, cause a more remarkable loss of neuronal dendrites suggesting the activation of microglial inflammasome to limit neuronal destruction. 94
Following uptake of Aβ in the astrocytes, IL‐1β is produced through inflammasome signalling. 95 According to Liu et al, 90 palmitate increases NLRC4 inflammasome activation in astrocytes of rat and causes maturation of IL‐1β. In an IL‐1 and MyD88 knockout rat model, microglia were not activated. This suggests that Aβ activates MYD88, which results in activation of NLRP3 inflammasome and ultimately cleavage of caspase 1.
In PD, aggregation of α‐syn stimulates TLR, which further contributes to the assembly of NLRP3 inflammasome and conversion of its downstream marker pro‐IL‐1β to IL‐1β. 96 Fyn kinase enables the transfer of α‐syn to microglia via PKC‐delta‐dependent NF‐κB‐p65 nuclear translocation. This transfer further contributes to mitochondrial ROS generation and an increase in the level of cathepsin B, which therefore activates NLRP3 inflammasome 97 , 98 . Apart from oxidative stress, mitochondrial DNA disruption can stimulate pro‐inflammatory mediators to activate NLRP3 inflammasome. 99 Lee et al 100 found that microglial NLRP3 inflammasome activation in mouse model of MPTP‐induced PD is essential for the motor deficit and dopaminergic neuronal loss. Elevated levels of IL‐1β were also found in CSF and blood of the PD patients. 101 In addition to this, increased NLRP3, caspase‐1 and IL‐1β were detected in transgenic α‐syn‐A53T overexpressed mouse model of PD. 12 The serine–threonine kinase cyclin‐dependent kinase 5 (CDK‐5) is responsible for phosphorylating NLRP, ASC and POP1 ASC‐2, which ultimately leads to inflammasome activation. 102
Immunotherapeutics for age‐related neurodegenerative diseases
Hitherto, FDA‐approved drugs for symptomatic treatment targeting Aβ and Tau tangle are, donepezil, galantamine, rivastigmine and AADvac1 (a vaccine developed by Axon Neuroscience (NCT02579252), currently in clinical trial phase II) are in practice. 103 Now, targeting immune cells such as microglia and macrophages for modulation of neuroinflammation is of great interest. Aβ plaque burden can also be reduced by injections of anti‐Aβ monoclonal antibodies (mAbs) into the systemic circulation. 103 , 104 , 105 , 106 Tyrosine kinase inhibitors, which are currently in phase III clinical trial, can inhibit mast cell differentiation and degranulation, exhibiting beneficial effects as an adjunct with conventional therapy (NCT01872598). Endocannabinoids inhibit the neuronal microglial activation by promoting the receptor interaction of CD200. 107
Vaccination
In April 2020, Elan/Wyeth's group is planning to launch a dose‐escalation, multiple‐dose, randomized and double‐blind phase II clinical trial for the AN1792 vaccine (NCT00021723) against AD. It comprises QS21 and preaggregated Ab1–42 as an adjuvant. 108 Recently, a phase I clinical trial on active vaccine CAD106 was launched by Novartis Pharmaceuticals (NCT00411580). This vaccine targets the small Aβ fragment (Aβ 1‐6), which is a B‐cell epitope. 109 It showed marked improvement of cognitive behaviour in phase I trials, but the result of phase II clinical trial is yet to be disclosed (NCT02565511). Janssen and Pfizer conducted a phase II clinical trial on ACC‐001 (NCT01284387). It also targets Aβ(1–6) fragment, which is coupled to a carrier protein and the adjuvant QS‐21, a surface‐active saponin. 110
Passive immunization using monoclonal antibody is the recent trend seen in immunotherapy, in which administration of exogenous anti‐Aβ monoclonal antibody is performed without the chance of Th1‐mediated antibody production. 111 Janssen/Pfizer conducted phase II clinical trial on bapineuzumab AAB‐001 (NCT00606476) and bapineuzumab AAB‐003, 112 while Eli Lilly conducted phase III clinical trial on solanezumab (NCT02760602). 111 However, these drugs failed to show any significant clinical improvement in phase III trial. The limitation of passive immunization is the unavailability of appropriate antigen targets, repeated injections in chronic diseases, haemorrhagic risk, blood‐brain barrier (BBB) penetration, high costs and generation of the immune response against injected antibodies.
In PD, immunotherapeutics such as peroxisome‐proliferated receptor gamma (PPAR‐γ) agonists such as fenofibrate help to reduce inflammation and oxidative stress, and simultaneously elevate the dopamine level. 113 NSAIDs such as aspirin, an irreversible COX‐1 inhibitor, decrease inflammation and oxidative stress by elevation of lipoxin. 114 Statins inhibit TNF‐α, IL‐1β and IL‐6 and lead to a decline in inflammation. 115 In an experiment by Gordon et al., it has been observed that daily oral administration of MCC950 (20 mg/kg) can inhibit NLRP3 inflammasome‐mediated caspase‐1 activation in striatum. 101 Han et al. 116 revealed that kaempferol inhibits the formation of the NLRP3 inflammasome by activating autophagy in microglia, which further reduced the NLRP3 protein expression. Bushen‐Yizhi formula suppresses NLRP3 inflammasome activation in the SN‐ and MPP+‐stimulated BV‐2 microglia cell lines. 117 PMX‐205, the antagonist of C5a, protects against dopaminergic neurodegeneration. Anti‐α‐synuclein immunotherapy not only can be a promising approach, but it acts by inhibiting the movement of extracellular α‐synuclein to other neurons. 118 Other vaccinations such as 9E4 (humanized mouse monoclonal antibody) and AFF‐1 (short peptides–AFFITOPEs) minimize the aggregation of calpain cleaved α‐synuclein and oligomers of α‐synuclein, respectively. 13 There are many limitations regarding the development of immunotherapy, one of them is the direct administration of α‐synuclein antibody to the brain, which hinders α‐synuclein homeostasis. Moreover, these clinical trials are costly and take a longer duration to develop new approaches for synucleinopathy 119 (Table 1).
TABLE 1.
Immunotherapies for various neurodegenerative diseases
| Sr. No. | Disease | Target | Treatment | |
|---|---|---|---|---|
| Approved | Clinical trials | |||
| AD | Reduce Tau oligomer accumulation | — | AADvac1 103 | |
| Microglial activation inhibitor | Endocannabinoids 107 | — | ||
| ALS | Suppress glutamate release | Riluzole 171 | — | |
| Inhibit nitration of tyrosine residue | Edaravone 172 | — | ||
| Inhibit TNF‐α and FasL | Lenalidomide and thalidomide 173 | Cyclosporin 174 | ||
| Inflammasome | Cyclo(His‐pro) 178 | — | ||
| MS | Halt humoral and cell‐mediated immunity | IFN‐β, glatiramer acetate and natalizumab 181 | — | |
| Sphingosine 1 phosphate modulator | Fingolimod, teriflunomide and dimethyl fumarate 184 | Laquinimod, ponesimod, siponimod and ceralifimod 183 | ||
| PD | PPAR‐α agonist | Fenofibrate 113 | — | |
| Inhibit TNF‐α, IL−6 and IL−1β | Statins and aspirin 114 , 115 . | — | ||
| Reducing the accumulation of α‐synuclein oligomers and calpain cleaved α‐synuclein. | — | 9E4 and AFF−1 13 | ||
| Stroke | NLRP3 inflammasome inhibitor | — | Beta‐hydroxy butyrate and MCC‐950 244 , 249 | |
| Treg cell activator | — | Trichostatin 252 | ||
| HD | Semaphorin 4D inhibitor | — | VX−15 218 | |
STIMULATION OF THE IMMUNE SYSTEM IN MULTIPLE SCLEROSIS AND AMYOTROPHIC LATERAL SCLEROSIS
Complement system
Experimental autoimmune encephalomyelitis (EAE) is the best model for studying the role of the complement system in MS. 120 Binding of C3d to CR3 receptor mediates phagocytosis of myelin by microglia. 121 Further, it leads to the activation of TNF‐α and the generation of NO, which culminate in demyelination. 122 At 2 h post‐formation, terminal complex C5b‐9 in lytic dose in macrophages, neurons and oligodendrocyte progenitor cells induces demyelination. 123 In contrast, the sublytic dose of C5b‐9 can prevent oligodendrocyte apoptosis via phosphatidylinositol 3‐kinase/Akt pathway (PI3K/Akt). 124 Watkins et al. 125 revealed clinical data of 22 MS patients, which confirm the upregulation of C1q protein of the classical complement system and fragment Bb of the alternative complement pathway in the grey matter of cortex. To validate the role of complement system in MS, transgenic mice deficient in C3 or factor B were used in an antibody‐independent model of EAE, which showed less severity of disease. 126 A marked decrease in the infiltration of T cells and macrophages, along with a reduced expression of P‐selectin, was detected in PVG/C6‐deficient rats. 127 Therefore, it can be observed that the complement system has a dualistic role. Hence, its modulation instead of complete inhibition can improve the quality of life in MS patients. 27
In the case of ALS, the upregulation of various components of the complement system, such as C1q, C3, is reported. 128 Activation of its components such as MBL, MASP‐1, C3, C4a and C5a leads to the destruction of BBB integrity. 128 One of the critical elements is C5a peptide, which binds to the C5aR receptor of neurons and glia. 129 It also links to another G protein‐coupled receptor C5a‐like receptor 2 (C5L2), which leads to the activation of mitogen‐activated protein kinase (MAPK) and protein kinase B (PKB/Akt) pathways. 130 C5a is responsible for the entry of macrophages in the skeletal muscles of the hSOD1G93A mouse model, contributing to muscle denervation in ALS. 131 SOD1G93A C5aR1 knockout mouse model has shown to increase the survival rate, which indicates that C5aR1 activation exacerbates the deleterious effect of ALS. 132 Another mouse model, TDP‐43Q331K, shows similar activation of complement C5a in the lumbar region of the spinal cord and tibialis anterior muscle. 133 This indicates that C5a can be a potential therapeutic target for ALS. Microarray analysis and laser‐capture microdissection in the transgenic animal model revealed that the levels of C1q and C4 were elevated throughout the progression of the disease. 134 Moreover, mRNA and protein of classical components C1q and C4 in addition to terminal complements C3 and C5b‐9 were elevated in 16 ALS patients. 135 Farber et al. found that activated C1q is responsible for converting resting microglia into reactive microglia via Ca2+ signalling, which further stimulates pro‐inflammatory markers such as TNF‐α and IL‐6, which are detrimental for neurons. 136 Thus, modulating complement system activation can be a promising approach for neuroprotection in ALS (Figure 4).
FIGURE 4.
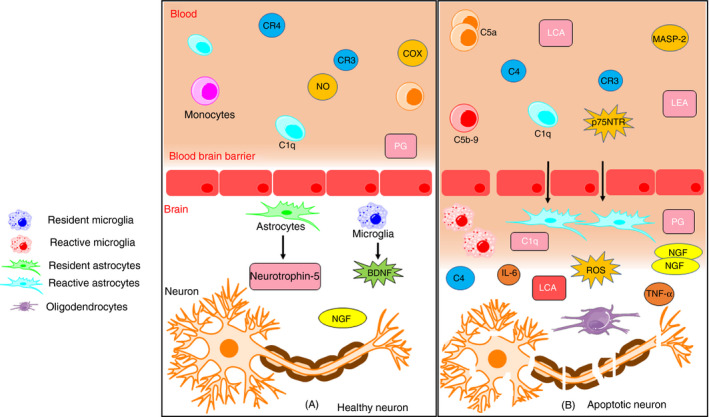
Neuroinflammation in ALS. (A) Components of the complement system, microglia and astrocytes reside near the blood‐brain barrier and help in homeostasis and protection of CNS. It promotes neurogenesis by releasing BDNF, neurotrophin‐5, etc. It also helps in removing debris and suppressing inflammation and therefore leads to neuroprotection. (B) On tissue injury, activation of C4a, C5a and C3 of the complement system is responsible for the entry of microglia and astrocytes to promote its conversion from resting to its activated form. Reactive microglia and astrocytes release pro‐inflammatory markers such as TNF‐α, IL‐1β and iNOS. These markers further lead to the generation of ROS and culminate in oxidative stress. In addition to this, reactive microglia causes upregulation of LFA‐1, LCA, complement receptor CR3 and CR4, immunoglobulin receptor FcγR1 and MHC‐II complex. Chronic activation of C3a and C5a promotes entry of peripheral immune cells, which secretes chemokines and cytokines on endothelial cells and disrupts the integrity of BBB. It also causes induction of NGF correlated with p75NTR in an area of degenerating motor neurons, which leads to apoptosis (BDNF – brain‐derived neurotrophic factor, TNF‐α – tumour necrosis factor, IL – interleukin, iNOS – inducible nitric oxide synthase, ROS – reactive oxygen species, LFA‐1 – lymphocyte function‐associated molecule‐1, LCA – leucocyte common antigen, NGF – nerve growth factor, p75NTR‐p75 – neurotrophin factor, FcγR1 – fragment crystallizable gamma receptor 1, BBB – blood‐brain barrier, MHC – major histocompatibility complex) (Adapted from Servier Medical Art by Servier, licensed under a Creative Commons Attribution 3.0 Unported Licence (http://smart.servier.com/))
Microglia activation in ALS
Microglia play the role of a double‐edged sword. In rNLS8 mouse model, human TAR DNA binding protein 43 (hTDP‐43) pathology‐related ALS microglia exhibit a protective role by reducing inflammatory markers. Inhibitors of microgliosis such as PLZ3397, CSF1R and c‐kit failed to recover motor function in rNLS8 mice. 137 Moreover, after microglial activation, the M2 phenotype secretes neuroprotective factors such as BDNF, while M1 releases inflammatory cytokines to detrimental effect. 138 Fractalkine, a chemo‐attractant bound to surface receptor CX3CR1 of microglia, which helps them in intercellular signalling of impaired neurons. 139 It acts as neuroprotective by stimulating PI3K/Akt, which leads to activation Bcl‐xL(antiapoptotic protein) and quelling of BAD (pro‐apoptotic protein). 140
The SOD1G93A transgenic mouse model is extensively used for studying human ALS. 141 In the SOD1G93A mouse model, reactive microglia and astrocytes cause induction of nerve growth factor (NGF) expression correlated with p75 neurotrophin factor (p75NTR) expression in the area of degenerating motor neurons, which lead to apoptosis. 142 , 143 Reactive microglia causes upregulation of lymphocyte function‐associated molecule‐1 (LFA‐1), leucocyte common antigen (LCA), complement receptor CR3 and CR4, immunoglobulin receptor FcγR1 and MHC‐II complex. 144 These reactive microglia generate the superoxide anions (O2−) to promote oxidative stress, which worsens the ALS condition. 33 In addition to this, reactive microglia secretes pro‐inflammatory markers such as IL‐1, IL‐6 and TNF‐α. IL‐1 further recruits IL‐6, colony‐stimulating factor (CSF), IL‐8 and IFN‐α/β. 145 Apart from these inflammatory markers, there is a threefold elevation in the expression of COX‐2 protein and mRNA in astrocytes, microglia, neurons and in the spinal cord of the transgenic mouse model during the progression of disease. 146
Microglia and T‐cell interaction in MS
The induction of the immunological response against CNS antigen in MS is still paradoxical. 147 Activated microglia contribute to oxidative stress following oligodendrocytes, and neuronal and axonal injury‐associated demyelination. 148 Two contrary hypotheses are involved in describing the functions of the immune system in lesions of MS. First, the pathology starts in the periphery by stimulation of innate and adaptive immunity and progress towards the CNS. 149 The second hypothesis states that it begins from CNS and then shifts to the periphery. 150 Activated microglia helps in the infiltration of T cells responsible for adaptive immunity such as Th1, Th2 and Th17. Th1 releases pro‐inflammatory markers (IFN‐γ and TNF‐α), Th‐2 secretes anti‐inflammatory markers (IL‐4, IL‐6, and IL‐13), and Th17 releases IL‐17 and granulocyte‐macrophage colony‐stimulating factor (GM‐CSF). 151 Both MHC‐I and MHC‐II are expressed by activated microglia, which later stimulate CD8+ T cells and CD4+ T cells, respectively. 152 CD4+ release antibodies against myelin sheath and oligodendrocyte antigens result in demyelination. 153 In contrast, stimulated CD4+ cells can modulate T cells into iTreg (regulatory T cells), which suppress the immune system by inhibiting the antigen‐presenting cell (APC) and induce neurotrophic factors such as BDNF and GDNF, but impaired activation of iTreg leads to autoimmune disease. 154 , 155 , 156
Inflammasome activation
Dysregulation of NLRP3 inflammasome can be detrimental and eventually end up with degeneration of neurons. 157 In the SOD1G93A mouse model, increased expression of TLR‐4 and NF‐κB in the diseased brain suggests that NLRP3 inflammasome activation culminates in pyroptosis. 158 Scientists have also observed an increased expression and co‐localization of NLRP3 inflammasome in the spinal cord of SOD‐1G93A mouse model. 159 Co‐localization and activation of NLRP3 and upregulation of IL‐18 in the serum of ALS patients were reported by Kadhim et al., which then further activates caspase‐1. 160 Increased expression of the NLRP3 inflammasome, along with aggregation of p62 and damaged mitochondria, is indicative of diminished autophagy in ALS. 161 According to Meissner et al., in the SOD‐1G93A mouse model, activation of microglial IL‐1β and caspase‐1 occurs without activation of NLRP3. 162 In contrast, protein aggregates such as TDP‐43Q331K, TDP‐43A315T, TDP‐43WT, and SOD‐1G93A showed microglial NLRP3‐dependent inflammasome activation in TDP‐43Q331K and a SOD‐1G93A mouse model of ALS, respectively, by using Nlrp3−/− knockout and NLRP3 inhibitor MCC950. 163
In MS, elevated levels of IL‐1β and caspase‐1 in peripheral blood mononuclear cells (PBMC) indicate the contribution of NLRP3 inflammasome in experimental mouse model. 164 NLRP3 inflammasome stimulates the migration of Th cells and APC in CNS to increase the expression of chemotactic proteins such as osteopontin, CXCR6 and CCR2. 164 Co‐localization and activation of NLRP3 through IL‐1β aid in demyelination, while stimulation of NLCR4 helps in neuroprotection. 165 Moreover, NF‐κB facilitates pro‐IL‐1β transcription, while expression of pro‐IL‐18 is constituted, which increases after cellular activation. 166 Progression of relapse‐onset MS was marked by the upregulation of IL‐1β in CSF. 167 In addition to this, MS patients showed a significant increase in serum and CSF levels of IL‐18 levels. 168 IL‐18 is responsible for the stimulation of CD8+ mucosal‐associated invariant T (MAIT) cells along with activation of integrin, which enables the infiltration of CD8+T in CNS. 169 Increased rare genetic variants in NLRP1/3 and CASP1 lead to its inactivation through autophagy, mitophagy and type 1 interferons. 170
Immunotherapeutics for modulating the immune system in ALS and MS
To date, US FDA‐approved drug, riluzole, can increase life expectancy for about 2‐3 months. 171 The mechanism of action is to suppress the glutamate release at synaptic cleft. 171 Recently in May 2017, edaravone becomes the second FDA‐approved drug. It shows functional recovery by preventing the nitration of a tyrosine residue in CSF. 172 Inflammatory markers such as TNF‐α and FasL expression are suppressed by COX‐2 inhibitors, thalidomide and its congener lenalidomide in the SOD‐1G93A mouse model. 173 Another COX‐2 inhibitor cyclosporin has been proven effective in the transgenic mouse model, and currently, it is in clinical trial phase IV (NCT01795872). 174 , 175 A clinical trial of glatiramer acetate at a dose of 40 mg/day failed due to inappropriate regime to modulate ALS. 176 Anakinra is an antagonist of IL‐1, which then further inhibits activation of IL‐6 and other pro‐inflammatory markers in ALS. 177 PMX205, the antagonist of C5aR, shows extended survival and slow progression of disease. 133 Cyclo(His‐pro) helps in the reduction of ROS generation and NF‐κB and therefore inhibits NLRP3 inflammasome in ALS. 178 Two vaccines, tgG‐DSE2lim and tgG‐DSE5b, were tested in hSOD1G37R mouse, which resulted in late‐onset and increased life expectancy. 179
NEDA (no evidence of disease activity) is a broadly accepted approach for early treatment of MS that starts with injectables followed by oral doses and infusions. 180 Injectable disease‐modifying therapies include interferon‐βs (IFN‐βs), glatiramer acetate (GA) and natalizumab (NTZ). 181 Immunomodulating drugs such as mitoxantrone intercalate with DNA and halt humoral immunity and T‐cell count. 182 Infusion of rituximab and ocrelizumab that are anti‐CD‐20 antibodies is approved for relapsing–remitting multiple sclerosis (RRMS) and primary progressive multiple sclerosis (PPMS). 183 Sphingosine 1 phosphate (S1P) modulator, fingolimod, is the first oral approved drug that is phosphorylated by sphingosine kinase to form fingolimod phosphate, impeding with circulating immune cells. 184 Other orally approved drugs are teriflunomide and dimethyl fumarate for RRMS. 185 , 186 The drugs that are in clinical trials are laquinimod, ponesimod, siponimod and ceralifimod. 183 A combination of two antibodies trastuzumab and pertuzumab can be used for regulating complement system. 187 Intranasal administration of genetically engineered CAR/FoxP3 Treg cells can quell the EAE model of MS. 188 As myriads of treatment are emerging, these therapies are expected to improve the livelihood of MS patients (Figure 5).
FIGURE 5.
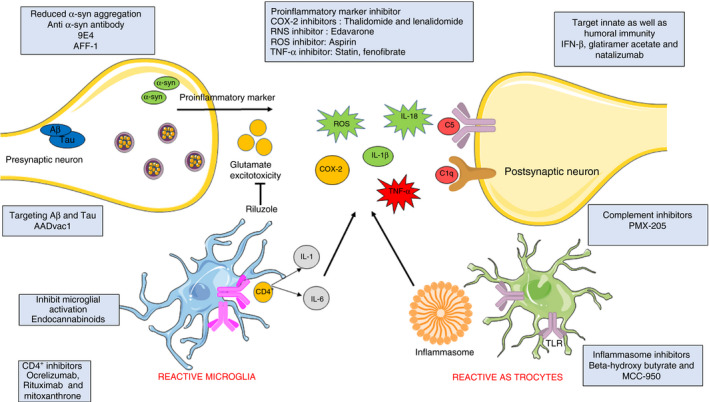
Different therapeutic targets of neurodegenerative diseases. Various therapeutic approaches targeting glutamate excitotoxicity, humoral and cell‐mediated immunity and aggregation of Tau and α‐synuclein. Other components of the immune system such as the complement system, inflammasome, reactive microglia and astrocytes, and pro‐inflammatory markers are modulated by different immunotherapies (Adapted from Servier Medical Art by Servier, licensed under a Creative Commons Attribution 3.0 Unported Licence (http://smart.servier.com/))
IMMUNE MODULATORS OF HUNTINGTON'S DISEASE
Complement system
HD is an inherited, dominant and autosomal neurodegenerative disease that is triggered by an expansion of three‐base‐pair (CAG) repeat in exon 1 of the HTT gene. 189 This expansion repeats translate into a polyglutamine tract at the N‐terminus of the protein. 190 , 191 Experimental data suggest that in HD, microglia activate the classical pathway of the complement system via pathological peptide‐like mutant huntingtin (mHtt). Mutation in the gene of chromosome 4p 16.3 leads to the generation of mHtt. 191 , 192 In situ hybridization study reveals that the striatum, neurons, astrocytes and myelin sheath of the HD brain are strongly positive for C1q, C1r, C3, C4, C9‐neoepitope and iC3b‐neoepitope deposition. 190 , 193 In the early disease stages, negative outcome for staining of complement component suggests that early neuronal damage precedes local synthesis and activation of complement components. Microarray analysis reports of HD brain tissue indicate that in the caudate nucleus and motor cortex area, complement components C3, C4A and C4B get highly expressed. 194 Researchers have worked on various animal models of HD. Among them, the R6/2 transgenic mouse model was proven to be a more realistic model despite having the limitations to demonstrate the significance of complement system in HD. 195
Microglial activation
Marked microgliosis has been found in the post‐mortem human HD brains. 193 It was reported that medium spiny GABA neurons (MSNs) of the striatum are most vulnerable to the neurotoxicity mediated by mHtt. 196 mHtt mostly accumulates in nerve terminals and neuronal dendrites rather than in soma and other cells, leading to microglial activation. 196 Activated and proliferated microglia contributes to neurodegeneration by upregulating inflammatory cytokines. 197 , 198 There are several pathways through which microglial activation occurs in HD, which are as follows:
NF‐κB pathway
Microglia mostly express TL‐2 receptors, which are accountable for IL‐10 and IL‐6 secretions. 199 , 200 It also expresses TLR‐3 and TLR‐4 receptors that are linked with the production of pro‐inflammatory cytokines such as IL‐6, IL‐10, IL‐12, TNF‐α and CXCL‐10. 200 A group of inhibitory proteins, IκBs, sequestered NF‐κB in the cytoplasm. 199 When IκB kinase (IKK) phosphorylates IκBs, it leads to the dissociation of NF‐κB from IκBs and translocation of NF‐κB to the nucleus. 201 IKKα, IKKβ and IKKγ are the subunits of IKK that is activated by soluble mHtt, ultimately leading to NF‐κB pathway activation. 202 Trager et al. reported that the interaction of mHtt with IKK causes transcriptional changes in the NF‐κB signalling with an induction in the level of IL‐6, IL‐8, IL‐1β and TNF‐α. siRNA lowers the mHtt level and causes attenuation of the NF‐κB pathway with concurrent downregulation of pro‐inflammatory cytokine levels in HD. 203
Kynurenine pathway
Studies on the R6/2 HD mouse model have shown that peripheral macrophages exacerbate the HD pathogenic condition through the Kynurenine mono‐oxygenase pathway (KP). 204 KP is the major route of metabolism of L‐tryptophan in mammals and the principal pathway for the formation of nicotinamide adenine dinucleotide (NAD+). 205 Levels of two neurotoxic metabolites of the kynurenine pathway such as 3‐hydroxykynurenine (3‐HK) and quinolinic acid (QUIN) are increased in neocortex and striatum in the early stage of HD. 204 These QUIN and 3‐HK, in addition to astrocytic deficiency and altered glutamate reuptake, have a role in N‐methyl D‐aspartate (NMDA) receptor overstimulation (excitotoxicity), free radical formation, lipid peroxidation and decrease in dopamine release. 206 , 207 This will ultimately contribute to neurodegeneration and cognitive dysfunction.
Cannabinoid pathway
Cannabinoid receptors are GPCR and mainly consist of CB1 and CB2 types of receptor. 208 CB2 plays a role in cell survival, differentiation and proliferation by adenylyl cyclase, cyclic AMP‐protein kinase A (PKA), extracellular signal‐regulated kinase 1 (ERK1) and ERK2, p38 mitogen‐activated protein kinase and JUN N‐terminal kinase (JNK) pathways. 209 CB1 receptors are mostly present in striatal MSNs in R6/2 HD mouse model. 210 It plays a neuroprotective role by increasing the expression of brain‐derived neurotrophic factor (BDNF). 211 In the R6/2 mouse model, the genetic knockdown of CB2 receptors shows microglial activation along with exacerbation of behavioural abnormalities and reduction in life span. 212
Inflammasome activation
The mHtt aggregates in the caudate and putamen area and acts as DAMPs to cause neostriatal atrophy. 213 Although recent studies have observed that in the absence of any stimuli, mHtt overexpression in the primary murein microglia induces inflammatory gene expression, 214 studies on the R6/2 mouse model suggest that caspase‐1 and caspase‐3 get activated and further aggravate neurodegeneration. 215 Another research shows that inhibition of ATP‐sensitive P2 purinergic receptor (P2X7) leads to the alleviation of HD through the suppression of NLRP3 inflammasome signalling. 216 An increase in expression, as well as activity of the major myeloid transcription factors such as CCAAT/enhancer‐binding protein alpha/beta (C/EBPα/β) and Pu.1, may cause the production of pro‐inflammatory cytokines, whereas the aggregation of mHtt triggers activation of inflammasome, but the mechanism to activate Pu.1 and C/EBP activity in microglia is yet to be explored. 214 , 217
Immunotherapeutics in HD
Immunotherapy is a promising area for the treatment of HD. Recently, Vaccinex – ‘a Rochester’ – a New York‐based biotechnology company, is conducting a double‐blinded clinical trial on antibody VX‐15. 218 It inhibits Semaphorin 4D, which is a transmembrane protein causing neuroinflammation and neurodegeneration. 219 Apart from this, an antisense oligonucleotide (ASO) therapy is also in phase I clinical trial. 220 The limitation of ASO therapy is that it can be administered through lumbar puncture only. 220
INVOLVEMENT OF THE IMMUNE SYSTEM IN EXACERBATION OF ISCHAEMIC STROKE
Complement system
The complement system plays a significant role in the pathogenesis of ischaemic stroke. 221 , 222 After an ischaemic attack, the levels of C3a, C4d and soluble C5b‐9 in plasma are increased instantly, while the elevation of C5a occurs within 7‐14 days. 222 , 223 The role of C1q, initiator of the classical pathway in the pathogenesis of ischaemic stroke, is quite complex. Reports suggest that the accumulation of C1q occurs at 3 to 6 h post‐ischaemic stroke. 224 After 24 h of ischaemic insult, biosynthesis and functional activity of C1q by microglia get drastically increased. 224 However, C1q‐deficient transgenic mice do not show any protective role in ischaemic stroke. 223 Conversely, neonatal C1q‐deficient mice show less functional deficit. 225 This discrepancy between neonatal and adult C1q is mainly due to the differences in the presence of C1q in the CNS of adult and neonatal mice. 226 MBL, the initiator of the lectin pathway, also exacerbates ischaemic stroke pathology in transgenic mice and exhibits less cerebral infarct with better functional outcome post‐stroke. 227
Compared with the above two pathways, the alternative pathway has also been reported in ischaemic stroke pathology. 221 , 222 , 223 Factor B‐deficient mice, when treated with CR2‐fH, an alternative pathway inhibitor, show better improvement 24 h post‐reperfusion. 228 CR2‐fH inhibits the complement activation site by recognizing C3 opsonin. 229
Microglial responses after ischaemic stroke
Microglial activation shows characteristic temporal and spatial patterns in the peri‐infarct zone. 230 In the case of focal cerebral ischaemia, microgliosis is prominent in the core region, accumulation region and marginal regions. 230 Within the cortical region where blood flow is normal, ramified microglia are majorly observed, whereas microglia with few stout and short processes are observed in the marginal zone. Finally, the hypertrophic cell body containing amoeboid microglia is found in the accumulation region and in the core region. 231 The distribution pattern signifies the transformation of morphology microglia that reflects the changes of its function and also a different pathological state of the tissue. 51 After global transient ischaemia, the hippocampal CA1 region has an extensive microglial expansion. 232 Immediately after an ischaemic episode, the release of IFN‐γ shifts the microglial polarization towards pro‐inflammatory M1 phenotype, whereas at a later stage, CXCL‐16 and IL‐4 lead to anti‐inflammatory M2 polarization. 233 , 234 In the early stage, activation of pro‐inflammatory M1 microglia leads to the production of cytokine, TNF‐α, IL‐1, ROS, RNS and protease, while at the reparative phase, anti‐inflammatory M2 microglia helps in attenuation of inflammation by producing IL‐10 and TGF‐β. 39 , 235 Prolonged overexpression of microglia further leads to peripheral immunosuppression that may predispose to bacterial infection, pneumonia and urinary tract infection (UTI). 236 Hence, microglial activation following ischaemic stroke leads to exacerbation of pathology, whereas at the late stage, microglia contribute to neuronal protection. 51
Role of inflammasome signalling
The activation of inflammasome signalling is involved in atherogenesis, a major risk factor for stroke. 237 , 238 Macrophages are transformed into foam cells, and these can activate NLRP3 inflammasome. Atherogenesis causes the formation of plaque that leads to lysosomal rupturing of foam cells. 238 It further causes the release of cathepsin, various proenzyme and ROS, which ultimately activate NLRP1 and NLRP3. 239 TLR‐4 and TLR‐12 identify minimally oxidized LDL and FFA, and increase myeloid differentiation primary response protein MyD88 following activation of TIR‐domain‐containing adaptor‐inducing interferon‐β (TRIF) to induce the expression of NF‐κB. 240 Further, an increase in the expression of NLRP‐3, ASC and pro‐caspase‐1 contributes to inflammasome assembly. 241 , 242 Inflammatory cytokines such as IL‐1β and IL‐18 activation also induce neutrophil, macrophage and lymphocyte infiltration, followed by their activation. 243 Release of MMPs 1, 2, 3, 9 and 12 is triggered by the inflammatory cytokines. 244 These MMPs play an important role in extracellular matrix remodelling and also trigger plaque stability. 245 This cyclic mechanism activates NLRP3 and aggravates atherosclerosis, which therefore contributes to the development and exacerbation of stroke pathology.
Post‐stroke activation of inflammasome signalling
After ischaemic stroke, damaged cells release DAMPs, which leads to activation of PRRs such as TLR‐2, TLR‐4, IL‐1β and RAGE, which may further activate NLRP1 and NLRP3 inflammasome via MAPK and NF‐κB pathways. 45 , 246 . In the core area, depletion of ATP leads to activation of NLRP1 by conversion of ATP‐bound inactive inflammasome to ADP‐bound active inflammasome. 247 Increased expression of NLRP3 or NLRP1, along with ASC and pro‐caspase‐1, leads to inflammasome assembly, and thus maturation of pro‐inflammatory cytokines into inflammatory cytokines that further leads to neuronal cell death via pyroptosis. 248
Immunotherapy in ischaemic stroke
Recent studies reported that β‐hydroxybutyrate and MCC950 could inhibit the NLRP3 inflammasome by reducing the secretion of pro‐inflammatory cytokine (IL‐1b) by macrophage. 244 , 249 , 250 Treg cells are suppressors of neuroinflammation. 251 Forkhead box transcription factor (FOXP3) is a marker of Treg cells, and FoxP3+ Treg cells are reported to attenuate inflammation. 251 Trichostatin A activates FoxP3 and reduces the proliferation of Th1 cell, which suppress the production of IFN‐γ by promoting the Treg cells. 252
MICROGLIAL MODULATION VIA GUT MICROFLORA AFFECTING CNS DISORDERS
As discussed previously, microglial activation leads to generation of NO. Aberrant NO generation has been linked with various neurodegenerative diseases, specifically AD and PD. 253 Studies have reported that gut commensal bacteria produce short‐chain fatty acids (SCFA) to a certain extent, which are particularly responsible for microglial maturation and functional regulation. 254 Disruption of gut microflora has been observed to contribute to different CNS disorders via microglial dysfunction. 254 , 255 Hence, targeting the link between microglia and host gut microflora is a promising therapeutic approach. The Kallyope company is conducting a study for investigating the communication pathways of the gut/brain axis for developing novel therapies for various CNS disorders. However, this study is still in its infancy, which requires further research to determine how probiotics can be utilized to improve microglial function and finally be beneficial in CNS disorders. 256
CHALLENGES TO TREAT NEURODEGENERATIVE DISORDERS VIA IMMUNE SYSTEM MANIPULATION
Manipulation of immune system as a novel therapeutic approach for neurodegenerative disorders possesses few challenges. Studies have shown that there are major gaps in understanding the disease mechanism, which often restricts bedside translation of promising in vivo results. 257 In vivo studies for neurodegenerative diseases are extensively based on genetic models, which reflect the familial forms of the disease. Hence, development of effective therapies is limited with the use of such models. 257 Along with this, age‐related factors and effect of lifestyle are not considered extensively in preclinical studies. As discussed in the previous section, host gut microflora can affect both brain function and immune response. 257 It has been recently found that heterogeneity among individuals can also be a result of latent CNS infections causing neurodegenerative disease such as AD. 258 This appears consistent with ageing‐related IFN‐1 expression at the choroid plexus. 259 , 260 Therefore, common immunological factors contributing to different neurodegenerative diseases can be modified by systemic immunomodulation and microglial phenotype modulation.
CONCLUSION
This review summarizes the neuroimmune crosstalk with fundamental mechanisms and potential therapeutic approaches. As discussed in the article, the innate immune reaction can be detrimental to neurons and oligodendrocytes, and at the same time, it can also be beneficial for recovery. This response depends upon the sources of cytokines and on their cognate receptor's nature. The production of innate immune protein, as well as the presence of adaptive immune cells in the brain environment, is an important feature of neurodegeneration. Dysregulation of components of the immune systems such as microglia, T cells, complement system and inflammasome signalling plays a vital role in degenerating neurons, but the genetic background, gender and environment also contribute towards progression of the pathology. Singlet therapeutic approaches, in addition to the present choices of therapy, can suffice to reduce neuroinflammation and toxicity, thus preventing the induction and exacerbation of neurodegenerative diseases such as AD, PD, ALS, MS, HD and ischaemic stroke.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
AUTHOR CONTRIBUTIONS
FB, MB, PB, XW, KRD and DRY conceived and designed the study. FB, MB, AD, DS and PB outlined the performed rigorous literature search. FB, MB, BS, PJ, SR, AS and US conceived and designed the figures and images. FB, MB, AD, AB, KK, PB, KRD and DRY wrote the manuscript.
ACKNOWLEDGMENTS
The authors acknowledge Department of Pharmaceuticals, Ministry of Chemical and Fertilizers, Govt. of India, and National Institute of Pharmaceutical Education and Research (NIPER) Ahmedabad, Gandhinagar, India.
Falguni Baidya and Mariya Bohra are contributed equally to this work.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Chen WW, Zhang XI, Huang WJ. Role of neuroinflammation in neurodegenerative diseases. Mol Med Rep. 2016;13(4):3391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurogibol. 2014;112:24–49. [DOI] [PubMed] [Google Scholar]
- 3. Feigin VL, Nichols E, Alam T, Bannick MS, Beghi E, Blake N, et al. Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(5):459–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carroll WM. The global burden of neurological disorders. Lancet Neurol. 2019;18(5):418–9. [DOI] [PubMed] [Google Scholar]
- 5. Chauhan NB, Mehla J. Ameliorative effects of nutraceuticals in neurological disorders In Watson RR, Preedy VR. (eds.), Bioactive Nutraceuticals and Dietary Supplements in Neurological and Brain Disease. Cambridge, MA: Academic Press; 2015:245–60. [Google Scholar]
- 6. Meisel C, Meisel A. Suppressing immunosuppression after stroke. N Engl J Med. 2011;365(22):2134–6. [DOI] [PubMed] [Google Scholar]
- 7. Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147(S1):S232–S240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Block ML, Hong JS. Microglia and inflammation‐mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurogibol. 2005;76(2):77–98. [DOI] [PubMed] [Google Scholar]
- 9. Bennett ML, Bennett FC. The influence of environment and origin on brain resident macrophages and implications for therapy. Nat Neurosci. 2019;23:157–66. [DOI] [PubMed] [Google Scholar]
- 10. Krakauer T. Inflammasomes, autophagy, and cell death: the trinity of innate host defense against intracellular bacteria. Mediators Inflamm. 2019;2019:2471215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 2018;21(10):1359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Voet S, Srinivasan S, Lamkanfi M, van Loo G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med. 2019;11(6):e10248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liang Z, Zhao Y, Ruan L, Zhu L, Jin K, Zhuge Q, et al. Impact of aging immune system on neurodegeneration and potential immunotherapies. Prog Neurogibol. 2017;157:2–8. [DOI] [PubMed] [Google Scholar]
- 14. Bonifati DM, Kishore U. Role of complement in neurodegeneration and neuroinflammation. Mol Immunol. 2007;44(5):999–1010. [DOI] [PubMed] [Google Scholar]
- 15. Orsini F, De Blasio D, Zangari R, Zanier ER, De Simoni MG. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front Cell Neurosci. 2014;8:380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Veerhuis R, Nielsen HM, Tenner AJ. Complement in the brain. Mol Immunol. 2011;48(14):1592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wagner E, Frank MM. Therapeutic potential of complement modulation. Nat Rev Drug Discovery. 2010;9(1):43–56. [DOI] [PubMed] [Google Scholar]
- 18. Carpanini SM, Torvell M, Morgan BP. Therapeutic inhibition of the complement system in diseases of the central nervous system. Front Immunol. 2019;10:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walport MJ. Complement. N Engl J Med. 2001;344(14):1058–66. [DOI] [PubMed] [Google Scholar]
- 20. Morgan BP. Complement in the pathogenesis of Alzheimer’s disease. Semin Immunopathol. 2018;40(1):113–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ehrnthaller C, Ignatius A, Gebhard F, Huber‐Lang M. New insights of an old defense system: structure, function, and clinical relevance of the complement system. Mol Med. 2011;17(3):317–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Veerhuis R, Janssen I, De Groot CJ, Van Muiswinkel FL, Hack CE, Eikelenboom P. Cytokines associated with amyloid plaques in Alzheimer's disease brain stimulate human glial and neuronal cell cultures to secrete early complement proteins, but not C1‐inhibitor. Exp Neurol. 1999;160(1):289–99. [DOI] [PubMed] [Google Scholar]
- 23. Garcia BL, Zwarthoff SA, Rooijakkers SH, Geisbrecht BV. Novel evasion mechanisms of the classical complement pathway. J Immunol. 2016;197(6):2051–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dahl MR, Thiel S, Matsushita M, Fujita T, Willis AC, Christensen T, et al. MASP‐3 and its association with distinct complexes of the mannan‐binding lectin complement activation pathway. Immunity. 2001;15(1):127–35. [DOI] [PubMed] [Google Scholar]
- 25. Müller‐Eberhard HJ. Molecular organization and function of the complement system. Annu Rev Biochem. 1988;57(1):321–47. [DOI] [PubMed] [Google Scholar]
- 26. Harboe M, Mollnes TE. The alternative complement pathway revisited. J Cell Mol Med. 2008;12(4):1074–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alexander JJ, Anderson AJ, Barnum SR, Stevens B, Tenner AJ. The complement cascade: Yin‐Yang in neuroinflammation–neuro‐protection and‐degeneration. J Neurochem. 2008;107(5):1169–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Merle NS, Church SE, Fremeaux‐Bacchi V, Roumenina LT. Complement system part I–molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Norris GT, Kipnis J. Immune cells and CNS physiology: microglia and beyond. J Exp Med. 2019;216(1):60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Song WM, Colonna M. The microglial response to neurodegenerative disease. Adv Immunol. 2018;139:1–50. [DOI] [PubMed] [Google Scholar]
- 31. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA‐DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38(8):1285. [DOI] [PubMed] [Google Scholar]
- 32. Walter L, Neumann H. Role of microglia in neuronal degeneration and regeneration. Semin Immunopathol. 2009;31(4):513–25. [DOI] [PubMed] [Google Scholar]
- 33. Colton CA, Gilbert DL. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett. 1987;223(2):284–8. [DOI] [PubMed] [Google Scholar]
- 34. Sawada M, Kondo N, Suzumura A, Marunouchi T. Production of tumor necrosis factor‐alpha by microglia and astrocytes in culture. Brain Res. 1989;491(2):394–7. [DOI] [PubMed] [Google Scholar]
- 35. Tan HY, Wang N, Li S, Hong M, Wang X, Feng Y. The reactive oxygen species in macrophage polarization: reflecting its dual role in progression and treatment of human diseases. Oxid Med Cell Longev. 2016;2016:2795090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Subhramanyam CS, Wang C, Hu Q, Dheen ST. Microglia‐mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol. 2019;94:112–20. [DOI] [PubMed] [Google Scholar]
- 37. Frakes AE, Ferraiuolo L, Haidet‐Phillips AM, Schmelzer L, Braun L, Miranda CJ, et al. Microglia induce motor neuron death via the classical NF‐κB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81(5):1009–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6(4):193. [DOI] [PubMed] [Google Scholar]
- 39. Amantea D, Micieli G, Tassorelli C, Cuartero MI, Ballesteros I, Certo M, et al. Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front Neurosc. 2015;9:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019;10(2):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Song L, Pei L, Yao S, Wu Y, Shang Y. NLRP3 inflammasome in neurological diseases, from functions to therapies. Front Cell Neurosci. 2017;11:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327(5963):296–300. [DOI] [PubMed] [Google Scholar]
- 43. Van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LA. Inflammasome activation and IL‐1β and IL‐18 processing during infection. Trends Immunol. 2011;32(3):110–6. [DOI] [PubMed] [Google Scholar]
- 44. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–5. [DOI] [PubMed] [Google Scholar]
- 45. Fann DY, Lim YA, Cheng YL, Lok KZ, Chunduri P, Baik SH, et al. Evidence that NF‐κB and MAPK signaling promotes NLRP inflammasome activation in neurons following ischemic stroke. Mol Neurobiol. 2018;55(2):1082–96. [DOI] [PubMed] [Google Scholar]
- 46. Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, et al. CLICs‐dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun. 2017;8(1):1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Samary CS, Pelosi P, Silva PL, Rocco PR. Immunomodulation after ischemic stroke: potential mechanisms and implications for therapy. Crit Care. 2016;20(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kawai T, Akira S. The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol. 2010;11(5):373. [DOI] [PubMed] [Google Scholar]
- 49. Evavold CL, Kagan JC. How inflammasomes inform adaptive immunity. J Mol Biol. 2018;430(2):217–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hornung V, Ablasser A, Charrel‐Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase‐1‐activating inflammasome with ASC. Nature. 2009;458(7237):514–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Taylor RA, Sansing LH. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin Dev Immunol. 2013;2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Burns A, Iliffe S. Alzheimer’s disease. BMJ. 2009;338:b158. [DOI] [PubMed] [Google Scholar]
- 53. Lambert JC, Ibrahim‐Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45(12):1452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41(10):1094–9. [DOI] [PubMed] [Google Scholar]
- 55. Crehan H, Holton P, Wray S, Pocock J, Guerreiro R, Hardy J. Complement receptor 1 (CR1) and Alzheimer's disease. Immunobiology. 2012;217(2):244–50. [DOI] [PubMed] [Google Scholar]
- 56. Yu JT, Tan L. The role of clusterin in Alzheimer’s disease: pathways, pathogenesis, and therapy. Mol Neurobiol. 2012;45(2):314–26. [DOI] [PubMed] [Google Scholar]
- 57. Webster S, Lue LF, Brachova L, Tenner AJ, McGeer PL, Terai K, et al. Molecular and cellular characterization of the membrane attack complex, C5b‐9, in Alzheimer’s disease. Neurobiol Aging. 1997;18(4):415–21. [DOI] [PubMed] [Google Scholar]
- 58. Fonseca MI, McGuire SO, Counts SE, Tenner AJ. Complement activation fragment C5a receptors, CD88 and C5L2, are associated with neurofibrillary pathology. J Neuroinflammation. 2013;10(1):803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, et al. Complement C3 deficiency protects against neurodegeneration in aged plaque‐rich APP/PS1 mice. Sci Transl Med. 2017;9(392):eaaf6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nizami S, Hall‐Roberts H, Warrier S, Cowley SA, Di Daniel E. Microglial inflammation and phagocytosis in Alzheimer's disease: Potential therapeutic targets. Br J Pharmacol. 2019;176(18):3515–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Konishi H, Kiyama H. Microglial TREM2/DAP12 signaling: a double‐edged sword in neural diseases. Front Cell Neurosci. 2018;12:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Loeffler DA, Camp DM, Conant SB. Complement activation in the Parkinson's disease substantia nigra: an immunocytochemical study. J Neuroinflammation. 2006;3(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yamada T, McGeer PL, McGeer EG. Lewy bodies in Parkinson's disease are recognized by antibodies to complement proteins. Acta Neuropathol. 1992;84(1):100–4. [DOI] [PubMed] [Google Scholar]
- 64. Cho K. Emerging roles of complement protein C1q in neurodegeneration. Aging Dis. 2019;10(3):652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Depboylu C, Schäfer MK, Arias‐Carrión O, Oertel WH, Weihe E, Höglinger GU. Possible involvement of complement factor C1q in the clearance of extracellular neuromelanin from the substantia nigra in Parkinson disease. J Neuropathol Exp Neurol. 2011;70(2):125–32. [DOI] [PubMed] [Google Scholar]
- 66. McGeer PL, McGeer EG. Inflammation and neurodegeneration in Parkinson's disease. Parkinsonism Relat Disord. 2004;10:S3–7. [DOI] [PubMed] [Google Scholar]
- 67. Hou L, Wang K, Zhang C, Sun F, Che Y, Zhao X, et al. Complement receptor 3 mediates NADPH oxidase activation and dopaminergic neurodegeneration through a Src‐Erk‐dependent pathway. Redox Biol. 2018;14:250–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang Y, Hancock AM, Bradner J, Chung KA, Quinn JF, Peskind ER, et al. Complement 3 and factor h in human cerebrospinal fluid in Parkinson's disease, Alzheimer's disease, and multiple‐system atrophy. Am J Pathol. 2011;178(4):1509–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rozemuller AJ, Eikelenboom P, Theeuwes JW, Steur EJ, De Vos RA. Activated microglial cells and complement factors are unrelated to cortical Lewy bodies. Acta Neuropathol. 2000;100(6):701–8. [DOI] [PubMed] [Google Scholar]
- 70. McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008;5(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16(6):358–72. [DOI] [PubMed] [Google Scholar]
- 72. Yeh FL, Hansen DV, Sheng M. TREM2, microglia, and neurodegenerative diseases. Trends Mol Med. 2017;23(6):512–33. [DOI] [PubMed] [Google Scholar]
- 73. Zheng H, Cheng B, Li Y, Li X, Chen X, Zhang YW. TREM2 in Alzheimer’s disease: microglial survival and energy metabolism. Front Aging Neurosci. 2018;10:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013;368(2):107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jin SC, Benitez BA, Karch CM, Cooper B, Skorupa T, Carrell D, et al. Coding variants in TREM2 increase risk for Alzheimer's disease. Hum Mol Genet. 2014;23(21):5838–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Song W, Hooli B, Mullin K, Jin SC, Cella M, Ulland TK, et al. Alzheimer's disease‐associated TREM2 variants exhibit either decreased or increased ligand‐dependent activation. Alzheimers Dementia. 2017;13(4):381–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hirsch EC, Hunot S. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol. 2009;8(4):382–97. [DOI] [PubMed] [Google Scholar]
- 78. Ferreira SA, Romero‐Ramos M. Microglia response during Parkinson’s disease: alpha‐synuclein intervention. Front Cell Neurosci. 2018;12:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, et al. Aggregated α‐synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J. 2005;19(6):533–42. [DOI] [PubMed] [Google Scholar]
- 80. Daher JP, Volpicelli‐Daley LA, Blackburn JP, Moehle MS, West AB. Abrogation of α‐synuclein–mediated dopaminergic neurodegeneration in LRRK2‐deficient rats. Proc Natl Acad Sci USA. 2014;111(25):9289–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Smith JA, Das A, Ray SK, Banik NL. Role of pro‐inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 2012;87(1):10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor‐α (TNF‐α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. 1994;165(1–2):208–10. [DOI] [PubMed] [Google Scholar]
- 83. Mosley RL, Hutter‐Saunders JA, Stone DK, Gendelman HE. Inflammation and adaptive immunity in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2(1):a009381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Barcia C, Ros CM, Annese V, Gomez A, Ros‐Bernal F, Aguado‐Llera D, et al. IFN‐γ signaling, with the synergistic contribution of TNF‐α, mediates cell specific microglial and astroglial activation in experimental models of Parkinson's disease. Cell Death Dis. 2012;3(8):e379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tangestani Fard M, Stough C. A review and hypothesized model of the mechanisms that underpin the relationship between inflammation and cognition in the elderly. Front Aging Neurosci. 2019;11:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Moore AH, Wu M, Shaftel SS, Graham KA, O'Banion MK. Sustained expression of interleukin‐1β in mouse hippocampus impairs spatial memory. Neuroscience. 2009;164(4):1484–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Vane JR, Bakhle YS, Botting RM. CYCLOOXYGENASES 1 AND 2. Annu Rev Pharmacol Toxicol. 1998;38(1):97–120. [DOI] [PubMed] [Google Scholar]
- 88. Radomski MW, Palmer RM, Moncada S. Glucocorticoids inhibit the expression of an inducible, but not the constitutive, nitric oxide synthase in vascular endothelial cells. Proc Natl Acad Sci USA. 1990;87(24):10043–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hoogendam JP, Kalleveen IM, de Castro CS, Raaijmakers AJ, Verheijen RH, van Den Bosch MA, et al. High‐resolution T 2‐weighted cervical cancer imaging: a feasibility study on ultra‐high‐field 7.0‐T MRI with an endorectal monopole antenna. Eur Radiol. 2017;27(3):938–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Heneka MT, McManus RM, Latz E. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci. 2018;19(10):610–21. [DOI] [PubMed] [Google Scholar]
- 91. Freeman L, Guo H, David CN, Brickey WJ, Jha S, Ting JP. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med. 2017;214(5):1351–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E, et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener. 2016;11(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. 2013;14(8):812–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cho MH, Cho K, Kang HJ, Jeon EY, Kim HS, Kwon HJ, et al. Autophagy in microglia degrades extracellular β‐amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014;10(10):1761–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Liu L, Chan C. IPAF inflammasome is involved in interleukin‐1β production from astrocytes, induced by palmitate; implications for Alzheimer's disease. Neurobiol Aging. 2014;35(2):309–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lang Y, Chu F, Shen D, Zhang W, Zheng C, Zhu J, et al. Role of inflammasomes in neuroimmune and neurodegenerative diseases: a systematic review. Mediators Inflamm. 2018;2018:1549549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Panicker N, Sarkar S, Harischandra DS, Neal M, Kam TI, Jin H, et al. Fyn kinase regulates misfolded α‐synuclein uptake and NLRP3 inflammasome activation in microglia. J Exp Med. 2019;216(6):1411–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Bai H, Yang B, Yu W, Xiao Y, Yu D, Zhang Q. Cathepsin B links oxidative stress to the activation of NLRP3 inflammasome. Exp Cell Res. 2018;362(1):180–7. [DOI] [PubMed] [Google Scholar]
- 99. Wang S, Yuan YH, Chen NH, Wang HB. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson's disease. Int Immunopharmacol. 2019;67:458–64. [DOI] [PubMed] [Google Scholar]
- 100. Lee E, Hwang I, Park S, Hong S, Hwang B, Cho Y, et al. MPTP‐driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2019;26(2):213–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Gordon RA, Albornoz EC, Christie DR, Langley MAB, Kumar VS, Mantovani S, et al. Inflammasome inhibition prevents αsynuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med. 2018;10(465):eaah4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhang P, Shao XY, Qi GJ, Chen Q, Bu LL, Chen LJ, et al. C dk5‐D ependent A ctivation of neuronal inflammasomes in Parkinson's disease. Mov Disord. 2016;31(3):366–76. [DOI] [PubMed] [Google Scholar]
- 103. Lemere CA. Immunotherapy for Alzheimer’s disease: hoops and hurdles. Mol Neurodegener. 2013;8(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid β‐peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6(8):916–9. [DOI] [PubMed] [Google Scholar]
- 105. DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti‐Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98(15):8850–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Wisniewski T, Goni F. Immunotherapy for Alzheimer's disease. Biochem Pharmacol. 2014;88(4):499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hernangómez M, Carrillo‐Salinas JF, Mecha M, Correa F, Mestre L, Loría F, Feliú A, Docagne F, Guaza C. Brain innate immunity in the regulation of neuroinflammation: therapeutic strategies by modulating CD200‐CD200R interaction involve the cannabinoid system. Curr Pharm Des. 2014;20(29):4707–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wisniewski T, Frangione B. Immunological and anti‐chaperone therapeutic approaches for Alzheimer disease. Brain Pathol. 2005;15(1):72–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bossaller L, Marshak‐Rothstein A. The innate immune system in SLE In Wallace D, Hahn B. (Eds.), Dubois' Lupus Erythematosus and Related Syndromes. Philadelphia: WB Saunders; 2013:57–61. [Google Scholar]
- 110. Ryan JM, Grundman M. Anti‐amyloid‐β immunotherapy in Alzheimer's disease: ACC‐001 clinical trials are ongoing. J Alzheimers Dis. 2009;17(2):243. [DOI] [PubMed] [Google Scholar]
- 111. Wisniewski T, Goñi F. Immunotherapeutic approaches for Alzheimer’s disease. Neuron. 2015;85(6):1162–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. Phase 3 trials of solanezumab for mild‐to‐moderate Alzheimer's disease. N Engl J Med. 2014;370(4):311–21. [DOI] [PubMed] [Google Scholar]
- 113. Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator‐activated receptors α and γ are activated by indomethacin and other non‐steroidal anti‐inflammatory drugs. J Biol Chem. 1997;272(6):3406–10. [DOI] [PubMed] [Google Scholar]
- 114. Lyman M, Lloyd DG, Ji X, Vizcaychipi MP, Ma D. Neuroinflammation: the role and consequences. Neurosci Res. 2014;79:1–2. [DOI] [PubMed] [Google Scholar]
- 115. Terblanche M, Almog Y, Rosenson RS, Smith TS, Hackam DG. Statins and sepsis: multiple modifications at multiple levels. Lancet Infect Dis. 2007;7(5):358–68. [DOI] [PubMed] [Google Scholar]
- 116. Han X, Sun S, Sun Y, Song Q, Zhu J, Song N, et al. Small molecule‐driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: implications for Parkinson disease. Autophagy. 2019;15(11):1860–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Mo Y, Xu E, Wei R, Le B, Song L, Li D, et al. Bushen‐yizhi formula alleviates neuroinflammation via inhibiting NLRP3 inflammasome activation in a mouse model of Parkinson’s disease. Evid Based Complement Alternat Med. 2018;2018:3571604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lee JS, Lee SJ. Mechanism of anti‐α‐synuclein immunotherapy. J Mov Disord. 2016;9(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Brundin P, Dave KD, Kordower JH. Therapeutic approaches to target alpha‐synuclein pathology. Exp Neurol. 2017;298:225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ingram G, Hakobyan S, Robertson NP, Morgan BP. Complement in multiple sclerosis: its role in disease and potential as a biomarker. Clin Exp Immunol. 2009;155(2):128–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Prineas JW, Kwon EE, Cho ES, Sharer LR, Barnett MH, Oleszak EL, et al. Immunopathology of secondary‐progressive multiple sclerosis. Ann Neurol. 2001;50(5):646–57. [DOI] [PubMed] [Google Scholar]
- 122. van der Laan LJ, Ruuls SR, Weber KS, Lodder IJ, Döpp EA, Dijkstra CD. Macrophage phagocytosis of myelin in vitro determined by flow cytometry: phagocytosis is mediated by CR3 and induces production of tumor necrosis factor‐α and nitric oxide. J Neuroimmunol. 1996;70(2):145–52. [DOI] [PubMed] [Google Scholar]
- 123. Tatomir A, Talpos‐Caia A, Anselmo F, Kruszewski AM, Boodhoo D, Rus V, et al. The complement system as a biomarker of disease activity and response to treatment in multiple sclerosis. Immunol Res. 2017;65(6):1103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Soane L, Cho HJ, Niculescu F, Rus H, Shin ML. C5b–9 terminal complement complex protects oligodendrocytes from death by regulating Bad through phosphatidylinositol 3‐kinase/Akt pathway. J Immunol. 2001;167(4):2305–11. [DOI] [PubMed] [Google Scholar]
- 125. Watkins LM, Neal JW, Loveless S, Michailidou I, Ramaglia V, Rees MI, et al. Complement is activated in progressive multiple sclerosis cortical grey matter lesions. J Neuroinflammation. 2016;13(1):161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Nataf S, Carroll SL, Wetsel RA, Szalai AJ, Barnum SR. Attenuation of experimental autoimmune demyelination in complement‐deficient mice. J Immunol. 2000;165(10):5867–73. [DOI] [PubMed] [Google Scholar]
- 127. Tran GT, Hodgkinson SJ, Carter N, Killingsworth M, Spicer ST, Hall BM. Attenuation of experimental allergic encephalomyelitis in complement component 6‐deficient rats is associated with reduced complement C9 deposition, P‐selectin expression, and cellular infiltrate in spinal cords. J Immunol. 2002;168(9):4293–300. [DOI] [PubMed] [Google Scholar]
- 128. Kjaeldgaard AL, Pilely K, Olsen KS, Pedersen SW, Lauritsen AØ, Møller K, et al. Amyotrophic lateral sclerosis: The complement and inflammatory hypothesis. Mol Immunol. 2018;102:14–25. [DOI] [PubMed] [Google Scholar]
- 129. Woodruff TM, Costantini KJ, Crane JW, Atkin JD, Monk PN, Taylor SM, et al. The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J Immunol. 2008;181(12):8727–34. [DOI] [PubMed] [Google Scholar]
- 130. Chen NJ, Mirtsos C, Suh D, Lu YC, Lin WJ, McKerlie C, et al. C5L2 is critical for the biological activities of the anaphylatoxins C5a and C3a. Nature. 2007;446(7132):203–7. [DOI] [PubMed] [Google Scholar]
- 131. Wang HA, Lee JD, Lee KM, Woodruff TM, Noakes PG. Complement C5a–C5aR1 signalling drives skeletal muscle macrophage recruitment in the hSOD1 G93A mouse model of amyotrophic lateral sclerosis. Skelet Muscle. 2017;7(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Woodruff TM, Lee JD, Noakes PG. Role for terminal complement activation in amyotrophic lateral sclerosis disease progression. Proc Natl Acad Sci USA. 2014;111(1):E3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Lee JD, Levin SC, Willis EF, Li R, Woodruff TM, Noakes PG. Complement components are upregulated and correlate with disease progression in the TDP‐43 Q331K mouse model of amyotrophic lateral sclerosis. J Neuroinflammation. 2018;15(1):1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ferraiuolo L, Heath PR, Holden H, Kasher P, Kirby J, Shaw PJ. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J Neurosci. 2007;27(34):9201–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Sta M, Sylva‐Steenland RM, Casula M, de Jong JM, Troost D, Aronica E, et al. Innate and adaptive immunity in amyotrophic lateral sclerosis: evidence of complement activation. Neurobiol Dis. 2011;42(3):211–20. [DOI] [PubMed] [Google Scholar]
- 136. Färber K, Cheung G, Mitchell D, Wallis R, Weihe E, Schwaeble W, et al. C1q, the recognition subcomponent of the classical pathway of complement, drives microglial activation. J Neurosci Res. 2009;87(3):644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Spiller KJ, Restrepo CR, Khan T, Dominique MA, Fang TC, Canter RG, et al. Microglia‐mediated recovery from ALS‐relevant motor neuron degeneration in a mouse model of TDP‐43 proteinopathy. Nat Neurosci. 2018;21(3):329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Geloso MC, Corvino V, Marchese E, Serrano A, Michetti F, D’Ambrosi N. The dual role of microglia in ALS: mechanisms and therapeutic approaches. Front Aging Neurosci. 2017;9:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1‐expressing microglia. Proc Natl Acad Sci USA. 1998;95(18):10896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Boehme SA, Lio FM, Maciejewski‐Lenoir D, Bacon KB, Conlon PJ. The chemokine fractalkine inhibits Fas‐mediated cell death of brain microglia. J Immunol. 2000;165(1):397–403. [DOI] [PubMed] [Google Scholar]
- 141. Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano‐Sanchez A, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185(2):232–40. [DOI] [PubMed] [Google Scholar]
- 142. Pehar M, Cassina P, Vargas MR, Castellanos R, Viera L, Beckman JS, et al. Astrocytic production of nerve growth factor in motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. 2004;89(2):464–73. [DOI] [PubMed] [Google Scholar]
- 143. Krenz NR, Weaver LC. Nerve growth factor in glia and inflammatory cells of the injured rat spinal cord. J Neurochem. 2000;74(2):730–9. [DOI] [PubMed] [Google Scholar]
- 144. Kawamata T, Akiyama H, Yamada T, McGeer PL. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol. 1992;140(3):691. [PMC free article] [PubMed] [Google Scholar]
- 145. Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344(22):1688–700. [DOI] [PubMed] [Google Scholar]
- 146. Yasojima K, Tourtellotte WW, McGeer EG, McGeer PL. Marked increase in cyclooxygenase‐2 in ALS spinal cord: implications for therapy. Neurology. 2001;57(6):952–6. [DOI] [PubMed] [Google Scholar]
- 147. Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622–36. [DOI] [PubMed] [Google Scholar]
- 148. Luo C, Jian C, Liao Y, Huang Q, Wu Y, Liu X, et al. The role of microglia in multiple sclerosis. Neuropsychiatr Dis Treat. 2017;13:1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Palmer AM. Multiple sclerosis and the blood‐central nervous system barrier. Cardiovasc Psychiatry Neurol. 2013;2013:530356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 2015;14(4):406–19. [DOI] [PubMed] [Google Scholar]
- 151. Kasper LH, Shoemaker J. Multiple sclerosis immunology: the healthy immune system vs the MS immune system. Neurology. 2010;74(Supplement 1):S2–8. [DOI] [PubMed] [Google Scholar]
- 152. Strachan‐Whaley M, Rivest S, Yong VW. Interactions between microglia and T cells in multiple sclerosis pathobiology. J Interferon Cytokine Res. 2014;34(8):615–22. [DOI] [PubMed] [Google Scholar]
- 153. Henderson AP, Barnett MH, Parratt JD, Prineas JW. Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann Neurol. 2009;66(6):739–53. [DOI] [PubMed] [Google Scholar]
- 154. Khattar M, Chen W, Stepkowski SM. Expanding and converting regulatory T cells: a horizon for immunotherapy. Arch Immunol Ther Exp (Warsz). 2009;57(3):199–204. [DOI] [PubMed] [Google Scholar]
- 155. Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, et al. Nitrated α–synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS One. 2008;3(1):e1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Costantino CM, Baecher‐Allan CM, Hafler DA. Human regulatory T cells and autoimmunity. Eur J Immunol. 2008;38(4):921–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Morrice JR, Gregory‐Evans CY, Shaw CA. Necroptosis in amyotrophic lateral sclerosis and other neurological disorders. Biochim Biophys Acta Mol Basis Dis. 2017;1863(2):347–53. [DOI] [PubMed] [Google Scholar]
- 158. Gugliandolo A, Giacoppo S, Bramanti P, Mazzon E. NLRP3 inflammasome activation in a transgenic amyotrophic lateral sclerosis model. Inflammation. 2018;41(1):93–103. [DOI] [PubMed] [Google Scholar]
- 159. Johann S, Heitzer M, Kanagaratnam M, Goswami A, Rizo T, Weis J, et al. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia. 2015;63(12):2260–73. [DOI] [PubMed] [Google Scholar]
- 160. Kadhim H, Deltenre P, Martin JJ, Sebire G. In‐situ expression of Interleukin‐18 and associated mediators in the human brain of sALS patients: hypothesis for a role for immune‐inflammatory mechanisms. Med Hypotheses. 2016;86:14–7. [DOI] [PubMed] [Google Scholar]
- 161. Debye B, Schmülling L, Zhou L, Rune G, Beyer C, Johann S. Neurodegeneration and NLRP3 inflammasome expression in the anterior thalamus of SOD1 (G93A) ALS mice. Brain Pathol. 2018;28(1):14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Meissner F, Molawi K, Zychlinsky A. Mutant superoxide dismutase 1‐induced IL‐1β accelerates ALS pathogenesis. Proc Natl Acad Sci USA. 2010;107(29):13046–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Deora V, Lee JD, Albornoz EA, McAlary L, Jagaraj CJ, Robertson AA, et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia. 2020;68(2):407–21. [DOI] [PubMed] [Google Scholar]
- 164. Keane RW, Dietrich WD, de Rivero Vaccari JP. Inflammasome proteins as biomarkers of multiple sclerosis. Front Neurol. 2018;9:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Soares JL, Oliveira EM, Pontillo A. Variants in NLRP3 and NLRC4 inflammasome associate with susceptibility and severity of multiple sclerosis. Mult Scler Relat Disord. 2019;29:26–34. [DOI] [PubMed] [Google Scholar]
- 166. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2012;109(26):10480–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Losy J, Niezgoda A. IL‐18 in patients with multiple sclerosis. Acta Neurol Scand. 2001;104(3):171–3. [DOI] [PubMed] [Google Scholar]
- 169. Willing A, Leach OA, Ufer F, Attfield KE, Steinbach K, Kursawe N, et al. CD 8+ MAIT cells infiltrate into the CNS and alterations in their blood frequencies correlate with IL‐18 serum levels in multiple sclerosis. Eur J Immunol. 2014;44(10):3119–28. [DOI] [PubMed] [Google Scholar]
- 170. Vidmar L, Maver A, Drulović J, Sepčić J, Novaković I, Ristič S, et al. Multiple Sclerosis patients carry an increased burden of exceedingly rare genetic variants in the inflammasome regulatory genes. Sci Rep. 2019;9(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Dharmadasa T, Kiernan MC. Riluzole, disease stage and survival in ALS. Lancet Neurol. 2018;17(5):385–6. [DOI] [PubMed] [Google Scholar]
- 172. Yoshino H. Edaravone for the treatment of amyotrophic lateral sclerosis. Expert Rev Neurother. 2019;19(3):185–93. [DOI] [PubMed] [Google Scholar]
- 173. Kiaei M, Petri S, Kipiani K, Gardian G, Choi DK, Chen J, et al. Thalidomide and lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2006;26(9):2467–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Karlsson J, Fong KS, Hansson MJ, Elmàr E, Csiszar K, Keep MF. Life span extension and reduced neuronal death after weekly intraventricular cyclosporin injections in the G93A transgenic mouse model of amyotrophic lateral sclerosis. J Neurosurg. 2004;101(1):128–37. [DOI] [PubMed] [Google Scholar]
- 175. Appel SH, Stewart SS, Appel V, Harati Y, Mietlowski W, Weiss W, et al. A double‐blind study of the effectiveness of cyclosporine in amyotrophic lateral sclerosis. Arch Neurol. 1988;45(4):381–6. [DOI] [PubMed] [Google Scholar]
- 176. Meininger V, Drory VE, Leigh PN, Ludolph A, Robberecht W, Silani V. Glatiramer acetate has no impact on disease progression in ALS at 40 mg/day: a double‐blind, randomized, multicentre, placebo‐controlled trial. Amyotroph Lateral Scler. 2009;10(5–6):378–83. [DOI] [PubMed] [Google Scholar]
- 177. Maier A, Deigendesch N, Müller K, Weishaupt JH, Krannich A, Röhle R, et al. Interleukin‐1 antagonist anakinra in amyotrophic lateral sclerosis—a pilot study. PLoS One. 2015;10(10):e0139684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Grottelli S, Mezzasoma L, Scarpelli P, Cacciatore I, Cellini B, Bellezza I. Cyclo (His‐Pro) inhibits NLRP3 inflammasome cascade in ALS microglial cells. Mol Cell Neurosci. 2019;94:23–31. [DOI] [PubMed] [Google Scholar]
- 179. Zhao B, Marciniuk K, Gibbs E, Yousefi M, Napper S, Cashman NR. Therapeutic vaccines for amyotrophic lateral sclerosis directed against disease specific epitopes of superoxide dismutase 1. Vaccine. 2019;37(35):4920–7. [DOI] [PubMed] [Google Scholar]
- 180. Smith AL, Cohen JA, Hua LH. Therapeutic targets for multiple sclerosis: current treatment goals and future directions. Neurotherapeutics. 2017;14(4):952–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Goodin DS. Disease‐modifying therapy in multiple sclerosis: update and clinical implications. Neurology. 2008;71(24 suppl 3):S8–13. [DOI] [PubMed] [Google Scholar]
- 182. Edan G, Miller D, Clanet M, Confavreux C, Lyon‐Caen O, Lubetzki C, et al. Therapeutic effect of mitoxantrone combined with methylprednisolone in multiple sclerosis: a randomised multicentre study of active disease using MRI and clinical criteria. J Neurol Neurosurg Psychiatry. 1997;62(2):112–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. TaŞKapilioĞLu Ö. Recent advances in the treatment for multiple sclerosis; Current new drugs specific for multiple sclerosis. Arch Neuropsychiatry. 2018;55(Suppl 1):S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Eckstein C, Bhatti MT. Currently approved and emerging oral therapies in multiple sclerosis: An update for the ophthalmologist. Surv Ophthalmol. 2016;61(3):318–32. [DOI] [PubMed] [Google Scholar]
- 185. Miller AE, Wolinsky JS, Kappos L, Comi G, Freedman MS, Olsson TP, et al. Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis (TOPIC): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Neurol. 2014;13(10):977–86. [DOI] [PubMed] [Google Scholar]
- 186. Mills EA, Ogrodnik MA, Plave A, Mao‐Draayer Y. Emerging understanding of the mechanism of action for dimethyl fumarate in the treatment of multiple sclerosis. Front Neurol. 2018;9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Melis JP, Strumane K, Ruuls SR, Beurskens FJ, Schuurman J, Parren PW. Complement in therapy and disease: regulating the complement system with antibody‐based therapeutics. Mol Immunol. 2015;67(2):117–30. [DOI] [PubMed] [Google Scholar]
- 188. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3‐engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation. 2012;9(1):112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Walker FO. Huntington's disease. Lancet. 2007;369(9557):218–28. [DOI] [PubMed] [Google Scholar]
- 190. Nayak A, Ansar R, Verma SK, Bonifati DM, Kishore U. Huntington's disease: an immune perspective. Neurol Res Int. 2011;2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72(6):971–83. [DOI] [PubMed] [Google Scholar]
- 192. Rawlins MD, Wexler NS, Wexler AR, Tabrizi SJ, Douglas I, Evans SJ, et al. The prevalence of Huntington's disease. Neuroepidemiology. 2016;46(2):144–53. [DOI] [PubMed] [Google Scholar]
- 193. Singhrao SK, Neal JW, Morgan BP, Gasque P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington's disease. Exp Neurol. 1999;159(2):362–76. [DOI] [PubMed] [Google Scholar]
- 194. Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet. 2006;15(6):965–77. [DOI] [PubMed] [Google Scholar]
- 195. Larkin PB, Muchowski PJ. Genetic deficiency of complement component 3 does not alter disease progression in a mouse model of Huntington's disease. J Huntingtons Dis. 2012;1(1):107–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Zhao T, Hong Y, Li S, Li XJ. Compartment‐dependent degradation of mutant huntingtin accounts for its preferential accumulation in neuronal processes. J Neurosci. 2016;36(32):8317–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Wang N, Gray M, Lu XH, Cantle JP, Holley SM, Greiner E, et al. Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington's disease. Nat Med. 2014;20(5):536–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Ochaba J, Monteys AM, O’Rourke JG, Reidling JC, Steffan JS, Davidson BL, et al. PIAS1 regulates mutant huntingtin accumulation and Huntington’s disease‐associated phenotypes in vivo. Neuron. 2016;90(3):507–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Ghosh S, May MJ, Kopp EB. NF‐κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16(1):225–60. [DOI] [PubMed] [Google Scholar]
- 200. Yang HM, Yang S, Huang SS, Tang BS, Guo JF. Microglial activation in the pathogenesis of Huntington’s disease. Front Aging Neurosci. 2017;9:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Ghosh S, Karin M. Missing pieces in the NF‐κB puzzle. Cell. 2002;109(2):S81–96. [DOI] [PubMed] [Google Scholar]
- 202. Khoshnan A, Ko J, Watkin EE, Paige LA, Reinhart PH, Patterson PH. Activation of the IκB kinase complex and nuclear factor‐κB contributes to mutant huntingtin neurotoxicity. J Neurosci. 2004;24(37):7999–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Träger U, Andre R, Lahiri N, Magnusson‐Lind A, Weiss A, Grueninger S, et al. HTT‐lowering reverses Huntington’s disease immune dysfunction caused by NFκB pathway dysregulation. Brain. 2014;137(3):819–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Raison CL, Dantzer R, Kelley KW, Lawson MA, Woolwine BJ, Vogt G, et al. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN‐α: relationship to CNS immune responses and depression. Mol Psychiatry. 2010;15(4):393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Pocivavsek A, Wu HQ, Potter MC, Elmer GI, Pellicciari R, Schwarcz R. Fluctuations in endogenous kynurenic acid control hippocampal glutamate and memory. Neuropsychopharmacology. 2011;36(11):2357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Staal RGW, Möller T. Neuroinflammation in Huntington’s disease In Peterson PK, Toborek M. (Eds.), Neuroinflammation and Neurodegeneration. New York, NY: Springer; 2014:179–97. [Google Scholar]
- 207. Sathyasaikumar KV, Stachowski EK, Amori L, Guidetti P, Muchowski PJ, Schwarcz R. Dysfunctional kynurenine pathway metabolism in the R6/2 mouse model of Huntington’s disease. J Neurochem. 2010;113(6):1416–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Glass M, Northup JK. Agonist selective regulation of G proteins by cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1999;56(6):1362–9. [DOI] [PubMed] [Google Scholar]
- 209. Bisogno T, Oddi S, Piccoli A, Fazio D, Maccarrone M. Type‐2 cannabinoid receptors in neurodegeneration. Pharmacol Res. 2016;111:721–30. [DOI] [PubMed] [Google Scholar]
- 210. Chiarlone A, Bellocchio L, Blázquez C, Resel E, Soria‐Gómez E, Cannich A, et al. A restricted population of CB1 cannabinoid receptors with neuroprotective activity. Proc Natl Acad Sci USA. 2014;111(22):8257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Blázquez C, Chiarlone A, Bellocchio L, Resel E, Pruunsild P, García‐Rincón D, et al. The CB 1 cannabinoid receptor signals striatal neuroprotection via a PI3K/Akt/mTORC1/BDNF pathway. Cell Death Differ. 2015;22(10):1618–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Palazuelos J, Aguado T, Pazos MR, Julien B, Carrasco C, Resel E, et al. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington's disease excitotoxicity. Brain. 2009;132(11):3152–64. [DOI] [PubMed] [Google Scholar]
- 213. Ellrichmann G, Reick C, Saft C, Linker RA. The role of the immune system in Huntington’s disease. Clin Dev Immunol. 2013;2013:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106(3):255–8. [DOI] [PubMed] [Google Scholar]
- 215. Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, et al. Minocycline inhibits caspase‐1 and caspase‐3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. 2000;6(7):797–801. [DOI] [PubMed] [Google Scholar]
- 216. Sperlágh B, Illes P. P2X7 receptor: an emerging target in central nervous system diseases. Trends Pharmacol Sci. 2014;35(10):537–47. [DOI] [PubMed] [Google Scholar]
- 217. Labzin LI, Heneka MT, Latz E. Innate immunity and neurodegeneration. Annu Rev Med. 2018;69:437–49. [DOI] [PubMed] [Google Scholar]
- 218. Rodrigues FB, Wild EJ. Huntington’s disease clinical trials corner: August 2018. J Huntingtons Dis. 2018;7(3):279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Southwell AL, Franciosi S, Villanueva EB, Xie Y, Winter LA, Veeraraghavan J, et al. Anti‐semaphorin 4D immunotherapy ameliorates neuropathology and some cognitive impairment in the YAC128 mouse model of Huntington disease. Neurobiol Dis. 2015;76:46–56. [DOI] [PubMed] [Google Scholar]
- 220. Soulet D, Cicchetti F. The role of immunity in Huntington's disease. Mol Psychiatry. 2011;16(9):889–902. [DOI] [PubMed] [Google Scholar]
- 221. Ma Y, Liu Y, Zhang Z, Yang GY. Significance of complement system in ischemic stroke: a comprehensive review. Aging Dis. 2019;10(2):429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Széplaki G, Szegedi R, Hirschberg K, Gombos T, Varga L, Karádi I, et al. Strong complement activation after acute ischemic stroke is associated with unfavorable outcomes. Atherosclerosis. 2009;204(1):315–20. [DOI] [PubMed] [Google Scholar]
- 223. Niemela M, Tulamo R, Hernesniemi J, Scozzafava J, Findlay JM, Macdonald RL, Dempsey RJ.Alterations in plasma complement levels after human ischemic stroke‐Comments.
- 224. Schäfer MK, Schwaeble WJ, Post C, Salvati P, Calabresi M, Sim RB, et al. Complement C1q is dramatically up‐regulated in brain microglia in response to transient global cerebral ischemia. J Immunol. 2000;164(10):5446–52. [DOI] [PubMed] [Google Scholar]
- 225. Ten VS, Sosunov SA, Mazer SP, Stark RI, Caspersen C, Sughrue ME, et al. C1q‐deficiency is neuroprotective against hypoxic‐ischemic brain injury in neonatal mice. Stroke. 2005;36(10):2244–50. [DOI] [PubMed] [Google Scholar]
- 226. Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, et al. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci. 2013;33(33):13460–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Cervera A, Planas AM, Justicia C, Urra X, Jensenius JC, Torres F, et al. Genetically‐defined deficiency of mannose‐binding lectin is associated with protection after experimental stroke in mice and outcome in human stroke. PLoS One. 2010;5(2):e8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Elvington A, Atkinson C, Zhu H, Yu J, Takahashi K, Stahl GL, et al. The alternative complement pathway propagates inflammation and injury in murine ischemic stroke. J Immunol. 2012;189(9):4640–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Huang Y, Qiao F, Atkinson C, Holers VM, Tomlinson S. A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in ischemia/reperfusion injury. J Immunol. 2008;181(11):8068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Perego C, Fumagalli S, De Simoni MG. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J Neuroinflammation. 2011;8(1):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Li T, Pang S, Yu Y, Wu X, Guo J, Zhang S. Proliferation of parenchymal microglia is the main source of microgliosis after ischaemic stroke. Brain. 2013;136(12):3578–88. [DOI] [PubMed] [Google Scholar]
- 232. Ju F, Ran Y, Zhu L, Cheng X, Gao H, Xi X, et al. Increased BBB permeability enhances activation of microglia and exacerbates loss of dendritic spines after transient global cerebral ischemia. Front Cell Neurosci. 2018;12:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Lepore F, D'Alessandro G, Antonangeli F, Santoro A, Esposito V, Limatola C, et al. CXCL16/CXCR6 axis drives microglia/macrophages phenotype in physiological conditions and plays a crucial role in glioma. Front Immunol. 2018;9:2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Vay SU, Flitsch LJ, Rabenstein M, Rogall R, Blaschke S, Kleinhaus J, et al. The plasticity of primary microglia and their multifaceted effects on endogenous neural stem cells in vitro and in vivo. J Neuroinflammation. 2018;15(1):226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Aloisi F. Immune function of microglia. Glia. 2001;36(2):165–79. [DOI] [PubMed] [Google Scholar]
- 236. Vogelgesang A, Dressel A. Immunological consequences of ischemic stroke: immunosuppression and autoimmunity. J Neuroimmunol. 2011;231(1–2):105–10. [DOI] [PubMed] [Google Scholar]
- 237. Jin C, Flavell RA. Innate sensors of pathogen and stress: linking inflammation to obesity. J Allergy Clin Immunol. 2013;132(2):287–94. [DOI] [PubMed] [Google Scholar]
- 238. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Hua KF, Yang SM, Kao TY, Chang JM, Chen HL, Tsai YJ, et al. Osthole mitigates progressive IgA nephropathy by inhibiting reactive oxygen species generation and NF‐κB/NLRP3 pathway. PLoS One. 2013;8(10):e77794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Lin M, Tang SC. Toll‐like receptors: sensing and reacting to diabetic injury in the kidney. Nephrol Dial Transplant. 2014;29(4):746–54. [DOI] [PubMed] [Google Scholar]
- 241. Palová‐Jelínková L, Dáňová K, Drašarová H, Dvořák M, Funda DP, Fundová Pe, et al. Pepsin digest of wheat gliadin fraction increases production of IL‐1β via TLR4/MyD88/TRIF/MAPK/NF‐κB signaling pathway and an NLRP3 inflammasome activation. PLoS One. 2013;8(4):e62426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Segovia J, Sabbah A, Mgbemena V, Tsai SY, Chang TH, Berton MT, et al. TLR2/MyD88/NF‐κB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS One. 2012;7(1):e29695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, et al. Activated platelets mediate inflammatory signaling by regulated interleukin 1β synthesis. J Cell Biol. 2001;154(3):485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite β‐hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat Med. 2015;21(3):263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Zheng F, Xing S, Gong Z, Xing Q. NLRP3 inflammasomes show high expression in aorta of patients with atherosclerosis. Heart Lung Circ. 2013;22(9):746–50. [DOI] [PubMed] [Google Scholar]
- 246. Fann DY, Lee SY, Manzanero S, Chunduri P, Sobey CG, Arumugam TV. Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res Rev. 2013;12(4):941–66. [DOI] [PubMed] [Google Scholar]
- 247. Liao KC, Mogridge J. Activation of the Nlrp1b inflammasome by reduction of cytosolic ATP. Infect Immun. 2013;81(2):570–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Ismael S, Zhao L, Nasoohi S, Ishrat T. Inhibition of the NLRP3‐inflammasome as a potential approach for neuroprotection after stroke. Sci Rep. 2018;8(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz‐Planillo R, Inserra MC, et al. A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Saver JL, Goyal M, Bonafe A, Diener HC, Levy EI, Pereira VM, et al. Stent‐retriever thrombectomy after intravenous t‐PA vs. t‐PA alone in stroke. N Engl J Med. 2015;372(24):2285–95. [DOI] [PubMed] [Google Scholar]
- 251. Hernandez AL, Kitz A, Wu C, Lowther DE, Rodriguez DM, Vudattu N, et al. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J Clin Investig. 2015;125(11):4212–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Okoye IS, Coomes SM, Pelly VS, Czieso S, Papayannopoulos V, Tolmachova T, et al. MicroRNA‐containing T‐regulatory‐cell‐derived exosomes suppress pathogenic T helper 1 cells. Immunity. 2014;41(1):89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Asiimwe N, Yeo SG, Kim MS, Jung J, Jeong NY. Nitric oxide: exploring the contextual link with Alzheimer’s disease. Oxid Med Cell Longev. 2016;2016:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Erny D, de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18(7):965–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Carrillo‐Salinas FJ, Mestre L, Mecha M, Feliú A, Del Campo R, Villarrubia N, et al. Gut dysbiosis and neuroimmune responses to brain infection with Theiler’s murine encephalomyelitis virus. Sci Rep. 2017;7:44377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Wang Y, Wang Z, Wang Y, Li F, Jia J, Song X, et al. The gut‐microglia connection: implications for central nervous system diseases. Front Immunol. 2018;9:2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Deczkowska A, Schwartz M. Targeting neuro–immune communication in neurodegeneration: Challenges and opportunities. J Exp Med. 2018;215(11):2702–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Readhead B, Haure‐Mirande JV, Funk CC, Richards MA, Shannon P, Haroutunian V, et al. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron. 2018;99(1):64–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, et al. Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer’s disease pathology. Nat Commun. 2015;6(1):1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Keren‐Shaul H, Spinrad A, Weiner A, Matcovitch‐Natan O, Dvir‐Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169(7):1276–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.