

Abstract
Inflammasomes are large cytosolic multiprotein complexes assembled in response to infection and cellular stress, and are crucial for the activation of inflammatory caspases and the subsequent processing and release of pro-inflammatory mediators. While caspase-1 is activated within the canonical inflammasome, the related caspase-4 (also known as caspase-11 in mice) and caspase-5 are activated within the non-canonical inflammasome upon sensing of cytosolic lipopolysaccharide (LPS) from Gram-negative bacteria. However, the consequences of canonical and non-canonical inflammasome activation are similar. Caspase-1 promotes the processing and release of the pro-inflammatory cytokines interleukin (IL)-1β and IL-18 and the release of danger signals, as well as a lytic form of cell death called pyroptosis, whereas caspase-4, caspase-5 and caspase-11 directly promote pyroptosis through cleavage of the pore-forming protein gasdermin D (GSDMD), and trigger a secondary activation of the canonical NLRP3 inflammasome for cytokine release. Since the presence of the non-canonical inflammasome activator LPS leads to endotoxemia and sepsis, non-canonical inflammasome activation and regulation has important clinical ramifications. Here we discuss the mechanism of non-canonical inflammasome activation, mechanisms regulating its activity and its contribution to health and disease.
Keywords: caspase-4, caspase-5, caspase-11, non-canonical inflammasome, LPS, pyroptosis, sepsis, guanylate-binding protein
1. Introduction
Innate immunity relies on a coordinated response upon detection of conserved microbial- or pathogen-associated molecular patterns (MAMPs and PAMPs) and host-derived danger associated molecular patterns (DAMPs) by multiple pattern recognition receptors (PRRs). Transmembrane PRRs belonging to the Toll-like receptor (TLR) family respond to a diverse repertoire of DAMPs and PAMPs within the extracellular environment and endosomes. For instance, TLR4 specifically senses the major outer cell wall component of Gram-negative bacteria Lipopolysaccharide (LPS) (Hoshino et al., 1999; Poltorak et al., 1998). TLR receptor engagement leads to the activation of MyD88 or TRIF-dependent cytosolic signaling pathways, culminating in the activation of nuclear factor-kappa B (NF-κB), mitogen-activated protein kinases (MAPKs) and interferon regulatory factors (IRFs), and subsequently the transcription of inflammatory cytokines, interferons, and antimicrobial factors (Kawai and Akira, 2010). In addition to membrane bound PRRs, there are intracellular PRRs which may assemble signaling platforms referred to as inflammasomes. Canonical inflammasomes are assembled by the nucleotide-binding oligomerization domain (NOD) leucine-rich repeat (LRR)-containing protein receptors (NLRs), including NLRP1, NLRP3, NLRC4, NLRP6, NLRP7, NLRP9b, the absent in melanoma 2 (AIM2)-like receptor (ALR) AIM2, as well as Pyrin (Broz and Dixit, 2016; Dorfleutner et al., 2015; Martinon et al., 2002). Several other related proteins have also been implicated as inflammasome sensors, but yet with limited evidence, including NLRP2, NLRC5, NLRP12, and Interferon-γ inducible protein 16 (IFI16), which all have other, more established cellular functions (Davis et al., 2011; Kerur et al., 2011; Matsuoka et al., 2019; Minkiewicz et al., 2013; Vladimer et al., 2012). Ligand recognition and inflammasome sensor activation results in sensor oligomerization and recruitment of the essential adaptor protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), which nucleates ASC polymerization and consequently, proximity-induced clustering and activation of caspase-1. Active caspase-1 then proteolytically matures pro-IL-1β and pro-IL-18 into their biologically active, secreted forms (Cerretti et al., 1992; Dinarello, 1998; Thornberry et al., 1992). Maximal IL-1β and IL-18 secretion occurs after inflammasome-mediated pore formation or lytic cell death known as pyroptosis, which is also influenced by membrane repair mechanisms (Evavold et al., 2018; Fink and Cookson, 2006; Ruhl et al., 2018). To prevent improper canonical NLRP3 inflammasome activation, a two-step activation mechanism is employed. First, “priming”, for example by TLRs, to arm the system, which involves activation of NF-κB to upregulate inflammasome components and cytokine targets and posttranslational modifications of inflammasome components and second, “activation”, which then promotes inflammasome assembly (Khare et al., 2010; Swanson et al., 2019). Unlike other canonical inflammasomes, NLRP3 appears to not directly recognize a specific PAMP or DAMP, but is rather activated by molecular events associated with microbial infection and cellular stress, including potassium efflux, calcium signaling, lysosomal rupture and mitochondrial defects, including reactive oxygen species (ROS) production and the release of oxidized mitochondrial DNA. NLRP3 and several other canonical inflammasomes are regulated by a family of small endogenous inhibitors belonging to the PYRIN domain-only proteins (POPs) and caspase recruitment and activation domain (CARD)-only proteins (COPs) (Chu et al., 2015; Dorfleutner et al., 2015; Indramohan et al., 2018; Swanson et al., 2019). The NLRP3 inflammasome has also been implicated in cellular responses to LPS (Mariathasan et al., 2006). However, more recently LPS has been found to trigger the activation of a distinct type of inflammasome, known as the “non-canonical” inflammasome (Hagar et al., 2013; Kayagaki et al., 2011; Kayagaki et al., 2013; Matikainen et al., 2020; Rathinam et al., 2019; Shi et al., 2014; Yi, 2017). Rather than activating caspase-1, the non-canonical inflammasome results in the activation of the related caspase-4 and caspase-5 in humans and caspase-11 in mice (Fig. 1). However, the functional consequences are reminiscent to canonical inflammasome responses, featuring pyroptosis, as well as IL-1β and IL-18 release through secondary activation of the canonical NLRP3 inflammasome, referred to as non-canonical NLRP3 inflammasome activation (Kayagaki et al., 2015; Matikainen et al., 2020; Rathinam et al., 2019; Yi, 2017, 2020). Unlike canonical inflammasomes where ligand sensing, assembly, and effector functions are carried out by multiple protein components, the non-canonical caspases function as both sensor and effector molecules for LPS. However, recent evidence also implicates the guanylate-binding protein (GBP) 1 as an LPS sensor upstream of the non-canonical inflammasome, which triggers GBP hetero-oligomerization and recruitment of caspase-4 (Fisch et al., 2020; Kutsch et al., 2020; Santos et al., 2020; Wandel et al., 2020). Although pyroptosis has largely been considered a form of cell death restricted to innate immune cells, this pathway is also active in epithelial cells, hepatocytes, keratinocytes, endothelial cells and other non-immune cells (Cheng et al., 2017; Knodler et al., 2014; Liu et al., 2020; Shi et al., 2014).
Figure 1.

Schemata of human (left) and mouse (right) inflammatory caspases. The yellow asterisk marks the catalytic center and red arrows mark proteolytic cleavage sites involved in processing the caspase zymogens.
2. The non-canonical inflammasome
2.1. Discovery of LPS as activator of the non-canonical inflammasome
LPS is a prototypic PAMP and a potent mediator of sepsis and septic shock, which remain a major cause of mortality and therefore, identifying the cellular response triggered by LPS has been the focus of intense investigations. LPS is comprised of three main parts: the most conserved lipid A moiety, a core oligosaccharide chain, and a variable polysaccharide chain known as O-antigen (Raetz and Whitfield, 2002). Specifically, the lipid A moiety of LPS is responsible for the immunostimulatory activity of LPS, which is recognized by the TLR4/MD2 complex in conjunction with its co-receptor CD14 following interaction with the LPS-binding protein (LBP) (Gegner et al., 1995; Zanoni et al., 2011). Excessive signaling by TLR4 plays a major role in LPS-induced shock or sepsis, and accordingly, Tlr4−/− mice are highly resistant to endotoxic shock upon injection of a high dose of LPS (Hoshino et al., 1999; Poltorak et al., 1998). However, mice deficient in caspase-1 (Casp1−/−) also show a comparable resistance to LPS-induced shock, as do mice deficient in other inflammasome components, such as ASC and NLRP3 (Li et al., 1995; Mariathasan et al., 2004; Mariathasan et al., 2006; Yamamoto et al., 2004). Furthermore, deletion of caspase-11 (Casp11−/−) phenocopies this LPS resistance of caspase-1 and other canonical inflammasome component deficiencies (Wang et al., 1998). Hence, LPS mediated sepsis is also linked to inflammasome activation. Investigating the response of murine macrophages to LPS and cholera toxin B (CTB), as well as other bacterial inflammasome activators, led to the discovery of this non-canonical inflammasome pathway, which relies on caspase-11 and is independent of classical TLR4 signaling (Aachoui et al., 2013; Kayagaki et al., 2011; Kayagaki et al., 2013; Shi et al., 2014). The ability of CTB to activate caspase-11 in LPS primed bone marrow macrophages was eventually determined to be due to the intracellular delivery of LPS by CTB. The originally used Casp1−/− mice were generated from 129S6 strain ES cells, which carry a five-base pair deletion in Casp11 and therefore encode a nonfunctional protein, and were unknowingly double knock out for Casp1 and Casp11 (Casp1/11−/−) (Kayagaki et al., 2011). Unfortunately, the close proximity of Casp1 and Casp11 loci prevents segregation by recombination. However, transgenic expression of caspase-11 in those original Casp1/11−/− mice (Casp1/11−/−/Casp11TG) reverse their phenotype and render these mice susceptible to LPS-induced shock, which demonstrates that caspase-11 rather than caspase-1 is the critical component in this pathway (Kayagaki et al., 2011). Furthermore, loss of caspase-11 alone (Casp11−/−) in BMDMs does not affect IL-1β secretion when stimulated with canonical NLRP3, AIM2 and NLRC4-dependent inflammasome activators, but impairs IL-1β secretion in response to infection with Gram-negative bacteria Escherichia coli, Citrobacter rodentium and Vibrio cholerae, which elicits an LPS/caspase-11-driven response (Kayagaki et al., 2011). Ultimately, this distinct response of caspase-1 and caspase-11 was verified in Casp1−/− mice generated on the C57BL/6 strain, which encodes a functional caspase-11 (Kayagaki et al., 2015; Man et al., 2017). Caspase-11 deficiency but not canonical inflammasome component deficiency, protects mice against lethality in response to a high LPS dose (Kayagaki et al., 2011). Interestingly, Nlrp3 or Asc deficiency is able to ameliorate lethality in response to lower doses of LPS, implying that the canonical inflammasome may play a role in the amplification of the shock response (Mariathasan et al., 2004; Mariathasan et al., 2006).
After the discovery of caspase-11 in mice (Wang et al., 1996; Wang et al., 1998), efforts were made to identify human caspase-11. However, two homologous proteins, caspase-4 and caspase-5, were identified instead, which share approximately 55% protein identity to murine caspase-11 and cluster on chromosome 4 with caspase-1 and the caspase-1-regulatory COPs (Dorfleutner et al., 2015; Man and Kanneganti, 2016; Stehlik and Dorfleutner, 2007; Van Opdenbosch and Lamkanfi, 2019). Both caspases appear to redundantly mediate activation of the non-canonical inflammasome. However, caspase-4 exhibits a higher resemblance in size and sequence to caspase-11, which may also suggest a closer functional role. In human macrophages pyroptosis and NLRP3 inflammasome-mediated cytokine release in response to cytosolic LPS and invasive Gram-negative bacteria is impaired upon deletion of either caspase-4 or caspase-5, reminiscent to caspase-11 deletion (Casson et al., 2015; Kajiwara et al., 2014; Knodler et al., 2014; Lagrange et al., 2018; Platnich et al., 2018; Schmid-Burgk et al., 2015; Shi et al., 2014; Sollberger et al., 2012; Vigano et al., 2015). However, a recent study found that heme induced pyroptosis in human macrophages is predominantly mediated by caspase-4, but heme induced release of IL-1β is mediated by caspase-4 and caspase-5, as well as non-canonical activation of caspase-1. Hence, non-canonical inflammasome caspases can exhibit unique and non-redundant functions under certain circumstances (Bolívar et al., 2020).
2.2. Intracellular sensing of LPS and Gram-negative bacteria
A paradigm shift in innate immunity happened when murine caspase-11, the human ortholog caspase-4 and the related caspase-5 were identified as direct receptors for intracellular LPS (Shi et al., 2014). Similar to caspase-1 they are initiator caspases and are comprised of an N-terminal CARD, which is responsible for its oligomerization, and the p10 and the p20 caspase domains (Man and Kanneganti, 2016). Since canonical inflammasomes require an adaptor to link sensors to caspase-1 activation, investigators anticipated a CARD-containing protein may similarly serve as an LPS upstream sensor and activator of caspase-11. However, activation of caspase-11 was not observed after co-expression of 18 different CARD-containing proteins, suggesting that such a protein may not be necessary (Shi et al., 2014). However, a caspase bimolecular fluorescence complementation (BiFC) assay revealed the possibility of upstream sensors in caspase-4 and caspase-5 oligomerization, including NLRP1, NLRP3 and NLRC4 and NLRP1, respectively (Sanders et al., 2015). NLRP1 has also been earlier implicated in caspase-5 activation (Martinon et al., 2002). However, the hypothesis that caspase-11 itself might act as an LPS receptor arose from the observation that caspase-11 purified from E.coli, but not insect cells, was oligomerized under non-denaturing conditions, suggesting that bacterial components may induce caspase-11 oligomerization, which is an essential hallmark for its activation (Chang et al., 2003; Salvesen and Dixit, 1999; Shi et al., 2014). Indeed, LPS is responsible for this oligomerization, as insect cells incubated with LPS produced oligomeric caspase-11. Several positively charged motifs in the CARD of caspase-4, caspase-5 and caspase-11 are required for LPS binding (Shi et al., 2014). However, a subsequent cell-based analysis proposed a modified mode of interaction, as not only positively charged residues in the CARD of caspase-11 are critical for LPS binding but also positively charged amino acids in the caspase domain between amino acid 220 to 294 (Chu et al., 2018). Binding of caspase-11 to LPS not only triggers its oligomerization but also induces catalytic activity (Shi et al., 2014). Thus, unlike canonical inflammasome activation that requires a multiprotein scaffold for activation, non-canonical inflammasomes directly serve as receptors for intracellular LPS (Hagar et al., 2013; Kayagaki et al., 2013). However, recently several members of the GBP protein family have been implicated in the formation of a caspase-4 signaling platform (Fisch et al., 2020; Kutsch et al., 2020; Santos et al., 2020; Wandel et al., 2020).
2.3. The role of GBPs and LPS entry to the cytosol in non-canonical inflammasome activation
IFN inducible GTPases, including GBPs and immunity related GTPases (IRGs), have several important functions in the immune defense against pathogens. They can directly attach to bacteria and cause bacteriolysis, they can disrupt the outer membrane of pathogen-containing vacuoles so that pathogens are freely accessible for cytosolic PRR recognition, including the non-canonical inflammasome, and most recently they have been described to function as LPS receptors that recruit the non-canonical inflammasome caspases to cytosolic bacteria (Meunier et al., 2014; Pilla et al., 2014; Santos et al., 2018). Accordingly, deletion of all GBPs on chromosome 3 (Gbpchr3−/−) results in attenuated activation of caspase-11 (Balakrishnan et al., 2018; Meunier et al., 2014; Pilla et al., 2014; Tang et al., 2018). GBPs also recruit IRGB10 to the membranes of cytosolic bacteria in macrophages resulting in permeabilization and liberation of bacterial products, including LPS (Balakrishnan et al., 2018; Man et al., 2016). However, more recent studies in IFN-γ activated epithelial cells demonstrate that liberation of Salmonella Typhimurium from vacuolar membranes does not depend on GBP1, but that GBP1 instead acts as a cytosolic LPS receptor indispensable for non-canonical inflammasome activation (Kutsch et al., 2020; Santos et al., 2020; Wandel et al., 2020). After targeting the Gram-negative bacterial surface, GBP1 quickly recruits GBP2, GBP3 and GBP4 to create a stabilized GBPs oligomeric structure, which then sequesters caspase-4 to the bacterial surface, thus facilitating the activation of caspase-4 by LPS (Santos et al., 2020; Wandel et al., 2020). However, two studies found opposing results for the function of GBP2 and GBP3 in caspase-4 recruitment and activation (Santos et al., 2020; Wandel et al., 2020). While Santos et al. showed GBP1/3/4, but not GBP2, are adequate for caspase-4 recruitment and activation, Wandel et al. demonstrated that GBP1/2/4, but not GBP3, are essential for caspase-4 recruitment, while all four are required for caspase-4 activation. Comparable results were obtained using a Shigella flexneri mutant deficient in the virulence factor OspC3, which inhibits caspase-4 (Wandel et al., 2020). Further analysis revealed that GBP1 associates with LPS by electrostatic attraction of the negatively charged phosphate groups on lipid A and the inner core of LPS to the positively charged surface patch (KKK61-63) in the GTPase domain of GBP1, and is also dependent on its GTPase activity. Mutation of this patch impairs GBP1 recruitment to S. Typhimurium surfaces and GBP1 oligomerization (Santos et al., 2020). Other mutations within the GTPase domain, such as mutation of the Mg2+ binding site (GBP1S52N), GTP binding site (GBP1K51A) and GTP hydrolysis site (GBP1R48A) as well as a prenylation deficient mutant (GBP1C589S) also fail to target GBP1 to the bacterial surface and to recruit caspase-4 (Wandel et al., 2020). In agreement, LPS deficient in the O-antigen can still facilitate GBP1 coating and caspase-4 binding (Wandel et al., 2020). Caspase-4 subsequently binds to oligomerized GBP1 (Santos et al., 2020; Wandel et al., 2020). Furthermore, while LPS strongly binds GBP1, some LPS binding is also observed with GBP3, suggesting that GBP3 may also act as an LPS sensor (Santos et al., 2020). In vivo, infection with S. Typhimurium or S. flexneri ΔospC3, resulted in higher bacterial burden and lethality in Gbp1−/− mice (Wandel et al., 2020). Interestingly, the function of human GBP1 in cell-intrinsic immunity is microbe-specific, as GBP1 contributes to the disruption of both vacuolar and parasite plasma membranes of Toxoplasma gondii, and escape out of vacuoles in human macrophages, leading to AIM2 inflammasome activation and atypical apoptosis (Fisch et al., 2020). However, in contrast to S. Typhimurium infection, this response does not recruit caspase-4 and raises the possibility that GBP1 may recognize other MAMPs beyond LPS (Fisch et al., 2020).
Activation of caspase-4 leads to GSDMD cleavage, IL-18 maturation and release, and subsequent pyroptosis. Similarly, the GTPase domain of GBP1 is essential for IL-18 processing and release during infection (Wandel et al., 2020). However, purified but not membrane integrated LPS elicits caspase-4-dependent responses, which may be due to inaccessibility of the embedded lipid A moiety (Shi et al., 2014; Wandel et al., 2020). GBPs do not only concentrate caspase-4 at sufficiently high concentration on bacterial surfaces, but also make lipid A more accessible by potentially destabilizing the O-antigen barrier on bacterial surfaces (Kutsch et al., 2020). Nevertheless, bacterial host cell invasion is not a prerequisite for non-canonical inflammasome activation, as outer membrane vesicles (OMVs) generated by Gram-negative bacteria can also facilitate the entry of LPS to the cytosol. OMVs are membrane-bound structures released during bacterial growth and/or stress and contain an abundance of LPS (Ryu et al., 2017). They are internalized by endocytosis, and OMV-bound LPS is subsequently released into the cytosol from early endocytic compartments via GBPs and IRGB10 and activate non-canonical inflammasomes in human and mouse macrophages (Bitto et al., 2018; Cecil et al., 2017; Chen et al., 2018; Chu et al., 2018; Finethy et al., 2017; Santos et al., 2018; Vanaja et al., 2016; Wacker et al., 2017). In bone marrow-derived macrophages, GBP1, GBP2, and GBP5 are recruited to internalized OMVs and bind to LPS (Kim et al., 2016; Santos et al., 2018). Under conditions in which no pathogenic bacteria are present, such as injection of free LPS into mice, activation of the non-canonical inflammasome can also occur, however, the mechanism through which LPS gains access to the cytosol is not fully understood.
Besides vacuole disruption by cellular GBPs, some Gram-negative bacteria have their own escape mechanism, which consequently enables their cytosolic detection by the non-canonical inflammasome. Hence, no efficient immune response can be mounted in the absence of caspase-11 in mice, and Casp11−/− mice are more susceptible to infection with cytosol-invasive Gram-negative bacteria, including Burkholderia thailandensis, Burkholderia pseudomallei, Klebsiella pneumoniae, S. Typhimurium, Legionella pneumophila, S. flexneri and Acinetobacter baumannii (Aachoui et al., 2015; Aachoui et al., 2013; Broz et al., 2012; Knodler et al., 2014; Wang et al., 2017). Experimentally, some Gram-negative bacteria can be forced to enter the cytosol by mutating/deleting their phagosome stabilizing factors. For instance, S. Typhimurium mutants lacking the phagosome stabilizing factor SifA or the Salmonella pathogenicity island 2 (SPI2), as well as L. pneumophila lacking the vacuole stabilizing SdhA are primarily cytosolic (Aachoui et al., 2013). LPS internalization can also occur by endocytosis through direct binding to CD14 (Gegner et al., 1995; Zanoni et al., 2011). An additional route of LPS internalization is the direct binding of LPS to high-mobility group box 1 (HMGB1), which triggers receptor mediated endocytosis of LPS through the receptor for glycation end products (RAGE) (Andersson et al., 2000; Andersson et al., 2018; Xu et al., 2014; Youn et al., 2008). Leakage of LPS into the cytosol by this mechanism activates the non-canonical inflammasome in humans and mice and contributes to LPS-induced lethality in mice (Deng et al., 2018; Wang et al., 1999; Wang et al., 2004; Xu et al., 2014). HMGB1 combined with LPS, but not LPS alone, induces significant caspase-11 and caspase-4 dependent pyroptosis in both murine peritoneal macrophages and human THP-1 monocytes, suggesting a direct role for HMGB1 in the delivery of LPS to the cytosol (Deng et al., 2018). Pull-down experiments have confirmed that the HMGB1 A and B box motif bind to the LPS polysaccharide and lipid A regions, respectively (Youn et al., 2011). Delivery of the HMGB1/LPS complex to the cytosol is mediated by RAGE, as its genetic deficiency or blockage protects mice from LPS-induced septic shock (Deng et al., 2018).
Recently, the small protein secretoglobin (SCGB) 3A2 has been identified as a chaperone for delivery of LPS to the cytosol in macrophages, as co-treatment of LPS with SCGB3A2 synergistically induces activation of caspase-11 and caspase-11-mediated pyroptosis (Yokoyama et al., 2018). Initial evidence came from observed differences between different recombinant SCG3A2 preparations and its growth inhibitory potency in LLC cells, which was subsequently determined to be associated with LPS contamination. Using biotinylated LPS the authors confirmed direct binding of LPS to recombinant SCG3A2 by streptavidin affinity purification. Subsequent LPS/SCGB3A2 entry into the cytosol occurs independently of TLR4, but is inhibited by Dynamin and the GTPase inhibitor Dynasore, suggesting that clathrin-mediated endocytosis is involved in SCGB3A2-mediated delivery of LPS into the cytosol (Yokoyama et al., 2018). of Protein microarray analysis subsequently identified syndecan-1 as the SCG3A2/LPS receptor (Yokoyama et al., 2018).
2.4. Alternative activation of the non-canonical inflammasome
In addition to LPS, some evidence also supports certain other lipids as caspase-11 activators. Arachidonic acid-derived oxidized phospholipids, including oxPAPC, elicit a caspase-11 mediated pro-inflammatory response specifically in dendritic cells (DCs). However, while cytosolic LPS triggers caspase-11 mediated IL-1β release as well as pyroptosis, oxPAPC only induces IL-1β release, putting cells in a hyperactivated state without undergoing cell death (Zanoni et al., 2016). These differences might be due to the different binding sites within caspase-11 that are targeted by LPS compared to oxPAPC and to their differences in promoting caspase-11 enzyme activity. While LPS binds to the CARD of caspase-11 and induces the enzymatic activity of caspase-11, oxPAPC binds to, and blocks the catalytic domain. Consequently, and contrary to LPS-mediated IL-1β release, the catalytic activity of caspase-11 is dispensable for oxPAPC mediated IL-1β release. Hence, LPS and oxPAPC utilize two distinct mechanisms for caspase-11 mediated IL-1β release (Zanoni et al., 2016). On the cell surface LPS bind to CD14 and induces endocytosis (Zanoni et al., 2011). Comparable to LPS, oxPAPC also binds to CD14 and is also able to induce CD14 internalization for delivering oxPAPC into cells. Interestingly, oxPAPC is only able to induce inflammasome activation and IL-1β release in DCs, but not in macrophages. However, oxPAPC is a mixture of lipids and stimulation with pure PGPC or POVPC, two minor components within the oxPAPC mixture, induce the release of IL-1β from both macrophages and DCs (Zanoni et al., 2017). POVPC has previously been implicated in the activation of the canonical NLRP3 inflammasome in macrophages, inducing ASC oligomerization, caspase-1 activation and IL-1β secretion (Yeon et al., 2017). In macrophages, PGPC and POVPC induce GSDMD-dependent IL-1β release and DNA intercalating dye uptake, indicating pore formation, but not LDH release, which is a hallmark of pyroptosis. Hence, caspase-11 and caspase-1 can have a trigger-dependent pyroptotic or non-pyroptotic function, but GSDMD is responsible for IL-1β release in both scenarios (Evavold et al., 2018).
The glycolipid lipophosphoglycan (LPG) present on the surface of Leishmania parasites activates canonical and non-canonical inflammasomes in macrophages (de Carvalho et al., 2019). Cytosolic delivery of Leishmania, but not LPG deficient Leishmania into macrophages, causes the activation of caspase-11 and subsequent activation of NLRP3 and caspase-1. Infection of Nlrp3−/− and Casp11−/− macrophages and Casp11−/− and Nlrp3−/− mice with Leishmania results in increased parasite loads and reduced host resistance, supporting a role for both canonical and non-canonical inflammasome activation during Leishmania infection (de Carvalho et al., 2019). However, in contrast to LPS and oxPAPC, LPG does not bind to caspase-11 and caspase-4, and fails to activate the non-canonical inflammasome in vitro (de Carvalho et al., 2019), suggesting the requirement of additional factors.
Secreted aspartyl proteinases Sap2 and Sap6 from yeast have previously been implicated in the activation of the canonical NLRP3 inflammasome, and recently in the activation of caspase-11 (Gabrielli et al., 2015; Pietrella et al., 2013). However, this response was mediated by regulating expression of caspase-11 by type I IFN production, rather than caspase-11 activation. Similarly, the addictive drug, methamphetamine, has been reported to activate caspase-11 to drive neuronal cell death, but also affects caspase-11 expression rather than activation (Huang et al., 2015).
Furthermore, some evidence also supports non-lipid molecules in the activation of the non-canonical inflammasome. As already mentioned above, heme has been identified as a DAMP activating caspase-4-dependent IL-1b release and pyroptosis as well as caspase-5-dependent release of IL-1β in human macrophages, but whether heme directly binds to these caspases is still unknown (Bolívar et al., 2020). The relevance of this response is emphasized by the observation that heme-induced IL-1β release is elevated in macrophages from sickle cell disease patients (Bolívar et al., 2020).
2.5. Consequences of non-canonical inflammasome activation
Caspase-1, but not caspase-11, is able to directly proteolytically mature pro-IL-1β and pro-IL-18, (Ramirez et al., 2018). Nevertheless, caspase-4 and caspase-5 have been implicated in the processing and release of IL-1b and IL-18, since silencing of caspase-4 and caspase-5, but not caspase-1, impaired cytokine release in response to S. Typhimurium and E. coli infection (Knodler et al., 2014). However, to our knowledge, compelling evidence for a direct cleavage of these cytokines by either caspase-4 or caspase-5 is still lacking. In vitro peptide analysis recently determined that in contrast to most caspases, including caspase-1, where the substrate specificity is determined by the P1-P4 tetrapeptide, the non-canonical caspase specificity is influenced by the prime-side amino acids P1’-P4’ and strongly prefer GSDMD over pro-IL-1β and pro-IL-18 (Bibo-Verdugo et al., 2020). However, while caspase-4 can cleave GSDMD and pro-IL-18 similar to caspase-1, it cannot cleave pro-IL-1p. On the other hand, caspase-5 also cleaves GSDMD but has only very weak activity for pro-IL-18 and pro-IL-1β (Bibo-Verdugo et al., 2020). The major function of non-canonical inflammasomes is the induction of pyroptosis (Fig. 2). Comparable to caspase-1, caspase-4, caspase-5 and caspase-11 also cleave GSDMD into an N-terminal (GSDMD-N) and C-terminal (GSDMD-C) fragment after aspartic acid residue 276 (Asp276). GSDMD-N is then sufficient to promote cell lysis as well as canonical inflammasome activation (He et al., 2015; Kayagaki et al., 2015; Shi et al., 2015). Site-specific auto-processing of caspase-4 and caspase-11 is required and sufficient for the cleavage of GSDMD and induction of pyroptosis (Wang et al., 2020). GSDMD-N is separated by a linker from GSDMD-C, and proteolytic cleavage releases the autoinhibitory GSDMD-C, which remains cytosolic (Wang et al., 2020). On the other hand, GSDMD-N inserts into the lipid bilayer through interaction with inner membrane glycerophospholipids, such as phosphatidylinositol phosphates, phosphatidic acid and phosphatidylserine, thus generating approximately 20 nm pores, which results in cell swelling and lysis (Aglietti et al., 2016; Chen et al., 2016; Ding et al., 2016; Liu et al., 2016; Russo et al., 2016; Sborgi et al., 2016). The importance of GSDMD-N is supported by the observation that expression of GSDMD-N alone is sufficient to induce pyroptosis (Aglietti et al., 2016; Chen et al., 2016; Ding et al., 2016; Kayagaki et al., 2015; Liu et al., 2016; Sborgi et al., 2016; Shi et al., 2015; Wang et al., 2020). Pyroptotic pores also function as channels for the release of cytosolic contents, including danger signals and IL-1β (Evavold et al., 2018; Heilig et al., 2018; Kayagaki et al., 2015; Shi et al., 2015). GSDMD-N also interacts with Cardiolipin, which is present in the inner leaflet of the mitochondrial membrane and in bacterial membranes. Indeed, cell free studies demonstrated that GSDMD-N can kill Gram-positive and Gram-negative bacteria (Liu et al., 2016). Furthermore, comparable to caspase-3 generated GSDME-N, also inflammasome-generated GSDMD-N permeabilizes mitochondria, thereby linking inflammasomes to apoptosome activation through inducing cytochrome-c release and caspase-3 activation (Rogers et al., 2019). Further proof for acting within the same pathway comes from the observation that Gsdmd−/− mice were as protected as Casp11−/− mice during LPS-induced shock (Hagar et al., 2013; Kayagaki et al., 2015; Kayagaki et al., 2011; Kayagaki et al., 2013; Wang et al., 1998). Further, caspase-11 cleavage and activation, as well as GSDMD cleavage, are crucial for LPS-induced shock, since mice harboring a caspase-11 mutation inactivating its enzymatic activity (caspase-11Cys254) , a mutation preventing caspase-11 auto-processing at the inter-subunit linker (caspase-11Asp285) or a mutation preventing GSDMD processing (GsdmdD276) are all protected from LPS-induced lethality and phenocopy Casp11−/− and Gsdmd−/− mice (Lee et al., 2018).
Figure 2.
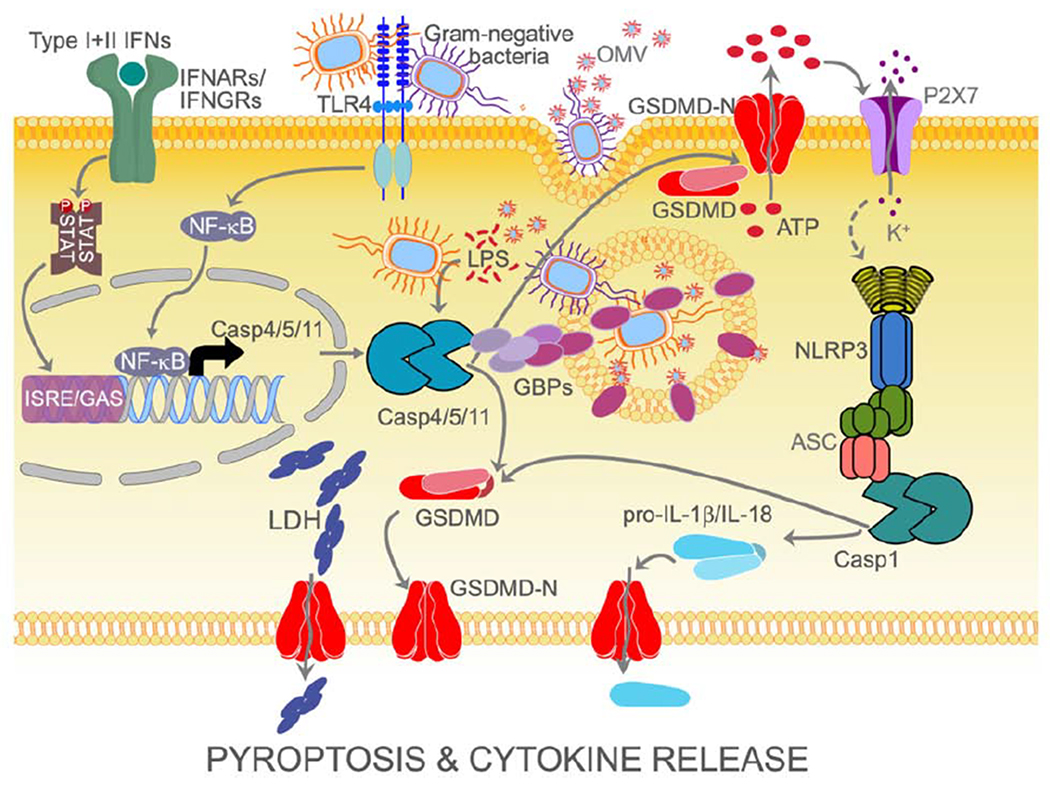
Overview of non-canonical inflammasome activation leading to pyroptosis and subsequent activation of the canonical NLRP3 inflammasome for cytokine maturation and secretion.
2.6. Regulation of the non-canonical inflammasome
Inducible expression of non-canonical inflammasome components is a prerequisite for non-canonical inflammasome activation. While mouse caspase-1 is constitutively expressed, human caspase-1 is inducible by IFN-γ in monocytes. Caspase-4 has been reported to be constitutively expressed in monocytes, but recently shown by genome wide screen to require IRF2 or IFN-γ-induced IRF1 for inducible expression (Benaoudia et al., 2019; Lin et al., 2000). Caspase-5 expression is induced by type I and type II IFNs and LPS in monocytes (Lin et al., 2000). However, cell type-specific differences in inducible expression of human inflammatory caspases have been reported (Christgen et al., 2020). Similar to caspase-5, also caspase-11 expression is undetectable in resting macrophages, but LPS, IFNα/β and IFN-γ elevate expression. LPS-induced caspase-11 expression via TLR4-TRIF signaling is dependent on NF-κB and IFNs/STAT1 signaling (Rathinam et al., 2012; Schauvliege et al., 2002). Furthermore, the complement component-related carboxypeptidase B1 (Cbp1) transcriptionally regulates caspase-11 via enhancement of TLR4-TRIF-IFNAR signaling (Napier et al., 2016). Importantly, Tlr4−/− mice are rendered susceptible to LPS induced septic shock by priming with a TLR3 agonist, such as poly(I:C), suggesting that TLR3 priming can bypass the lack of TLR4 to increase caspase-11 expression (Hagar et al., 2013; Kayagaki et al., 2013). Crucially, GBPs are strongly up-regulated by type I IFNs and IFN-γ (Kim et al., 2012; Li et al., 2009). However, exceptions to the requirement for type I IFN have been reported in the case of L. pneumophila and Yersinia pseudotuberculosis, where the priming signal can be bypassed for rapid non-canonical inflammasome activation, indicating that some basal expression of caspase-11 is sufficient for this response (Casson et al., 2013). In addition to transcriptional priming and the above discussed upstream acting GBPs, further regulation of the non-canonical inflammasome has been discovered. This high level of regulation is not surprising, considering the crucial role of the non-canonical inflammasome in cell death and eliminating bacterial infections. Inhibition can occur by cellular as well as bacterial factors that either directly interact with caspase-4, caspase-5 and caspase-11, or utilize a yet unknown mechanism (Fig. 3). As described above, oxPAPC activates non-canonical inflammasome-mediated IL-1β secretion, which requires canonical NLRP3 inflammasome activation, CD14 and TLR4. However, multiple pro- and anti-inflammatory activities have been reported for oxPAPC in a context-dependent and concentration-dependent manner (Miller and Shyy, 2017). For instance, it inhibits several key steps in LPS-mediated TLR4 activation by competing with LPS for LBP, CD14 and MD-2 binding (Erridge, 2009).
Figure 3.
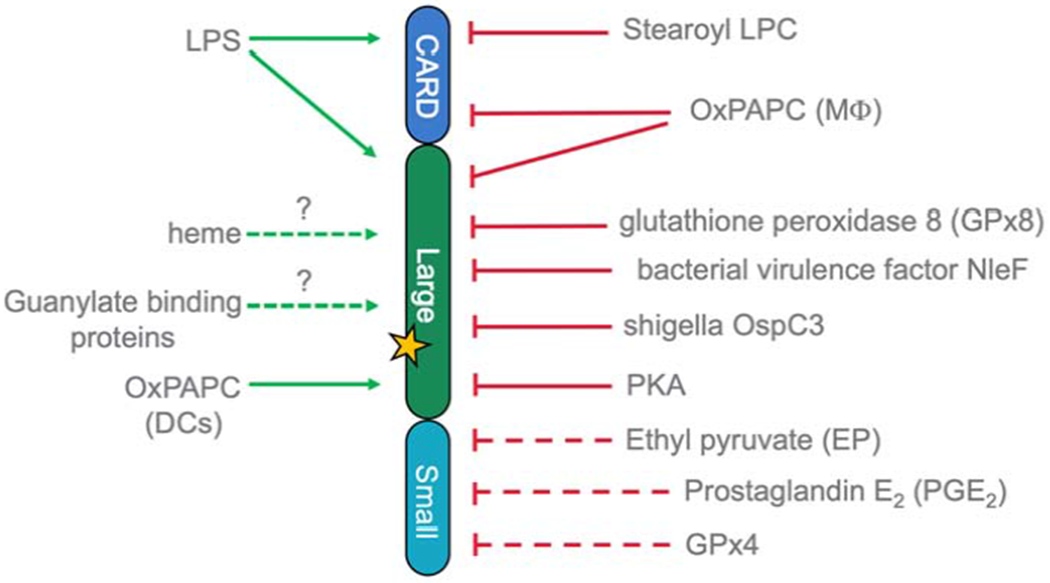
Schemata of activators and inhibitors that directly- (solid line) or indirectly (dashed line) interact with non-canonical inflammasome caspases. The yellow asterisk marks the catalytic center.
Physiological concentrations present in serum are generally considered to be anti-inflammatory (Oskolkova et al., 2010), and accordingly, oxPAPC prevents TLR4 activation and LPS-induced shock in vivo by preventing and inhibiting DC activation by LPS (Bluml et al., 2005; Bochkov et al., 2002). Considering that oxPAPC is able to interfere with LPS binding and signaling of TLR4, and that the lipid A moiety of LPS similarly interacts with TLR4 and caspase-11, it is not surprising that intracellular oxPAPC, as well as its sub-components PGPC and POVPC, also inhibit intracellular LPS-mediated activation of the non-canonical inflammasome (Chu et al., 2018). The study by Chu et al. utilizes primed macrophages, which are transfected with LPS for intracellular delivery of LPS in the presence or absence of oxPAPC and pyroptosis is determined by LDH release, GSDMD cleavage and HMGB1 release. Notably, TLR2 or TLR3 ligands rather than LPS were used for priming to avoid interference from TLR4. To further rule out any effects of oxPAPC on TLR4, the inhibitory effect of oxPAPC on intracellular LPS mediated pyroptosis was also observed in Tlr4−/− cells. Under these conditions, oxPAPC protects from LPS-induced pyroptosis only in macrophages and not in DCs. As expected, the subsequent canonical inflammasome activation and IL-1β release is also inhibited by oxPAPC in human and mouse macrophages. The molecular mechanism is based on competitive binding of LPS and oxPAPC to caspase-11 and caspase-4. While LPS has been shown to interact with the CARD of caspase-11 (Shi et al., 2014) and DC activating oxPAPC to the caspase domain (Zanoni et al., 2016), Chu et al. find that positively charged residues in the CARD and the catalytic domain contribute to LPS binding (Chu et al., 2018). While oxPAPC binding also involves positively charged residues in the CARD and the catalytic domain, oxPAPC and LPS compete for partially overlapping binding sites (Chu et al., 2018). Consequently, LPS and oxPAPC seemingly are unable to simultaneously bind to caspase-11 or caspase-4, similar to what has now also been shown for Lysophosphatidylcholine and ethyl pyruvate (Li et al., 2018b; Qiu et al., 2020). Significantly, in a caspase-11 dependent in vivo model of LPS-induced shock, oxPAPC was able to rescue mice in a TLR4-independent manner, comparable to Casp11−/− mice (Chu et al., 2018). Contrary to the anti-inflammatory function of oxPAPC described by Chu et al., Zanoni et al. described a pro-inflammatory function of oxPAPC and further investigations are needed. While Zanoni et al. initially reported that oxPAPC induces IL-1β release from DC but fails to induce IL-1β release from macrophages, they later found that PGPC and POVPC induce IL-1β release from macrophages. In vivo, LPS primed and LPS, oxPAPC and PGPC challenged mice exhibit an increase in IL-1β, IL-6 and TNFa over LPS primed mice without challenge, but while LPS challenged mice do not survive past 30h, oxPAPC and PGPC challenged mice do not display any lethality (Zanoni et al., 2016; Zanoni et al., 2017). Conflicting results about pro- and anti-inflammatory functions of oxPAPC have also been reported for TLR4 activation and are believed to be connected to concentration and cell type specific effects. A similar explanation is possible for caspase-11 activation in addition to experimental differences particularly in the precise oxPAPC composition and the extra- or intra-cellular delivery of LPS. However, further support for an anti-inflammatory activity of oxPAPC comes from the observation that also Lysophosphatidylcholine, a major component of oxidized low-density lipoproteins with a structural similarity to oxPAPC, directly binds to caspase-11 and significantly impedes the interaction between LPS and caspase-11 and consequently blocks caspase-11-mediated pyroptosis (Li et al., 2018b).
Glutathione peroxidase 4 (GPX4) is an antioxidant enzyme which catalyzes the reduction of lipid peroxides and protects the cells against oxidative stress. In the absence of GPX4, lipid peroxides accumulate and promote ferroptotic cell death. GPX4 expression is elevated in septic mice and patients and negatively regulates pyroptosis mediated by both canonical- and non-canonical inflammasomes in response to cytosolic LPS and E. coli infection by a phospholipase C gamma 1 (PLCG1) and Ca+-dependent mechanism (Kang et al., 2018). Mice with myeloid-specific Gpx4 deletion are more susceptible to polymicrobial sepsis and this phenotype can be rescued by deletion or inhibition of caspase-11 or GSDMD (Kang et al., 2018). While details on the molecular mechanism are currently elusive, altered caspase-11 expression has been ruled out (Kang et al., 2018).
GPx8 lacks enzymatic activity, but inhibits caspase-4 and caspase-11 activation by forming a disulfide bridge between cysteine 79 in GPx8 and the conserved cysteine 118 in caspase-4 and caspase-11 (Hsu et al., 2020). Accordingly, GPx8−/− mice are more susceptible to colitis and endotoxic shock through impaired regulation of the non-canonical inflammasome (Hsu et al., 2020). Upon stimulation with intracellular LPS, GPx8−/− macrophages release elevated IL-1β and LDH compared to WT macrophages, and ulcerative colitis patients display reduced GPx8 and up-regulated caspase-4 levels (Hsu et al., 2020).
The metabolite ethyl pyruvate, an aliphatic ester derived from pyruvic acid, has anti-inflammatory activity and protects from LPS-induced shock and polymicrobial sepsis (Qiu et al., 2020; Yang et al., 2016). One of its mechanisms involves blocking the non-canonical inflammasome, preventing GSDMD cleavage and pyroptosis and consequently, IL-1α and IL-1β secretion. Ethyl pyruvate does not interfere with the cytosolic translocation of LPS, but interferes with the interaction between LPS and caspase-11. The underlying mechanism is still unknown, but at higher concentrations, ethyl pyruvate impairs also expression of pro-caspase-11 (Qiu et al., 2020). Ethyl pyruvate has been previously shown to also prevent canonical NLRP3 inflammasome activation (Li et al., 2018a).
The stress hormone and neurotransmitter L-adrenaline (epinephrine) has been identified in a cell-based screen for compounds promoting survival in response to electroporation of LPS in a library of FDA-approved drugs (Chen et al., 2019b). L-adrenaline prevents caspase-4 and caspase-11 activation, GSDMD and IL-1β cleavage, and the release of LDH and HMGB1 in response to cytosolic LPS or infection with E. coli in macrophages (Chen et al., 2019b). In response to cytosolic LPS and L-adrenaline, the L-adrenaline receptor Adrenoceptor α 2B (ADRA2B) and the adenylyl cyclase 4 (ADCY4), the enzyme responsible for cyclic adenosine monophosphate (cAMP) synthesis, are up-regulated, which leads to the accumulation of intracellular cAMP and consequently, activation of protein kinase A (PKA). PKA in turn prevents caspase-11-driven pyroptosis and IL-1β release by directly binding and phosphorylating caspase-11. Limiting cAMP hydrolysis by phosphodiester 8A (PDE8A) consequently impairs non-canonical inflammasome activation, providing evidence for a previously unknown metabolic regulation of this pathway by the ADRA2B-ADCY4-PDE8A-PKA axis (Chen et al., 2019b). Importantly, systemic administration of L-adrenaline or 8-Br-cAMP to increase cAMP, or blocking cAMP hydrolysis with the PDE8A inhibitor PF-04957325, protects mice from LPS-induced lethality. Comparable protection is observed following increased systemic cAMP levels by acute starvation stress. This screen further identified the antipsychotic Levosulpiride, the anesthetic Tetracaine, the synthetic glucocorticoid Prednisolone and the angiotensin-converting enzyme (ACE) inhibitor Quinapril as inhibitors of cytosolic LPS-induced pyroptosis, and Qinapril and Prednisolone, but not Levosulpiride were also protective in vivo (Chen et al., 2019b).
Prostaglandin E2 (PGE2) exhibits both pro- and anti-inflammatory activities, inhibits the canonical NLRP3 inflammasome, and has recently been identified to prevent caspase-11 activation during human asthma and mouse allergic airway inflammation (Sokolowska et al., 2015; Zaslona et al., 2020). Protection is conferred not by blocking non-canonical inflammasome activation, but instead PGE2 prevents IFN-b-mediated transcription of caspase-4 and caspase-11 (Zaslona et al., 2020). Importantly, caspase-4 and caspase-11 are also elevated in alveolar macrophages of asthma patients and in the lungs of mice with allergic airway inflammation, respectively (Zaslona et al., 2020). This finding provides a potential explanation for why NSAIDs, which block PGE2 production, can worsen asthma.
Beside cell intrinsic regulators, pathogens also employ multiple strategies to avoid caspase-11-mediated host defense to facilitate infection. The simplest, most direct approach is modification of LPS, such as the tetra-acylated lipid A from Helicobacter pylori, as caspase-11 binds to hexa-acylated LPS (Aachoui et al., 2013; Casson et al., 2013). Similarly, S. flexneri usually contains hexa-acylated LPS, which is hypoacylated when growing intracellularly, resulting in vastly reduced immunostimulation (Paciello et al., 2013). However, caspase-4 is also activated by tetra-acylated LPS from Francisella novicida, suggesting that the human non-canonical inflammasomes may have a broader reactivity than those of mice (Lagrange et al., 2018).
Enteropathogenic E. coli in human- and C. rodentium in mouse epithelial cells induce caspase-4 and caspase-11-mediated non-canonical inflammasome activation. However, the virulence factor NleF, which translocates into host cell via T3SS, will eventually suppress this response by directly binding to, and blocking the catalytic domain of caspase-4 and caspase-11, resulting in impaired IL-18-mediated host defense and neutrophil influx to aid colonization (Blasche et al., 2013; Pallett et al., 2017).
The S. flexneri T3SS effector protein, OspC3, is another example of a bacterial virulence factor targeting the non-canonical inflammasome in epithelial cells to prevent pyroptosis, as infection with ΔospC3 S. flexneri decreases host cell viability (Kobayashi et al., 2013; Wandel et al., 2020). OspC3 specifically interacts with caspase-4, preventing heterodimerization and formation of the catalytic pocket, but does not interact with caspase-11 (Kobayashi et al., 2013).
3. Crosstalk between non-canonical and canonical inflammasome activation
Although the non-canonical inflammasome cannot or only weakly directly cleave pro-IL-1β and pro-IL-18 (Bibo-Verdugo et al., 2020; Ramirez et al., 2018), non-canonical inflammasome activation nevertheless leads to their secretion (Kayagaki et al., 2011). In contrast to LPS-induced pyroptosis, which does not require caspase-1, cytokine release is dependent on caspase-1 activation by the canonical NLRP3 inflammasome (Fig. 2) (Kayagaki et al., 2011; Rathinam et al., 2012; Ruhl and Broz, 2015; Schmid-Burgk et al., 2015; Yang et al., 2015). Direct binding of caspase-11 to caspase-1 has been initially observed, which could contribute to caspase-1 activation (Wang et al., 1998). However, more recent studies demonstrated that caspase-11 functions further upstream and requires NLRP3 and ASC for caspase-11-mediated caspase-1 activation (Kayagaki et al., 2011; Rathinam et al., 2012; Ruhl and Broz, 2015; Schmid-Burgk et al., 2015; Yang et al., 2015). LPS-mediated activation of caspase-4 and caspase-11 results in potassium efflux, which is the common NLRP3 inflammasome activating event (Munoz-Planillo et al., 2013; Ruhl and Broz, 2015; Schmid-Burgk et al., 2015). Initially, pannexin-1 has been implicated in this cross-talk, following proteolytical cleavage by active caspase-11 after Asp378, thereby promoting ATP release and activation of the ATP-gated ion channel P2X7, which leads to potassium and sodium efflux and feeds into the well-established activation mechanism of the canonical NLRP3 inflammasome (Yang et al., 2015). Consequently, Panx1−/−, P2x7−/− and Casp11−/− mice are equally protected from LPS-induced shock and the cleavage resistant pannexin-1D378A mutant prevents caspase-11-mediated activation of the canonical NLRP3 inflammasome (Yang et al., 2015). However, a more recent analysis failed to reproduce a requirement for pannexin-1 in the cross talk between the non-canonical and the canonical NLRP3 inflammasome when using pharmacological and genetic tools, but rather identified that it mediates NLRP3 activation during intrinsic and extrinsic apoptosis and that non-canonical NLRP3 activation proceeds as a consequence of GSDMD pores (Chen et al., 2020; Chen et al., 2019a; Qu et al., 2011). During L. pneumophila infection, this cross-talk is amplified by activation of the canonical AIM2 inflammasome and the resulting caspase-1-mediated membrane damage promotes potassium efflux and non-canonical NLRP3 inflammasome activation (Cunha et al., 2017). This membrane damage may reflect GSDMD pores, as IL-1β and IL-18 release is impaired in Gsdmd−/− macrophages (Evavold et al., 2018; He et al., 2015; Kayagaki et al., 2015; Russo et al., 2016; Shi et al., 2015). Hence, potassium efflux may be a consequence of ATP release and P2X7 activation or may occur directly through GSDMD pores. Another piece of the puzzle comes from the finding that caspase-11 also cleaves the transient receptor potential channel 1 (TRPC1), which leads to its degradation. TRPC1 inhibits LPS-induced IL-1β release and consequently Trpc1−/− enhances LPS-induced IL-1β release without affecting caspase-1 cleavage or pyroptosis. Hence, TRPC1 degradation during an inflammatory response may alter the properties of TRPC1 containing channels in order to promote unconventional protein secretion (Py et al., 2014). This finding is in agreement with another report demonstrating that unprocessed caspase-1 mediates membrane damage for non-canonical NLRP3 activation (Cunha et al., 2017). Furthermore, while the precise role of ROS in the activation of the canonical NLRP3 inflammasome is still controversial, caspase-4 activation by Shiga toxin expressed by enteric pathogens requires activation of ROS downstream of caspase-4 for NLRP3 activation and release of IL-1β (Platnich et al., 2018). Yet another layer of crosstalk between the non-canonical and the canonical NLRP3 inflammasome exists at the level of caspase-1. GBP1, which initiates GBP oligomerization to recruit and activate caspase-4, can be cleaved and consequently inactivated by caspase-1 (Fisch et al., 2020). Cleaved GBP1 fragments are unable to target the S. Typhimurium surface and therefore fail to sequester caspase-4 and elicit pyroptosis. Accordingly, the non-cleavable GBP1D192E mutant enhances caspase-4-mediated pyroptosis in S. Typhimurium infected cells (Fisch et al., 2020).
4. The non-canonical inflammasome in health and disease
The non-canonical inflammasome’s major role is defense against Gram-negative bacteria that escape the phagosome and invade the cytosol (Aachoui et al., 2013). In addition, human and mouse non-canonical inflammasomes directly target pathogen-containing phagosomes by binding to the actin interacting protein 1 (Aip1) or through RhoA, thereby altering cofilin phosphorylation and actin polymerization. This results in efficient trafficking, phagosome-lysosome fusion and bacterial killing, as well as the chemotaxis of macrophages and neutrophils (Caution et al., 2015; Caution et al., 2019; Li et al., 2007; Paciello et al., 2013). Hence, a major contribution of the non-canonical inflammasome is clearance of invaded pathogens and alerting neighboring cells through the release of alarmins, DAMPs and canonical NLRP3 inflammasome-dependent cytokines, and if the threat persists, eventually removal of infected cells by pyroptosis (Zanoni et al., 2017). Consequently, the non-canonical inflammasome contributes to sepsis, when the initial immune response fails to clear an infection and a sustained inflammatory response develops, which has been well-established in mice (Chu et al., 2018; Hagar et al., 2017; Hagar et al., 2013; Kayagaki et al., 2011; Kayagaki et al., 2013; Vanaja et al., 2016; Wang et al., 1998; Yang et al., 2015). This also extends to the human non-canonical inflammasome, as caspase-4 transgenic mice are more susceptible to LPS-induced shock, even in the absence of caspase-1 and caspase-11, and caspase-4 restores protection against B. thailandensis in Casp11−/− mice (Aachoui et al., 2015; Kajiwara et al., 2014). HMGB1 release by pyroptosis, rather than IL-1β and IL-18 mediate acute LPS-induced lethality (Lamkanfi et al., 2010; Vande Walle et al., 2011; Wang et al., 1999; Willingham et al., 2009).
The non-canonical inflammasome also contributes to lung pathologies, and while most studies focus on myeloid cells, LPS can also enter human and mouse endothelial cells and activate the non-canonical inflammasome. Hence, conventional and endothelial cell-specific Casp11−/− mice are protected from LPS-induced lung injury (Cheng et al., 2017). Moreover, Casp11−/− mice are protected from allergic lung inflammation, as evident from decreased levels of bronchoalveolar lavage fluid-infiltrating CD4 T cells and eosinophils. As discussed above, PGE2-suppresses caspase-11 expression in allergic airways (Zaslona et al., 2020). Importantly, alveolar macrophages from asthma patients also show elevated caspase-4 expression (Zaslona et al., 2020). Caspase-11 also contributes to cigarette smoke-induced airway inflammation, as Casp11−/− mice show reduced IL-1β and IL-18 secretion and neutrophil infiltration into bronchoalveolar lavage fluid (Eltom et al., 2014). Indeed, evidence supports a role of caspase-11 in neutrophil and macrophage migration during K. pneumoniae infection and MSU-induced arthritis, at least in part due to defect KC-dependent chemotaxis, and defect cofilin phosphorylation (Caution et al., 2019; Wang et al., 2017). In addition, Casp11−/− neutrophils fail to produce neutrophil extracellular traps (NETs) (Caution et al., 2019). Similarly, Casp11−/− mice are also protected from LPS-induced renal injury (Ye et al., 2019).
Altered microbiota associated with pathogen invasion and colonization contributes to intestinal dysfunction, and increasing evidence supports a role of the non-canonical inflammasome in regulating intestinal inflammation and development of inflammatory bowel disease. Caspase-11 expression in the intestinal mucosa is elevated after dextran sodium sulfate (DSS)-induced colitis, and caspase-4 and caspase-5 expression is significantly increased in colonic biopsies from ulcerative colitis patients (Demon et al., 2014; Williams et al., 2015). Similar to Casp1−/− mice, also Casp11−/− mice are more susceptible to DSS-induced colitis with increased mortality and accelerated disease severity, which is at least in part due to defective IL-18-mediated epithelial cell proliferation and barrier function (Demon et al., 2014; Oficjalska et al., 2015; Williams et al., 2015). Silencing of caspase-4 in cultured epithelial cells shows a comparable IL-18-mediated protection in response to colonization with S. Typhimurium and E. coli (Knodler et al., 2014). Furthermore, disruption of colonic function by high-fat diet is linked to the non-canonical inflammasome, as palmitate promotes the intracellular transport of LPS into enteric neurons, thereby linking high-fat diet to enteric neuronal cell death. In agreement, Casp11−/− mice are protected from western-diet induced colonic dysmotility associated with decreased enteric neuronal pyroptosis (Ye et al., 2020).
Chronic inflammation is also implicated in the etiology of neurodegenerative disease, and evidence exists for a contribution of the non-canonical inflammasome. In the autoimmune encephalomyelitis (EAE) mouse model of Multiple Sclerosis, Casp11−/− mice show reduced activation of caspase-3, correlating with less oligodendrocyte cell death and increased resistance to EAE (Hisahara et al., 2001). A similar observation has been made in a genetic mouse model of amyotrophic lateral sclerosis and in an induced model of Parkinson’s Disease (Furuya et al., 2004; Kang et al., 2003). In all instances, an increased expression of caspase-11 was observed in neurons of diseased mice, preceding neuronal defects (Furuya et al., 2004; Hisahara et al., 2001; Kang et al., 2003).
5. Perspective
Canonical inflammasomes were first described 18 years ago and ample progress has been made to dissect their precise role in physiology and pathology (Martinon et al., 2002). Although, caspase-11 has been discovered even before that, the non-canonical inflammasome has only been identified nine years ago, and we learned even more recently about its role in LPS recognition (Hagar et al., 2013; Kayagaki et al., 2011; Kayagaki et al., 2013). Nevertheless, as discussed throughout this article, significant progress has been made in discovering more details about its activation by GBPs, its role in host defense, and its contribution to human disease. However, substantial effort is nevertheless needed to fill in many of the still existing gaps. While this review focuses on mammalian non-canonical inflammasomes, a related pathway has been characterized in zebrafish, further emphasizing the importance of this host defense system. Interestingly, the zebrafish genome contains 5 functional inflammatory caspases with 3 showing greater than 50% homology to human caspase-1, caspase-4, and caspase-5 (Sakamaki and Satou, 2009). Indeed, zebrafish caspy2, which shares the highest homology to caspase-5, induces pyroptosis and its knockdown protects larvae from LPS-induced lethality (Yang et al., 2018). Reminiscent to the canonical inflammasome effector zCaspy, also zCaspy2 contains a PYRIN domain (PYD), rather than the CARD found in mammalian inflammatory caspases (Masumoto et al., 2003; Yang et al., 2018). Yet, the PYD of zCaspy2 also directly binds LPS, resulting in its oligomerization, activation and cleavage of a GSDMD homolog, GSDME, to induce pyroptosis and to promote shock in vivo (Yang et al., 2018). This provides opportunities for the discovery of novel aspects of non-canonical inflammasome biology in the simpler zebrafish system. Although, caspase-4 and caspase-5 have largely been considered to be redundant in many aspects and we are just beginning to understand some of the functional differences between caspase-4 and caspase-5, as well as the crosstalk with the canonical inflammasome. For example, NLRP1 can recruit both, canonical caspase-1 and non-canonical caspase-5, and under experimental conditions of lysing cells in the presence of LPS, both are necessary for full NLRP1 inflammasome activation (Martinon et al., 2002). Recent evidence shows that IL-1α is specifically cleaved and released by the human non-canonical caspase-5 inflammasome and mouse non-canonical caspase-11 inflammasome (Wiggins et al., 2019). Although the downstream effector functions of IL-1α are still not fully elucidated, it is required for the IL-1α-dependent senescence-associated secretory phenotype (SASP) and deficiency of these caspases impairs SASP-mediated immune surveillance (Wiggins et al., 2019). We can expect to continue elucidating additional unique aspects for each of these inflammatory caspases. While the pyroptotic role is best characterized, non-canonical inflammasomes contribute to other cellular aspects, such as non-lytic GSDMD activation, caspase-3 activation in neurodegenerative disease, phagosome and autophagosome maturation, actin remodeling and leukocyte migration. It is most likely that novel cellular functions and substrates have yet to be discovered (Akhter et al., 2012; Caution et al., 2015; Evavold et al., 2018; Furuya et al., 2004; Hisahara et al., 2001; Kang et al., 2003; Krause et al., 2018; Li et al., 2007; Wang et al., 2017; Zanoni et al., 2017). While LPS is the best described ligand for the non-canonical inflammasome, several other activators have been identified, including oxPAPC, Leishmania lipophosphoglycan and heme, which therefore may provide scenarios for identifying novel non-canonical inflammasome components (Bolívar et al., 2020; de Carvalho et al., 2019; Zanoni et al., 2016). Numerous posttranslational modifications have been identified for canonical inflammasome components (Christgen et al., 2020), and evidence exists for PKA-mediated phosphorylation of caspase-11 (Chen et al., 2019b), and therefore it is very likely that additional modifications will be discovered. While we begin to unravel cell type specific roles, availability of conditional Casp11−/− mice will surely enable the discovery of context specific non-canonical inflammasome functions. And most importantly, initial discovery of compounds targeting the non-canonical inflammasome or downstream effectors, including already FDA-approved ones, will extend these findings to the clinic (Chen et al., 2019b; Chu et al., 2018; Hu et al., 2020; Li et al., 2018b; Qiu et al., 2020). Therefore, there are many opportunities for discovering novel aspects of non-canonical inflammasome biology ahead.
Acknowledgments
Funding
This work was supported by the National Institutes of Health (AI099009 and AR064349 to C.S., AI134030, AI140702 and AI120625 to C.S. and A.D., and AR066739 to A.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
References
- Aachoui Y, Kajiwara Y, Leaf IA, Mao D, Ting JP, Coers J, Aderem A, Buxbaum JD, Miao EA, 2015. Canonical Inflammasomes Drive IFN-gamma to Prime Caspase-11 in Defense against a Cytosol-Invasive Bacterium. Cell Host Microbe 18 (3), 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, Miao EA, 2013. Caspase-11 protects against bacteria that escape the vacuole. Science 339 (6122), 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC, 2016. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A 113 (28), 7858–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhter A, Caution K, Abu Khweek A, Tazi M, Abdulrahman BA, Abdelaziz DH, Voss OH, Doseff AI, Hassan H, Azad AK, Schlesinger LS, Wewers MD, Gavrilin MA, Amer AO, 2012. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity 37 (1), 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, Tracey KJ, 2000. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med 192 (4), 565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson U, Yang H, Harris H, 2018. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin Immunol 38, 40–48. [DOI] [PubMed] [Google Scholar]
- Balakrishnan A, Karki R, Berwin B, Yamamoto M, Kanneganti TD, 2018. Guanylate binding proteins facilitate caspase-11-dependent pyroptosis in response to type 3 secretion system-negative Pseudomonas aeruginosa. Cell Death Discov 4, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benaoudia S, Martin A, Puig Gamez M, Gay G, Lagrange B, Cornut M, Krasnykov K, Claude JB, Bourgeois CF, Hughes S, Gillet B, Allatif O, Corbin A, Ricci R, Henry T, 2019. A genome-wide screen identifies IRF2 as a key regulator of caspase-4 in human cells. EMBO Rep 20 (9), e48235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibo-Verdugo B, Snipas SJ, Kolt S, Poreba M, Salvesen GS, 2020. Extended subsite profiling of the pyroptosis effector protein gasdermin D reveals a region recognized by inflammatory caspase-11. J Biol Chem 295 (32), 11292–11302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitto NJ, Baker PJ, Dowling JK, Wray-McCann G, De Paoli A, Tran LS, Leung PL, Stacey KJ, Mansell A, Masters SL, Ferrero RL, 2018. Membrane vesicles from Pseudomonas aeruginosa activate the noncanonical inflammasome through caspase-5 in human monocytes. Immunol Cell Biol 96 (10), 1120–1130. [DOI] [PubMed] [Google Scholar]
- Blasche S, Mortl M, Steuber H, Siszler G, Nisa S, Schwarz F, Lavrik I, Gronewold TM, Maskos K, Donnenberg MS, Ullmann D, Uetz P, Kogl M, 2013. The E. coli effector protein NleF is a caspase inhibitor. PLoS One 8 (3), e58937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluml S, Kirchberger S, Bochkov VN, Kronke G, Stuhlmeier K, Majdic O, Zlabinger GJ, Knapp W, Binder BR, Stockl J, Leitinger N, 2005. Oxidized phospholipids negatively regulate dendritic cell maturation induced by TLRs and CD40. J Immunol 175 (1), 501–508. [DOI] [PubMed] [Google Scholar]
- Bochkov VN, Kadl A, Huber J, Gruber F, Binder BR, Leitinger N, 2002. Protective role of phospholipid oxidation products in endotoxin-induced tissue damage. Nature 419 (6902), 77–81. [DOI] [PubMed] [Google Scholar]
- Bolívar BE, Brown AN, Rohrman BA, Charendoff CI, Yazdani V, Belcher JD, Vercellotti GM, Flanagan JM, Bouchier-Hayes L, 2020. Non-canonical roles of caspase-4 and caspase-5 in heme driven-IL-1β release and cell death. bioRxiv 2020.02.28.969899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, Dixit VM, 2016. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16 (7), 407–420. [DOI] [PubMed] [Google Scholar]
- Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, Monack DM, 2012. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490 (7419), 288–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casson CN, Copenhaver AM, Zwack EE, Nguyen HT, Strowig T, Javdan B, Bradley WP, Fung TC, Flavell RA, Brodsky IE, Shin S, 2013. Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog 9 (6), e1003400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casson CN, Yu J, Reyes VM, Taschuk FO, Yadav A, Copenhaver AM, Nguyen HT, Collman RG, Shin S, 2015. Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc Natl Acad Sci U S A 112 (21), 6688–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caution K, Gavrilin MA, Tazi M, Kanneganti A, Layman D, Hoque S, Krause K, Amer AO, 2015. Caspase-11 and caspase-1 differentially modulate actin polymerization via RhoA and Slingshot proteins to promote bacterial clearance. Sci Rep 5, 18479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caution K, Young N, Robledo-Avila F, Krause K, Abu Khweek A, Hamilton K, Badr A, Vaidya A, Daily K, Gosu H, Anne MNK, Eltobgy M, Dakhlallah D, Argwal S, Estfanous S, Zhang X, Partida-Sanchez S, Gavrilin MA, Jarjour WN, Amer AO, 2019. Caspase-11 Mediates Neutrophil Chemotaxis and Extracellular Trap Formation During Acute Gouty Arthritis Through Alteration of Cofilin Phosphorylation. Front Immunol 10, 2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecil JD, O’Brien-Simpson NM, Lenzo JC, Holden JA, Singleton W, Perez-Gonzalez A, Mansell A, Reynolds EC, 2017. Outer Membrane Vesicles Prime and Activate Macrophage Inflammasomes and Cytokine Secretion In Vitro and In Vivo. Front Immunol 8, 1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, March CJ, Kronheim SR, Druck T, Cannizzaro LA, et al. , 1992. Molecular cloning of the interleukin-1 beta converting enzyme. Science 256 (5053), 97–100. [DOI] [PubMed] [Google Scholar]
- Chang DW, Ditsworth D, Liu H, Srinivasula SM, Alnemri ES, Yang X, 2003. Oligomerization is a general mechanism for the activation of apoptosis initiator and inflammatory procaspases. J Biol Chem 278 (19), 16466–16469. [DOI] [PubMed] [Google Scholar]
- Chen KW, Demarco B, Broz P, 2020. Pannexin-1 promotes NLRP3 activation during apoptosis but is dispensable for canonical or noncanonical inflammasome activation. Eur J Immunol 50 (2), 170–177. [DOI] [PubMed] [Google Scholar]
- Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, Pelczar P, Broz P, 2019a. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J 38 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Zeng L, Zhu S, Liu J, Zeh HJ, Kroemer G, Wang H, Billiar TR, Jiang J, Tang D, Kang R, 2019b. cAMP metabolism controls caspase-11 inflammasome activation and pyroptosis in sepsis. Sci Adv 5 (5), eaav5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Yang D, Wen Y, Jiang Z, Zhang L, Jiang J, Chen Y, Hu T, Wang Q, Zhang Y, Liu Q, 2018. Dysregulated hemolysin liberates bacterial outer membrane vesicles for cytosolic lipopolysaccharide sensing. PLoS Pathog 14 (8), e1007240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, He WT, Hu L, Li J, Fang Y, Wang X, Xu X, Wang Z, Huang K, Han J, 2016. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res 26 (9), 1007–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, Gao X, An S, Mittal M, Vogel SM, Miao EA, Rehman J, Malik AB, 2017. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christgen S, Place DE, Kanneganti TD, 2020. Toward targeting inflammasomes: insights into their regulation and activation. Cell Res 30 (4), 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu LH, Gangopadhyay A, Dorfleutner A, Stehlik C, 2015. An updated view on the structure and function of PYRIN domains. Apoptosis 20 (2), 157–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu LH, Indramohan M, Ratsimandresy RA, Gangopadhyay A, Morris EP, Monack DM, Dorfleutner A, Stehlik C, 2018. The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nat Commun 9 (1), 996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha LD, Silva ALN, Ribeiro JM, Mascarenhas DPA, Quirino GFS, Santos LL, Flavell RA, Zamboni DS, 2017. AIM2 Engages Active but Unprocessed Caspase-1 to Induce Noncanonical Activation of the NLRP3 Inflammasome. Cell Rep 20 (4), 794–805. [DOI] [PubMed] [Google Scholar]
- Davis BK, Roberts RA, Huang MT, Willingham SB, Conti BJ, Brickey WJ, Barker BR, Kwan M, Taxman DJ, Accavitti-Loper MA, Duncan JA, Ting JP, 2011. Cutting edge: NLRC5-dependent activation of the inflammasome. J Immunol 186 (3), 1333–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho RVH, Andrade WA, Lima-Junior DS, Dilucca M, de Oliveira CV, Wang K, Nogueira PM, Rugani JN, Soares RP, Beverley SM, Shao F, Zamboni DS, 2019. Leishmania Lipophosphoglycan Triggers Caspase-11 and the Non-canonical Activation of the NLRP3 Inflammasome. Cell Rep 26 (2), 429–437 e425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demon D, Kuchmiy A, Fossoul A, Zhu Q, Kanneganti TD, Lamkanfi M, 2014. Caspase-11 is expressed in the colonic mucosa and protects against dextran sodium sulfate-induced colitis. Mucosal Immunol 7 (6), 1480–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, Zhao X, Liu J, Tang C, Liu Z, Huang Y, Peng H, Xiao L, Tang D, Scott MJ, Wang Q, Liu J, Xiao X, Watkins S, Li J, Yang H, Wang H, Chen F, Tracey KJ, Billiar TR, Lu B, 2018. The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 49 (4), 740–753 e747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA, 1998. Interleukin-1 beta, interleukin-18, and the interleukin-1 beta converting enzyme. Ann N Y Acad Sci 856, 1–11. [DOI] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F, 2016. Poreforming activity and structural autoinhibition of the gasdermin family. Nature 535 (7610), 111–116. [DOI] [PubMed] [Google Scholar]
- Dorfleutner A, Chu L, Stehlik C, 2015. Inhibiting the inflammasome: one domain at a time. Immunol Rev 265 (1), 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltom S, Belvisi MG, Stevenson CS, Maher SA, Dubuis E, Fitzgerald KA, Birrell MA, 2014. Role of the inflammasome-caspase1/11-IL-1/18 axis in cigarette smoke driven airway inflammation: an insight into the pathogenesis of COPD. PLoS One 9 (11), e112829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge C, 2009. Oxidized phospholipid inhibition of LPS-signaling: a good side to the bad guys? Arterioscler Thromb Vasc Biol 29 (3), 337–338. [DOI] [PubMed] [Google Scholar]
- Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC, 2018. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48 (1), 3544 e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finethy R, Luoma S, Orench-Rivera N, Feeley EM, Haldar AK, Yamamoto M, Kanneganti TD, Kuehn MJ, Coers J, 2017. Inflammasome Activation by Bacterial Outer Membrane Vesicles Requires Guanylate Binding Proteins. mBio 8 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink SL, Cookson BT, 2006. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol 8 (11), 1812–1825. [DOI] [PubMed] [Google Scholar]
- Fisch D, Clough B, Domart MC, Encheva V, Bando H, Snijders AP, Collinson LM, Yamamoto M, Shenoy AR, Frickel EM, 2020. Human GBP1 Differentially Targets Salmonella and Toxoplasma to License Recognition of Microbial Ligands and Caspase-Mediated Death. Cell Rep 32 (6), 108008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya T, Hayakawa H, Yamada M, Yoshimi K, Hisahara S, Miura M, Mizuno Y, Mochizuki H, 2004. Caspase-11 mediates inflammatory dopaminergic cell death in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. J Neurosci 24 (8), 1865–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielli E, Pericolini E, Luciano E, Sabbatini S, Roselletti E, Perito S, Kasper L, Hube B, Vecchiarelli A, 2015. Induction of caspase-11 by aspartyl proteinases of Candida albicans and implication in promoting inflammatory response. Infect Immun 83 (5), 1940–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegner JA, Ulevitch RJ, Tobias PS, 1995. Lipopolysaccharide (LPS) signal transduction and clearance. Dual roles for LPS binding protein and membrane CD14. J Biol Chem 270 (10), 5320–5325. [DOI] [PubMed] [Google Scholar]
- Hagar JA, Edin ML, Lih FB, Thurlow LR, Koller BH, Cairns BA, Zeldin DC, Miao EA, 2017. Lipopolysaccharide Potentiates Insulin-Driven Hypoglycemic Shock. J Immunol 199 (10), 3634–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA, 2013. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341 (6151), 1250–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J, 2015. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 25 (12), 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P, 2018. The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur J Immunol 48 (4), 584–592. [DOI] [PubMed] [Google Scholar]
- Hisahara S, Yuan J, Momoi T, Okano H, Miura M, 2001. Caspase-11 mediates oligodendrocyte cell death and pathogenesis of autoimmune-mediated demyelination. J Exp Med 193 (1), 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S, 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162 (7), 3749–3752. [PubMed] [Google Scholar]
- Hsu JL, Chou JW, Chen TF, Hsu JT, Su FY, Lan JL, Wu PC, Hu CM, Lee EY, Lee WH, 2020. Glutathione peroxidase 8 negatively regulates caspase-4/11 to protect against colitis. EMBO Mol Med 12 (1), e9386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, Ruan J, Luo X, Lou X, Bai Y, Wang J, Hollingsworth LR, Magupalli VG, Zhao L, Luo HR, Kim J, Lieberman J, Wu H, 2020. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol 21 (7), 736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Xie WB, Qiao D, Qiu P, Huang E, Li B, Chen C, Liu C, Wang Q, Lin Z, Wang H, 2015. Caspase-11 plays an essential role in methamphetamine-induced dopaminergic neuron apoptosis. Toxicol Sci 145 (1), 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indramohan M, Stehlik C, Dorfleutner A, 2018. COPs and POPs Patrol Inflammasome Activation. J Mol Biol 430 (2), 153–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiwara Y, Schiff T, Voloudakis G, Gama Sosa MA, Elder G, Bozdagi O, Buxbaum JD, 2014. A critical role for human caspase-4 in endotoxin sensitivity. J Immunol 193 (1), 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, Cao L, Xie M, Ran Q, Kroemer G, Wang H, Billiar TR, Jiang J, Tang D, 2018. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe 24 (1), 97–108 e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SJ, Sanchez I, Jing N, Yuan J, 2003. Dissociation between Neurodegeneration and Caspase-11-Mediated Activation of Caspase-1 and Caspase-3 in a Mouse Model of Amyotrophic Lateral Sclerosis. The Journal of Neuroscience 23 (13), 5455–5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S, 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11 (5), 373–384. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM, 2015. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526 (7575), 666–671. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM, 2011. Non-canonical inflammasome activation targets caspase-11. Nature 479 (7371), 117–121. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW, Dixit VM, 2013. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341 (6151), 1246–1249. [DOI] [PubMed] [Google Scholar]
- Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, Chandran B, 2011. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 9 (5), 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare S, Luc N, Dorfleutner A, Stehlik C, 2010. Inflammasomes and their activation. Crit Rev Immunol 30 (5), 463–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BH, Chee JD, Bradfield CJ, Park ES, Kumar P, MacMicking JD, 2016. Interferon-induced guanylate-binding proteins in inflammasome activation and host defense. Nat Immunol 17 (5), 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BH, Shenoy AR, Kumar P, Bradfield CJ, MacMicking JD, 2012. IFN-inducible GTPases in host cell defense. Cell Host Microbe 12 (4), 432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, Ernst RK, Steele-Mortimer O, Celli J, Vallance BA, 2014. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 16 (2), 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Ogawa M, Sanada T, Mimuro H, Kim M, Ashida H, Akakura R, Yoshida M, Kawalec M, Reichhart JM, Mizushima T, Sasakawa C, 2013. The Shigella OspC3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe 13 (5), 570–583. [DOI] [PubMed] [Google Scholar]
- Krause K, Caution K, Badr A, Hamilton K, Saleh A, Patel K, Seveau S, Hall-Stoodley L, Hegazi R, Zhang X, Gavrilin MA, Amer AO, 2018. CASP4/caspase-11 promotes autophagosome formation in response to bacterial infection. Autophagy 14 (11), 1928–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsch M, Sistemich L, Lesser CF, Goldberg MB, Herrmann C, Coers J, 2020. Direct binding of polymeric GBP1 to LPS disrupts bacterial cell envelope functions. EMBO J 39 (13), e104926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagrange B, Benaoudia S, Wallet P, Magnotti F, Provost A, Michal F, Martin A, Di Lorenzo F, Py BF, Molinaro A, Henry T, 2018. Human caspase-4 detects tetra-acylated LPS and cytosolic Francisella and functions differently from murine caspase-11. Nat Commun 9 (1), 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, Tracey KJ, Kanneganti TD, Dixit VM, 2010. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol 185 (7), 4385–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BL, Stowe IB, Gupta A, Kornfeld OS, Roose-Girma M, Anderson K, Warming S, Zhang J, Lee WP, Kayagaki N, 2018. Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. J Exp Med 215 (9), 2279–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Zhang J, Sun Y, Wang H, Wang Y, 2009. The evolutionarily dynamic IFN-inducible GTPase proteins play conserved immune functions in vertebrates and cephalochordates. Mol Biol Evol 26 (7), 1619–1630. [DOI] [PubMed] [Google Scholar]
- Li J, Brieher WM, Scimone ML, Kang SJ, Zhu H, Yin H, von Andrian UH, Mitchison T, Yuan J, 2007. Caspase-11 regulates cell migration by promoting Aip1-Cofilin-mediated actin depolymerization. Nat Cell Biol 9 (3), 276–286. [DOI] [PubMed] [Google Scholar]
- Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, Towne E, Tracey D, Wardwell S, Wei F-Y, Wong W, Kamen R, Seshadri T, 1995. Mice deficient in IL-1β-converting enzyme are defective in production of mature IL-1β and resistant to endotoxic shock. Cell 80, 401–411. [DOI] [PubMed] [Google Scholar]
- Li S, Liang F, Kwan K, Tang Y, Wang X, Tang Y, Li J, Yang H, Chavan SS, Wang H, Andersson U, Lu B, Tracey KJ, 2018a. Identification of ethyl pyruvate as a NLRP3 inflammasome inhibitor that preserves mitochondrial integrity. Mol Med 24 (1), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhang W, Deng M, Loughran P, Tang Y, Liao H, Zhang X, Liu J, Billiar TR, Lu B, 2018b. Stearoyl Lysophosphatidylcholine Inhibits Endotoxin-Induced Caspase-11 Activation. Shock 50 (3), 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin XY, Choi MS, Porter AG, 2000. Expression analysis of the human caspase-1 subfamily reveals specific regulation of the CASP5 gene by lipopolysaccharide and interferon-gamma. J Biol Chem 275 (51), 39920–39926. [DOI] [PubMed] [Google Scholar]
- Liu J, Du S, Kong Q, Zhang X, Jiang S, Cao X, Li Y, Li C, Chen H, Ding Z, Liu L, 2020. HSPA12A attenuates lipopolysaccharide-induced liver injury through inhibiting caspase-11-mediated hepatocyte pyroptosis via PGC-1alpha-dependent acyloxyacyl hydrolase expression. Cell Death Differ 27 (9), 2651–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J, 2016. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535 (7610), 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Kanneganti TD, 2016. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol 16 (1), 7–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Karki R, Briard B, Burton A, Gingras S, Pelletier S, Kanneganti TD, 2017. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci Rep 7, 45126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, Frase S, Zhu Q, Malireddi RKS, Kuriakose T, Peters JL, Neale G, Brown SA, Yamamoto M, Kanneganti TD, 2016. IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell 167 (2), 382–396 e317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM, 2004. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430 (6996), 213–218. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM, 2006. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440 (7081), 228–232. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J, 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10 (2), 417–426. [DOI] [PubMed] [Google Scholar]
- Masumoto J, Zhou W, Chen FF, Su F, Kuwada JY, Hidaka E, Katsuyama T, Sagara J, Taniguchi S, Ngo-Hazelett P, Postlethwait JH, Nunez G, Inohara N, 2003. Caspy, a zebrafish caspase, activated by ASC oligomerization is required for pharyngeal arch development. J Biol Chem 278 (6), 4268–4276. [DOI] [PubMed] [Google Scholar]
- Matikainen S, Nyman TA, Cypryk W, 2020. Function and Regulation of Noncanonical Caspase-4/5/11 Inflammasome. J Immunol 204 (12), 3063–3069. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Yamashita A, Matsuda M, Kawai K, Sawa T, Amaya F, 2019. NLRP2 inflammasome in dorsal root ganglion as a novel molecular platform that produces inflammatory pain hypersensitivity. Pain 160 (9), 2149–2160. [DOI] [PubMed] [Google Scholar]
- Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, Roose-Girma M, Bumann D, Kayagaki N, Takeda K, Yamamoto M, Broz P, 2014. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 509 (7500), 366–370. [DOI] [PubMed] [Google Scholar]
- Miller YI, Shyy JY, 2017. Context-Dependent Role of Oxidized Lipids and Lipoproteins in Inflammation. Trends Endocrinol Metab 28 (2), 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkiewicz J, de Rivero Vaccari JP, Keane RW, 2013. Human astrocytes express a novel NLRP2 inflammasome. Glia 61 (7), 1113–1121. [DOI] [PubMed] [Google Scholar]
- Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G, 2013. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38 (6), 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napier BA, Brubaker SW, Sweeney TE, Monette P, Rothmeier GH, Gertsvolf NA, Puschnik A, Carette JE, Khatri P, Monack DM, 2016. Complement pathway amplifies caspase-11-dependent cell death and endotoxin-induced sepsis severity. J Exp Med 213 (11), 2365–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oficjalska K, Raverdeau M, Aviello G, Wade SC, Hickey A, Sheehan KM, Corr SC, Kay EW, O’Neill LA, Mills KH, Creagh EM, 2015. Protective role for caspase-11 during acute experimental murine colitis. J Immunol 194 (3), 1252–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskolkova OV, Afonyushkin T, Preinerstorfer B, Bicker W, von Schlieffen E, Hainzl E, Demyanets S, Schabbauer G, Lindner W, Tselepis AD, Wojta J, Binder BR, Bochkov VN, 2010. Oxidized phospholipids are more potent antagonists of lipopolysaccharide than inducers of inflammation. J Immunol 185 (12), 7706–7712. [DOI] [PubMed] [Google Scholar]
- Paciello I, Silipo A, Lembo-Fazio L, Curcuru L, Zumsteg A, Noel G, Ciancarella V, Sturiale L, Molinaro A, Bernardini ML, 2013. Intracellular Shigella remodels its LPS to dampen the innate immune recognition and evade inflammasome activation. Proc Natl Acad Sci U S A 110 (46), E4345–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallett MA, Crepin VF, Serafini N, Habibzay M, Kotik O, Sanchez-Garrido J, Di Santo JP, Shenoy AR, Berger CN, Frankel G, 2017. Bacterial virulence factor inhibits caspase-4/11 activation in intestinal epithelial cells. Mucosal Immunol 10 (3), 602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrella D, Pandey N, Gabrielli E, Pericolini E, Perito S, Kasper L, Bistoni F, Cassone A, Hube B, Vecchiarelli A, 2013. Secreted aspartic proteases of Candida albicans activate the NLRP3 inflammasome. Eur J Immunol 43 (3), 679–692. [DOI] [PubMed] [Google Scholar]
- Pilla DM, Hagar JA, Haidar AK, Mason AK, Degrandi D, Pfeffer K, Ernst RK, Yamamoto M, Miao EA, Coers J, 2014. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci U S A 111 (16), 6046–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platnich JM, Chung H, Lau A, Sandall CF, Bondzi-Simpson A, Chen HM, Komada T, Trotman-Grant AC, Brandelli JR, Chun J, Beck PL, Philpott DJ, Girardin SE, Ho M, Johnson RP, MacDonald JA, Armstrong GD, Muruve DA, 2018. Shiga Toxin/Lipopolysaccharide Activates Caspase-4 and Gasdermin D to Trigger Mitochondrial Reactive Oxygen Species Upstream of the NLRP3 Inflammasome. Cell Rep 25 (6), 1525–1536 e1527. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B, 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282 (5396), 2085–2088. [DOI] [PubMed] [Google Scholar]
- Py BF, Jin M, Desai BN, Penumaka A, Zhu H, Kober M, Dietrich A, Lipinski MM, Henry T, Clapham DE, Yuan J, 2014. Caspase-11 controls interleukin-1beta release through degradation of TRPC1. Cell Rep 6 (6), 1122–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Cheng X, Zhang J, Yuan C, Zhao M, Yang X, 2020. Ethyl pyruvate confers protection against endotoxemia and sepsis by inhibiting caspase-11-dependent cell pyroptosis. Int Immunopharmacol 78, 106016. [DOI] [PubMed] [Google Scholar]
- Qu Y, Misaghi S, Newton K, Gilmour LL, Louie S, Cupp JE, Dubyak GR, Hackos D, Dixit VM, 2011. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J Immunol 186 (11), 6553–6561. [DOI] [PubMed] [Google Scholar]
- Raetz CR, Whitfield C, 2002. Lipopolysaccharide endotoxins. Annu Rev Biochem 71, 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez MLG, Poreba M, Snipas SJ, Groborz K, Drag M, Salvesen GS, 2018. Extensive peptide and natural protein substrate screens reveal that mouse caspase-11 has much narrower substrate specificity than caspase-1. J Biol Chem 293 (18), 7058–7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA, 2012. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150 (3), 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VAK, Zhao Y, Shao F, 2019. Innate immunity to intracellular LPS. Nat Immunol 20 (5), 527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES, 2019. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun 10 (1), 1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhl S, Broz P, 2015. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol 45 (10), 2927–2936. [DOI] [PubMed] [Google Scholar]
- Ruhl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P, 2018. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362 (6417), 956–960. [DOI] [PubMed] [Google Scholar]
- Russo HM, Rathkey J, Boyd-Tressler A, Katsnelson MA, Abbott DW, Dubyak GR, 2016. Active Caspase-1 Induces Plasma Membrane Pores That Precede Pyroptotic Lysis and Are Blocked by Lanthanides. J Immunol 197 (4), 1353–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu JK, Kim SJ, Rah SH, Kang JI, Jung HE, Lee D, Lee HK, Lee JO, Park BS, Yoon TY, Kim HM, 2017. Reconstruction of LPS Transfer Cascade Reveals Structural Determinants within LBP, CD14, and TLR4-MD2 for Efficient LPS Recognition and Transfer. Immunity 46 (1), 38–50. [DOI] [PubMed] [Google Scholar]
- Sakamaki K, Satou Y, 2009. Caspases: evolutionary aspects of their functions in vertebrates. J Fish Biol 74 (4), 727–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvesen GS, Dixit VM, 1999. Caspase activation: the induced-proximity model. Proc Natl Acad Sci U S A 96 (20), 10964–10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders MG, Parsons MJ, Howard AG, Liu J, Fassio SR, Martinez JA, Bouchier-Hayes L, 2015. Single-cell imaging of inflammatory caspase dimerization reveals differential recruitment to inflammasomes. Cell Death Dis 6, e1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JC, Boucher D, Schneider LK, Demarco B, Dilucca M, Shkarina K, Heilig R, Chen KW, Lim RYH, Broz P, 2020. Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat Commun 11 (1), 3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JC, Dick MS, Lagrange B, Degrandi D, Pfeffer K, Yamamoto M, Meunier E, Pelczar P, Henry T, Broz P, 2018. LPS targets host guanylate-binding proteins to the bacterial outer membrane for non-canonical inflammasome activation. EMBO J 37 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Muller DJ, Broz P, Hiller S, 2016. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J 35 (16), 1766–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauvliege R, Vanrobaeys J, Schotte P, Beyaert R, 2002. Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. J Biol Chem 277 (44), 41624–41630. [DOI] [PubMed] [Google Scholar]
- Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V, 2015. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol 45 (10), 2911–2917. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F, 2015. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526 (7575), 660–665. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F, 2014. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514 (7521), 187–192. [DOI] [PubMed] [Google Scholar]
- Sokolowska M, Chen LY, Liu Y, Martinez-Anton A, Qi HY, Logun C, Alsaaty S, Park YH, Kastner DL, Chae JJ, Shelhamer JH, 2015. Prostaglandin E2 Inhibits NLRP3 Inflammasome Activation through EP4 Receptor and Intracellular Cyclic AMP in Human Macrophages. J Immunol 194 (11), 5472–5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollberger G, Strittmatter GE, Kistowska M, French LE, Beer HD, 2012. Caspase-4 is required for activation of inflammasomes. J Immunol 188 (4), 1992–2000. [DOI] [PubMed] [Google Scholar]
- Stehlik C, Dorfleutner A, 2007. COPs and POPs: modulators of inflammasome activity. J Immunol 179 (12), 7993–7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson KV, Deng M, Ting JP, 2019. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19 (8), 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Zhang R, Xue Q, Meng R, Wang X, Yang Y, Xie L, Xiao X, Billiar TR, Lu B, 2018. TRIF signaling is required for caspase-11-dependent immune responses and lethality in sepsis. Mol Med 24 (1), 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, et al. , 1992. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356 (6372), 768–774. [DOI] [PubMed] [Google Scholar]
- Van Opdenbosch N, Lamkanfi M, 2019. Caspases in Cell Death, Inflammation, and Disease. Immunity 50 (6), 1352–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, Rathinam VAK, 2016. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 165 (5), 1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Walle L, Kanneganti TD, Lamkanfi M, 2011. HMGB1 release by inflammasomes. Virulence 2 (2), 162–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigano E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A, 2015. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat Commun 6, 8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, Conlon JE, Burbage JJ, Proulx MK, Liu Q, Reed G, Mecsas JC, Iwakura Y, Bertin J, Goguen JD, Fitzgerald KA, Lien E, 2012. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 37 (1), 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker MA, Teghanemt A, Weiss JP, Barker JH, 2017. High-affinity caspase-4 binding to LPS presented as high molecular mass aggregates or in outer membrane vesicles. Innate Immun 23 (4), 336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandel MP, Kim BH, Park ES, Boyle KB, Nayak K, Lagrange B, Herod A, Henry T, Zilbauer M, Rohde J, MacMicking JD, Randow F, 2020. Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat Immunol 21 (8), 880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ, 1999. HMG-1 as a late mediator of endotoxin lethality in mice. Science 285 (5425), 248–251. [DOI] [PubMed] [Google Scholar]
- Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al-Abed Y, Wang H, Metz C, Miller EJ, Tracey KJ, Ulloa L, 2004. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 10 (11), 1216–1221. [DOI] [PubMed] [Google Scholar]
- Wang J, Shao Y, Wang W, Li S, Xin N, Xie F, Zhao C, 2017. Caspase-11 deficiency impairs neutrophil recruitment and bacterial clearance in the early stage of pulmonary Klebsiella pneumoniae infection. Int J Med Microbiol 307 (8), 490–496. [DOI] [PubMed] [Google Scholar]
- Wang K, Sun Q, Zhong X, Zeng M, Zeng H, Shi X, Li Z, Wang Y, Zhao Q, Shao F, Ding J, 2020. Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell 180 (5), 941–955 e920. [DOI] [PubMed] [Google Scholar]
- Wang S, Miura M, Jung Y, Zhu H, Gagliardini V, Shi L, Greenberg AH, Yuan J, 1996. Identification and characterization of Ich-3, a member of the interleukin-1beta converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J Biol Chem 271 (34), 20580–20587. [DOI] [PubMed] [Google Scholar]
- Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J, 1998. Murine caspase-11, an ICEinteracting protease, is essential for the activation of ICE. Cell 92 (4), 501–509. [DOI] [PubMed] [Google Scholar]
- Wiggins KA, Parry AJ, Cassidy LD, Humphry M, Webster SJ, Goodall JC, Narita M, Clarke MCH, 2019. IL-1alpha cleavage by inflammatory caspases of the noncanonical inflammasome controls the senescence-associated secretory phenotype. Aging Cell 18 (3), e12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TM, Leeth RA, Rothschild DE, McDaniel DK, Coutermarsh-Ott SL, Simmons AE, Kable KH, Heid B, Allen IC, 2015. Caspase-11 attenuates gastrointestinal inflammation and experimental colitis pathogenesis. Am J Physiol Gastrointest Liver Physiol 308 (2), G139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ, Duncan JA, Ting JP, 2009. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol 183 (3), 2008–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z, Li Y, Scott MJ, Xiao G, Li S, Fan L, Billiar TR, Wilson MA, Fan J, 2014. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ 21 (8), 1229–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Yaginuma K, Tsutsui H, Sagara J, Guan X, Seki E, Yasuda K, Yamamoto M, Akira S, Nakanishi K, Noda T, Taniguchi S, 2004. ASC is essential for LPS-induced activation of procaspase-1 independently of TLR-associated signal adaptor molecules. Genes Cells 9 (11), 1055–1067. [DOI] [PubMed] [Google Scholar]
- Yang D, He Y, Munoz-Planillo R, Liu Q, Nunez G, 2015. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 43 (5), 923–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Zheng X, Chen S, Wang Z, Xu W, Tan J, Hu T, Hou M, Wang W, Gu Z, Wang Q, Zhang R, Zhang Y, Liu Q, 2018. Sensing of cytosolic LPS through caspy2 pyrin domain mediates noncanonical inflammasome activation in zebrafish. Nat Commun 9 (1), 3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Zhu S, Tonnessen TI, 2016. Ethyl pyruvate is a novel anti-inflammatory agent to treat multiple inflammatory organ injuries. J Inflamm (Lond) 13, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Li G, Goebel A, Raju AV, Kong F, Lv Y, Li K, Zhu Y, Raja S, He P, Li F, Mwangi SM, Hu W, Srinivasan S, 2020. Caspase-11-mediated enteric neuronal pyroptosis underlies Western diet-induced colonic dysmotility. J Clin Invest 130 (7), 3621–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z, Zhang L, Li R, Dong W, Liu S, Li Z, Liang H, Wang L, Shi W, Malik AB, Cheng KT, Liang X, 2019. Caspase-11 Mediates Pyroptosis of Tubular Epithelial Cells and Septic Acute Kidney Injury. Kidney Blood Press Res 44 (4), 465–478. [DOI] [PubMed] [Google Scholar]
- Yeon SH, Yang G, Lee HE, Lee JY, 2017. Oxidized phosphatidylcholine induces the activation of NLRP3 inflammasome in macrophages. J Leukoc Biol 101 (1), 205–215. [DOI] [PubMed] [Google Scholar]
- Yi YS, 2017. Caspase-11 non-canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology 152 (2), 207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi YS, 2020. Functional crosstalk between non-canonical caspase-11 and canonical NLRP3 inflammasomes during infection-mediated inflammation. Immunology 159 (2), 142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama S, Cai Y, Murata M, Tomita T, Yoneda M, Xu L, Pilon AL, Cachau RE, Kimura S, 2018. A novel pathway of LPS uptake through syndecan-1 leading to pyroptotic cell death. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn JH, Kwak MS, Wu J, Kim ES, Ji Y, Min HJ, Yoo JH, Choi JE, Cho HS, Shin JS, 2011. Identification of lipopolysaccharide-binding peptide regions within HMGB1 and their effects on subclinical endotoxemia in a mouse model. Eur J Immunol 41 (9), 2753–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn JH, Oh YJ, Kim ES, Choi JE, Shin JS, 2008. High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide-mediated TNF-alpha production in human monocytes. J Immunol 180 (7), 5067–5074. [DOI] [PubMed] [Google Scholar]
- Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, Granucci F, Kagan JC, 2011. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 147 (4), 868–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, Donado CA, Shao F, Wu H, Springstead JR, Kagan JC, 2016. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352 (6290), 1232–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I, Tan Y, Di Gioia M, Springstead JR, Kagan JC, 2017. By Capturing Inflammatory Lipids Released from Dying Cells, the Receptor CD14 Induces Inflammasome-Dependent Phagocyte Hyperactivation. Immunity 47 (4), 697–709 e693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaslona Z, Flis E, Wilk MM, Carroll RG, Palsson-McDermott EM, Hughes MM, Diskin C, Banahan K, Ryan DG, Hooftman A, Misiak A, Kearney J, Lochnit G, Bertrams W, Greulich T, Schmeck B, McElvaney OJ, Mills KHG, Lavelle EC, Wygrecka M, Creagh EM, O’Neill LAJ, 2020. Caspase-11 promotes allergic airway inflammation. Nat Commun 11 (1), 1055. [DOI] [PMC free article] [PubMed] [Google Scholar]