

Abstract
Declining mitochondrial function and homeostasis is a hallmark of aging. It is appreciated that the role of mitochondria is much more complex than generating reactive oxygen species to cause aging-related tissue damage. More recent literature describes that the ability of mitochondria to undergo fission or fusion events with each other impacts aging processes. A dynamic balance of mitochondrial fission and fusion events is required to sustain critical cellular functions including cell cycle. Specifically, cell cycle regulators modulate molecular activities of the mitochondrial fission (and fusion) machinery towards regulating cell cycle progression. In this review, we discus literature leading to our understanding on how shifts in the dynamic balance of mitochondrial fission and fusion can modulate progression through, exit from, and re-entry to the cell cycle or in undergoing senescence. Importantly, core regulators of mitochondrial fission or fusion are emerging as crucial stem cell regulators. We discuss the implication of such regulation in stem cells in the context of aging, given that aberrations in adult stem cells promote aging. We also propose a few hypotheses that may provide direction for further understanding about the roles of mitochondrial fission-fusion dynamics in aging biology.
1. Introduction
Mitochondria perform the crucial task of energy metabolism in eukaryotic cells, and are also critically involved in cell signaling and organismal homeostasis. Declining mitochondrial function in various tissues is considered to be a major hallmark of aging. According to the mitochondrial free radical theory of aging, the cumulative tissue damage by mitochondrial reactive oxygen species (ROS) is the primary cause for aging. However, attenuating mitochondrial ROS generation sometimes produces unexpected or inconsistent effects on aging, suggesting complex roles for ROS in aging processes (Balaban et al., 2005). In contrast to the cellular damage associated with ROS overproduction, finely regulated production of mitochondrial ROS contributes to normal signaling (Reczek and Chandel, 2015). Furthermore, low levels of mitochondrial ROS have been implicated in adaptive anti-aging responses, a phenomenon termed as mitohormesis (Ristow, 2014). Therefore, a broader outlook on the mitochondrial contribution to aging is required given mitochondria contributes to almost all the 9 identified hallmarks of aging (Lopez-Otin et al., 2013).
Tissues primarily consist of differentiated cells that have exited proliferating cycle and must maintain chronological survival. Cells of certain lineages, like the hematopoietic lineage, can re-enter proliferating cycle after activation. Additionally, various tissues harbor adult stem cells that can regenerate tissues. When activated, the quiescent adult stem cells enter a self-renewal and proliferating cycle and further differentiate into their lineage specific counterparts (Oh et al., 2014). Adult stem cell exhaustion or their senescence (inability to exit cell cycle quiescence) has been linked to aging. Mitochondrial properties, including their fission and fusion ability, have been demonstrated to contribute to cell cycle regulation, cellular quiescence and senescence, and also organismal aging. Here, we will review literature related to the role of mitochondrial fission and fusion in regulating the cell cycle and how that can possibly impact stem cells in the context of organismal aging. Given, this review covers research from non-mammalian and mammalian model organisms as well as human, our discussion would pertain to mammalian system unless otherwise mentioned.
2. Mitochondrial fission-fusion dynamics
In proliferating cells mitochondria are not static structures as they rapidly undergo the processes of fission or fusion with each other. Mitochondrial fission divides larger mitochondria into smaller elements, and mitochondrial fusion joins smaller mitochondria to form larger ones. Also, mitochondria can transiently fuse and share their contents without altering mitochondrial shape or size. Cells maintain a given mitochondrial shape or size architecture by maintaining a dynamic balance between the countering processes of mitochondrial fission and fusion (Fig. 1). The details of the mitochondrial fission and fusion processes have been reviewed elsewhere (Kraus and Ryan, 2017; Labbe et al., 2014; Pernas and Scorrano, 2016). Here we introduce only the essential mechanistic principles involving the core machinery of mitochondrial fission and fusion (Fig. 1 and Table 1).
Fig. 1.
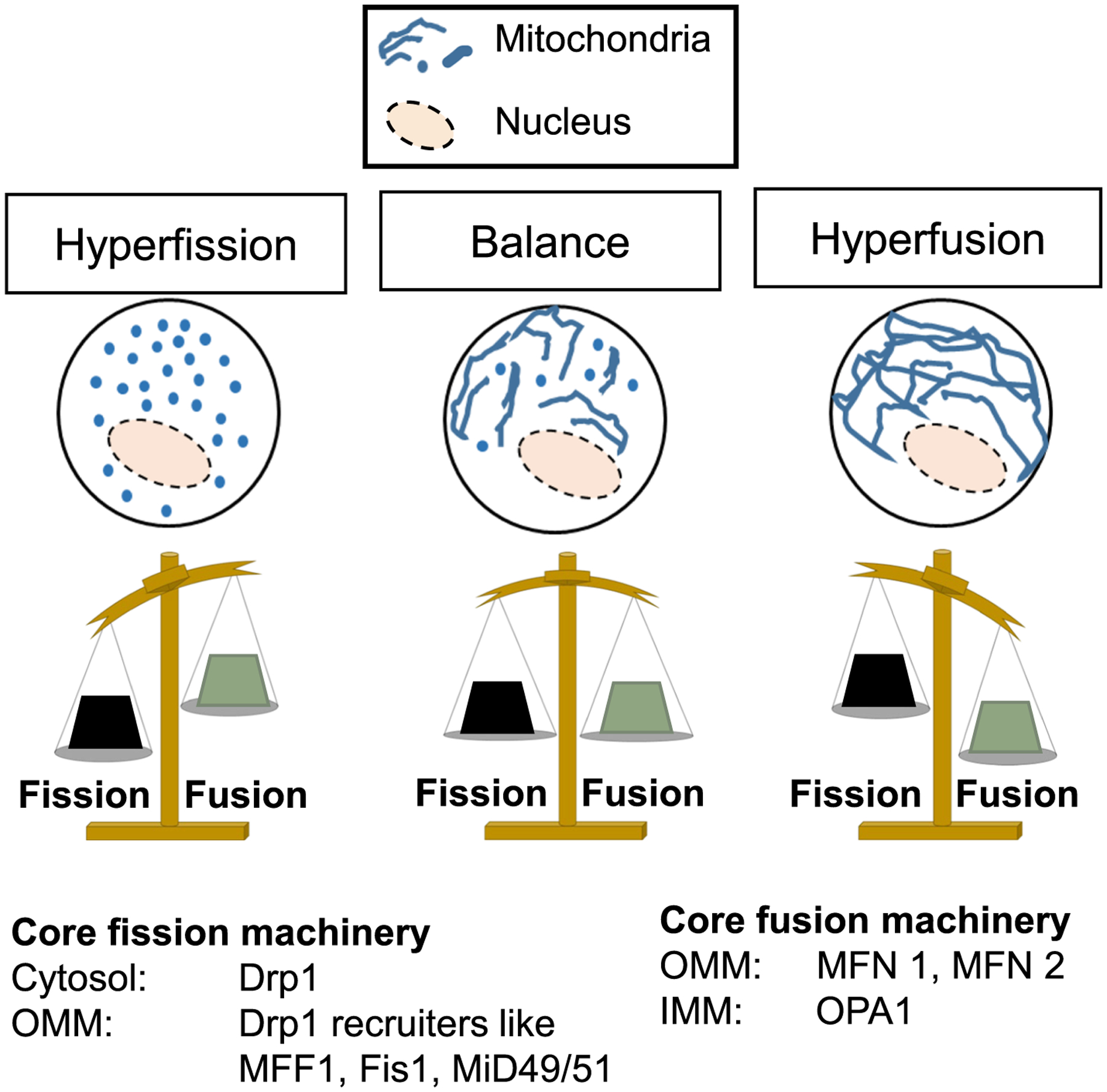
Mitochondrial fission-fusion dynamics. The dynamic balance between mitochondrial fission and fusion lies between mitochondrial hyperfission (left) and hyperfusion (right). The core proteins of the mitochondrial fission and fusion machinery are indicated. OMM: Outer mitochondrial membrane; IMM: Inner mitochondrial membrane. See Table 1 for gene names of homologous machinery across different species.
Table 1.
Known homologs encoding the core proteins of mitochondrial fission-fusion machinery and the relevant cell cycle genes (described in Fig. 2), across different species.
Mitochondria are double membrane bound organelles with an intermembrane space between the outer and the inner mitochondrial membranes. The inner mitochondrial membrane bounds the mitochondrial matrix and folds into specialized structures called cristae where mitochondrial respiratory complexes reside. The inner and outer mitochondrial membranes meet at certain contact sites called the cristae junctions at the base of individual cristae. Mitochondrial fission and fusion processes involve both the inner and outer mitochondrial membranes. The core regulatory proteins of mitochondrial fission and fusion processes are members of the dynamin related protein family. Mitochondrial fission is brought about by the GTPase action of the multimerized Dynamin-Related Protein 1 (Drp1) when it is recruited from the cytosol to the mitochondrial outer membrane (Kageyama et al., 2011). The outer mitochondrial membrane proteins, mitochondrial fission factor 1 (MFF1), mitochondrial dynamics proteins of 49 and 51 KDa (MID49/MID51), or fission protein 1 (Fis1) act as receptors for Drp1 on the mitochondria (Kraus and Ryan, 2017). Although the Drp1 regulatory role of MID49/MID51 remains under active investigation, they can co-assemble with Drp1 to form the protein ring that causes mitochondrial fission (Kalia et al., 2018). Fis1 recruits Drp1 to mitochondria in yeast, but in mammalian cells Fis1 is thought to recruit Drp1 to the mitochondria to cause mitochondrial fission only in certain stress related situations (Qi et al., 2013; Shen et al., 2014). Fis1 may also inhibit mitochondrial fusion machinery independent of Drp1 (Yu et al., 2019). Recent evidence suggests that Drp1 driven mitochondrial fission is regulated through interaction with the endoplasmic reticulum membranes and the cytoskeletal elements (Chakrabarti et al., 2018; Friedman et al., 2011; Moore et al., 2016; Rehklau et al., 2017), and may also involve Dynamin 2 that primarily promotes endocytosis (Lee et al., 2016). The mitochondrial fusion apparatus is split between the outer membrane GTPases mitofusins 1 and 2 (Mfn1/2), and the inner membrane GTPase Optic Atrophy protein 1 (OPA1). The mitofusins of apposing mitochondria interact and oligomerize in a heterotypic or homotypic manner to dock mitochondria together. Subsequent fusion depends on Mfn1/2 driven destabilization of the mitochondrial outer membrane in a way that is not fully characterized (Daste et al., 2018; Huang et al., 2017; Pernas and Scorrano, 2016). The inner membrane fusion protein Opa1 is either tethered to the mitochondrial inner membrane or localizes to mitochondrial inter membrane space. Opa1 gets processed by membrane bound proteases Yme1 or Oma1 into a long (L-Opa1) and a short form (S-Opa1), though the long form is sufficient for the fusogenic activity of Opa1(Song et al., 2007). Other than driving mitochondrial fusion, Opa1 is also responsible for maintaining the organization of the mitochondrial cristae (Cogliati et al., 2013; Frezza and Cipolat, 2006). The molecular machinery supporting the cristae junctions and their contact with the outer mitochondrial membrane is called MICOS (mitochondrial contact site and cristae organization system) (Rampelt et al., 2017). Interaction of Opa1 with components of MICOS may contribute to Opa1 driven regulation of mitochondrial fusion and cristae organization (Darshi et al., 2011). An in vitro study suggests that mitochondrial inner membrane fusion is brought about by interaction of L-Opa1 with the inner membrane specific lipid cardiolipin, while S-Opa1 modulates this interaction (Ban et al., 2017). The same study suggests that cardiolipin independent action of Opa1 may contribute to mitochondrial cristae maintenance. Thus far, no fission machinery has been identified for mitochondrial inner membrane. Moreover, the mechanism for coordination of fission and fusion of the outer and inner membranes remains to be elucidated, although some candidates have been proposed. The yeast protein Ugo1 has been demonstrated to mediate mitochondrial inner and outer membrane fusion events (Hoppins et al., 2009). Recently, a potential mammalian homologue has been identified with similar function to Ugo1, loss of which maintains mitochondrial hyperfused state (Abrams et al., 2015). Notably, regulation of mitochondrial inner and outer membrane fusion events appears to be distinct. For example, mitochondrial fusion of the inner membranes in mammals is ATP dependent, while the outer membrane fusion is not (Mishra and Chan, 2016).
Mitochondrial fission and fusion activities counteract each other. Tilting of the balance towards fission leads to maintenance of individual punctate mitochondria (hyperfission), while tilting of the balance towards fusion maintains a singular fused mitochondrial element (hyperfusion) (Kraus and Ryan, 2017; Labbe et al., 2014; Pernas and Scorrano, 2016) (Fig. 1). We recently designed quantitative metrics for the steady state levels of mitochondrial fission and fusion that contribute to the maintenance of mitochondrial shape in any given cell. Using these metrics we found that although counteracting fission and fusion status are inversely related to each other, their quantitative relationship may be influenced by other factor(s). (Spurlock et al., 2019). One such factor could be mitophagy, which is the process of clearance of mitochondria through autophagosomal machinery. Mitochondrial fission-fusion processes clearly crosstalk with mitophagy (Twig and Shirihai, 2011; Song and Dorn, 2015). It has been hypothesized that fusion of damaged and undamaged mitochondria may serve as a protective measure by diluting damage. On the other hand, mitochondrial fission segregates damaged mitochondria to allow their degradation by mitophagy. Moreover, certain mitochondria derived vesicles carrying specific cargo have been found to be targeted for degradation (Pickles et al., 2018). It is important to note that the abundance of punctate mitochondria after lowering of levels of mitochondrial fusion proteins is a result of lowering of both mitochondrial fusion and mitophagic clearance of the damaged punctate mitochondria (Chen and Dorn, 2013; Lee et al., 2012; Sebastian et al., 2016). Our analyses of the quantitative relationship between steady state mitochondrial fission and fusion status generated the testable hypothesis that reduction of mitochondrial fusion only below threshold leads to linear increase in mitophagy (Spurlock et al., 2019).
There is an ongoing debate about the rate of mitochondrial fission-fusion dynamics in different tissues in intact animals. While measurement of mitochondrial fission and fusion rate has been challenging, several efforts have been made to measure the rate of mitochondrial fusion-fusion dynamics using various live cell microscopy based approaches. The state of the art method of measurement of mitochondrial fission-fusion dyanmics is by the use of a live cell pulse chase assay using photoconvertible probes targeted to the mitochondrial matrix. This assay has been successfully performed in intact cells and in ex vivo tissue from animals expressing a photoconvertible mitochondrial probe (Karbowski et al., 2004; Mishra et al., 2015; Spurlock et al., 2019; Twig et al., 2008). However, the absolute rate of mitochondrial fission-fusion dynamics in an in vivo system has not been measured yet, but has been estimated. Based on the rate of detectable decrease in mitochondrial size in fusion deficient conditions, it has been estimated that mitochondrial fission/fusion cycle in adult heart may be as slow once in two weeks (Song and Dorn, 2015). However, mitochondrial fission-fusion dynamics could be detected within 30 mins in excised muscle tissues ex vivo, using live cell pulse chase assay, where rate of mitochondrial fission-fusion dynamics was shown to vary between different muscle fibers (Mishra et al., 2015). It has been hypothesized that the slower mitochondrial fission/fusion cycle in differentiated tissues, like cardiomyocytes, are primarily important for mitochondrial quality control through regulation of mitophagy (Song and Dorn, 2015).
Ablation of the various core proteins regulating mitochondrial fission or fusion can result in embryonic lethality across various model organisms (Kraus and Ryan, 2017; Labbe et al., 2014; Pernas and Scorrano, 2016), demonstrating the critical role of mitochondrial fission-fusion and their regulatory proteins. However, the exact role of mitochondrial fission and fusion events may vary between proliferating cells and differentiated tissues, as well as in different molecular context in any tissue type.
3. Regulation of mitochondrial fission-fusion dynamics during cell cycle
Cell cycle is one of the various physiological stimuli that have been shown to impact mitochondrial fission or fusion abilities (Chen and Chan, 2017; Labbe et al., 2014). In a symmetric division cycle, the two daughter cells resulting from mitotic division of the mother cell undergo a long growth (G1) phase followed by a DNA synthesis phase (S) that is followed by a short growth phase (G2) before the next round of mitotic division (M). The asymmetric division of stem cells is driven by the intrinsic or extrinsic (niche based) cues that influence the mitotic machinery (Venkei and Yamashita, 2018). The core cell cycle regulation depends on the cyclins, which oscillate in their expression and/or activity in a cell cycle stage-specific manner. They bind and activate their cognate Cyclin Dependent Kinases (CDKs) to phosphorylate and modulate various target proteins in the respective phase of the cell cycle. The core cell cycle regulators regulate mitochondrial functions including biogenesis, energy metabolism, ROS production and fission-fusion dynamics (Lee and Finkel, 2013; Lopez-Mejia and Fajas, 2015). Here, we will focus only on regulation of mitochondrial fission-fusion dynamics by canonical Cyclins and CDKs (for the sake of relevance, the cell cycle independent regulation by any non-canonical Cyclin/CDKs (Lopez-Mejia and Fajas, 2015) will not be covered here).
Work from various laboratories, including ours, revealed cell cycle associated change in mitochondrial shape in various mammalian cells (Chen and Chan, 2017; Mitra, 2013). In normal proliferating cells, mitochondria undergo hyperfusion during G1 to S transition, and hyperfission occurs during mitotic transition (Fig. 2, red in Mitotic Cycle). The majority of cell cycle driven regulation of mitochondrial fission/fusion reportedly happens at the level of the mitochondrial fission protein Drp1. Enhanced mitochondrial fission in mitosis is brought about by activation of Drp1 by phosphorylation at its S616 residue by the mitotic Cyclin-B-CDK1 complex (Taguchi et al., 2007). The mitotic kinase Aurora kinase A activates the RalA/RalBP1 complex to mediate the Cyclin-B-CDK1 driven activation of Drp1 on the mitochondrial outer membrane (Kashatus et al., 2011) (Fig. 2). Ectopic overexpression of Cyclin B1 in the mitochondrial matrix was not found to impact Drp1 in spite of impacting mitochondrial energetics during mitotic transition (Wang et al., 2014b). Although Aurora kinase A can support mitochondrial fission at endogenous levels, when overexpressed in cancer cells Aurora kinase A localizes to mitochondria to possibly promote mitochondrial fusion in a RalA independent manner (Bertolin et al., 2018). Microtubule reorganization is a crucial structural component for segregation of chromosomes during mitosis and division of cytosol during cytokinesis. In mammalian cells, a particular isoform of Drp1 (Drp1-x01) particularly associates with microtubules and not mitochondria (Strack et al., 2013). However, phosphorylation of Drp1 at S616 disrupts the Drp1-microtubule interaction and recruits Drp1 to mitochondria causing mitochondrial hyperfission in mitosis (Strack et al., 2013). In fission yeast, a model has been proposed where association of microtubules with mitochondria prevents assembly of mitochondrial fission apparatus driven by Dnm1 (yeast homologue of Drp1) (Mehta et al., 2019). This competition model proposes that engagement of microtubules to form mitotic spindle precludes micro-tubule association with mitochondria, thus allowing assembly of the Drp1 driven fission apparatus on mitochondria during mitosis.
Fig. 2.
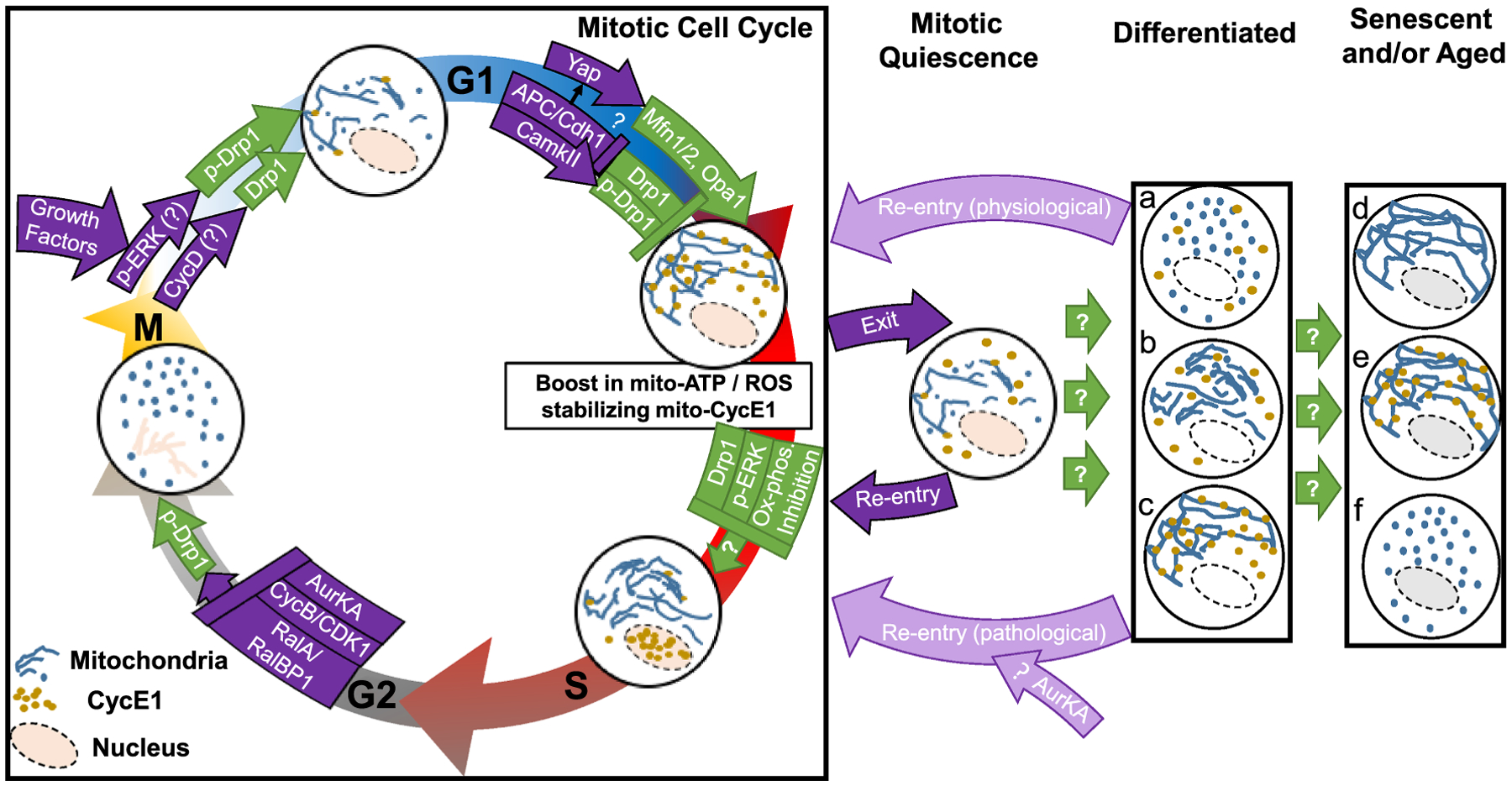
Model depicting integration of mitochondrial fission-fusion dynamics within mitotic cell cycle, which may impact mitotic quiescence, differentiation, senescence and aging. Cell cycle regulators modulate Drp1 (and Mfn1/2) function to alter the dynamic balance of mitochondrial fission-fusion towards regulating cell cycle progression. Lowering of Drp1 levels by APC/Cdh1 driven degradation during the G1-S transition, promotes mitochondrial hyperfusion, supported by Yap-mediated transcription of Mitofusin and Opa1. Mitochondrial hyperfusion boosts mitochondrial ATP and/or ROS levels, leading to recruitment and stabilization of Cyclin E on mitochondria in late G1. During the G1-S transition Cyclin E may translocate from mitochondria to the nucleus to drive cell cycle progression through S phase and G2 into mitosis. Release of mitochondrial Cyclin E can happen by activation of Drp1, lowering of mitochondrial energetics (ox-phos), and/or activation of p-ERK. Mitotic activation of Drp1 promotes mitochondrial hyperfission to possibly allow symmetric segregation of mitochondria. The inability of released mitochondrial Cyclin E to translocate to the nucleus may lead to its degradation or inactivity and mitotic cell cycle exit into mitotic quiescence. Differentiation cues act on quiescent cells to confer lineage specificity so that mitochondrial fission-fusion status and mitochondrial Cyclin E pool can vary between lineages (a, b, c). Physiological or pathological signals can cause differentiated cells to re-enter mitotic cell cycle. Moreover, senescence and/or aging of various differentiated cells can be supported by altered mitochondrial fission-fusion status (d, e, f). (?) Signifies proposed hypothesis to be tested; p-Drp1 specifically signifies the activating p-S16-Drp1.
Mitochondrial hyperfusion at the G1-S transition may occur by activating the degradation of Drp1 by the cell cycle specific degradation machinery, APC-Cdh1 (Horn et al., 2011). However, the general degradation properties of Drp1, including its turnover rate, remain to be studied. Additionally, a recent study proposed that cytosolic calcium/CaMKII driven Drp1-S616 phosphorylation can prevent hyperfusion of mitochondria during G1 to S transition and block this cell cycle transition (Koval et al., 2019). The G1 phase is regulated by distinct set of molecules before and after the restriction point beyond which the cell commits to the G1-S transition (Johnson and Skotheim, 2013). The early G1 phase leading to the restriction point is marked by growth factor driven induction of MAPK and/or other signaling pathways. Importantly, growth factors and MAPK pathway-induced ERK activation activates Drp1 in multiple mammalian cells, including stem cells (Fu et al., 2017; Kashatus et al., 2015; Koval et al., 2019; Prieto et al., 2016; Serasinghe et al., 2015). It is tempting to speculate that such MAPK kinase dependent activation of Drp1 can potentially suppress precocious hyperfusion of mitochondria in the early G1 phase (before restriction point), ensuring mitochondrial hyperfusion happens only during G1 to S transition at late G1(after restriction point) (Fig. 2). Drp1 was identified as a major interacting protein of the growth factor induced Cyclin D1 (Jirawatnotai et al., 2011), while Cyclin D1 knockout mice were found to harbor long tubular mitochondria reminiscent of the hyperfused mitochondria in Drp1 repressed conditions (Wang et al., 2006). These findings lead to the hypothesis that Cyclin-D1 interacts with Drp1 to sustain its activity to prevent transition to late G1, which remains to be tested (Fig. 2).
Direct evidence of cell cycle dependent regulation of the core mitochondrial fusion proteins, MFN1/2 and Opa1, is limited. Degradation of fzo1 protein (yeast homologue of MFN1) was proposed to be cell cycle dependent (Neutzner and Youle, 2005). Circumstantial evidence links MFN1/2 and Opa1 to cell cycle driven regulation of the Hippo signaling pathway as follows: The upstream Mst1/2 (mammalian homologue of Drosophila Hippo kinase) activates LATS kinase to repress the downstream transcriptional regulator Yap or Yorkie (Drosophila homologue of Yap) (Yu et al., 2015). APC-Cdh1 driven degradation of the LATS kinase activates Yap/Yorkie in the G1 phase of cell cycle (Kim et al., 2019). Activation of Yap/Yorkie has been shown to drive transcription of MFN1 and its Drosophila homologue Marf1 as well as Opa1 to regulate cell proliferation (Nagaraj et al., 2012). Therefore, it is possible that the APC-Cdh1 driven activation of Yap contributes to mitochondrial fusion during G1 to S transition by transcriptional activation of Mfn1/2 and Opa1 genes (Fig. 2). It is worth testing this hypothesis in stem cells given Hippo pathway regulates stemness (Yu et al., 2015). The Hippo kinase itself has also been proposed to regulate Drp1, but the mechanism remains unclear (Ouyang et al., 2019; Wang and Song, 2018).
In summary, data from several laboratories indicate that regulation of mitochondrial fission-fusion status is integrated within the cell cycle regulation (Fig. 2). Such integration remains to be specifically validated in stem cells, which would advance understanding how integration of mitochondria and cell cycle modules can impact cell cycle exit, maintenance of quiescence, senescence and differentiation of stem cells.
4. Impact of mitochondrial fission-fusion on cell cycle regulation and cell proliferation
Propagation of errors in cell cycle regulation is minimized by specific check points. Various mitochondrial stresses activate metabolic check points in cell cycle, which reflects passive involvement of mitochondria in cell cycle regulation. On the other hand, active involvement of mitochondria in regulation of cell cycle involves modulation of cell cycle activities (e.g. DNA synthesis, chromosomal segregation) by mitochondria derived metabolites, ATP, ROS etc. Depletion of mitochondrial supplies that actively control cell cycle may also activate cell cycle blocks or delay cell cycle. Mitochondrial ATP production is modulated according to the energetic demand of different cell cycle stages, maximizing during the G1/S and/or G2/M transitions, likely depending on the cell context (Mitra et al., 2009; Wang et al., 2014b; Harbauer et al., 2014; Koval et al., 2019). Also, mitochondria derived ROS contributes to the redox regulation of the cell cycle (Lee and Finkel, 2013).
Mitochondrial ATP and ROS production are linked through the mitochondrial energetics circuitry, and are also subject to nutrient availability (Murphy, 2009). Mitochondrial ATP and ROS production and nutrient utilization crosstalk in a bi-directional manner with mitochondrial fission and fusion dynamics, although the mechanistic details of these interactions are being investigated (Galloway et al., 2012; Willems et al., 2015). Multiple lines of evidence lead to the consensus that in nutrient-rich conditions, mitochondrial hyperfission maintains energetically less efficient mitochondria, whereas in nutrient-deprived conditions, energetically efficient hyperfused mitochondria are maintained (Liesa and Shirihai, 2013; Schrepfer and Scorrano, 2016). In this regard, we and others have shown in non-neoplastic mammalian cells that hyperfusion achieved by repression of mitochondrial fission protein Drp1 at G1-S transition (or in other physiological conditions) is associated with stimulation of mitochondrial respiration (Mitra et al., 2009). In budding yeast, it has been proposed that import of key mitochondrial fusion protein fzo1 (yeast homologue of mitofusins) is required to maintain enhanced fusion to support mitochondrial respiration in mitosis (Harbauer et al., 2014). Similarly in cancer cells, Aurora kinase A overexpression has been proposed to support mitochondrial respiration in mitosis (Bertolin et al., 2018).
It remains to be determined whether hyperfusion promotes efficient nutrient/metabolite utilization, electron transport chain activity, ATP synthesis or optimal ROS generation. Ablation of the mitochondrial fission protein Drp1, which sustains hyperfused mitochondria, enhances maximal respiratory capacity of mitochondria, suggesting a possible regulation of the mitochondrial electron transport efficiency in the hyperfused mitochondria (Parker et al., 2015). Furthermore, such increase in respiratory capacity took place only in the presence of exogenous factors that support cell proliferation, suggesting additional factors regulate respiratory efficiency (Parker et al., 2015). Ablation of the inner membrane fusion protein Opa1 impacts various aspects of mitochondrial energetics, including organization of the mitochondrial respiratory chain super-complexes by maintaining proper mitochondrial cristae structure (Cogliati et al., 2013; Del Dotto et al., 2018). However, the fusion ability of Opa1 is thought to be independent of its ability to sustain energetics (Frezza and Cipolat, 2006). This distinction in Opa1 function is likely achieved by its multiple isoforms (Del Dotto et al., 2018). For example, the S-Opa1 isoform supports mitochondrial energetics in spite of not being able to support fusion (Lee et al., 2017). Nonetheless, Opa1 inactivity reduces cell proliferation (Chen et al., 2005; Merkwirth et al., 2008). Similarly, ablation of mitochondrial fusion proteins MFN1/2 dramatically compromises mitochondrial respiration and slows down proliferation (Chen et al., 2003). This may be due to defects in mtDNA maintenance (Chen et al., 2003), reduction of coenzyme Q pools (Mourier et al., 2015) or autophagic regulation (Chen and Dorn, 2013; Lee et al., 2012; Sebastian et al., 2016). Over-expression of MFN2, can also reduce cell proliferation in certain cellular contexts (Chen et al., 2004; Cheng et al., 2013). Drp1, Mitofusins and Opa1 have all been implicated in the control of cellular metabolism in proliferating and/or differentiated cells (Del Dotto et al., 2018; Liesa and Shirihai, 2013; Schrepfer and Scorrano, 2016). How these alterations in metabolism are functionally linked to the impact of mitochondrial fission-fusion dynamics on energetics needs to be tested. In this regard, multiparametric analyses of mitochondrial energetics would shed additional insight on the mechanistic regulation of the bi-directional crosstalk of mitochondrial energetics and mitochondrial fission-fusion dynamics. Towards this goal, we recently developed the mito-SinCe2 approach to quantitatively analyze mitochondrial ATP or redox status and fission/fusion status in single cells (Spurlock et al., 2019). Mito-SinCe2 analyses of tumor initiating stem cells revealed that mitochondrial fusion status and ATP increase linearly with each other only once mitochondrial hyperfusion is achieved. The analyses also revealed that the status of mitochondrial fission-fusion changes linearly either with ATP or with redox, but not simultaneously with both.
What is the significance of the mitochondrial ATP synthesis in proliferating cells? We found active mitochondrial ATP synthesis is required for G1-S transition of cell cycle, while not for mitosis at least in non-neoplastic mammalian cells tested (Mitra et al., 2009). Given ATP can also be synthesized through the (faster) glycolytic pathway in the cytosol, what may be the advantage of mitochondrial ATP production during G1 to S transition? The answer may lie in Cyclin E that governs the G1 to S transition (Siu et al., 2012). We and others found that Cyclin E, is uniquely linked to mitochondrial energetics and metabolism at least in mammalian cells and Drosophila (Mandal et al., 2010; Mandal et al., 2005; Mitra et al., 2009; Ohhara et al., 2017; Parker et al., 2015; Qian et al., 2012; Xu et al., 2014). In Drosophila, genetic perturbation of mitochondrial energetics prevents G1-S transition by specifically degrading Cyclin E (Mandal et al., 2010; Mandal et al., 2005). Consistent with this we showed that boost in mitochondrial respiration supported by ablation of Drp1 driven mitochondrial fission prevents the normal Cyclin E degradation in mitosis in mouse embryonic fibroblasts (MEFs) (Parker et al., 2015). This mitochondrial regulation of Cyclin E happens by its recruitment to the mitochondria that we detected in MEFs and in Drosophila (Parker et al., 2015). Specifically, repression of Drp1 enhances the mitochondrial pool of Cyclin E that can be released by perturbing mitochondrial energetics, as tested in MEFs (Parker et al., 2015). Therefore, we proposed that Drp1 repression increases mitochondrial respiration to recruit Cyclin E on energetically active mitochondria to prevent its degradation and facilitate its build up required for G1-S transition (Fig. 2). Cyclin E abundance throughout the cell cycle causes various cell cycle defects and aberrant cell proliferation (Siu et al., 2012), as well as stem cell maintenance (Orford and Scadden, 2008). Consistent with this, repression of Drp1 driven mitochondrial fission, which maintains Cyclin E throughout the cell cycle, promotes various cell cycle defects including the following: a) cell cycle blocks (Mitra et al., 2009; Qian et al., 2012; Rehman et al., 2012; Tanwar et al., 2016; Westrate et al., 2014), b) context dependent aberrant cell proliferation and inhibition of differentiation (Mitra et al., 2012; Mitra et al., 2009; Parker et al., 2015), and c) maintenance of stem cell properties (Khacho and Slack, 2017; Parker et al., 2015; Todd et al., 2010). Aberrant cell proliferation resulting from Drp1 repression is dependent on EGFR-MAPK signaling in Drosophila and in MEFs (Mitra et al., 2012; Parker et al., 2015; Tomer et al., 2018). Importantly, aberrant cell proliferation and inhibition of differentiation in cells ablated for Drp1 can be prevented by genetic perturbation of mitochondrial energetics in Drosophila (Tomer et al., 2018). Inappropriate Cyclin E activity in mitosis leads to aneuploidy and genomic instability (Siu et al., 2012). Indeed, aneuploidy induced by Drp1 repression can be attenuated by Cyclin E downregulation in certain cancer cells tested (Qian et al., 2012). Interestingly, the Drp1 ablated cells escape contact inhibition and proliferate aberrantly when the mitochondrial Cyclin E pool is released by ERK activation in both MEFs and Drosophila (Parker et al., 2015). From the above findings, we propose that modulation of mitochondrial fission-fusion dynamics controls critical aspects of mitochondrial energetics to promote G1-S transition and initiation of DNA replication by stabilizing Cyclin E on the mitochondrial surface (Fig. 2). Four critical questions arising from this line of thought are: a) Does mitochondrial Cyclin E, when released, translocate to the nucleus (Fig. 2)? b) Is Cyclin E regulated by ATP or ROS generated from mitochondria? c) Are there specific metabolites that maintain the energetic state of mitochondria towards regulating Cyclin E? d) Is a feedback loop established because Cyclin E has been reported to regulate mitochondrial energetics in erythroid lineage in mice (Xu et al., 2014)?
The increase in Drp1 driven mitochondrial fission in M phase, as described in the previous section, can be hypothesized to promote cell proliferation, and possibly important for cell proliferative disorders like cancer. From analyses of publically available cancer genomics data, we found Drp1 co-expresses with crucial genes controlling mitosis in a wide variety of cancer tissues, demonstrating the importance of Drp1 in neoplastic cell cycle (Tanwar et al., 2016). Moreover, enhanced Drp1 activity in mitosis can be envisioned to be important for segregation of mitochondria between daughter cells during mitosis. However, partitioning of mitochondria happens in Drp1 ablated mouse embryonic fibroblasts, although there may be asymmetry in mitochondrial partitioning in daughter cells (Ishihara et al., 2009).
Cell cycle regulatory events guide the determination of whether a cell will remain quiescent, proliferate or differentiate. It is clear that genetic disruption of fission or fusion machinery alters cell proliferation at specific points during the cell cycle. The mechanistic details of such regulatory processes are being investigated (Fig. 2). Tight regulation of mitochondrial energetics by mitochondrial fission and/or fusion events actively regulates normal progression through the cell cycle. It will, therefore, be important to better understand the crosstalk between mitochondrial fission/fusion and energetics to elucidate regulation of the energetics of cell proliferation and cell fate determination.
5. Can mitochondrial fission-fusion influence stem cell self-renewal and differentiation by regulating cell cycle events?
Asymmetric mitotic division of adult stem cells results in one self-renewing cell and one differentiating cell. However, adult stem cells are largely maintained in a state of mitotic-quiescence. Maintenance of stem cell quiescence is a necessary step for preventing stem cell exhaustion and/or senescence (Oh et al., 2014). Thus, stringent regulation of re-entry of quiescent stem cells into self-renewal and proliferative cycle is critical for maintenance of healthy tissues. The cell cycle status of the stem cells critically contributes to their cell fate determination between self-renewal, differentiation and quiescence (Liu et al., 2019; Julian et al., 2016; Orford and Scadden, 2008). Proliferating stem cells have distinctly different cell cycle profile from their differentiated progenies, including a shorter G1 phase and prolonged S and/or G2-M phase, while induction of differentiation prolongs the length of G1 (Julian et al., 2016). Importantly, prolonging the S and/or G2-M phase (involving Cyclin E and Cyclin B, respectively) maintains self-renewal (Gonzales et al., 2015), while levels of the growth factor Cyclin D determine the lineage of differentiation (Pauklin and Vallier, 2013).
Modulation of mitochondrial fission and fusion is emerging as critical for regulation of both pluripotent and adult stem cells. The consensus is that the balance of mitochondrial fission-fusion is tilted more towards unopposed fission in embryonic stem cells or induced pluripotent stem cells (Fu et al., 2019) (covered more detail in a relevant review in this special issue). However, transition from naïve to primed pluripotent cells involves tilting the balance towards mitochondrial fusion (Bahat et al., 2018). Interestingly, a longer G1 is maintained in naïve pluripotency state than a primed pluripotency state (Ter Huurne et al., 2017), raising the interesting possibility that mitochondrial fusion may be linked to shortening the overall length of G1 in primed pluripotency state. We showed that Drp1 ablated MEFs, which maintain hyperfused mitochondria, have reduced G1 length and elevated levels of the stem cell marker Sox-2, in comparison to the wild type controls (Parker et al., 2015). Consistently, the Sox-2 positive adult mouse neural stem cells harbor tubular mitochondria (indicating elevated fusion or reduced fission) relative to their differentiated progeny (Khacho et al., 2016). The self-renewal of these neural stem cells is sustained by redox balance maintained by mitochondrial fusion proteins Mfn1 and Opa1 (Khacho et al., 2016). Involvement of any cell cycle regulation by mitochondrial fusion in maintenance of these neural stem cells has not been tested yet. Importantly, neural progenitor cells derived in vitro from pluripotent stem cells also harbor more tubular mitochondria in comparison to the parental pluripotent stem cells (Lorenz et al., 2017). The mitochondrial fusion proteins have been shown to sustain self-renewal ability of Drosophila male germline stem cells by facilitating lipid utilization, where involvement of cell cycle has not been tested (Senos Demarco et al., 2019). Sustaining mitochondrial hyperfusion by repressing mitochondrial fission protein Drp1 maintains elevated Cyclin E levels and enhanced cell proliferation in the Drosophila ovarian epithelial cells (Mitra et al., 2012), including their stem cells (our unpublished results).
Maintenance of mitochondrial hyperfusion throughout the cell cycle reportedly disturbs the partitioning of mitochondria into daughter cells (Ishihara et al., 2009), raising the possibility that sustained hyperfusion regulates stem cell properties by modulating mitochondrial partitioning during asymmetric division of stem cells. Consistent with this idea, Mfn1 driven fusion proficient mitochondria preferentially partition in stem cells during asymmetric division (Wu et al., 2019). Moreover, depletion of Mfn1 resulted in symmetric division with loss of stemness and undue induction of differentiation in a normal mammary stem cell model (Wu et al., 2019). Similarly, appropriate partitioning of specific mitochondria during asymmetric division has been proposed as a way to maintain stemness in a neoplastic mammary stem cell model (Katajisto et al., 2015). The asymmetric division of budding yeast serves as model for studying asymmetric stem cell division (Pernice et al., 2017). In this model, the partitioning of mitochondria into daughter cells happens in proportion with the cytosolic volume (Jajoo et al., 2016) and involves interactions of the mitochondrial fission/fusion machinery, cytoskeletal elements, endoplasmic reticulum and plasma membrane (Pernice et al., 2017). The details of these interactions continue to be investigated. Towards such an endeavor, interaction between a myosin motor and mitochondrial fission and fusion genes has been proposed as a mechanism of mitochondrial partitioning to the bud during asymmetric division (Bockler et al., 2017).
Taken together, the literature suggests that mitochondrial fusion supports maintenance of various adult stem cells. Adult stem cells can undergo neoplastic transformation leading to initiation of tumor formation (Magee et al., 2012). Enhanced Drp1 driven mitochondrial fission can sustain self-renewal properties of the tumor initiating cells (Xie et al., 2015). However, repression of Drp1, that sustains mitochondrial hyperfusion, can enhance certain markers for tumor initiating cells (Parker et al., 2015). We recently reported that tumor initiating cells with hyperfused mitochondria and enhanced mitochondrial ROS and can be triggered to self-renew and proliferate more efficiently (at least in vitro) than tumor initiating cells that do not have those mitochondrial properties (Spurlock et al., 2019). Interestingly, triggering self-renewal converts the mitochondrial hyperfusion state to a hyperfission state. Therefore, we proposed that the tumor initiating cells with mitochondrial hyperfusion status in quiescence are ‘primed’ to self-renew more efficiently than others (Spurlock et al., 2019). This priming appears to result from alterations in cell cycle (our unpublished data).
Exit from the mitotic cell cycle to a mitotic quiescence state is a prerequisite for induction of differentiation. Differentiated cells can re-enter mitotic cycle when stimulated by physiological (ex: immune cell proliferation) or pathological cues (ex: cancer cell proliferation). Undue induction of differentiation observed with depletion of Mfn1 in human mammary stem cells (Wu et al., 2019) is consistent with our observation of precocious induction of differentiation induced by depletion of Marf 1 (Drosophila homologue of mitofusins) in the Drosophila ovarian epithelial cell layer (Mitra et al., 2012). Based on our data, we proposed mitochondrial fission sustained by reduced mitofusin activity is required for cell cycle exit, which may allow increase in mitochondrial biogenesis during differentiation (Mitra, 2013). Indeed, in a muscle differentiation model, enhanced mitochondrial fission was observed early in differentiation that was followed by increase in PGC1-alpha driven mitochondrial biogenesis (Sin et al., 2016). We further showed that mitochondrial fusion may resume after cell cycle exit and initiation of differentiation depending on the cell type (Mitra et al., 2012). Opa1 driven mitochondrial fusion has been proposed to be required for inducing differentiation in Drosophila intestinal epithelial progenitor cells (Deng et al., 2018) and for cardiomyocyte differentiation from mouse embryonic stem cells (Kasahara et al., 2013). Differentiation of pluripotent cells to various other lineages associates with mitochondrial fusion and boost in mitochondrial energetics (Fu et al., 2019).
Naïve embryonic stem cells differentiate into prime embryonic stem cells that further differentiate into lineage specific adult stem/progenitor cells, ultimately giving rise to the terminally differentiated cells of the respective lineage. Shifts in mitochondrial fission-fusion dynamics appears to contribute at various levels of stem cell maintenance and differentiation. Based on the above discussed evidence, we have developed the following hypothesis to be experimentally tested in future (Fig. 3). We hypothesize that the level of mitochondrial fusion gradually increases from naïve embryonic cells to the adult stem cells in a lineage specific manner. At each level, modulating mitochondrial fission-fusion dynamics guides the maintenance of quiescent G0/G1 state and their entry into a proliferating state towards maintaining self-renewal and differentiation.
Fig. 3.
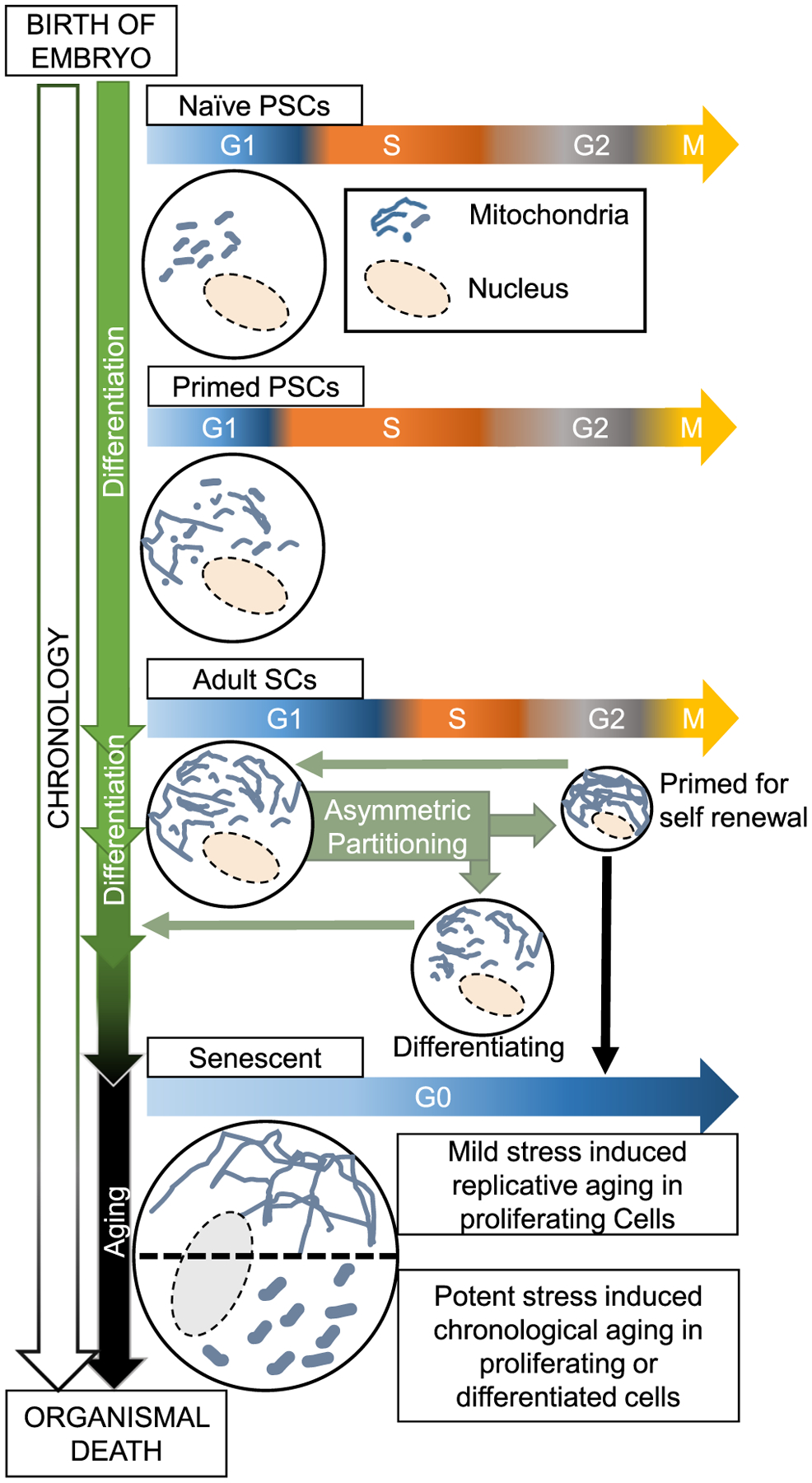
Proposed hypothesis about how interaction of cell cycle modules with mitochondrial fission-fusion dynamics in stem cells and their differentiated counterparts may impact senescence and aging. During development, a gradual overall increase in mitochondrial fusion is associated with differentiation of embryonic naïve pluripotent stem cells (PSCs) to adult stem cells (SCs). At each level of differentiation, the length of the G1 phase may vary with mitochondrial fusion activity. Various adult SCs (or their progenitors) may be self-renewed by cell cycle-regulated mitochondrial hyperfusion, which when inhibited leads to cell cycle exit and differentiation. Sustained mitochondrial hyperfusion by mild stresses may lead to permanent cell cycle exit and cause senescence. Also, different potent stresses during the lifespan may sustain a mitochondrial hyperfission state in adult SCs and/or their differentiated counterparts in association with senescence and aging.
6. Aging and mitochondrial fission-fusion dynamics
Aging is a complex multifactorial process (Lopez-Otin et al., 2013). Imbalance in mitochondrial fission or fusion contributes to both chronological aging (decline in function of non-proliferating cells) and replicative aging (loss of proliferative capacity i.e. senescence). Moreover, mitochondrial fission and fusion dynamics is linked to dysregulation of nutrient sensing, genomic instability, cellular senescence and stem cell exhaustion, all of which contribute to the biology of aging. Here, we will discuss the current literature on the contribution of mitochondrial fission and fusion dynamics in both replicative and chronological aging, although any connection to cell cycle remains speculative.
Replicative senescence is defined as an irreversible exit from the cell division cycle, in contrast quiescent cells are able to re-establish a proliferative state. Importantly, elimination of senescent cells increases life span (McHugh and Gil, 2018). Senescence is primarily caused by the stress induced DNA damage response, which brings about permanent cell cycle block by activating the CDK inhibitors p16 and p21 (Chandler and Peters, 2013). The senescent state has been proposed to resist oncogenic induction of cell proliferation. Paradoxically, the senescence associated secretory phenotype (SASP) provides cytokines to support proliferation of oncogenic cells by paracrine regulation (McHugh and Gil, 2018). In various cell culture systems including certain adult stem cells, β-gal positive senescent cells maintain hyperfused mitochondria. Certain senescent cells were found to have reduction of mitochondrial fission proteins Drp1 and/or Fis1 and down-regulation of Drp1 or Fis 1 was sufficient to induce senescent like features, including increase in β-gal positive staining (Lin et al., 2015; Mai et al., 2010; Zhang et al., 2017). Moreover, over activation of mitochondrial fusion protein Mfn1 resulting from its reduced turn over (driven by ubiquitin ligase MARCH5) could promote senescent like phenotypes (Park et al., 2010). Also, chemotherapy induced senescence in melanoma cells has been shown to involve Mfn1 driven induction of the interleukin IL-6 that contributes to SASP (Martinez et al., 2019). However, SASP induced by various forms of mitochondrial dysfunction in non-neoplastic cells appears to be distinct from IL-6 induction (Wiley et al., 2016). The mitochondrial fusion protein Mfn2 has been proposed to be involved in maintenance of senescence triggered by reduction of FGF21 in mesenchymal stem cells (Li et al., 2019). Various forms of mild stress can induce mitochondrial hyperfusion, which has been proposed to be a protective measure to boost mitochondrial respiration to counteract the stress (Eisner et al., 2018; Youle and van der Bliek, 2012). Mitochondrial hyperfusion sustained by Drp1 repression brings about cell cycle dependent genomic instability to elicit the ATR driven DNA damage response in cancer cells tested (Qian et al., 2012). Also, persistent Drp1 repression can bring about cell cycle block in various cells (Mitra et al., 2009; Qian et al., 2012; Tanwar et al., 2016; Westrate et al., 2014). Therefore, it can be hypothesized that such cell cycle block triggered by sustained mitochondrial hyperfusion causing a DNA damage response can potentially underlie the senescence induction in the above proliferating cell culture systems. In contrast, endothelial cell senescence in the context of diabetes is associated with activation of mitochondrial fission by sulphenylation of Drp1 (Kim et al., 2018). Similarly, senescence occurring with myocardial infarction related hypoxia involves Drp1 driven mitochondrial fission (Nishimura et al., 2018). In the single cellular budding yeast model system, replicative aging is studied by the number of daughter cells produced by the mother cell that ages and dies (Steinkraus et al., 2008). replicative aging in yeast is associated with maintenance of mitochondria in hyperfission state (Scheckhuber et al., 2007; Wang et al., 2014a).Moreover, the quality and quantity of mitochondria retained in the mother yeast cell are reduced with age (Hughes and Gottschling, 2012; McFaline-Figueroa et al., 2011; Pernice et al., 2016). Additionally, distinct shapes of mitochondria in non-proliferating yeast cells, characterized as globular vs vesicular, were found to indicate cell quiescence or apparent senescence in chronological aging (Laporte et al., 2018). Although this study did not find any involvement of mitochondrial fission in determining the vesicular or globular mitochondrial phenotype, the effects of mitochondrial fusion remain to be tested.
A shift towards mitochondrial hyperfission has been observed with aging of differentiated tissues in several model organisms and in humans. Repression of mitochondrial fission has been found to enhance life span in various model organisms, strongly suggesting a regulatory role of mitochondrial fission in causing aging-related phenotypes. Deletion of the mitochondrial fission protein Dnm1 (yeast homologue of Drp1) increases chronological lifespan in yeast as assessed by the number of yeast cells surviving in cultures for an extended period of time (Scheckhuber et al., 2007). Moreover, deletion of the mitochondrial fusion protein mgm1 (yeast homologue of Opa1) reduces lifespan in yeast (Scheckhuber et al., 2011). The simple multicellular model, C elegans, has been extensively used in chronological aging studies primarily due to the ease of quantification of lifespan and aging-related pathologies in this model (Kenyon, 2010). Interestingly, aging-related mitochondrial fission has been reported in multiple tissues of C elegans (Chaudhari and Kipreos, 2017; Jiang et al., 2015). Age related increase in drp1 driven mitochondrial fission in certain neurons of C elegans has been linked to lack of neuronal activity (Jiang et al., 2015). Regulated mitochondrial fission in neurons has been proposed to contribute to trafficking of mitochondria for synaptic activity (Morsci et al., 2016). Due to the substantial evolutionary conservation of the aging-related signaling pathways with mammals, C. elegans model has been also extensively used to characterize the signaling pathways in aging and lifespan. In fact, critical aging pathways were first identified by analyzing long or short lived C elegans mutants (Kenyon, 2010). One example is the extension of lifespan with attenuation of insulin signaling pathway, which has been widely confirmed in other models. Interestingly, attenuation of mitochondrial fission protein drp1 further prolongs the lifespan of insulin signaling mutants of C elegans, but not in the wild type (Yang et al., 2011). This synergistic epistatic interaction between the insulin pathway and drp1 remains to be studied further in the context of aging. Increase in longevity achieved with genetic modulation of various pathways associates with enhanced mitochondrial fusion state as detected in body wall muscle in C elegans (Chaudhari and Kipreos, 2017). Moreover, downregulation of eat-3 (C elegans homologue of Opa1) reversed the longevity phenotype of a majority of the longevity pathways tested, including repressed insulin signaling. This suggests different longevity pathways may share a requirement of optimal levels of eat-3/opa1 to maintain mitochondrial fusion and/or energetics. In this study, the authors proposed that altered proteolytic processing of eat-3 protein causes the age-related decline in mitochondrial fusion, leading to mitochondrial hyperfission. Nonetheless, the possibility remains that reduction of eat-3 levels would induce overall apoptosis which could explain the reversal of longevity.
Dietary restriction (DR) consistently prolongs lifespan in model organisms ranging from C elegans to Rhesus macaques (Riera et al., 2016). The full molecular mechanisms underlying DR mediated longevity are not fully understood but the role of mitochondrial fission-fusion dynamics is currently receiving attention. Activation of the AMP-activated protein kinase (AMPK) in response to DR contributes to DR mediated longevity in C elegans. Genetic deletion of mitochondrial fusion proteins fzo-1 (C elegans homologue of mitofusin) or eat-3 reverses this effect, suggesting DR mediated extension of lifespan is mediated by mitochondrial fusion at least in C elegans (Weir et al., 2017). In this report, deletion of either fzo-1 or drp1 did not enhance normal longevity, likely due to disturbance of mitochondrial homeostasis. Interestingly, simultaneous deletion of drp1 and fzo-1 maintained mitochondrial homeostasis and markedly increased longevity in fed animals. This is consistent with the prediction generated from computational modeling that reducing rates of mitochondrial fission-fusion cycles may prolong healthy lifespan by preventing propagation of mitochondrial damage (Figge et al., 2012). However, the prolonged lifespan in the drp1/fzo-1 double knockout C elegans was not observed with intermittent fasting, likely because fasting involves remodeling of mitochondria by either fission and/or fusion processes (Weir et al., 2017). Indeed, starvation promotes mitochondrial hyperfusion in mammalian models, which is proposed to sustain mitochondrial respiration and evade mitophagy (Gomes et al., 2011; Rambold et al., 2011).
The Sirtuin family of proteins, which modulate acetylation status of certain proteins, are major regulators of aging and metabolism (Houtkooper et al., 2012). Aging is associated with reduction of Sirtuin levels in some tissues. Some Sirtuins specifically modulate mitochondrial proteins and may also localize to the mitochondria. Action of Sirt3, one of the mitochondrial Sirtuins, leads to boost in mitochondrial energy metabolism (Ahn et al., 2008; Hirschey et al., 2010). Also, Sirt3 overexpression can improve regenerative capacity of at least aged hematopoietic stem cells as tested in a mouse model (Brown et al., 2013). Interestingly, Sirt3 can suppress the induction of Drp1 driven mitochondrial fission in protecting against acute kidney injury in mouse model (Morigi et al., 2015). It remains to be tested whether anti-aging effects of the Sirtuins involves modulating mitochondrial fission-fusion dynamics.
In mice, age related decline in muscle functionality due to ablation of the mitochondrial fusion protein Mfn2 is accompanied by altered mitophagic clearance of dysfunctional mitochondria (Sebastian et al., 2016). Interestingly, exercise promotes mitochondrial fusion in muscles in multiple model organisms including humans (Chaudhari and Kipreos, 2017; Huertas et al., 2019; Picard et al., 2013) and has been proposed to delay age related decline in mitochondrial fusion proteins Mfn1/2 (Halling et al., 2017). A recent report demonstrated age related decline in the levels of Drp1, Opa1 and Mfn1/2 in muscle biopsies of ‘sedentary’ senior human subjects but not in ‘sportsmen’ senior human subjects (Tezze et al., 2017). However, decrease in muscle activity only correlated with decrease in Opa1. Importantly, muscle-specific ablation of Opa1 in mice led to systemic metabolic changes, senescence in multiple tissues and caused premature organismal aging and death. However, in this study it was not tested if the fusogenic activity of Opa1 can restore the function of Opa1 ablation. In another study, S-nitrosylation mediated activation of the mitochondrial fission protein Drp1 and impairment of mitophagy were proposed as the underlying causes of aging mouse tissues (Rizza et al., 2018). These changes are brought about by age related reduction of GSNOR denitrosylase activity, which was observed in aging humans but not in centenarians. Therefore, it was proposed that preventing such mitochondrial changes may prolong lifespan. A study of normal human fibroblasts (NHFs) obtained from healthy subjects demonstrated age-dependent increase in oxidative respiration that was mediated by Mfn1 and Opa1 (Son et al., 2017). However, it is not clear whether these changes lead to aging or protect from age related changes. Taken together, it can be hypothesized that mitochondrial fusion promotes health and longevity, and that mitophagic clearance of damaged mitochondria incapable of fusion is also beneficial. Interestingly, using a Drosophila model, it was demonstrated that activating Drp1 (or reducing mitofusin) to promote mitochondrial fission particularly in midlife increased healthspan while improving mitophagic clearance and proteostasis (Rana et al., 2017).
Elucidation of complexities of organismal aging is a challenging endeavor. Experiments with model organisms like yeast, C elegans, Drosophila and mouse together demonstrate that timely regulation of mitochondrial fission-fusion dynamics contributes to the complexities of replicative aging (senescence) of proliferating cells and chronological aging in in non-proliferating differentiated tissue. Some of these aspects have also been verified in humans. Given mitochondria perform distinct functions in proliferating and differentiated cells (Vander Heiden et al., 2009), it is not clear if and how the impact of modulation of mitochondrial fission/fusion differs in proliferating cells undergoing cell cycle and in cells that are in mitotic quiescence including adult stem cells. Work to establish mechanistic links between mitochondrial fission-fusion and the hallmarks of aging will require determining where their optimal fission-fusion balance lies for different tissues and how it shifts as cells proliferate and commit to cell fates over a lifespan.
7. Conclusion
Progression through, exit from, and re-entry to the cell cycle are actively guided by the shifts in the dynamic balance of mitochondrial fission and fusion. Such mitochondrial structural changes impact mitochondrial function of cells as they self-renew, proliferate, differentiate, and age. Alteration of mitochondrial fission and fusion balance also contributes to various age related disorders including cancer, diabetes, neurodegenerative, cardiovascular and musculoskeletal diseases (discussed elsewhere (Sebastian et al., 2017)). The mechanistic molecular details of such an integrated control of mitochondrial fission-fusion and cell cycle have only begun to emerge. The understanding of the impact of the shifting balance of mitochondrial fission-fusion dynamics would be further aided by gene manipulation techniques that are more sophisticated than current gene knockout/knockdown approaches. Moreover, screening of drugs for specifically impacting the activities of mitochondrial fission or fusion proteins would provide additional tools for both basic understanding of the processes they impact and for prolonging healthspan. It is important to highlight that mitochondria are extremely heterogeneous: they are wired differently to achieve distinct function in distinct tissues, while mitochondria perform distinct functions in proliferating and differentiated cells. Therefore, the role of mitochondrial fission and fusion may be distinct in the different cellular context of the differentiated tissue and their proliferating cells including their stem cells.
Acknowledgements
This work was supported by the National Institutes of Health (NIH) [R33ES025662] to B.S., K. M, and R01 AG043076 and R56 AG059590 to JLH, and a BBSRC New Investigator Grant BB/R003629/1 to JMAT.
References
- Abrams AJ, Hufnagel RB, Rebelo A, Zanna C, Patel N, Gonzalez MA, Campeanu IJ, Griffin LB, Groenewald S, Strickland AV, Tao F, Speziani F, Abreu L, Schule R, Caporali L, La Morgia C, Maresca A, Liguori R, Lodi R, Ahmed ZM, Sund KL, Wang X, Krueger LA, Peng Y, Prada CE, Prows CA, Schorry EK, Antonellis A, Zimmerman HH, Abdul-Rahman OA, Yang Y, Downes SM, Prince J, Fontanesi F, Barrientos A, Nemeth AH, Carelli V, Huang T, Zuchner S, Dallman JE, 2015. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat Genet. 47, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T, 2008. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 105, 14447–14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahat A, Goldman A, Zaltsman Y, Khan DH, Halperin C, Amzallag E, Krupalnik V, Mullokandov M, Silberman A, Erez A, Schimmer AD, Hanna JH, Gross A, 2018. MTCH2-mediated mitochondrial fusion drives exit from naive pluripotency in embryonic stem cells. Nat Commun. 9, 5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T, 2005. Mitochondria, oxidants, and aging. Cell. 120, 483–495. [DOI] [PubMed] [Google Scholar]
- Ban T, Ishihara T, Kohno H, Saita S, Ichimura A, Maenaka K, Oka T, Mihara K, Ishihara N, 2017. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat Cell Biol. 19, 856–863. [DOI] [PubMed] [Google Scholar]
- Bertolin G, Bulteau AL, Alves-Guerra MC, Burel A, Lavault MT, Gavard O, Le Bras S, Gagne JP, Poirier GG, Le Borgne R, Prigent C, Tramier M, 2018. Aurora kinase A localises to mitochondria to control organelle dynamics and energy production. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockler S, Chelius X, Hock N, Klecker T, Wolter M, Weiss M, Braun RJ, Westermann B, 2017. Fusion, fission, and transport control asymmetric inheritance of mitochondria and protein aggregates. J Cell Biol. 216, 2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K, Xie S, Qiu X, Mohrin M, Shin J, Liu Y, Zhang D, Scadden DT, Chen D, 2013. SIRT3 reverses aging-associated degeneration. Cell Rep. 3, 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti R, Ji WK, Stan RV, de Juan Sanz J, Ryan TA, Higgs HN, 2018. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J Cell Biol. 217, 251–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler H, Peters G, 2013. Stressing the cell cycle in senescence and aging. Curr Opin Cell Biol. 25, 765–771. [DOI] [PubMed] [Google Scholar]
- Chaudhari SN, Kipreos ET, 2017. Increased mitochondrial fusion allows the survival of older animals in diverse C. elegans longevity pathways. Nat Commun. 8 (182). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan DC, 2017. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. 26, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dorn GW 2nd., 2013. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 340, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC, 2003. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 160, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KH, Guo X, Ma D, Guo Y, Li Q, Yang D, Li P, Qiu X, Wen S, Xiao RP, Tang J, 2004. Dysregulation of HSG triggers vascular proliferative disorders. Nat Cell Biol. 6, 872–883. [DOI] [PubMed] [Google Scholar]
- Chen H, Chomyn A, Chan DC, 2005. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 280, 26185–26192. [DOI] [PubMed] [Google Scholar]
- Cheng X, Zhou D, Wei J, Lin J, 2013. Cell-cycle arrest at G2/M and proliferation inhibition by adenovirus-expressed mitofusin-2 gene in human colorectal cancer cell lines. Neoplasma. 60, 620–626. [DOI] [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, Perales-Clemente E, Salviati L, Fernandez-Silva P, Enriquez JA, Scorrano L, 2013. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 155, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darshi M, Mendiola VL, Mackey MR, Murphy AN, Koller A, Perkins GA, Ellisman MH, Taylor SS, 2011. ChChd3, an inner mitochondrial membrane protein, is essential for maintaining crista integrity and mitochondrial function. J Biol Chem. 286, 2918–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daste F, Sauvanet C, Bavdek A, Baye J, Pierre F, Le Borgne R, David C, Rojo M, Fuchs P, Tareste D, 2018. The heptad repeat domain 1 of Mitofusin has membrane destabilization function in mitochondrial fusion. EMBO Rep. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Dotto V, Fogazza M, Carelli V, Rugolo M, Zanna C, 2018. Eight human OPA1 isoforms, long and short: What are they for? Biochim Biophys Acta Bioenerg. 1859, 263–269. [DOI] [PubMed] [Google Scholar]
- Deng H, Takashima S, Paul M, Guo M, Hartenstein V, 2018. Mitochondrial dynamics regulates Drosophila intestinal stem cell differentiation. Cell Death Discov. 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner V, Picard M, Hajnoczky G, 2018. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol. 20, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figge MT, Reichert AS, Meyer-Hermann M, Osiewacz HD, 2012. Deceleration of fusion-fission cycles improves mitochondrial quality control during aging. PLoS Comput Biol. 8, e1002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L, 2006. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 126, 177–189. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK, 2011. ER tubules mark sites of mitochondrial division. Science. 334, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Dong Q, He J, Wang X, Xing J, Wang E, Qiu X, Li Q, 2017. SIRT4 inhibits malignancy progression of NSCLCs, through mitochondrial dynamics mediated by the ERK-Drp1 pathway. Oncogene. 36, 2724–2736. [DOI] [PubMed] [Google Scholar]
- Fu W, Liu Y, Yin H, 2019. Mitochondrial dynamics: biogenesis, fission, fusion, and mitophagy in the regulation of stem cell behaviors. Stem Cells Int. 9757201, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway CA, Lee H, Yoon Y, 2012. Mitochondrial morphology-emerging role in bioenergetics. Free Radic Biol Med. 53, 2218–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L, 2011. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 13, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales KA, Liang H, Lim YS, Chan YS, Yeo JC, Tan CP, Gao B, Le B, Tan ZY, Low KY, Liou YC, Bard F, Ng HH, 2015. Deterministic restriction on pluripotent state dissolution by cell-cycle pathways. Cell. 162, 564–579. [DOI] [PubMed] [Google Scholar]
- Halling JF, Ringholm S, Olesen J, Prats C, Pilegaard H, 2017. Exercise training protects against aging-induced mitochondrial fragmentation in mouse skeletal muscle in a PGC-1alpha dependent manner. Exp Gerontol. 96, 1–6. [DOI] [PubMed] [Google Scholar]
- Harbauer AB, Opalinska M, Gerbeth C, Herman JS, Rao S, Schonfisch B, Guiard B, Schmidt O, Pfanner N, Meisinger C, 2014. Mitochondria. Cell cycle-dependent regulation of mitochondrial preprotein translocase. Science. 346, 1109–1113. [DOI] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV Jr., Alt FW, Kahn CR, Verdin E, 2010. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 464, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S, Horner J, Song C, McCaffery JM, Nunnari J, 2009. Mitochondrial outer and inner membrane fusion requires a modified carrier protein. J Cell Biol. 184, 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn SR, Thomenius MJ, Johnson ES, Freel CD, Wu JQ, Coloff JL, Yang CS, Tang W, An J, Ilkayeva OR, Rathmell JC, Newgard CB, Kornbluth S, 2011. Regulation of mitochondrial morphology by APC/CCdh1-mediated control of Drp1 stability. Mol Biol Cell. 22, 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, Auwerx J, 2012. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 13, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Zhou X, Hu X, Joshi AS, Guo X, Zhu Y, Chen Q, Prinz WA, Hu J, 2017. Sequences flanking the transmembrane segments facilitate mitochondrial localization and membrane fusion by mitofusin. Proc Natl Acad Sci U S A. 114, E9863–E9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas JR, Ruiz-Ojeda FJ, Plaza-Diaz J, Nordsborg NB, Martin-Albo J, Rueda-Robles A, Casuso RA, 2019. Human muscular mitochondrial fusion in athletes during exercise. FASEB J 33 (11), 12087–12098. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Gottschling DE, 2012. An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature. 492, 261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, Mihara K, 2009. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 11, 958–966. [DOI] [PubMed] [Google Scholar]
- Jajoo R, Jung Y, Huh D, Viana MP, Rafelski SM, Springer M, Paulsson J, 2016. Accurate concentration control of mitochondria and nucleoids. Science. 351, 169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HC, Hsu JM, Yen CP, Chao CC, Chen RH, Pan CL, 2015. Neural activity and CaMKII protect mitochondria from fragmentation in aging Caenorhabditis elegans neurons. Proc Natl Acad Sci U S A. 112, 8768–8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YE, Clark AB, Kunkel TA, van Harn T, Xia B, Correll M, Quackenbush J, Livingston DM, Gygi SP, Sicinski P, 2011. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature. 474, 230–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A, Skotheim JM, 2013. Start and the restriction point. Curr Opin Cell Biol. 25, 717–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julian LM, Carpenedo RL, Rothberg JL, Stanford WL, 2016. Formula G1: Cell cycle in the driver’s seat of stem cell fate determination. Bioessays. 38, 325–332. [DOI] [PubMed] [Google Scholar]
- Kageyama Y, Zhang Z, Sesaki H, 2011. Mitochondrial division: molecular machinery and physiological functions. Curr Opin Cell Biol. 23, 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia R, Wang RY, Yusuf A, Thomas PV, Agard DA, Shaw JM, Frost A, 2018. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature. 558, 401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Arnoult D, Chen H, Chan DC, Smith CL, Youle RJ, 2004. Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J Cell Biol. 164, 493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara A, Cipolat S, Chen Y, Dorn GW 2nd, Scorrano L, 2013. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science. 342, 734–737. [DOI] [PubMed] [Google Scholar]
- Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD, Counter CM, 2011. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol. 13, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, Kashatus DF, 2015. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell. 57, 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katajisto P, Dohla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, Zoncu R, Chen W, Weinberg RA, Sabatini DM, 2015. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science. 348, 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ, 2010. The genetics of ageing. Nature. 464, 504–512. [DOI] [PubMed] [Google Scholar]
- Khacho M, Slack RS, 2017. Mitochondrial activity in the regulation of stem cell self-renewal and differentiation. Curr Opin Cell Biol. 49, 1–8. [DOI] [PubMed] [Google Scholar]
- Khacho M, Clark A, Svoboda DS, Azzi J, MacLaurin JG, Meghaizel C, Sesaki H, Lagace DC, Germain M, Harper ME, Park DS, Slack RS, 2016. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell. 19, 232–247. [DOI] [PubMed] [Google Scholar]
- Kim YM, Youn SW, Sudhahar V, Das A, Chandhri R, Cuervo Grajal H, Kweon J, Leanhart S, He L, Toth PT, Kitajewski J, Rehman J, Yoon Y, Cho J, Fukai T, Ushio-Fukai M, 2018. Redox regulation of mitochondrial fission protein Drp1 by protein disulfide isomerase limits endothelial senescence. Cell Rep. 23, 3565–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Cho YS, Wang X, Park O, Ma X, Kim H, Gan W, Jho EH, Cha B, Jeung YJ, Zhang L, Gao B, Wei W, Jiang J, Chung KS, Yang Y, 2019. Hippo signaling is intrinsically regulated during cell cycle progression by APC/C(Cdh1). Proc Natl Acad Sci U S A. 116, 9423–9432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koval OM, Nguyen EK, Santhana V, Fidler TP, Sebag SC, Rasmussen TP, Mittauer DJ, Strack S, Goswami PC, Abel ED, Grumbach IM, 2019. Loss of MCU prevents mitochondrial fusion in G1-S phase and blocks cell cycle progression and proliferation. Sci Signal. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus F, Ryan MT, 2017. The constriction and scission machineries involved in mitochondrial fission. J Cell Sci. 130, 2953–2960. [DOI] [PubMed] [Google Scholar]
- Labbe K, Murley A, Nunnari J, 2014. Determinants and functions of mitochondrial behavior. Annu Rev Cell Dev Biol. 30, 357–391. [DOI] [PubMed] [Google Scholar]
- Laporte D, Gouleme L, Jimenez L, Khemiri I, Sagot I, 2018. Mitochondria reorganization upon proliferation arrest predicts individual yeast cell fate. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IH, Finkel T, 2013. Metabolic regulation of the cell cycle. Curr Opin Cell Biol. 25, 724–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sterky FH, Mourier A, Terzioglu M, Cullheim S, Olson L, Larsson NG, 2012. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum Mol Genet. 21, 4827–4835. [DOI] [PubMed] [Google Scholar]
- Lee JE, Westrate LM, Wu H, Page C, Voeltz GK, 2016. Multiple dynamin family members collaborate to drive mitochondrial division. Nature. 540, 139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Smith SB, Yoon Y, 2017. The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J Biol Chem. 292, 7115–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Hong Y, He H, Jiang G, You W, Liang X, Fu Q, Han S, Lian Q, Zhang Y, 2019. FGF21 mediates mesenchymal stem cell senescence via regulation of mitochondrial dynamics. Oxid Med Cell Longev. 4915149, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M, Shirihai OS, 2013. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 17, 491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JR, Shen WL, Yan C, Gao PJ, 2015. Downregulation of dynamin-related protein 1 contributes to impaired autophagic flux and angiogenic function in senescent endothelial cells. Arterioscler Thromb Vasc Biol. 35, 1413–1422. [DOI] [PubMed] [Google Scholar]
- Liu L, Michowski W, Kolodziejczyk A, Sicinski P, 2019. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat Cell Biol. 21, 1060–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Mejia IC, Fajas L, 2015. Cell cycle regulation of mitochondrial function. Curr Opin Cell Biol. 33, 19–25. [DOI] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G, 2013. The hallmarks of aging. Cell. 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz C, Lesimple P, Bukowiecki R, Zink A, Inak G, Mlody B, Singh M, Semtner M, Mah N, Aure K, Leong M, Zabiegalov O, Lyras EM, Pfiffer V, Fauler B, Eichhorst J, Wiesner B, Huebner N, Priller J, Mielke T, Meierhofer D, Izsvak Z, Meier JC, Bouillaud F, Adjaye J, Schuelke M, Wanker EE, Lombes A, Prigione A, 2017. Human iPSC-derived neural progenitors are an effective drug discovery model for neurological mtDNA disorders. Cell Stem Cell. 20, 659–674 e659. [DOI] [PubMed] [Google Scholar]
- Magee JA, Piskounova E, Morrison SJ, 2012. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 21, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai S, Klinkenberg M, Auburger G, Bereiter-Hahn J, Jendrach M, 2010. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J Cell Sci. 123, 917–926. [DOI] [PubMed] [Google Scholar]
- Mandal S, Guptan P, Owusu-Ansah E, Banerjee U, 2005. Mitochondrial regulation of cell cycle progression during development as revealed by the tenured mutation in Drosophila. Dev Cell. 9, 843–854. [DOI] [PubMed] [Google Scholar]
- Mandal S, Freije WA, Guptan P, Banerjee U, 2010. Metabolic control of G1-S transition: cyclin E degradation by p53-induced activation of the ubiquitin-protea-some system. J Cell Biol. 188, 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Tarallo D, Martinez-Palma L, Victoria S, Bresque M, Rodriguez-Bottero S, Marmisolle I, Escande C, Cassina P, Casanova G, Bollati-Fogolin M, Agorio C, Moreno M, Quijano C, 2019. Mitofusins modulate the increase in mitochondrial length, bioenergetics and secretory phenotype in therapy-induced senescent melanoma cells. Biochem J. 476, 2463–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFaline-Figueroa JR, Vevea J, Swayne TC, Zhou C, Liu C, Leung G, Boldogh IR, Pon LA, 2011. Mitochondrial quality control during inheritance is associated with lifespan and mother-daughter age asymmetry in budding yeast. Aging Cell. 10, 885–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D, Gil J, 2018. Senescence and aging: causes, consequences, and therapeutic avenues. J Cell Biol. 217, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta K, Chacko LA, Chug MK, Jhunjhunwala S, Ananthanarayanan V, 2019. Association of mitochondria with microtubules inhibits mitochondrial fission by precluding assembly of the fission protein Dnm1. J Biol Chem. 294, 3385–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Lower B, Wunderlich FT, von Kleist-Retzow JC, Waisman A, Westermann B, Langer T, 2008. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 22, 476–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Chan DC, 2016. Metabolic regulation of mitochondrial dynamics. J Cell Biol. 212, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Varuzhanyan G, Pham AH, Chan DC, 2015. Mitochondrial dynamics is a distinguishing feature of skeletal muscle fiber types and regulates organellar compartmentalization. Cell Metab. 22, 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra K, 2013. Mitochondrial fission-fusion as an emerging key regulator of cell proliferation and differentiation. Bioessays. 35, 955–964. [DOI] [PubMed] [Google Scholar]
- Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J, 2009. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A. 106, 11960–11965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra K, Rikhy R, Lilly M, Lippincott-Schwartz J, 2012. DRP1-dependent mitochondrial fission initiates follicle cell differentiation during Drosophila oogenesis. J Cell Biol. 197, 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AS, Wong YC, Simpson CL, Holzbaur EL, 2016. Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat Commun. 7, 12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morigi M, Perico L, Rota C, Longaretti L, Conti S, Rottoli D, Novelli R, Remuzzi G, Benigni A, 2015. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest. 125, 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsci NS, Hall DH, Driscoll M, Sheng ZH, 2016. Age-related phasic patterns of mitochondrial maintenance in adult Caenorhabditis elegans neurons. J Neurosci. 36, 1373–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourier A, Motori E, Brandt T, Lagouge M, Atanassov I, Galinier A, Rappl G, Brodesser S, Hultenby K, Dieterich C, Larsson NG, 2015. Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J Cell Biol. 208, 429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, 2009. How mitochondria produce reactive oxygen species. Biochem J. 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj R, Gururaja-Rao S, Jones KT, Slattery M, Negre N, Braas D, Christofk H, White KP, Mann R, Banerjee U, 2012. Control of mitochondrial structure and function by the Yorkie/YAP oncogenic pathway. Genes Dev. 26, 2027–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neutzner A, Youle RJ, 2005. Instability of the mitofusin Fzo1 regulates mitochondrial morphology during the mating response of the yeast Saccharomyces cerevisiae. J Biol Chem. 280, 18598–18603. [DOI] [PubMed] [Google Scholar]
- Nishimura A, Shimauchi T, Tanaka T, Shimoda K, Toyama T, Kitajima N, Ishikawa T, Shindo N, Numaga-Tomita T, Yasuda S, Sato Y, Kuwahara K, Kumagai Y, Akaike T, Ide T, Ojida A, Mori Y, Nishida M, 2018. Hypoxia-induced interaction of filamin with Drp1 causes mitochondrial hyperfission-associated myocardial senescence. Sci Signal. 11. [DOI] [PubMed] [Google Scholar]
- Oh J, Lee YD, Wagers AJ, 2014. Stem cell aging: mechanisms, regulators and therapeutic opportunities. Nat Med. 20, 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohhara Y, Kobayashi S, Yamanaka N, 2017. Nutrient-dependent endocycling in steroidogenic tissue dictates timing of metamorphosis in Drosophila melanogaster. PLoS Genet. 13, e1006583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orford KW, Scadden DT, 2008. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 9, 115–128. [DOI] [PubMed] [Google Scholar]
- Ouyang H, Zhou E, Wang H, 2019. Mst1-Hippo pathway triggers breast cancer apoptosis via inducing mitochondrial fragmentation in a manner dependent on JNKDrp1 axis. Onco Targets Ther. 12, 1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Park YY, Lee S, Karbowski M, Neutzner A, Youle RJ, Cho H, 2010. Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J Cell Sci. 123, 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker DJ, Iyer A, Shah S, Moran A, Hjelmeland AB, Basu MK, Liu R, Mitra K, 2015. A new mitochondrial pool of cyclin E, regulated by Drp1, is linked to cell-density-dependent cell proliferation. J Cell Sci. 128, 4171–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauklin S, Vallier L, 2013. The cell-cycle state of stem cells determines cell fate propensity. Cell. 155, 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernas L, Scorrano L, 2016. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol. 78, 505–531. [DOI] [PubMed] [Google Scholar]
- Pernice WM, Vevea JD, Pon LA, 2016. A role for Mfb1p in region-specific anchorage of high-functioning mitochondria and lifespan in Saccharomyces cerevisiae. Nat Commun. 7, 10595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernice WM, Swayne TC, Boldogh IR, Pon LA, 2017. mitochondrial tethers and their impact on lifespan in budding yeast. Front Cell Dev Biol. 5, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Gentil BJ, McManus MJ, White K, St Louis K, Gartside SE, Wallace DC, Turnbull DM, 2013. Acute exercise remodels mitochondrial membrane interactions in mouse skeletal muscle. J Appl Physiol (1985) 115, 1562–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickles S, Vigie P, Youle RJ, 2018. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 28, R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto J, Leon M, Ponsoda X, Sendra R, Bort R, Ferrer-Lorente R, Raya A, Lopez-Garcia C, Torres J, 2016. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat Commun. 7, 11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Qvit N, Su YC, Mochly-Rosen D, 2013. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci. 126, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Choi S, Gibson GA, Watkins SC, Bakkenist CJ, Van Houten B, 2012. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J Cell Sci. 125, 5745–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J, 2011. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A. 108, 10190–10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampelt H, Zerbes RM, van der Laan M, Pfanner N, 2017. Role of the mitochondrial contact site and cristae organizing system in membrane architecture and dynamics. Biochim Biophys Acta Mol Cell Res. 1864, 737–746. [DOI] [PubMed] [Google Scholar]
- Rana A, Oliveira MP, Khamoui AV, Aparicio R, Rera M, Rossiter HB, Walker DW, 2017. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat Commun. 8, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reczek CR, Chandel NS, 2015. ROS-dependent signal transduction. Curr Opin Cell Biol. 33, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehklau K, Hoffmann L, Gurniak CB, Ott M, Witke W, Scorrano L, Culmsee C, Rust MB, 2017. Cofilin1-dependent actin dynamics control DRP1-mediated mitochondrial fission. Cell Death Dis. 8, e3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C, Archer SL, 2012. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 26, 2175–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riera CE, Merkwirth C, De Magalhaes Filho CD, Dillin A, 2016. signaling networks determining life span. Annu Rev Biochem. 85, 35–64. [DOI] [PubMed] [Google Scholar]
- Ristow M, 2014. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat Med. 20, 709–711. [DOI] [PubMed] [Google Scholar]
- Rizza S, Cardaci S, Montagna C, Di Giacomo G, De Zio D, Bordi M, Maiani E, Campello S, Borreca A, Puca AA, Stamler JS, Cecconi F, Filomeni G, 2018. S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc Natl Acad Sci U S A. 115, E3388–E3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheckhuber CQ, Erjavec N, Tinazli A, Hamann A, Nystrom T, Osiewacz HD, 2007. Reducing mitochondrial fission results in increased life span and fitness of two fungal ageing models. Nat Cell Biol. 9, 99–105. [DOI] [PubMed] [Google Scholar]
- Scheckhuber CQ, Wanger RA, Mignat CA, Osiewacz HD, 2011. Unopposed mitochondrial fission leads to severe lifespan shortening. Cell Cycle. 10, 3105–3110. [DOI] [PubMed] [Google Scholar]
- Schrepfer E, Scorrano L, 2016. Mitofusins, from mitochondria to metabolism. Mol Cell. 61, 683–694. [DOI] [PubMed] [Google Scholar]
- Sebastian D, Sorianello E, Segales J, Irazoki A, Ruiz-Bonilla V, Sala D, Planet E, Berenguer-Llergo A, Munoz JP, Sanchez-Feutrie M, Plana N, Hernandez-Alvarez MI, Serrano AL, Palacin M, Zorzano A, 2016. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 35, 1677–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian D, Palacin M, Zorzano A, 2017. Mitochondrial dynamics: coupling mitochondrial fitness with healthy aging. Trends Mol Med. 23, 201–215. [DOI] [PubMed] [Google Scholar]
- Senos Demarco R, Uyemura BS, D’Alterio C, Jones DL, 2019. Mitochondrial fusion regulates lipid homeostasis and stem cell maintenance in the Drosophila testis. Nat Cell Biol. 21, 710–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, Jabado O, Hoehn K, Kageyama Y, Sesaki H, Chipuk JE, 2015. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell. 57, 521–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Yamano K, Head BP, Kawajiri S, Cheung JT, Wang C, Cho JH, Hattori N, Youle RJ, van der Bliek AM, 2014. Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol Biol Cell. 25, 145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin J, Andres AM, Taylor DJ, Weston T, Hiraumi Y, Stotland A, Kim BJ, Huang C, Doran KS, Gottlieb RA, 2016. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy. 12, 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu KT, Rosner MR, Minella AC, 2012. An integrated view of cyclin E function and regulation. Cell Cycle. 11, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son JM, Sarsour EH, Kakkerla Balaraju A, Fussell J, Kalen AL, Wagner BA, Buettner GR, Goswami PC, 2017. Mitofusin 1 and optic atrophy 1 shift metabolism to mitochondrial respiration during aging. Aging Cell. 16, 1136–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Dorn GW 2nd, 2015. Mitoconfusion: noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab. 21, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Chen H, Fiket M, Alexander C, Chan DC, 2007. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 178, 749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurlock B, Gupta P, Basu MK, Mukherjee A, Hjelmeland AB, Darley-Usmar V, Parker D, Foxall ME, Mitra K, 2019. New quantitative approach reveals heterogeneity in mitochondrial structure-function relations in tumor initiating cells. J Cell Sci 132 (9), jcs230755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinkraus KA, Kaeberlein M, Kennedy BK, 2008. Replicative aging in yeast: the means to the end. Annu Rev Cell Dev Biol. 24, 29–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, Wilson TJ, Cribbs JT, 2013. Cyclin-dependent kinases regulate splice-specific targeting of dynamin-related protein 1 to microtubules. J Cell Biol. 201, 1037–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K, 2007. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 282, 11521–11529. [DOI] [PubMed] [Google Scholar]
- Tanwar DK, Parker DJ, Gupta P, Spurlock B, Alvarez RD, Basu MK, Mitra K, 2016. Crosstalk between the mitochondrial fission protein, Drp1, and the cell cycle is identified across various cancer types and can impact survival of epithelial ovarian cancer patients. Oncotarget. 7, 60021–60037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter Huurne M, Chappell J, Dalton S, Stunnenberg HG, 2017. Distinct cell-cycle control in two different states of mouse pluripotency. Cell Stem Cell. 21, 449–455 e444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, Ciciliot S, Soriano ME, Morbidoni V, Cerqua C, Loefler S, Kern H, Franceschi C, Salvioli S, Conte M, Blaauw B, Zampieri S, Salviati L, Scorrano L, Sandri M, 2017. Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab. 25, 1374–1389 e1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd LR, Damin MN, Gomathinayagam R, Horn SR, Means AR, Sankar U, 2010. Growth factor erv1-like modulates Drp1 to preserve mitochondrial dynamics and function in mouse embryonic stem cells. Mol Biol Cell. 21, 1225–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomer D, Chippalkatti R, Mitra K, Rikhy R, 2018. ERK regulates mitochondrial membrane potential in fission deficient Drosophila follicle cells during differentiation. Dev Biol. 434, 48–62. [DOI] [PubMed] [Google Scholar]
- Twig G, Shirihai OS, 2011. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal. 14, 1939–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS, 2008. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB, 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkei ZG, Yamashita YM, 2018. Emerging mechanisms of asymmetric stem cell division. J Cell Biol. 217, 3785–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Song Q, 2018. Mst1 regulates post-infarction cardiac injury through the JNKDrp1-mitochondrial fission pathway. Cell Mol Biol Lett. 23, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Li Z, Lu Y, Du R, Katiyar S, Yang J, Fu M, Leader JE, Quong A, Novikoff PM, Pestell RG, 2006. Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc Natl Acad Sci U S A. 103, 11567–11572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IH, Chen HY, Wang YH, Chang KW, Chen YC, Chang CR, 2014a. Resveratrol modulates mitochondria dynamics in replicative senescent yeast cells. PLoS One. 9, e104345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Fan M, Candas D, Zhang TQ, Qin L, Eldridge A, Wachsmann-Hogiu S, Ahmed KM, Chromy BA, Nantajit D, Duru N, He F, Chen M, Finkel T, Weinstein LS, Li JJ, 2014b. Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev Cell. 29, 217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir HJ, Yao P, Huynh FK, Escoubas CC, Goncalves RL, Burkewitz K, Laboy R, Hirschey MD, Mair WB, 2017. Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab. 26, 884–896 e885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westrate LM, Sayfie AD, Burgenske DM, MacKeigan JP, 2014. Persistent mitochondrial hyperfusion promotes G2/M accumulation and caspase-dependent cell death. PLoS One. 9, e91911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, Gerencser AA, Verdin E, Campisi J, 2016. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 23, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems PH, Rossignol R, Dieteren CE, Murphy MP, Koopman WJ, 2015. redox homeostasis and mitochondrial dynamics. Cell Metab. 22, 207–218. [DOI] [PubMed] [Google Scholar]
- Wu MJ, Chen YS, Kim MR, Chang CC, Gampala S, Zhang Y, Wang Y, Chang CY, Yang JY, Chang CJ, 2019. Epithelial-mesenchymal transition directs stem cell polarity via regulation of mitofusin. Cell Metab. 29, 993–1002 e1006. [DOI] [PubMed] [Google Scholar]
- Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, Dombrowski SM, Huang Z, Fang X, Shi Y, Ferguson AN, Kashatus DF, Bao S, Rich JN, 2015. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci. 18, 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Swartz KL, Siu KT, Bhattacharyya M, Minella AC, 2014. Fbw7-dependent cyclin E regulation ensures terminal maturation of bone marrow erythroid cells by restraining oxidative metabolism. Oncogene. 33, 3161–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CC, Chen D, Lee SS, Walter L, 2011. The dynamin-related protein DRP-1 and the insulin signaling pathway cooperate to modulate Caenorhabditis elegans longevity. Aging Cell. 10, 724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM, 2012. Mitochondrial fission, fusion, and stress. Science. 337, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Guan KL, 2015. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 163, 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Jin SB, Lendahl U, Nister M, Zhao J, 2019. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JJ, Liu WQ, Peng JJ, Ma QL, Peng J, Luo XJ, 2017. miR-21–5p/203a-3p promote ox-LDL-induced endothelial cell senescence through down-regulation of mitochondrial fission protein Drp1. Mech Ageing Dev. 164, 8–19. [DOI] [PubMed] [Google Scholar]