

Abstract
Regulation of glial activation and neuroinflammation are critical factors in the pathogenesis of Alzheimer Disease (AD). YKL-40, a primarily astrocytic protein encoded by the gene Chi3l1, is a widely-studied cerebrospinal fluid biomarker which increases with aging and early in AD. However, the function of Chi3l1/YKL-40 in AD is unknown. In a cohort of AD patients, we observed that a variant in the human CHI3L1 gene, which results in decreased CSF YKL-40 expression, was associated with slower AD progression. At baseline, Chi3l1 deletion in mice had no effect on astrocyte activation while modestly promoting microglial activation. In a mouse APP/PS1 model of AD, Chi3l1 deletion decreased amyloid plaque burden and increased peri-plaque expression of the microglial lysosomal marker CD68, suggesting that Chi3l1 may suppress glial phagocytic activation and promote amyloid accumulation. Accordingly, Chi3l1 knockdown increased phagocytosis of zymosan particles and of amyloid-beta peptide in both astrocytes and microglia in vitro. We further observed that expression of Chi3l1 is regulated by the circadian clock, as deletion of the core clock proteins BMAL1 or CLOCK/NPAS2 strongly suppresses basal Chi3l1 expression, whereas deletion of the negative clock regulators PER1/PER2 increased Chi3l1 expression. Basal Chi3l1 mRNA was non-rhythmic due to a long mRNA half-life in astrocytes. However, inflammatory induction of Chi3l1 was gated by the clock. Our findings reveal Chi3l1/YKL-40 as a modulator of glial phagocytic activation and AD pathogenesis in both mice and humans, and suggest that the astrocyte circadian clock regulates inflammatory Chi3l1 induction.
One Sentence Summary:
Chi3l1/YKL-40is regulated by BMAL1 in astrocytes and influences AD pathogenesis in both mouse models and humans.
Introduction
Neuroinflammation plays a critical role in the pathogenesis of most neurodegenerative diseases, including Alzheimer Disease (AD) (1, 2). In AD, triggering of the brain’s innate immune response, characterized in part by activation of astrocytes and microglia, can exert both degenerative and protective effects in a context-dependent manner (1, 3). Although astrocytes and microglia surround amyloid plaques and limit plaque growth through amyloid-beta phagocytosis, degradation, and clearance, they can also exacerbate pathology via dysfunctional inflammatory responses (1, 2). Thus, a deeper understanding of factors affecting glial function and neuroinflammation in neurodegenerative conditions is needed.
YKL-40, a secreted glycoprotein encoded by the Chi3l1 gene, is a well-described human cerebrospinal fluid biomarker of neuroinflammation, which is elevated in AD (4–6), as well as other neurologic diseases including multiple sclerosis, ALS, and frontotemporal dementia (7–9). CSF YKL-40 expression steadily increases with age starting in middle age, even in amyloid-negative individuals (5). Chi3l1/YKL-40 is expressed primarily in astrocytes in the brain, in macrophages in the periphery, and is induced in the setting of inflammation (4, 10, 11). Animal studies suggest that it may mitigate inflammation and protect cells from oxidative stress in the periphery (12–14). In AD, numerous studies show that CSF Chi3l1/YKL-40 increases in parallel with tau and other markers of inflammation and neurodegeneration, and that elevated Chi3l1/YKL-40 predicts disease progression (4, 5). Although Chi3l1/YKL-40 has been studied extensively as a biomarker, little is known about its function in the brain and in AD.
Many patients with neurodegenerative diseases also exhibit circadian system dysfunction, including those with preclinical AD (15). This dysfunction has been implicated as a potential contributor to AD pathogenesis (16, 17). The circadian system orchestrates 24-hour rhythms in many physiological and behavioral parameters, including sleep, activity, and endocrine function (18). The core circadian clock consists of a transcription-translation feedback loop, driven by the master circadian transcription factor BMAL1, which regulates circadian rhythms in transcription in a cell type-specific manner. The circadian clock regulates immune responses in a variety of peripheral innate and adaptive immune cell types (19). Astrocytes and microglia also possess functioning circadian clocks, and circadian timing can impact their inflammatory responses (20–22). Our group has shown that disruption of core circadian clock transcription via deletion of Bmal1 can promote neuroinflammation, and that BMAL1 regulates astrocyte activation in a cell-autonomous manner (23, 24). However, the mechanisms linking glial clocks to neuroinflammation are poorly understood.
Here, we report data from humans and mouse models suggesting that Chi3l1/YKL-40 accelerates AD pathogenesis, potentially by altering glial function in the brain. We also show evidence from mice that the core circadian clock in astrocytes strongly regulates Chi3l1 transcription and gates its inflammatory induction. Together, our results reveal Chi3l1/YKL-40 as a modulator of AD pathogenesis, and illuminate a link between glial circadian clocks and Chi3l1 expression.
Results
A human genetic variant causing decreased CHI3L1/YKL-40 is associated with slower disease progression in human AD patients
We sought to determine if CHI3L1/YKL-40 influences AD pathogenesis in humans. First, we examined a human single-nucleus RNAseq dataset derived from cortical brain tissue from 3 patients with AD(25) and observed that CHI3L1 expression was strongly associated with the astrocyte cell cluster (Fig 1A). Although a few CHI3L1-expressing microglia were present, differential gene expression analysis shows that CHI3L1 is enriched in astrocytes and minimally expressed in neurons (Fig 1B). We have previously reported that CSF YKL-40 expression in humans is strongly controlled by genetic variation in the CHI3L1 locus, and have described a common genetic variant in the CHI3L1 locus (rs10399931) associated with decreased CSF YKL-40 concentrations (26). Using the Knight Alzheimer’s Disease Research Center at Washington University clinical database, we next examined if this rs10399931 SNP was associated with any changes in AD progression. We examined data from 778 participants enrolled in longitudinal observational studies and included only participants with AD (confirmed by clinical assessment and CSF biomarker profile), 26% of whom carried the CC_TT polymorphism at rs10399931. We examined clinical progression of AD as assessed by the rate of increase in the Clinical Dementia Rating Sum-of-Boxes score (CDR-SB). We found that the rs10399931 SNP was significantly associated (p=0.031) with slower rate of AD progression, suggesting that people with genetically lower CHI3L1/YKL-40 expression have 16% slower disease progression (Fig. 1C. For comparison, pathogenic variants in the Trem2 gene were previously associated with a 23% increased rate of progression using the same methods (27). This initial human data suggested that CHI3L1/YKL-40 might be a modulator of human disease pathogenesis. Thus, we sought to examine the mechanisms by which CHI3L1/YKL-40 might exacerbate AD pathogenesis, including glial activation, in mouse models.
Figure 1. A polymorphism in human CHI3L1 impacts rate of AD progression.

A. Single nucleus RNAseq tSNE plot showing cell clusters (top panel) or CHI3L1 expression (purple, bottom panel). Ex = excitatory neuron, In = Inhibitory neuron, OPC = oligodendrocyte precursor cells, Endo = endothelial cells. Data available at http://ngi.pub/snuclRNA-seq/.
B. CHI3L1 expression by cell cluster from data in (A). ****P=3×10−280.
C. AD progression (change in CDR-SB) from human patients with and without the CC_TT polymorphism of the Rs10399931 SNP in CHI3L1.
Chi3l1 modulates the astrocyte and microglial inflammatory response in vivo
Our data (Fig. 1A) and other studies show that Chi3l1 is highly enriched in human astrocytes (28) and only negligibly expressed in other cell types, whereas in the mouse CNS at baseline it is highly expressed in astrocytes and OPCs, but less so in microglia (29). Thus, we sought to investigate the role of astrocytic Chi3l1 in regulating neuroinflammation. We used siRNA to knock down Chi3l1 (siChi3l1) in mouse primary astrocyte cultures, achieving a 91% decrease in Chi3l1 mRNA (Fig. 2A). Loss of Chi3l1 induced a small decrease in Gfap, and a small increase in Il6. This suggests that Chi3l1 suppression may exert differential effects on astrocyte activation and cytokine expression. In vivo, constitutive Chi3l1 KO (30) had no effects on expression of several inflammatory transcripts (Fig. 2B, S1C). As a group, transcripts related to astrocyte activation were modestly altered at baseline by Chi3l1 KO, though no individual transcripts were changed (Fig. 2C, S2A). Similarly, microglial activation genes (including Trem2, Spp1, Gpnmb1, and Apoe) were slightly increased at baseline as a group in Chi3l1 KO mice, though no individual genes were different (Fig. 2D, S2B). Chi3l1 did not alter baseline GFAP immunoreactivity, a marker of astrocyte activation, in the hippocampus (Fig. 2E, F). However, Chi3l1 deletion subtly increased staining for the microglial marker IBA1 in the cortex (Fig. 2G, H), suggesting that Chi3l1 differentially modulates astrocyte and microglial activation at baseline.
Figure 2. Loss of Chi3l1 mildly shifts glial activation.
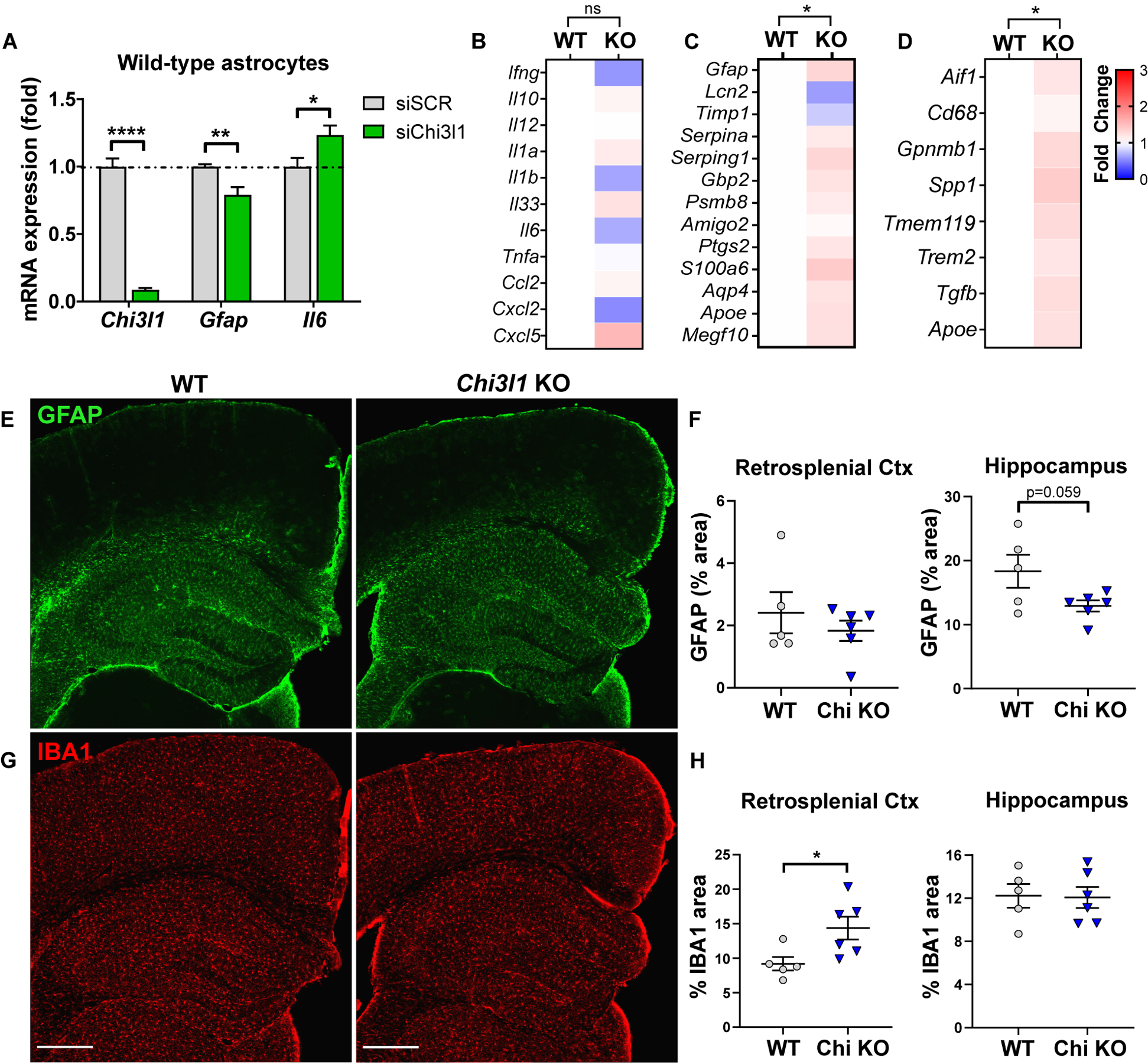
A. qPCR gene expression from primary astrocytes transfected with control (siScr) or Chi3l1 (siChi3l1) siRNA. n = 6–10 replicates from 3 independent experiments.
B–D. Cytokine and chemokine (B), astrocyte activation marker (C) or microglia activation marker (D) expression from fluidigm qPCR of 2–5mo Chi3l1−/− and WT control mouse cortex 6 hours after i.p. PBS. Mean of 6 mice/group normalized to WT. Two-way ANOVA with Tukey correction for multiple comparisons.
E–H. Representative images depicting GFAP (astrocyte) staining (E) and associated quantification (F) or IBA1 (microglia) staining (G) and associated quantification (H) in Chi3l1−/− and WT control mice. Scale bar = 400μm
All data represent mean +/− SEM. *p < 0.05, **p < 0.01, ****p < 0.0001 by two-tailed students t-test with Holm-Sidak correction for multiple comparisons when appropriate.
We next examined the effect of Chi3l1 on acute neuroinflammation induced by the inflamogen lipopolysaccharide (LPS). In primary astrocyte cultures, Chi3l1 siRNA exacerbated LPS-induced cytokine expression (Fig. S1A). Chi3l1 KO mice exhibited a general exacerbation of the inflammatory response with increased hippocampal expression of several inflammatory transcripts, including several microglia-specific transcripts such as Cybb and Nlrp3 following i.p. LPS injection (Fig. S1B–E). However, Chi3l1 KO did not alter LPS-induced expression of astrocyte activation markers or AD-associated microglial activation markers (Fig. S2A, B). Together, our data suggest that Chi3l1/YKL-40 deletion did not affect astrocyte activation, but mildly enhances microglial activation at baseline, and modestly potentiates LPS-induced inflammatory cytokine expression in astrocytes and microglia.
Deletion of Chi3l1 reduces amyloid plaque deposition in a mouse model of AD
As YKL-40 is increased in the CSF of patients with AD and is used as a biomarker of AD (4, 6), we next sought to test the hypothesis that Chi3l1/YKL-40 could also influence pathology in a mouse model of AD-related β-amyloidosis. We crossed APP/PS1-21 mice, which express the human amyloid precursor protein (APP) gene with the KM670/671NL (Swedish) mutation and human presenilin 1 (PS1) with the L166P mutation (31), with WT or Chi3l1 KO mice. All mouse brain tissue was harvested at 8 months of age when mice had developed substantial plaque pathology in both the cortex and hippocampus (31). Staining with X-34, which selectively labels β–pleated sheet fibrillar Aβ plaques (32), revealed that loss of Chi3l1 reduced fibrillar plaque number by 21% (Fig. 3A, C) and plaque area by 17% (Fig. 3A, D) in the hippocampus, but did not alter these measures in the cortex (Fig. S3A–C). Staining with an Aβ antibody (HJ3.4) revealed a much more pronounced 55% reduction in plaque burden in the hippocampus (Fig. 3A, E) as well as a 42% decrease in the cortex (Fig. S3A, D). This discrepancy between staining methodologies led us to hypothesize that Chi3l1 deletion results in the selective reduction of non-fibrillar Aβ. Subtraction of X-34 signal from total plaque (HJ3.4) staining indeed revealed a halo of aggregated, non-fibrillar Aβ surrounding the fibrillar plaque core in a majority of plaques in control APP/PS1 mice, as well as some X34-negative, HJ3.4+ plaques (Fig. 3A, B, S3A). We observed an 85% loss of this non-fibrillar Aβ in the hippocampus of Chi3l1 KO mice (Fig. 3F), along with a 54% reduction in the cortex (Fig. S3E). In order to more closely investigate this selective loss of non-fibrillar Aβ, regions with similar amounts of fibrillar (X-34+) plaque load (Fig. S3F, G) were imaged using confocal microscopy and 3D reconstruction in a subset of mice. Plaque volume measurements again revealed a loss in antibody (HJ3.4) positive Aβ (Fig. S3H) and confirmed the selective loss of non-fibrillar Aβ in the absence of Chi3l1 (Fig. 3G, H). Moreover, loss of Chi3l1 altered the distribution of antibody (HJ3.4) stained plaques in APP/PS1 mice (Fig. S4A, B) with a substantial decrease in plaque number and a small decrease in average plaque area (Fig S4C, D). This pool of aggregated but non-fibrillar Aβ could not be separated biochemically via sequential fractionation with increasing concentrations of guanidine (Fig. S4E). Loss of Chi3l1 did not result in changes in the amount of APP protein, (Fig. S4F), or in the expression of several key amyloid processing/metabolic genes, including App, Bace1, Ide, Mmp2 or 9, Ldlr, Lrp1, Mme, Klk7, and Apoe (Fig. S4G). These data, in combination with the fact that Chi3l1 is generally not expressed in neurons, indicates that YKL-40 likely does not alter the production of Aβ. Taken together, these data suggest that the loss of Chi3l1 mitigates the accumulation of Aβ plaque pathology, particularly non-fibrillar plaque material.
Figure 3. Loss of Chi3l1 mitigates amyloid pathology.

A. Representative hippocampal images from 8mo Chi3l1−/−:APP/PS1+ and APP/PS1+ control mice depicting staining by X34 (fibrillar plaques), HJ3.4 antibody (total Aβ), and subtraction of fibrillar (X34) from total (HJ3.4) Aβ. Hippocampus outlined in yellow. Yellow rectangle denotes region of inset in (B). Scale bar = 300μm
B. Representative higher magnification images depicting stains from (A). “Halo” of non-fibrillar Aβ surrounding fibrillar plaque core (X34) substantially reduced in Chi3l1−/− mice. Scale bar = 50μm
C–F. Quantification of X34+ puncta (fibrillar plaque number) (C), X34+ area (fibrillar Aβ) (D), HJ3.4+ area (total Aβ) (E), or area covered by non-fibrillar Aβ (total Aβ in HJ3.4 - fibrillar X34) (F) in hippocampal staining from (A).
G. X34 and HJ3.4 colocalization (top) with 3D surface rendering of X34 (red) and HJ3.4 (green) staining (middle) with 20μm shells (pink) around each fibrillar plaque. Scale bar = 30μm
H. Quantification of non-fibrillar plaque volume as ratio of HJ3.4 to X34 (top) or total Aβ in HJ3.4 - fibrillar X34 (bottom) in 20μm shell surrounding each X34+ plaque in subset of mice from (C)-(F).
Data points represent average of 2–4 sections/mouse, 4–6 (Chi3l1+/+) and 4–12 (Chi3l1−/−:) mice per group. All data represent mean +/− SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by two-tailed students t-test.
Peri-plaque astrocyte activation is suppressed by Chi3l1 deletion
As astrocytes and microglia have been shown to play integral roles in both the clearance of and neuroinflammation induced by Aβ (1, 2), we next examined the consequences of Chi3l1 deletion on the glial response to Aβ plaques. While previous reports disagree about whether impairing astrocyte activation increases or decreases Aβ plaque burden (33, 34), we discovered that Chi3l1 deletion resulted in decreased astrocyte activation as measured by GFAP staining in the hippocampus and throughout the cortex in APP/PS1+ mice (Fig. 4A, B, Fig. S5A). Modest peri-plaque GFAP reductions were also observed (Fig. 4A, C), as controlling for the decreased fibrillar plaque area (Fig. 4C) or number (Fig. S5B) could not fully account for the lessened GFAP, particularly in the hippocampus. Thus, the amount of GFAP staining per plaque area is reduced with Chi3l1 deletion. In concordance with our staining data, transcriptional profiling of astrocyte activation markers revealed a slight attenuation of hippocampal astrocyte activation in Chi3l1 KO; APP/PS1 mice (Fig. 4D, S5C). Notably, Chi3l1 mRNA was increased in APP/PS1 cortex at 8 months and absent in Chi3l1 KO mice (Fig. S5C).
Figure 4. Loss of Chi3l1 mitigates astrogliosis, but facilitates phagocytosis in the presence of Aβ.

A. Representative high magnification images from hippocampi of 8 mo Chi3l1−/−:APP/PS1+ and APP/PS1+ control mice stained for X34 (fibrillar plaques) and GFAP (astrocytes). Scale bar = 20μm
B–C. Quantification of GFAP coverage (B) or GFAP coverage normalized to X34+ area in same section (C) from mice in (A). Quantified from widefield image in Fig. S5. RS = retrosplenial. n = 6 (Chi3l1+/+) and 12 (Chi3l1−/−:) mice per group.
D. Astrocyte activation marker gene expression from fluidigm qPCR of 8 mo Chi3l1−/−:APP/PS1−, WT:APP/PS1−, Chi3l1−/−:APP/PS1+, and APP/PS1+ control mouse hippocampus. Mean of 4 mice (APP/PS1−) or 6–10 (APP/PS1+) mice per group normalized to WT:APP/PS1−. Two-way ANOVA with Tukey correction for multiple comparisons.
E–F. pHrodo-labeled zymosan bead (E) or TAMRA-Aβ (F) uptake by primary astrocyte cultures transfected with control (siSCR) or Chi3l1 (siChi3l1) siRNA, +/− cytochalasin D to inhibit phagocytosis (+cytoD). Each point represents one field of view with an average of 804 (E) or 517 (F) cells/field. Data from 2 independent experiments.
All data represent mean +/− SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Analyzed by two-tailed students t-test (B–C) or one-way ANOVA (E-F). All data subjected to Sidak correction for multiple comparisons unless otherwise noted.
As astrocyte activation state can affect phagocytosis (3) and astrocytes are known to phagocytose Aβ plaque material (35), we next sought to evaluate whether Chi3l1 may be regulating phagocytosis in astrocytes. siRNA-mediated Chi3l1 knockdown, which suppresses Chi3l1 mRNA expression in primary astrocyte cultures by ~91% (see Fig. 2A), increased the phagocytosis of zymosan-coated pHrodo-labeled beads by 13% (Fig. 4E) and fluorescent (TAMRA-labeled) Aβ42 peptide by 50% (Fig. 4F), each in two separate experiments. In combination, these data support the idea that loss of Chi3l1 tempers astrocyte activation while potentially increasing astrocytic phagocytosis in response to Aβ plaques.
Chi3l1 deletion promotes plaque-related microglial CD68 expression
To address the possibility that Chi3l1 could also be regulating the microglial response to Aβ we utilized IBA1 to label microglia and the microglial lysosomal marker CD68 to assess microglial phagosome expression (Fig. 5A, S6A). When normalized to plaque area, we did not observe any changes in absolute coverage of IBA1 in the hippocampus, motor cortex, or retrosplenial cortex (Fig. S6A, B). However, the amount of CD68 staining normalized to X34+ plaque area was elevated across all regions examined in Chi3l1 KO;APP/PS1 mice (Fig. 5B) indicating increased microglial phagocytic activation relative to plaque burden. This effect appeared to be driven by increased CD68 staining around amyloid plaques, as colocalized IBA1/CD68 area per X34+ plaque area was also increased in Chi3l1 KO mice (Fig. 5A, C). These changes seem to be CD68-specific as there were no differences observed in transcript expression of other lysosomal markers (Fig. 5D, S6C). Transcriptional analysis of hippocampal tissue for a selection of known microglia activation markers revealed an increase in the activation marker Spp1 in 8mo Chi3l1 KO mice without plaques. We also observed a slight overall dampening of microglial activation marker expression (Fig. 5D, S6C) and inflammatory markers (Fig. S7A, B) with Chi3l1 deletion in the presence of Aβ pathology. These changes are very likely due to reduced plaque burden in Chi3l1 KO;APP/PS1 mice.
Figure 5. Loss of Chi3l1 alters microglial activation and enhances Aβ phagocytosis.
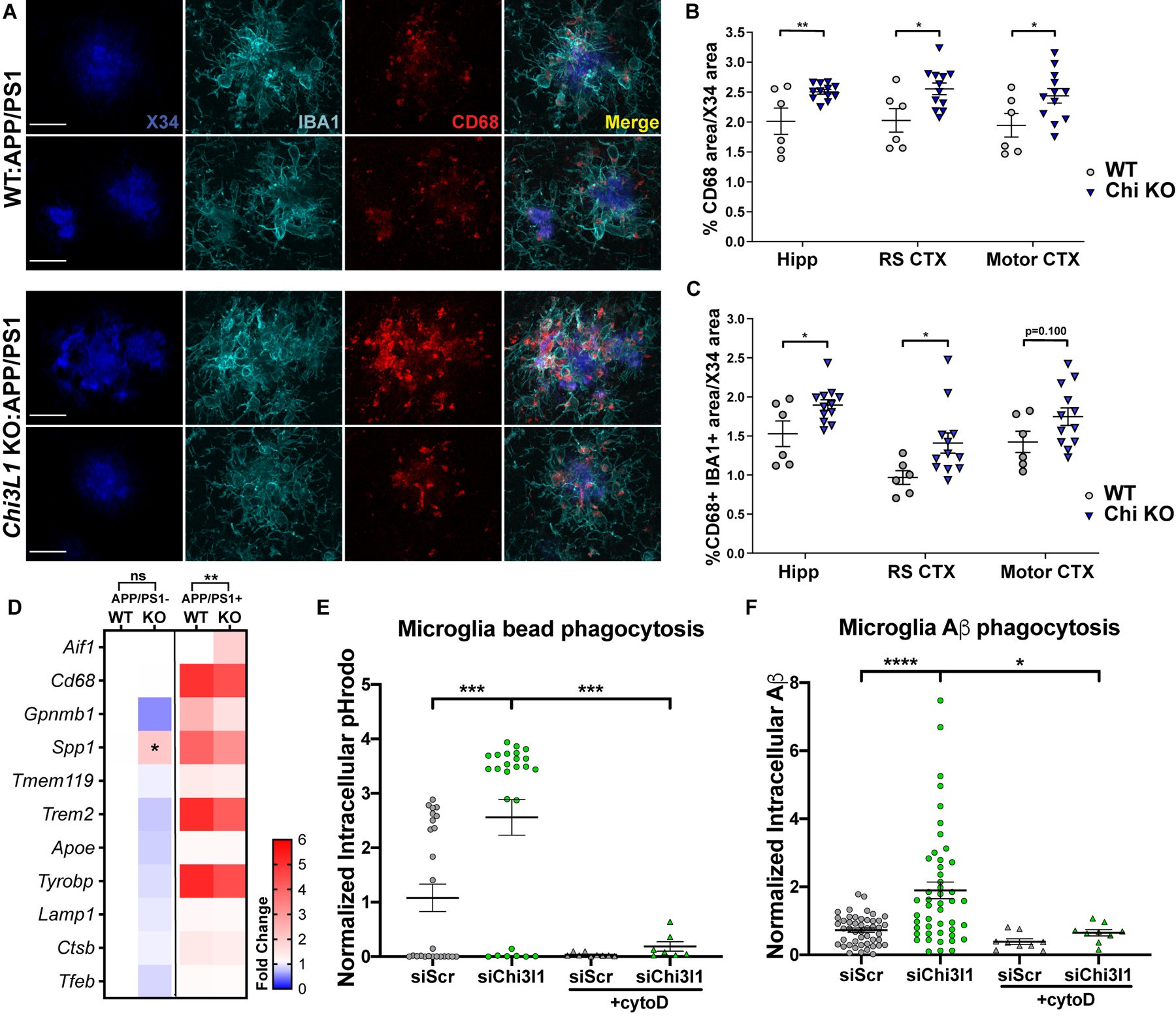
A. Representative high magnification images from hippocampi of 8 mo Chi3l1−/−:APP/PS1+ and APP/PS1+ control mice stained for X34 (fibrillar plaques), IBA1 (microglia), and CD68 (phagocytic microglia). Scale bar = 20μm
B–C. Quantification of CD68 (B) or colocalized IBA1/CD68 (C) from mice in (A) normalized to X34+ area in same section. Quantified from widefield image in Fig. S6. n = 6 (Chi3l1+/+) and 12 (Chi3l1−/−) mice per group. RS = retrosplenial
D. Microglia-associated gene expression from Fluidigm qPCR of 8 mo Chi3l1−/−:APP/PS1−, WT:APP/PS1−, Chi3l1−/−:APP/PS1+ and APP/PS1+ control mouse hippocampus. Mean of 4 mice (APP/PS1−) or 6–10 (APP/PS1+) mice per group normalized to WT:APP/PS1−. Two-way ANOVA with Tukey correction for multiple comparisons
E–F. pHrodo-labeled zymosan bead (E) or TAMRA-Aβ (F) uptake by primary microglia cultures transfected with control (siSCR) or Chi3l1 (siChi3l1) siRNA, +/− cytochalasin D to inhibit phagocytosis (+cytoD). Each point represents one field of view with an average of 209 (E) or 56 (F) cells/field. Data from 2 (E) or 1 (F) independent experiments.
All data represent mean +/− SEM. *p < 0.05, **p < 0.01, ***p < 0.001. Analyzed by two-tailed students t-test (B–C) or one-way ANOVA (E–F). All data subjected to Sidak correction for multiple comparisons unless otherwise noted.
Due to the increase in microglial CD68 expression in Chi3l1 KO;APP/PS1 mice and the observed increase in astrocytic phagocytosis with loss of Chi3l1, we next examined phagocytosis in cultured primary mouse microglia. Chi3l1 siRNA (siChi3l1) suppressed Chi3l1 mRNA in microglial cultures by 97% (Fig. S7C). Loss of Chi3l1 in microglia had an even greater effect than in astrocytes, with Chi3l1 knockdown increasing phagocytosis of pHrodo-labeled zymosan beads by 148% (Fig. 5E) and fluorescent (TAMRA-labeled) Aβ by 100% (Fig. 5F) in microglia in vitro. Taken together, our data demonstrate that loss of Chi3l1 leads to decreased amyloid plaque burden and increased microglial CD68 expression in vivo, and enhances phagocytosis of Aβ by both astrocytes and microglia in vitro.
Chi3l1 expression is non-rhythmic, but controlled by the circadian clock
We next explored possible molecular mechanisms regulating Chi3l1 expression and potential link to glial activation. We previously reported that deletion of the master circadian clock gene Bmal1 caused astrocyte activation in a cell-autonomous manner (24). While examining an existing transcriptional dataset from control and brain-specific Bmal1 KO (Nestin-Cre;Bmal1f/f) cortex to identify circadian clock targets which may regulate astrocyte activation, we noticed Chi3l1 to be among the most downregulated transcripts in Bmal1 KO mice. Indeed, while Chi3l1 is reported to be among the 50 most-upregulated transcripts in activated astrocytes following in vivo inflammation (LPS injection) (36), our transcriptomic data showed that Chi3l1 was markedly downregulated in Bmal1 KO brain (−89%) and strongly upregulated in Per1mut;Per2mut (+230%) mice across circadian timepoints (Fig. 6A, B). This unexpected finding of reciprocal expression changes in mice with mutation of the positive limb (Bmal1) and negative limb (Per1/2) of the circadian clock suggested that Chi3l1 might be a clock-controlled gene. However, basal Chi3l1 mRNA did not show circadian oscillation in control mice in this array data (Fig. 6B). Follow-up qPCR analysis of cortex tissue collected every 4 hours in constant darkness confirmed a lack of circadian oscillation in Chi3l1 mRNA in control mice, but again showed an average of a 91% loss in Chi3l1 transcript in Bmal1 KO brain (Fig. S8A). Moreover, we found that, similar to Bmal1 KO tissue, Chi3l1 mRNA was decreased in cortex from Clock/Npas2 double KO mice, as compared to Clock KO alone (Fig. 6C). Clock/Npas2 double KO mice lack a binding partner for Bmal1 and thus have a dysfunctional positive limb of the clock. This decrease in Chi3l1 expression mirrored that of the BMAL1-CLOCK/NPAS2 target, Nr1d1 (Fig. S8B), and occurred despite a compensatory increase in Bmal1 expression (Fig. S8C). These data strongly support the requirement of a functioning positive limb of the clock (consisting of BMAL1/CLOCK or BMAL1/NPAS2 heterodimers) for Chi3l1 expression.
Figure 6. Chi3l1 is regulated by the circadian clock.

A. Microarray in Nestin-Cre;Bmal1f/f and Per1/2mut vs control, Bmal1f/f cortex (Lananna et al, 2018) cross referenced with 50 genes most upregulated in astrocytes with in vivo LPS (Zamanian et al, 2012). CT = clock time.
B. Microarray data from (A) with 2 additional timepoints for Cre- and Per1/2mut mice.
C. qPCR depicting gene expression in global Npas2 KO, Clock KO, Clock/Npas2 double KO or Bmal1 KO mouse cortex. n = 2 mice/group, normalized to WT control.
D–F. qPCR showing Chi3l1 expression from Aldh1l1-Cre;Bmal1f/f (ALC) hippocampus (D) or Cx3cr1-Cre;Bmal1f/f (CX3) cortex (E), or gene expression from WT primary astrocytes 4–8 days after transfection with control (siSCR) or Bmal1 (siBmal1) siRNA (F). n = 3 mice/group (D), 6–8 mice/group (E), or 15 biological replicates from 5 independent experiments (F).
G. ELISA of Chi3l1 protein (YKL-40) in culture medium of WT primary astrocytes 6 days after transfection with control (siSCR) or Bmal1 (siBmal1) siRNA. n = 3 biological replicates/group.
H. ChIP-qPCR from mouse primary astrocytes with anti-BMAL1 antibody or IgG control. Three distinct E-box-containing regions in the Chi3l1 promoter were assayed. A known BMAL1 binding E-box-containing region in the Dbp promoter was assayed as positive control.
I. qPCR showing expression of Chi3l1 and Bmal1 mRNA in primary mouse astrocytes treated with actinomycin D at 0 hours, then harvested at intervals thereafter. n = 3 wells/timepoint, normalized to Actb mRNA. *p<0.05 compared to timepoint 0
All data represent mean +/− SEM. *p < 0.05, **p < 0.01, ****p < 0.0001 by two-tailed students t-test with Holm-Sidak correction for multiple comparisons when appropriate.
To test whether the circadian clock is regulating Chi3l1 in astrocytes, we measured Chi3l1 in inducible astrocyte-specific Bmal1 KO (Aldh1l1-CreERT2:Bmal1f/f) mice, which we have previously shown results in the loss of approximately 70% of astrocytic BMAL1 (24). Chi3l1 expression was reduced by 52% in the cortex of Cre+ animals (Fig. 6D), while we did not observe any change in Chi3l1 expression in microglia-specific Bmal1 KO, (Cx3cr1-CreERT2:Bmalf/f) mice (Fig. 6E). Treating primary mouse astrocyte cultures with siRNA targeting Bmal1 resulted in a 64% loss in the direct BMAL1 transcriptional target Nr1d1 and a 71% loss in Chi3l1 (Fig. 6F), whereas the secretion of CHI3L1 protein (YKL-40) decreased by 72% (Fig. 6G). Knockdown of Bmal1 in primary microglia cultures resulted in a 79% loss in Nr1d1, but no change in Chi3l1 (Fig. S8D). To determine if BMAL1 directly regulates Chi3l1, we examined the putative Chi3l1 promoter (within 700 bp of the Chi3l1 transcriptional start site), and identified 6 E-boxes, 3 of which previously displayed weak binding of BMAL1 in an existing liver ChIP-seq dataset (37) (Fig. S8E). ChIP-qPCR for BMAL1 binding to 3 of these E-boxes (Fig. S8E) revealed enrichment when compared to IgG controls at all 3 sites, as well as at a known BMAL1 binding site in the Dbp promoter (Fig. 6H). While Chi3l1 transcription is directly regulated by BMAL1, it is not rhythmic at baseline (Fig. 6B, Fig. S8A, F). One possibility for the lack of Chi3l1 oscillation in astrocytes is that its mRNA may have a long half-life. We measured mRNA degradation kinetics in primary astrocyte cultures following inhibition of transcription with actinomycin D, and observed that the half-life of Chi3l1 transcript was much longer than that of Bmal1, a rhythmic gene (Fig. 6I). Thus, while the positive limb of the circadian clock directly regulates Chi3l1 expression, basal Chi3l1 mRNA is not rhythmic likely due to its long half-life.
Induction of Chi3l1 in astrocytes is gated by the circadian clock
As Chi3l1/YKL-40 has been shown to increase during inflammatory conditions (10, 38) and is regulated by NF-kappaB (11), we next sought to evaluate the regulation of Chi3l1 induction by BMAL1 in the setting of inflammation. In cultured astrocytes, Chi3l1 was induced by LPS stimulation, but both basal and LPS-stimulated Chi3l1 expression was suppressed in Bmal1 KO cells (though some LPS-induced increase in Chi3l1 was still observed) (Fig. 7A). To more closely examine the possibility that the inflammatory induction of Chi3l1 is gated by the astrocyte circadian clock, we measured basal and LPS-induced Chi3l1 expression in synchronized primary astrocytes across circadian timepoints. Consistent with our in vivo data, Chi3l1 transcript did not oscillate in synchronized primary astrocytes (Fig. S8F). However, LPS-mediated induction of Chi3l1 was highly dependent on circadian phase, as LPS-induced Chi3l1 expression varied anti-phase to Bmal1 mRNA expression and closely mirrored the expression pattern of Nr1d1, which is dependent on BMAL1 transcriptional activity (Fig. 7B, C). This time-of-day variation in Chi3l1 induction was not observed in cells treated with Bmal1 siRNA, indicating a requirement for a functioning circadian clock (Fig. 7D). These data suggest that circadian oscillations in BMAL1 transcriptional activity gate the inflammatory induction of Chi3l1 in astrocytes (Fig. 7E). Importantly, the induction of Chi3l1 in astrocytes was not limited to LPS as exposure of primary astrocyte cultures to amyloid-beta-42 (Aβ42) fibrils resulted in an approximately 4.5-fold increase in Chi3l1 (Fig. 7F).
Figure 7. Chi3l1 is induced during inflammation in a Bmal1 dependent manner.
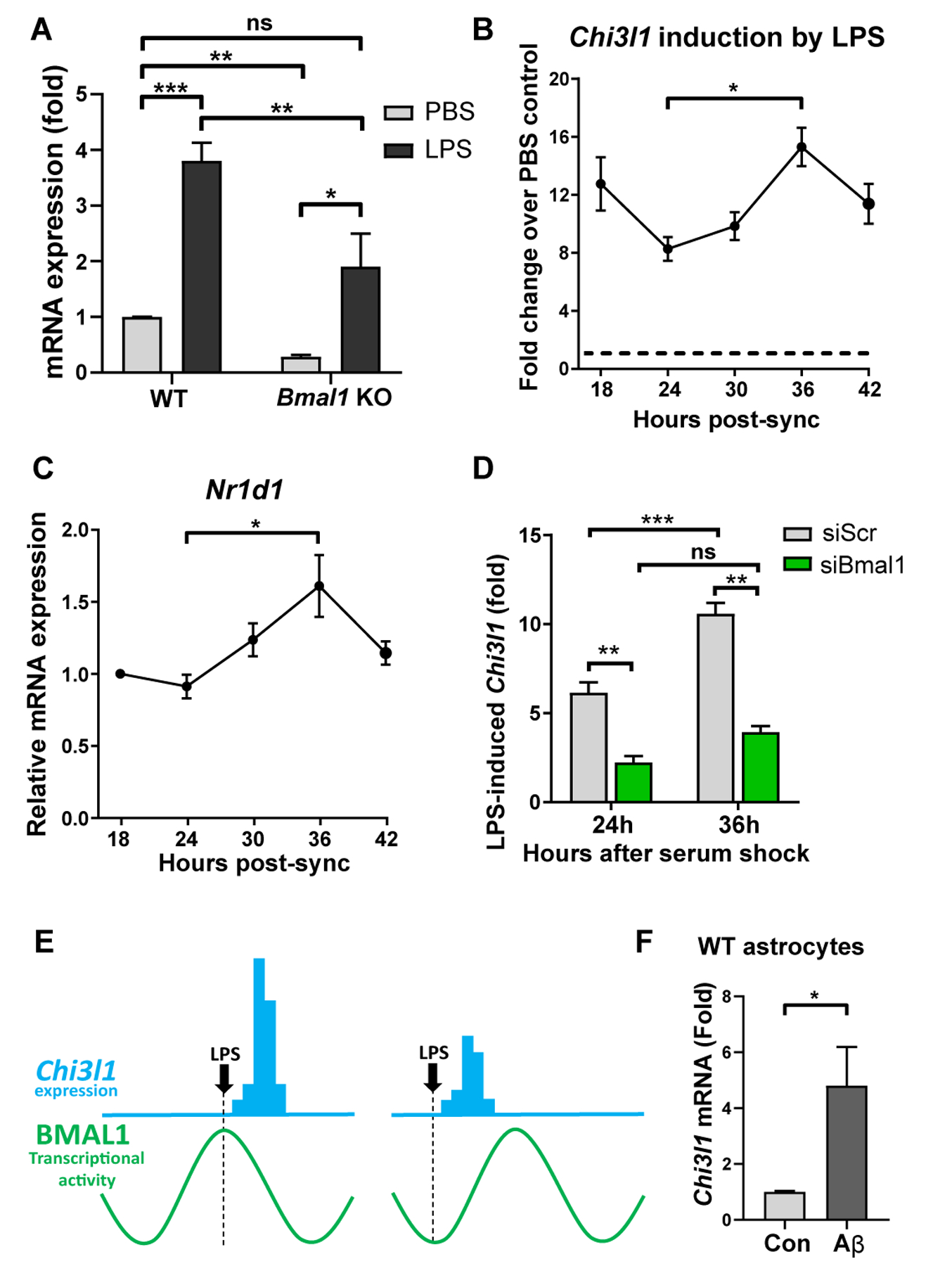
A. qPCR showing gene expression from WT and Bmal1 KO primary astrocytes +/− 500ng/ml LPS for 6 hours. n = 5 independent experiments.
B–C. qPCR showing LPS-induced Chi3l1 gene expression (B) or expression of Nr1d1 (C) in WT primary astrocytes synchronized with high serum shock. Cells were treated with 500 ng/ml LPS (B) or PBS (C) at designated timepoints and collected 3 hours after treatment. n = 6–9 replicates from 2–3 independent experiments per timepoint. Data normalized to basal Chi3l1 expression in PBS control cells (depicted by dashed line) (B) or expression at 18 hours (C). Main effect p = 0.0150 (B), p = 0.0013 (C). Multiple comparison tests depicted on graphs.
D. qPCR showing LPS-induced Chi3l1 expression in WT primary astrocytes after transfection with control (siSCR) or Bmal1 (siBmal1) siRNA, treated with 500ng/ml LPS at 24h or 36h post-synchronization (as in (B)). n = 3 replicates/genotype/timepoint.
E. Diagram depicting hypothesis that induction of Chi3l1 expression (blue) is dependent on the circadian phase of BMAL1 transcriptional activity (green curve).
F. qPCR showing gene expression from WT primary astrocytes +/− 10μM Aβ fibrils for 48–72 hours. n = 6 replicates from 2 independent experiments.
All data represent mean +/− SEM. *p < 0.05, **p < 0.01, ***p < 0.001. Analyzed by two-way ANOVA (A, D) or one-way ANOVA (B, C) with Tukey correction for multiple comparisons or two-tailed students t-test (F).
Discussion
Because glia can exert either protective or degenerative influences on the brain, striking a delicate balance of glial activation and inflammatory signaling is critical for maintaining brain health. Thus, factors that alter glial activation state may disrupt this balance and promote neurodegeneration. Although Chi3l1/YKL-40 is an AD biomarker, its role in the progression of AD remains unknown. Here, we show that in mice and cells, Chi3l1 deletion alters glial inflammatory responses, promotes astrocyte and microglial Aβ phagocytosis, and mitigates amyloid plaque formation. Furthermore, we show that a genetic polymorphism associated with lower CSF CHI3L1/YKL-40 concentrations in humans is associated with slower AD progression. These data suggest that increases in Chi3l1/YKL-40 that occur during aging and AD (4–6) may have a detrimental impact on AD pathogenesis by altering glial function and plaque deposition.
Glial activation is thought to be a double-edged sword in AD, as activated glia can phagocytose Aβ and tau and prevent proteopathy, whereasexcessive inflammatory activation can accelerate plaque accumulation and synapse loss (2). Several studies find Chi3l1 to be anti-inflammatory and/or neuroprotective in the setting of bacterial infection (39), traumatic brain injury, (14) and EAE (13). However, pharmacologic inhibition of Chi3l1 has been reported to suppress Aβ deposition in a rat Aβ infusion model, albeit through a purported anti-inflammatory mechanism (40). We observed that Chi3l1 KO enhances inflammation in response to LPS but not amyloid plaques. Moreover, the transcriptional signature of microglial activation in response to LPS versus amyloid plaques is very different, with opposite regulation of key mediators such as Trem2 (1). Thus, the effect of Chi3l1/YKL-40 on the balance of glial activation and neuroinflammation appears to be context dependent such that loss of Chi3l1 could be neuroprotective in AD, but destructive in settings of acute inflammation.
In APP/PS1 mice, we observed that Chi3l1 deletion causes decreased peri-plaque astrocyte clustering. Attenuation of astrocyte activation in APP/PS1 mice has previously yielded mixed results, as Gfap:Vimentin double KO increases (33), while Stat3 KO decreases (34) plaque burden. Our finding that Chi3l1 knockdown enhances phagocytosis of zymosan beads and Aβ by cultured astrocytes suggests that Chi3l1/YKL-40 is a general regulator of astrocyte phagocytosis and reveals a potential mechanism for the decrease in amyloid plaques in vivo. Interestingly, attenuated astrocyte activation in APP/PS1 models has previously been associated with increased microglial plaque clustering (33, 34) and in one case with increased microglial phagocytosis of Aβ (34), which is consistent with our data. Decreased astrocyte activation in Chi3l1 KO mice (and in these other models) could relent a physical barrier that previously limited microglial access to the plaque. Alternatively, astrocyte-secreted YKL-40 may signal to restrain microglial phagocytic activation.
The exacerbation of LPS-induced microglial transcriptional changes with Chi3l1 deletion match previous reports showing that Chi3l1 can cell-autonomously suppress the macrophage inflammatory response in the periphery (39). Together, these data suggest that Chi3l1 could also be important in microglia. Recently, a single-nucleus RNAseq study identified Chi3l1 as a gene that is strongly upregulated in microglia in the brains of human AD patients (41). While our single-nucleus RNAseq data shows clear expression of Chi3l1 in astrocytes, a small number of microglia do appear to express Chi3l1 and this could increase in the setting of disease. Thus, a cell-autonomous effect of Chi3l1 in microglia is highly possible. Indeed, our observation that Chi3l1 knockdown in cultured microglia increases phagocytosis of beads and Aβ suggests that the increase in peri-plaque CD68 expression in Chi3l1 KO;APP/PS1 mice may be due to a cell-autonomous effect in microglia. In this case, increased Chi3l1 expression in microglia in AD (as reported by Zhou et al (41)) would be expected to suppress microglial – and potentially astrocytic - Aβ phagocytosis and accelerate plaque growth, in keeping with our data.
In patients with AD, we observed that a common variant in the CHI3L1 gene that causes decreased CSF YKL-40 concentrations is associated with slower AD progression. This observation was made in a cohort of extremely well-characterized individuals confirmed to have AD by both clinical evaluation and biomarkers (based on CSF Aβ/tau or amyloid PET imaging). Because CSF YKL-40 increases in response to aging, inflammation, and neurodegeneration, it is difficult to determine how YKL-40 itself might be impacting these processes simply by measuring it in CSF. By examining disease progression in carriers of this genetic variant, which lowers CHI3L1/YKL-40 expression throughout life, we can assume that changes in progression are likely caused by reduced CHI3L1/YKL-40 signaling, providing a unique opportunity to assess a possible causal relationship between Chi3l1/YKL-40 expression and AD. Our results suggest that inhibition of Chi3l1/YKL-40 may be a potential future therapeutic target for limiting plaque accumulation, optimizing the glial phagocytic response to plaques, and slowing progression of AD.
Research into the relationship between circadian disruption and neurodegenerative disease has begun to uncover a role for the clock in regulating astrogliosis (24), microgliosis (22) and plaque deposition (17). Our findings provide an example of a clock-controlled gene (Chi3l1) that does not oscillate at the mRNA level, likely due to a long mRNA halflife. However, because Chi3l1 induction by LPS is suppressed with Bmal1 deletion and is greatest at times when BMAL1 transcriptional activity is highest, it appears that BMAL1 transcriptional activity gates the induction of Chi3l1 in astrocytes, revealing a role for circadian timing in Chi3l1 regulation.
As Chi3l1 is strongly suppressed in Bmal1 KO astrocytes, it is important to reconcile the reduction in Aβ plaque burden we have observed in Chi3l1 KO mice with our previous data showing that global Bmal1 deletion increased fibrillar plaque burden (17). Global Bmal1 deletion affects every cell type in the brain while also disrupting peripheral clocks, sleep-wake cycles, and whole-animal rhythmicity. The resultant phenotype in global Bmal1 KO mice is thus a summation of many smaller and possibly divergent effects. It is likely that in global Bmal1 KO mice, the potentially beneficial effect of decreased Chi3l1 on plaques is overwhelmed by effects on sleep and other processes, resulting in a net increase in plaques. Astrocyte-specific effects of Bmal1 deletion on amyloid plaque deposition remain to be explored. Elucidating the intricacies of competing cell-specific pathways regulated by the clock in the brain is vital in understanding how circadian dysfunction may impact the course of neurodegeneration (42).
There are several limitations to this study. Our experiments do not differentiate the relative effects of Chi3l1 in astrocytes versus microglia in vivo. Such disentanglement would require cell type-specific Chi3l1 manipulation. The utilized APP/PS1 mouse models amyloid plaque formation due to rare familial mutations and does not recapitulate all aspects of human AD, especially tau aggregation. Future studies in alternate models, such as tau transgenic mice, will be needed. The effects of Chi3l1 on phagocytosis of various Aβ and tau species, such as oligomers, was not investigated. Finally, our clinical data does not establish how CHI3L1 polymorphisms impact different aspects of AD pathology or glial activation in humans. Human pathological and biomarker data will need to integrated with CHI3L1 genotyping in future studies.
In summary, we have provided evidence that Chi3l1 regulates glial activation, Aβ phagocytosis and amyloid plaque depostion in mice, and influences AD progression in humans. These findings idenitfy Chi3l1/YKL-40 as a potential therapeutic target for slowing disease progression in AD and provide insights into regulation of neuroinflammation by the astrocyte circadian clock.
Materials and Methods
Study Design
The goal of this study was to elucidate the role of the well-known biomarker of Alzheimer Disease (AD), Chi3l1/YKL-40, in neuroinflammation and AD pathogenesis. We used data from a large observation study of AD to determine if a known genetic variant in the CHI3l1 gene in humans, which was associated with lower CSF YKL-40 levels, might influence the rate of AD progression. This analysis method was based on previous work by members of our group in identifying the variant in CHI3L1 which affects YKL-40 levels and in developing a method to accurately detect single-gene influences on clinical AD progression(26). Mouse studies were then carried out using constitutive Chi3l1−/− mice, which were crossed to an APP/PS1 model of β-amyloidosis for some experiments. Finally, the regulation of Chi3l1 expression by the circadian clock was investigated using a variety of tissue-specific Bmal1 KO mice, as well as other circadian clock gene mutant mice. Cell culture experiments using primary mouse glial cultures and siRNA were also used for mechanistic studies. Sample size for APP/PS1-21 experiments was determined at the outset and based on power calculations derived from previous analysis of amyloid plaque pathology in this line from our lab. Mice were not randomized but were grouped based on genotype with groups containing roughly equal numbers of male and female animals. Mouse tissue samples were all processed together and analyzed to prevent batch effects. Investigators were blinded to genotype throughout the data analysis process. All mice were housed in the same facility, and cohorts of mice were bred at the same time such that they aged together. The number of biological replicates is indicated in the figure legends.
Mice
All mouse experiments were conducted in accordance with protocols approved by the Washington University IACUC. Bmal1−/−, as well as Aldh1L1-CreERT2+, CX3CR1-CreERT2+, and Bmal1f/f mice were obtained from The Jackson Laboratory and bred at Washington University. Tissue from NPAS2 KO, CLOCK KO and CLOCK/NPAS2 double KO was kindly provided by Dr. David Weaver (University of Massachusetts, Worcester, MA). Chi3l1−/− mice were obtained from Dr. Jack Elias (Brown University, Providence, RI). APP/PS1-21 mice were obtained from Dr. Mathias Jucker (University of Tübingen, Tübingen, Germany). Timed-pregnant wildtype CD1 mice for culture experiments were obtained from Charles River (Wilmington, MA). All mice except those used for perinatal cultures were maintained on a C57Bl6 background and housed under 12-hour light/12-hour dark conditions, unless otherwise specified. All mice expressing any Cre or APP/PS1 transgene were heterozygous for these transgenes. Aldh1L1-CreERT2+;Bmal1f/f and CX3CR1-CreERT2+; Bmal1f/f mice were given tamoxifen (Sigma, dissolved in corn oil, 2mg/mouse/day for 5 days) by oral gavage at 1mo or 2mo, respectively, to induce Bmal1 deletion in the applicable tissue. Cre-, Bmal1f/f control littermates were given identical tamoxifen treatment.
Human Studies
For human studies, data was collected as part of a several ongoing longitudinal observational studies of aging and dementia carried out at the Knight Alzheimer’s Disease Research Center. Participants are evaluated annually by clinical staff who are blinded to the participant’s previous diagnosis and all previously collected data, allowing an unbiased assessment of AD diagnosis Clinical Dementia Rating each year (43). The inclusion/exclusion criteria for our analyses were pre-determined and only data from participants meeting these criteria were queried. Inclusion required a clinical AD diagnosis at the last visit, available CSF biomarkers with a profile compatible with AD (based on CSF Aβ/tau profiles with established cutoffs), a Clinical Dementia Rating >0 at last assessment, and at least 1.5 years of follow-up. Exclusions included a clinical diagnosis of a non-AD form of dementia, or diagnoses of another co-existent neurological diseases. In total, 778 participants were included.
Statistical Analysis
For mouse and cell experiments, statistical analyses were performed using GraphPad Prism v8.02. When multiple t-tests were performed, Holm-Sidak correction test for multiple comparisons was applied unless otherwise noted. Fluidigm qPCR data was analyzed by two-way (individual gene or gene groups provided no baseline change in PBS or APP/PS1− mice) or three-way (if a significant main effect of genotype was found by two-way ANOVA at baseline in PBS or APP/PS1− mice) analysis of variance (ANOVA) with Tukey correction for multiple comparisons. Other tests are noted in figure legends. In mouse and cell experiments, data points were determined to be outliers (and thus excluded) based on the ROUT method in Prism 8, Q=1%, performed post-hoc where appropriate.
Statistical analysis of human AD progression was carried out using R statistical software and the package nlme was used for a linear mixed model. A linear mixed-model repeated measure framework was used to account for correlation between repeated measures in the same individual. Disease progression was modeled as follows:
Where: Y was CDR-SB, the change in CDR Sum of Boxes per year baseline CDR, baseline Age, Gender, follow-up time, level of education, and, to avoid the possibility of spurious association due to population substructure, the two first principal components scores were included as covariates.
Supplementary Material
Materials and Methods
Fig. S1. Loss of Chi3l1 exacerbates the LPS-induced inflammatory response
Fig. S2. Loss of Chi3l1 does not change astrocyte reactivity gene signature, but does modulate the microglial response to LPS.
Fig. S3. Loss of Chi3l1 mitigates amyloid pathology.
Fig. S4. Plaque number and size reduced with Chi3l1 deletion without affecting APP processing.
Fig. S5. Loss of Chi3l1 mitigates plaque-related astrogliosis.
Fig. S6. Loss of Chi3l1 alters plaque-related microglial activation.
Fig. S7. Loss of Chi3l1 reduces inflammation in APP/PS1+ mouse hippocampus.
Fig. S8. Chi3l1 is regulated by the circadian clock and expressed in astrocytes.
Acknowledgements:
The authors thank Dr. David Weaver (UMass Medical School) for Clock and Npas2 KO mouse tissue, and the Washington University Center for Cellular Imaging (WUCCI) for imaging assistance.
Funding was provided by NIH grants R01AG054517 (E.S.M) and R01AG044546, P01AG003991, RF1AG053303, R01AG058501, U01AG058922, U01AG052411, and R01AG05777 (C.C.). WUCCI is supported by Washington University School of Medicine, The Children’s Discovery Institute of WU and St. Louis Children’s Hospital (CDI-CORE-2015-505) and the Foundation for Barnes-Jewish Hospital (3770). The Knight ADRC at Washington University is funded by NIA grants P50-AG05681, P01-AG03991, and P01-AG026276.
Footnotes
Competing interests: The authors report no financial competing interests. ESM has consulted for Eisai Pharmacauticals. No patents related to this work are pending.
Data availability: All the data are present in the main text or supplementary material. Microarray data is freely available on the EMBL-EBI ArrayExpress database, accession # E-MTAB-7151.
References and Notes:
- 1.Colonna M, Butovsky O, Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol 35, 441–468 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arranz AM, De Strooper B, The role of astroglia in Alzheimer’s disease: pathophysiology and clinical implications. Lancet Neurol, (2019). [DOI] [PubMed] [Google Scholar]
- 3.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA, Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craig-Schapiro R, Perrin RJ, Roe CM, Xiong C, Carter D, Cairns NJ, Mintun MA, Peskind ER, Li G, Galasko DR, Clark CM, Quinn JF, D’Angelo G, Malone JP, Townsend RR, Morris JC, Fagan AM, Holtzman DM, YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biological psychiatry 68, 903–912 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sutphen CL, Jasielec MS, Shah AR, Macy EM, Xiong C, Vlassenko AG, Benzinger TL, Stoops EE, Vanderstichele HM, Brix B, Darby HD, Vandijck ML, Ladenson JH, Morris JC, Holtzman DM, Fagan AM, Longitudinal Cerebrospinal Fluid Biomarker Changes in Preclinical Alzheimer Disease During Middle Age. JAMA Neurol 72, 1029–1042 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llorens F, Thune K, Tahir W, Kanata E, Diaz-Lucena D, Xanthopoulos K, Kovatsi E, Pleschka C, Garcia-Esparcia P, Schmitz M, Ozbay D, Correia S, Correia A, Milosevic I, Andreoletti O, Fernandez-Borges N, Vorberg IM, Glatzel M, Sklaviadis T, Torres JM, Krasemann S, Sanchez-Valle R, Ferrer I, Zerr I, YKL-40 in the brain and cerebrospinal fluid of neurodegenerative dementias. Mol Neurodegener 12, 83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malmestrom C, Axelsson M, Lycke J, Zetterberg H, Blennow K, Olsson B, CSF levels of YKL-40 are increased in MS and replaces with immunosuppressive treatment. J Neuroimmunol 269, 87–89 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Illan-Gala I, Alcolea D, Montal V, Dols-Icardo O, Munoz L, de Luna N, Turon-Sans J, Cortes-Vicente E, Sanchez-Saudinos MB, Subirana A, Sala I, Blesa R, Clarimon J, Fortea J, Rojas-Garcia R, Lleo A, CSF sAPPbeta YKL -40, and NfL along the ALS-FTD spectrum. Neurology 91, e1619–e1628 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Alcolea D, Vilaplana E, Suarez-Calvet M, Illan-Gala I, Blesa R, Clarimon J, Llado A, Sanchez-Valle R, Molinuevo JL, Garcia-Ribas G, Compta Y, Marti MJ, Pinol-Ripoll G, Amer-Ferrer G, Noguera A, Garcia-Martin A, Fortea J, Lleo A, CSF sAPPbeta, YKL-40, and neurofilament light in frontotemporal lobar degeneration. Neurology 89, 178–188 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Querol-Vilaseca M, Colom-Cadena M, Pegueroles J, San Martin-Paniello C, Clarimon J, Belbin O, Fortea J, Lleo A, YKL-40 (Chitinase 3-like I) is expressed in a subset of astrocytes in Alzheimer’s disease and other tauopathies. J Neuroinflammation 14, 118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhardwaj R, Yester JW, Singh SK, Biswas DD, Surace MJ, Waters MR, Hauser KF, Yao Z, Boyce BF, Kordula T, RelB/p50 complexes regulate cytokine-induced YKL-40 expression. J Immunol 194, 2862–2870 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He CH, Lee CG, Dela Cruz CS, Lee CM, Zhou Y, Ahangari F, Ma B, Herzog EL, Rosenberg SA, Li Y, Nour AM, Parikh CR, Schmidt I, Modis Y, Cantley L, Elias JA, Chitinase 3-like 1 regulates cellular and tissue responses via IL-13 receptor alpha2. Cell Rep 4, 830–841 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonneh-Barkay D, Wang G, Laframboise WA, Wiley CA, Bissel SJ, Exacerbation of experimental autoimmune encephalomyelitis in the absence of breast regression protein 39/chitinase 3-like 1. Journal of neuropathology and experimental neurology 71, 948–958 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiley CA, Bonneh-Barkay D, Dixon CE, Lesniak A, Wang G, Bissel SJ, Kochanek PM, Role for mammalian chitinase 3-like protein 1 in traumatic brain injury. Neuropathology 35, 95–106 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Musiek ES, Bhimasani M, Zangrilli MA, Morris JC, Holtzman DM, Ju YE, Circadian Rest-Activity Pattern Changes in Aging and Preclinical Alzheimer Disease. JAMA Neurol 75, 582–590 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Musiek ES, Holtzman DM, Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science (New York, N.Y 354, 1004–1008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kress GJ, Liao F, Dimitry J, Cedeno MR, FitzGerald GA, Holtzman DM, Musiek ES, Regulation of amyloid-beta dynamics and pathology by the circadian clock. J Exp Med 215, 1059–1068 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohawk JA, Green CB, Takahashi JS, Central and Peripheral Circadian Clocks in Mammals. Annual review of neuroscience 35, 445–462 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Curtis AM, Bellet MM, Sassone-Corsi P, O’Neill LA, Circadian clock proteins and immunity. Immunity 40, 178–186 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Prolo LM, Takahashi JS, Herzog ED, Circadian rhythm generation and entrainment in astrocytes. J Neurosci 25, 404–408 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fonken LK, Frank MG, Kitt MM, Barrientos RM, Watkins LR, Maier SF, Microglia inflammatory responses are controlled by an intrinsic circadian clock. Brain Behav Immun 45, 171–179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakazato R, Hotta S, Yamada D, Kou M, Nakamura S, Takahata Y, Tei H, Numano R, Hida A, Shimba S, Mieda M, Hinoi E, Yoneda Y, Takarada T, The intrinsic microglial clock system regulates interleukin-6 expression. Glia 65, 198–208 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Musiek ES, Lim MM, Yang G, Bauer AQ, Qi L, Lee Y, Roh JH, Ortiz-Gonzalez X, Dearborn JT, Culver JP, Herzog ED, Hogenesch JB, Wozniak DF, Dikranian K, Giasson BI, Weaver DR, Holtzman DM, Fitzgerald GA, Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J Clin Invest 123, 5389–5400 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lananna BV, Nadarajah CJ, Izumo M, Cedeno MR, Xiong DD, Dimitry J, Tso CF, McKee CA, Griffin P, Sheehan PW, Haspel JA, Barres BA, Liddelow SA, Takahashi JS, Karatsoreos IN, Musiek ES, Cell-Autonomous Regulation of Astrocyte Activation by the Circadian Clock Protein BMAL1. Cell Rep 25, 1–9 e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Del-Aguila JL, Li Z, Dube U, Mihindukulasuriya KA, Budde JP, Fernandez MV, Ibanez L, Bradley J, Wang F, Bergmann K, Davenport R, Morris JC, Holtzman DM, Perrin RJ, Benitez BA, Dougherty J, Cruchaga C, Harari O, A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimers Res Ther 11, 71 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deming Y, Black K, Carrell D, Cai Y, Del-Aguila JL, Fernandez MV, Budde J, Ma S, Saef B, Howells B, Bertelsen S, Huang KL, Sutphen CL, Tarawneh R, Fagan AM, Holtzman DM, Morris JC, Goate AM, Dougherty JD, Cruchaga C, Chitinase-3-like 1 protein (CHI3L1) locus influences cerebrospinal fluid levels of YKL-40. BMC Neurol 16, 217 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Del-Aguila JL, Fernandez MV, Schindler S, Ibanez L, Deming Y, Ma S, Saef B, Black K, Budde J, Norton J, Chasse R, Harari O, Goate A, Xiong C, Morris JC, Cruchaga C, Assessment of the Genetic Architecture of Alzheimer’s Disease Risk in Rate of Memory Decline. J Alzheimers Dis 62, 745–756 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, Duncan JA 3rd, Cheshier SH, Shuer LM, Chang EF, Grant GA, Gephart MG, Barres BA, Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89, 37–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ, An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee CG, Hartl D, Lee GR, Koller B, Matsuura H, Da Silva CA, Sohn MH, Cohn L, Homer RJ, Kozhich AA, Humbles A, Kearley J, Coyle A, Chupp G, Reed J, Flavell RA, Elias JA, Role of breast regression protein 39 (BRP-39)/chitinase 3-like-1 in Th2 and IL-13-induced tissue responses and apoptosis. J Exp Med 206, 1149–1166 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jaggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Holscher C, Mathews PM, Jucker M, Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7, 940–946 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Styren SD, Hamilton RL, Styren GC, Klunk WE, X-34, a fluorescent derivative of Congo red: a novel histochemical stain for Alzheimer’s disease pathology. J Histochem Cytochem 48, 1223–1232 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Kraft AW, Hu X, Yoon H, Yan P, Xiao Q, Wang Y, Gil SC, Brown J, Wilhelmsson U, Restivo JL, Cirrito JR, Holtzman DM, Kim J, Pekny M, Lee JM, Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J 27, 187–198 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reichenbach N, Delekate A, Plescher M, Schmitt F, Krauss S, Blank N, Halle A, Petzold GC, Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer’s disease model. EMBO Mol Med 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM, Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nature medicine 10, 719–726 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA, Genomic analysis of reactive astrogliosis. J Neurosci 32, 6391–6410 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, Takahashi JS, Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science (New York, N.Y 338, 349–354 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janelidze S, Mattsson N, Stomrud E, Lindberg O, Palmqvist S, Zetterberg H, Blennow K, Hansson O, CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology 91, e867–e877 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dela Cruz CS, Liu W, He CH, Jacoby A, Gornitzky A, Ma B, Flavell R, Lee CG, Elias JA, Chitinase 3-like-1 promotes Streptococcus pneumoniae killing and augments host tolerance to lung antibacterial responses. Cell Host Microbe 12, 34–46 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi JY, Yeo IJ, Kim KC, Choi WR, Jung JK, Han SB, Hong JT, K284–6111 prevents the amyloid beta-induced neuroinflammation and impairment of recognition memory through inhibition of NF-kappaB-mediated CHI3L1 expression. J Neuroinflammation 15, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, Cella M, Ulland TK, Zaitsev K, Miyashita A, Ikeuchi T, Sainouchi M, Kakita A, Bennett DA, Schneider JA, Nichols MR, Beausoleil SA, Ulrich JD, Holtzman DM, Artyomov MN, Colonna M, Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nature medicine 26, 131–142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lananna BV, Musiek ES, The wrinkling of time: Aging, inflammation, oxidative stress, and the circadian clock in neurodegeneration. Neurobiology of disease 139, 104832 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morris JC, Weintraub S, Chui HC, Cummings J, Decarli C, Ferris S, Foster NL, Galasko D, Graff-Radford N, Peskind ER, Beekly D, Ramos EM, Kukull WA, The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord 20, 210–216 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and Methods
Fig. S1. Loss of Chi3l1 exacerbates the LPS-induced inflammatory response
Fig. S2. Loss of Chi3l1 does not change astrocyte reactivity gene signature, but does modulate the microglial response to LPS.
Fig. S3. Loss of Chi3l1 mitigates amyloid pathology.
Fig. S4. Plaque number and size reduced with Chi3l1 deletion without affecting APP processing.
Fig. S5. Loss of Chi3l1 mitigates plaque-related astrogliosis.
Fig. S6. Loss of Chi3l1 alters plaque-related microglial activation.
Fig. S7. Loss of Chi3l1 reduces inflammation in APP/PS1+ mouse hippocampus.
Fig. S8. Chi3l1 is regulated by the circadian clock and expressed in astrocytes.