

Abstract
Background & Aims
Cell stress signaling pathways result in phosphorylation of the eukaryotic translation initiation factor 2 subunit alpha (EIF2S1 or EIF2A), which affects regulation of protein translation. Translation reprogramming mitigates stress by activating pathways that result in autophagy and cell death, to eliminate damaged cells. Actin is modified during stress and EIF2A is dephosphorylated to restore homeostasis. It is not clear how actin affects EIF2A signaling. We studied the actin-binding proteins villin 1 (VIL1) and gelsolin (GSN) in intestinal epithelial cells (IECs) to determine whether they respond to cell stress response and affect signaling pathways.
Methods
We performed studies with mice with disruptions in Vil1 and Gsn (double-knockout mice). Wild-type mice either were or were not (controls) exposed to cell stressors such as tumor necrosis factor and adherent-invasive Escherichia coli. Distal ileum tissues were collected from mice; IECs and enteroids were cultured and analyzed by histology, immunoblots, phalloidin staining, immunohistochemistry, electron microscopy, and flow cytometry. HT-29 cells were incubated with cell stressors such as dithiothreitol, interferon, and adherent-invasive E coli or control agents; cells were analyzed by immunoblots and quantitative PCR. Full-length and mutant EIF2A were expressed from a lentiviral vector. The mouse immunity related GTPase (IRGM1) was overexpressed in embryonic fibroblasts from dynamin1 like (DNML1) protein-knockout mice or their wild-type littermates. IRGM1 was overexpressed in embryonic fibroblasts from receptor interacting serine/threonine kinase 1-knockout mice or their wild-type littermates. Human IRGM was overexpressed in human epithelial cell lines incubated with the DNML1-specific inhibitor Mdivi-1. Mitochondria were analyzed by semi-quantitative confocal imaging. We performed immunohistochemical analyses of distal ileum tissues from 6–8 patients with Crohn’s disease (CD) and 6–8 individuals without CD (controls).
Results
In IECs exposed to cell stressors, EIF2A signaling reduced expression of VIL1 and GSN. However, VIL1 and GSN were required for dephosphorylation of EIF2A and recovery from cell stress. In mouse and human IECs, prolonged, unresolved stress was accompanied by continued downregulation of VIL1 and GSN, resulting in constitutive phosphorylation of EIF2A and overexpression of IRGM1 (or IRGM), which regulates autophagy. Overexpression of IRGM1 (or IRGM) induced cell death by necroptosis, accompanied by release of damage associated molecular patterns (DAMPs). In double-knockout mice, constitutive phosphorylation of EIF2A and over-expression of IRGM1 resulted in spontaneous ileitis that resembled human CD in symptoms and histology. Distal ileum tissues from patients with CD had lower levels of VIL1 and GSN, increased phosphorylation of EIF2A, increased levels of IRGM and necroptosis, and increased release of nuclear DAMPs compared to controls.
Conclusions
In studies of intestinal epithelial tissues from patients with CD and embryonic fibroblasts from mice, along with enteroids and human IEC lines, we found that induction of cell stress alters the cytoskeleton in IECs, via changes in the actin-binding proteins VIL1 and GSN. Acute changes in actin dynamics increase IEC survival, whereas long-term changes in actin dynamics lead to IEC death and intestinal inflammation. IRGM regulates necroptosis and release of DAMPs to induce gastrointestinal inflammation, linking IRGM activity with CD.
Keywords: TNF, cytoskeleton, IBD, immune response
Introduction
Multicellular organisms respond rapidly, and adapt to cellular stress to maximize cell survival. The cellular stress response, also called integrated stress response (ISR), is universally conserved and independent of the stressor.1 ISR induces rapid, transient reprogramming of cellular protein translation to decrease growth-related and increase stress-related proteins. ISR also changes cellular response to subsequent stress which affects the long-term adaptation of the cell to chronic stress. Cell stress response is initiated by one of four stress sensing kinases all of which phosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 (EIF2A).2 This converts EIF2A from a substrate to an inhibitor of its own guanine nucleotide exchange factor EIF2B, leading to a decrease in global protein synthesis.3 ISR is terminated by dephosphorylation of EIF2A by a protein phosphatase PP1 which requires the direct association of its regulatory subunits protein phosphatase 1 regulatory subunit 15A and 15B (PPP1R15A and PPP1R15B) with globular (G) actin.4 Paradoxically, phosphorylation of EIF2A also favors the translation of the activating transcription factor 4 (ATF4) which activates the pro-survival effects of autophagy in the short term, but prolonged activation of ATF4 activates the DNA-damage inducible transcript 3 protein (DDIT3) to induce cell death.5 Depending on the severity and the duration of stress, the final outcome of cellular stress therefore, can be cell survival or cell death.6 Additionally, the cell must elect to promote apoptosis or immunogenic cell death. The control switch that modulates cell stress response between cell survival and cell death is key to multiple diseases since excessive cell death is linked to inflammation.
Actin is a major component of cellular homeostatic functions and an early target of cell stress response.7, 8 EIF2A signaling regulates the actin cytoskeleton such as by the direct binding of the stress sensing kinase eukaryotic translation initiation factor 2 alpha kinase 2 (EIF2AK2) with gelsolin, which inhibit gelsolin’s actin-severing function.8 Actin is also a major component of the protein translational machinery.9 G-actin binds the regulatory subunits of PP1 to dephosphorylate pEIF2A which, restores cellular homeostasis.4 Downstream of EIF2A, DDIT3 induced cell death is also regulated by actin-binding proteins including the villin-1/gelsolin homolog advillin.4, 10
The pEIF2A pathway is activated upon infection with most CD associated pathogens.11 Marked decrease in autophagy and increased susceptibility to ileal bacterial infection is also observed in mice with IEC-specific expression of non-phosphorylatable EIF2A.12 Similarly, in patients with CD, pEIF2A is increased in mucosal tissue with active disease.13 Ablation of cell death pathways following pEIF2A signaling alleviates colitis in mice.14 Despite this, the pathophysiological relevance of EIF2A signaling in CD is unknown. Immunity-related GTPase family M protein (IRGM) plays a role in innate immunity by regulating autophagy in response to several intracellular pathogens.15 IRGM has been identified in genome wide association studies and meta-analysis as a genetic risk factor in CD.16 IRGM polymorphism confers susceptibility to CD but not ulcerative colitis and is associated with more frequent need for surgery.17 EIF2A signaling is essential for stress-induced autophagy gene expression including ATG16L1, another autophagy gene linked to the pathogenesis of CD.18 Even though IRGM is the strongest autophagy gene linked to CD, its regulation by EIF2A signaling remains unknown. Overexpression of IRGM in CD suggests that its role in CD pathogenesis is independent of its effect on xenophagy but its link to intestinal inflammation has not been identified.19 Increased cell death by RIPK1–RIPK3 (receptor interacting serine/threonine kinase 1, 3) mediated necroptosis and the release of DAMPs such as HMGB1 (high mobility group box 1) are associated with intestinal inflammation and pathogenesis of CD but the molecular basis for their activation is unknown.19, 20 Cellular stress can induce necroptosis and release of DAMPs, but the role of EIF2A signaling in CD associated necroptosis and HMGB1 release has not been determined. In view of that, it is clear that discovering the detailed mechanisms of IEC stress response could improve assessment and treatment of CD patients.
Methods
All authors had access to the study data and reviewed and approved the final manuscript
Villin-1/Gelsolin Double knockout (DKO mice) and DSS administration
DKO mice were generated as described previously. DKO mice and their WT littermates were given DSS in drinking water as described previously.21 The animal research related to this article was approved by the University of Houston Institutional Animal Care and Use Committee.
In vitro and in vivo bacterial infection
HT-29 cells were infected with the AIEC reference strain O83:H1 as well as a non-pathogenic K12 strain at 40 bacteria per cell. The AIEC reference strain O83:H1 was a kind gift from Dr. Alfredo G. Torres (The University of Texas Medical Branch). The K12 strain was purchased from New England Biolabs. Mice were orally given gentamicin (3mg/Kg/d) and vancomycin (40mg/Kg/d) once a day for 3 days and then orally challenged with 109 CFU of AIEC once a day for 4 days.
Tumor necrosis factor (TNF) injection of mice and isolation of epithelial cells
WT mice were either injected intra-peritoneally or not, with 1μg TNF and epithelial cells were isolated from the small intestine after 48 hours as described previously.21
Human tissue collection
Study of archived human CD samples was approved by the Institutional Research Board of Baylor College of Medicine and the University of Houston.
Intestinal enteroid culture
Enteroid cultures were derived from distal ileum of mice as previously described.20 Cell imaging of enteroids was acquired using the Olympus Fluoview FV1200 Laser scanning confocal microscope with a 60X NA 1.35 objective.
Cell lines, cell culture, shRNA knockdown and cellular stress experiments
Hela Tet-Off cells with stable knock down of gelsolin were generated by lentiviral shRNA transduction (Sigma) and selected using Puromycin (2μg/ml). Cells stably expressing Mito-DsRed1 and full-length human villin-1 (WT-villin-1) were generated by selection in G418 (100μg/ml) and Hygromycin (500μg/ml) essentially as described previously.22 Dnm1l wild-type (WT) and knockout (KO) mouse embryonic fibroblasts (MEFs) were a kind gift from Dr. Hiromi Sesaki (Johns Hopkins University). Ripk1 WT and KO MEFs were kindly provided by Dr. Alexei Degterev (Tufts University). Dnm1l WT and KO MEFs were cultured in Iscove’s Modified Dulbecco’s Medium supplemented with 10% heat inactivated serum. Ripk1 WT and KO MEFs, Hela and HT-29 cell lines were culture in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum. For cellular stress experiments HT-29 cells were incubated with 2mM DTT for 3 hours or 100ng/ml Interferon (IFN) for 24 hours. Cellular recovery was measured essentially as described before.23 For the cellular recovery experiment Hela Tet-Off cells expressing or not full-length human villin-1 and gelsolin were incubated with 2mM DTT for 1 hour. The DTT containing media was replaced with 10% FBS containing DMEM to allow recovery. Cell lysates were collected every 2 hours after that to measure the phosphorylation levels of EIF2A.
Measurement of mitochondrial lengths
Mitochondrial morphology was quantified as described previously.24 To visualize mitochondria, the MEFs were stained with 10μM Mitotracker Red for 30 minutes. Alternatively, Hela Tet-Off cells stably transfected with Mito-DsRed1 were used to quantitate changes in mitochondrial morphologies. Images were acquired with the Olympus FV1200 confocal microscope and mitochondrial lengths were measured using the Fluoview software. Hela cells were incubated with 10uM Mdivi1 to measure DNM1L independent mitochondrial fission.
Measurement of F-actin levels
Cells were fixed and stained with Phalloidin and cellular F-actin levels were measured by flow cytometry as previously described.21
Cell death analysis
Cells transfected with EGFP-IRGM or IRGM1-EGFP were incubated with 50μM Necrostatin-1 (Nec1) or 20μM Z-VAD-FMK for up to 24 hours and cell death measured essentially as described previously.25 Rounded cells were quantified by visualizing the morphology of 100 or more transfected cells in at least 10 fields per group using confocal microscopy. Additionally, cell viability was measured with the Guava EasyCyte flow cytometer (EMD Millipore) using either the Viacount assay for cell viability according to the manufacturer’s protocol or by staining for dead cells with 1ug/ml propidium iodide.
Immunoprecipitation of PP1
Immunoprecipitation of PP1 was carried out as described earlier.4 Briefly cells were lysed with lysis buffer (20mM Tris/Cl pH 8, 150mM NaCl, 0.5mM EDTA, 0.5% Triton X-100, 1mM PMSF and Protease inhibitor cocktail) and incubated for 2 hours with protein A magnetic beads (Biorad) bound to PP1 antibody. Beads were then washed 3 times with the same lysis buffer and proteins were eluted by boiling in 1X laemmli buffer.
Electron Microscopy
Electron microscopy was performed as described previously.21 Images were collected on a JEOL 1200EX Electron microscope (JEOL USA, Peabody, MA, USA) with an AMT 2K digital camera.
Statistical analysis
Statistical analysis was performed using the two-tailed Student’s t-test for experiments comparing two groups. For experiments comparing more than two groups one-way ANOVA with Turkey’s multiple comparisons test was performed. The error bars are the measured standard error of mean (SEM).
Results
Villin-1 and gelsolin are targets of cellular stress response
EIF2A pathway is activated in gastrointestinal (GI) epithelium in response to bacterial infection, and defects in this pathway are linked to pathogenesis of CD.11, 13 Loss of two homologous actin severing proteins villin-1 and gelsolin, both of which are expressed in very significant amount in IECs, is also linked to pathogen entry and activation of innate immunity.8, 26 We hypothesized that downregulation of villin-1 and gelsolin expression could be regulated by EIF2A signaling to promote host defense against pathogens. For these studies HT-29 cells were incubated without or with different stressors including DTT or IFN. Alternatively HT-29 cells were infected with the CD associated bacteria adherent-invasive E. coli (AIEC) O83:H1 or a non-pathogenic E. coli strain K12.27 Phosphorylation of EIF2A and a decrease in villin-1 and gelsolin expression levels were noted with all cellular stressors (Fig 1A–C). Autophagy a tightly controlled homeostatic pathway regulated by EIF2A signaling was also activated as revealed by a significant increase in the expression of IRGM (Fig 1A–C). Likewise, wild-type (WT) B6/129 mice injected with TNF or orally administered AIEC showed increased phosphorylation of EIF2A and decrease in the levels of both villin-1 and gelsolin (Fig 1D, 1F). Repeated injection of mice with TNF resulted in sustained, significant increase in IRGM1 (the mouse ortholog) levels (Fig 1E). These changes in villin-1 and gelsolin levels were not due to changes in mRNA levels (Supplementary Figure 1A–D). To validate our findings HT-29 cells were infected with lentiviral particles to express GFP or GFP-tagged phosphorylation mutant of EIF2A (GFP-EIF2A-S52A; Supplementary Figure 2). As expected, the expression of non-phosphorylatable EIF2A protein prevents changes in villin-1, gelsolin and IRGM proteins (Fig 1G). Moreover, in the presence of endogenous EIF2A, the EIF2A-S52A mutant functions as a dominant negative protein. These data demonstrate that villin-1 and gelsolin are the direct targets of EIF2A signaling during cellular stress.
Figure 1. Villin-1 and gelsolin levels decrease in response to diverse cellular stressors.

(A–C) Western blots of HT-29 cells either incubated or not with DTT (A), human IFN (B), non-pathogenic (K12) and pathogenic E. Coli (AIEC) (C). (D–F) Western blots of epithelial cells isolated from small intestine of WT mice injected or not with mouse recombinant TNF (D, E) and orally administered or not pathogenic AIEC (F). Chronic (48–72 h) treatment of WT mice with mouse recombinant TNF shows up-regulation of IRGM1 (E). (G) HT-29 cells expressing either GFP or the non-phosphorylatable mutant GFP-EIF2A-S52A were incubated or not with DTT. Each protein was normalized against tubulin or actin. Data shown are representative of at least three independent experiments. Student’s t-test was used for 1A, 1B, 1D, 1F and 1G. One-way ANOVA was used for 1C and 1E: *, P<0.05; **, P<0.005.
Villin-1 and Gelsolin regulate the phosphorylation of EIF2A
To characterize the relevance of villin-1 and gelsolin in the regulation of ISR, we used epithelial cells that express either both or neither of these proteins (Fig 2A). Villin-1 and gelsolin are actin severing proteins with significant functional redundancy. In the absence of both proteins, there is a change in total cellular actin dynamics resulting in an increase in F-actin levels (Fig 2B). Since total cellular actin levels do not change, this implies a decrease in cellular G-actin. More importantly, while the presence of villin-1 and gelsolin promoted recovery from cellular stress by regulating the dephosphorylation of EIF2A, their absence resulted in constitutive phosphorylation of EIF2A (Fig 2C). A recovery of cellular F-actin simultaneously with recovery of IRGM, gelsolin and villin-1 levels is noted in HT-29 cells following removal of DTT (Supplementary Figure 3A–B). EIF2A phosphorylation occurs as early as 1.5h post DTT addition, while loss of villin-1 and gelsolin proteins occurs much later around 3h post DTT addition (Fig 2D). To validate these findings, we used the Vil1−/−, Gsn−/− double knock out (DKO) mouse described in our previous studies.21 Genetic deletion of villin-1 and gelsolin in mice was also associated with sustained increase in F-actin levels in IECs (Fig 2E). Since total cellular actin levels do not change, this implies a decrease in cellular G-actin. More importantly, loss of villin-1 and gelsolin in mice was associated with constitutive phosphorylation of EIF2A which was seen in whole tissue as well as in enteroids derived from distal ileum of DKO mice (Fig 2F–G). No significant phosphorylation of EIF2A was seen in IEC of either Vil1−/− or Gsn−/− single knockout mice (Supplementary Figure 3C). F-actin distribution in DKO mice has been described in our previous study.21 These findings demonstrate that changes in cellular actin dynamics as a result of loss of both villin-1/gelsolin proteins prevents the dephosphorylation of pEIF2A. Previous studies have shown that association with G-actin is required by the phosphatase PP1, to stabilize its interaction with its regulatory subunits allowing the dephosphorylation of pEIF2A.4 We hypothesized a function for villin-1 and gelsolin in restoring cellular G-actin levels to promote dephosphorylation of EIF2A by PP1. To test this, we measured the association of PP1 with G-actin in cells that express villin-1 and gelsolin compared to cells that lack both proteins. As noted in Fig 2H, in the presence of villin-1 and gelsolin there is increased association of PP1 with G-actin. Constitutive phosphorylation of EIF2A in DKO mice was also associated with increased expression of the autophagy gene, IRGM1 (Fig 2I). These data reveal that the intestinal epithelial cell cytoskeleton is both the target and regulator of EIF2A signaling.
Figure 2. EIF2A signaling is constitutively activated in the absence of villin-1 and gelsolin.

(A) Western blots of cells expressing either both villin-1 and gelsolin (VIL1+GSN+) or neither proteins (VIL1−GSN−). Protein levels were normalized against tubulin. (B) Phalloidin stained cells show increased F-actin in cells that lack both villin-1 and gelsolin (VIL1−GSN−) compared to cells expressing both (VIL1+GSN+). Total F-actin levels were measured using Flow Cytometry. (C) Western blot analysis of VIL1+GSN+ and VIL1−GSN− cells that were incubated with DTT and then allowed to recover following DTT removal. (D) Western blot of HT-29 cells that were either incubated or not with DTT for 1.5h/3h. Each protein was normalized against tubulin. Data shown are representative of three independent experiments (One-way ANOVA: *, P<0.05; **, P<0.005; ***, P<0.0005). (E) Total F-actin levels were measured in IECs isolated from DKO mice and their WT littermates using Flow Cytometry. (F) Immunohistochemistry for pEIF2A in SI of DKO and WT littermates. Arrowheads indicate increase in pEIF2A in DKO mice. (G) Immunofluorescence staining for pEIF2A in small intestinal enteroids from WT and DKO mice. (H) PP1 was immunoprecipitated from lysates of VIL1+GSN+ and VIL1−GSN− cells. The samples were analyzed by western blot using PP1 and actin antibody for pulldown and input lysate (lower panel). (I) Immunohistochemistry for IRGM1 in SI of WT and DKO mice. Arrowheads indicate increase in IRGM1 expression. Data shown are representative of n=6 animals.
Loss of villin-1 and gelsolin induces mitochondrial stress and cell death by necroptosis
We have previously reported that genetic deletion of both villin-1 and gelsolin in mice results in mitochondrial stress and cellular damage with high rates of cell death.21 We documented abnormally excessive IEC death in DKO mice that were not seen in either the Vil1−/− or Gsn−/− mice or their respective WT littermates.21 This suggested to us that loss of both proteins is required for this effect on IEC homeostasis.21 A previous study by Singh et al noted that overexpression of IRGM is associated with mitochondrial depolarization, mitochondrial fission and induction of cell death accompanied by the release of DAMPs.25 Based on that we hypothesized that the increased cell death and mitochondrial stress in DKO could be linked to IRGM1 overexpression. To test this we used transmission electron microscopy (TEM) to analyze IECs of DKO mice which identified mitochondria with variably disorganized cristae, including complete loss of cristae and the appearance of homogenous electron dense aggregates within them (Fig 3A–C).21 Collapsing mitochondrial internal membranes were accompanied by loss of mitochondrial outer membrane integrity (Fig 3C; arrowhead). There is an accumulation of lipofuscin (L) derived from these degenerating mitochondria (M) and the presence of numerous secondary lysosomes (Avi: autophagosomes and Avd: autophagolysosomes) indicating sub-lethal insult selectively affecting the mitochondria (Fig 3D–E). Increased IRGM1 expression was also associated with mitochondrial hyper-fission (Fig 3F; arrowheads). These mitochondrial defects were not seen in the Vil1−/− or Gsn−/− mice.21 TEM showed IEC with chromatin margination, undergoing karyorrhexis (KH), karyolysis (KL) and pyknosis (PK) and clear loss of nuclear membrane all indicative of cell death by necroptosis (Fig 3G–I). Areas of microerosions (loss of one or more IECs) indicated loss of epithelial barrier integrity (Fig 3I, arrowhead). These observations were validated immunohistologically by examining the necroptosis marker, RIPK3 which was significantly increased in IECs of DKO mice (Fig 3J). Loss of membrane integrity due to necroptosis resulted in the release of nuclear HMGB1 in DKO but not WT mice (Fig 3K). The increase in mouse IRGM1 and RIPK3 was also noted in Western blots of isolated IECs from DKO mice compared to their WT littermates (Supplementary Figure 4). Absence of active caspase-8 in DKO mice confirmed cell death by necroptosis and not apoptosis (Fig 3L). These studies identify the function of villin-1 and gelsolin in regulating immunogenic IEC death.
Figure 3. Genetic deletion of villin-1 and gelsolin results in mitochondrial defects and necroptosis.

(A–C) Electron micrographs showing mitochondria in DKO IECs with disorganized cristae, homogenous electron dense aggregates, collapsing mitochondrial membrane and loss of outer mitochondrial membrane integrity (C; arrowhead). (D) Shows accumulation of lipofuscin (L) from degenerating mitochondrial (M). (E) DKO IECs with autophagosome (Avi), autophagolysosome (Avd) and secondary lysosomes. (F) Electron micrographs showing mitochondrial hyper-fission in IECs of DKO mice (red, blue and green arrowheads indicate multiple fission events with single mitochondria). (G) IEC with loss of nuclear membrane (arrowheads). (H) Chromatin margination, karyorrhexis (KH), karyolysis (KL), and pyknotic cells (PK). (I) Shows microerosion with complete loss of IEC. (J–K) Immunohistochemistry and quantification for RIPK3 (J) and HMGB1 (K) in SI of WT and DKO mice. Arrowheads in J indicate increase in RIPK3 expression in DKO mice and arrowheads in K indicate the presence of HMGB1 in the nuclei of WT mice and the absence of nuclear HMGB1 in DKO mice. Data shown are representative of n=6 animals (Student’s t-test: *, P<0.05; **, P<0.005). (L) Shows lack of caspase-8 activation in WT and DKO mice.
IRGM1/IRGM overexpression induces mitochondrial hyper-fission and necroptosis
The nature of IRGM-induced cell death has not been characterized in previous studies.25, 28 We hypothesized that overexpression of IRGM1 is directly linked to mitochondrial hyper-fission and induction of necroptosis independent of the related fission protein DNM1L. To test this we overexpressed mouse IRGM1 and its human ortholog, IRGM and identified its effect on mitochondrial division and cell survival. Live cell imaging of Dnm1l WT and null MEFs was done by labeling mitochondria with Mitotracker red and transfection with EGFP or EGFP-tagged IRGM1. Indeed overexpression of mouse IRGM1 induces mitochondrial hyper-fission that resembles the hyper-fission noted in the IECs of DKO mice and this effect was independent of DNM1L (Fig 4A). Overexpression of IRGM1 induces cell death (Fig 4B) that requires RIPK1 (Fig 4C). More importantly, mouse IRGM1 induced cell death (Fig 4D) and mitochondrial hyper-fission (Fig 4E) can be inhibited with the necroptosis-specific inhibitor necrostatin-1(Nec1). However, mouse IRGM1 induced cell death is resistant to the pan-caspase inhibitor Z-VAD-FMK (Fig 4D). Live cell imaging of human epithelial cells stably transfected with Mito-DsRed1 (to label the mitochondria) and transiently transfected with EGFP or EGFP-IRGM was done using the mitochondrial fission protein and dynamin related protein DNM1L inhibitor Mdivi1. Similar to the IRGM1, overexpression of human IRGM in the absence of DNM1L activity induced mitochondrial hyper-fission (Fig 5A). Inhibiting DNM1L in the absence of IRGM increased mitochondrial fusion but in the presence of IRGM caused mitochondrial hyper-fission (Fig 5B). Overexpression of IRGM but not overexpression of DNM1L, induced cell death and the absence or presence of DNM1L had no effect on IRGM-induced cell death (Fig 5C). Inhibiting necroptosis prevented IRGM induced mitochondrial hyper-fission (Fig 5D) and cell death (Fig 5E). Inhibiting apoptosis had no effect on IRGM-induced cell death (Fig 5E). The dead cells were positive for RIPK3 and extracellular HMGB1 (Supplementary Figure 5). These findings highlight the DNM1L-independent well conserved effects of mouse and human IRGM1/IRGM on mitochondrial hyper-fission and necroptosis.
Figure 4. Overexpression of IRGM1 causes DNM1L independent mitochondrial hyper-fission and necroptosis.

(A) Live cell imaging and quantification of mean mitochondrial length in Dnm1l null MEFs. Mitochondria were labeled with Mitotracker Red and cells were transiently transfected with either EGFP alone or EGFP-tagged IRGM1. Bottom panels show higher magnification of boxed areas indicating overexpression of EGFP-IRGM1 induces mitochondrial hyper-fission unlike only EGFP. Arrowheads indicate multiple fission events. Data shown are representative of n=500 to 600 mitochondria. (B) Overexpression of IRGM1 in Dnm1l WT MEFs significantly increases cell death. (C) Overexpression of IRGM1 in Ripk1 WT but not Ripk1 null MEFs increases cell death measured by flow cytometry using the Viacount assay. (D) IRGM1-induced cell death in Ripk1 WT MEFs is inhibited by the necroptosis specific inhibitor Necrostatin-1(Nec1) but not by the pan-caspase inhibitor, Z-VAD-FMK. Flow cytometry was also used to quantify viable cells using PI staining. (E) IRGM1 induced mitochondrial hyper-fission in Dnm1l WT MEFs is prevented by Nec1. Data shown are representative of n=500 to 600 mitochondria. Student’s t-test was used for 4A, 4B and One-way ANOVA was used for 4C, 4E: *, P<0.05; ***, P<0.0005.
Figure 5. Overexpression of IRGM causes DNM1L independent mitochondrial hyper-fission and necroptosis.

(A–B) Live cell imaging and quantification of mean mitochondrial length in cells that were incubated or not with Mdivi1 followed by transfection with either EGFP or EGFP-IRGM. Overexpression of IRGM in the presence of Mdivi1 induces mitochondrial hyper-fission (A, arrowheads). Data shown are representative of n=500 to 600 mitochondria. (C) Cells transfected with EGFP, EYFP-DNM1L or EGFP-IRGM were incubated or not with Mdivi1 and the percentage of rounded cells were quantified. (D) Live cell imaging and quantification of mean mitochondrial length in cells transfected with either EGFP or EGFP-IRGM followed by incubation with Nec1. Data shown are representative of n=500–600 mitochondria. (E) Cell death measured in cells overexpressing IRGM treated without or with either the pan-caspase inhibitor Z-VAD-FMK or Nec1. Flow cytometry was also used to quantify viable cells using PI staining. Data shown are representative of at least three independent experiments. One-way ANOVA was used for 4B–4E: ***, P<0.0005.
Genetic deletion of villin-1 and gelsolin is associated with GI inflammation and ileocolitis resembling human CD
Systematic characterization of DKO mice between 6–40 weeks of age showed that at 30 week of age, DKO mice have a 25% decrease in total body weight, compared to their WT littermates and compared to the Vil1−/− and Gsn−/− single knockouts (p<0.05, n=8).21 Blood biochemistry of 30 week DKO and WT littermates identified loss of vectorial transport functions and defects in nutrient absorption indicated by changes in plasma protein and albumin levels (data not shown). Loss of villin-1 and related brush border proteins in mice is associated with loss of apical enzymes and transporters involved in nutrient absorption resulting in growth delays in mice.29 Between 30–40 weeks of age, DKO mice showed purulent discharge from rectum, chronic diarrhea and significant dilation of the SI with necrotic areas in both the mid- and distal-ileum (Supplementary Figure 6A). The DKO mice developed ileocolitis and showed segmental injury with regions of inflammation in both the ileum and the colon (Fig 6A). SI morphology demonstrated villus architectural distortions including irregular blunted, fused and thickened villi (Fig 6B–G), crypt elongation and crypt branching (Fig 6B–D; 6G), atrophic mucosa with a complete loss of villi (Fig 6D); hypertrophy of the muscularis propria and fibrosis (Fig 6B–G); Paneth cell hyperplasia; displacement of Paneth cells and an increase in the number of intermediate cells which exhibit Paneth cell phenotype containing eosinophilic granules were also noted (Fig 6B–E; 6G). This was accompanied by inflammatory cell infiltration of the gut including transmural inflammation (Fig 6F). A similar increase in Paneth cell number was noted in enteroids isolated from DKO mice compared to their WT littermates (Supplementary Figure 6D). This phenotype is similar to that of SAMP1/YitFc and TNFΔARE mice and consistent with histomorphological changes seen in human CD.30, 31 The DKO mice exhibit an early onset, increased severity and symptoms of colitis in response to DSS including a much higher death probability compared to WT littermates (Kaplan Meier transform p=0.007; n=7; Supplementary Figure 6B). The histology of SI in Vil1−/− or Gsn−/− mice in contrast was normal and comparable to that of WT littermates (Supplementary Figure 6C).
Figure 6. Genetic deletion of villin-1 and gelsolin is associated with GI inflammation and ileocolitis resembling human CD.
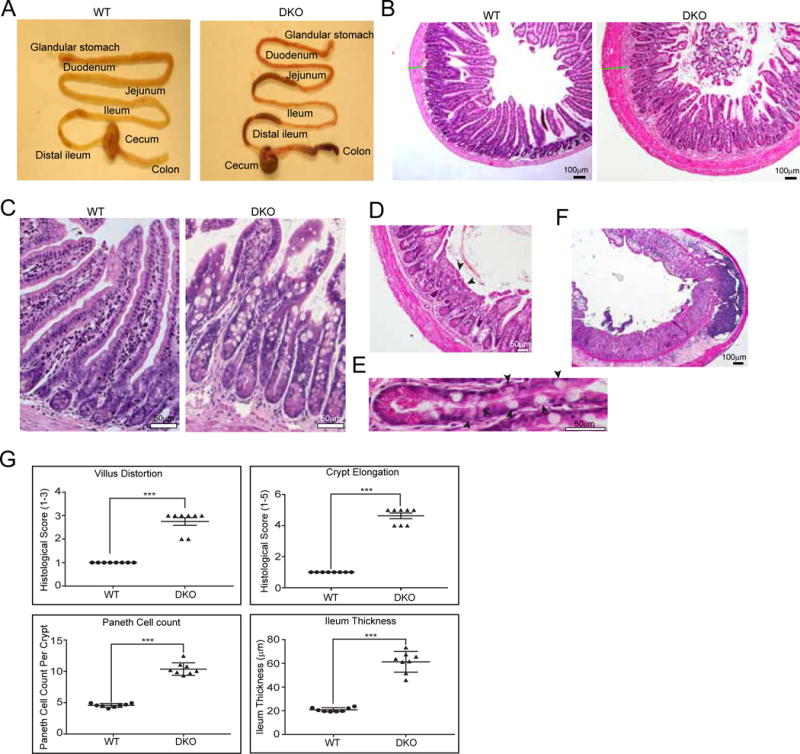
(A) Segmental injury observed in DKO mice showing regions of inflammation in both ileum and colon. (B–F) H & E staining of terminal ileum from DKO mice showing villus architectural distortions (B), crypt elongation and branching (C), atrophic mucosa with complete loss of villi (D, arrowheads), hypertrophy of the muscularis propria and fibrosis (B–D), Paneth cell hyperplasia, displacement of Paneth cells and increase in the number of intermediate cells (E, arrowheads), inflammatory cell infiltration including transmural inflammation (F). (G) Quantification of histological scores. Data shown are representative of n=8 animals per group (Student’s t-test: ***, P<0.001).
Chronic loss of villin-1 and gelsolin expression in CD patients is associated with necroptosis and release of DAMPs
Loss of villin-1 expression in enterocytes from CD patients relative to healthy controls has been reported previously.32 Loss of gelsolin is also associated with inflammatory diseases, however much less is known about its expression in IECs.33 We evaluated distal ileal samples from Crohn’s patients and noted a significant decrease including complete loss of villin-1 (Fig 7A; Supplementary Figure 7A) and gelsolin (Fig 7B; Supplementary Figure 7B) expression from the epithelium of all CD patient samples analyzed but in none of the non-CD controls. No changes in gelsolin expression were noted in the lamina propria of CD or non-CD controls, suggesting specific role of IEC gelsolin in EIF2A signaling (Fig 7B). Loss of villin-1 and gelsolin was also associated with mitochondrial stress accompanied by a significant increase in pEIF2A levels (Fig 7C; Supplementary Figure 7C), increased levels of IRGM (Fig 7D; Supplementary Figure 7D), mitochondrial hyper-fission (Fig 7E), increased levels of necroptosis marker RIPK3 (Fig 7F; Supplementary Figure 7E) and loss of nuclear HMGB1 (Fig 7G; Supplementary Figure 7F). No changes were noted in active caspase-8 (Supplementary Figure 6D). Our cell, mouse and human studies highlight the function of the actin cytoskeleton and specifically villin-1 and gelsolin at the convergence of cellular stress, autophagy, cell autonomous and innate immunity all of which are mechanisms of genetically affected pathways in CD (Supplementary Figure 8).
Figure 7. Chronic loss of villin-1 and gelsolin in CD patients is associated with necroptosis and release of DAMPs.

(A–D) Immunohistochemistry for villin-1 (A), gelsolin (B) pEIF2A (C) and IRGM (D) in SI of CD patients and non-CD controls. Arrowheads in A and B indicate high expression of villin-1 and gelsolin respectively in epithelial cells of non-CD controls. Arrowheads in C indicate an increase in pEIF2A in CD patients. (E) Electron micrographs showing mitochondrial hyper-fission in IECs of CD patients (red arrowheads). (F–G) Immunohistochemistry for RIPK3 (F) and HMGB1 (G) in SI of CD patients and non-CD controls. Arrowheads in F indicate an increase in RIPK3 expression in CD patients. Data shown are representative of n=6 or more human samples per group.
Discussion
In this study we make the novel observation that villin-1 and gelsolin are downregulated in response to diverse cellular stressors including bacteria associated with the pathogenesis of CD. The high plasticity and dynamics of the actin cytoskeleton are exploited by bacterial and viral pathogens to enter host cells.34 Not surprisingly then, changes in the expression and/or loss of actin severing functions of villin-1 and gelsolin have been shown to bolster the immune response by limiting pathogen entry into mammalian cells.8, 26, 35 We provide a molecular basis for this function of villin-1 and gelsolin by identifying them as targets of EIF2A signaling which we suggest serves to achieve broad anti-viral and anti-bacterial protection. Our study establishes that changes in actin dynamics are crucial determinants of cell fate during cellular adaptation to stress. Villin-1 and gelsolin serve as biosensors of cellular stress to integrate environmental sensing pathways with determinants of cell fate. The dynamic conversion of cellular G- and F-actin levels allows rapid restoration of cellular homeostasis through the regulation of PP1.21, 36 G-actin binding to PP1 and its regulatory subunits stabilizes the phosphatase activity of PP1 which dephosphorylates pEIF2A and restores homeostasis.4, 37 We confirm the role of villin-1 and gelsolin in regulating the association of G-actin with the PP1. A good measure of cell fate during cell stress may be this ability of PP1 to bind G-actin or not. We also demonstrate that stabilization of F-actin is the cue for initiating cell death, and we suggest that a cell measures the amount of damage during stress by whether cellular F-actin levels exceed their tolerance limits. The limits of F-actin levels required to initiate cell death may be unique to different cell types and may be achieved by tissue-specific actin regulatory proteins. We suggest that changes in cellular F-actin levels could provide a quantitative marker of cellular damage. This also confirms that actin modifying proteins have important signaling properties that influence cell fate. It may be noted that while advillin has been identified as a specific target of DDIT3, its function in EIF2A signaling remains unknown. We propose that like its homologs, villin-1 and gelsolin, changes in advillin activity in response to cellular stress could regulate constitutive phosphorylation of EIF2A to prevent recovery from cellular stress. Advillin is a actin bundling protein and pEIF2A signaling increases advillin expression which is expected to increase cellular F-actin levels to induce cell death downstream of DDIT3 activation.10 Similarly, downregulation of cofilin-1 increases cellular F-actin levels and induces cell death downstream of DDIT3 activation.38 Changes in cellular actin dynamics thus, may be more widely used as an adaptive response to cellular stress. More importantly, this regulatory EIF2A-actin pathway is highly conserved and intersects with signaling pathways pivotal to inflammation and disease pathogenesis.
IRGM plays a role in innate immunity by regulating autophagy in response to several intracellular pathogens.39 Exposure to microbial products or bacterial invasion increases IRGM expression and IRGM guides autophagic clearance of these bacteria.40 Based on that, the thinking has been that by direct elimination of intracellular bacteria and preventing the activation of pattern recognition receptor signaling IRGM regulates gut homeostasis. In CD a single nucleotide polymorphism (SNP) lying upstream of the IRGM gene in the non-coding region results in sustained increase not decrease in IRGM protein expression and more importantly, despite the high levels of IRGM in CD patients, it is accompanied by an increase in intracellular bacteria.19 So the role of IRGM in the etiology of CD is unknown.41 Our studies provide for the first time a molecular basis for the paradoxical function of IRGM in CD. Our study highlights the autophagy-independent function of IRGM in inducing necroptosis, releasing DAMPs and initiating intestinal inflammation thus providing a molecular basis for IRGM in the pathogenesis of CD.
We propose that unlike other autophagy genes, the specific increase in the expression of IRGM by pEIF2A plays a major role as a checkpoint allowing host cell survival by autophagy or host cell elimination by necroptosis. The localization of IRGM to the mitochondrial inner membrane, its ability to induce mitochondrial fission, mitochondrial depolarization, autophagy-independent cell death and the release of HMGB1 results in the induction of necroptosis an important part of the innate immune signaling.25 This suggests to us that IRGM can modulate cell stress response but also that overexpression of IRGM can induce excessive immunogenic cell death which may be key to the pathogenesis of CD.44,45 ER stress has been linked to the pathogenesis of CD.42 We now show that mitochondrial stress could be linked to the pathogenesis of CD. Furthermore, both ER and mitochondrial stress converge at the ISR pathway with the phosphorylation of EIF2A.43
Necroptosis is an inflammatory response that promotes mitochondrial fragmentation by unregulated fission and is accompanied by the release of alarmins and DAMPs.44 Necroptosis is therefore an effective mechanism to contain pathogen replication and spread while facilitating the clearance of infected cells using the immune system. Many pathogens have evolved evasive strategies to inhibit apoptosis by encoding inhibitors of caspase-8.45 In such instances the host induces caspase-independent cell death by necroptosis through the activation of the RIPK1–RIPK3 complex. Additionally, inhibition of necroptosis prevents IEC death and inflammation.46 We propose that IRGM-induced release of HMGB1 is linked to necroptosis rather than apoptosis, as shown previously.25 HMGB1 has a weak interaction with chromatin and can readily move outside the cell when membrane integrity is lost.47 In apoptosis, membrane integrity is maintained and persists; besides HMGB1 has been shown to be retained in the nuclear membrane during apoptosis. Additionally, apoptotic cells are rapidly degraded and removed by phagocytic cells which would serve to limit the access of immune cells to HMGB1. Necrotic cells that lack HMGB1 fail to induce cytokine production confirming our observation that HMGB1 is a dominant immune player during cell death.48
Mucosal healing is a major treatment outcome in CD because it is associated with reduction of relapse rates, decreased hospitalization requirements, reduced need for surgery, and decreased risk of CD associated cancer.49 Most current therapies are unable to consistently achieve or maintain mucosal healing in CD patients. Our study identifies the cellular mechanisms that advance mucosal injury which could be used to identify new improved therapeutic strategies without suppressing the immune system. We propose that small molecule inhibitors of ISR such as the ISRIB which render cells insensitive to EIF2A phosphorylation and thus, inhibit ISR downstream of EIF2A phosphorylation could result in attenuating some of the deleterious effects of the stress response pathway in CD.50 By improving cellular adaptation to stress, the survival of IECs could be improved. Our findings imply that chronic inflammatory diseases can also be ameliorated by manipulating the actin cytoskeleton, revealing additional new therapeutic targets. We suggest that this approach could be broadly useful in the treatment of other inflammatory diseases.
Supplementary Material
Acknowledgments
This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases (grant number DK-98120 to S.K.); the Public Health Service (grant number DK-56338).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
S.R., S.P., Y.W., A.E., S.P.G., S.K. conceived the experiments and analyzed the data. S.R., S.P., Y.W., A.E., S.P.G., A.A. conducted the experiments. J.K.H. provided human CD tissue samples and provided intellectual input. A.J.H. analyzed the histology data. H.S. provided the Dnm1l null MEFs. All authors reviewed the manuscript.
There is no conflict of interest to declare.
Author names in bold designate shared co-first authorship.
References
- 1.Pakos-Zebrucka K, Koryga I, Mnich K, et al. The integrated stress response. EMBO Rep. 2016;17:1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams BR. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–20. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- 3.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–4. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 4.Chambers JE, Dalton LE, Clarke HJ, et al. Actin dynamics tune the integrated stress response by regulating eukaryotic initiation factor 2alpha dephosphorylation. Elife. 2015;4 doi: 10.7554/eLife.04872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fulda S, Gorman AM, Hori O, et al. Cellular stress responses: cell survival and cell death. Int J Cell Biol. 2010;2010:214074. doi: 10.1155/2010/214074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muralidharan S, Mandrekar P. Cellular stress response and innate immune signaling: integrating pathways in host defense and inflammation. J Leukoc Biol. 2013;94:1167–84. doi: 10.1189/jlb.0313153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desouza M, Gunning PW, Stehn JR. The actin cytoskeleton as a sensor and mediator of apoptosis. Bioarchitecture. 2012;2:75–87. doi: 10.4161/bioa.20975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irving AT, Wang D, Vasilevski O, et al. Regulation of actin dynamics by protein kinase R control of gelsolin enforces basal innate immune defense. Immunity. 2012;36:795–806. doi: 10.1016/j.immuni.2012.02.020. [DOI] [PubMed] [Google Scholar]
- 9.Kim S, Coulombe PA. Emerging role for the cytoskeleton as an organizer and regulator of translation. Nat Rev Mol Cell Biol. 2010;11:75–81. doi: 10.1038/nrm2818. [DOI] [PubMed] [Google Scholar]
- 10.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–9. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 11.Bretin A, Carriere J, Dalmasso G, et al. Activation of the EIF2AK4-EIF2A/eIF2alpha-ATF4 pathway triggers autophagy response to Crohn disease-associated adherent-invasive Escherichia coli infection. Autophagy. 2016;12:770–83. doi: 10.1080/15548627.2016.1156823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao SS, Wang M, Harrington JC, et al. Phosphorylation of eIF2alpha is dispensable for differentiation but required at a posttranscriptional level for paneth cell function and intestinal homeostasis in mice. Inflamm Bowel Dis. 2014;20:712–22. doi: 10.1097/MIB.0000000000000010. [DOI] [PubMed] [Google Scholar]
- 13.Hu S, Ciancio MJ, Lahav M, et al. Translational inhibition of colonic epithelial heat shock proteins by IFN-gamma and TNF-alpha in intestinal inflammation. Gastroenterology. 2007;133:1893–904. doi: 10.1053/j.gastro.2007.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Namba T, Tanaka K, Ito Y, et al. Positive role of CCAAT/enhancer-binding protein homologous protein, a transcription factor involved in the endoplasmic reticulum stress response in the development of colitis. Am J Pathol. 2009;174:1786–98. doi: 10.2353/ajpath.2009.080864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCarroll SA, Huett A, Kuballa P, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet. 2008;40:1107–12. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–17. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu XC, Tao Y, Wu C, et al. Association between Variants of the Autophagy Related Gene-IRGM and Susceptibility to Crohn’s Disease and Ulcerative Colitis: A Meta-Analysis. PLoS ONE. 2013;8:e80602. doi: 10.1371/journal.pone.0080602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.B’Chir W, Maurin AC, Carraro V, et al. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41:7683–99. doi: 10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brest P, Lapaquette P, Souidi M, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet. 2011;43:242–5. doi: 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- 20.Gunther C, Martini E, Wittkopf N, et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–9. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, George SP, Srinivasan K, et al. Actin reorganization as the molecular basis for the regulation of apoptosis in gastrointestinal epithelial cells. Cell Death Differ. 2012;19:1514–24. doi: 10.1038/cdd.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomar A, Wang Y, Kumar N, et al. Regulation of cell motility by tyrosine phosphorylated villin. Mol Biol Cell. 2004;15:4807–17. doi: 10.1091/mbc.E04-05-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kojima E, Takeuchi A, Haneda M, et al. The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: elucidation by GADD34-deficient mice. FASEB J. 2003;17:1573–5. doi: 10.1096/fj.02-1184fje. [DOI] [PubMed] [Google Scholar]
- 24.Rintoul GL, Filiano AJ, Brocard JB, et al. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci. 2003;23:7881–8. doi: 10.1523/JNEUROSCI.23-21-07881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh SB, Ornatowski W, Vergne I, et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol. 2010;12:1154–65. doi: 10.1038/ncb2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Athman R, Fernandez MI, Gounon P, et al. Shigella flexneri infection is dependent on villin in the mouse intestine and in primary cultures of intestinal epithelial cells. Cell Microbiol. 2005;7:1109–16. doi: 10.1111/j.1462-5822.2005.00535.x. [DOI] [PubMed] [Google Scholar]
- 27.Cieza RJ, Hu J, Ross BN, et al. The IbeA invasin of adherent-invasive Escherichia coli mediates interaction with intestinal epithelia and macrophages. Infect Immun. 2015;83:1904–18. doi: 10.1128/IAI.03003-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antonioli M, Di Rienzo M, Piacentini M, et al. Emerging Mechanisms in Initiating and Terminating Autophagy. Trends Biochem Sci. 2017;42:28–41. doi: 10.1016/j.tibs.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 29.Revenu C, Ubelmann F, Hurbain I, et al. A new role for the architecture of microvillar actin bundles in apical retention of membrane proteins. Mol Biol Cell. 2012;23:324–36. doi: 10.1091/mbc.E11-09-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012 doi: 10.1016/S0140-6736(12)60026-9. [DOI] [PubMed] [Google Scholar]
- 31.Pizarro TT, Arseneau KO, Bamias G, et al. Mouse models for the study of Crohn’s disease. Trends Mol Med. 2003;9:218–22. doi: 10.1016/s1471-4914(03)00052-2. [DOI] [PubMed] [Google Scholar]
- 32.Kersting S, Bruewer M, Schuermann G, et al. Antigen transport and cytoskeletal characteristics of a distinct enterocyte population in inflammatory bowel diseases. Am J Pathol. 2004;165:425–37. doi: 10.1016/S0002-9440(10)63308-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osborn TM, Verdrengh M, Stossel TP, et al. Decreased levels of the gelsolin plasma isoform in patients with rheumatoid arthritis. Arthritis Res Ther. 2008;10:R117. doi: 10.1186/ar2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cossart P. Perspective Series: Host/Pathogen Interactions. J Clin Invest. 2016;99:2307–2311. doi: 10.1172/JCI119409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lhocine N, Arena ET, Bomme P, et al. Apical invasion of intestinal epithelial cells by Salmonella typhimurium requires villin to remodel the brush border actin cytoskeleton. Cell Host Microbe. 2015;17:164–77. doi: 10.1016/j.chom.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Srinivasan K, Siddiqui MR, et al. A novel role for villin in intestinal epithelial cell survival and homeostasis. J Biol Chem. 2008;283:9454–64. doi: 10.1074/jbc.M707962200. [DOI] [PubMed] [Google Scholar]
- 37.Chen R, Rato C, Yan Y, et al. G-actin provides substrate-specificity to eukaryotic initiation factor 2alpha holophosphatases. Elife. 2015;4 doi: 10.7554/eLife.04871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nukazuka A, Fujisawa H, Inada T, et al. Semaphorin controls epidermal morphogenesis by stimulating mRNA translation via eIF2alpha in Caenorhabditis elegans. Genes Dev. 2008;22:1025–36. doi: 10.1101/gad.1644008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh SB, Davis AS, Taylor GA, et al. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–41. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 40.Lapaquette P, Bringer MA, Darfeuille-Michaud A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell Microbiol. 2012;14:791–807. doi: 10.1111/j.1462-5822.2012.01768.x. [DOI] [PubMed] [Google Scholar]
- 41.Pilla-Moffett D, Barber MF, Taylor GA, et al. Interferon-Inducible GTPases in Host Resistance, Inflammation and Disease. J Mol Biol. 2016;428:3495–513. doi: 10.1016/j.jmb.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hosomi S, Kaser A, Blumberg RS. Role of endoplasmic reticulum stress and autophagy as interlinking pathways in the pathogenesis of inflammatory bowel disease. Curr Opin Gastroenterol. 2015;31:81–8. doi: 10.1097/MOG.0000000000000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malhotra JD, Kaufman RJ. ER stress and its functional link to mitochondria: role in cell survival and death. Cold Spring Harb Perspect Biol. 2011;3:a004424. doi: 10.1101/cshperspect.a004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maeda A, Fadeel B. Mitochondria released by cells undergoing TNF-alpha-induced necroptosis act as danger signals. Cell Death Dis. 2014;5:e1312. doi: 10.1038/cddis.2014.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat Rev Immunol. 2011;12:79–88. doi: 10.1038/nri3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao H, Jaffer T, Eguchi S, et al. Role of necroptosis in the pathogenesis of solid organ injury. Cell Death Dis. 2015;6:e1975. doi: 10.1038/cddis.2015.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gerlitz G, Hock R, Ueda T, et al. The dynamics of HMG protein-chromatin interactions in living cells. Biochem Cell Biol. 2009;87:127–37. doi: 10.1139/O08-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Urbonaviciute V, Furnrohr BG, Meister S, et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205:3007–18. doi: 10.1084/jem.20081165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neurath MF, Travis SP. Mucosal healing in inflammatory bowel diseases: a systematic review. Gut. 2012;61:1619–35. doi: 10.1136/gutjnl-2012-302830. [DOI] [PubMed] [Google Scholar]
- 50.Sidrauski C, McGeachy AM, Ingolia NT, et al. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife. 2015;4 doi: 10.7554/eLife.05033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.