

Indomethacin, naproxen, and ibuprofen inhibited nociceptive trigeminocervical neurons activated by stimulation of the dura mater, whereas only indomethacin inhibited responses activated by a nitric oxide donor.
Keywords: Trigeminal, Nonsteroidal anti-inflammatory drugs, Cyclooxygenase inhibitor, Primary headache
Abstract
Nonsteroidal anti-inflammatory drugs, cyclooxygenase inhibitors, are used routinely in the treatment of primary headache disorders. Indomethacin is unique in its use in the diagnosis and treatment of hemicrania continua and paroxysmal hemicrania. The mechanism of this specific action is not fully understood, although an interaction with nitric oxide (NO) signaling pathways has been suggested. Trigeminovascular neurons were activated by dural electrical stimulation, systemic administration of an NO donor, or local microiontophoresis of L-glutamate. Using electrophysiological techniques, we subsequently recorded the activation of trigeminovascular neurons and their responses to intravenous indomethacin, naproxen, and ibuprofen. Administration of indomethacin (5 mg·kg−1), ibuprofen (30 mg·kg−1), or naproxen (30 mg·kg−1) inhibited dural-evoked firing within the trigeminocervical complex with different temporal profiles. Similarly, both indomethacin and naproxen inhibited L-glutamate-evoked cell firing suggesting a common action. By contrast, only indomethacin was able to inhibit NO-induced firing. The differences in profile of effect of indomethacin may be fundamental to its ability to treat paroxysmal hemicrania and hemicrania continua. The data implicate NO-related signaling as a potential therapeutic approach to these disorders.
1. Introduction
Primary headache disorders represent a substantial component of neurological practice. One important group is indomethacin-sensitive headaches, notably paroxysmal hemicrania and hemicrania continua.27 The question of what is unique about indomethacin compared to other nonsteroidal anti-inflammatory drugs (NSAIDs) is a crucial question for developing new therapies for these disorders.
Nonsteroidal anti-inflammatory drugs, which are cyclooxygenase (COX) inhibitors, are used in headache therapy, such as in migraine,26 in addition to their use in pain more broadly. They are distinguished by different chemical structures, eg, indomethacin is an acetic acid derivative, whereas ibuprofen is a propionic acid derivative; and by their profiles of absorption, metabolism, and excretion. Remarkably, NSAIDs do not act equally in all headache disorders. A particularly striking example is the indomethacin-sensitive trigeminal-autonomic cephalalgias: paroxysmal hemcrania14,57 and hemicrania continua.13,58 Indeed, the current diagnostic criteria for these disorders use an indomethacin response as a defining characteristic.27 An animal model recapitulating aspects of the trigeminal-autonomic cephalalgias has reported that indomethacin was significantly more effective than naproxen,1 suggesting an alternate mechanism of action from that previously demonstrated in animal models of migraine mechanisms.6 Taken together, it seems likely that indomethacin has a unique action that is not yet clarified.
Here, the effects of indomethacin, naproxen, and ibuprofen were compared in an established animal model of trigeminovascular nociception, which uses activation of dural nociceptive inputs in the trigeminocervical complex (TCC), believed to be important in the pathophysiology of a range of primary headache disorders.25 This model has proven effective at predicting antimigraine clinical efficacy.6 We also compared the local effects of microiontophoretically applied indomethacin and naproxen on iontophoresed glutamate-activated dural-responsive second-order neurons in the TCC. It has been suggested that indomethacin may modulate nitric oxide (NO) signaling pathways. Nitric oxide is known to induce headache and delayed migraine in patients.2,32 In preclinical studies, indomethacin is able to inhibit NO-induced dural vasodilation,3 and this effect is unique because naproxen or ibuprofen were ineffective in this regard.61 Nitrergic mechanisms may be involved in the pathophysiology of headache disorders and therefore indomethacin may target pathways of NO metabolism and signaling as its therapeutic action. We thus hypothesized that indomethacin may demonstrate differential modulation of second-order neurons with trigeminovascular nociceptive inputs and further that indomethacin may be differentially responsive at inhibiting NO-induced neuronal activity compared to naproxen or ibuprofen.
2. Materials and methods
All experiments were conducted under license of the University of California, San Francisco Institutional Animal Care and Use Committee and conforming to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.49 Experiments adhered to the guidelines of the Committee for Research and Ethical Issues of the International Association for the Study of Pain68 and the ARRIVE guidelines.35
2.1. General
Male Sprague-Dawley rats (n = 65, 275-359 g) were used in all experiments, randomized to experimental groups, and analyzed by an observer blinded to their grouping. The selection was limited to male animals so that no interference of the estrous cycle was obtained and the number of animals used could be minimized for ethical reasons, while offering the opportunity of observing the effects of the tested drugs in steady conditions.4 Anesthesia was induced by intraperitoneal application of sodium pentobarbital (Nembutal, 60 mg·kg−1). After reaching a sufficient level of anesthesia, the animal was then placed on a thermostatically controlled homeothermic blanket and kept within physiological ranges. The femoral veins and left femoral artery were cannulated for intravenous administration of subsequent anesthesia, experimental drugs, and blood pressure monitoring (CT-1000 +ALM 932; CWE, Inc, Ardmore, PA). Anesthesia was maintained by intravenous application of sodium pentobarbital (25-30 mg·kg−1·h−1). The trachea was cannulated for mechanical ventilation with oxygen-enriched air (2-3 mL, 80-100 strokes·min−1, small rodent ventilator, Model 683; Harvard Instruments, Kent, United Kingdom). Adequate ventilation was monitored through end-tidal CO2 analysis (limit: 3.5%-4.5%, Capstar-100; CWE, Inc). For further procedures, animals were positioned in a stereotactic frame. The blood pressure, end-tidal CO2, and temperature were electronically displayed online, and together with the repeated observation of the animal's corneal and noxious withdrawal reflexes, used for monitoring suitable depth of anesthesia, and dose of sodium pentobarbitone was adjusted accordingly within the given range. Upon conclusion of electrophysiological recording protocols, all animals were euthanized by an i.v. dose of pentobarbital followed by central nervous tissue collection.
2.2. Electrophysiological recordings
For all electrophysiological recordings, a midline incision was made to expose the skull above the middle meningeal artery (MMA), and the appropriate area of the spine above the first and second cervical (C1/C2) levels. A small craniotomy above the MMA was then performed using a saline cooled dental drill, and a hemilaminectomy of C1 was performed, followed by a small incision of the dura mater so the recording electrode (either a 0.5 MΩ tungsten recording electrode, World Precision Instruments, United Kingdom, tip diameter 0.5 μm or microiontophoresis combination electrode: Carbostar 7s; Kation Scientific, Minneapolis, MN) could be lowered (piezoelectric motor/controller system: IW-811; Burleigh Instruments, Harpenden, United Kingdom; 8200 Controller; EXFO, Plano, TX) into the dorsal horn (5 µm steps) of the exposed TCC. Wide-dynamic-range neurons, identified by noxious pinch, and innocuous brush, responding to electrical stimulation of the MMA/dura mater (0.5 Hz, 0.1-0.2 ms, 5-16 V), were identified and recorded as described.7 For microiontophoretic experiments, the cells had to show stable baselines of increased firing rate in response to microiontophoretic L-glutamate ejections and stable baselines of increased firing rate in response to electrical stimulation of the MMA. The criteria for intravenous experiments did not include microiontophoretic L-glutamate ejections. Poststimulus histograms (PSTH) were established for sequences of 20 stimulations. A mean firing rate of 30% above baseline was required,48 within a 7 to 10 ms period of the main firing episode, corresponding to Aδ fibers. Poststimulus histograms were collected with 1-ms bin sizes over a poststimulus period of 100 ms. The action potential firing of the neurons recorded in response to microiontophoresis of L-glutamate were collected in successive 1-second bins and analyzed as cumulative rate histograms.
To study the intravenous effect of indomethacin, naproxen, and ibuprofen on electrical stimulation of the MMA, a baseline response was evaluated before administration of the drug/control baseline (mean out of 4 stimulation series of 20 sweeps). One of the tested drugs or the vehicle control (H2O for injection, pH 8-8.3) 1 ml·kg−1 was then administered intravenously. In all electrophysiological studies, animals received only a single dose of an NSAID intravenously. Further PSTHs were collected 5, 10, 15, 20, 25, 30, and 45 minutes after administration.
According to anatomical measurements and nerve conduction velocities, all recorded responses were meeting criteria for classification as A fibers (response 4-20 ms after stimulation).
All experimental and physiological data were acquired, displayed, and saved on a personal computer using an online data analysis system (Power 1401plus, CED and Spike5 software, United Kingdom).
At the end of the experiment, the recording site was electrically lesioned or marked by microiontophoretic ejection of pontamine sky blue (PSB), and tissue collected for further histological processing. Topological localization of lesion sites was then identified according to the Paxinos and Watson brain atlas.52
2.3. Microiontophoresis
By application of holding currents between 5 and 7 nA, with a polarity opposite to the charge of the respective ion, all ions were retained in the barrels.60 Ejection currents of the same polarity as the molecule's polarity were used for ejection of indomethacin and naproxen, and the charges ranged from 70 to 100 nA. The negative ejection currents for L-glutamate microiontophoresis ranged from 30 to 80 nA. To receive a response similar to receptive field stimulation of the first and second trigeminal dermatomes, the ejection current for L-glutamate was established individually for each recorded cell.
To test the effect of indomethacin and naproxen on L-glutamate-evoked firing, L-glutamate was microiontophoresed, using a current generator (Dagan 6400; Dagan Corporation, MN), in ejection/retaining cycles with ejection period of 7 to 9 seconds and retaining period of 30 seconds of the ejection time to avoid desensitization of the L-glutamate response. Once 5 stable baseline responses were achieved, the testing compounds or control (OH−) were coejected over a period of 3 to 5 minutes of L-glutamate ejection, followed by recovery. For statistics, the first 5 cycles after the ejection of the test-compound ceased were analyzed (Fig. 1A).
Figure 1.
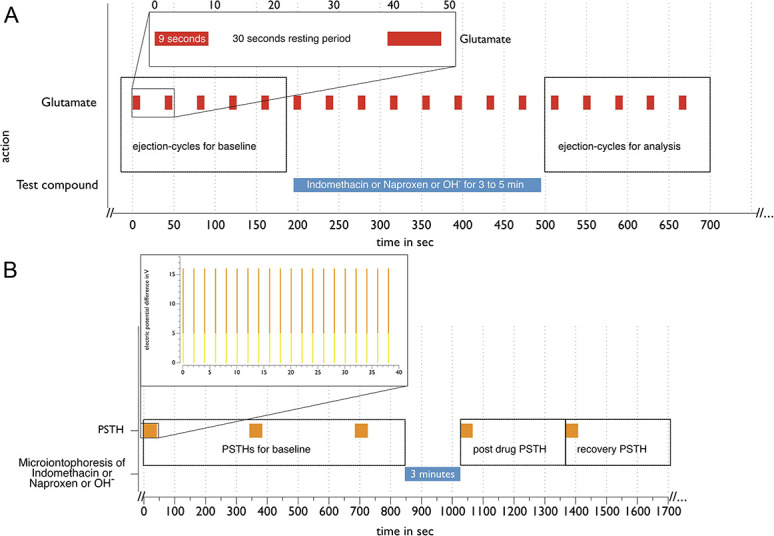
Timeline of microiontophoresis experiments. (A) One cycle of microiontophoresis establishing a baseline with repetitive L-Glutamate ejection-cycles, followed by microiontophoretic ejection of a test compound, and then an additional series. (B) One PSTH experiment, establishing a baseline with repetitive stimulation cycles of the MMA/dura for recordings followed by microiontophoretic ejection of one of the test compounds as indicated by horizontal bars. Further PSTHs are recorded at the time of termination of the ejection of the test compound, and a following PSTH during the recovery episode. The enlargement is displaying the timeline of a single PSTH recording, with the orange top of vertical bars indicating the range of voltage used for different experiments (5-16 V). The axis of abscissae displays time in sec within the timeline and enlargement. PSTH, poststimulus histograms.
For testing of the effect of the microiontophoresed compounds on MMA stimulation, a baseline response of 3 baseline PSTHs separated by five-minute recovery intervals was established. After further 2 minutes, this was followed by 3 minutes of microiontophoresis (−70 to −100 nA) of one of the drugs and a PSTH at the end of the microiontophoresis episode after further 5 minutes of recovery. After full recovery, this process was repeated with the other substances used (Fig. 1B). The drugs were given in pseudorandomized order. For full recovery, cells had to display evoked firing rates as before the application of the first compound tested and they were left for 30 minutes without further testing and establishing the according baseline. Resistances for the individual barrels ranged from 20 to 100 MΩ.
Pontamine sky blue was ejected (4 µA, 10 minutes) at the end of the experiment for later localization of the recording sites, and for reconstruction of further recording sites in compliance with the microdrive readings. After termination of each experiment, the brain tissue was collected and fixed in 10% formalin for histological processing.
2.4. Nitric oxide-induced trigeminal firing
In a separate group, all the animals received additional cannulation of the carotid artery, ipsilateral to the TCC recording site, for NO donor infusion. Having identified stable cells fulfilling the criteria for electrophysiological measurements: stable dural responsiveness and receptive field in the V1 branch of the trigeminal nerve, we observed the response of wide-dynamic-range neurons to intra-arterial administration of sodium nitroprusside (SNP), 2 µg·kg−1·min−1 dissolved in 0.9% saline solution, over 5 minutes. After a recovery period of 15 minutes, SNP infusion was repeated and the recorded firing rates of the duration of SNP infusion were averaged; the calculated value equals the SNP-induced activity at baseline. After a resting period of 3 minutes' control, indomethacin (5 mg·kg−1), naproxen (30 mg·kg−1), or ibuprofen (30 mg·kg−1) was slowly infused intravenously. Ten minutes after infusion, SNP injection cycles of 5-minute infusion time and recovery periods of 15 minutes were started. Sodium nitroprusside infusions were repeated at 10, 30, 50, and 70 minutes after drug administration.
2.5. Drugs
Microiontophoresis barrels of the combination electrode were filled with 200 mM L-glutamate monosodium, (Sigma, St. Louis, MO), pH 8.0; 15 mM indomethacin (Sigma), pH 8.0 to 8.3; 50 mM naproxen (Sigma), pH 8.0 to 8.3; distilled Water (dH20), pH 8 to 8.5 as control; and 2.5% PSB (Gurr 6BX, BDH Laboratory Supplies, Poole, United Kingdom) in 100 mM sodium acetate and 200 mM NaCl for current balance.8 L-glutamate, indomethacin, naproxen, and PSB were ionized as anions. OH anions were microiontophoresed as control.
2.6. Data analysis
The experiments, recording the effect of the intravenous administered drugs on the electrically elicited firing rate in the TCC, were analyzed by comparison of the recorded PSTHs. The firing rate in response to SNP infusion was analyzed as the mean firing rate over 180 seconds pre-SNP infusion, and was subtracted from the mean firing rate during SNP infusion, resulting in the SNP induced firing rate. Analysis of the effect of microiontophoresed indomethacin and naproxen on L-glutamate-evoked firing was performed by calculation of the mean firing rate of 5 successive epochs of L-glutamate ejection predrug ejection. Testing of reliability was performed using Cronbach α. The mean response for each drug was then calculated by averaging the firing rate of 5 successive pulses during each drugs microejection. After ejection, 5 further pulses were averaged as the postejection response. The mean firing rate of spontaneous activity over 150 seconds was calculated and compared with the mean spontaneous firing during and after the microiontophoresis of each drug. For statistical analysis, we used IBM-SPSS (v20.0, New York, NY). The data sets of intravenous experiments and the effect of electrical stimulation and SNP, as well as microiontophoresis experiments (background and elicited activity) were analyzed by performing a mixed-model repeated-measures analysis of variance. Greenhouse–Geisser corrections were applied if the assumption of sphericity was violated. For all multiple comparisons, Bonferroni correction was applied. In the case of P values <0.05, post hoc comparisons were made using paired-sample t test for the effect of each intervention. The effect size r has been calculated using Pearson correlation coefficient. Results are expressed as percentages of baseline ± SE. Significance was assessed at the P < 0.05 level unless otherwise stated.
3. Results
3.1. General
Slow intravenous administration of vehicle control, indomethacin, naproxen, and ibuprofen had no effect on any physiological parameter recorded (cell firing rate at rest, blood pressure, and end-tidal CO2). The cells recorded in the experiments were identified being located within the TCC located in the dorsal horn within layers III-V at the level of C1.
3.2. Intravenous administration of test drugs
The effect of intravenously administered drugs on PSTHs recorded in the TCC was tested in 33 animals. Administration of indomethacin (n = 9; F7,56 = 4.072; P = 0.001) led to a 17 ± 3% inhibition of cell response 10 minutes after administration (t8 = 5.442, P = 0.001, r = 0.89) when compared to baseline. Naproxen (n = 8) demonstrated a slower profile, significantly inhibiting cell firing (F2,14 = 7.756; P = 0.006) after 20 to 30 minutes with a maximum inhibition of 26 ± 6% after 30 minutes (t7 = 3.828, P = 0.006, r = 0.82). Neuronal activation was also inhibited by ibuprofen (n = 8; F7,49 = 2.524; P = 0.027) over a prolonged period (10-30 minutes), with a maximum of 20 ± 5% 30 minutes after administration (t7 = 4.093, P = 0.005, r = 0.84; Fig. 2). Administration of vehicle control (n = 8) had no significant effect on neuronal activation (F7,49 = 0.738; P = 0.641).
Figure 2.
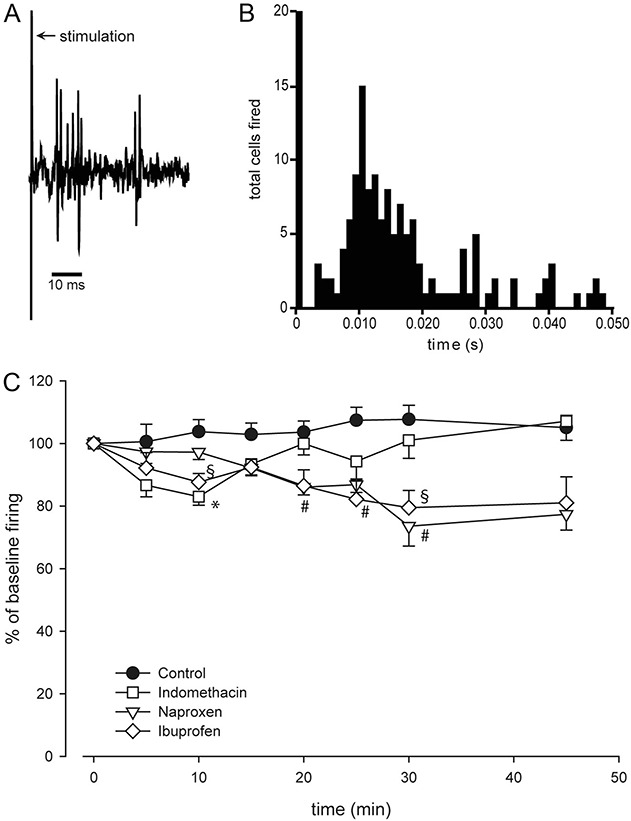
Display of responses to electrical stimulation of the dura mater/middle meningeal artery (A and B) and in relation to time after administration of indomethacin, ibuprofen, naproxen, and vehicle control (C). (A) Example single-unit trace of one of the recorded second-order neurons within the trigeminocervical complex, displaying the cluster response in reaction to electrical stimulation of the dura mater/middle meningeal artery. (B) Poststimulus histogram summarizing 20 responses of the second-order neuron shown in (A) (ordinate displaying the number of units firing due to electrical stimulation). (C) After the baseline response was established, one of the drugs (indomethacin 5 mg·kg−1, naproxen 30 mg·kg−1, ibuprofen 30 mg·kg−1, or control) was applied intravenously. All the tested drugs showed an inhibitory effect on the evoked firing with separate time point of maximum inhibition. Although indomethacin was demonstrated to induce early inhibition, naproxen induces a long-lasting inhibition at a later time point. Ibuprofen is shown to have early-onset inhibitory effect and a second inhibitory effect starting later and reaching its maximum parallel to naproxen (values for 0 indicate mean baseline responses). *, §, #P < 0.05 when compared to baseline response.
3.3. Trigeminocervical complex microiontophoresis
The responses of 14 cells from 8 animals were investigated using the protocols for L-glutamate-evoked firing in conjunction with dural stimulation.
3.3.1. L-Glutamate-evoked firing
The current for microiontophoresis ranged from 60 to 80 nA, and drugs were microiontophoresed for 5 to 6 cycles with ejection periods of 5 to 10 seconds and retaining periods of 15 to 22 seconds. The baseline responses did not show significant differences (F4,112 = 1.401, P = 0.238) and were highly reliable (Cronbach α value ≥0.96). Application of control currents (cells n = 8) had no effect on L-glutamate-evoked cell firing in the TCC (F2,16 = 1.739, P = 0.207). Indomethacin (cells n = 9) significantly inhibited neuronal activity (F2,16 = 8.123, P = 0.004) by 22 ± 8% (t15 = 2.739, P < 0.015, r = 0.58) compared to control. After ejection ceased, L-glutamate cycles continued to be inhibited by 27 ± 10% (t10 = 2.748, P < 0.020, r = 0.66). This effect lasted for up to 15 minutes. Naproxen (cells n = 7) also showed an effect on L-glutamate-evoked cell firing in the TCC (F2,12 = 10.525, P = 0.002). However, the onset of the effect was not significant during the ejection of the drug but during the recovery period, displaying a 21 ± 9% (t13 = 2.298, P = 0.039, r = 0.54) inhibition (Fig. 3). The microiontophoresis of control ions, indomethacin, or naproxen had no significant effect on the spontaneous neuronal activity in the TCC (all P ≥ 0.28).
Figure 3.
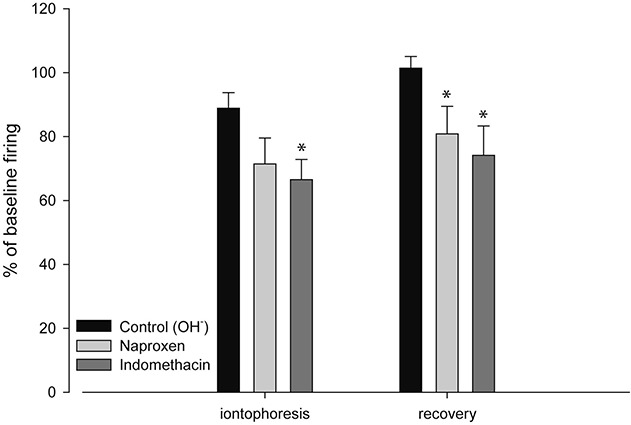
Effect of microiontophoresis of indomethacin, naproxen, and control on trigeminal neuronal firing elicited by repetitive microiontophoresis of L-glutamate. Summary of changes by microiontophoresis of indomethacin (−60 to 80 nA) vs naproxen vs control (OH−) at the same current. The bars representing the recovery period were calculated in the early recovery phase of the first 5 ejection cycles of L-glutamate after the ejection of the testing compound has ceased. *P < 0.05.
3.3.2. Dural stimulation and microiontophoresis
The microiontophoresis of control ions, indomethacin, or naproxen had no effect on neuronal firing due to electrical stimulation of the dura mater (cells n = 11; P ≥ 0.27).
3.4. Sodium nitroprusside infusion induced activity
The SNP protocol was conducted in 24 animals. The infusion of SNP increased the background firing rate of the tested cells (t23 = 8.775; P = 0.000, r = 0.88) by 26 ± 3%, returning to baseline levels within 2 minutes after each SNP infusion (Fig. 4A). The background activity between SNP infusions remained unchanged in the absence of drug treatment throughout the experiments, and no changes in the expansion of dural and cutaneous receptive fields were observed. Intravenous administration of indomethacin significantly altered TCC activity induced by SNP infusion (F4,20 = 3.19, P = 0.035; Figs. 4A and B). By contrast, control (F2,11 = 1.24, P = 0.331), naproxen (F4,20 = 0.67, P = 0.62), or ibuprofen (F1,7 = 1.12, P = 0.354) did not alter the increase of background activity in response to SNP infusions (n = 6 per group; Fig. 4B).
Figure 4.
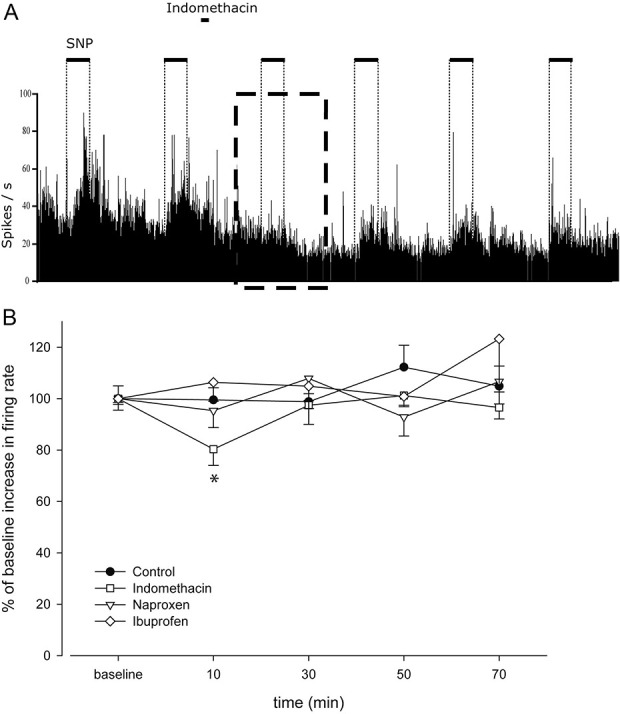
Representative recordings of firing rates (A) in the trigeminocervical complex from experiments investigating the effect of indomethacin on second-order neuronal activity, induced by repeated sodium nitroprusside (SNP) infusions. The neuronal activity increases shortly after SNP (2 µg·kg−1·min−1) administration. The box in A indicates the decreased neuronal response due to infusion of SNP 10 minutes after intravenous administration of indomethacin. After the baseline response was established, one of the drugs (indomethacin 5 mg·kg−1, naproxen 30 mg·kg−1, ibuprofen 30 mg·kg−1, or control) was applied intravenously, followed by SNP infusions (each for a 5-minute duration, followed by a 15-minute recovery period, starting 10 minutes after the administration of one of the drugs). An inhibitory effect on the SNP-induced firing was registered exclusively for indomethacin (B). *P < 0.05 when compared to baseline response.
In line with results of a previous study, we monitored a minor drop in the baseline blood pressure when SNP infusion was started, and the reading returned to baseline level within 3 minutes after SNP infusion.
4. Discussion
The current study provides evidence of a substantial difference of the tested NSAIDs' capabilities for blocking the SNP infusion-induced effects in the TCC. When comparing the effects of the NSAIDs on electrically induced cell firing within the TCC, it becomes clear that they have individual time-dependent profiles of action. Here, we demonstrate a transient inhibitory effect after application of indomethacin on NO-induced, as well as electrically induced trigeminal firing in the TCC, that is evident only 10 minutes after drug infusion, whereas the inhibitory effect on NO-induced vasodilation has been shown to outlast 70 minutes.61 In line with the effects of naproxen on electrically induced vasodilation, it had a much slower onset of effect on electrically induced firing in the TCC and reached its peak inhibition near the end of the experiment, a possible sign of a delayed passage of the blood–brain barrier as has been described earlier.21,22 In a similar in vivo model, acetylsalicylic acid and ketorolac, which is structurally related to indomethacin, were demonstrated to have a time course of inhibition matching the one of naproxen measured in our experiments,34 although these experiments were conducted in cat.
The unique effects of indomethacin demonstrating inhibitory modulation of NO-induced vasodilation and trigeminal activity, with a rapid onset, do not define whether this is a central or peripheral effect, although experimental data suggest a limited penetration through the blood–brain barrier.64 A significant central action of indomethacin is supported by the diversity in the time course of indomethacin's effect in this and our previous experiments. The recorded activity after SNP infusion is unlikely to result from peripheral vascular effects over 70 minutes.61 Because hemicrania continua is a long-lasting headache, it remains unclear, however, to what extent the central NO-activated mechanisms investigated here are relevant to hemicrania continua and paroxysmal hemicrania and if it is the central or peripheral NO-induced effect. As stated before, indomethacin elicited a clear potential in inhibiting dural-evoked activation and interestingly a long-lasting inhibitory effect on the activation recorded in the TCC after stimulation of the superior salivatory nucleus.1 Combining these results involvement of NO mechanisms, modifiable by indomethacin, at the level of the superior salivatory nucleus is a possible mechanism. Indomethacin may activate modulatory mechanisms within the central nervous system not investigated here. Interestingly, indomethacin can modulate nociceptive signaling in a model of trigeminal autonomic cephalalgias,1 which is in line with the data presented here.
The dose of indomethacin, ibuprofen, and naproxen used was based on the knowledge of similar high absorption rates and plasma protein binding if taken orally, as well as the ratio of their maximum daily dose for the treatment of headache, viz., indomethacin: ibuprofen: naproxen = >225 mg·d−1:1200 mg·d−1:1200 mg·d−1 = 1:6:6. We used the same doses as previously reported61 because these doses have been shown to be well tolerated; the dose for indomethacin is slightly lower than its oral LD10.50 There is a broad knowledge from in vitro experiments about NSAIDs' action on the different cyclooxygenases, including their kinetic profiles23 and their capabilities in the inhibition of COX1/COX2 mechanisms, resulting in specific COX1/COX2 ratios.45 COX1 and COX2 are prominent, not only in the dura mater as a key structure of trigeminal innervation,42,67 but COX2 activity has been shown to be modifiable at the level of the caudal trigeminal trigeminocervical nucleus.65 COX1/COX2 inhibition has been shown to alter trigeminocervical neurons responding to nociceptive dural activation.34 Interestingly, the inhibitory effect of the tested drugs at the level of the TCC seems to be rather limited compared to the effect of triptans, such as rizatriptan and naratriptan.15,16 This is in contrast to the greater use of NSAIDs in migraine, although it is consistent with severe migraine attacks being generally better treated with triptans. Certainly, in clinical trials, fewer patients respond to NSAIDs than triptans.10
The microiontophoretic data demonstrate a central effect of indomethacin and naproxen on postsynaptic second-order neurons within the TCC. However, in our experimental setup, no effect was seen when we studied the microiontophoretic modulation of TCC firing. This might be due to a comparatively low potency of the tested drugs in the TCC. The effect of ibuprofen was not investigated microiontophoretically for technical reasons regarding the electrical properties of the compound.54
Investigating the interaction of indomethacin with NO and NO-mediated mechanisms, indomethacin has been shown to reduce NO production from rat microglia.17 Indomethacin also inhibits the expression of endothelial NO synthase in vivo measured in the kidneys47 and inducible NO synthase (iNOS) in macrophages in vitro,30 a mechanism that is most likely due to decreased PGE2 production through COX2, as it has been shown that PGE2 is able to upregulate iNOS expression in macrophages in vitro.11,51 Yet, this mechanism is unlikely to be essential for our results because SNP is a direct donor of NO without iNOS involvement and the selective iNOS inhibitor GW274150 failed in prophylactic treatment of migraine.28 The NO donor glyceryl trinitrate has been known to trigger migraine for some time.29 The delayed headache has the typical clinical phenotype of a migraine attack.2,31,55 Glyceryl trinitrate is also capable of triggering a cluster headache attack with a delay of about 10 minutes after infusion, if it is used during an active cluster period.20 Given this context, the modulation of NO-induced early activation of trigeminal activity, as demonstrated here, suggests that NO might play a significant role in the pathophysiology of paroxysmal hemicrania and hemicrania continua. Interestingly, NO also demonstrated modulatory effects through activation of Nav1.9 channels in a mouse model of triptan-induced medication overuse headache,9 and indeed indomethacin has been reported to produce a migraine-like headache when used in hemicrania continua.33
Nitric oxide release facilitates the parasympathetic craniofacial pathways24 as one key structure within the pathophysiological pathway of trigeminal autonomic cephalagias.44 Nitric oxide donors have previously been described to cause activation of neurons within the spinal trigeminal nucleus, midbrain, and forebrain structures when tested in animal studies.53,62 Direct activation of trigeminovascular neurons as well as sensitization was found performing electrophysiological work using GTN in an animal in vivo study.2,39 This suggests the activation, driven by SNP infusion, may be at least partly caused by local NO effects in the TCC or other central structures with modulatory connectivity to the TCC. This includes cervical inputs5 and structures feeding the downstream modulatory pathway to the TCC such as the periaqueductal gray,36 rostral ventromedial medulla,19 and hypothalamic nuclei such as the A11.12,59 Nitric oxide is known to activate cells through indirect elevation of cyclic guanosine-monophosphate (cGMP) levels,46 causing vasodilation through subsequent decrease of Ca2+. Independent from cGMP levels, it is also capable of modulating the level of CGRP release within the trigeminal ganglion.18 In addition to these effects, the microiontophoresis of an NOS inhibitor at the level of the TCC showed inhibitory effects on L-glutamate and electrical-induced activity of second-order neurons, thereby demonstrating a direct effect on second-order neurons.40 Nitric oxide/nNOS activity has also been suggestive of antinociceptive capabilities in chronic inflammation,63 yet these effects are limited to chronic pain and rely on nNOS activity.
In our experiments, there was no sensitization as determined by altered baseline activity, change of dural, or cutaneous receptive fields. Although NO has been described to induce central sensitization in a rat model, infusion of SNP intravenously did not produce a significant immediate facilitation of trigeminal firing.37 Nitric oxide is a highly volatile molecule with a very short half-life46; so, we may have achieved higher levels of NO at relevant structures, although the dose of SNP per injection period was lower in our experimental setup (10 vs 50 µg·kg−1), when compared to that of Koulchitsky et al,37 and higher than in their study where they investigated sensitization at the level of trigeminal nucleus.38 A comparison of these studies is, however, challenging because SNP was infused into the carotid artery ipsilateral to the electrophysiological recording sites, with similar direct action to what has been reported.39 Studies of the effect of NO donors on neuronal activity in the trigeminal ganglion, but not the TCC, found a delayed activation.41,66 This might also be attributed to an increase in local CGRP levels, as well as the receptor activity modulating protein 1 (RAMP-1) component of the CGRP receptor because it was immunohistochemically demonstrated after i.v. administration of GTN to rats.56 Application of CGRP itself on trigeminal ganglia cultures, however, demonstrated increased iNOS expression and NO release.43
In summary, the data, taken with previous work, suggest a possible central action of the NSAIDs indomethacin and naproxen. An inhibitory effect on electrically induced trigeminal firing is demonstrated for indomethacin, ibuprofen, and naproxen; by contrast, the effect on NO-evoked trigeminal firing can be seen for only indomethacin. The results offer new insights into NSAID mechanisms in primary headache disorders and highlight alternative signaling pathways involved in the particular pathophysiology of hemicrania continua and paroxysmal hemicrania. Moreover, the data are consistent with the substantially central nervous system pathogenesis of paroxysmal hemicrania and hemicrania continua. Taken together with our previous studies, perhaps therapies directed at nitrergic mechanisms may be a promising target for the treatment of paroxysmal hemicrania and hemicrania continua.
Conflict of interest statement
O. Summ has no conflicts of interest to declare; and reports a fellowship from MSD Sharp & Dohme GmbH not related to the work. A.P. Andreou declares no direct conflicts; and reports equipment grant from eNeura, honoraria and travel expenses from Eli Lilly and eNeura, in relation to educational duties and advisory boards, as well as sponsorships for educational purposes from Autonomic Technologies, eNeura, Allergan, Eli Lilly, and Novartis. S. Akerman declares no conflicts; and reports personal fees from Allergan, Amgen, GSK, Novartis, and A&O unrelated to the submitted work. P.R. Holland declares no direct conflicts; and reports research grants from Amgen, Celgene, and Eli Lilly, as well as honoraria and travel expenses in relation to educational duties and advisory boards from Allergan, Novartis, and Almirall. J. Hoffmann declares no direct conflicts and reports honoraria for consulting activities and/or serving on advisory boards from Allergan, Autonomic Technologies Inc, Chordate Medical AB, Eli Lilly, Hormosan Pharma, Novartis, and Teva. He received personal fees for medicolegal work as well as from Sage Publishing, Springer Healthcare, and Quintessence Publishing. All these activities are unrelated to the submitted work. J. Hoffmann reports a research grant from Celgene. P.J. Goadsby declares no direct conflicts, and reports, over the last 36 months, grants and personal fees from Amgen and Eli-Lilly and Company, grant from Celgene, and personal fees from Alder Biopharmaceuticals, Allergan, Aeon Bopharma, Biohaven Pharmaceuticals Inc, Clexio, Electrocore LLC, eNeura, Epalex, Impel Neuropharma, MundiPharma, Novartis, Santara Therapeutics, Teva Pharmaceuticals, Trigemina Inc, and WL Gore, and personal fees from medicolegal work, Massachusetts Medical Society, Up-to-Date, Oxford University Press, and Wolters Kluwer; and a patent magnetic stimulation for headache assigned to eNeura without fee.
Acknowledgements
The authors thank Michele Lasalandra for excellent technical assistance.
Author contributions: O. Summ designed and conducted experiments, collated the data, performed the analysis, and wrote the first draft of the manuscript. A.P. Andreou assisted with the conduct of experiments and edited the manuscript. S. Akerman designed and assisted with the conduct of experiments and edited the manuscript. P. R. Holland designed and assisted with the conduct of experiments and edited the manuscript. J. Hoffmann assisted with the conduct of experiments and edited the manuscript. P.J. Goadsby designed the experiments, reviewed the data, and edited the manuscript in detail.
Supported by the Sandler Foundation, The Wellcome Trust (104033/Z/14/Z), the Medical Research Council (Grant number MR/P006264/1) and by EUROHEADPAIN European Union FP7 (602633). O. Summ received a grant by the deanship of the faculty of medicine of the University of Muenster and has been supported by a fellowship of the European Federation of Neurological Societies. J. Hoffmann was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft (DFG; HO4369/1-1).
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
References
- [1].Akerman S, Holland PR, Summ O, Lasalandra MP, Goadsby PJ. A translational in vivo model of trigeminal autonomic cephalalgias: therapeutic characterization. Brain 2012;135(pt 12):3664–75. [DOI] [PubMed] [Google Scholar]
- [2].Akerman S, Karsan N, Bose P, Hoffmann JR, Holland PR, Romero-Reyes M, Goadsby PJ. Nitroglycerine triggers triptan-responsive cranial allodynia and trigeminal neuronal hypersensitivity. Brain 2019;142:103–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Akerman S, Williamson DJ, Kaube H, Goadsby PJ. The effect of anti-migraine compounds on nitric oxide-induced dilation of dural meningeal vessels. Eur J Pharmacol 2002;452:223–8. [DOI] [PubMed] [Google Scholar]
- [4].Ayata C, Jin H, Kudo C, Dalkara T, Moskowitz MA. Suppression of cortical spreading depression in migraine prophylaxis. Ann Neurol 2006;59:652–61. [DOI] [PubMed] [Google Scholar]
- [5].Bartsch T, Goadsby PJ. Stimulation of the greater occipital nerve induces increased central excitability of dural afferent input. Brain 2002;125:1496–509. [DOI] [PubMed] [Google Scholar]
- [6].Bergerot A, Holland PR, Akerman S, Bartsch T, Ahn AH, MaassenVanDenBrink A, Reuter U, Tassorelli C, Schoenen J, Mitsikostas DD, van den Maagdenberg AMJM, Goadsby PJ. Animal models of migraine. Looking at the component parts of a complex disorder. Eur J Neurosci 2006;24:1517–34. [DOI] [PubMed] [Google Scholar]
- [7].Bergerot A, Storer RJ, Goadsby PJ. Dopamine inhibits trigeminovascular transmission in the rat. Ann Neurol 2007;61:251–62. [DOI] [PubMed] [Google Scholar]
- [8].Bloom FE. To spritz or not to spritz: the doubtful value of aimless iontophoresis. Life Sci 1974;14:1819–34. [DOI] [PubMed] [Google Scholar]
- [9].Bonnet C, Hao J, Osorio N, Donnet A, Penalba V, Ruel J, Delmas P. Maladaptive activation of Nav1.9 channels by nitric oxide causes triptan-induced medication overuse headache. Nat Commun 2019;10:4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Brandes JL, Kudrow D, Stark SR, O'Carroll CP, Adelman JU, O'Donnell FJ, Alexander WJ, Spruill SE, Barrett PS, Lener SE. Sumatriptan-naproxen for acute treatment of migraine: a randomized trial. JAMA 2007;297:1443–54. [DOI] [PubMed] [Google Scholar]
- [11].Chang YH, Lee ST, Lin WW. Effects of cannabinoids on LPS-stimulated inflammatory mediator release from macrophages: involvement of eicosanoids. J Cell Biochem 2001;81:715–23. [DOI] [PubMed] [Google Scholar]
- [12].Charbit AR, Akerman S, Holland PR, Goadsby PJ. Neurons of the dopaminergic/calcitonin gene-related peptide A11 cell group modulate neuronal firing in the trigeminocervical complex: an electrophysiological and immunohistochemical study. J Neurosci 2009;29:12532–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cittadini E, Goadsby PJ. Hemicrania Continua: a clinical study of 39 patients with diagnostic implications. Brain 2010;133:1973–86. [DOI] [PubMed] [Google Scholar]
- [14].Cittadini E, Matharu MS, Goadsby PJ. Paroxysmal hemicrania: a prospective clinical study of thirty-one cases. Brain 2008;131:1142–55. [DOI] [PubMed] [Google Scholar]
- [15].Cumberbatch MJ, Hill RG, Hargreaves RJ. Rizatriptan has central antinociceptive effects against durally evoked responses. Eur J Pharmacol 1997;328:37–40. [DOI] [PubMed] [Google Scholar]
- [16].Cumberbatch MJ, Hill RG, Hargreaves RJ. Differential effects of the 5HT1B/1D receptor agonist naratriptan on trigeminal versus spinal nociceptive responses. Cephalalgia 1998;18:659–64. [DOI] [PubMed] [Google Scholar]
- [17].Du ZY, Li XY. Inhibitory effects of indomethacin on interleukin-1 and nitric oxide production in rat microglia in vitro. Int J Immunopharmacol 1999;21:219–25. [DOI] [PubMed] [Google Scholar]
- [18].Eberhardt M, Neeb L, Vogel EM, Tiegs G, Reuter U, Messlinger K, Fischer MJ. Glyceroltrinitrate facilitates stimulated CGRP release but not gene expression of CGRP or its receptor components in rat trigeminal ganglia. Neuropeptides 2009;43:483–9. [DOI] [PubMed] [Google Scholar]
- [19].Edelmayer RM, Vanderah TW, Majuta L, Zhang ET, Fioravanti B, De Felice M, Chichorro JG, Ossipov MH, King T, Lai J, Kori SH, Nelsen AC, Cannon KE, Heinricher MM, Porreca F. Medullary pain facilitating neurons mediate allodynia in headache-related pain. Ann Neurol 2009;65:184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ekbom K. Nitroglycerin as a provocative agent in cluster headache. Arch Neurol 1968;19:487–93. [DOI] [PubMed] [Google Scholar]
- [21].Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest 2003;112:440–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fan W, Wu Y, Li XK, Yao N, Li X, Yu YG, Hai L. Design, synthesis and biological evaluation of brain-specific glucosyl thiamine disulfide prodrugs of naproxen. Eur J Med Chem 2011;46:3651–61. [DOI] [PubMed] [Google Scholar]
- [23].Gierse JK, Koboldt CM, Walker MC, Seibert K, Isakson PC. Kinetic basis for selective inhibition of cyclo-oxygenases. Biochem J 1999;339:607–14. [PMC free article] [PubMed] [Google Scholar]
- [24].Goadsby PJ, Adner M, Edvinsson L. Characterisation of endothelin ETA receptors in the cerebral vasculature and their lack of effect upon spreading depression. J Cereb Blood Flow Metab 1996;16:698–704. [DOI] [PubMed] [Google Scholar]
- [25].Goadsby PJ, Holland PR, Martins-Oliveira M, Hoffmann J, Schankin C, Akerman S. Pathophysiology of Migraine-A disorder of sensory processing. Physiol Rev 2017;97:553–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Goadsby PJ, Sprenger T. Current practice and future directions in the management of migraine: acute and preventive. Lancet Neurol 2010;9:285–98. [DOI] [PubMed] [Google Scholar]
- [27].Headache Classification Committee of the International Headache Society (IHS). The international classification of headache disorders, 3rd edition. Cephalalgia 2018;38:1–211. [DOI] [PubMed] [Google Scholar]
- [28].Hoivik HO, Laurijssens BE, Harnisch LO, Twomey CK, Dixon RM, Kirkham AJ, Williams PM, Wentz AL, Lunnon MW. Lack of efficacy of the selective iNOS inhibitor GW274150 in prophylaxis of migraine headache. Cephalalgia 2010;30:1458–67. [DOI] [PubMed] [Google Scholar]
- [29].Horton BT. Headache; clinical varieties and therapeutic suggestions. Med Clin North Am 1949;33:973–1005. [DOI] [PubMed] [Google Scholar]
- [30].Hrabak A, Vercruysse V, Kahan IL, Vray B. Indomethacin prevents the induction of inducible nitric oxide synthase in murine peritoneal macrophages and decreases their nitric oxide production. Life Sci 2001;68:1923–30. [DOI] [PubMed] [Google Scholar]
- [31].Iversen H. Human migraine models. Cephalalgia 2001;21:781–5. [DOI] [PubMed] [Google Scholar]
- [32].Iversen HK, Olesen J. Nitroglycerin-induced headache is not dependent on histamine release: support for a direct nociceptive action of nitric oxide. Cephalalgia 1994;14:437–42. [DOI] [PubMed] [Google Scholar]
- [33].Jurgens TP, Schulte LH, May A. Indomethacin-induced de novo headache in hemicrania continua—fighting fire with fire? Cephalalgia 2013;33:1203–5. [DOI] [PubMed] [Google Scholar]
- [34].Kaube H, Hoskin KL, Goadsby PJ. Intravenous acetylsalicylic acid inhibits central trigeminal neurons in the dorsal horn of the upper cervical spinal cord in the cat. Headache 1993;33:541–50. [DOI] [PubMed] [Google Scholar]
- [35].Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG; NC3Rs Reporting Guidelines Working Group. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 2010;160:1577–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Knight YE, Goadsby PJ. The periaqueductal gray matter modulates trigeminovascular input: a role in migraine? Neuroscience 2001;106:793–800. [DOI] [PubMed] [Google Scholar]
- [37].Koulchitsky S, Fischer MJ, De Col R, Schlechtweg PM, Messlinger K. Biphasic response to nitric oxide of spinal trigeminal neurons with meningeal input in rat—possible implications for the pathophysiology of headaches. J Neurophysiol 2004;92:1320–8. [DOI] [PubMed] [Google Scholar]
- [38].Koulchitsky S, Fischer MJ, Messlinger K. Calcitonin gene-related peptide receptor inhibition reduces neuronal activity induced by prolonged increase in nitric oxide in the rat spinal trigeminal nucleus. Cephalalgia 2009;29:408–17. [DOI] [PubMed] [Google Scholar]
- [39].Lambert GA, Donaldson C, Boers PM, Zagami AS. Activation of trigeminovascular neurons by glycerol trinitrate. Brain Res 2000;887:253–9. [DOI] [PubMed] [Google Scholar]
- [40].Lambert GA, Hoskin KL, Zagami AS. Nitrergic and glutamatergic neuronal mechanisms at the trigeminovascular first-order synapse. Neuropharmacology 2004;47:92–105. [DOI] [PubMed] [Google Scholar]
- [41].Levy D, Strassman AM. Modulation of dural nociceptor mechanosensitivity by the nitric oxide-cyclic GMP signaling cascade. J Neurophysiol 2004;92:766–72. [DOI] [PubMed] [Google Scholar]
- [42].Levy D, Zhang XC, Jakubowski M, Burstein R. Sensitization of meningeal nociceptors: inhibition by naproxen. Eur J Neurosci 2008;27:917–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Li J, Vause CV, Durham PL. Calcitonin gene-related peptide stimulation of nitric oxide synthesis and release from trigeminal ganglion glial cells. Brain Res 2008;1196:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].May A, Goadsby PJ. The trigeminovascular system in humans: pathophysiological implications for primary headache syndromes of the neural influences on the cerebral circulation. J Cereb Blood Flow Metab 1999;19:115–27. [DOI] [PubMed] [Google Scholar]
- [45].Mitchell JA, Warner TD. Cyclo-oxygenase-2: pharmacology, physiology, biochemistry and relevance to NSAID therapy. Br J Pharmacol 1999;128:1121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev 1991;43:109–42. [PubMed] [Google Scholar]
- [47].Nagappan AS, Varghese J, Pranesh GT, Jeyaseelan V, Jacob M. Indomethacin inhibits activation of endothelial nitric oxide synthase in the rat kidney: possible role of this effect in the pathogenesis of indomethacin-induced renal damage. Chem Biol Interact 2014;221:77–87. [DOI] [PubMed] [Google Scholar]
- [48].Nagler J, Conforti N, Feldman S. Alterations produced by cortisol in the spontaneous activity and responsiveness to sensory stimuli of single cells in the tuberal hypothalamus of the rat. Neuroendocrinology 1973;12:52–66. [DOI] [PubMed] [Google Scholar]
- [49].National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals. Washington (DC): National Academies Press (US), 2011. [PubMed] [Google Scholar]
- [50].Omogbai EK, Ozolua RI, Idaewor PE, Isah AO. Some studies on the rodenticidal action of indomethacin. Drug Chem Toxicol 1999;22:629–42. [DOI] [PubMed] [Google Scholar]
- [51].Panaro MA, Brandonisio O, Sisto M, Acquafredda A, Leogrande D, Fumarola L, Mitolo V. Nitric oxide production by Leishmania-infected macrophages and modulation by prostaglandin E2. Clin Exp Med 2001;1:137–43. [DOI] [PubMed] [Google Scholar]
- [52].Paxinos G, Watson C. The rat brain in stereotaxic coordinates. London, United Kingdom: Academic Press, 1998. [Google Scholar]
- [53].Ramachandran R, Bhatt DK, Ploug KB, Hay-Schmidt A, Jansen-Olesen I, Gupta S, Olesen J. Nitric oxide synthase, calcitonin gene-related peptide and NK-1 receptor mechanisms are involved in GTN-induced neuronal activation. Cephalalgia 2014;34:136–47. [DOI] [PubMed] [Google Scholar]
- [54].Ruiz MA, Gallardo V, Ouazzani N, Lopez-Viota J, Lopez-Duran JD. Electrophoretic properties of acrylic latex suspensions (Kollicoat MAE 30 D) and ibuprofen. Farmaco 2004;59:657–62. [DOI] [PubMed] [Google Scholar]
- [55].Sances G, Tassorelli C, Pucci E, Ghiotto N, Sandrini G, Nappi G. Reliability of the nitroglycerin provocative test in the diagnosis of neurovascular headaches. Cephalalgia 2004;24:110–19. [DOI] [PubMed] [Google Scholar]
- [56].Seiler K, Nusser JI, Lennerz JK, Neuhuber WL, Messlinger K. Changes in calcitonin gene-related peptide (CGRP) receptor component and nitric oxide receptor (sGC) immunoreactivity in rat trigeminal ganglion following glyceroltrinitrate pretreatment. J Headache Pain 2013;14:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sjaastad O, Dale I. Evidence for a new (?) treatable headache entity. Headache 1974;14:105–8. [DOI] [PubMed] [Google Scholar]
- [58].Sjaastad O, Spierings EL. Hemicrania continua: another headache absolutely responsive to indomethacin. Cephalalgia 1984;4:65–70. [DOI] [PubMed] [Google Scholar]
- [59].Skagerberg G, Bjorklund A, Lindvall O, Schmidt RH. Origin and termination of the diencephalo-spinal dopamine system in the rat. Brain Res Bull 1982;9:237–44. [DOI] [PubMed] [Google Scholar]
- [60].Stone TW. Microiontophoresis and pressue ejection. In: Smith AD, editor. Methods in the Neurosciences. Vol. 8 Chichester: John Wiley and Sons, 1985. [Google Scholar]
- [61].Summ O, Andreou AP, Akerman S, Goadsby PJ. A potential nitrergic mechanism of action for indomethacin, but not of other COX inhibitors—relevance to indomethacin-sensitive headaches. J Headache Pain 2010;11:477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tassorelli C, Joseph SA. NADPH-diaphorase activity and Fos expression in brain nuclei following nitroglycerin administration. Brain Res 1995;695:37–44. [DOI] [PubMed] [Google Scholar]
- [63].Tesser-Viscaino SA, Denadai-Souza A, Teixeira SA, Ervolino E, Cruz-Rizzolo RJ, Costa SK, Muscara MN, Casatti CA. Putative antinociceptive action of nitric oxide in the caudal part of the spinal trigeminal nucleus during chronic carrageenan-induced arthritis in the rat temporomandibular joint. Brain Res 2009;1302:85–96. [DOI] [PubMed] [Google Scholar]
- [64].Upton RN, Rasmussen M, Grant C, Martinez AM, Cold GE, Ludbrook GL. Pharmacokinetics and pharmacodynamics of indomethacin: effects on cerebral blood flow in anaesthetized sheep. Clin Exp Pharmacol Physiol 2008;35:317–23. [DOI] [PubMed] [Google Scholar]
- [65].Varga H, Pardutz A, Vamos E, Plangar I, Egyud E, Tajti J, Bari F, Vecsei L. Cox-2 inhibitor attenuates NO-induced nNOS in rat caudal trigeminal nucleus. Headache 2007;47:1319–25. [DOI] [PubMed] [Google Scholar]
- [66].Zhang X, Kainz V, Zhao J, Strassman AM, Levy D. Vascular extracellular signal-regulated kinase mediates migraine-related sensitization of meningeal nociceptors. Ann Neurol 2013;73:741–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Zhang X, Levy D, Kainz V, Noseda R, Jakubowski M, Burstein R. Activation of central trigeminovascular neurons by cortical spreading depression. Ann Neurol 2011;69:855–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. PAIN 1983;16:109–10. [DOI] [PubMed] [Google Scholar]