

ABSTRACT
Immune checkpoint blockade (ICB) expands the therapeutic options for metastatic lung cancer nowadays representing a standard frontline strategy as monotherapy or combination therapy, as well as an option in oncogene-addicted NSCLC after exhaustion of targeted therapies. Predictive markers are urgently needed, since only a minority of patients benefits from ICB, while serious adverse effects of immunotoxicity may occur. The study cohort included 43 ICB-treated metastatic lung adenocarcinoma showing long-term response (n = 16), rapid progression (n = 21) or intermediate patterns of response (n = 6). Lung biopsies acquired before initiation of ICB were analyzed by targeted mRNA expression profiling of 770 genes. Level and proportions of 14 immune cell types were estimated using characteristic gene expression signatures. Abundance of B cells (HR = 0.66, p = .00074), CD45+ cells (HR = 0.61, p = .01) and total TILs (HR = 0.62, p = .025) was associated with prolonged progression-free survival after ICB treatment. In a ROC analysis, B cells (AUC = 0.77, p = .0055) and CD45+ cells (AUC = 0.73, p = .019) predicted benefit of ICB, which was not the case for PD-L1 mRNA (AUC = 0.54, p = .72) and PD-L1 protein expression (AUC = 0.68, p = .082). Clustering of 79 candidate predictive markers identified among 770 investigated genes revealed two distinct predictive clusters which included cytotoxic cell or macrophage markers, respectively. In summary, targeted gene expression profiling was feasible using routine diagnostics biopsies. This study proposes B cells and total TILs as complementary predictors of ICB benefit in NSCLC. While further preferably prospective validation is required, gene expression profiling could be integrated in the routine diagnostic work-up complementing existing NGS protocols.
KEYWORDS: Lung adenocarcinoma, immune checkpoint blockade, mRNA expression, B cells, tumor-infiltrating lymphocytes, response prediction
Background
Immune checkpoint inhibitors targeting the programmed cell death protein 1 (PD-1) – programmed death-ligand 1 (PD-L1) axis1,2 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)3,4 have dramatically improved the treatment options for a growing number of tumor entities including advanced non-small cell lung cancer (NSCLC). However, only some patients respond to immune checkpoint blockade (ICB) and several mechanisms of de novo or acquired resistance are progressively being described,5–8 so that durable disease control is achieved only in a minority of ICB-treated patients.
Hence, predictive biomarkers are urgently needed for identification of the most likely responding patients and to offer better suited treatment alternatives for the others. While numerous biomarkers were evaluated, PD-L1 protein expression analyzed by immunohistochemistry (IHC)9,10 and microsatellite instability (MSI)/mismatch repair deficiency are the only ones approved so far.11 However, both of them are far away from being perfect: PD-L1 IHC is limited in both sensitivity and specificity – as well as its heterogeneity, while MSI will identify only a very restricted subset of all ICB responders. In addition, in retrospective molecular analyses complementing clinical trials, tumor mutational burden (TMB) showed promising results as a predictive marker, 1–4,12 but so far this has not been validated in prospective trials. Furthermore, we and others delineated biological, technological, and bioinformatical parameters impairing correct measurement of TMB in a clinical setting.13–15 Hence, the suitability of TMB as a predictive marker is currently discussed controversially and remains practically challenging.
ICB-related mRNA expression profiling was advocated in several studies as a promising approach enabling comprehensive interrogation of the ICB effector compartment.16,17 The characterization of tumor-infiltrating immune cell populations18 plays a key role in understanding potential antitumor effects and the response mediated by the tumor microenvironment (TME). In this context, gene signatures that were described to correlate with the occurrence of specific immune cell populations19 are of particular interest from a clinical perspective. In a growing number of clinical laboratories, mRNA analysis is performed in parallel to DNA analysis as part of the routine NGS diagnostic work-up of lung adenocarcinoma for the detection of oncogenic gene fusions.20 Gene expression based immune profiling could be easily integrated in this workflow without the need for additional tissue and would represent an excellent opportunity to incorporate comprehensive TME characterization into the clinical work-up.
In the present study, we demonstrate that targeted mRNA expression profiling in routine diagnostic biopsies of metastatic lung adenocarcinoma is feasible. We confirm the hypothesis that gene signatures related to specific immune cell types – in particular B cell and macrophages – can contribute to the prediction of long-term ICB benefit.
Material and methods
Patient and public involvement
Neither patients nor the public were involved in the design of the study. The results of the study will be disseminated in public patient Information events.
Study cohort
The study cohort included 43 patients diagnosed with stage IV lung adenocarcinoma according to the 8th TNM staging and the current WHO classification and treated with ICB at the Thoraxklinik of Heidelberg University Hospital (Supplement 1 and 2). To analyze the difference of the TME in cases with a significantly divergent clinical course, the study cohort was intentionally enriched for patients showing durable response (of at least 12 months) and showing early progress (within 2 months). Only patients with lung biopsies taken prior to initiation of ICB available and suitable for mRNA expression analysis were included. The majority of patients received ICB as monotherapy (n = 31, 72%), while the remaining patients received ICB together with a platinum doublet (n = 12, 28%). Patients were closely followed up for response with chest-abdomen CT and brain MRI at baseline and every 6–8 weeks (for patients without brain involvement, brain MRI was repeated every 6 months). Clinical data were systematically collected from the patients’ records, and the date of tumor progression for each case was verified by review of radiological images by the investigators. All patients provided signed informed consent for inclusion of their clinical data and specimens in our Lung Biobank and use in research projects, according to the recommendation of the Heidelberg University ethics committee (S-270/2001). The retrospective study presented here was approved separately by the ethics committee of Heidelberg University (S-145/2017).
TCGA lung adenocarcinoma cohort
Gene expression and clinical data of the TCGA were downloaded from the PanCanAtlas webpage at Genomic Data Commons repository (https://gdc.cancer.gov/about-data/publications/pancanatlas). Samples diagnosed as lung adenocarcinoma (LUAD) and of the type “primary solid tumor” (code: 01) were included in the analysis. With the exception of two recurrent tumors, all LUAD tumors analyzed in the TCGA could be included (515/517 = 99.6%).
Nucleic acid extraction and semiconductor sequencing
Starting with formalin-fixed and paraffin-embedded (FFPE) biopsy samples, tumor tissue was macrodissected to achieve a histological tumor cell content of at least 30%. DNA and RNA extraction, library preparation, and semiconductor sequencing were performed as described previously.21 Data analysis was performed using the Ion Torrent Suite Software (Thermo Fisher Scientific Inc., Waltham, MA). Variant calling was performed with the variant caller plugin (version 5.0 up to 5.6) within the Torrent Suite Software and the IonReporter package using a corresponding bed-file containing the coordinates of the amplified regions.22
Targeted mRNA expression profiling
RNA extracts passing the following steps of quality control were considered as suitable for gene expression analysis: RNA concentration of at least 10 ng/µl, sufficient RNA purity with a A260/A280 in the range 1.7–2.3 and sufficient RNA integrity with at least 90% of the fragments longer than 100 nucleotides. Targeted mRNA expression profiling was performed using the NanoString nCounter gene expression platform (NanoString Technologies, Seattle, WA) using a 770-gene panel (PanCancer Human IO360 Panel) focused on the complex interplay between the tumor, the tumor microenvironment, and the immune response in cancer. Per sample, 100 ng of total RNA in a final volume of 5 μl were mixed with a 3′ biotinylated capture probe and a 5′ reporter probe tagged with a fluorescent barcode from the PanCancer IO360 gene expression code set. Probes and target transcripts were hybridized at 65°C for 18 hours according to the manufacturer’s recommendations. Hybridized samples were run on the NanoString nCounter preparation station using the high-sensitivity protocol, in which excess capture and reporter probes are removed and transcript-specific complexes are immobilized on a streptavidin-coated cartridge. The samples were scanned at maximum resolution on the nCounter Digital Analyzer.
Immunohistochemistry
For immunohistochemical staining of PD-L1, CD3, CD30, and CD21, 3 μm thick paraffin sections were prepared. Samples used for IHC and mRNA were consecutive sections from the same FFPE block minimizing a possible inteference with tumor heterogeneity. Deparaffinization and tissue staining were performed using a Benchmark Ultra IHC Staining module according to standard protocols (Ventana PD-L1 assay, clone SP263; CONFIRM anti-CD3 Primary Antibody, clone 2GV6; CONFIRM anti-CD20 Primary Antibody, clone L26; Rabbit Monoclonal Primary Antibody anti CD21, clone EP3093; all four Roche, Mannheim, Germany). Hematoxylin was used for counterstaining of cell nuclei. IHC stainings were evaluated by a specialist in pathology and scoring of PD-L1 was performed according to standardized criteria.9 CD20 expression was evaluated semiquantitatively and the number of tertiary lymphoid structures (TLS) was reported by a pathologist.
Data processing
Statistical analysis and graphics generation were performed using the programming language R. Expression data were preprocessed by background subtraction and subsequent sample normalization. For sample normalization, the 20 panel genes with the lowest coefficient of variation and an expression level of at least 100 in The Cancer Genome Atlas (TCGA) lung adenocarcinoma (LUAD) dataset were used as housekeepers (AKT1, API5, DNAJC14, EIF2B4, ELA, ERCC3, GLUD1, HDAC3, HMGB1, IFNAR1, MLH1, OAZ1, PUM1, RIPK1, SF3A1, STAT3, TBC1D10B, TLK2, TMUB2, and UBB). The gene expression profile of each sample was scaled by the median expression level of these housekeeping genes. Gene expression data were log2-transformed prior to statistical analysis.
Absolute and relative scores of immune cell infiltration
The abundance of 14 immune cell populations (B cells, CD45+ cells, CD56dim, CD8+ T cells, cytotoxic cells, dendritic cells, exhausted CD8+ T cells, macrophages, mast cells, neutrophils, NK cells, T cells, Th1 cells, and Treg cells) was estimated from the mRNA expression of marker genes as described before.19 Fifty-four of the 60 genes described there (90%) were covered by the 770-gene panel. RNA markers for other immune cell types were not available or could not be validated.19 The expression of the marker genes in the study cohort was clustered and visualized in a heatmap (Supplement 3). The B cell expression signature included the eight marker genes BLK, CD19, FCRL2, MA4A1, TNFRSF17, TCL1A, SPIB, and PNOC, the macrophage gene expression signature included the four marker genes CD68, CD84, CD163, and MS4A4A.19 A total score of tumor-infiltrating lymphocytes (total TILs) was calculated as the average of the scores of 11 immune cell populations (all populations, but excluding dendritic cells, mast cells, and Treg cells). In addition to the absolute scores of the 14 cell populations, relative scores of the 11 cell populations with respect to the total level of immune activity were calculated as residuals with respect to a linear regression fit against the total TIL score.
Heatmaps and hierarchical clustering
For heatmap displays, each of the cell populations (or genes) was centered (but not scaled) with respect to the mean abundance (or mean mRNA expression) over the samples. Abundances (or expression levels) above the mean appear in red, abundances below the mean in green. Hierarchical clustering was performed using Pearson correlations as similarity measure and the average linkage as measure of distances between clusters.
Immune gene signatures
The T cell-inflamed gene expression profile (GEP) was calculated as average of the expression of the 16 genes of the 18 signature genes that were interrogated by the NanoString assay (IL2RG, CXCR6, CD3D, CD2, TAGAP, HLA-DRA, CCL5, NKG7, CD3E, HLA-E, GZMB, GZMK, CXCL13, CXCL10, IDO1, LAG3, STAT1, CIITA) as described.23 The T effector and IFNγ signature24 were calculated as average of the expression of the eight signature genes (CD8A, GZMA, GZMB, IFNG, EOMES, CXCL9, CXCL10, and TBX21), cytolytic activity25 was calculated as average of granzyme A (GZMA) and perforin (PRF1) expression.
Statistical analysis
The suitability of cell populations as prognostic markers was analyzed using Cox regression with respect to progression-free survival (PFS) after ICB initiation. The suitability of cell populations as predictive markers was analyzed using logistic regression based on ICB benefit (long term response vs. rapid progress). Markers were considered as continuous variables and both analyses were performed without cutpoints. Hazard ratios (HRs) and odds ratios (ORs) per doubling of marker levels were reported including 95% confidence intervals (CIs). Multiple testing was accounted for by correcting p-values using the Benjamini–Hochberg method. Candidate markers were further analyzed by receiver operator characteristic (ROC) curves including significance assessment with the unpaired Wilcoxon test.
Correlation of the mRNA marker signature for B cells with the level of CD20-positive immune cell infiltrates was assessed using the Jonckheere-Terpstra test for trends as implemented in the R package clinfun. Correlation the of mRNA marker signature for B cells with the presence of TLS was assessed using the Wilcoxon test.
A list of candidate marker genes was compiled using the threshold p < .05 for uncorrected p-values.
Results
The retrospective study cohort included 43 metastatic lung adenocarcinoma patients that were treated with immune checkpoint inhibitors either as monotherapy or combination therapy at the Heidelberg University Hospital, most of them between 2016 and 2020 (Figure 1). There were 16 long-term responders (LTR; durable response of 12 months or more), 21 rapid progressors (RP; disease progression within 2 months) and 6 patients with an intermediate duration of response (IR). The study cohort was intentionally enriched for LTR and RP by inclusion of all patients that met the predefined inclusion criteria (see Material and Methods). Gene expression profiles including 770 genes were generated from lung biopsies using the NanoString nCounter technology (Supplement 4). The focus of the gene expression assay on immune biology was reflected by annotation of 430 of the investigated genes (55.8%) to the Gene Ontology (GO) category “immune system process” (GO: 0009987).
Figure 1.
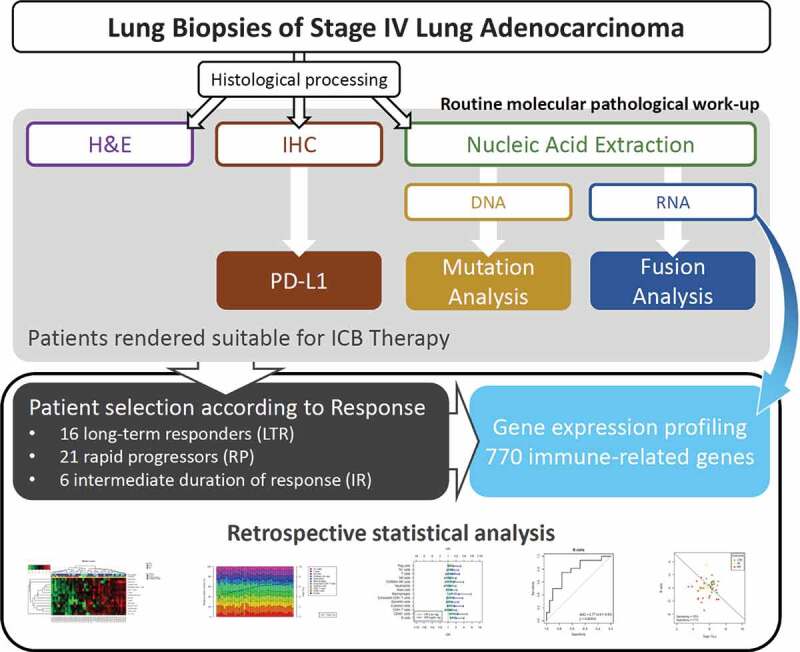
Study cohort, routine molecular pathology work-up and mRNA expression profiling of immune-related genes
Absolute and relative quantification of immune cell populations
The abundance of 14 specific immune cell populations (“absolute scores”) was estimated from the mRNA expression profiles as recently described.19 Hierarchical clustering separated samples into immunological “hot” tumors versus immunological “cold” tumors (Figure 2a). Response to ICB was significantly different (p = .046) with a higher percentage of LTR within the group of “hot” tumors (52%) compared to “cold” tumors (20%). Pairwise correlations between the different immune cell types were either significantly positive or non-significant. Highly positive pairwise correlations were detected between cytotoxic cells, T cells, CD8+ T cells and exhausted CD8+ T cells (all R > 0.71) as well as between macrophages and CD45+ cells (R = 0.75).
Figure 2.
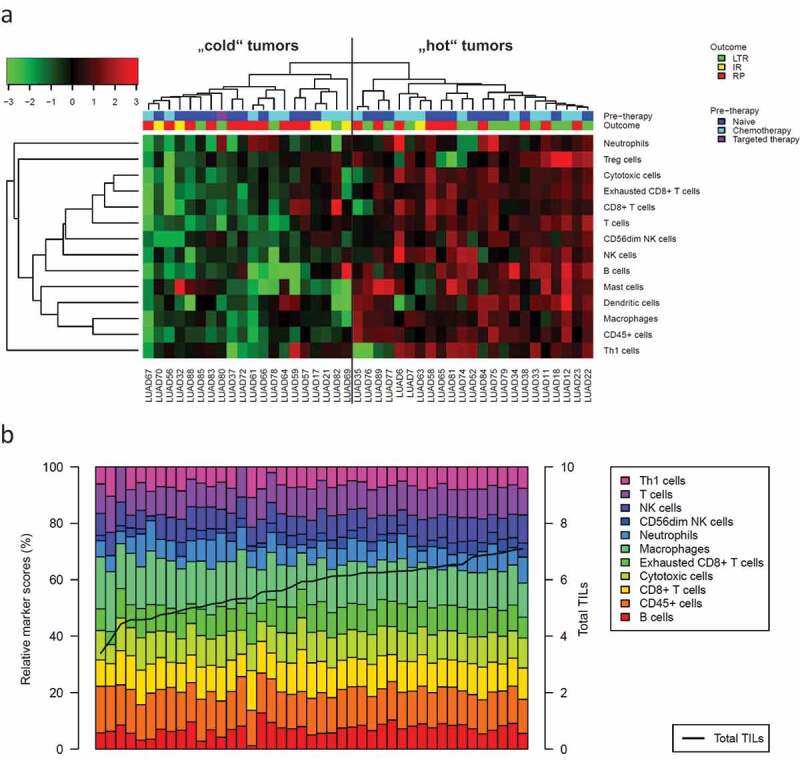
Composition of the tumor immune microenvironment in the study cohort of metastatic adenocarcinoma
The abundance of 14 specific immune cell populations and total TILs was estimated from mRNA expression profiles. (a) Hierarchical clustering separated n = 23 immunological “hot” tumors from n = 20 immunological “cold” tumors. Response to ICB (LTR/IR/RP) was significantly different in these clusters (p = .046) with a higher percentage of LTR in the cluster of “hot” tumors (52%) compared to the cluster of “cold” tumors (20%). (b) Barplot showing the contribution of specific immune cell types (in %, bars) to total TILs (logarithmic score, line). LTR = long-term responder, IR = intermediate responder, RP = rapid progressor.
In a second analysis, the TME was characterized by a total TIL score plus relative immune cell scores instead of absolute immune cell scores. Following a concept similar to “partial correlations,” we calculated relative scores of each immune cell population as residual in the linear regression of the absolute scores against total TILs. Figure 2b shows the contribution of the different immune cell types to total TILs in each of the tumors.
Immune cell populations as predictive markers for response to immunotherapy
Immune cell populations were correlated with PFS after ICB using univariate Cox proportional hazard models and with ICB response using univariate logistic regression. In general, high absolute immune scores were associated with a lower risk of progression (Figure 3a). Controlling the false discovery rate (FDR < 10%) two of the absolute scores showed a significant association: B cells with HR = 0.66 (0.52–0.84, p = .00074) and CD45+ cells with HR = 0.61 (0.42–0.89, p = .01). Furthermore, high absolute scores of B cells, CD45+ cells and macrophages were associated with better ICB response, OR = 2.1 (1.2–4.0, p = .012), OR = 2.3 (1.2–5.4, p = .029) and OR = 2.6 (1.1–7.5, p = .046), respectively.
Figure 3.
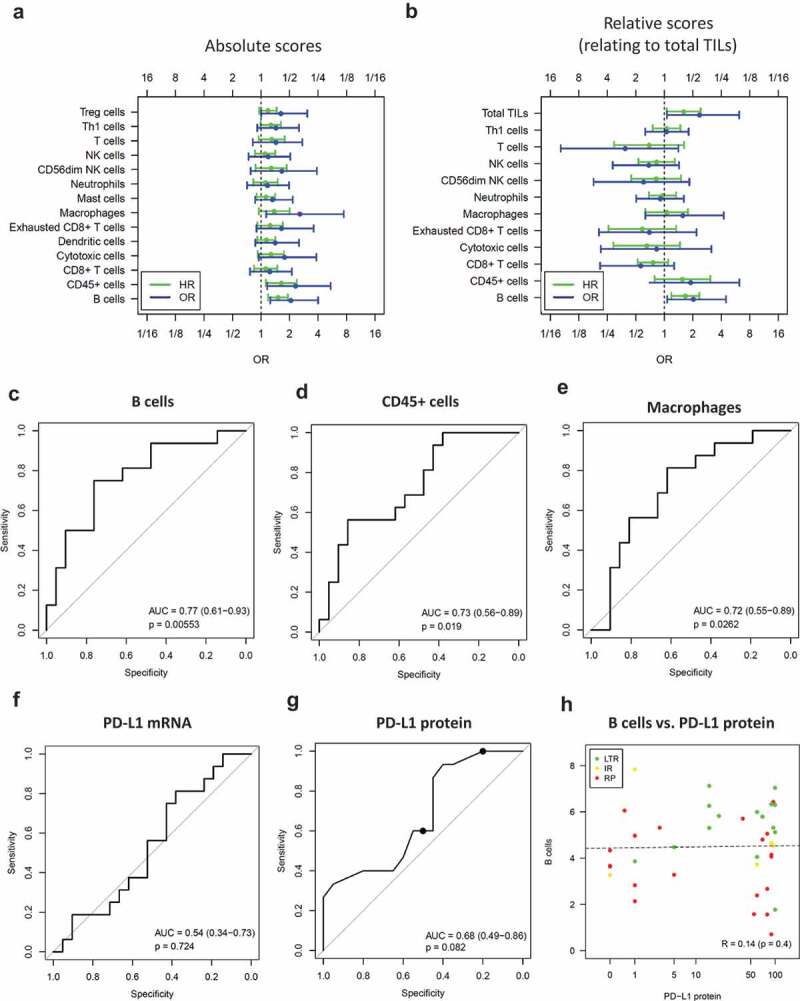
Immune cell scores as positive predictive markers for ICB benefit
Absolute quantification (a,c-e,h) and relative quantification of immune cell populations (b) from mRNA expression data. (a,b) Odds ratios (OR) of long-term responders (LTR, n = 16) versus rapid progressors (RP, n = 21) and hazard ratios (HR) of PFS (n = 43). ORs and HRs relate to a doubling of the immune cell abundance. (c-e) ROC analysis of B cells, CD45+ cells and macrophages. (f) ROC analysis of PD-L1 mRNA. (g) ROC analysis of PD-L1 protein including cutpoints relating to positivity of 50% and 1% of tumor cells (black dots). (h) Correlation analysis of B cells and PD-L1 protein. An increase of one on the y-axis corresponds to doubling of the B cell abundance. HR = hazard ratio per doubling of abundance, OR = odds ratio per doubling of abundance.
Next, we correlated relative immune cell and total TIL scores with PFS and response to therapy (Figure 3b). High relative scores of B cells and high total TILs were associated with significantly longer PFS with HR = 0.6 (0.43–0.84, p = .0033) and HR = 0.62 (0.41–0.94, p = .025), respectively. Furthermore, high relative scores of B cells were associated with significantly better response with OR = 2.0 (1.1–4.5, p = .048), while high total TILs were associated with better response without reaching significance with OR = 2.4 (1.1–6.2, p = .052).
Up-regulation of the B cell signature was associated with favorable clinical outcome after ICB treatment, an observation that could be either therapy-independent or ICB-specific. To differentiate between the two possibilities, we additionally investigated immune cell populations as prognostic markers in a large cohort of conventionally treated lung adenocarcinoma patients (TCGA LUAD, n = 506, Supplement 5). In the TCGA cohort, high absolute scores of B cells and high relative scores were associated with only slightly better PFS, HR = 0.91 (0.85–0.98, p = .0098) and HR = 0.90 (0.81–1.01, p = .067), respectively. By contrast, the association in ICB-treated patients was much stronger, supporting a role of B cells as predictive marker specifically linked to ICB treatment.
ROC curve analysis and comparison with established gene expression signatures and PD-L1 expression
Absolute scores of B cells, CD45+ cells and macrophages were significantly predictive with AUC = 0.77 (95% CI 0.61–0.93, p = .0055), AUC = 0.73 (0.56–0.89, p = .019) and AUC = 0.72 (0.55–0.89, p = .026), respectively (Figure 3c-e). Immune gene signatures such as the T cell-inflamed GEP23 and T-effector/INFγ signature24 were developed to predict the response to ICB but neither of them nor cytolytic activity showed a significant association with long-term ICB benefit in the study cohort (Supplement 6).
PD-L1 protein expression evaluated by IHC represents an approved companion diagnostic test for pembrolizumab monotherapy in NSCLC.10 However, neither PD-L1 mRNA nor PD-L1 protein expression predicted ICB response in the study cohort (Figure 3f-g). In a stratified analysis for therapy type, PD-L1 protein expression was significantly associated with response in the subgroup of patients receiving combination therapy, but not in the subgroup of patients receiving monotherapy (Supplement 7). The latter negative result can potentially be explaied by a selection bias concerning PD-L1 expression and a very low number of PD-L1-negative tumors in the monotherapy subcohort as a consequence: Of the 30 tumors treated with monotherapy and with PD-L1 staining available, 22 (73%) had 50% or more PD-L1-positive tumors cells, while 29 (97%) had 1% or more PD-L1-positive tumor cells. Interestingly, correlation between B cells and PD-L1 protein was absent in the study cohort (Figure 3h) supporting a role of B cells as independent biomarker for the prediction of immune therapy response complementary to PD-L1.
Validation of B-cell infiltration using immunohistochemistry
To validate the B cell infiltrates detected by the gene expression analysis, all cases from the study cohort with sufficient tissue available (n = 26) were stained for CD20 (Figure 4). Both, the presence of TLS as well as levels of CD20-positive cells correlated significantly with the mRNA marker of B cell infiltration (p = 9.6e-05 and p = .0013). Two example cases (one LTR and one RP) with very high and very low levels of the mRNA marker were additional stained for CD3 and CD21. The patterns of immune cell populations observed in the first case included CD21-positive networks of follicular dendritic cells and were indicative of TLS, while CD20-positive B cells were low in the second case (Supplement 8).
Figure 4.
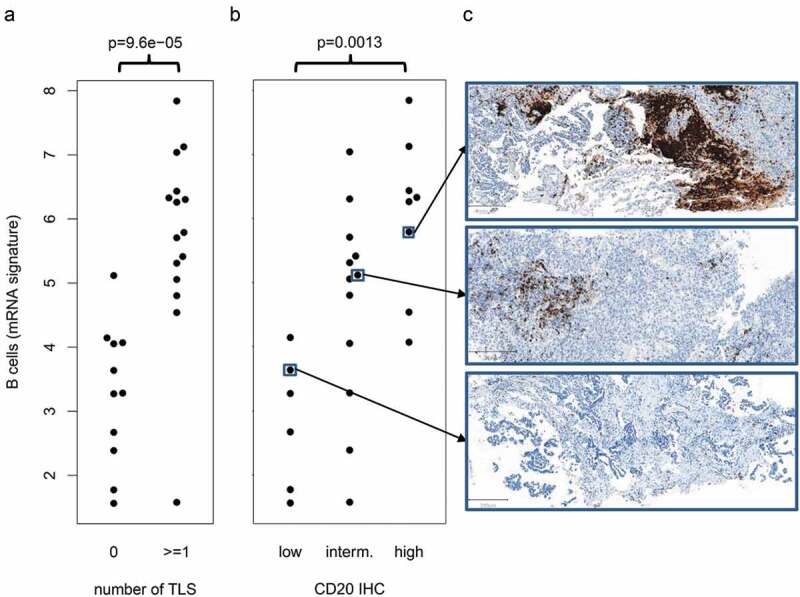
Validation of the B cell mRNA signature by CD20 IHC
(a) Significant correlation of the mRNA signature with the presence of tertiary lymphoid structures (TLS). (b) Significant correlation of the mRNA signature with the abundance of CD20-positive immune cells. (c) Example cases showing high, intermediate and low infiltration of CD20-positive B cells.
Bivariate analysis of specific immune cell types and total TILs
Relative B cell scores and total TILs were combined to separate LTR from RP. To this end both cell scores were included as independent variables in a bivariate logistic regression model to predict the response status (LTR or RP). Separation by the model was significant (p = .012) with OR = 2.0 (1.0–4.7) for B cells and OR = 2.3 (1.0–6.3) for total TILs. Also, prediction of PFS was significant (p = .0031) in bivariate Cox regression with HR = 0.63 (0.44–0.89) for B cells and HR = 0.68 (0.44–1) for total TILs. The performance of the model in the study cohort (which also served as training set) is shown in Figure 5a,b. Prediction sensitivity was 75% at a specificity of 71% for the cutpoint suggested by the logistic model. Varying of the cutpoint (corresponding to a parallel shift of the decision horizon in Figure 4a) resulted in an AUC of 0.78 (0.62–0.93) in an ROC analysis. A model combining total TILs and relative macrophagescores instead of B cells performed slightly inferior (Figure 5c,d). While the results of the bivariate analyses support the feasibility of prediction models including a specific immune cell marker (such as B cells or macrophages) together with a marker of overall immune reaction (such as total TILs), we are aware that the sample size in the study cohort is too small for fine tuning of a cutpoint and for valid estimation of prediction sensitivity and specificity.
Figure 5.
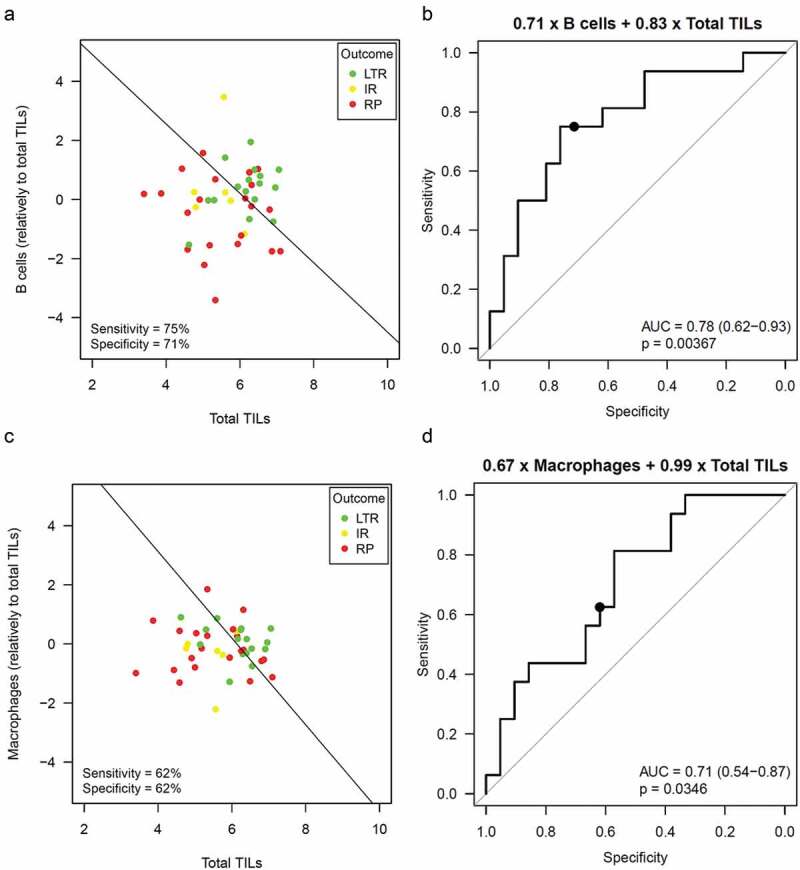
Prediction of ICB benefit by combining B cells or macrophages with total TILs
Bivariate logistic regression to separate LTR from RP combining total TILs and the relative B cell score or the relative macrophage score. (a,c) Separating LTR from RP using the logistic regression model. An increase of one on the x-axis or y-axis corresponds to a double abundance of the corresponding cell population. (b,d) ROC curve showing the performance of the logistic model when varying the cutpoint.
Multivariate analysis of B cells
Absolute B cell scores were analyzed in a multivariate analysis including sex, age, prior therapy and therapy type (Table 1). B cells remained a significant prognostic factor in both multivariate Cox regression and multivariate logistic regression, HR = 0.64 (0.47–0.87, p = .0046) and OR = 2.9 (1.3–5.2, p = .016). Also, relative B cells scores remained a prognostic factor in both multivariate Cox regression and multivariate logistic regression (data not shown).
Table 1.
Multivariate analysis of absolute B cell scores. Cox regression was used to analyze PFS after immunotherapy. Logistic regression to analyze response to immunotherapy (LTR vs. RP)
| Cox regression |
Logistic regression |
|||
|---|---|---|---|---|
| Variable | HR | p | OR | p |
| B cells | 0.64 (0.47–0.87) | 0.0046 | 2.3 (1.3–5.2) | 0.016 |
| Sex (female vs. male) | 2.2 (0.91–5.1) | 0.082 | 0.18 (0.019–1.2) | 0.092 |
| Age (per year) | 0.95 (0.9–1) | 0.033 | 1.1 (1–1.3) | 0.029 |
| Prior therapy (treated vs. naive) | 0.34 (0.12–0.98) | 0.045 | 4.6 (0.61–54) | 0.17 |
| Therapy type (combination vs. mono) | 0.9 (0.36–2.2) | 0.81 | 0.35 (0.027–3) | 0.37 |
Potentially confounding factors
The association of B cells and total TILs with ICB benefit was investigated in a subgroup analysis (Figure 6). Both markers were predictive in patients treated by ICB monotherapy (OR = 2.3, p = .017 and OR = 3.2, p = .042), while no significant association was observed in patients treated with combination therapy (OR = 1.5, p = .52 and OR = 0.9, p = .9). Neither ICB benefit (Supplement 9A) nor the abundance of specific immune cell types (data not shown) was associated with prior therapies. Furthermore, ICB benefit was not associated with therapy type (Supplement 9B). Status of driver mutations had been determined during routine molecular diagnostics using targeted NGS: Twenty-three tumors of the study cohort had activating KRAS mutations, 19 had deleterious or probably deleterious TP53 mutations, 9 had non-synonymous KEAP1 mutations. Neither ICB benefit (Supplement 9C-E) nor the abundance of specific immune cell types (data not shown) was associated with mutation status. Thus, neither prior therapies nor driver mutation status interfered with the association between ICB benefit and the immune cell markers.
Figure 6.
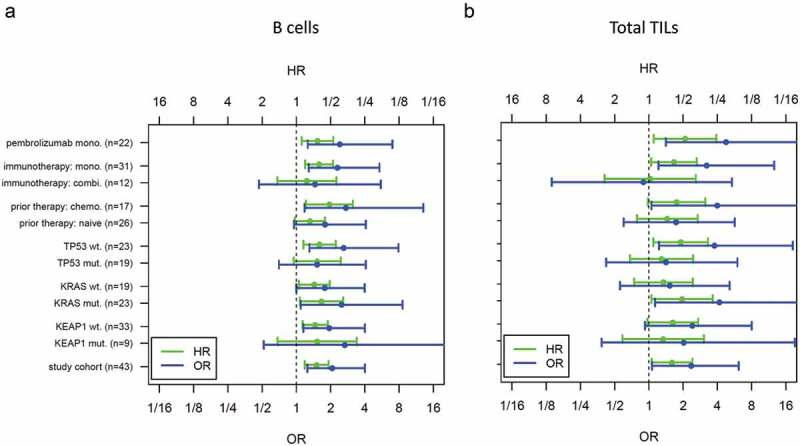
Subgroup analysis of B cells (a) and total TILs (b)
Genes associated with ICB benefit
We analyzed the suitability of the 770 genes as predictive markers using ROC curves and significance assessment with the unpaired Wilcoxon test. A list of 79 significant (p < .05) candidate markers emerged and was analyzed in a heatmap (Figure 7, AUCs between 0.69 and 0.81). The majority of these genes (n = 76) correlated positively with ICB benefit, while three genes (SOX2, HDAC4, and G6PD) correlated negatively. The gene list partitioned the tumors into three clusters, T1 including 17% LTR, T2a including 25% LRT and T2b including 65% LTR. The expression patterns in these tumor clusters were different in the two gene clusters G1 and G2: The cluster T2b with the highest proportion of LTR was characterized by high expression of cytotoxic cell markers (cluster G1) and macrophage markers (cluster G2). The cluster T2a with a moderate number of LTR was characterized by high expression of cytotoxic cell markers, while the cluster T1 with the lowest number of LTR was characterized by low expression of cytotoxic cell and macrophage markers. The highest prediction accuracy was reached for the genes IGF2R (AUC = 0.8, 95% CI 0.65–0.95), IL7R (AUC = 0.77, 95% CI 0.61–0.92) and HCK (AUC = 0.76, 95% CI 0.6–0.92), see Supplement 10. The confidence intervals of the genes included the AUC values for B cells, macrophages, and total TILs. Thus, there was no evidence that a gene-based stratification could outperform an immune cell-type-based stratification.
Figure 7.
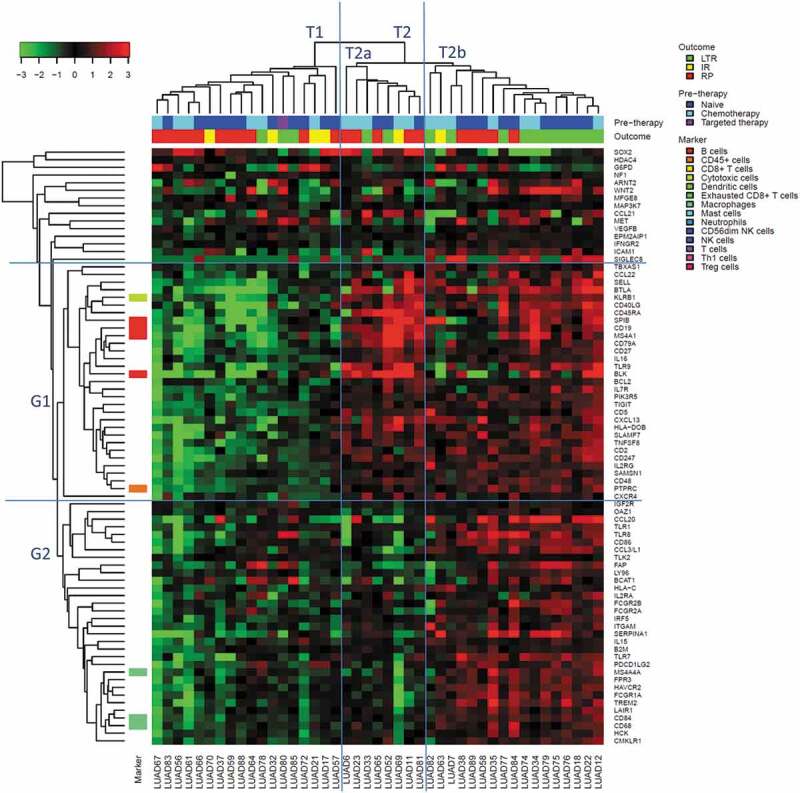
Heatmap of the top 79 genes predicting ICB benefit in the study cohort (16 LTR vs. 21 RP)
Sample clusters (T1, T2, T2a and T2b) and gene clusters (G1 and G2).
Discussion
Investigating the immune TME26 of NSCLC from LTR and RP we observed that the total level of immune cell infiltration25,27,28 defined a group of immunological “hot” tumors and was associated with a higher probability of response to ICB. Cytotoxic cell including CD8+ T cells and CD8+ exhausted T cells contributed with about 30% to total TILs (Figure 2b), in line with previous reports which assign a major role to these cell types in biological concepts29 and clinical response prediction.30 Additionally, we observed that macrophages and specifically the abundance of B cells were associated with improved PFS of patients. The identification of macrophages fits well into the current concept that macrophages appear to mediate T cell recruitment and ICB response.26,31–33 B cells remained an independent predictor of PFS when analyzed in a bivariate model together with total TILs and when analyzed in a multivariate model together with key clinico-pathological characteristics of the patients.
While B cells are known to be associated with autoimmune events during ICB treatment, 34 they were only very recently reported to play a role in ICB response prediction and data on NSCLC patients have not yet been published until now. After antigen exposure, B cells can be broadly subdivided into those that contribute to antibody-mediated immune response and those that regulate immune response similar to the concept of regulatory T cells. These regulatory B cells (Breg)35 characterized by IL10 production were shown to interfere with immune responses in several diseases including cancer.36,37 IGF2 whose receptor IGF2R was upregulated and predictive in our study, appears to be one of the factors that can influence Breg function.38 By contrast, there are also data attributing an antigen-presenting39 and pro-inflammatory role to B cells.35,40,41 This is in line with a recent report of Petitprez and coauthors42 that B cells, partially located in TLS, were strong positive predictors of ICB response in sarcoma patients receiving pembrolizumab in a phase 2 clinical trial. Similar observations were made by Griss et al.43 and Cabrita et al.44 who investigated the role of B cells in melanoma. Griss and coauthors identified a specific B cell population with similarities to Bregs, which they termed TIPB and which appears to recruit CD8-positive T cells in melanomas. Their exploratory analysis in a small cohort of patients who received anti-CD20 antibodies showed that reduction of B cells in melanoma was associated with attenuated immune response and reduced TLS. Cabrita and coauthors investigating samples of metastatic melanoma noted that the spatial distribution of co-occurring CD8-positive T cells and CD20-positive B cells resembled TLS and was associated with improved PFS. Of note, the majority of their samples in this study were derived from lymph node metastases which might influence results. Nevertheless, consistent with our data, they also observed B cells and a TLS-related signature in non-lymph node samples.
Up-regulation of the B cell signature was associated with a higher probability of long-term response and prolonged progression-free survival after ICB treatment. Since two previous studies identified a prognostic impact of B-cells and TLS in small cohorts of lung cancer patients of various disease stages, 45,46 we employed the TCGA dataset of lung adenocarcinomas for an independent analysis. In the TCGA LUAD cohort, which was sequenced at times when ICB treatment was still in development and not standard of care, the influence of B cells on PFS was weak (HR per doubling of score = 0.91). By contrast, the influence of B cells on PFS after ICB treatment was much stronger (HR per doubling of score = 0.66) supporting a possible role of B cells as predictive biomarker for ICB efficacy beyond a prognostic value only. However, differences in the clinico-pathological characteristics – advanced tumors in the study cohort, but surgically resected, mostly early-stage tumors in the TCGA cohort – represent a limitation for a comparison. Thus, further investigation of biomarkers for B cell infiltration, optimally in the setting of randomized clinical trials and including an interaction analysis, is recommended.
A limitation of our study is its single-center character and the limited number of samples available for analysis which was due to careful selection of patients and the associated samples. Analyzed cases had to adhere to clinical and technical inclusion criteria (see Material and Methods) and we did not include samples with lymph node metastases as they may influence data derived from profiling of the tumor immune microenvironment. In addition, a careful analysis of potential confounding factors was performed. We did not note an influence of any mutated genes analyzed in our sample set including mutated KEAP1, 47 a genomic marker48 that was reported to influence ICB treatment outcome. B cells remained a significant prognostic factor in a multivariate analysis including sex, age, prior therapy, and therapy type.
In summary, our data suggest that, in addition to the molecular profile of the tumor cell compartment, 48 analysis of the effector compartment, i.e. the tumor immune microenvironment, is important to identify NSCLC patients who likely benefit from ICB treatment. According to our results, B cells appear to influence clinical outcome and further studies are warranted to validate these findings. Since our study used routine diagnostic FFPE material from lung biopsies and did not require a specific clinical and molecular work-up beyond already established workflows, implementation in routine diagnostic procedures would be feasible if confirmed clinically relevant in the future.
Supplementary Material
Funding Statement
This work was supported by the German Center for Lung Research (Deutsches Zentrum für Lungenforschung, DZL) and by the German Cancer Consortium (Deutsches Konsortium für Translationale Krebsforschung, DKTK).
Abbreviations
- AUC
area under the curve
- CI
confidence interval
- FFPE
formalin-fixed and paraffin-embedded
- GEP
gene expression profile
- HR
hazard ratio
- ICB
immune checkpoint blockade
- IR
intermediate responder
- LTR
long-term responder
- LUAD
the lung adenocarcinoma data set of TCGA
- MSI
microsatellite instability
- NSCLC
non small cell lung cancer
- OR
odds ratio
- ROC
receiver operator characteristics
- RP
rapid progressor
- TCGA
the cancer genome atlas
- TILs
tumor infiltrating lymphocytes
- TLS
tertiary lymphoid structure
- TME
tumor microenvironment
Authors’ contributions
Jan Budczies: Conceptualization, Methodology, Software, Formal analysis, Writing – Original Draft, Writing – Review & Editing, Visualization, Supervision, Project administration Martina Kirchner: Investigation, Writing – Original Draft, Writing – Review & Editing Klaus Kluck: Software, Formal analysis, Writing – Review & Editing, Visualization Daniel Kazdal: Writing – Original Draft, Writing – Review & Editing, Visualization Julia Glade: Investigation, Writing – Review & Editing Michael Allgäuer: Writing – Original Draft, Writing – Review & Editing Mark Kriegsmann: Investigation, Writing – Review & Editing Claus-Peter Heußel: Investigation, Writing – Review & Editing Felix J. Herth: Resources, Writing – Review & Editing Hauke Winter: Resources, Writing – Review & Editing Michael Meister: Resources, Writing – Review & Editing Thomas Muley: Resources, Writing – Review & Editing Stefan Fröhling: Conceptualization, Writing – Review & Editing Solange Peters: Conceptualization, Writing – Review & Editing Barbara Seliger: Conceptualization, Writing – Review & Editing Peter Schirmacher: Conceptualization, Writing – Review & Editing Michael Thomas: Conceptualization, Resources, Writing – Review & Editing, Funding acquisition Petros Christopoulos: Conceptualization, Methodology, Investigation, Resources, Writing – Review & Editing, Supervision, Funding acquisition Albrecht Stenzinger: Conceptualization, Methodology, Writing – Original Draft, Writing – Review & Editing, Supervision, Project administration, Funding acquisition.
Disclosure of interests
Dr. Budczies has nothing to disclose.
Dr. Kirchner reports personal fees from QuIP, outside the submitted work.
Mr. Kluck has nothing to disclose.
Dr. Kazdal reports personal fees from AstraZeneca, personal fees from Bristol-Myers Squibb GmbH, personal fees from Pfizer Pharma GmbH, outside the submitted work.
Dr. Glade has nothing to disclose.
Dr. Allgäuer has nothing to disclose.
Dr. Kriegsmann has nothing to disclose.
Dr. Heußel reports personal fees from Schering-Plough, personal fees from Pfizer, personal fees from Basilea, personal fees from Boehringer Ingelheim, personal fees from Novartis, personal fees from Roche, personal fees from Astellas, personal fees from Gilead, personal fees from MSD, personal fees from Lilly, personal fees from Intermune, personal fees from Fresenius, personal fees from Essex, personal fees from AstraZeneca, personal fees from Bracco, personal fees from MEDA Pharma, personal fees from Chiesi, personal fees from Siemens, personal fees from Covidien, personal fees from Pierre Fabre, personal fees from Grifols, personal fees from Bayer, personal fees from Siemens, personal fees from Pfizer, personal fees from MeVis, personal fees from Boehringer Ingelheim, personal fees from German Cancer for Lung Research, outside the submitted work.
Dr. Herth has nothing to disclose.
Dr. Winter has nothing to disclose.
Dr. Meister reports grants from German Center for Lung Research, during the conduct of the study.
Dr. Muley reports grants, personal fees and non-financial support from Roche Diagnostics, grants from Chugai, grants from Hummingbird Diagnostics GmbH, outside the submitted work; In addition, Dr. Muley has patents Roche Diagnostics pending.
Dr. Fröhling reports personal fees from Amgen, grants from AstraZeneca, personal fees from Bayer, personal fees from Eli Lilly, grants from Pfizer, grants and personal fees from PharmaMar, grants and personal fees from Roche, outside the submitted work.
Dr. Peters has received education grants, provided consultation, attended advisory boards, and/or provided lectures for: Abbvie, Amgen, AstraZeneca, Bayer, Biocartis, Bioinvent, Blueprint Medicines, Boehringer-Ingelheim, Bristol-Myers Squibb, Clovis, Daiichi Sankyo, Debiopharm, Eli Lilly, F. Hoffmann-La Roche, Foundation Medicine, Illumina, Janssen, Merck Sharp and Dohme, Merck Serono, Merrimack, Novartis, Pharma Mar, Pfizer, Regeneron, Sanofi, Seattle Genetics, Takeda and Vaccibody, from whom she has received honoraria (all fees to institution).
Dr. Seliger has nothing to disclose.
Dr. Schirmacher has nothing to disclose.
Dr. Thomas reports grants, personal fees and non-financial support from AstraZeneca, grants, personal fees and non-financial support from Bristol-Myers Squibb, personal fees and non-financial support from Boehringer Ingelheim, personal fees and non-financial support from Celgene, personal fees and non-financial support from Chugai, personal fees and non-financial support from Lilly, personal fees and non-financial support from MSD, personal fees and non-financial support from Novartis, personal fees and non-financial support from Pfizer, grants, personal fees and non-financial support from Roche, grants, personal fees and non-financial support from Takeda, outside the submitted work.
Dr. Christopoulos reports grants and personal fees from AstraZeneca, grants and personal fees from Novartis, grants and personal fees from Roche, grants and personal fees from Takeda, personal fees from Boehringer, personal fees from Chugai, personal fees from Pfizer, outside the submitted work.
Dr. Stenzinger reports personal fees from AstraZeneca, personal fees from Bayer, personal fees from BMS, personal fees from Illumina, personal fees from Novartis, personal fees from Seattle Genetics, personal fees from Takeda, personal fees from ThermoFisher, personal fees from AstraZeneca, personal fees from Bayer, personal fees from BMS, personal fees from Illumina, personal fees from MSD, personal fees from Novartis, personal fees from Roche, personal fees from Seattle Genetics, personal fees from Takeda, personal fees from ThermoFisher, personal fees from Chugai, personal fees from BMS, personal fees from Bayer, personal fees from Ventana Roche, personal fees from Lilly, during the conduct of the study.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–14. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909–1920. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Geukes Foppen MH, Goldinger SM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim JM, Chen DS.. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol. 2016;27(8):1492–1504. doi: 10.1093/annonc/mdw217. [DOI] [PubMed] [Google Scholar]
- 6.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin J-R, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175(4):984–97 e24. doi: 10.1016/j.cell.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buttner R, Gosney JR, Skov BG, Adam J, Motoi N, Bloom KJ, Dietel M, Longshore JW, López-Ríos F, Penault-Llorca F, et al. Programmed death-ligand 1 immunohistochemistry testing: a review of analytical assays and clinical implementation in non-small-cell lung cancer. J Clin Oncol. 2017;35(34):3867–3876. doi: 10.1200/JCO.2017.74.7642. [DOI] [PubMed] [Google Scholar]
- 10.Lantuejoul S, Sound-Tsao M, Cooper WA, Girard N, Hirsch FR, Roden AC, Lopez-Rios F, Jain D, Chou T-Y, Motoi N, et al. PD-L1 testing for lung cancer in 2019: perspective from the IASLC pathology committee. J Thorac Oncol. 2020;15(4):499–519. doi: 10.1016/j.jtho.2019.12.107. [DOI] [PubMed] [Google Scholar]
- 11.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, Peters S. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30(1):44–56. doi: 10.1093/annonc/mdy495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kazdal D, Endris V, Allgauer M, Kriegsmann M, Leichsenring J, Volckmar AL, Harms A, Kirchner M, Kriegsmann K, Neumann O, et al. Spatial and temporal heterogeneity of panel-based tumor mutational burden in pulmonary adenocarcinoma: separating biology from technical artifacts. J Thorac Oncol. 2019;14(11):1935–1947. doi: 10.1016/j.jtho.2019.07.006. [DOI] [PubMed] [Google Scholar]
- 14.Stenzinger A, Endris V, Budczies J, Merkelbach-Bruse S, Kazdal D, Dietmaier W, Pfarr N, Siebolts U, Hummel M, Herold S, et al. Harmonization and standardization of panel-based tumor mutational burden measurement: real-world results and recommendations of the quality in pathology study. J Thorac Oncol. 2020;15(7):1177–1189. doi: 10.1016/j.jtho.2020.01.023. [DOI] [PubMed] [Google Scholar]
- 15.Budczies J, Kazdal D, Allgauer M, Christopoulos P, Rempel E, Pfarr N, Weichert W, Fröhling S, Thomas M, Peters S, et al. Quantifying potential confounders of panel-based tumor mutational burden (TMB) measurement. Lung Cancer. 2020;142:114–119. doi: 10.1016/j.lungcan.2020.01.019. [DOI] [PubMed] [Google Scholar]
- 16.Catacchio I, Scattone A, Silvestris N, Mangia A. Immune prophets of lung cancer: the prognostic and predictive landscape of cellular and molecular immune markers. Transl Oncol. 2018;11(3):825–835. doi: 10.1016/j.tranon.2018.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Talla SB, Rempel E, Endris V, Jenzer M, Allgauer M, Schwab C, Kazdal D, Stögbauer F, Volckmar A-L, Kocsmar I, et al. Immuno-oncology gene expression profiling of formalin-fixed and paraffin-embedded clear cell renal cell carcinoma: performance comparison of the NanoString nCounter technology with targeted RNA sequencing. Genes Chromosomes Cancer. 2020;59(7):406–416. doi: 10.1002/gcc.22843. [DOI] [PubMed] [Google Scholar]
- 18.Backman M, La Fleur L, Kurppa P, Djureinovic D, Elfving H, Brunnstrom H, Mattsson JSM, Pontén V, Eltahir M, Mangsbo S, et al. Characterization of patterns of immune cell infiltration in NSCLC. J Thorac Oncol. 2020. doi: 10.1016/j.jtho.2019.12.127. [DOI] [PubMed] [Google Scholar]
- 19.Danaher P, Warren S, Dennis L, D’Amico L, White A, Disis ML, Geller MA, Odunsi K, Beechem J, Fling SP, et al. Gene expression markers of tumor infiltrating leukocytes. J Immunother Cancer. 2017;5(1):18. doi: 10.1186/s40425-017-0215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirchner M, Neumann O, Volckmar AL, Stogbauer F, Allgauer M, Kazdal D, Budczies J, Rempel E, Brandt R, Talla SB, et al. RNA-based detection of gene fusions in formalin-fixed and paraffin-embedded solid cancer samples. Cancers (Basel). 2019;11(9):1309. doi: 10.3390/cancers11091309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Endris V, Penzel R, Warth A, Muckenhuber A, Schirmacher P, Stenzinger A, Weichert W. Molecular diagnostic profiling of lung cancer specimens with a semiconductor-based massive parallel sequencing approach: feasibility, costs, and performance compared with conventional sequencing. J Mol Diagn. 2013;15(6):765–775. doi: 10.1016/j.jmoldx.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 22.Volckmar AL, Leichsenring J, Kirchner M, Christopoulos P, Neumann O, Budczies J, Morais de Oliveira CM, Rempel E, Buchhalter I, Brandt R, et al. Combined targeted DNA and RNA sequencing of advanced NSCLC in routine molecular diagnostics: analysis of the first 3,000 Heidelberg cases. Int J Cancer. 2019;145(3):649–661. doi: 10.1002/ijc.32133. [DOI] [PubMed] [Google Scholar]
- 23.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fehrenbacher L,Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, Park K, Smith D, Artal-Cortes A, Lewanski C, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387(10030):1837–1846. doi: 10.1016/S0140-6736(16)00587-0. [DOI] [PubMed] [Google Scholar]
- 25.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. 2006;90:297–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dammeijer F, Lau SP, van Eijck CHJ, van der Burg SH, Aerts J. Rationally combining immunotherapies to improve efficacy of immune checkpoint blockade in solid tumors. Cytokine Growth Factor Rev. 2017;36:5–15. doi: 10.1016/j.cytogfr.2017.06.011. [DOI] [PubMed] [Google Scholar]
- 31.House IG, Savas P, Lai J, Chen AXY, Oliver AJ, Teo ZL, Todd KL, Henderson MA, Giuffrida L, Petley EV, et al. Macrophage-derived CXCL9 and CXCL10 are required for antitumor immune responses following immune checkpoint blockade. Clin Cancer Res. 2020;26(2):487–504. doi: 10.1158/1078-0432.CCR-19-1868. [DOI] [PubMed] [Google Scholar]
- 32.Hwang S, Kwon AY, Jeong JY, Kim S, Kang H, Park J, Kim J-H, Han OJ, Lim SM, An HJ, et al. Immune gene signatures for predicting durable clinical benefit of anti-PD-1 immunotherapy in patients with non-small cell lung cancer. Sci Rep. 2020;10(1):643. doi: 10.1038/s41598-019-57218-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel SA, Minn AJ. Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity. 2018;48(3):417–433. doi: 10.1016/j.immuni.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liudahl SM, Coussens LM. B cells as biomarkers: predicting immune checkpoint therapy adverse events. J Clin Invest. 2018;128(2):577–579. doi: 10.1172/JCI99036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Gallastegui N, Rosenblatt JD. Regulatory B cells in anti-tumor immunity. Int Immunol. 2015;27(10):521–530. doi: 10.1093/intimm/dxv034. [DOI] [PubMed] [Google Scholar]
- 36.Mauri C, Menon M. The expanding family of regulatory B cells. Int Immunol. 2015;27(10):479–486. doi: 10.1093/intimm/dxv038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mauri C, Menon M. Human regulatory B cells in health and disease: therapeutic potential. J Clin Invest. 2017;127(3):772–779. doi: 10.1172/JCI85113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geng XR, Yang G, Li M, Song JP, Liu ZQ, Qiu S, Liu Z, Yang P-C. Insulin-like growth factor-2 enhances functions of antigen (Ag)-specific regulatory B cells. J Biol Chem. 2014;289(25):17941–17950. doi: 10.1074/jbc.M113.515262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nielsen JS, Sahota RA, Milne K, Kost SE, Nesslinger NJ, Watson PH, Nelson BH. CD20+ tumor-infiltrating lymphocytes have an atypical CD27- memory phenotype and together with CD8+ T cells promote favorable prognosis in ovarian cancer. Clin Cancer Res. 2012;18(12):3281–3292. [DOI] [PubMed] [Google Scholar]
- 40.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549–555. doi: 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amaria RN, Reddy SM, Tawbi HA, Davies MA, Ross MI, Glitza IC, Cormier JN, Lewis C, Hwu W-J, Hanna E, et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat Med. 2018;24(11):1649–1654. doi: 10.1038/s41591-018-0197-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petitprez F, de Reynies A, Keung EZ, Chen TW, Sun CM, Calderaro J, Jeng Y-M, Hsiao L-P, Lacroix L, Bougoüin A, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577(7791):556–560. doi: 10.1038/s41586-019-1906-8. [DOI] [PubMed] [Google Scholar]
- 43.Griss J, Bauer W, Wagner C, Simon M, Chen M, Grabmeier-Pfistershammer K, Maurer-Granofszky M, Roka F, Penz T, Bock C, et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat Commun. 2019;10(1):4186. doi: 10.1038/s41467-019-12160-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561–565. doi: 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 45.Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, Rabbe N, Laurans L, Tartour E, de Chaisemartin L, et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol. 2008;26(27):4410–4417. doi: 10.1200/JCO.2007.15.0284. [DOI] [PubMed] [Google Scholar]
- 46.Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, Griss J, Bauer W, Wagner C, Simon M, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med. 2014;189(7):832–844. doi: 10.1164/rccm.201309-1611OC. [DOI] [PubMed] [Google Scholar]
- 47.Goeman F, De Nicola F, Scalera S, Sperati F, Gallo E, Ciuffreda L, Pallocca M, Pizzuti L, Krasniqi E, Barchiesi G, et al. Mutations in the KEAP1-NFE2L2 pathway define a molecular subset of rapidly progressing lung adenocarcinoma. J Thorac Oncol. 2019;14(11):1924–1934. doi: 10.1016/j.jtho.2019.07.003. [DOI] [PubMed] [Google Scholar]
- 48.Keenan TE, Burke KP, Van Allen EM. Genomic correlates of response to immune checkpoint blockade. Nat Med. 2019;25(3):389–402. doi: 10.1038/s41591-019-0382-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.