

ABSTRACT
Lipids are complex organic compounds made up of carbon, oxygen, and hydrogen. These play a diverse and intricate role in cellular processes like membrane trafficking, protein sorting, signal transduction, and bacterial infections. Both Gram-positive bacteria (Staphylococcus sp., Listeria monocytogenes, etc.) and Gram-negative bacteria (Chlamydia sp., Salmonella sp., E. coli, etc.) can hijack the various host-lipids and utilize them structurally as well as functionally to mount a successful infection. The pathogens can deploy with various arsenals to exploit host membrane lipids and lipid-associated receptors as an attachment for toxins’ landing or facilitate their entry into the host cellular niche. Bacterial species like Mycobacterium sp. can also modulate the host lipid metabolism to fetch its carbon source from the host. The sequential conversion of host membrane lipids into arachidonic acid and prostaglandin E2 due to increased activity of cPLA-2 and COX-2 upon bacterial infection creates immunosuppressive conditions and facilitates the intracellular growth and proliferation of bacteria. However, lipids’ more debatable role is that they can also be a blessing in disguise. Certain host-lipids, especially sphingolipids, have been shown to play a crucial antibacterial role and help the host in combating the infections. This review shed light on the detailed role of host lipids in bacterial infections and the current understanding of the lipid in therapeutics. We have also discussed potential prospects and the need of the hour to help us cope in this race against deadly pathogens and their rapidly evolving stealthy virulence strategies.
KEYWORDS: Gram-positive, gram-negative, bacterial infection, lipid rafts, cholesterols, lipid droplet, invasion, intracellular survival, bacterial toxin, therapeutics, extracellular bacteria, immunomodulation, bacterial clearance, proinflammatory, anti-inflammatory, sphingolipids, signaling, virulence
Introduction
Lipids are a class of complex organic compounds usually made up of carbon, oxygen, and hydrogen (nitrogen and phosphorous are present as well). These are soluble in an organic solvent, but they are either insoluble or partially soluble in water due to their hydrophobic or amphipathic nature. Lipids comprise fatty acids, glycerophospholipids, sphingolipids, glycolipids, prenol lipids, sterols, polyketides, and ether-linked glycerophospholipids. They have diverse cellular functions such as cell-membrane components, energy storage, signaling, and cellular trafficking. Moreover, it is well known that lipid homeostasis is lost during various cardiovascular and other metabolic diseases, indicating that they are crucial in maintaining a healthy state [1].
In correlation to host lipid interactions and hijacking, the pathogen’s invasion into the host cells to maintain the intracellular niches has been well documented. The cell membrane harbors distinct lipid microdomains, composed of cholesterols and sphingolipids and membrane proteins known as Lipid Rafts. On the membrane, these rafts guide protein-protein and lipid-protein interactions, based on their properties to determine interacting proteins and the ability to knit together to generate a broader domain. Physiologically, they are reported to play a role in diverse cellular processes such as membrane trafficking, protein sorting, and signal transduction events. Thus, it also interplays with apoptosis, proliferation, adhesion, and migration [2]. A substantial amount of literature is available on lipid rafts’ role in viral infection [3,4]. There has been an increase in reports relating to bacterial, fungal, and protozoan infections as well.
Successful adhesion of bacteria to the host cell membrane leads to colonization of host tissue, followed by invasion, intracellular multiplication, and ultimately dissemination [5,6]. The adhesion and uptake of bacteria by the host cell membrane depends upon the structural modification of membrane lipids. Most of the gram-negative pathogenic bacteria synthesize and transport numerous virulent factors, effector proteins into the cytoplasm of the host cells by T3SS. These effectors change the host cell membrane’s morphology by modulating actin filaments, etc [5,6]. The extracellular bacteria use cholesterols or GM1 receptors present on the eukaryotic membrane to bind and form pores on the host cell membrane. One of the notable examples includes AB toxins that use GM1 ganglioside receptors for attachment. Lipid microdomains also mediate bacterial entry into the host cells. Bacteria being a prokaryote, do not possess cellular tools to synthesize cholesterols and sphingolipids but have evolved to acquire the same from the eukaryotic host. These host lipids impart various roles in prokaryotic systems such as escaping host defense mechanisms, maintaining cell permeability, membrane fluidity, energy or nutrient sources, and signaling processes. Intracellular bacteria such as Chlamydiaceae sp., Salmonella sp., Shigella sp., Mycobacterium sp., Listeria sp., and Enteropathogenic E. coli acquire host lipids to avoid the deleterious defense mechanism of the host and thus helps to maintain intracellular niches [7–13]. Host lipids are not only hijacked, but their metabolism is also altered during infection. One of the classic examples remains Mycobacterium spp., which modulates host lipid metabolism and manages to use it as their sole carbon source during chronic infections [14]. Extracellular bacteria such as Mycoplasma sp., H. pylori, on the other hand, have also been reported to incorporate host lipids, primarily cholesterols, and convert them to glycol-cholesterol complex [15–17].
Lipids also have major functions as first and second messenger molecules in host defense signaling. Thus, bacteria usually modulate these to escape host immune signaling and thereby promoting their survival inside the host. One of the most important lipid mediators of the cell, PGE2, elicits an immune response during chronic and acute infections [18]. It also promotes host protection against mitochondrial inner membrane disruption and limits bacterial spreading because of necrosis [19]. PGE2 plays a role in the clearance of bacteria from the system by increasing the apoptosis of infected host cells and protecting against inner mitochondrial damage [20]. Another example of lipid in host protection remains the redistribution of glycerophospholipids in the membrane to mediate the Mycobacterium infected cells’ efferocytosis and limit the growth [21]. Various host sphingolipids enhance bacteria’s maturation containing phagosome into acidified lysosome upon its phosphorylation [22]. Active metabolites of vitamin D also help prevent bacteria’s clearance by inducing paracrine signaling of macrophage and epithelial cells to enhance the release of antimicrobial peptides [22]. Hence, it can be concluded that host lipids are among the major targets of bacterial invasion among other host molecules.
Macrophages, the professional phagocyte, and antigen-presenting cells are the host immune system’s front-line defense and a link between innate and adaptive immunities. They are well known to be polarized either into inflammatory or anti-inflammatory macrophages. Increasingly, it is evident that lipids play an equitably important role in macrophages’ function during polarization, thus influencing the outcome [23]. At different stages of infection, the inflammatory and anti-inflammatory responses are strictly regulated and pathogen-specific [24–26]. Perturbance of this equilibrium usually results in either excessive inflammation or failure to activate an immune response. Hence, various intracellular bacteria perturb this to thrive inside these otherwise bactericidal cells successfully.
Lately, a major therapeutic strategy domain has been explored using lipids as a delivery agent. The amphipathic nature of lipids has been harnessed to encapsulate drugs inside the lipid layer called liposomes. These liposomes show better drug retention and excellent circulation of encapsulated drugs in the body. As a substitute for conventional lipid carriers like liposomes and emulsions, solid lipid nanoparticles (SLNs) have been developed recently. They have several advantages that confer them better stability [27]. Apart from SLN, there is another set of lipid carriers with a more structured framework called nanostructured lipid carriers (NLC), enhancing the drug’s oral bioavailability [28]. Besides being an excellent carrier, lipids of bacterial origin and host origin (hijacked by bacteria) can be effective targets for antibiotics. So, therapeutic ventures have been made to tweak the in-vivo scenario, preventing pathogens [29]. One of the lipids playing inhibitory roles is sphingosine-1-phosphate, which has been extensively studied for potential therapeutic aspects [30]. There is also a marked difference between virus-infected individuals’ lipid profile than that of bacteria-infected ones; these have been used as markers for quick probing and detection of the diseases and further analysis [31].
This review will be dissecting various roles of lipids, both during facultative and obligate intracellular bacterial infection. It is interesting to know that lipids exploitation by these two groups of bacteria is quite diverse; intracellular bacteria use host lipids to avoid phagosome maturation while extracellular bacteria use host lipids for their membranes’ integrity. However, bacterial species’ host lipid acquisition, especially cholesterol, is increasingly important in bacterial pathogenesis in recent literature.
Bacterial and host lipids
Lipids are the most common yet one of the differentiating biomolecules across all life domains; Eukarya, Archaea, and Bacteria. They are essentially and broadly two kinds: storage lipids (triglycerides-TAG) and membrane lipids (glycerophospholipids). Just a handful of bacteria can only synthesize storage lipids and, thus, are mostly limited to eukaryotes. On the contrary, it has been shown by several reports that those handfuls of bacteria such as Mycobacterium, Nocardia, Rhodococcus, Micromonospora, Dietzia, and Gordonia are often associated with harboring even neutral lipid bodies (LD), mainly comprising of TAG and wax esters (WE) [32,33]. On the other hand, glycerophospholipids or membrane lipids are part of every life domain [34]. However, there are striking differences between the membrane lipids of these three domains. In Eukarya and Bacteria, the fatty acyl side chains are linked to the sn-glycerol-3-phosphate backbone via an ester bond. In Archaea, there are ether linkages between isoprenoid hydrocarbon side chains and sn-glycerol-1-phosphate backbone [35]. In bacterial cell membranes, there are three major classes of phospholipids; phosphatidylethanolamine (PE), phosphatidylglycerol (PG), and cardiolipins (CL). Zwitterionic PE accounts for about 75% of the membrane lipids, anionic PG about 20%, and the rest 5% is CL [36]. However, these percentage contributions often vary, depending on which growth stage bacteria are in; for example, during the stationary phase in E. coli, the CL percentage is higher [37]. Apart from glycerophospholipids, various bacteria are capable of synthesizing diacylglycerols (DAG)-derived membrane lipids; these lack phosphorus in their structure, typical examples of these are glycolipids (GL), sulfolipids (sulfoquinovosyl diacylglycerol -SQDG), and diacyl-glyceryl trimethyl-homoserine (DGTS) lipids [38]. There are also membrane lipids which completely lack glycerol backbones, such as isoprenoids and hopanoids; these serve a specialized function in bacterial membranes and are a structural surrogate for sterols. Necessarily suggesting that bacteria have immense lipid diversity and all kinds of lipids exist in various bacterial species. It is widely known that apart from proteinaceous virulence factors, bacteria also harbor a set of lipids accounting for their virulence. These lipids are generally expressed on their surface, capable of modulating the host lipid metabolism and triggering an immune response. A major component of the Gram-negative bacterial cell wall is lipopolysaccharide (LPS), which comprises lipids A, core oligosaccharide region, and variable carbohydrate [39]. LPS has been extensively studied concerning orchestrating host lipid biosynthesis dynamics upon infection, discussed in detail later [40]. Also, it has been reported that LPS is often self-modulated by various pathogenic bacteria as a part of escaping strategies from the host immune system [41]. Apart from LPS, another bacterial lipid capable of host immune and metabolism modulating function is Trehalose-6,6ʹ- di-mycolate (TDM). TDM is expressed copiously on the cell wall of bacteria containing mycolic acid such as Mycobacterium, Nocardia, Corneybacterium species. TDM harbors trehalose as sugar esterified to two residues of mycolic acid, and the length of mycolic acid residue varies from 20–80, based on the bacterial species. It has been reported in several studies done in Mycobacterium that TMD is responsible for upregulating multiple cytokines (TNF-α, IL-1β, IL-1 m, IL-10, and MCSF-1) and chemokines (CCL3, CCL4, CCL7, CCL12, CCR12, CXCL1, CXCL2, and CXCL10). TMD also elicits the foamy cell formation and trigger activated foreign body and hypersensitivity-type granulomas in mice. There are vast literature and many detailed reviews available on the synthesis, structures, and functions of these lipids [37,38,42]
Like bacterial lipids, host or eukaryotes also have lipids such as PE, PG, and CL, along with phosphatidylserine (PS). However, PG and CL are synthesized and confined to the mitochondrial membrane, primarily due to its prokaryotic origin. Apart from these, eukaryotic membranes also harbor phosphatidylcholine (PC) and phosphatidyl-inositol (PI) as glycerophospholipids. The cellular distribution of these membrane lipids is also based on individual lipid species’ curvature and the given sub-cellular organelle requirement. Besides this, the endoplasmic reticulum (ER) serves as the main site for the synthesis of lipids, producing a lot of structural phospholipids and cholesterols, along with a decent amount of TAG and cholesteryl esters (having non-structural roles) [43,44]. ER also make ceramide (Cer), the precursor of sphingolipids, and GalCer is only produced in the epithelial and myelinating cell’s ER. Apart from all of these lipids, ER also harbors various minor lipids playing a pivotal role in intermediates and end products of certain pathways (this includes: DAG, lysophospholipids, and dolichol) [45]. The other class of eukaryotic lipids, called sphingolipids, has an amino-alcohol backbone. Previously, these were only associated with membrane microdomains or rafts; however, more recent scientific development in the last decade shows that these are also involved in regulating several cellular functions such as autophagy and apoptosis. Sphingolipids are synthesized in the Golgi complex, producing sphingomyelin (SM) GlcCer and lactosylceramide (LacCer) and other higher-order glycerol-sphingolipids (GSL) [46,47]. The plasma membrane majorly comprises higher density glycerophospholipids, sphingolipids, sterols, and PI for various cell signaling cascades to sustain mechanical stress [48]. Besides this, there is a lot of dynamic fission and fusion of eukaryotic membranes in the endocytic pathways; those are mostly regulated by a dedicated system of phosphoinositide specific kinases and phosphatases. Essentially this manages the pool of phosphatidylinositol phosphates such as phosphatidylinositol 3-phosphate (PI3P), phosphatidylinositol 4-phosphate (PI4P), and phosphatidylinositol (4,5)- bisphosphate [PI(4,5)P2] on the target and vesicular membranes. Examples are PI4P in the Golgi, phosphatidylinositol 3-phosphate (PI3P) in endosomes, and [PI(4,5)P2] in plasma membranes [48,49].
Host lipids and bacterial toxins
Toxins is a soluble protein known to have receptor-specific interactions present on the putative rafts [2]. Bacterial exotoxins are broad of two classes; (1) extracellularly acting and (2) intracellularly acting. Further, the extracellularly acting bacterial exotoxin are divided into two broad categories: a) non-membrane damaging and b) membrane damaging; there is a further division into different sub-categories based on the enzymatic activity possessed by the toxin. Since membrane damaging and intracellular acting toxins are involved in the interaction with host lipids, this section mainly focuses on them. The membrane damaging bacterial toxins, also known as pore-forming toxins (PFT), has three classes a) hemolysins, b) leukocidins, and c) phospholipases. PFTs account for 20–30% of all bacterial exotoxins, predominantly function to perforate the plasma membrane but can also act on intracellular organelle membranes [50,51]. The preliminary step for any toxin to act on the host cell is to identify and interact with its cell’s either membrane-spanning or GPI-anchored-proteins or lipids microdomains. It has been well demonstrated that bacterial toxins have exploited the lipid microdomain of host cell membranes as binding and concentrating devices for toxins (Refer to Table 1) [52,53]. Lipid rafts help in inducing toxin oligomerization to form pores in the host cell membrane. Aerolysin from Aeromonas hydrophila is a water-dwelling, gram-negative rod-shaped bacterium, majorly causing diarrheal illness and sometimes necrotizing skin and soft tissue infections in individuals with immunocompromised conditions [54]. Upon binding to lipid rafts, Aerolysin undergoes the heptamerization process followed by pore formation [55,56]. Similar mechanisms of toxin oligomerization on lipid rafts of the host cell membrane have also been found in Staphylococcus sp. leukotoxin (octameric pore) and E. coli. ClyA (13-meric pore) [57]. The alpha-toxin produced by Clostridium sp., most potent of them is produced by Clostridium perfringens, harbors phospholipase C (PLC) activity [58], and the N-terminal domain retains only lecithinase activity and has a lower potency than the holoenzyme [59].
Cholera toxin (CT) is an AB toxin, which possesses a single A subunit and five B subunits in a pentameric ring fashion [60]. CT has been shown to interact with these lipid microdomains cluster of GM1 ganglioside receptors for attachment like Velcro mechanisms [61]. The A subunit’s catalytic activity solely depends upon internalization into the host cell, resulting in the efflux of ions and water from the cells [62,63]. H. pylori classified as a group 1 carcinogen and one of the handfuls of bacteria that are directly linked to cancer [64]. VacA, from H. pylori, has been known to affect host cells by inducing cytoplasmic vacuoles, which support the survival of bacteria by increasing permeability of the host cell [65]. It has also been reported that the host sphingomyelin and depletion of cholesterol might have a role in toxin functionality, thus proposing strong pieces of evidence for the role of host lipid rafts in the activity of VacA [66]. Consequently, the selection of receptors harbored in rafts is critical in bacterial toxin-induced hyper-inflammatory responses, host defense, or endotoxin tolerance [67]. The diphtheria toxin is reported to enter the cell via clathrin-coated pits (COP-I), classically known to function independently of lipid rafts [68–70]. On the contrary, it has been shown that the depletion of sphingolipid facilitates the entry of Diphtheria toxins [71]. Anthrax, another toxin, also invades the cell via the clathrin-dependent process. This is mediated by a receptor called tumor endothelial marker 8 (TEM8, ANTXR1) and capillary morphogenesis gene 2 (CMG2, ANTXR2) but, the receptor-toxin cluster in the lipid raft and disruption of the same leads to a subsequent defect in toxin internalization [72,73]. Shiga toxin (Stx1/2), another AB5 toxin, interacts with amphipathic glycosphingolipid (GSL), which are globotriaosylceramide, and globotetraosylceramide. These are embedded in membrane lipid microdomains or lipid rafts [53,74].
Other sets of toxins have also been seen to utilize the lipid microdomains for attachment and entry into the cell; these are given as a tabulated form (Refer table 1). Few toxins help in survival inside the intracellular niches. Upon entry into the host cell, bacteria end up having two options: either to escape the vacuole or to deviate phagolysosome maturation, thereby securing an intracellular replicative niche. One classic example is employed by L. monocytogenes. To escape the vacuole and come into the cytoplasm, L. monocytogenes secretes pore-forming cytolysin, called listeriolysin O. It has been shown to interact with lipid rafts, and it belongs to the cholesterol-dependent cytolysin(CDC) family [75]. Besides this, Listeriolysin O also heightens the release of proinflammatory cytokines via the NF-KB pathway in macrophages [76]. This helps recruit a larger number of prospective host cells to the infection site and further helps the pathogen disseminate. Targeting the toxins’ binding to the host cell by inhibiting the host lipid-binding sites serves as a potent therapeutic strategy against the toxins known to cause life-threatening conditions in the host. Recent literature in this field is vast, and many excellent reviews focus on each toxin’s detailed mechanism [77–79]. This review has limited our focus to a more holistic aspect of the host-lipids race during bacterial infection.
Host lipids and bacterial endocytosis
Plasma membrane domains (enriched in cholesterol/sphingosine) function to sort the protein (based on the apical and basolateral side) of epithelial cells that mediate selective bacterial entry into the cell [80]. CD55, located in the apical membrane, acts as a receptor for various bacteria and viruses, causing mucosal infections [81–84]. Bacteria exclusively enter through the raft domain even when the entry receptors are available throughout the host cell membrane. Mañes et al. proposed that raft-mediated entry has two advantages: firstly, it activates cellular modulation for cytoskeleton remodeling along with membrane ruffling, and secondly, to circumvent intracellular degradative pathway [85]. Acquiring host lipid rafts to form phagosome also helps survive various intracellular bacteria, notably obligate intracellular such as Chlamydiaceae, and facultative intracellular such as Salmonella sp., Shigella sp., Mycobacterium sp. inside phagocytic cells [61]. E. coli K1 mediated infection causes meningitis. The route of access of E. coli K1 into the cells depends upon the integrity and cholesterol content of lipid rafts [86]. The Chlamydia trachomatis is the most common cause of all sexually transmitted diseases (causing urogenital trachoma and conjunctivitis infection) and has been reported to have lipid-mediated entry into the cell [85,87,88]. Lipid rafts mediate attachments, followed by the entry in the host cells of C. trachomatis. Some reports show that C. trachomatis serovars E and F enter through a membrane area rich in GM1, a lipid raft marker, along with caveolin1 and caveolin2 (Refer Figure 1)[89–92]. A study done using chelator of cellular cholesterol has shown that the elementary body (infecting stage) of C. trachomatis serovars L2, D, E, and K attaches to the cholesterol-rich site in the membrane of epithelial cells [93]. On the contrary, C. trachomatis serovars A and C enter the cell through clathrin-mediated endocytosis and are uninhibited by cholesterol depletion [91]. Apart from this, Chlamydia spp. also traffic to the Golgi apparatus to hijack sphingolipids rich vesicles, thereby preventing lysosomal fusion (Refer Figure 1)[90]. However, recent reports suggest that Chlamydia sp. utilizes heparan sulfate proteoglycans (HSPGs) on the host cell membrane for initial attachment followed by high-affinity binding to a battery of host cell receptors and thereby mediating entry into the host cells [94–96].
Figure 1.
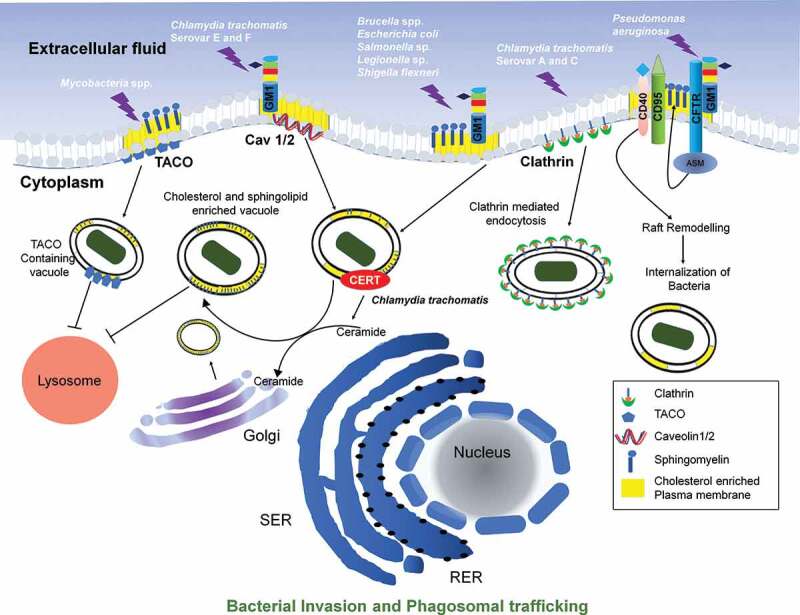
Host lipid raft in bacterial entry and phagosomal trafficking: Mycobacterial spp. incorporated raft-associated TACO (tryptophan-aspartate-containing coat protein) of the host cell and thus inhibit lysosomal fusion. Chlamydia trachomatis serovars E and F enter the host cell through lipid rafts associated with GM1 ganglioside receptor, caveolin 1/2 heteromeric complex. Upon entry, it recruits ceramide transfer to facilitate the transfer of ceramide to the Golgi bodies. Further upon conversion of ceramide to sphingolipid and cholesterol, it is recruited onto bacteria containing phagosome and thus escape fusion with Lysosome. Other species of Chlamydia, Brucella spp., E. coli, Salmonella sp., and Legionella sp. enters through lipid raft region rich in cholesterol. Chlamydia trachomatis 2 serovars A and C enters the cell through clathrin-dependent endocytosis which functions exclusive of lipid rafts. Pseudomonas aeruginosa enters and mediates an uptake by raft remodeling via CFTR, CD40, and CD95 dependent pathway
Salmonella sp. and Shigella sp. are two Enterobacteriaceae that utilize type 3 secretion system (T3SS), a molecular syringe to inject effector protein into the membrane and cytosol the host cell that facilitates the entry in non-phagocytic cells and survival. T3SS has two effector proteins, which are part of these apparatus both in Shigella (IpaB and IpaC, invasion plasmid antigens) and in Salmonella (SipB and SipC) that are encoded by Salmonella pathogenicity islands (SPI). They are preassembled, but the T3SS does not get activated to translocate the effectors until the bacterium is in contact with the host lipid raft. IpaB and SipB are cholesterol-binding proteins, and it has been shown that membrane insertion of IpaB relies on lipid raft domains(Refer Figure 1)[97,98]. However, experiments that were done using cells lacking the ability to form lipid rafts have shown that invasion and intracellular survival of Salmonella enterica serovar Typhimurium and C. trachomatis (certain serovars) is not dependent on cholesterol, unlike Coxiella burnetii [99]. A bacterial invasion is a complex event, and thus, not only lipids but also cholesterol/sphingosine associated membrane proteins play equally important roles.
Similarly, in Mycobacterium spp. it has been reported that to hide from adaptive immune responses, they have incorporated or modified host lipids and proteins from rafts in their phagosome, thereby escaping antigen processing and presentation apparatus-phagosome containing Mycobacterium spp. The host cell actively binds TACO (tryptophan-aspartate-containing coat protein), which prevents the fusion of bacteria-containing phagosome to the lysosome [100]. This interaction of TACO with phagosome is cholesterol-dependent(Refer Figure 1)[101].
Brucella abortus, which is Gram-negative, intracellular, zoonotic infection-causing bacteria, harbors Vir-B that requires macrophages’ plasma membrane cholesterol for mediating internalization(Refer Figure 1)[102]. Class A scavenger receptor, cholesterol, and ganglioside GM1 facilitated entry in macrophages is reported for B. ovis and B. canis (Refer Figure 1)[103]. Another intracellular, zoonotic, Gram-negative, and facultative pathogen, Francisella tularensis, has been reported to have inhibited entry upon removal of GPI-anchored protein due to the destabilization of cholesterol/sphingosine [104].
Pseudomonas aeruginosa, a Gram-negative, opportunistic pathogen, causing nosocomial infection apart from causing chronic pulmonary infection in immunocompromised or cystic fibrosis patients [105–108]. Studies on primary human nasal and murine tracheal epithelial cells showed that it triggers lipid microdomain transformation into large platforms of insoluble membranes to stimulate its internalization [109], the stimulation of CD95 and CD40 trigger membrane reorganization subjecting acid sphingomyelinase (ASM) toward the extracellular leaflet. Thus, leading to the breakdown of sphingomyelin to ceramide [110,111]. This is further required to reorganize these microdomains into larger signaling platforms [112]. A T3SS mediated upregulation of CD95 is reported in Pseudomonas infection, which helps to internalize bacteria, induce apoptosis, and regulate cytokines release; and mice that are deficient in CD95 show higher susceptibility to Pseudomonas infection [109]. The other role of sphingosine and derivatives in infection with Mycobacterium sp. has been discussed later. Asialylated ganglioside receptor, namely asialoGM1, another sphingolipid, expressed on the surface of cystic fibrosis patients’ respiratory epithelial cells enhances binding and invasion of Pseudomonas aeruginosa [113]. It can also be concluded that cholesterol/sphingosine and overall membrane integrity is vital for host defense mechanisms. Lipid rafts also cluster a chlorine channel CFTR (nonfunctional in Cystic Fibrosis), which colocalizes with GM1 marker [114]; a study using human corneal epithelial cells shows that P. aeruginosa enters via CFTR [7]. CFTR-mediated Pseudomonas infection activates the NF-KB pathway followed by cytokine release; however, methyl-β-cyclodextrin administration inhibits this NF-KB activation (Refer Figure 2)[114]. Macrophage-mediated bacterial endocytosis by CFTR dependent lipid rafts is inhibited in stress conditions, thus challenging one of the immune system’s potent arsenals [115].
Figure 2.
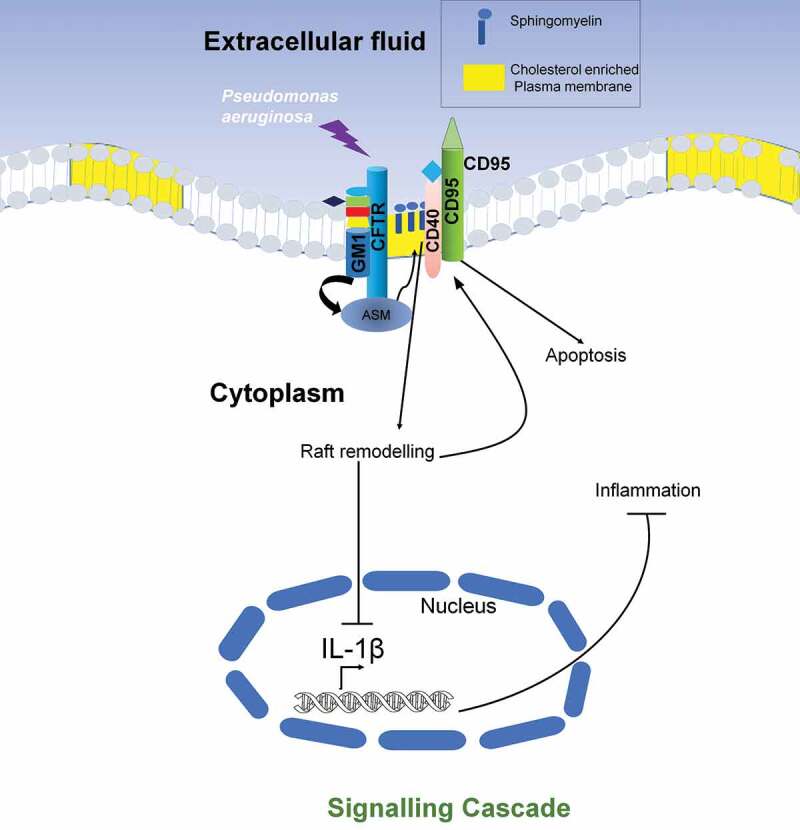
Host lipid raft mediated Signaling modulation: Pseudomonas aeruginosa upon binding to cystic fibrosis transmembrane conductance regulator (CFTR) and triggers activity of acid sphingomyelinase (ASM). ASM then leads to the production of ceramide in a CD40 and CD95 mediated pathway, which remains knitted with lipid rafts clustering and activation of downstream signaling to inhibit IL-1β expression and thus again inducing CD95 dependent apoptosis
Host lipids and intracellular bacteria
Host lipid remodeling on to phagosome of intracellular bacteria helps to maintain growth and viability. A considerable amount of investigation regarding the same has been done. The fusion of pathogen containing vacuole with lipid droplets which are enriched with neutral lipids like TAG, sterol esters (SE) along with proteins like perilipin family proteins, adipose differentiation-related proteins (ADRP), tail interacting protein (TIP47), etc and surrounded by phospholipid hemimembrane facilitates the nourishment of bacterial pathogens such as Chlamydia trachomatis, M. tuberculosis, M. leprae, etc by supplying adequate amount of nutrients [116]. In the case of Chlamydiaece, upon entry, the elementary body, which is bound to the inclusion bodies derived from the host cell membrane, differentiates into reticulate bodies and establishes a replicative niche [117]. During this phase, the inclusion bodies avoid lysosomal fusion and are transported to the peri-Golgi region, where it interacts with various cellular components and trafficking routes to gain access to nutrients. Even though C. trachomatis can synthesize their membrane lipids; they have been found to harbor host-derived lipids such as sphingomyelin and cholesterol on their membranes. Chlamydia inclusion hijacks sphingomyelin and cholesterol-containing exocytic vesicles by a vesicular trafficking pathway which is Brefeldin A(inhibits activation of Arf1- crucial in vesicle formation) sensitive [8,118]. Ceramide is a precursor of sphingomyelin. Conversion from ceramide to sphingomyelin takes place in the Golgi. This further accumulates within the inclusion membrane at the early time points of infection [118,119]. Nonetheless, not all lipids are acquired from Golgi dependent pathway, and it is limited to only particular lipids. Rab-GTPase and SNARE proteins, which are key players of vesicle fusion in physiological conditions, could be involved in the acquisition of lipid from the host [95,120–124]. Apart from these mechanisms, Chlamydia also recruits neutral lipids from either de novo synthesis or lipid droplets through IncA (effector protein) rich site [8,125,126].
Chlamydia inclusion protein IncD facilitates the recruitment of ceramide-transfer protein (CERT) and channelizes ceramide trafficking from the endoplasmic reticulum into the inclusion membrane [127–130]. The CERT also executes the transfer of ceramide to the Golgi. Inhibiting the CERT leads to lesser recruitment of sphingomyelin to the inclusion membrane [130]. Disruption of host cellular organelles (such as Golgi fragmentation) can heighten lipids’ uptake into the Chlamydia inclusion, resulting in increased C. trachomatis replication. This process is also governed by both host (Golgin-84) and Chlamydia proteases (chlamydial protease-like activity factor-CPFA) [131].
Abundant intracellular cholesterol levels trigger the esterification and packaging of Cholesterol ester into lipid droplets (LD). Chlamydia translocates various host proteins such as perilipin, Rab18, and ATGI, present on LD’s membrane to the inclusion membrane. Conversely, many bacterial proteins on the inclusion membrane are trafficked to adjacent LD membranes [125]. This results in organelle mimicry and thus deceiving host machinery of lysosomal fusion.
Mycobacterium spp. which are facultative intracellular pathogens, efficiently thrive in macrophages, also require cholesterol for their survival and uptake [101]. High polarity lipid derived from M. avium has been shown to interact with lipid rafts present on murine macrophages [132]. The ability to induce lipid droplet formation inside host cells is an important virulence factor for pathogenic Mycobacterium sp [133–135]. M. bovis BCG or M. tuberculosis derived LAM’s cell wall component can induce lipid droplet formation in time and the dose-dependent manner by mimicking the host.
Salmonella sp. upon entry mediated by T3SS encoded by SPI-1 in the non-phagocytic cell, maintain its intracellular replicative niche inside a vacuole called Salmonella containing vacuole (SCV) with the help of Salmonella pathogenicity island-1 and 2 (SPI-1 & 2) encoded T3SS and effector molecules both in non-phagocytic and phagocytic cells [136,137]. Remodeling of the outer membrane (OM) during Gram-negative bacterial infection helps intracellular survival and replications in tissue [10,138]. S. enterica serovar Typhi and S. enterica serovar Typhimurium both survive in acidified phagosomes facilitated by PhoPQ, ArcAB, mgtCB, and other regulons, a two-component regulator [139]. The genes are responsible for modifying OM in terms of increasing hydrophobicity and decreasing negative charge, resulting in the prevention of cationic antimicrobial peptide (CAMP) binding [9]. One recent report has shown that PbgA, under the control of the PhoPQ two-component system, traffics the cardiolipin from the inner membrane to the OM of Salmonella Typhimurium by binding to cardiolipin itself [140]. SseJ, another effector of SPI-2 having a glycerophospholipid cholesterol acyltransferase activity, localizes to the cytoplasmic face of SCV and esterifies the cholesterols. SseJ activity, along with localization, depends on the host protein, RhoA (GTPase) [141]. Cholesterol accumulation and CD55 protein recruitment on SCV suggests an important role for cholesterol in intracellular survival [11]. Another molecular complex of host CD44, the hyaluronan receptors, and IpaB (bacterial invasion) in the case of enteroinvasive Shigella infection partitions in the lipid rafts. Invasion of bacteria is reduced in the cells deficient for sphingolipids as it is utterly predictive of the possible roles lipid rafts play in shigellosis [142].
In L. monocytogenes, cholesterol is not required for entry into the cells. However, cholesterol plays a vital role in the event downstream to invasion, such as F-actin polymerization [12]. One of the reports demonstrates that, during enterohemorrhagic E. coli (EHEC) and enteropathogenic E. coli (EPEC) infection, cytoskeleton rearrangement was inhibited upon cholesterol depletion. Also, cholesterol depletion hampered adherence to epithelial cells in EPEC infection but not EHEC infection [13]. Coxiella burnetii genome has been reported to encode enzymes for the synthesis of fatty acids and phospholipids. Still, it lacks enzymes for de-novo synthesis of cholesterol [143]. However, it also encodes sterol reductase homolog, reducing double bonds of sterol to cholesterol in the final step. One of the studies showed that this enzyme was active in the yeast model system. Thus, it was proposed that C. burnetii could hijack and act on host sterols for their intracellular growth [144].
Contrary to the host lipid’s role in the survival of intracellular pathogens trapped inside the vacuole, it also restricts bacterial proliferation. The attachment of ER-resident immune-related GTPase Irgm3 (47 kDa) with ADRP helps the dendritic cells cross-present phagocytosed exogenous antigen exogenous CD8+ T cells [145]. High-resolution single-cell Raman microscopy has revealed the association of arachidonate enriched lipid bodies with phagosomes in neutrophils, which finally activates NADPH phagocytic oxidase [146]. The derivatives of glycerophospholipid, phosphatidylinositol (PI) of host cells play a divergent role in the maturation of phagolysosome during bacterial infection [147]. Salmonella enterica uses SopB protein to dephosphorylate PI (3,5) P2, PI (4,5) P2, and PI (3,4,5) P3 of the host cell to prevent the maturation of phagolysosome [148,149]. The engagement of phosphatidylinositol-3-phosphate or PI (3) P on the phagosomal membrane recruits p40phox that further activates NADPH phagocytic oxidase during FcɣIIA receptor-mediated phagocytosis of IgG coated RBCs in COS7 cell line [150]. M. tuberculosis can restrict the acidification of the phagosome by secreting SapM phosphatase that dephosphorylates PI (3) P of host cells [151–153]. The intracellular proliferation of Mycobacterium strain lacking PIP phosphatase MptpB that dephosphorylates PI (3) P, PI (4) P, PI (5) P, and PI (3,5) P2 is significantly attenuated in guinea pig macrophage [154,155]. Host cell lipids such as arachidonic acid, ceramide, sphingosine, sphingomyelin, and phosphatidylinositol-4,5-bisphosphate can restrict the intracellular proliferation of Mycobacterium tuberculosis and Mycobacterium avium by triggering the actin assembly, phagosome maturation, phagolysosome fusion, and acidification in J774A.1 macrophage cell [156].
Host lipids and extracellular bacteria
Extracellular bacteria have also been reported to incorporate host lipids. Specifically, Mycoplasma spp. has been known to acquire free cholesterol from serum lipoprotein and modify it to glycolipids, cholesteryl ester to be specific [15–17]. Although lipid auxotrophy is not a surprising concept in biology, the spirochete also depends on external cholesterol reservoirs for replication and multiplication because it cannot synthesize cholesterols. However, they show hallmark characteristics of eukaryotic lipid rafts (containing phosphatidylcholine, phosphatidylglycerols, and lipoproteins) on their cell membrane [157–160]. Besides these, it also has free cholesterols and two cholesterol glycolipids, acylated cholesteryl galactoside (ACGal) and cholesteryl-galactoside (CGal), along with non-cholesterol glycolipid and mono-galactosyl diacylglycerol(MGalD) [161–164]. Sterols that help in lipid raft formation and many detergent resistance membranes are vital for B. burgdorferi membrane’s integrity [157]. Although the mechanism of cholesterol acquisition from the host cell to spirochetes is not very clear, it is proposed to be mediated by lipid rafts-lipid rafts interactions [165].
Extracellular bacterial species H. pylori also incorporates cholesterols and converts it into the glucosyl-cholesterol complex. Glycolipids identified in cholesteryl glucosides are cholesteryl-α-D-glucopyranoside, cholesteryl-6-O-tetra-decanoyl-α-D-glucopyranoside, and plausibly cholesteryl-6-O-phosphatidyl-a-D-glucopyranoside. It has been known for a long time that H. pylori have a preference specific to cholesterols [166]. One study found that bacteria grown in the absence of cholesterol were more susceptible to antimicrobial peptides and antibiotics [167]. The presence of cholesterol on the H. pylori membrane helps modify the membrane to facilitate the colonization of bacteria in gastric mucosa without hampering the Lewis antigen’s expression [168].
Host lipid serving as nutrients for bacteria
Bacterial pathogens can use host lipid as a carbon source, and for energy as well, they efficiently hijack the host lipid metabolism to develop convenient reservoirs that help the pathogen to survive in the host during infection. Mycobacterium sp. is well known to establish infection via hijacking host lipids out of all intracellular pathogens. It is well reported that Mycobacterium sp. uses host lipid as a primary source of carbon in- vivo [14,169]. Based on their lipid composition and metabolism, M. tuberculosis and other genus members are one of a kind. They harbor a variety of lipids such as glycolipids, mycolic acids, and polyketides in the cell wall [170], the genes for biosynthesis is encoded in their genome. They possess approximately 250 enzymes, which are linked to fatty acid metabolism [171]. In different mouse models, it has been shown that the enzyme that governs the direction of degraded fatty acid product to a pathway also dictates the phenotype of the bacterium [172–174]. Interestingly M. tuberculosis does not depend on a single lipid source, shows plasticity in lipid utilization, and can co-catabolize different lipids as carbon sources [175]. M. tuberculosis makes use of host fatty acids as 1) substrates for β-oxidation, 2) acyl-primers for polyketide synthesis, and 3) incorporation into phospholipids and triacylglycerol (TAG) [176,177].M. tuberculosis has a dedicated cholesterol import system that allows the bacterium to use lipid as a sole source of carbon and energy during persistent chronic infection inside activated macrophages. M. tuberculosis gene cluster mce4 encodes an importer of cholesterol, which serves as the sole carbon source for the pathogen inside the host cell [178].
The cholesterol usage comes with an added drawback for the bacterium, the terminal product of cholesterol catabolism being propionyl-CoA [179], a toxic compound that requires further metabolism. To ameliorate this toxic effect, M. tuberculosis has acquired various strategies to metabolize propionyl-CoA as an energy source and as building blocks for cell wall synthesis. They convert propionyl CoA into pyruvate and succinate via methyl citrate cycle and subsequently incorporate these into the tricarboxylic acid (TCA) cycle to produce energy [180]. It is perhaps due to the presence of Methyl-isocitrate lyase enzymatic activity. Another pathway utilized by Mycobacterium sp. is the conversion of propionyl- CoA into methyl-malonyl-CoA by the action of propionyl-CoA carboxylase, and further assimilated into the vitamin B12 mediated methyl-malonyl pathway that results in the production of succinyl CoA, that is used in the TCA cycle as well [181]. Finally, they have polyketide synthase, which can convert propionyl CoA into methyl-malonyl CoA and incorporate it into the bacterium’s cell wall [182].
M. tuberculosis encodes for four phospholipase C proteins among them plcA, plcB and plcC are in an operon, and plcD is placed in a distant genomic region. These enzymes hydrolyze phospholipids, resulting in free fatty acids [171], and are essential for the pathogens’ virulence [183]. They metabolize fatty acids via two pathways: β -oxidation or glyoxylate shunts. Even though the β -oxidation pathway involves only five enzymes, M. tuberculosis is known to have approximately 100 genes encoded for enzymes having a redundant function in this cycle [184]. The glyoxylate shunt has been long known as an anabolic pathway essential for carbon synthesis among prokaryotes. It has been reported in a proteomic and genomic-based study on Mycobacterium sp. that one of the key enzymes isocitrate lyase (ICLs) of glyoxylate shunt is upregulated during infection in macrophages [185,186]. There are two homologous genes for ICL, namely icl1 and icl2; studies on mutant lacking icl1 have demonstrated that it affects bacterium survival inside the activated macrophages. The double mutant of the icl mice model has also shown rapid clearance from the lungs of infected mice, thus, providing evidence for the essentiality of the icl gene and glyoxylate pathway in M. tuberculosis pathogenesis [187].
It is well known that M. tuberculosis can persist as a dormant infection within mononuclear cells with a characteristic and pathological hallmark. Granulomas are generally an assemblage of immune cells, comprising of different types of macrophages such as multinucleated enlarge cells, epithelioid cells, and foamy cells, enclosed by a border of lymphocytes. Foamy cells are one of the granulomas hallmarks and can be defined as macrophages enriched with lipid droplets [134,172]. To maintain the latency in host M. tuberculosis derives host triacylglycerol from these foamy cells and utilizes it as a carbon source [176]. In foamy cells, bacteria are responsible for diverting the glycolytic pathway toward ketone body synthesis [188], and various mycobacterial molecules are involved in accumulating lipids in foamy cells [189].
Lipid- a blessing in disguise
Besides their roles in the augmentation of bacterial infection in host eukaryotic cells, which majorly include adhesion, invasion, and survival of bacterial pathogens, many times host lipids such as sphingolipids orchestrate significant inhibitory role during bacterial infection. Tuberculosis caused by Mycobacterium tuberculosis has been challenging humankind with its wrath since time immemorial. After several decades of decline, at present, tuberculosis cases are on the rise again. One of the reasons is its co-infection with HIV, and another reason is the emergence of MDR-strains. Also, TB treatments are prolonged (for six months), complex and expensive. At present, there is a dire need for new therapies to treat TB. M tuberculosis’s success as an intracellular pathogen is attributed to its property to resist killing by fusion of lysosome with phagosome containing it. It has been found to employ multiple mechanisms for this purpose, including inhibition of phagosomal localization of vacuolar H+/ATPase that acidifies the phagolysosome, modulations of the vesicular soluble N-ethylmaleimide attachment protein cellubrevin, and alterations of the regulatory GTPases of the Rab family and its early endosomal marker Ag1. Sphingosine-1-phosphate, which is produced from ceramide by the consecutive catalytic activity of ceramidase and sphingosine kinase, has been reported to exert an inhibitory effect on the intracellular proliferation of nonpathogenic Mycobacterium smegmatis and pathogenic Mycobacterium tuberculosis H37Rv strains in human monocyte-derived macrophages (hMDM) and THP-1 cells [30,190,191]. The translocation of sphingosine kinase to the phagosomal membrane is inhibited by Mycobacterium tuberculosis, further diminishing the intracellular level of sphingosine-1-phosphate and ultimately resulting in the reduction of intracellular Ca2+ level required for the acidification of phagolysosome [192,193]. Sphingosine-1-phosphate can express iNOS in macrophages, differentiate the latter toward M1 phenotype, secretion of IFN-ɣ during infection, and enhance pulmonary infiltration CD11b+ macrophages. Also, treatment with Sphingosine-1-phosphate enhances the expression of key signaling proteins of inflammatory response like phosphor-MAPK(pp38), phospho-NF-ĸβ, phospho-STAT3 in the lungs of 6–8 week old C57BL6 mice infected with Mtb [194]. Overexpression of Sphingosine Kinase (SphK-1) conferred resistance against M. smegmatis infection due to the enhanced generation of NO, iNOS species, pp38 LAMP-2 [195]. Burkholderia pseudomallei, the causative agent of melioidosis, improves its intracellular proliferation in murine macrophage by avoiding fusion with lysosome with the help of sphingosine-1-phosphate lyase, which is secreted into the host cytosol upon infection to degrade cellular pool of Sphingosine 1 Phosphate [196]. Shigella flexneri can evade the NFKB dependent proinflammatory immune response in adult mice’s gut by reducing the level of sphingosine-1-phosphate with heightened activity S1P lyase and S1P phosphatase [197]. A recent study has illustrated that hydrolysis of sphingomyelin into ceramide and phosphorylcholine activates NADPH dependent phagocytic oxidase to generate ROS in macrophage and helps in the clearance of Salmonella Typhimurium and Pseudomonas aeruginosa [198,199].
Despite their bactericidal functions, a wide array of pro-bacterial roles of sphingolipids have also been extensively studied. The mice’s enhanced resistance against the infection caused by Pseudomonas aeruginosa upon deleting sphingosine kinase 2, the enzyme required to synthesize sphingosine-1-phosphate, clearly indicates the contribution of sphingosine to the establishment of bacterial pathogenesis [200].
Legionella pneumophila lacking LegS2 gene coding for a novel sphingosine-1-phosphate lyase becomes hyper-proliferative in bone marrow-derived macrophages and hMDMs by downregulating the expression of proinflammatory cytokines [201].
Apart from sphingolipids, cholesterol has also been reported to possess antibacterial activity. As discussed earlier, cholesterol has a substantial role in supporting the survival of intracellular pathogens. On the contrary to this, it has been demonstrated that a high level of cholesterol in the parasitophorous vacuole (PV) either by supplementation or by treatment of U18666A, a drug that traps cholesterol in the parasitophorous vacuole, enhances the acidity of PV and successfully inhibits intracellular bacterial growth in both mouse embryonic fibroblast and THP1 macrophage cell lines [202]. A recent study has proved that oxysterol 25 hydroxycholesterol (25HC) can rapidly internalize the accessible fraction of cholesterol from the plasma membrane by activating acyl-CoA: cholesterol acyltransferase (ACAT) enzyme and builds up protection against invading bacterial pathogens such as Shigella flexneri, Listeria monocytogenes, etc [203]. As already discussed, ceramides are a group of sphingolipids that have been shown to have an antibacterial effect.
Macrophages are front line soldiers associated with host defenses against foreign pathogens. They are one of the professional phagocytes and antigen-presenting cells of the host immune system and harbor various pathogen recognition and signaling receptors on their outer membrane’s surface. They can efficiently uptake pathogens, digest them, and present them to T cells. Macrophages are generally classified into proinflammatory macrophages (classically activated, M1 macrophages) or anti-inflammatory macrophages (alternatively activated, M2 macrophages) based on their physiological features and state of polarization. They undergo polarization upon encountering foreign bodies/invading bacteria [204]. Proinflammatory cytokines such as IFN-ɣ and TNF-α are responsible for the induction of the M1 phenotype, further characterized by higher antigen presentation and heightened expression of IL-12, IL-23, reactive nitrogen species, and reactive oxygen species, etc.
On the other hand, macrophages’ M2 phenotype is augmented by anti-inflammatory cytokines like IL-4, IL-10, and IL-13 [205]. An increasing amount of research is going on, which demonstrates dynamic lipid remodeling during host defense mechanisms. M1 and M2 macrophages show two distinct metabolic pathways for energy generation; the former uses aerobic glycolysis, whereas the latter is skewed toward fatty acid oxidation.
There is also a plethora of genes associated with the metabolism of fatty acids, which are upregulated and downregulated in each of these polarized states. For M1 macrophages, the upregulation of COX-2 and downregulation of COX-1, leukotriene A4 hydrolase, thromboxane A synthase 1, and arachidonate 5-lipoxygenase (5-LO) have been reported (Refer Figure 3). Contrary to this, in the M2 state, there is an upregulation of COX-1 and arachidonate 15 lipoxygenases (15-LO) [39]. Generally, it has been observed that the effects of eicosanoids in the intimate relation of host-pathogen can be categorized into two parts: proinflammatory and anti-inflammatory. Eicosanoids such as PGE2 and lipoxin A4 (LXA4) are the products from the oxidation reaction of arachidonic acid and other polyunsaturated fatty acids and extremely active lipid mediators of the immune response during bacterial infection [206,207]. These lipid mediators help the host by different mechanisms. During the infection of avirulent M. tuberculosis, PGE2 has been found to modulate host survival strategies against the perturbation of the inner mitochondrial membrane via EP2 receptors, which further restrict the pathogen’s spread [19].
Figure 3.
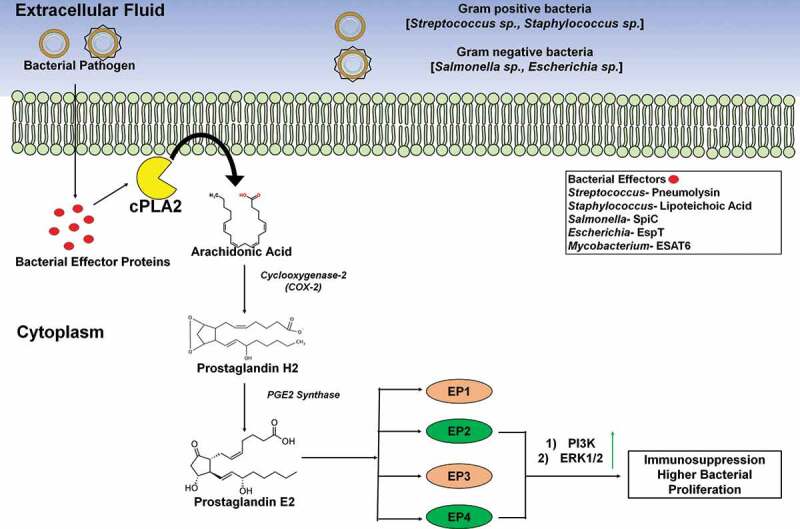
Role of host membrane lipid in immunomodulation during bacterial infection. After invading the host cell, bacteria (both gram positive and gram negative) synthesize and secrete effector proteins. Bacterial effector proteins/virulent factors aggravate the catalytic activity of cytosolic calcium-dependent phospholipase A2 (cPLA-2). As a result of which Arachidonic Acid is released from membrane glycerophospholipids into the cytoplasm. Subsequently, host cellular Cyclooxygenase-2 (COX-2) present in the cytoplasm 5 converts this AA into Prostaglandin H2 (PGH2) which represent a precursor of PGE2. Ultimately, PGH2 is transformed into PGE2 by the catalytic activity of the enzyme PGE2 Synthase. PGE2 performs a pivotal role in cytokine secretion & activation of several innate immune cells (like- macrophage, neutrophil, etc.). The production of PGE2 can elicit an immune response via any one of the four eicosanoid receptors termed as EP1, EP2, EP3, EP4. Interestingly, EP2 and EP4 represent the key players in developing the immunosuppressive response of PGE2, thus helping in survival and proliferation of bacteria inside immune cells. Downstream signaling by both EP2 and EP4, activate phosphatidylinositol 3- Kinase (PI3K) pathway. PI3K, in turn, activates ERK-1/2 to initiate immunosuppression
Interestingly it has also been reported that PGE2 activates synaptotagmin 7, a calcium-sensing protein that plays a pivotal role in lysosome-mediated membrane repair to reseal the host plasma membrane [208]. This re-joining process is vital to prevent necrosis and promote apoptosis, leading to pathogen’s clearance, thus helping the host. Strikingly the opposite role of PGE2, which potentiates the survival of intracellular pathogens in the host, has also been documented in great detail. PGE2 has been reported to acquire the ability to inhibit macrophage maturation and reduce the activation of NADPH oxidase and thus facilitates mycobacterial survival [209,210]. With the help of SPI-2 encoded virulent factor SpiC Salmonella Typhimurium can upregulate the expression of COX-2, which produces PGE2 in macrophages. PGE2 can enhance the production of IL-10 in a protein kinase A-dependent manner, which further accelerates Salmonella’s intracellular survival [210]. During infection, the virulent strain of Mycobacterium sp. abrogates the expression of PGE2 and aggravates the expression of lipoxin A4 (LXA4), which is anti-inflammatory and produced by the catalytic activities of 5-LO and 15-LO [207]. The enhanced expression of lipoxin A4 promotes necrosis and mediates the pathogen’s rapid spread [211].
Lipid and therapeutics
Since the 20th century, the liposome or the phospholipid bilayer capsule has received quite some attention due to its potential to carry bio-active molecules into the living system. Liposomes are formed by allowing phospholipids to swell in an aqueous solution, whereby they form concentric layers of lipids. A liposome can entrap an aqueous phase inside it, and if a solute remains dissolved in the aqueous phase, it can also entrap it. The initial experimentation had started with the administration of enzymes using liposomes, but later the work was extended by using liposomes as carriers for drugs and other therapeutic materials [212], including actinomycin D [213] and methotrexate [3]. When these drugs were administered via liposome in rats, they were retained more in the body than when injected in free solution. Also, a larger fraction of the drugs reached the liver and spleen. Solid Lipid nanoparticles (SLNs) are colloidal carriers developed in the past decade as an alternative to traditional lipid carriers like emulsions and liposomes. They constitute a new generation of submicron-sized emulsions where the solid lipid has replaced the liquid lipid (oil). The use of SLNs is advantageous because they offer better stability and upgradability to the production scale than liposomes [27]. The use of SLNs to administer antitubercular drugs like rifampicin, isoniazid, pyrazinamide reduced their dosing frequency and increased patient compliance [214]. SLNs have also been used to transfer mRNA to the cells, a promising strategy for vaccine development. Entrapping RNA within SLNs acts to protect it from degradation by RNases [215]. However, few disadvantages regarding SLNs include poor drug loading capacity, drug expulsion after polymeric transition during storage periods, etc. The lipid matrix also governs the drug loading capacity of SLNs. If the lipid matrix is made of similar lipids, a perfect crystal structure with few imperfections is formed. Drugs are incorporated in the crystal imperfections, between lipid layers, and between fatty acid chains. Due to the less amount of crystal imperfections, drug loading capacity decreases. To address these problems, nanostructured lipid carriers (NLC) were developed. The idea was to mix different lipids with varying fatty acid chains, such that there is quite some distance between the fatty acid chains, which leads to the formation of imperfect crystals and can accommodate more drug molecules, thereby improving the drug loading scenario. In recent years, research is being conducted to characterize NLCs.
NLCs demonstrated the faster release of the entrapped drug clotrimazole than low drug-loading SLNs, although there was no significant difference at high-drug loading [216]. NLCs were more stable than SLNs at 25°C. Also, the drug release profile of NLCs did not change after three months of storage, whereas there was a decline for SLNs [217]. NLCs have been tested to enhance the oral bioavailability of drugs like clotrimazole, apomorphine, ketoprofen, and lovastatin [28]. NLCs can be used for the targeted delivery of anti-tubercular drugs by the pulmonary route [218]. In addition to the oral route and pulmonary route, drug administration using NLCs is possible using the topical and intravenous routes. Administration through each route has its own set of advantages and shortcomings. Although very few NLCs have been used in clinical practice, they are promising candidates for lipid-nanocarrier mediated drug delivery [219].
Till now, we have dealt with how lipids can be used as carriers for drugs. From now, we will deal with how lipids can be used as targets for drugs. Lipid synthesis has been a significant target for antibiotic classes like lantibiotics, mannopeptimycins, and ramoplanins (Refer table 2) [220]. Lipid II has an extensively characterized role in cell wall synthesis. The cell wall of bacteria is composed of alternating units of n-acetylglucosamine (GlcNAc) and n-acetylmuramic acid (MurNAc), with these glycan chains cross-linked with a pentapeptide sequence [221]. The pentapeptide crosslinking confers strength and rigidity to the cell wall. The formation of a cell wall occurs in an extensive process involving the cytoplasmic face and the plasma membrane’s periplasmic face. The assembly of the cell wall subunits starts from the plasma membrane’s cytoplasmic side, where the assembly of UDP- MurNAc-pentapeptide is coupled to bactoprenol phosphate, yielding Lipid I. Next, the coupling of GluNAc occurs by peripherally membrane-attached protein MurG to yield Lipid II, the precursor of the cell wall, which is then translocated to the plasma membrane’s exterior by an unknown mechanism. Different types of antibiotics form different types of interactions with lipid II [220]. Their roles have been summarized in (Table 2).
Two new peptide antibiotics, katanosins, and plusbacins have been isolated from different bacterial strains Cytophaga and Pseudomonas, and they are found to have potent antibacterial activity against MRSA and Vancomycin-resistant Enterococci. Although their function has not been extensively characterized, they function Lipid-II mediated [222,223]. However, almost all these antibiotics have developed resistance against them, which raises the need to identify novel drug targets, and most importantly, identify novel therapeutic molecules. Lipid A is present mainly in Gram-negative bacteria where it is present as a hydrophobic anchor to lipopolysaccharide present exterior to the outer membrane. Inhibition of lipid A in LPS-deficient mutants led to growth defects, which indicate that lipid A is essential for the bacterium. Picomolar lipid A levels are seen to trigger TLR4/MD activation leading to acute inflammation in the mammalian immune system, thereby proving its potency as an endotoxin [224,225]. The biosynthetic pathway of Lipid A can be used to develop drugs. One such drug target is LpxC, the second enzyme in the biosynthetic pathway [226]. Several hydroxamates containing candidates are being developed, and clinical trials are on. This pathway can be further used for therapeutic purposes. As seen in the Kdo2-lipid molecule from E.coli, there are two phosphate groups, which are important for TLR4/MD2 activation [227]. Mono-phospholipid A can be prepared and used as an adjuvant due to its capability to partially activate TLR4/MD2 [228].
Some species of bacteria, like Francisella tularensis, produce amino-phospholipid A due to phosphatases like LpxE and LpxF, which remove the phosphates of Lipid A. The LpxE gene was chemically engineered into a Salmonella strain that acts as a potent oral vaccine candidate [229]. Lipid A itself cannot have any therapeutic application, although its modified form, like Lipid IV-A, can have therapeutic applications [230]. Lipid IV-A is an antagonist of TLR4 in humans and agonists in murine cells. Lipid IV-A could not be utilized due to its instability upon storage, leading to synthetic analogs like E5531 and E5564 [231]. Although E5564 was not able to reduce the mortality of septic patients, it protected mice from lethal influenza infection, showing that TLR4 can be one possible drug target in viral infection [232]. Numerous sphingolipids extracted and purified from natural sources have been known to have bactericidal activity against various microorganisms, including Gram-positive bacteria, Gram-negative bacteria, fungi, or microalgae. Ceramides, as discussed earlier too, are a group of sphingolipids that are structurally heterogeneous and complex, containing derivatives of fatty acids in amide linkage with a variety of fatty acids. Short-chain C6- ceramides and a functionalized ω-azido C6-ceramide were found to have antimicrobial properties against Neisseria meningitidis and Neisseria gonorrhoeae.
Further studies have shown that these molecules were incorporated into the bacterial cell membrane and its functionality by dissipating its membrane potential [233]. As we know, Cystic fibrosis patients are predisposed to infection by Pseudomonas aeruginosa. Inhalation of mice with sphingosine or acid ceramidase helped in the recovery of this condition. This treatment successfully inhibited other pathogens that affected cystic fibrosis patients, including Acinetobacter baumannii, Moraxella catarrhalis, Hemophilus influenzae, Burkholderia cepacia opening a new horizon for treatment of lung infection by inhalation of sphingosine or ceramidase [234]. Administration of myriocin in intestinal epithelial SW480 cells, an inhibitor of de novo biosynthesis of sphingolipids, facilitates the intracellular proliferation of wildtype Salmonella Typhimurium by inhibiting autophagy and HBD-2 dependent response [235]. Also, ceramide analogs with modified 1-position, e.g., 1-O-methyl modification, have been found to have potent inhibition on Chlamydia trachomatis L2 strain with IC50 being in the micromolar or sub-micromolar range. The exact mechanism is yet to be elucidated, but it is speculated that these compounds may target a host molecule that the bacteria is using for its sphingosine acquisition [236]. The coating of hydroxyapatite surfaces with sphingosine and its derivatives decreased the adherence and biofilm-forming potential of Streptococcus mutans [237]. The supplementation of susceptible cystic fibrosis mice with C18-sphingosines by inhalation provided them a survival advantage against the infection caused by pulmonary Staphylococcus aureus [238]. As we already know, sphingosine-1 phosphate plays a critical role in inhibiting Mtb pathogenesis. Enhanced levels of Sphingosine-1-phosphate can be used as a novel therapeutic strategy to combat mycobacterial infections by boosting overall host immunity.
Another common application of lipids is in sanitizers used for fresh poultry animal carcass sanitization to decrease human pathogens’ bacterial burden, including Salmonella enterica. Cetyl pyridinium chloride (CPC) is a quaternary sanitizer that produces loss of membrane integrity in Salmonella, leading to loss of electrostatic repulsion between cells cytoplasmic content leakage and emulsification of the cytoplasmic contents by membrane lipids [239]. Lipids as emerging as a potential biomarker to differentiate between bacterial and viral infections. Detecting these infections molecular and culture-based techniques is available, but they take 24–48 hours to give results and are associated with many false-positive and false-negative cases. Also, antibiotics are administered as a precautionary measure, leading to the rampant misuse of these drugs. Hence, a more reliable and faster diagnostic test is the necessity of the moment. Comparing the lipidomic profile of two groups of febrile children with confirmed bacterial and viral infection revealed some glycerophosphoinositol species, sphingomyelin, lysophosphatidylcholine, and cholesterol sulfate that were higher in the virus-infected group. In contrast, some species of glycerophosphocholine, fatty acids, lactosylceramide, and bilirubin were higher in the confirmed bacterial group. The increase in cholesterol sulfate during viral infection might reflect its necessity in cellular lipid biogenesis and T cell signaling during viral infection. The elevated levels of lysophosphatidylcholine might be attributed to its role inducing membrane curvature needed for virus budding. Lactosylceramide LacCer (d18:1/24:1) and LacCer (d18:1/16:0) were higher in a bacteria-infected group than in a virally infected group. This might be due to lactosyl-ceramides’ property to act as Pathogen Recognition Receptors (PRRs) to Pathogen Associated Molecular Patterns (PAMPs). Lactosyl-ceramide with long fatty acid chains, i.e., LacCer(d18:1/24:1), increased as it is essential for the formation of LacCer-Lyn complexes on neutrophils, which happen to be responsible for αMβ2- mediated phagocytosis. Sulfatides were higher in the group with bacterial infection as they are known for immune system regulation during infection. Five species of glycerophosphocholine, i.e., PC(16:0/18:2), PC(18:0/18:1), PC(18:0/18:2), PC (16:0/16:0), PC(16:0/18:1), and bilirubin were higher in case of bacterial infection, although their role in infection is still unclear. This study had been confined to children within the age of 1 month to 9 years, and as metabolism changes with age, further studies need to be done in adult groups [31]. Recent studies have also revealed that infection and inflammation can induce metabolic changes and changes in lipidomics. Infection and inflammation can induce the Acute-Phase Protein Response (APR) that can alter lipid and lipoprotein metabolism to neutralize invading microorganisms and participate in the local immune response. A notable increase in the plasma triglyceride levels can result from increased VLDL secretion due to adipose tissue lipolysis, suppression of fatty acid oxidation, and increased de novo fatty acid synthesis. In animals, this hyper-triglyceridemic effect is brought about by LPS and Lipoteichoic acid (LTA, also a component of the cell wall in Gram-positive bacteria) and is influenced by multiple cytokines. LPS and cytokines’ hyper-triglyceridemic effect is rapid, start within 2 hours of administration, and continue till 24 hours. Also, it is seen that more severe infection leads to decreased VLDL clearance following decreased lipoprotein lipase and apolipoprotein E in VLDL. In rodents, increased hepatic cholesterol synthesis and decreased LDL clearance, conversion of cholesterol to bile acids, and cholesterol secretion to bile attribute to hypercholesterolemia. There are marked alterations in proteins important for HDL metabolism, leading to decreased reverse cholesterol transport and cholesterol delivery to immune cells increase. There is an increase in the oxidation of LDL and VLDL. HDL becomes a proinflammatory molecule. Lipoproteins become enriched in ceramide, glucosylceramide, and sphingomyelin, enhancing uptake by macrophages. Thus, it can be understood that molecular mechanisms that decrease the synthesis of many proteins during APR decrease several nuclear hormone receptors like liver X receptor and farnesoid X receptor. Thus, it can be understood that APR acts through lipid metabolism to protect the host cell from bacteria and viruses. Animals showed improved survival when infusions of synthetic chylomicrons or triglyceride-proteins were administered 30 mins after exposure to LPS, indicating that lipoprotein may have a therapeutic role during endotoxemia. Hypolipidemic mice were more sensitive to LPS-induced lethality, which was reversed by increasing serum lipid concentration to the physiological range [240]. Once fully characterized, these studies hold immense potential to be applied in laboratories as one important and reliant diagnostic test.
Conclusion and future perspective
Lipid plays many crucial roles in maintaining the cellular structure and function homeostasis, including cell membrane fluidity, endocytosis, signaling, energy reserve, and membrane trafficking. A vast amount of literature suggests distinct microdomains (enriched with cholesterols and sphingosine) that maintain proper receptor interaction, subsequently inducing signaling cascade inside the cell, as we know that these are time and again exploited by various pathogens. Various bacterial toxins utilize these plasma membrane microdomains to aid in its entry into the host cells. Pathogens that survive intracellularly employ various host-derived lipids for their entry into the cell. Also, Mycobacteria sp. is foremost to exploit host lipids because they often feed off host lipids as their sole carbon source. Few small lipid molecules play a role in immune signaling to escape the immune surveillance of bacteria such as Mycobacteria sp., Salmonella enterica, Enteropathogenic E. coli, Streptococcus pneumoniae, and Pseudomonas aeruginosa have developed stealthy strategies which bypasses the signaling via EP receptor-mediated pathway and hence mediate immunosuppression which in turn increases the bacterial proliferation. However, lipids such as sphingosine-1-phosphate have a much more debatable role in limiting or flaring up bacterial infections and can act in both ways. So, in cases where limiting the infection can be used as a therapeutic enhancement of the S1P function. There has been an increase in research to deduce the exact molecular mechanism employed by bacteria to execute these above-stated functions. This would not only help us to understand molecular pathogenesis but would also have implications on therapeutic target designing. As the antibiotic resistance era is a current emerging concern, targeting these host lipids hijacked by bacteria can be an excellent antibacterial molecule [241,242]. However, these will pose a greater overall risk to the host because targeting any host’s intrinsic homeostatic biomolecules might significantly enhance side effects. Therefore, research around host lipid- bacterial infection dynamics is all the more important and demands more attention to the open-ended questions in this field. Hence, this review will be of benefit and interest to this field of research.
Supplementary Material
Acknowledgments
We thank the financial support from Department of Biotechnology (DBT), Ministry of Science and Technology; Department of Science and Technology (DST), Ministry of Science and Technology. DC acknowledges DAE for the SRC outstanding investigator award and funds and ASTRA Chair Professorship funds. The authors jointly acknowledge the DBT-IISc partnership programme. Infrastructure support from ICMR (Center for Advanced Study in Molecular Medicine), DST (FIST), and UGC-CAS (special assistance) is acknowledged. RC duly acknowledges CSIR-SRF fellowship. ARC acknowledges Shamrao M. Kaikini and Krishna S. Kaikini fellowship. DM acknowledges IISc fellowship.
Funding Statement
This work was supported by the Department of Atomic Energy, Government of India [DAE0137].
Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Jackson CL, Walch L, Verbavatz JM.. Lipids and their trafficking: an integral part of cellular organization. Dev Cell. 2016;39(2):139–153. [DOI] [PubMed] [Google Scholar]
- [2].Manes S, Del Real G, Martinez AC. Pathogens: raft hijackers. Nat Rev Immunol. 2003;3(7):557–568. [DOI] [PubMed] [Google Scholar]
- [3].Nicola AV, Aguilar HC, Mercer J, et al. Virus entry by endocytosis. Advances in virology 2013; 2013:469538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cossart P, Helenius A. Endocytosis of viruses and bacteria. Cold Spring Harb Perspect Biol. 2014;6(8):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rottner K, Stradal TE, Wehland J. Bacteria-host-cell interactions at the plasma membrane: stories on actin cytoskeleton subversion. Dev Cell. 2005;9(1):3–17. [DOI] [PubMed] [Google Scholar]
- [6].Pizarro-Cerda J, Cossart P. Bacterial adhesion and entry into host cells. Cell. 2006;124(4):715–727. [DOI] [PubMed] [Google Scholar]
- [7].Zaidi T, Bajmoczi M, Zaidi T, et al. Disruption of CFTR-dependent lipid rafts reduces bacterial levels and corneal disease in a murine model of Pseudomonas aeruginosa keratitis. Investigative ophthalmology & visual science 2008; 49:1000–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Carabeo RA, Mead DJ, Hackstadt T. Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proc Natl Acad Sci U S A. 2003;100(11):6771–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dalebroux ZD, Miller SI. Salmonellae PhoPQ regulation of the outer membrane to resist innate immunity. Curr Opin Microbiol. 2014;17:106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pages JM, James CE, Winterhalter M. The porin and the permeating antibiotic: a selective diffusion barrier in Gram-negative bacteria. Nat Rev Microbiol. 2008;6(12):893–903. [DOI] [PubMed] [Google Scholar]
- [11].Catron DM, Sylvester MD, Lange Y, et al. The Salmonella-containing vacuole is a major site of intracellular cholesterol accumulation and recruits the GPI-anchored protein CD55. Cellular microbiology 2002; 4:315–28. [DOI] [PubMed] [Google Scholar]
- [12].Seveau S, Bierne H, Giroux S, et al. Role of lipid rafts in E-cadherin-- and HGF-R/Met--mediated entry of Listeria monocytogenes into host cells. The Journal of cell biology 2004; 166:743–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Riff JD, Callahan JW, Sherman PM. Cholesterol-enriched membrane microdomains are required for inducing host cell cytoskeleton rearrangements in response to attaching-effacing Escherichia coli. Infect Immun. 2005;73(11):7113–7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bloch H, Segal W. Biochemical differentiation of Mycobacterium tuberculosis grown in vivo and in vitro. J Bacteriol. 1956;72(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Smith PF. Biosynthesis of cholesteryl glucoside by Mycoplasma gallinarum. J Bacteriol. 1971;108(3):986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Smith PF, Mayberry WR. Identification of the major glycolipid from Mycoplasma sp., strain J as 3,4,6-triacyl-beta-Glucopyranose. Biochemistry. 1968;7(8):2706–2710. [DOI] [PubMed] [Google Scholar]
- [17].Slutzky GM, Razin S, Kahane I, et al. Cholesterol transfer from serum lipoproteins to mycoplasma membranes. Biochemistry 1977; 16:5158–63. [DOI] [PubMed] [Google Scholar]
- [18].Nagamatsu T, Schust DJ. The immunomodulatory roles of macrophages at the maternal-fetal interface. Reprod Sci. 2010;17(3):209–218. [DOI] [PubMed] [Google Scholar]
- [19].Chen M, Divangahi M, Gan H, et al. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. The Journal of experimental medicine 2008; 205:2791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bansal K, Narayana Y, Patil SA, et al. bovis BCG induced expression of COX-2 involves nitric oxide-dependent and -independent signaling pathways. Journal of leukocyte biology 2009; 85:804–16. [DOI] [PubMed] [Google Scholar]
- [21].Callahan MK, Williamson P, Schlegel RA. Surface expression of phosphatidylserine on macrophages is required for phagocytosis of apoptotic thymocytes. Cell Death Differ. 2000;7(7):645–653. [DOI] [PubMed] [Google Scholar]
- [22].Barnawi J, Tran H, Jersmann H, et al. Potential Link between the Sphingosine-1-Phosphate (S1P) System and Defective Alveolar Macrophage Phagocytic Function in Chronic Obstructive Pulmonary Disease (COPD). PloS one 2015; 10:e0122771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Parihar SP, Guler R, Lang DM, et al. Simvastatin enhances protection against Listeria monocytogenes infection in mice by counteracting Listeria-induced phagosomal escape. PloS one 2013; 8:e75490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2008;9(2):162–176. [DOI] [PubMed] [Google Scholar]
- [25].Wenk MR. Lipidomics of host-pathogen interactions. FEBS Lett. 2006;580(23):5541–5551. [DOI] [PubMed] [Google Scholar]
- [26].Helms B. Host-Pathogen interactions: lipids grease the way. Eur J Lipid Sci Technol. 2006;108(11):895–897. [Google Scholar]
- [27].Ghasemiyeh P, Mohammadi-Samani S. Solid lipid nanoparticles and nanostructured lipid carriers as novel drug delivery systems: applications, advantages and disadvantages. Res Pharm Sci. 2018;13(4):288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Haider M, Abdin SM, Kamal L, et al. Nanostructured Lipid Carriers for Delivery of Chemotherapeutics: A Review. Pharmaceutics 2020; 12 3 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Malin JJ, de Leeuw E. Therapeutic compounds targeting Lipid II for antibacterial purposes. Infect Drug Resist. 2019;12:2613–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Weigert A, Olesch C, Brune B. Sphingosine-1-phosphate and macrophage biology-how the sphinx tames the big eater. Front Immunol. 2019;10:1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang X, Nijman R, Camuzeaux S, et al. Plasma lipid profiles discriminate bacterial from viral infection in febrile children. Scientific reports 2019; 9:17714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Waltermann M, Steinbuchel A. Neutral lipid bodies in prokaryotes: recent insights into structure, formation, and relationship to eukaryotic lipid depots. J Bacteriol. 2005;187(11):3607–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang C, Liu P. The lipid droplet: a conserved cellular organelle. Protein Cell. 2017;8(11):796–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nelson DL, Cox MM, Lehninger AL. Lehninger principles of biochemistry. W.H. Freeman and Company, New York, 2019. [Google Scholar]
- [35].Woese CR, Fox GE. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A. 1977;74(11):5088–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Raetz CR, Dowhan W. Biosynthesis and function of phospholipids in Escherichia coli. J Biol Chem. 1990;265(3):1235–1238. [PubMed] [Google Scholar]
- [37].Raetz CR. Molecular genetics of membrane phospholipid synthesis. Annu Rev Genet. 1986;20(1):253–295. [DOI] [PubMed] [Google Scholar]
- [38].Lopez-Lara IM, Geiger O. Bacterial lipid diversity. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862(11):1287–1299. [DOI] [PubMed] [Google Scholar]
- [39].Teng O, Ang CKE, Guan XL. Macrophage-Bacteria Interactions-A Lipid-Centric Relationship. Front Immunol. 2017;8:1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Huang YL, Morales-Rosado J, Ray J, et al. Toll-like receptor agonists promote prolonged triglyceride storage in macrophages. J Biol Chem 2014; 289:3001–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Simpson BW, Trent MS. Pushing the envelope: LPS modifications and their consequences. Nat Rev Microbiol. 2019;17(7):403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].van Meer G, de Kroon AI. Lipid map of the mammalian cell. J Cell Sci. 2011;124(Pt 1):5–8. [DOI] [PubMed] [Google Scholar]
- [43].Sprong H, Kruithof B, Leijendekker R, et al. UDP-galactose:ceramide galactosyltransferase is a class I integral membrane protein of the endoplasmic reticulum. J Biol Chem 1998; 273:25880–8. [DOI] [PubMed] [Google Scholar]
- [44].Bell RM, Ballas LM, Coleman RA. Lipid topogenesis. J Lipid Res. 1981;22(3):391–403. [PubMed] [Google Scholar]
- [45].van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9(2):112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kunz TC, Kozjak-Pavlovic V. Diverse Facets of Sphingolipid Involvement in Bacterial Infections. Front Cell Dev Biol. 2019;7:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Futerman AH, Riezman H. The ins and outs of sphingolipid synthesis. Trends Cell Biol. 2005;15(6):312–318. [DOI] [PubMed] [Google Scholar]
- [48].Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443(7112):651–657. [DOI] [PubMed] [Google Scholar]
- [49].Bilanges B, Posor Y, Vanhaesebroeck B. PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev Mol Cell Biol. 2019;20(9):515–534. [DOI] [PubMed] [Google Scholar]
- [50].Los FC, Randis TM, Aroian RV, et al. Role of pore-forming toxins in bacterial infectious diseases. Microbiology and Molecular Biology Reviews 2013; 77:173–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Iacovache I, Bischofberger M, van der Goot FG. Structure and assembly of pore-forming proteins. Curr Opin Struct Biol. 2010;20(2):241–246. [DOI] [PubMed] [Google Scholar]
- [52].Tweten RK, Parker MW, Johnson AE. The cholesterol-dependent cytolysins. Curr Top Microbiol Immunol. 2001;257:15–33. [DOI] [PubMed] [Google Scholar]
- [53].Legros N, Pohlentz G, Runde J, et al. Colocalization of receptors for Shiga toxins with lipid rafts in primary human renal glomerular endothelial cells and influence of D-PDMP on synthesis and distribution of glycosphingolipid receptors. Glycobiology 2017; 27:947–65. [DOI] [PubMed] [Google Scholar]
- [54].Ugarte-Torres A, Perry S, Franko A, et al. Multidrug-resistant Aeromonas hydrophila causing fatal bilateral necrotizing fasciitis in an immunocompromised patient: a case report. J Med Case Rep 2018; 12:326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Abrami L, van Der Goot FG. Plasma membrane microdomains act as concentration platforms to facilitate intoxication by aerolysin. J Cell Biol. 1999;147(1):175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Podobnik M, Kisovec M, Anderluh G. Molecular mechanism of pore formation by aerolysin-like proteins. Philos Trans R Soc Lond B Biol Sci. 2017;372(1726):1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Eifler N, Vetsch M, Gregorini M, et al. Cytotoxin ClyA from Escherichia coli assembles to a 13-meric pore independent of its redox-state. EMBO J 2006; 25:2652–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Titball RW. Bacterial phospholipases C. Microbiol Rev. 1993;57(2):347–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Titball RW, Leslie DL, Harvey S, et al. Hemolytic and sphingomyelinase activities of Clostridium perfringens alpha-toxin are dependent on a domain homologous to that of an enzyme from the human arachidonic acid pathway. Infection and Immunity 1991; 59:1872–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Beddoe T, Paton AW, Le Nours J, et al. Structure, biological functions and applications of the AB5 toxins. Trends in biochemical sciences 2010; 35:411–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lafont F, Abrami L, van der Goot FG. Bacterial subversion of lipid rafts. Curr Opin Microbiol. 2004;7(1):4–10. [DOI] [PubMed] [Google Scholar]
- [62].Chinnapen DJ, Chinnapen H, Saslowsky D, et al. Rafting with cholera toxin: endocytosis and trafficking from plasma membrane to ER. FEMS microbiology letters 2007; 266:129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wernick NL, Chinnapen DJ, Cho JA, et al. Cholera Toxin: An Intracellular Journey into the Cytosol by Way of the Endoplasmic Reticulum. Toxins 2010; 2:310–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bouvard V, Baan R, Straif K, et al. A review of human carcinogens--Part B: biological agents. The Lancet Oncology 2009; 10:321–2. [DOI] [PubMed] [Google Scholar]
- [65].Montecucco C, Papini E, de Bernard M, et al. Molecular and cellular activities of Helicobacter pylori pathogenic factors. FEBS letters 1999; 452:16–21. [DOI] [PubMed] [Google Scholar]
- [66].Schraw W, Li Y, McClain MS, et al. Association of Helicobacter pylori vacuolating toxin (VacA) with lipid rafts. The Journal of biological chemistry 2002; 277:34642–50. [DOI] [PubMed] [Google Scholar]
- [67].Triantafilou M, Miyake K, Golenbock DT, et al. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. Journal of cell science 2002; 115:2603–11. [DOI] [PubMed] [Google Scholar]
- [68].Moya M, Dautry-Varsat A, Goud B, et al. Inhibition of coated pit formation in Hep2 cells blocks the cytotoxicity of diphtheria toxin but not that of ricin toxin. The Journal of cell biology 1985; 101:548–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sandvig K, van Deurs B. Membrane traffic exploited by protein toxins. Annu Rev Cell Dev Biol. 2002;18(1):1–24. [DOI] [PubMed] [Google Scholar]
- [70].Pitard I, Malliavin TE. Structural Biology and Molecular Modeling to Analyze the Entry of Bacterial Toxins and Virulence Factors into Host Cells. Toxins (Basel). 2019;11(6):369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Spilsberg B, Hanada K, Sandvig K. Diphtheria toxin translocation across cellular membranes is regulated by sphingolipids. Biochem Biophys Res Commun. 2005;329(2):465–473. [DOI] [PubMed] [Google Scholar]
- [72].Abrami L, Liu S, Cosson P, et al. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. The Journal of cell biology 2003; 160:321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Friebe S, van der Goot FG, Burgi J. The Ins and Outs of Anthrax Toxin. Toxins (Basel). 2016;8(3):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Legros N, Dusny S, Humpf HU, et al. Shiga toxin glycosphingolipid receptors and their lipid membrane ensemble in primary human blood-brain barrier endothelial cells. Glycobiology 2017; 27:99–109. [DOI] [PubMed] [Google Scholar]
- [75].Gekara NO, Weiss S. Lipid rafts clustering and signalling by listeriolysin O. Biochem Soc Trans. 2004;32(Pt 5):712–714. [DOI] [PubMed] [Google Scholar]
- [76].Sun R, Liu Y. Listeriolysin O as a strong immunogenic molecule for the development of new anti-tumor vaccines. Hum Vaccin Immunother. 2013;9(5):1058–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Do Vale A, Cabanes D, Sousa S. Bacterial Toxins as Pathogen Weapons Against Phagocytes. Front Microbiol. 2016;7:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Rudkin JK, McLoughlin RM, Preston A, et al. Bacterial toxins: Offensive, defensive, or something else altogether? PLoS Pathog 2017; 13:e1006452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Janik E, Ceremuga M, Saluk-Bijak J, et al. Biological Toxins as the Potential Tools for Bioterrorism. Int J Mol Sci 2019; 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1(1):31–39. [DOI] [PubMed] [Google Scholar]
- [81].Selvarangan R, Goluszko P, Popov V, et al. Role of decay-accelerating factor domains and anchorage in internalization of Dr-fimbriated Escherichia coli. Infection and immunity 2000; 68:1391–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Peiffer I, Servin AL, Bernet-Camard MF. Piracy of decay-accelerating factor (CD55) signal transduction by the diffusely adhering strain Escherichia coli C1845 promotes cytoskeletal F-actin rearrangements in cultured human intestinal INT407 cells. Infect Immun. 1998;66(9):4036–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Stuart AD, Eustace HE, McKee TA, et al. A novel cell entry pathway for a DAF-using human enterovirus is dependent on lipid rafts. Journal of virology 2002; 76:9307–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Pan J, Zhang L, Odenwald MA, et al. Expression of human decay-accelerating factor on intestinal epithelium of transgenic mice does not facilitate infection by the enteral route. Journal of virology 2015; 89:4311–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Rours GI, Duijts L, Moll HA, et al. Chlamydia trachomatis infection during pregnancy associated with preterm delivery: a population-based prospective cohort study. European journal of epidemiology 2011; 26:493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Loh LN, EMC McCarthy, Narang P, et al. Escherichia coli K1 utilizes host macropinocytic pathways for invasion of brain microvascular endothelial cells. Traffic 2017; 18:733–46. [DOI] [PubMed] [Google Scholar]
- [87].Munoz B, West S. Trachoma: the forgotten cause of blindness. Epidemiol Rev. 1997;19(2):205–217. [DOI] [PubMed] [Google Scholar]
- [88].Baneke A. Review: targeting trachoma: strategies to reduce the leading infectious cause of blindness. Travel Med Infect Dis. 2012;10(2):92–96. [DOI] [PubMed] [Google Scholar]
- [89].Subtil A, Wyplosz B, Balana ME, et al. Analysis of Chlamydia caviae entry sites and involvement of Cdc42 and Rac activity. Journal of cell science 2004; 117:3923–33. [DOI] [PubMed] [Google Scholar]
- [90].Norkin LC, Wolfrom SA, Stuart ES. Association of caveolin with Chlamydia trachomatis inclusions at early and late stages of infection. Exp Cell Res. 2001;266(2):229–238. [DOI] [PubMed] [Google Scholar]
- [91].Stuart ES, Webley WC, Norkin LC. Lipid rafts, caveolae, caveolin-1, and entry by Chlamydiae into host cells. Exp Cell Res. 2003;287(1):67–78. [DOI] [PubMed] [Google Scholar]
- [92].Webley WC, Norkin LC, Stuart ES. Caveolin-2 associates with intracellular chlamydial inclusions independently of caveolin-1. BMC Infect Dis. 2004;4(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Gabel BR, Elwell C, van Ijzendoorn SC, et al. Lipid raft-mediated entry is not required for Chlamydia trachomatis infection of cultured epithelial cells. Infection and immunity 2004; 72:7367–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Stallmann S, Hegemann JH. The Chlamydia trachomatis Ctad1 invasin exploits the human integrin beta1 receptor for host cell entry. Cell Microbiol. 2016;18(5):761–775. [DOI] [PubMed] [Google Scholar]
- [95].Elwell C, Mirrashidi K, Engel J. Chlamydia cell biology and pathogenesis. Nat Rev Microbiol. 2016;14(6):385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Witkin SS, Minis E, Athanasiou A, et al. Chlamydia trachomatis: the Persistent Pathogen. Clinical and vaccine immunology : CVI 2017; 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].van der Goot FG, Tran van Nhieu G, Allaoui A, et al. Rafts can trigger contact-mediated secretion of bacterial effectors via a lipid-based mechanism. The Journal of biological chemistry 2004; 279:47792–8. [DOI] [PubMed] [Google Scholar]
- [98].Mellouk N, Enninga J. Cytosolic Access of Intracellular Bacterial Pathogens: the Shigella Paradigm. Front Cell Infect Microbiol. 2016;6:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Gilk SD, Cockrell DC, Luterbach C, et al. Bacterial colonization of host cells in the absence of cholesterol. PLoS pathogens 2013; 9:e1003107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Ferrari G, Langen H, Naito M, Pieters J. A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell 1999; 97:435–47. [DOI] [PubMed] [Google Scholar]
- [101].Gatfield J, Pieters J. Essential role for cholesterol in entry of mycobacteria into macrophages. Science. 2000;288(5471):1647–1650. [DOI] [PubMed] [Google Scholar]
- [102].Watarai M, Makino S, Michikawa M, et al. Macrophage plasma membrane cholesterol contributes to Brucella abortus infection of mice. Infection and immunity 2002; 70:4818–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Martin-Martin AI, Vizcaino N, Fernandez-Lago L. Cholesterol, ganglioside GM1 and class A scavenger receptor contribute to infection by Brucella ovis and Brucella canis in murine macrophages. Microbes Infect. 2010;12(3):246–251. [DOI] [PubMed] [Google Scholar]
- [104].Tamilselvam B, Daefler S. Francisella targets cholesterol-rich host cell membrane domains for entry into macrophages. J Immunol. 2008;180(12):8262–8271. [DOI] [PubMed] [Google Scholar]
- [105].Chastre J, Fagon JY. Ventilator-associated pneumonia. Am J Respir Crit Care Med. 2002;165(7):867–903. [DOI] [PubMed] [Google Scholar]
- [106].Garau J, Gomez L. Pseudomonas aeruginosa pneumonia. Curr Opin Infect Dis. 2003;16(2):135–143. [DOI] [PubMed] [Google Scholar]
- [107].Hutchison ML, Govan JR. Pathogenicity of microbes associated with cystic fibrosis. Microbes Infect. 1999;1(12):1005–1014. [DOI] [PubMed] [Google Scholar]
- [108].Govan JR, Nelson JW. Microbiology of lung infection in cystic fibrosis. Br Med Bull. 1992;48(4):912–930. [DOI] [PubMed] [Google Scholar]
- [109].Grassme H, Jendrossek V, Riehle A, et al. Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nature medicine 2003; 9:322–30. [DOI] [PubMed] [Google Scholar]
- [110].Grassme H, Schwarz H, Gulbins E. Molecular mechanisms of ceramide-mediated CD95 clustering. Biochem Biophys Res Commun. 2001;284(4):1016–1030. [DOI] [PubMed] [Google Scholar]
- [111].Avota E, de Lira MN, Schneider-Schaulies S. Sphingomyelin Breakdown in T Cells: role of Membrane Compartmentalization in T Cell Signaling and Interference by a Pathogen. Front Cell Dev Biol. 2019;7:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Bibashi E, Memmos D, Kokolina E, et al. Fungal Peritonitis Complicating Peritoneal Dialysis during an 11‐Year Period: Report of 46 Cases. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 2003; 36:927–31. [DOI] [PubMed] [Google Scholar]
- [113].Saiman L, Prince A. Pseudomonas aeruginosa pili bind to asialoGM1 which is increased on the surface of cystic fibrosis epithelial cells. J Clin Invest. 1993;92(4):1875–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Kowalski MP, Pier GB. Localization of cystic fibrosis transmembrane conductance regulator to lipid rafts of epithelial cells is required for Pseudomonas aeruginosa-induced cellular activation. J Immunol. 2004;172(1):418–425. [DOI] [PubMed] [Google Scholar]
- [115].Ni I, Ji C, Vij N. Second-hand cigarette smoke impairs bacterial phagocytosis in macrophages by modulating CFTR dependent lipid-rafts. PLoS One. 2015;10(3):e0121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Melo RC, Dvorak AM. Lipid body-phagosome interaction in macrophages during infectious diseases: host defense or pathogen survival strategy? PLoS Pathog. 2012;8(7):e1002729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Scidmore MA. Recent advances in Chlamydia subversion of host cytoskeletal and membrane trafficking pathways. Microbes Infect. 2011;13(6):527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Hackstadt T, Rockey DD, Heinzen RA, et al. Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. The EMBO journal 1996; 15:964–77. [PMC free article] [PubMed] [Google Scholar]
- [119].Hackstadt T, Scidmore MA, Rockey DD. Lipid metabolism in Chlamydia trachomatis-infected cells: directed trafficking of Golgi-derived sphingolipids to the chlamydial inclusion. Proc Natl Acad Sci U S A. 1995;92(11):4877–4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Rejman Lipinski A, Heymann J, Meissner C, et al. Rab6 and Rab11 regulate Chlamydia trachomatis development and golgin-84-dependent Golgi fragmentation. PLoS pathogens 2009; 5:e1000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Capmany A, Damiani MT. Chlamydia trachomatis intercepts Golgi-derived sphingolipids through a Rab14-mediated transport required for bacterial development and replication. PLoS One. 2010;5(11):e14084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Rzomp KA, Scholtes LD, Briggs BJ, et al. Rab GTPases are recruited to chlamydial inclusions in both a species-dependent and species-independent manner. Infection and immunity 2003; 71:5855–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Cortes C, Rzomp KA, Tvinnereim A, et al. Chlamydia pneumoniae inclusion membrane protein Cpn0585 interacts with multiple Rab GTPases. Infection and immunity 2007; 75:5586–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Moore ER, Mead DJ, Dooley CA, et al. The trans-Golgi SNARE syntaxin 6 is recruited to the chlamydial inclusion membrane. Microbiology 2011; 157:830–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Kumar Y, Cocchiaro J, Valdivia RH. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr Biol. 2006;16(16):1646–1651. [DOI] [PubMed] [Google Scholar]
- [126].Cocchiaro JL, Kumar Y, Fischer ER, et al. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proceedings of the National Academy of Sciences of the United States of America 2008; 105:9379–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Elwell CA, Jiang S, Kim JH, et al. Chlamydia trachomatis co-opts GBF1 and CERT to acquire host sphingomyelin for distinct roles during intracellular development. PLoS pathogens 2011; 7:e1002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Derre I, Swiss R, Agaisse H. The lipid transfer protein CERT interacts with the Chlamydia inclusion protein IncD and participates to ER-Chlamydia inclusion membrane contact sites. PLoS Pathog. 2011;7(6):e1002092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Agaisse H, Derre I. Expression of the effector protein IncD in Chlamydia trachomatis mediates recruitment of the lipid transfer protein CERT and the endoplasmic reticulum-resident protein VAPB to the inclusion membrane. Infect Immun. 2014;82(5):2037–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Beatty WL. Late endocytic multivesicular bodies intersect the chlamydial inclusion in the absence of CD63. Infect Immun. 2008;76(7):2872–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Christian JG, Heymann J, Paschen SA, et al. Targeting of a chlamydial protease impedes intracellular bacterial growth. PLoS pathogens 2011; 7:e1002283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Maldonado-Garcia G, Chico-Ortiz M, Lopez-Marin LM, et al. High-polarity Mycobacterium avium-derived lipids interact with murine macrophage lipid rafts. Scandinavian journal of immunology 2004; 60:463–70. [DOI] [PubMed] [Google Scholar]
- [133].D’Avila H, Melo RC, Parreira GG, et al. Mycobacterium bovis Bacillus Calmette-Guérin Induces TLR2-Mediated Formation of Lipid Bodies: Intracellular Domains for Eicosanoid Synthesis In Vivo. The Journal of Immunology 2006; 176:3087–97. [DOI] [PubMed] [Google Scholar]
- [134].Peyron P, Vaubourgeix J, Poquet Y, et al. Foamy macrophages from tuberculous patients' granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS pathogens 2008; 4:e1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Almeida PE, Silva AR, Maya-Monteiro CM, et al. Mycobacterium bovis bacillus Calmette-Guerin infection induces TLR2-dependent peroxisome proliferator-activated receptor gamma expression and activation: functions in inflammation, lipid metabolism, and pathogenesis. Journal of immunology 2009; 183:1337–45. [DOI] [PubMed] [Google Scholar]
- [136].Waterman SR, Holden DW. Functions and effectors of the Salmonella pathogenicity island 2 type III secretion system. Cell Microbiol. 2003;5(8):501–511. [DOI] [PubMed] [Google Scholar]
- [137].Abrahams GL, Hensel M. Manipulating cellular transport and immune responses: dynamic interactions between intracellular Salmonella enterica and its host cells. Cell Microbiol. 2006;8(5):728–737. [DOI] [PubMed] [Google Scholar]
- [138].Needham BD, Trent MS. Fortifying the barrier: the impact of lipid A remodeling on bacterial pathogenesis. Nature Rev Microbiol. 2013;11(7):467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Haraga A, Ohlson MB, Miller SI. Salmonellae interplay with host cells. Nat Rev Microbiol. 2008;6(1):53–66. [DOI] [PubMed] [Google Scholar]
- [140].Dalebroux ZD, Edrozo MB, Pfuetzner RA, et al. Delivery of cardiolipins to the Salmonella outer membrane is necessary for survival within host tissues and virulence. Cell host & microbe 2015; 17:441–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Christen M, Coye LH, Hontz JS, et al. Activation of a bacterial virulence protein by the GTPase RhoA. Sci Signal 2009; 2:ra71. [DOI] [PMC free article] [PubMed]
- [142].Lafont F, Tran Van Nhieu G, Hanada K, et al. Initial steps of Shigella infection depend on the cholesterol/sphingolipid raft-mediated CD44-IpaB interaction. The EMBO journal 2002; 21:4449–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Duncan MJ, Li G, Shin JS, et al. Bacterial penetration of bladder epithelium through lipid rafts. The Journal of biological chemistry 2004; 279:18944–51. [DOI] [PubMed] [Google Scholar]
- [144].Gilk SD, Beare PA, Heinzen RA. Coxiella burnetii expresses a functional Delta24 sterol reductase. J Bacteriol. 2010;192(23):6154–6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Bougneres L, Helft J, Tiwari S, et al. A role for lipid bodies in the cross-presentation of phagocytosed antigens by MHC class I in dendritic cells. Immunity 2009; 31:232–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].van Manen HJ, Kraan YM, Roos D, et al. Single-cell Raman and fluorescence microscopy reveal the association of lipid bodies with phagosomes in leukocytes. Proc Natl Acad Sci U S A 2005; 102:10159–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Jeschke A, Haas A. Deciphering the roles of phosphoinositide lipids in phagolysosome biogenesis. Commun Integr Biol. 2016;9(3):e1174798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Weigele BA, Alto NM. Salmonella taking charge. Cell Host Microbe. 2010;7(6):421–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Bakowski MA, Braun V, Brumell JH. Salmonella-containing vacuoles: directing traffic and nesting to grow. Traffic. 2008;9(12):2022–2031. [DOI] [PubMed] [Google Scholar]
- [150].Suh CI, Stull ND, Li XJ, et al. The phosphoinositide-binding protein p40phox activates the NADPH oxidase during FcgammaIIA receptor-induced phagocytosis. J Exp Med 2006; 203:1915–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Vergne I, Chua J, Lee HH, et al. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 2005; 102:4033–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Puri RV, Reddy PV, Tyagi AK. Secreted acid phosphatase (SapM) of Mycobacterium tuberculosis is indispensable for arresting phagosomal maturation and growth of the pathogen in guinea pig tissues. PLoS One. 2013;8(7):e70514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Saikolappan S, Estrella J, Sasindran SJ, et al. The fbpA/sapM double knock out strain of Mycobacterium tuberculosis is highly attenuated and immunogenic in macrophages. PLoS One 2012; 7:e36198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Beresford N, Patel S, Armstrong J, et al. MptpB, a virulence factor from Mycobacterium tuberculosis, exhibits triple-specificity phosphatase activity. Biochem J 2007; 406:13–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Singh R, Rao V, Shakila H, et al. Disruption of mptpB impairs the ability of Mycobacterium tuberculosis to survive in guinea pigs. Mol Microbiol 2003; 50:751–62. [DOI] [PubMed] [Google Scholar]
- [156].Anes E, Kuhnel MP, Bos E, et al. Selected lipids activate phagosome actin assembly and maturation resulting in killing of pathogenic mycobacteria. Nat Cell Biol 2003; 5:793–802. [DOI] [PubMed] [Google Scholar]
- [157].LaRocca TJ, Pathak P, Chiantia S, et al. Proving lipid rafts exist: membrane domains in the prokaryote Borrelia burgdorferi have the same properties as eukaryotic lipid rafts. PLoS pathogens 2013; 9:e1003353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Belisle JT, Brandt ME, Radolf JD, et al. Fatty acids of Treponema pallidum and Borrelia burgdorferi lipoproteins. Journal of bacteriology 1994; 176:2151–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Jones JD, Bourell KW, Norgard MV, et al. Membrane topology of Borrelia burgdorferi and Treponema pallidum lipoproteins. Infection and immunity 1995; 63:2424–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Radolf JD, Goldberg MS, Bourell K, et al. Characterization of outer membranes isolated from Borrelia burgdorferi, the Lyme disease spirochete. Infection and immunity 1995; 63:2154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Stubs G, Fingerle V, Wilske B, et al. Acylated cholesteryl galactosides are specific antigens of borrelia causing lyme disease and frequently induce antibodies in late stages of disease. The Journal of biological chemistry 2009; 284:13326–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Stubs G, Fingerle V, Zahringer U, et al. Acylated cholesteryl galactosides are ubiquitous glycolipid antigens among Borrelia burgdorferi sensu lato. FEMS Immunology and Medical Microbiology 2011; 63:140–3. [DOI] [PubMed] [Google Scholar]
- [163].Ben-Menachem G, Kubler-Kielb J, Coxon B, et al. A newly discovered cholesteryl galactoside from Borrelia burgdorferi. Proceedings of the National Academy of Sciences of the United States of America 2003; 100:7913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Schroder NW, Schombel U, Heine H, et al. Acylated cholesteryl galactoside as a novel immunogenic motif in Borrelia burgdorferi sensu stricto. The Journal of biological chemistry 2003; 278:33645–53. [DOI] [PubMed] [Google Scholar]
- [165].Crowley JT, Toledo AM, LaRocca TJ, et al. Lipid exchange between Borrelia burgdorferi and host cells. PLoS pathogens 2013; 9:e1003109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Ansorg R, Müller KD, von Recklinghausen G, et al. Cholesterol binding of Helicobacter pylori. Zentralblatt für Bakteriologie: international journal of medical microbiology 1992; 276:323–9. [DOI] [PubMed] [Google Scholar]
- [167].McGee DJ, George AE, Trainor EA, et al. Cholesterol enhances Helicobacter pylori resistance to antibiotics and LL-37. Antimicrobial agents and chemotherapy 2011; 55:2897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Hildebrandt E, McGee DJ. Helicobacter pylori lipopolysaccharide modification, Lewis antigen expression, and gastric colonization are cholesterol-dependent. BMC Microbiol. 2009;9(1):258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Kotze LA, Young C, Leukes VN, et al. Mycobacterium tuberculosis and myeloid-derived suppressor cells: Insights into caveolin rich lipid rafts. EBioMedicine 2020; 53:102670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Kolattukudy PE, Fernandes ND, Azad AK, et al. Biochemistry and molecular genetics of cell-wall lipid biosynthesis in mycobacteria. Molecular microbiology 1997; 24:263–70. [DOI] [PubMed] [Google Scholar]
- [171].Cole ST, Brosch R, Parkhill J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998; 393:537–44. [DOI] [PubMed] [Google Scholar]
- [172].Liu K, Yu J, Russell DG. pckA-deficient Mycobacterium bovis BCG shows attenuated virulence in mice and in macrophages. Microbiology. 2003;149(Pt 7):1829–1835. [DOI] [PubMed] [Google Scholar]
- [173].Marrero J, Rhee KY, Schnappinger D, et al. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proceedings of the National Academy of Sciences of the United States of America 2010; 107:9819–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, et al. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 2000; 406:735–8. [DOI] [PubMed] [Google Scholar]
- [175].de Carvalho LP, Fischer SM, Marrero J, et al. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chemistry & biology 2010; 17:1122–31. [DOI] [PubMed] [Google Scholar]
- [176].Daniel J, Maamar H, Deb C, et al. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS pathogens 2011; 7:e1002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Trivedi OA, Arora P, Sridharan V, et al. Enzymic activation and transfer of fatty acids as acyl-adenylates in mycobacteria. Nature 2004; 428:441–5. [DOI] [PubMed] [Google Scholar]
- [178].Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A. 2008;105(11):4376–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Yang X, Nesbitt NM, Dubnau E, et al. Cholesterol metabolism increases the metabolic pool of propionate in Mycobacterium tuberculosis. Biochemistry 2009; 48:3819–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Munoz-Elias EJ, Upton AM, Cherian J, et al. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Molecular microbiology 2006; 60:1109–22. [DOI] [PubMed] [Google Scholar]
- [181].Savvi S, Warner DF, Kana BD, et al. Functional Characterization of a Vitamin B12-Dependent Methylmalonyl Pathway in Mycobacterium tuberculosis: Implications for Propionate Metabolism during Growth on Fatty Acids. Journal of bacteriology 2008; 190:3886–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Russell DG, VanderVen BC, Lee W, et al. Mycobacterium tuberculosis wears what it eats. Cell host & microbe 2010; 8:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Raynaud C, Guilhot C, Rauzier J, et al. Phospholipases C are involved in the virulence of Mycobacterium tuberculosis. Molecular microbiology 2002; 45:203–17. [DOI] [PubMed] [Google Scholar]
- [184].Williams KJ, Boshoff HI, Krishnan N, et al. The Mycobacterium tuberculosis beta-oxidation genes echA5 and fadB3 are dispensable for growth in vitro and in vivo. Tuberculosis 2011; 91:549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Graham JE, Clark-Curtiss JE. Identification of Mycobacterium tuberculosis RNAs synthesized in response to phagocytosis by human macrophages by selective capture of transcribed sequences (SCOTS). Proc Natl Acad Sci U S A. 1999;96(20):11554–11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Honer Zu Bentrup K, Miczak A, Swenson DL, et al. Characterization of Activity and Expression of Isocitrate Lyase in Mycobacterium avium and Mycobacterium tuberculosis. Journal of bacteriology 1999; 181:7161–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Munoz-Elias EJ, McKinney JD. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 2005;11(6):638–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Singh V, Jamwal S, Jain R, et al. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell host & microbe 2012; 12:669–81. [DOI] [PubMed] [Google Scholar]
- [189].Neyrolles O. Mycobacteria and the greasy macrophage: getting fat and frustrated. Infect Immun. 2014;82(2):472–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Proia RL, Hla T. Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J Clin Invest. 2015;125(4):1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Garg SK, Volpe E, Palmieri G, et al. Sphingosine 1-phosphate induces antimicrobial activity both in vitro and in vivo. The Journal of infectious diseases 2004; 189:2129–38. [DOI] [PubMed] [Google Scholar]
- [192].Malik ZA, Thompson CR, Hashimi S, et al. Cutting Edge: Mycobacterium tuberculosis Blocks Ca2+ 2+ Signaling and Phagosome Maturation in Human Macrophages Via Specific Inhibition of Sphingosine Kinase. The Journal of Immunology 2003; 170:2811–5. [DOI] [PubMed] [Google Scholar]
- [193].Thompson CR, Iyer SS, Melrose N, et al. Sphingosine kinase 1 (SK1) is recruited to nascent phagosomes in human macrophages: inhibition of SK1 translocation by Mycobacterium tuberculosis. Journal of immunology 2005; 174:3551–61. [DOI] [PubMed] [Google Scholar]
- [194].Prakash H, Luth A, Grinkina N, et al. Sphingosine kinase-1 (SphK-1) regulates Mycobacterium smegmatis infection in macrophages. PloS one 2010; 5:e10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Nadella V, Sharma L, Kumar P, et al. Sphingosine-1-Phosphate (S-1P) Promotes Differentiation of Naive Macrophages and Enhances Protective Immunity Against Mycobacterium tuberculosis. Frontiers in immunology 2019; 10:3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Custodio R, McLean CJ, Scott AE, et al. Characterization of secreted sphingosine-1-phosphate lyases required for virulence and intracellular survival of Burkholderia pseudomallei. Molecular microbiology 2016; 102:1004–19. [DOI] [PubMed] [Google Scholar]
- [197].Kim YI, Yang JY, Ko HJ, Kweon MN, Chang SY. Shigella flexneri Inhibits Intestinal Inflammation by Modulation of Host Sphingosine-1-Phosphate in Mice. Immune network 2014; 14:100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Zhang Y, Li X, Carpinteiro A, et al. Acid sphingomyelinase amplifies redox signaling in Pseudomonas aeruginosa-induced macrophage apoptosis. The Journal of Immunology 2008; 181:4247–54. [DOI] [PubMed] [Google Scholar]
- [199].McCollister BD, Myers JT, Jones-Carson J, et al. Constitutive acid sphingomyelinase enhances early and late macrophage killing of Salmonella enterica serovar Typhimurium. Infection and immunity 2007; 75:5346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Ebenezer DL, Fu P, Krishnan Y, et al. Genetic deletion of Sphk2 confers protection against Pseudomonas aeruginosa mediated differential expression of genes related to virulent infection and inflammation in mouse lung. BMC genomics 2019; 20: 984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Abu Khweek A, Kanneganti A, Guttridge DD, et al. The Sphingosine-1-Phosphate Lyase (LegS2) Contributes to the Restriction of Legionella pneumophila in Murine Macrophages. PloS one 2016; 11:e0146410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Mulye M, Samanta D, Winfree S, et al. Elevated Cholesterol in the Coxiella burnetii Intracellular Niche Is Bacteriolytic. mBio 2017; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Abrams ME, Johnson KA, Perelman SS, et al. Oxysterols provide innate immunity to bacterial infection by mobilizing cell surface accessible cholesterol. Nature microbiology 2020. [DOI] [PMC free article] [PubMed]
- [204].Orecchioni M, Ghosheh Y, Pramod AB, et al. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Frontiers in immunology 2019; 10: 1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Labonte AC, Tosello-Trampont AC, Hahn YS. The role of macrophage polarization in infectious and inflammatory diseases. Mol Cells. 2014;37(4):275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Chen EP, Smyth EM. COX-2 and PGE2-dependent immunomodulation in breast cancer. Prostaglandins Other Lipid Mediat. 2011;96(1–4):14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Planaguma A, Kazani S, Marigowda G, et al. Airway lipoxin A4 generation and lipoxin A4 receptor expression are decreased in severe asthma. American journal of respiratory and critical care medicine 2008; 178:574–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Divangahi M, Chen M, Gan H, et al. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nature immunology 2009; 10:899–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Serezani CH, Chung J, Ballinger MN, et al. Prostaglandin E2 suppresses bacterial killing in alveolar macrophages by inhibiting NADPH oxidase. American journal of respiratory cell and molecular biology 2007; 37:562–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Uchiya K, Nikai T. Salmonella enterica serovar Typhimurium infection induces cyclooxygenase 2 expression in macrophages: involvement of Salmonella pathogenicity island 2. Infect Immun. 2004;72(12):6860–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Divangahi M, Desjardins D, Nunes-Alves C, et al. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nature immunology 2010; 11:751–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Colley CM, Ryman BE. The liposome: from membrane model to therapeutic agent. Trends Biochem Sci. 1976;1(3):203–205. [Google Scholar]
- [213].Gregoriadis G. Drug entrapment in liposomes. FEBS Lett. 1973;36(3):292–296. [DOI] [PubMed] [Google Scholar]
- [214].Pandey R, Sharma S, Khuller GK. Oral solid lipid nanoparticle-based antitubercular chemotherapy. Tuberculosis (Edinb). 2005;85(5–6):415–420. [DOI] [PubMed] [Google Scholar]
- [215].Blakney AK, McKay PF, Yus BI, et al. Inside out: optimization of lipid nanoparticle formulations for exterior complexation and in vivo delivery of saRNA. Gene therapy 2019; 26:363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Mukherjee S, Ray S, Thakur RS. Solid lipid nanoparticles: a modern formulation approach in drug delivery system. Indian J Pharm Sci. 2009;71(4):349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Das S, Ng WK, Tan RB. Are nanostructured lipid carriers (NLCs) better than solid lipid nanoparticles (SLNs): development, characterizations and comparative evaluations of clotrimazole-loaded SLNs and NLCs? Eur J Pharm Sci. 2012;47(1):139–151. [DOI] [PubMed] [Google Scholar]
- [218].Salvi VR, Pawar P. Nanostructured lipid carriers (NLC) system: a novel drug targeting carrier. J Drug Delivery Sci Technol. 2019;51:255–267. [Google Scholar]
- [219].Fang CL, Al-Suwayeh SA, Fang JY. Nanostructured lipid carriers (NLCs) for drug delivery and targeting. Recent Pat Nanotechnol. 2013;7(1):41–55. [PubMed] [Google Scholar]
- [220].Breukink E, de Kruijff B. Lipid II as a target for antibiotics. Nat Rev Drug Discov. 2006;5(4):321–332. [DOI] [PubMed] [Google Scholar]
- [221].Harz H, Burgdorf K, Holtje JV. Isolation and separation of the glycan strands from murein of Escherichia coli by reversed-phase high-performance liquid chromatography. Anal Biochem. 1990;190(1):120–128. [DOI] [PubMed] [Google Scholar]
- [222].Shoji J, Hinoo H, Matsumoto K, et al. Isolation and characterization of katanosins A and B. The Journal of antibiotics 1988; 41:713–8. [DOI] [PubMed] [Google Scholar]
- [223].Shoji J, Hinoo H, Katayama T, et al. Isolation and characterization of new peptide antibiotics, plusbacins A1-A4 and B1-B4. The Journal of antibiotics 1992; 45:817–23. [DOI] [PubMed] [Google Scholar]
- [224].Joo SH. Lipid A as a drug target and therapeutic molecule. Biomol Ther (Seoul). 2015;23(6):510–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Ohto U, Fukase K, Miyake K, et al. Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proceedings of the National Academy of Sciences of the United States of America 2012; 109:7421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Onishi HR, Pelak BA, Gerckens LS, et al. Antibacterial agents that inhibit lipid A biosynthesis. Science 1996; 274:980–2. [DOI] [PubMed] [Google Scholar]
- [227].Rietschel ET, Kirikae T, Schade FU, et al. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 1994; 8:217–25. [DOI] [PubMed] [Google Scholar]
- [228].Persing DH, Coler RN, Lacy MJ, et al. Taking toll: lipid A mimetics as adjuvants and immunomodulators. Trends in microbiology 2002; 10:S32–7. [DOI] [PubMed] [Google Scholar]
- [229].Kong Q, Six DA, Roland KL, et al. Salmonella synthesizing 1-dephosphorylated [corrected] lipopolysaccharide exhibits low endotoxic activity while retaining its immunogenicity. Journal of immunology 2011; 187:412–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Golenbock DT, Hampton RY, Qureshi N, et al. Lipid A-like molecules that antagonize the effects of endotoxins on human monocytes. The Journal of biological chemistry 1991; 266:19490–8. [PubMed] [Google Scholar]
- [231].Christ WJ, Asano O, Robidoux AL, et al. E5531, a pure endotoxin antagonist of high potency. Science 1995; 268:80–3. [DOI] [PubMed] [Google Scholar]
- [232].Shirey KA, Lai W, Scott AJ, et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature 2013; 497:498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Becam J, Walter T, Burgert A, et al. Antibacterial activity of ceramide and ceramide analogs against pathogenic Neisseria. Scientific reports 2017; 7:17627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Pewzner-Jung Y, Tavakoli Tabazavareh S, Grassme H, et al. Sphingoid long chain bases prevent lung infection by Pseudomonas aeruginosa. EMBO molecular medicine 2014; 6:1205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Huang FC. De Novo sphingolipid synthesis is essential for Salmonella-induced autophagy and human beta-defensin 2 expression in intestinal epithelial cells. Gut Pathog. 2016;8(p):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Saied EM, Banhart S, Burkle SE, et al. A series of ceramide analogs modified at the 1-position with potent activity against the intracellular growth of Chlamydia trachomatis. Future medicinal chemistry 2015; 7:1971–80. [DOI] [PubMed] [Google Scholar]
- [237].Cukkemane N, Bikker FJ, Nazmi K, et al. Anti-adherence and bactericidal activity of sphingolipids against Streptococcus mutans. Eur J Oral Sci. 2015;123(4):221–227. [DOI] [PubMed] [Google Scholar]
- [238].Tavakoli Tabazavareh S, Seitz A, Jernigan P, et al. Lack of Sphingosine Causes Susceptibility to Pulmonary Staphylococcus Aureus Infections in Cystic Fibrosis. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 2016; 38:2094–102. [DOI] [PubMed] [Google Scholar]
- [239].Yegin Y, Oh JK, Akbulut M, et al. Cetylpyridinium chloride produces increased zeta-potential on Salmonella Typhimurium cells, a mechanism of the pathogen's inactivation. NPJ science of food 2019; 3:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Khovidhunkit W, Kim MS, Memon RA, et al. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res 2004; 45:1169–96. [DOI] [PubMed] [Google Scholar]
- [241].Poerio N, Bugli F, Taus F, et al. Liposomes loaded with bioactive lipids enhance antibacterial innate immunity irrespective of drug resistance. Scientific reports 2017; 7:45120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Yao J, Rock CO. How bacterial pathogens eat host lipids: implications for the development of fatty acid synthesis therapeutics. J Biol Chem. 2015;290(10):5940–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.