

Abstract
Rationale:
Long QT syndrome (LQTS) is an inheritable disease characterized by prolonged QT interval on the electrocardiogram. The pathogenesis of LQTS is related to mutations in LQTS-susceptible genes encoding cardiac ion channel proteins or subunits.
Patient concerns:
Here, we reported a 37-year-old female Uygur patient with palpitation and loss of consciousness.
Diagnoses:
At the time of admission, a 12-lead electrocardiogram showed a QTc interval of 514 ms. Genetic analysis revealed KCNQ1 G219E and TRPM4 T160M mutations.
Interventions:
Although beta-blockers remain the mainstay in treating LQTS, the patient underwent implantation of an automatic cardioverter defibrillator due to life-threatening arrhythmias.
Outcomes:
To explore the effect of the calcium ion antagonist verapamil on ion channels, we generated human induced pluripotent stem cell cardiomyocytes (hiPSC-CMs) from the peripheral blood mononuclear cells of the patient. The changes of action potential duration in response to verapamil were observed.
Lessons:
Our results showed that patient-derived hiPSC-CMs could recapitulate the electrophysiological features of LQTS and display pharmaceutical responses to verapamil.
Keywords: human induced pluripotent stem cells, long QT syndrome, potassium voltage-gated channel subfamily Q member 1, transient receptor potential cation channel subfamily M member 4
1. Introduction
Long QT syndrome (LQTS) is an inherited cardiac disorder characterized by QT interval prolongation on the electrocardiogram (ECG), leading to occurrence of episodic syncope or sudden cardiac death (SCD).[1,2] Proper treatment in patients with LQTS can dramatically reduce the mortality rate from ∼21% within 1 year since the first syncope to 1% at 15-year follow-up.[3,4] Thus, early diagnosis and intervention are critical in improving the overall survival of patients with LQTS.
The pathogenesis of LQTS has been related to the abnormal function of ion channels resulting from genetic alterations in LQTS-susceptibility genes.[5–7] Genetic testing provides valuable information that may facilitate early clinical intervention in LQTS. Over the past few decades, hundreds of LQTS-causing mutations have been identified in at least 15 LQTS-susceptible genes encoding cardiac ion channels or adaptor proteins.[8] Mutations in three major genes, including KCNQ1, KCNH2, and SCN5A, are responsible for nearly 80% of diagnosed cases,[9,10] whereas the mutations responsible for LQTS cases without these major ones remain elusive. Recently, transient potential melastatin 4 gene (TRPM4) encoding an intracellular Ca2+-activated non-selective cation channel has been involved in LQTS pathogenesis.[11] TRPM4 channels depolarize the membrane by allowing Na+ influx at negative membrane potentials, whereas repolarize the membrane by permitting K+ efflux at positive membrane potentials, playing an important role in the shape of action potential (AP).[12] TRPM4 participates in sensing intracellular Ca2+ by changing cell membrane potential, affecting voltage-dependent Ca2+ channels (VGCC) and non-voltage-dependent Ca2+ channels (NVDCC),[13,14] and the intracellular Ca2+ concentration is 0.4 to 9.8 At nmol/L, TRPM4 is activated, and the activation process is regulated by ATP, calcium-calmodulin and PKC.[15]
Due to the electrophysiological properties of cardiomyocytes (CMs) that vary from species to species, human induced pluripotent stem cells (hiPSCs) are helpful in testing patient-specific therapies for cardiovascular diseases. In previous studies, investigators have generated hiPSCs from fibroblasts and peripheral blood mononuclear cells (PBMCs) from individual patients and have induced them to differentiate into functional CMs[16–18] that can recapitulate the electrophysiological features of LQTS and respond to putative therapeutic agents.[16–20]
2. Case presentation
A 37-year-old female Uygur patient was referred to our hospital for palpitation and loss of consciousness. She had a family history of SCD, a history of intermittent palpitation, and 3 syncope events during the past 2 years. The patient was diagnosed with congenital heart disease based on electrocardiography and echocardiography in another hospital. She had been treated with atropine and propranolol for 1 year after the diagnosis of LOTS. The medications were discontinued in the past year, and the above-mentioned symptoms did not appear until a recent relapse. After this relapse, she was brought to a local hospital where ECG revealed a prolonged QT interval (500 ms). Then, she was referred to our hospital. The patient provided written informed consent.
In the emergency department before any treatment, ECG showed prolonged QT (506 ms) and QTc (514 ms) intervals (Fig. 1). Electrical cardioversion at 150J was performed twice for rhythm control, and continuous infusion of Esmolol was carried out to prevent arrhythmia. Then, the patient recovered from unconsciousness and was admitted to the ward with the diagnosis of LQTS and ventricular tachycardia (VT). One day after admission, she experienced a sudden loss of consciousness. ECG monitoring revealed recurrent episodes of premature ventricular contractions, VT, ventricular fibrillation, and apical torsional VT; 24-h dynamic ECG showed sinus rhythm, frequent ventricular premature beats, paired ventricular premature complexes, short bursts of VT, and ventricular fibrillation. Continuous chest compression and electrical defibrillation at 200J biphasic wave were performed immediately. The sinus rhythm was recovered 6 min later, and the patient regained consciousness. Due to life-threatening arrhythmias, the patient underwent implantation of an automatic cardioverter defibrillator.
Figure 1.
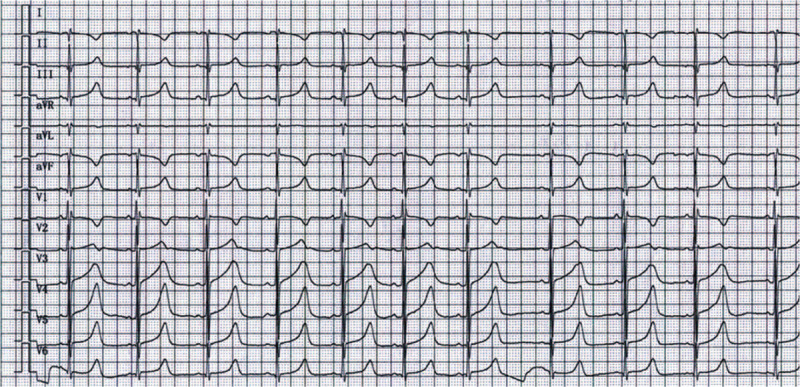
Electrocardiogram (ECG) of the proband with long QT syndrome (LQTS). A 37-year-old Uygur woman with dizziness and palpitations was diagnosed with LQT1 during the clinical evaluation. Surface ECG showed a prolonged QT interval (QT interval corrected for heart rate [QTc], 514 ms).
3. Genetic testing of the proband and her family members
This study was approved by the Ethics Committee of Xinjiang Medical University First Affiliated Hospital (Xinjiang, China). All participants provided written informed consent before blood collection. Whole exome sequencing of DNA from PBMCs of the patient showed that the TRPM4 gene had a T160M mutation resulting from a single base exchange in the fifth exon (479C→T). A G219E missense mutation was also found in the KCNQ1 gene resulting from a heterozygous single base exchange in the fourth exon (656G→A) (Fig. 2B). Subsequent screening of the members of the proband's family revealed prolonged QT in her mother (QTc 516 ms), son (QTc 506 ms), and elder sister's son (QTc 520 ms). The proband and her mother had recurrent syncope. The deceased elder sister experienced sudden death (Fig. 2A).
Figure 2.
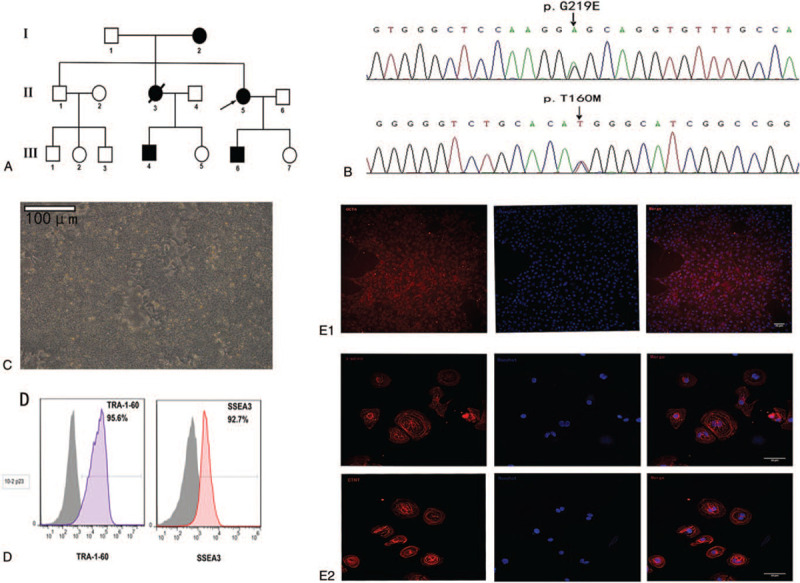
Generation and characterization of human induced pluripotent stem cells (hiPSCs). (A) Pedigree of the proband with LQTS. Screening of the proband's family revealed prolonged QT in her mother (Patient I-2), her son (Patient III-1), and her elder sister's son (Patient III-3). Squares and circles indicate male and female family members, respectively; solid, open, and line-crossed symbols indicate LQTS diagnosed, unaffected, and deceased family members, respectively. (B) Whole exome sequencing was performed on DNA isolated from peripheral blood samples of the proband and her family. (C) Embryonic stem cell-like clusters. (D) Flow cytometry analysis was performed to determine the percentage of iPSCs expressing the pluripotency surface markers SSEA3 and TRA-1-60. (E1 and E2) Immunofluorescent staining of the pluripotency marker OCT4 in iPSCs. Scale bar.
4. Generation and characterization of patient-specific iPSCs
Blood samples were obtained from the proband with LQT1 and her healthy brother. After generation of iPSCs, immunofluorescence staining, and flow cytometry were performed to examine the presence of pluripotency-associated proteins. The iPSC clones were further characterized by flow cytometry to determine the percentage of cells expressing the pluripotency surface markers SSEA and TRA-1–60 (Fig. 2D). The blood sample of the proband was reprogrammed with non-integrating Sendai virus vectors carrying Yamanaka factors (OCT4, SOX2, KLF4, and c-MYC). At day 15 post-transduction, embryonic stem cell (ESC)-like clusters started to appear (Fig. 2C). Immunofluorescent staining showed that the iPSC clones expressed the intracellular pluripotency marker OCT4 (Fig. 2E1). The positive staining of cardiomyocyte markers cTNT and α-actinin suggested that the iPSCs were differentiated into CMs (Fig. 2E2).
To evaluate whether the CMs derived from patient-specific iPSCs recapitulate the disease phenotype, we recorded the APs in single cells. Both spontaneously beating cells dissociated from LQTS and control explants responded to pacing and generated APs. Spontaneous and paced (1 Hz) APs were recorded in patient and control hiPSC derived-CMs. Pacing was conducted to standardize cell contraction rate and minimize the effects of varying cell contraction rates on AP duration (APD). Patient and control hiPSC-CMs were identified by their characteristic AP properties (Fig. 3A). We observed significantly prolonged APD90, APD50, and APD30 in patient hiPSC-CMs compared with control cells during spontaneous contraction (Fig. 3B), suggesting that patient hiPSC-CMs can recapitulate the electrophysiological features of LQTS as measured by patch clamp. In addition, verapamil exposure significantly decreased the QT interval in both control and patient hiPSC-CMs, indicating that hiPSC-CMs were responsive to therapeutic agents (Fig. 4A). The differences in APD between patient and control cells for A-like and N-like hiPSC-CMs were not statistically significant (Fig. 4B).
Figure 3.
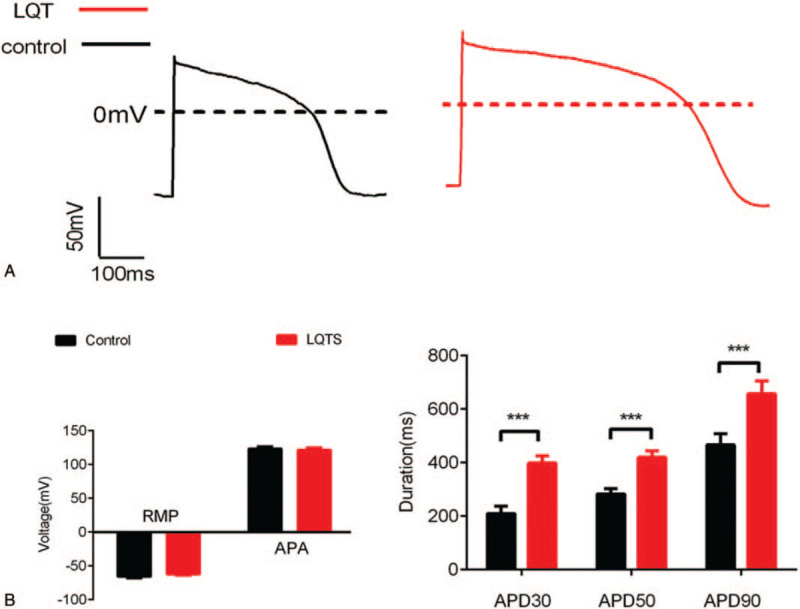
Electrophysiological characterization of hiPSC-derived cardiomyocytes (hiPSC-CMs). Spontaneous and paced (1 Hz) action potentials (AP) (A), resting membrane potential/AP amplitude, and APD90/APD50/APD30 (B) were determined in patient and control hiPSC derived-CMs, respectively. APA = action potential amplitude, APD = action potential duration, RMP = resting membrane potential.
Figure 4.
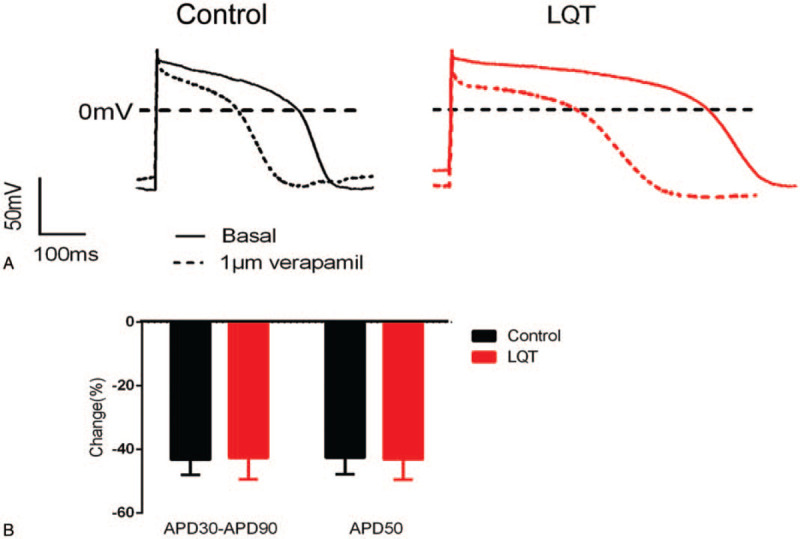
Drug response. HiPSC-MCs derived from the patient and control were treated with verapamil (1 μM). AP (A) and APD30/APD50/APD90 (B) in patient- and control-derived hiPSC-MCs at a 1-Hz stimulation rate were determined. Data are mean ± standard error of the mean (SEM).
5. Discussion
Mutations in KCNQ1, KCNH2, and SCN5A are responsible for ∼80% of cases diagnosed with LQTS, leaving room for the discovery of additional causal genes for this disease.[9,10] In this study, we identified missense mutations G219E and T160M in KCNQ1 and TRPM4, respectively, in a proband from a family affected by LQTS and reprogrammed PBMCs from the proband and one healthy family member to generate functional hiPSCs-CMs. These myocytes showed expression of specific markers and electrophysiological features. In addition, we observed disease-specific prolonged APD resulting from the trafficking defects of KCNQ1 and pharmacological response to verapamil (a calcium channel blocker) treatment in hiPSCs-CMs. Our data suggest that patient-specific hiPSC-CMs can be derived from somatic cells, recapitulate the electrophysiological features of the disorder, and display pharmaceutical responses to therapeutic drugs, serving as a predictive system for drug assessment in LQTS treatment.
The electrophysiological properties of CMs vary from species to species, and animal models can hardly simulate the electrophysiological properties of human cells. For example, downregulation of complementary rapid and slow delayed rectifier K+ currents has been observed in a transgenic animal model, but not in patient-specific myocytes with R190Q-KCNQ1 mutations. This discrepancy may be mutation-dependent or related to the model, highlighting the importance of using a model that can mimic the human condition. Patient-derived hiPSCs provide a promising approach for understanding the pathogenesis and testing patient-specific therapies for cardiovascular diseases in vitro. The pluripotency of hiPSCs and the unlimited number of induced CMs can facilitate a high-throughput drug screening.
KCNQ1 gene mutation reduces the main component of myocardial repolarization outward current Iks, delays ventricular repolarization, and thereby prolongs QT interval. Adrenergic stimulation in healthy individuals during vigorous exercise can increase repolarization outward current while reducing APD via increased Iks, Ica-1, Icl, and Icl-camp, leading to heart rate elevation and QTc shortening. In contrast, the Iks ion channel with structural abnormalities due to KCNQ1 mutation loses adrenaline responsiveness, leading to APD prolongation via increased inward current and decreased outward current. A previous study using a LQT1 model with LQT2 or rapid pacing showed that verapamil can significantly shorten QT interval and APD.[21] In this study, the APD of both normal control and LQT1 patient hiPSCs-derived CMs were significantly decreased in response to 1 μM verapamil, suggesting that these cells may display pharmaceutical responses to therapeutic drugs and could be used in drug screening for LQT therapy.
In conclusion, we reported a LQTS case with KCNQ1 G219E and TRPM4 T160M polymorphisms, and generated patient-specific hiPSCs from PBMCs. These cells were further directed to differentiate into functional hiPSCs-CMs. The AP changes in response to ion channel blockers were measured to evaluate pharmacological responses. Our data suggest that patient-specific hiPSC-CMs can be derived from somatic cells and may serve as a predictive system for drug assessment in LQTS treatment.
Glossary
Abbreviations: AP = action potential, APD = AP duration, CMs = cardiomyocytes, ECG = electrocardiogram, ESC = embryonic stem cell, hiPSCs = human induced pluripotent stem cells, LQTS = long QT syndrome, NVDCC = non-voltage-dependent Ca2 + channels, PBMCs = peripheral blood mononuclear cells, SCD = sudden cardiac death, TRPM4 = transient potential melastatin 4 gene, VT = ventricular tachycardia.
References
- [1].Alders M, Bikker H, Christiaans I. Long QT syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews ((R)). Seattle, WA; 1993. [Google Scholar]
- [2].Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol 2012;5:868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schwartz PJ. Idiopathic long QT syndrome: progress and questions. Am Heart J 1985;109:399–411. [DOI] [PubMed] [Google Scholar]
- [4].Schwartz PJ CL. Long QT and short QT syndromes. In: Zipes DP, Jalife J, eds. Cardiac Electrophysiology: From Cell to Bedside. 5th ed. Philadelphia, PA: Elsevier/Saunders. 2009:731–44. [Google Scholar]
- [5].Keating M, Atkinson D, Dunn C, et al. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science 1991;252:704–6. [DOI] [PubMed] [Google Scholar]
- [6].Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995;80:805–11. [DOI] [PubMed] [Google Scholar]
- [7].Curran ME, Splawski I, Timothy KW, et al. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995;80:795–803. [DOI] [PubMed] [Google Scholar]
- [8].Giudicessi JR, Ackerman MJ. Genotype- and phenotype-guided management of congenital long QT syndrome. Curr Probl Cardiol 2013;38:417–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tester DJ, Ackerman MJ. Novel gene and mutation discovery in congenital long QT syndrome: let's keep looking where the street lamp standeth. Heart Rhythm 2008;5:1282–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Campuzano O, Sarquella-Brugada G, Mademont-Soler I, et al. Identification of genetic alterations, as causative genetic defects in long QT syndrome, using next generation sequencing technology. PLoS One 2014;9:e114894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Demion M, Thireau J, Gueffier M, et al. Trpm4 gene invalidation leads to cardiac hypertrophy and electrophysiological alterations. PLoS One 2014;9:e115256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bianchi B, Ozhathil LC, Medeiros-Domingo A, et al. Four TRPM4 cation channel mutations found in cardiac conduction diseases lead to altered protein stability. Front Physiol 2018;9:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Barbet G, Demion M, Moura IC, et al. The calcium-activated nonselective cation channel TRPM4 is essential for the migration but not the maturation of dendritic cells. Nat Immunol 2008;9:1148–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cheng H, Beck A, Launay P, et al. TRPM4 controls insulin secretion in pancreatic beta-cells. Cell Calcium 2007;41:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nilius B, Prenen J, Tang J, et al. Regulation of the Ca2+ sensitivity of the nonselective cation channel TRPM4. J Biol Chem 2005;280:6423–33. [DOI] [PubMed] [Google Scholar]
- [16].Moretti A, Bellin M, Welling A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med 2010;363:1397–409. [DOI] [PubMed] [Google Scholar]
- [17].Hu J, Wang Y, Jiao J, et al. Patient-specific cardiovascular progenitor cells derived from integration-free induced pluripotent stem cells for vascular tissue regeneration. Biomaterials 2015;73:51–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Malan D, Zhang M, Stallmeyer B, et al. Human iPS cell model of type 3 long QT syndrome recapitulates drug-based phenotype correction. Basic Res Cardiol 2016;111:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Itzhaki I, Maizels L, Huber I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011;471:225–9. [DOI] [PubMed] [Google Scholar]
- [20].Zhang M, D’Aniello C, Verkerk AO, et al. Recessive cardiac phenotypes in induced pluripotent stem cell models of Jervell and Lange-Nielsen syndrome: disease mechanisms and pharmacological rescue. Proc Natl Acad Sci U S A 2014;111:E5383–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu Y, Xue Y, Wu S. Effect of verapamil in the treatment of type 2 long QT syndrome is not a dose-dependent pattern: a study from bedside to bench, and back. Eur Heart J Suppl 2016;18:A37–46. [Google Scholar]