

Abstract
Shiga toxin-producing Escherichia coli (STEC) contamination of agricultural water might be an important factor to recent foodborne illness and outbreaks involving leafy greens. Closed bacterial genomes from whole genome sequencing play an important role in source tracking. We aimed to determine the limits of detection and classification of STECs by qPCR and nanopore sequencing using 24 hour enriched irrigation water artificially contaminated with E. coli O157:H7 (EDL933). We determined the limit of STEC detection by qPCR to be 30 CFU/reaction, which is equivalent to 105 CFU/ml in the enrichment. By using Oxford Nanopore’s EPI2ME WIMP workflow and de novo assembly with Flye followed by taxon classification with a k-mer analysis software (Kraken2), E. coli O157:H7 could be detected at 103 CFU/ml (68 reads) and a complete fragmented E. coli O157:H7 metagenome-assembled genome (MAG) was obtained at 105−108 CFU/ml. Using a custom script to extract the E. coli reads, a completely closed MAG was obtained at 107−108 CFU/ml and a complete, fragmented MAG was obtained at 105−106 CFU/ml. In silico virulence detection for E. coli MAGs for 105−108 CFU/ml showed that the virulotype was indistinguishable from the spiked E. coli O157:H7 strain. We further identified the bacterial species in the un-spiked enrichment, including antimicrobial resistance genes, which could have important implications to food safety. We propose this workflow provides proof of concept for faster detection and complete genomic characterization of STECs from a complex microbial sample compared to current reporting protocols and could be applied to determine the limit of detection and assembly of other foodborne bacterial pathogens.
Introduction
Shiga toxin-producing Escherichia coli (STEC) is a foodborne pathogen capable of causing severe illness, notably hemolytic uremic syndrome (HUS), and death [1–4]. STEC-mediated foodborne illness cases and outbreaks are most commonly associated with the O157:H7 serotype; however, non-O157 STEC illnesses are increasingly being reported [5–8]. Foodborne transmission accounts for nearly 70% of O157:H7 incidents. Foodborne outbreaks have been increasingly produce-related, from 0.7% in the 1970s to 6% in the 1990s [9]. More recently (2004–2013) produce accounts for approximately 18% of foodborne outbreaks and E. coli is one of the most common bacterial sources [10–13]. Produce can become contaminated during production, packaging, or preparation; however, half of the produce-associated infections are linked to contamination prior to purchase [14]. As such, agricultural water, including water used for irrigation, is an important potential source of contamination [15–22]. While prevention and mitigation strategies are beyond the scope of this study, detection of STEC bacteria remains paramount for public health.
Current FDA reporting protocols for the detection of STECs in foods is based on a combined result from qPCR, a traditional microbiological method and whole genome sequencing (WGS) of a single isolate and is described in the FDA Bacteriological Analytical Manual (BAM) Chapter 4A [23]. It consists of a 24-hour sample enrichment in modified buffered peptone water with pyruvate at 42°C followed by qPCR detection of the main virulence genes (stx1 and stx2) and the wzy gene of the O157 antigen for a total analysis time of 2–3 days. While negative qPCR results are reported as negative for the presence of STECs, positive qPCR results undergo further analysis and O157:H7 STEC is confirmed by several rounds of selective plating on tellurite cefixime–sorbitol MacConkey agar (TC-SMAC), chromogenic agar, and trypticase soy agar with 0.6% yeast extract (TSAYE) plates for 2–4 more days of analysis time. Isolates confirmed to be pure cultures are assessed for toxigenic potential again by qPCR for stx1 and stx2. Further analysis by WGS is used to determine the complete scope of pathogenicity and antimicrobial resistance status of the isolated STEC strain which would add 3–5 days. In conclusion, approximately twelve business days are needed for STEC confirmation and characterization by the BAM method.
WGS is rapidly changing the approach to foodborne illnesses and outbreak investigations [24]. WGS is being used to monitor and identify foodborne pathogens [25, 26] and the presence of antimicrobial resistance or virulence genes [27, 28]. Specific information on serotype and pathogenicity as it relates to phylogenic relationships is increasingly important in outbreak scenarios [29, 30]. Some of the virulence genes detected by WGS mediate attachment and colonization of STECs and can be found in the locus of enterocyte effacement (LEE), including intimin (eae) and type 3 secretion system (TTSS) effector proteins (esp, esc, tir), non-LEE effectors (nleA, nleB, nleC), and other putative virulence genes (ehxA, etpD, subA, toxB, saa) [4, 28, 31, 32].
Metagenomics for sample microbial analysis and targeted detection have been used extensively in many sample types (e.g. spinach, chapati flour, and ice cream) [33–36]. Analysis is typically either by 16S rRNA gene profiling or by shotgun metagenomic sequencing [33–38]. Many studies have recently started using long read approaches [39–41] for metagenomic studies because it provides finished metagenome-assembled genomes (MAGs) for the most abundant species or bacteria in the microbiome sample [39–41]. Closed MAGs provide a better assessment of those genomes and their virulence potential or functionality in that ecosystem. Each metagenomic approach provides a different depth of analysis. 16S rRNA metagenomic approach is very sensitive and requires the lowest initial CFU sample concentration. However, because the 16S rRNA fragment is small and many species share nearly identical 16S rRNA, this approach cannot resolve species level identification and is limited to reporting at the genus level [42]. Shotgun metagenomic WGS provides a less sensitive detection limit (above 103 CFU/ml), but provides information from species to a strain level, including many functional genes in the microbiome sample analyzed [36, 39, 43, 44]. Metagenomic analyses can be made either by using short or long sequencing reads technologies. Short-read shotgun metagenomics was most commonly used for microbiome analyses [33–35, 38, 42, 45], while the use of long-read metagenomics is on the rise in the last few years [39–41, 43] for numerous reasons. These were summarized very concisely by Bertrand et al. (2019) [43], where the authors mention that short-read sequencing presents difficulty in accurately assembling the complex, highly repetitive regions that can range in sizes up to hundreds of kilobases [39], especially when multiple species are present. Classification of these short reads into species bins based on clustering relies on consensus genomes and is not precise enough for strain-level metagenomic assemblies that are necessary for outbreak and traceback scenarios [43].
Nanopore sequencing can produce completely closed genomes, while also offering affordability and portability [27, 46]. It does not employ a size selection process that limits the fragment length (unlike those seen with Illumina or Pacific Biosciences sequencers) resulting in longer reads that can help assemble highly repetitive and complex genomic regions, including phages. Furthermore, because nanopore sequencing can perform real-time base calling, it allows for semi-real-time analysis when paired with the Oxford Nanopore EPI2ME cloud service that has the “What’s in my pot” (WIMP) workflow [47]. WIMP identifies reads by taxa using an algorithm with the Centrifuge software [48] and the RefSeq sequence database at NCBI (https://www.ncbi.nlm.nih.gov/refseq/). It identifies the number of reads matching an organism of interest. These reads can potentially be retrieved from the total reads and analyzed separately. These extracted reads can then be de novo assembled and could result in a completely closed MAG for the organism of interest, in our case STEC O157:H7. This approach could dramatically reduce the time to detect and identify an STEC strain in a sample.
While current protocols utilize WGS after isolating individual colonies by the selective plating methods described, new methods for culture-independent detection and classification of O157:H7 STECs in irrigation water is increasingly important. Backflushed irrigation water samples that were FDA BAM-confirmed O157 STEC-positive were directly analyzed by Illumina MiSeq and Oxford Nanopore shotgun sequencing and produced negative results due to low concentrations of the organism of interest (Gonzalez-Escalona, unpublished results). However, instead of analyzing the water directly, we suggest that using samples after enrichment will have a sufficient STEC concentration to assemble their genome from nanopore sequencing, which will allow for identification of their serotype, virulence composition, and antimicrobial resistance gene (AMR) content [34].
Long-read nanopore sequencing has proven a useful tool to close bacterial genomes in metagenomic samples where the bacterial species are present in approximately equal proportions (approx. 12% or 107 CFU/ml) [39, 40]. Nanopore sequencing using a GridION with a FLO-MIN106 flow cell of mock microbial communities with no matrix background suggests that the technology is capable of detecting as few as 50 cells in the reaction (i.e. 4 reads) [40]. This, however, will be not enough reads to make an informative identification of STEC serotype or evaluation of potential risk to human health via presence of important virulence genes. The actual cell numbers in the sample needed for successful assembly of the genome of interest has not been determined yet.
In Leonard et al. (2015) [34] spinach samples were artificially contaminated prior to enrichment. The process of enrichment, however, is subject to microbial competition and inhibition and could lead to an imprecise final concentration in the enriched sample. While a known concentration added to the initial sample closely resembles a naturally occurring scenario, it more accurately measures the performance of the enrichment. The detection limit of the nanopore sequencing technique cannot be determined by an unknown final concentration in the enrichment. Therefore, we decided to spike a known concentration into the enrichment and test the limits of detection and assembly of completely closed or fragmented MAGs.
We aimed to utilize the current FDA BAM qPCR detection protocol for quantification of STECs in enriched irrigation water to predict when nanopore sequencing would generate a complete (closed or fragmented) MAG. To this end, the limits of detection and assembly of nanopore sequencing must be established. In order to test and empirically determine the limits of the technique, nanopore sequencing for: 1) detection, 2) characterization, and 3) closing genomes of STECs, we artificially contaminated STEC-negative enriched irrigation water with 10-fold dilutions of E. coli O157:H7 EDL933. We propose a workflow for detection and quantification of STECs in enriched samples by qPCR followed by identification and typing by nanopore sequencing. This workflow builds on existing reporting standards (qPCR) to inform appropriate implementation of sequencing technologies. We also developed a script to extract the desired reads by taxa from the total sequenced reads.
Materials and methods
Bacterial strains and media
We used a variant of the Shiga toxin-producing E. coli (STEC) EDL933 O157:H7 strain for all our experiments. We have named this variant strain EDL933_2. This strain was from our collection at CFSAN and is a variant of ATCC 43895 that after several passages in the lab has lost the stx2 phage. EDL933_2 was grown in static culture overnight in tryptic soy broth (TSB) at 37°C.
Preparation of E. coli EDL933_2 inocula for spiking experiments
For artificial contamination, overnight culture (109 CFU/ml) of E. coli EDL933_2 was serially diluted 10-fold in TSB. Dilutions containing approximately 109 CFU/ml through 101 CFU/ml were used for spiking studies. Dilutions of the overnight culture spread on tryptic soy agar (TSA) plates were used to calculate the number of CFUs per ml in the original culture.
Sample processing and artificial contamination
An STEC-negative irrigation water sample (200 ml) from the Southwestern US was enriched by adding an equal volume of 2X modified Buffered Peptone Water with pyruvate (mBPWp) and incubated at 37°C static for 5 hours. Antimicrobial cocktail [Acriflavin-Cefsulodin-Vancomycin (ACV)] was added and incubated at 42°C static overnight (18–24 h), according to Chapter 4A of the BAM. One milliliter of E. coli EDL933_2 dilutions (10^9–10^1) were added to 1 ml of the enriched irrigation water sample for a total of 9 samples (Water+Ecoli1-9). Additionally, a sample consisting of only the enriched irrigation water (Water) was used as a negative control for the presence of EDL933_2.
Nucleic acid extraction
DNA from artificially contaminated irrigation water enrichment was extracted by two methods for 1) qPCR and 2) nanopore sequencing. A 1ml fraction of each spiked enrichment sample was processed for qPCR analysis according to the FDA BAM Chapter 4A. Briefly, cells were pelleted by centrifugation at 12,000 x g for 3 minutes. The pellet was washed in 0.85% NaCl and resuspended in 1mL sterile, nuclease-free water. Samples were boiled at 100°C for 10 minutes then centrifuged to pellet debris. The DNA supernatant was saved. Another 1 ml portion of each spiked enrichment sample was extracted using the Maxwell RSC Cultured Cells DNA kit with a Maxwell RSC Instrument (Promega Corporation, Madison, WI) according to manufacturer’s instructions for Gram-negative bacteria with additional RNase treatment. DNA concentration was determined by Qubit 4 Fluorometer (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions. DNA quality was determined by Nanodrop (Thermo Fisher Scientific, Waltham, MA) according to manufacturer’s instructions.
STEC qPCR detection
The presence of STEC EDL933_2 was determined by qPCR as described in Chapter 4A of the BAM detecting stx1, stx2, and wzy. Briefly, the DNA recovered from boiled samples were diluted 1:10 in nuclease-free water and 2μl was added to 28μl master mix for a 1:5000 dilution. The master mix contained 0.25μM stx1 and stx2 primers, 0.3μM wzy primers, 0.2μM stx1 probe, 0.15μM stx2 and wzy probes, 1X Internal Positive Control Mix (Cat: 4308323, Applied Biosystems), 1X Express qPCR Supermix Universal Taq (Cat: 11785200, Invitrogen), and ROX passive dye. All primers and probes (S1 Table) employed in this study were purchased from IDT (Coralville, IA, USA).
Metagenomic sequencing, contigs assembly and annotation
DNA recovered from the E. coli EDL933_2 spiked water enrichment samples was sequenced using a GridION nanopore sequencer (Oxford Nanopore Technologies, Oxford, UK). We sequenced only 8 of the 9 samples (Water and Water+Ecoli1-7), including the negative control, because a minimum number of E. coli O157 reads were detected in the Water+Ecoli6 sample. The sequencing libraries for each individual sample were prepared with 1 μg starting material using the Genomic DNA by Ligation kit (SQK-LSK109) and each was run in a single FLO-MIN106 (R9.4.1) flow cell, according to the manufacturer’s instructions for 72 hours (Oxford Nanopore Technologies). The run was live base called using Guppy v3.2.10 included in the MinKNOW v3.6.5 (v19.12.6) software (Oxford Nanopore Technologies).
The initial classification of the reads for each run was done using the “What's in my pot” (WIMP) workflow (r3.2.2) contained in the EPI2ME cloud service (Oxford Nanopore Technologies). That workflow allows for taxonomic classification of the reads generated by the GridION sequencing in real time. Using the WIMP classification output, the reads that were identified as “Escherichia coli*” were extracted and saved in a single fastq file using a custom python script (v2.7.3) (S1 Note). The metagenome-assembled genomes (MAGs) for each spiked sample were obtained by de novo assembly using 1) all nanopore data output and 2) extracted E. coli reads using the Flye program v2.6 [49] with the meta parameter. The assembled contigs were classified by taxonomy by Kraken2 [50] using GalaxyTrakr [51] (Flye+Kraken). The presence of the complete genome and synteny of the completely closed genomes on the final assemblies was checked using Mauve genome aligner [52].
Closure of strain EDL933_2 genome by nanopore sequencing
For bioinformatic quality control purposes we generated a closed genome of the strain used in the artificial contamination studies. The long-read sequencing library was prepared and run as mentioned above for the spiked experiments. The nanopore output resulted in 144,000 reads for a total yield of 716 Mb. All reads below 5,000 base pairs in length were removed from further analysis. The genome was assembled using Flye v1.6 [49].
In silico serotyping
The major serotype present in each sample was determined by batch screening of the de novo assemblies in Ridom SeqSphere+ v 7.0.6 (Ridom, Münster, Germany) using the genes deposited in the Center for Genomic Epidemiology (https://cge.cbs.dtu.dk/services/) for E. coli as part of their web-based tool, SerotypeFinder 2.0 (https://cge.cbs.dtu.dk/services/SerotypeFinder/).
In silico identification of virulence genes
The presence of virulence genes from the de novo assemblies was determined by batch screening in Ridom SeqSphere+ software v 7.0.6 (Ridom) using the genes deposited in the NCBI Pathogen Detection Reference Gene Catalog (https://www.ncbi.nlm.nih.gov/pathogens/isolates#/refgene/) and described in Gonzalez-Escalona and Kase (2019) [28].
In silico identification of antimicrobial resistance genes
We identified the antimicrobial resistance genes present in our sequenced genomes using the EPI2ME Fastq Antimicrobial Resistance workflow (Oxford Nanopore Technologies). This workflow consists of three processes, including 1) quality control of the reads, 2) WIMP analysis (v2020.03.11) using centrifuge and NCBI RefSeq database (v88), and 3) detection of antimicrobial genes using the CARD database (v1.1.3) [53].
Metagenomic and EDL933_2 data accession numbers
The metagenomic sequence data from this study and the nanopore data for the EDL933_2 strain used in this study are available in GenBank under bioproject number PRJNA639799.
Results
Determination of the detection limit of the STEC qPCR method
The qPCR detection limit of E. coli EDL933_2 was determined by calculating CFUs per reaction. DNA was extracted from an E. coli EDL933_2 pure, overnight culture according to the boil method described in the BAM Chapter 4A for quantification. The starting CFU/ml concentration determined by plating on TSA plates was 1.5 x 109 CFU/ml. Several 10-fold dilutions (109–100 CFU/ml) of the original boil sample were tested in triplicate with the STEC qPCR assay. The wzy gene was detected over six orders of magnitude from 30 to 3 x 106 CFU per reaction (correlation coefficient (R2) = 0.99 and efficiency (E) = 98%, Fig 1A). Likewise, the stx1 gene was detected linearly over six orders of magnitude from 30 to 3 x 106 CFU per reaction (R2 = 0.99 and E = 96%, Fig 1B). The limit of the STEC qPCR detection, therefore, was 30 CFU per reaction. This means that the minimal number of detectable STEC cells in enrichment culture using this qPCR protocol will be approximately 105 CFU/ml.
Fig 1. Determination of the detection limit of the qPCR assay.
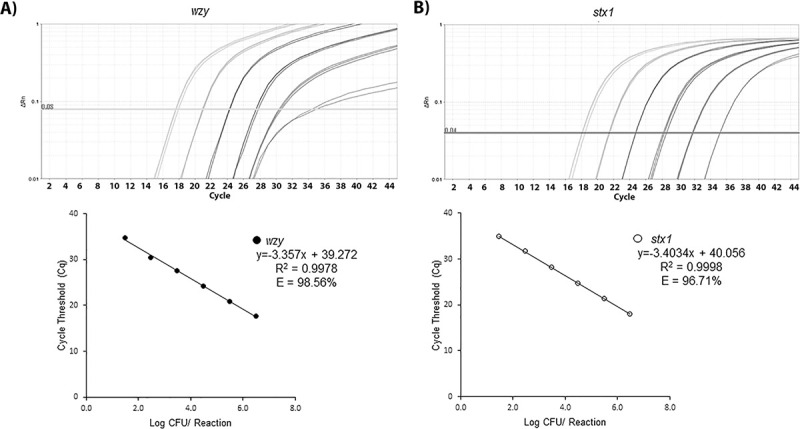
Calibration curves generated using 10-fold dilutions of DNA standards for E. coli EDL933_2 (top) detecting the wzy (A) and stx1 (B) genes. The Cq values were plotted against the log-scale CFU per reaction target concentration (bottom). The R2 values and reaction efficiency (E) are also shown.
STEC-spiked enriched sample preparation for limit of detection of nanopore sequencing
In order to assess the performance and detection limit of nanopore sequencing on STEC enrichment samples, we used previously analyzed irrigation water that was found to have nondetectable amounts of O157:H7 STEC by the FDA BAM method. Fractions of this enriched irrigation water sample were artificially contaminated with serial dilutions of E. coli EDL933_2 overnight culture. A schematic representation of the workflow is shown in Fig 2. The concentration of the stock E. coli EDL933_2 overnight culture was determined by agar plating to be 1.46 x 109 CFU/ml. Artificial contamination of the enriched field irrigation water sample (1:1), therefore, resulted in an approximate concentration of 7.3 x 108 CFU/ml for the sample referred to as Water+Ecoli1. We confirmed detection and quantification by the BAM qPCR method for this sample. The BAM qPCR method detected the presence of both wzy (Cq = 22) and stx1 (Cq = 23) genes in Water+Ecoli1. Using the standard curve determined above, we calculated 1.1 x 105 CFU/reaction using the wzy gene and 9.8 x 104 CFU/reaction using the stx1 gene. This approximates to 5.4 x 108 CFU/ml using the wzy gene and 4.9 x 108 CFU/ml using the stx1 gene in the Water+Ecoli1 sample. Therefore, the CFU/ml concentration measured by plating and qPCR was very similar.
Fig 2. Flow diagram of artificial contamination of enriched, STEC-negative irrigation water through analysis by nanopore sequencing.

Bacterial community associated with BAM enrichment of irrigation water
The bacterial community composition of the enriched irrigation water was determined by a metagenomic analysis using Oxford Nanopore sequencing of the DNA isolated from the sample. The nanopore output resulted in 5.75M reads in 16.9Gb total yield (S2 Table). The metagenomic bacterial composition of the sample was analyzed by two methods, Oxford Nanopore EPI2ME “What’s in my pot” (WIMP) workflow analysis and a de novo assembly using Flye of all the reads followed by a classification of all the contigs in the assembly by the k-mer software (Kraken2). We only reported the taxa accounting for greater than 1% of the total bacterial community. The total WIMP output can be found at https://epi2me.nanoporetech.com/workflow_instance/227465?token=D9EAC8AA-4839-11EA-99BC-71806BDB886C and the total Flye+Kraken output can be found in S3 Table.
WIMP analysis of the reads obtained by nanopore sequencing of the enriched water sample showed a highly diverse bacterial composition. Even though hundreds of bacterial species were identified in the sample, the majority of the sample was composed of nine bacterial species (>1% in Table 1). The nine bacterial species found in descending cumulative order of frequency were: Klebsiella pneumoniae (28.52%), Enterobacter cloacae (21.18%), Enterobacter sp. ODB01 (6.71%), Enterobacter kobei (6.53%), Pseudomonas putida (3.88%), Citrobacter freundii (3.86%), Acinetobacter baumannii (3.69%), Enterobacter hormaechei (3.42%), and Enterobacter xiangfangensis (1.22%). Additionally, there were 10,806 reads (0.25%) that were identified as belonging to Escherichia coli. These E. coli reads did not match STEC O157:H7.
Table 1. Bacterial community analysis of enriched irrigation water by WIMP.
Only species with frequencies above 1% are shown.
| Number of Reads | % Readsa | |
|---|---|---|
| Klebsiella pneumoniae | 1,215,922 | 28.52 |
| Enterobacter cloacae | 902,950 | 21.18 |
| Enterobacter sp. | 286,190 | 6.71 |
| Enterobacter kobei | 278,513 | 6.53 |
| Pseudomonas putida | 165,323 | 3.88 |
| Citrobacter freundii | 164,595 | 3.86 |
| Acinetobacter baumannii | 157,421 | 3.69 |
| Enterobacter hormaechei | 145,839 | 3.42 |
| Enterobacter xiangfangensis | 52,058 | 1.22 |
a% Reads were calculated as the proportion of total reads classified by WIMP software analysis.
All these microorganisms were identified in the Flye de novo assembly. Flye assembly resulted in 677 contigs of different sizes. Taxa identification by Kraken2 showed that the contigs of bigger sizes belonged to these taxa: Acinetobacter baumannii (3,784,399 bp), Citrobacter freundii (2,003,414 bp), Klebsiella pneumoniae (1,934,072 bp), Enterobacter cloacae (1,001,408 bp), Pseudomonas putida (438,653 bp), and Enterobacter kobei (365,044 bp). Enterobacter hormaechei and Enterobacter xiangfangensis were also represented in the contigs assembled by Flye, but in the Kraken2 database hormachei and xiangfangensis are listed as subspecies of E. hormachei. Many other microorganisms were also identified (S3 Table). Four of the 677 contigs were identified as matching E. coli with the largest being 30,837 bp in length.
AMR genes associated with BAM enrichment of irrigation water
We were also interested in testing the possibility that AMR genes could be identified. We have used the EPI2ME Fastq Antimicrobial Resistance workflow, which processes all nanopore reads in three stages: 1) reads are passed through a quality filter, 2) taxa are identified by the WIMP workflow, and 3) the classified reads are then analyzed for AMR genes using the CARD database (https://card.mcmaster.ca/home). The prior classification by WIMP permits identification of the AMR genes in each particular species. The AMR genes found in the field irrigation water sample include β-lactamase genes in Klebsiella pneumoniae (blaSHV, blaACT), Enterobacter cloacae (blaSHV, blaACT), Citrobacter freundii (blaSHV, blaCMY), Acinetobacter baumannii (blaOXA), and Enterobacter hormaechei (blaACT) (https://epi2me.nanoporetech.com/workflow_instance/232304?token=CCD817B6-742C-11EA-B9EA-1AEE73EF14E7). Several efflux pump genes that can be associated with antibiotic resistance were also found in Klebsiella pneumoniae (acr, ram), Enterobacter cloacae (acr, ram, vga), and Acinetobacter baumannii (abe, ade, mex). Lastly, a PhoP gene mutation was detected in Klebsiella pneumoniae (977 reads matching) conferring colistin resistance and the QnrB23 gene (29 reads matching) that confers fluoroquinolone resistance was detected in Citrobacter freundii.
Nanopore long-read detection limit for E. coli spiked into irrigation water enrichment
After establishing the bacterial community of the un-spiked enriched irrigation water sample, we sequenced DNA obtained from the artificially contaminated water enrichment at levels 7 x 108 CFU/ml (Water+Ecoli1) to 7 x 102 CFU/ml (Water+Ecoli7) by nanopore (Fig 2). The total nanopore output per sample can be found in S2 Table. On average, each run resulted in approximately 5 million reads with a total yield of 16 Gb.
The nanopore output for each sample/run was analyzed using the WIMP workflow and Flye+Kraken as described earlier for the enriched water sample. WIMP workflow classified between 1.7 million (Water+Ecoli1) and 6,300 (Water+Ecoli7) reads as E. coli for the serially diluted spiked irrigation water samples (Table 2). These reads accounted for 53 to 0.26% of the total reads for each run (Fig 3). The de novo Flye assembly of the reads for each of the spiked water samples produced assemblies with 555 to 780 contigs.
Table 2. Virulence genes assessment of the Flye assemblies obtained with all reads.
| Sample | Innoculation Level (CFU/ml)e | Serotypea | stx type | eae type | Contig No. (genome and plasmid)b | Percent EDL933_2 Genome Assembled |
|---|---|---|---|---|---|---|
| Water | 0 | O9 | - | - | 677 | - |
| Water+Ecoli1 | 7.3 x 108 | O157:H7 | 1a | gamma-1 | 555 (3+) | 100% |
| Water+Ecoli2 | 7.3 x 107 | O157:H7 | 1a | gamma-1 | 627 (4+) | 100% |
| Water+Ecoli3 | 7.3 x 106 | O157:H7/ O9 | 1a | gamma-1 | 753 (35+) | 100% |
| Water+Ecoli4c | 7.3 x 105 | O157:H7/ O9 | 1a | gamma-1 | 780 (80+) | 85% |
| Water+Ecoli5 | 7.3 x 104 | O9 | - | - | 640 | - |
| Water+Ecoli6d | 7.3 x 103 | O9 | - | - | 644 | - |
| Water+Ecoli7 | 7.3 x 102 | O9 | - | - | 526 | - |
ain silico serotype using genes defined by the Center for Genomic Epidemiology at the technical University of Denmark (DTU) (https://cge.cbs.dtu.dk/services/SerotypeFinder/).
bIn parenthesis, the number of contigs that contain the entire chromosome, a plus sign (+) indicates the presence of a contig that matched the EDL933_2 circular plasmid.
cfragmented genome assembly limit.
dE. coli O157 detection limit (4 reads matching O157 by BLAST classification).
eLevel of E. coli O157 inoculated (combination of 1 ml of enriched water and 1 ml of the E. coli O157 dilution).
Fig 3. Relative abundance of bacterial species associated with irrigation water un-spiked and spiked with E. coli EDL933_2.

Enriched irrigation water (Water) was artificially contaminated with 10-fold dilutions of E. coli EDL933_2 (+Ecoli) with a starting concentration of 7 x 108 CFU/ml (Water+Ecoli1). Reads were analyzed by the EPI2ME WIMP workflow. Bacterial species contributing more than 1% of the classified reads are shown and the sum of the remaining species identified are included as “Other”.
The enrichment samples with the highest E. coli EDL933_2 spiked concentrations, Water+Ecoli1 and 2, showed the highest number of reads classified as E. coli by WIMP (Table 2). As expected, the number of E. coli reads classified by WIMP decreased accordingly with dilution of spiked E. coli. Approximately 6,300 E. coli reads were identified in the lowest level of spiked EDL933_2 Water+Ecoli7 (7 x 102 CFU/ml), almost the same number of reads as the un-spiked enriched water sample (Table 2). In sample Water+Ecoli6, WIMP classified 68 reads as belonging to O157. However, a close analysis by BLAST showed that only 4 reads matched O157. In Water+Ecoli7, WIMP identified 22 O157 reads, but none matched the O157 genome by BLAST. Therefore, the detection limit for O157:H7 by nanopore sequencing was established at 7 x 103 CFU/ml in the enrichment.
Nanopore long-read genome assembly limit for E. coli spiked into irrigation water enrichment
After showing that the detection limit of nanopore sequencing was 7 x 103 CFU/ml, we sought to establish the genome assembly limit. A genome assembly limit is the minimum number of reads used in a de novo assembly that produces 1) a complete metagenome-assembled genome or MAG at 20X coverage (chromosome and plasmid, if present) or 2) a fragmented MAG from a complex bacterial background. Obtaining a complete or fragmented E. coli O157 MAG will allow us to perform a positive identification of the genome of interest, EDL933_2, as well as enabling a complete genomic characterization (determine serotype and presence of stx types, eae gene, virulence genes, and AMR genes). We expect the minimum number of reads for complete or fragmented assembly to be proportionally related to the initial number of CFU/ml in the sample. To test this hypothesis, we sequenced and completely closed genome of the E. coli EDL933_2 strain used in our spiking experiments. The complete closed circular genome resulted in one chromosome of 5,512,143 bp in length (50.2%GC) and a single plasmid of 93,248 bp in length (47.2%GC). The size of the wild type EDL933 (CP008957) chromosome (5,547,323 bp) is approximately 35 kb longer than our EDL933_2 variant, while the plasmid is very similar in size (92,076 bp). Our variant EDL933_2 lacks the stx2 gene. Our EDL933_2 strain is also serotype O157:H7 and has stx1a, eae gamma-1 and other virulence genes (toxB, etpD, tccP, etc), while missing the stx2a gene. We aligned both genomes and found that our variant (EDL933_2) has the entire stx2 phage missing (results not shown). By using this reference genome, we ensured the accuracy of our in silico analysis to detect the serotype and the entire virulence gene profile.
When the total nanopore sequencing output was de novo assembled by Flye, the total number of contigs was similar to the un-spiked water sample with 555 and 627 contigs for Water+Ecoli1 and 2, respectively. The E. coli EDL933_2 O157:H7 genome could be detected in 4 or 5 contigs, the plasmid was present as a single contig in each (Table 2). Serotype analysis accurately identified the E. coli as O157:H7. The presence of stx1a and eae gamma-1 were also detected. In fact, the serotype and stx and eae genes could be determined in Water+Ecoli3 and 4 (Table 2).
At the lowest spiked levels in samples Water+Ecoli5 and 6, the number of reads associated with E. coli was approximately the same as had been detected in the un-spiked water sample (Table 2). Only the O9 serotype was identified in the assemblies, which was the same as detected in the un-spiked water sample. Likewise, detection of the stx and eae genes was lost (Table 2). Therefore, we determined that the limit of fragmented assembly was approximately 7 x 105 CFU/ml, but we were not able to obtain a completely O157:H7 STEC closed MAG even at the highest level of 7 x 108 CFU/ml using this approach.
In order to improve the assembly, we decided to extract the E. coli reads and perform the de novo assembly again. Using WIMP classified reads allowed us to run a script (S1 Note) that extracted only the reads identified as E. coli and perform a similar analysis as above. Flye assembly of these filtered reads produced assemblies that contained fewer number of contigs with larger sizes. High inoculation levels in spiked samples Water+Ecoli1 and 2 resulted in assemblies with 44 and 40 contigs, respectively, each containing the completely closed E. coli O157 MAG (chromosome and plasmid each in a single contig) (Table 3). Serotype analysis accurately detected O157:H7 in assemblies from samples Water+Ecoli1 through 4. The same was observed for the recognition of stx1a and eae gamma-1 genes.
Table 3. Virulence of the Flye assemblies obtained with the WIMP E. coli extracted reads.
| Samplea | WIMP E. coli Reads | Serotypeb | stx type | eae type | Contig No. (genome and plasmid)c | Percent EDL933_2 Genome Assembled |
|---|---|---|---|---|---|---|
| Water | 10,806 | O9 | - | - | 31 | - |
| Water+Ecoli1 | 1,659,463 | O157:H7 | 1a | gamma-1 | 44 (1+) | 100% |
| Water+Ecoli2 | 432,649 | O157:H7 | 1a | gamma-1 | 40 (1+) | 100% |
| Water+Ecoli3 | 73,783 | O157:H7/ O9 | 1a | gamma-1 | 41 (8+) | 100% |
| Water+Ecoli4d | 17,203 | O157:H7/ O9 | 1a | gamma-1 | 92 (63+) | 85% |
| Water+Ecoli5 | 10,086 | O9 | - | - | 28 | - |
| Water+Ecoli6e | 8,515 | O9 | - | - | 24 | - |
aCFU/ml levels of EDL933_2 inoculation can be found in Table 2.
bin silico serotype using genes defined by the Center for Genomic Epidemiology at the technical University of Denmark (DTU) (https://cge.cbs.dtu.dk/services/SerotypeFinder/).
cIn parenthesis, the number of contigs that contain the entire chromosome, a plus sign (+) indicates the presence of a contig that matched the EDL933_2 circular plasmid.
dfragmented genome assembly limit.
eE. coli O157 detection limit (4 reads matching O157 by BLAST classification).
At the lowest spiked levels in samples Water+Ecoli5 and 6, Flye was able to still produce an assembly but with lower number of contigs (approximately 30 contigs). However, in this case no O157:H7, stx1a, or eae gamma-1 were detected by in silico analyses. Serotype analysis identified the presence of the O9 serotype (Table 3). Our fragmented assembly limit was still 7 x 105 CFU/ml, but the assembly improved as we were able to produce a completely circular closed chromosome in a single contig with a concentration of at least 7 x 107 CFU/ml.
Virulence gene identification
In addition to detecting and serotyping STECs, the detection of virulence genes provides necessary information in outbreak scenarios and is important to food safety. We again used the spiked E. coli EDL933_2 genome as reference for in silico analysis (Bioproject number PRJNA639799). We have previously reported a set of 94 virulence genes that can be used for differentiating E. coli pathotypes (STEC, ETEC, EAEC, UPEC, and EPEC) [28]. Of those 94 genes, 23 genes were present in the EDL933_2 genome (Table 4). Among those genes were esp, tccP, nle genes, tir, and toxB. The assemblies generated above were analyzed for the presence of all 23 virulence genes. Corresponding to the limits of MAG assembly (either completely closed or fragmented), all virulence genes were detected in Water+Ecoli1 and Water+Ecoli2 when using the total nanopore reads output or extracted reads. On the other hand, we failed to detect tccP in Water+Ecoli3 (7 x 106 CFU/ml) and pssA and air in Water+Ecoli4 (7 x 105 CFU/ml) samples in assemblies with the total nanopore output, but when the E. coli reads were extracted from the WIMP output, all virulence genes could be detected. Thus, our extracted E. coli reads script improved our genome assembly.
Table 4. In silico detection of virulence genes in Flye assemblies with all nanopore reads and E. coli extracted reads.
| All reads | E. coli extracted reads | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Water+Ecoli | Water+Ecoli | ||||||||||||||
| Virulence Gene | EDL933_2 Reference | Water | 1 | 2 | 3 | 4 | 5 | 6 | Water | 1 | 2 | 3 | 4 | 5 | 6 |
| astA | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| ehxA | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| espA | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| espB | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| espF | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| espJ | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| espK | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| espP | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| tccP | + | - | + | + | - | + | - | - | - | + | + | + | + | - | - |
| etpD | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| gad | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| iha | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| iss | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| nleA | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| nleB | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| nleC | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| tir | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| katP | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| pssA | + | - | + | + | + | - | - | - | - | + | + | + | + | - | - |
| air | + | - | + | + | + | - | - | - | - | + | + | + | + | - | - |
| toxB | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| ecf1 | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
| IEE | + | - | + | + | + | + | - | - | - | + | + | + | + | - | - |
+ Virulence gene detected.
- Virulence gene not detected.
Discussion
Considering the importance of irrigation water to food safety, accurate detection and classification of STECs potentially present is paramount, particularly during an outbreak incident. Current methods of detection include qPCR and extensive selective plating before WGS analysis. This method is a time-consuming process that only provides confirmation of an isolate after almost two weeks of labor. By combining qPCR and long-read metagenomic analysis of the enrichment we can definitively detect an STEC isolate, as well as characterize its virulence potential in 3–4 days. While this will not replace eventual confirmation by microbiological methods, this reduces the time for a prospective corrective measure by a complete week.
In our study we have empirically determined the in silico limits of detection, classification, and closing genomes of STECs in E. coli EDL933_2 artificially contaminated irrigation water using nanopore sequencing as a proof of concept. We have also developed a pipeline for determination of these limits that can be used for other foodborne or clinical bacteria (Fig 4). Our results showed that the level of STEC O157 needed for detection in the enrichment sample was 103 CFU/ml (Tables 2 and 3). While STEC O157 levels between 105 CFU/ml to 106 CFU/ml were enough to produce a fragmented MAG in a few contigs that allowed for complete characterization of the STEC genome, levels above 107 CFU/ml were enough to produce a completely closed STEC MAG (Table 3) with genome coverage of 385X. The complete plasmid was generated from STEC levels above 104 CFU/ml. These recovered MAGs (either fragmented or completely closed) from all spiked samples above 105 CFU/ml allowed us to comprehensively characterize the virulotype and genome synteny matching 100% to the spiked EDL933_2 strain (S1 Fig). The genome of the strain used in this study was sequenced and completely closed by us and used as reference for genome completeness for the de novo assemblies sourced from the artificially contaminated samples. Our variant strain, EDL933_2, was devoid of the stx2 phage by qPCR. A comparison of the EDL933 genome published earlier (GenBank accession AE005174) and ours showed that stx2 phage was completely missing in our strain, confirming the qPCR results.
Fig 4. STEC detection and classification by combined qPCR and nanopore sequencing approach.
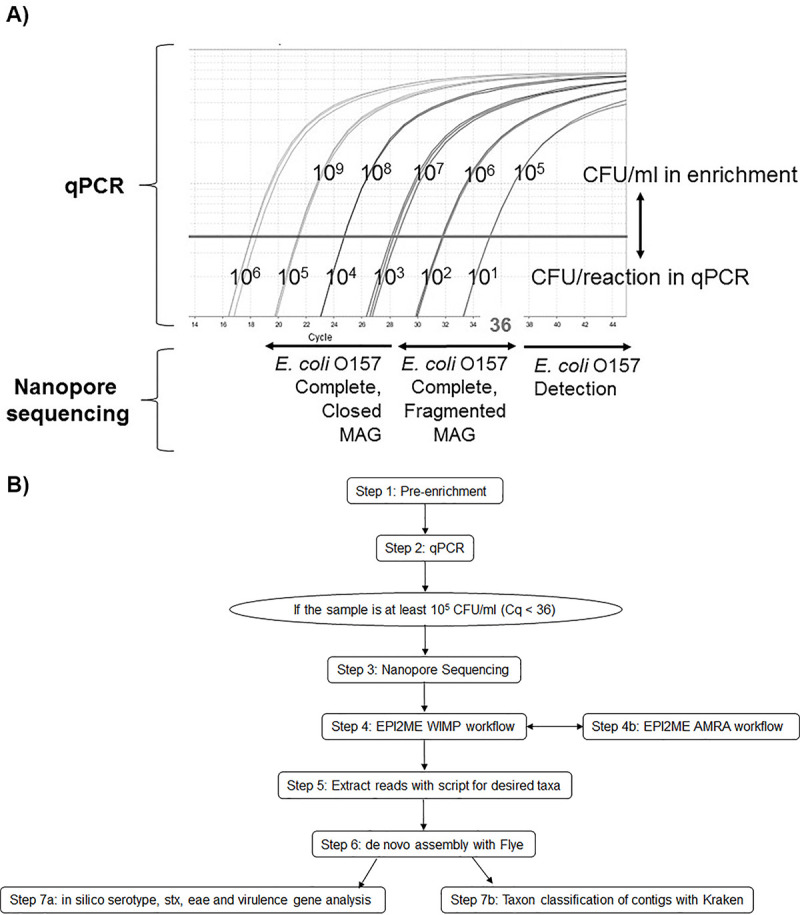
A) Direct comparison of quantitative qPCR detection with de novo assembly limits by nanopore sequencing informs detection and classification of STECs. B) Pipeline for detection and classification of STECs in enriched irrigation water using nanopore sequencing and EPI2ME cloud-based services to identify reads of a desired taxa for de novo assembly with Flye and in silico analysis.
In a previous study, Leonard et al. (2015) [34] used a different approach in which the STEC was spiked prior to enrichment. This process relies on the performance of the enrichment, which may be subject to microbial competition and inhibition. The final concentration in the enrichment is unknown. However, the implementation of our proposed pipeline is dependent on the final STEC concentration detected by the FDA BAM qPCR method in the enrichment. STEC qPCR quantification determines whether that concentration meets the minimum for the desired sequencing result, detection, complete, fragmented assembly or complete, closed assembly. That was the reason of why we spiked a known concentration (confirmed by qPCR) into the enrichment to test the limits of nanopore sequencing detection and assembly.
These limits of detection and assembly by nanopore sequencing in conjunction with the use of qPCR for screening the levels of STEC in the enrichments provide an obvious advantage. By combining the qPCR result and the likelihood of genome closure by nanopore sequencing, we have provided an excellent tool for predicting when to pursue sequencing DNA from a particular sample (Fig 4A). The tangible benefit of this combination will depend on the depth of analysis needed–detection versus characterization. While metagenomics has a lower detection limit than that of the BAM qPCR methodology (103 CFU/ml versus 105 CFU/ml) for STEC, we suggest that sequencing is best suited for STEC classification after qPCR detection. The sensitivity of nanopore can partially be attributed to the proportion of the initial sample that is used. Nanopore uses approximately 3% of the initial sample (approx. 200 ng/rxn of 6 ug DNA extracted), while qPCR uses 0.02% (2ul of a 1:10 dilution from 1 ml extraction, approx. 1ng/rxn). In the cases of Salmonella spp. or Listeria monocytogenes, their detection by qPCR or metagenomic analysis, is indicative as a presumptive positive as per FDA’s zero tolerance policy for these two microorganisms in foods and will be culturally confirmed before any regulatory action is taken to stop importation or interstate transport of a particular commodity [54–56]. In the case of STECs the entire genome is needed in order to make an informative decision of their potential health risk to humans. Many E. coli strains are not harmful to humans and will pose no risk for public health. Thus, obtaining the completely closed genome of any potential STEC will provide an accurate characterization of all virulence genes it possesses to allow a prediction of potential health risk [28]. In our study, the de novo assembly of the complete MAG for EDL933_2 (either in fragments or completely closed) was achieved in all samples with levels above 105 CFU/ml, which was almost equivalent to the limit of detection by qPCR, but with the added benefit of complete genome characterization which is critical during outbreak and traceback investigations. The methodology described herein will allow any laboratory to speed up detection and characterization (Fig 4B).
Mining for specific reads matching your organism of interest in metagenomic sequencing data is challenging and requires conducting assemblies using millions of reads with the consequent time and computing resources that can impact the accuracy of the genome assembler employed [33, 34, 40, 42, 46]. We took advantage of the WIMP workflow included in the EPI2ME cloud service (Oxford Nanopore) that classifies each single read in a.csv file and downloads those classified reads into a pass folder. A script was written that separated the desired reads by taxa into a new folder. Assemblies produced by using all reads versus using only filtered reads by taxa were compared and completely closed O157:H7 MAGs for 107 and 108 CFU/ml levels with filtered reads were obtained (Tables 2 and 3). As expected, the assemblies produced with taxa filtered reads were faster, more precise and consumed less resources.
Unlike other technologies, nanopore sequencing output is dependent on the quality of the DNA. Some nanopore metagenomics applications can be conducted directly from samples in which DNA extracts do not contain inhibitors and where the target organism(s) is in enough concentration to be detected [39, 40]. However, STECs in irrigation water require further processing due to low initial concentrations and the presence of considerable humic acid and other unknown inhibitors. Cleaning of those DNAs resulted in loss and shearing of the DNA (Gonzalez-Escalona, unpublished results), with the consequent loss of both resolution and capability of closing target genomes or MAGs. Hence, there is a need to increase the initial biomass of the target organism (STEC in our case) by an enrichment method that will also minimize the contaminants and improve the quality and quantity of the final DNA. By using the Oxford Nanopore Ligation Kit, we have maximized the potential output, which is preferable for metagenomic analyses. In the future we plan to validate these findings with the Rapid Kit (SQK-RAD004), which would further decrease time between sample processing and analysis with the expectation of completely closing MAGs of organisms above 107 CFU/ml. While we analyzed one sample per flow cell, we suggest that high STEC concentration samples (> 107 CFU/ml) could be sequenced with 3–4 samples per flow cell, reducing the sequencing cost per sample. During sample analysis, we noticed that 73% of the reads were below 5000 bp, this also could impact the closing of genomes of interest and also impact the microbial profile or MAGs from that sample. In our case after removing those reads, the same microbial profile was observed, albeit with fewer reads per organism (results not shown). Our future plans include finding a better method for DNA extraction that could provide higher DNA recovery with less shearing to maximize the potential of nanopore sequencing from enriched culture samples. Some authors have addressed DNA shearing when extracting the DNA and suggest gentler bead-beating steps or enzymatic lysis that may yield less sheared high molecular weight (HMW) DNA, but might fail in extracting DNA from most difficult organisms [39].
By using our proposed pipeline, we were not only able to improve detection and characterization of our desired organism (STEC), we were also able to identify the bacteria species that were present in the un-spiked enriched irrigation water. This analysis showed that the most common Gram-negative bacteria (>1% abundance) enriched by the BAM method belonged to the genera Enterobacter, Klebsiella, Pseudomonas, Acinetobacter, and Citrobacter. We also identified Salmonella, Escherichia, Serratia, Edwardsiella, Yersinia, and Cronobacter among the 0.1% abundance. De novo assembly of the long-read data resulted in 677 contigs with most of these MAGs in a fragmented stage, we were not able to recover a completely closed genome. Nevertheless, we were able to recover the complete genome for Acinetobacter baumannii (~3.9 Mb) in 17 contigs (with the longest contig 3,784,399 bp), Enterobacter cloacae in 167 contigs, Klebsiella pneumoniae in 35 contigs, Citrobacter freundii in 21 contigs, and Pseudomonas putida in 214 contigs (S4 Table). We could not recover the MAGs for Enterobacter sp., Enterobacter kobei, Enterobacter hormaechei subsp. Hormachei or Enterobacter hormaechei subsp. Xiangfangensis strains, even though they were present in higher abundance than Acinetobacter baumannii. The most plausible explanation could be that there were many different strains representing those species and therefore it was very hard to assemble their individual genomes. We did find some E. coli reads in the un-spiked enriched irrigation water sample (10,806 reads), suggesting the presence of E. coli in the original irrigation water sample at very low concentrations. However, the E. coli identified by in silico molecular serotyping matched to O9 serotype (Table 3), not the O157:H7 serotype of our spiked EDL933_2 strain, and no virulence genes were found by our in silico virulotyping [28]. As mentioned by Leonard et al. (2015), having found other non-pathogenic E. coli in the original water sample reinforces the need to obtain the complete genomes in order to assess the potential virulence of any E. coli strain [34]. If we applied the same script for other organisms with abundance above 3% but used different taxa to filter their reads, we could potentially close those genomes as well. This opens up a very attractive way of obtaining closed MAGs from metagenomic samples, similar to what was obtained previously for fecal samples [39].
While the enriched water sample provides a background matrix that would otherwise not be available with a mock microbial community, we did observe some distortion of the matrix at the highest concentration (Water+Ecoli1). The proportion of reads in the enriched water sample identified as Klebsiella was approximately 28%. Due to the high concentration of E. coli in Water+Ecoli1 (108 CFU/ml), the proportion of Klebsiella reads decreased to 11.5%. However, in Water+Ecoli2 (107CFU/ml) and the other subsequent 10-fold dilutions, the proportion of reads identified as Klebsiella (24%) returned to levels similar to the un-spiked enrichment.
In addition to taxa identification, another advantage of our proposed pipeline was that we surveyed the microbial community for the presence of AMR genes in the un-spiked enriched water sample. The Antimicrobial Resistance workflow in EPI2ME (Oxford Nanopore) provides AMR gene detection and identifies the organism carrying that AMR gene based on the WIMP classification of the read. This specific result will be hard to achieve when using short reads. AMR genes found in the irrigation water sample included several beta-lactamase and efflux pump genes that confer antibiotic resistance in Klebsiella pneumoniae, Enterobacter, Citrobacter freundii, Acinetobacter baumannii, and Enterobacter hormaechei. The qnrB23 gene variant (29 reads matching) that confers fluoroquinolone resistance was detected in Citrobacter freundii. Finally, besides AMR genes we observed the presence of point mutations which confers resistance to colistin in Klebsiella pneumoniae (PhoP gene mutation with 977 reads matching, 855X coverage). Antibiotic resistant bacteria in humans has been linked to food sources [57], making the presence of these AMR genes in known human pathogens such as Klebsiella pneumoniae and Acinetobacter baumannii worrisome. Acetinobacter has recently been shown to use killing-enhanced horizontal gene transfer [58], which suggests further study given the high number of AMR genes present in this sample. Additionally, as the soil filters and concentrates the bacteria in the irrigation water, the risk for human consumption increases [59]. National and international organizations, such as the National Antimicrobial Resistance Monitoring System (NARMS - https://www.cdc.gov/narms/index.html), One Health approach (https://www.cdc.gov/onehealth/index.html) and the Global AMR Surveillance System (GLASS - https://www.who.int/glass/en/) use the resources of the CDC, USDA, FDA, and WHO to monitor and report the prevalence of and distribute regulatory guidance on antimicrobial resistance in pathogenic and commensal bacteria in food and food animals [57, 60]. Our pipeline could be an important screening tool to enhance future testing.
Conclusions
Overall, we tested the limits of detection and assembly for EDL933_2 O157:H7 in enriched irrigation water using a shotgun long-read sequencing approach. We determined the detection limit of the BAM STEC qPCR (105 CFU/ml) coincided with our STEC assembly limit for a fragmented genome capable of STEC strain, serotype and virulotype identification by nanopore sequencing, aided by filtering reads by taxa. Therefore, we recommend a combination approach using qPCR and nanopore sequencing. In the screening stage, qPCR can provide both detection and an estimate of CFU/ml concentration which could predict if subsequent nanopore sequencing will produce enough data to obtain a complete MAG of the target organism, either closed or fragmented. We expect that the use of this pipeline could enhance the capacity of Public Health entities to respond faster and more accurately during outbreak and traceback investigations.
Supporting information
(DOCX)
(DOCX)
(XLSX)
(DOCX)
(DOCX)
Each sample extracted EDL933_2 matching contigs is laid out in a horizontal track and homologous segments are indicated in the same color and connected across genomes. Respective scales show the sequence coordinates in base pairs. A colored similarity plot is shown for each genome, the height of which is proportional to the level of sequence identity in that region. Contigs boundaries are represented by a red line. A) EDL933_2 vs all E. coli O157 MAGs from the different EDL933_2 spiking levels. From level Water+Ecoli4 we could not recover a complete fragmented O157 MAG. B) EDL933_2 vs levels where we could recover a completely closed O157 MAG, including the pO157 plasmid for visualization purposes.
(DOCX)
Data Availability
The metagenomic sequence data from this study and the nanopore data for the EDL933 strain used in this study are available in GenBank under bioproject number PRJNA639799.
Funding Statement
NGE research was supported by funding from the MCMi Challenge Grants Program Proposal #2018-646 and the FDA Foods Program Intramural Funds. MM was supported by funding from the MCMi Challenge Grants Program Proposal #2018-646. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Beutin L, Martin A. Outbreak of shiga toxin-producing Escherichia coli (STEC) O104:H4 infection in Germany causes a paradigm shift with regard to human pathogenicity of STEC strains. J. Food Prot. 2012; 75: 408–418. 10.4315/0362-028X.JFP-11-452 . [DOI] [PubMed] [Google Scholar]
- 2.Mellmann A, Bielaszewska M, Kock R, Friedrich AW, Fruth A, Middendorf B, et al. Analysis of collection of hemolytic uremic syndrome-associated enterohemorrhagic Escherichia coli. Emerg. Infect. Dis. 2008; 14: 1287–1290. 10.3201/eid1408.071082 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005; 365: 1073–1086. 10.1016/S0140-6736(05)71144-2 [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez-Escalona N, Meng J, Doyle MP (2019) Shiga toxin-producing Escherichia coli In: Doyle MP, Diez-Gonzalez F, Hill C, editors. Food Microbiology: Fundamentals And Frontiers. Washington DC: ASM press; pp. 289–315. 10.1128/9781555819972.ch11. [DOI] [Google Scholar]
- 5.Mead PS, Slutsker L, Dietz V, McCaig LF, Bresee JS, Shapiro C, et al. Food-related illness and death in the United States. Emerg. Infect. Dis. 1999; 5: 607–625. 10.3201/eid0505.990502 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allos BM, Moore MR, Griffin PM, Tauxe RV. Surveillance for sporadic foodborne disease in the 21st century: the FoodNet perspective. Clin. Infect. Dis. 2004; 38 Suppl 3: S115–S120. 10.1086/381577 [DOI] [PubMed] [Google Scholar]
- 7.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, et al. Foodborne illness acquired in the United States—major pathogens. Emerg. Infect. Dis. 2011; 17: 7–15. 10.3201/eid1701.p11101 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brooks JT, Sowers EG, Wells JG, Greene KD, Griffin PM, Hoekstra RM, et al. Non-O157 shiga toxin-producing Escherichia coli infections in the United States, 1983–2002. J. Infect. Dis. 2005; 192: 1422–1429. 10.1086/466536 [DOI] [PubMed] [Google Scholar]
- 9.Sivapalasingam S, Friedman CR, Cohen L, Tauxe RV. Fresh produce: a growing cause of outbreaks of foodborne illness in the United States, 1973 through 1997. J Food Prot. 2004; 67: 2342–2353. 10.4315/0362-028x-67.10.2342 [DOI] [PubMed] [Google Scholar]
- 10.Tack DM, Ray L, Griffin PM, Cieslak PR, Dunn J, Rissman T, et al. Preliminary incidence and trends of infections with pathogens transmitted commonly through food—Foodborne Diseases Active Surveillance Network, 10 U.S. Sites, 2016–2019. MMWR Morb Mortal Wkly Rep. 2020; 69: 509–514. 10.15585/mmwr.mm6917a1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bottichio L, Keaton A, Thomas D, Fulton T, Tiffany A, Frick A, et al. Shiga toxin producing Escherichia coli infections associated with romaine lettuce United States, 2018. Clin. Infect. Dis. 2019; e323–e330. 10.1093/cid/ciz1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer M, Bourner A, Plunkett G (2015) Outbreak alert! 2015: a review of foodborne illness in the US from 2004–2013. [Google Scholar]
- 13.Olaimat AN, Holley RA. Factors influencing the microbial safety of fresh produce: a review. Food Microbiol. 2012; 32: 1–19. 10.1016/j.fm.2012.04.016 [DOI] [PubMed] [Google Scholar]
- 14.Rangel JM, Sparling PH, Crowe C, Griffin PM, Swerdlow DL. Epidemiology of Escherichia coli O157:H7 outbreaks, United States, 1982–2002. Emerg. Infect. Dis. 2005; 11: 603–609. 10.3201/eid1104.040739 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.[Anonymous] (2018) https://www.fda.gov/food/outbreaks-foodborne-illness/environmental-assessment-factors-potentially-contributing-contamination-romaine-lettuce-implicated.
- 16.[Anonymous] (2019) https://www.fda.gov/food/outbreaks-foodborne-illness/investigation-summary-factors-potentially-contributing-contamination-romaine-lettuce-implicated-fall.
- 17.Steele M, Odumeru J (2004) Irrigation water as source of foodborne pathogens on fruit and vegetables. J. Food Prot 67: 2839–2849. 10.4315/0362-028x-67.12.2839 [DOI] [PubMed] [Google Scholar]
- 18.Uyttendaele M, Jaykus LA, Amoah P, Chiodini A, Cunliffe D, Jacxsens L, et al. Microbial hazards in irrigation water: standards, norms, and testing to manage use of water in fresh produce primary production. Compr. Rev. Food Sci. Food Saf. 2015; 14: 336–356. 10.1111/1541-4337.12133. [DOI] [Google Scholar]
- 19.Leaman S, Gorny J, Wetherington D, Bekris H (2014) Agricultural water. Center for Produce Safety Five Year Research Review. https://www.centerforproducesafety.org/amass/documents/document/247/CPS%20Ag%20Water%20Research%20Report%202014%20with%20corrections%201.1.pdf [Google Scholar]
- 20.Monaghan JM, Hutchison ML. Distribution and decline of human pathogenic bacteria in soil after application in irrigation water and the potential for soil-splash-mediated dispersal onto fresh produce. J. Appl. Microbiol. 2012; 112: 1007–1019. 10.1111/j.1365-2672.2012.05269.x [DOI] [PubMed] [Google Scholar]
- 21.Oliveira M, Vinas I, Usall J, Anguera M, Abadias M. Presence and survival of Escherichia coli O157:H7 on lettuce leaves and in soil treated with contaminated compost and irrigation water. Int. J. Food Microbiol. 2012; 156: 133–140. 10.1016/j.ijfoodmicro.2012.03.014 [DOI] [PubMed] [Google Scholar]
- 22.Allende A, Monaghan J. Irrigation water quality for leafy crops: a perspective of risks and potential solutions. Int. J. Environ. Res. Public Health. 2015; 12: 7457–7477. 10.3390/ijerph120707457 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.[Anonymous] (2018) https://www.fda.gov/food/laboratory-methods-food/bam-chapter-4a-diarrheagenic-escherichia-coli.
- 24.Allard MW, Bell R, Ferreira CM, Gonzalez-Escalona N, Hoffmann M, Muruvanda T, et al. Genomics of foodborne pathogens for microbial food safety. Curr. Opin. Biotechnol. 2018; 49: 224–229. 10.1016/j.copbio.2017.11.002 [DOI] [PubMed] [Google Scholar]
- 25.Bergholz TM, Moreno Switt AI, Wiedmann M. Omics approaches in food safety: fulfilling the promise? Trends Microbiol. 2014; 22: 275–281. 10.1016/j.tim.2014.01.006 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sekse C, Holst-Jensen A, Dobrindt U, Johannessen GS, Li W, Spilsberg B, et al. High throughput sequencing for detection of foodborne pathogens. Front. Microbiol. 2017; 8 10.3389/fmicb.2017.02029 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalez-Escalona N, Allard MA, Brown EW, Sharma S, Hoffmann M. Nanopore sequencing for fast determination of plasmids, phages, virulence markers, and antimicrobial resistance genes in Shiga toxin-producing Escherichia coli. PLoS One. 2019; 14: e0220494 10.1371/journal.pone.0220494 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gonzalez-Escalona N, Kase JA. Virulence gene profiles and phylogeny of shiga toxin-positive Escherichia coli strains isolated from FDA regulated foods during 2010–2017. PLoS One. 2019; 14: e0214620 10.1371/journal.pone.0214620 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffmann M, Luo Y, Monday SR, Gonzalez-Escalona N, Ottesen AR, Muruvanda T, et al. Tracing Origins of the Salmonella Bareilly Strain Causing a Food-borne Outbreak in the United States. J. Infect. Dis. 2016; 213: 502–508. 10.1093/infdis/jiv297 [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez-Escalona N, Toro M, Rump LV, Cao G, Nagaraja TG, Meng J. Virulence gene profiles and clonal relationships of Escherichia coli O26:H11 isolates from feedlot cattle as determined by whole-genome sequencing. Appl. Environ. Microbiol. 2016; 82: 3900–3912. 10.1128/AEM.00498-16 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaper JB, Nataro JP, Mobley HL. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004; 2: 123–140. 10.1038/nrmicro818 [DOI] [PubMed] [Google Scholar]
- 32.Garmendia J, Frankel G, Crepin VF. Enteropathogenic and enterohemorrhagic Escherichia coli infections: translocation, translocation, translocation. Infect Immun. 2005; 73: 2573–2585. 10.1128/IAI.73.5.2573-2585.2005 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leonard SR, Mammel MK, Lacher DW, Elkins CA. Strain-Level Discrimination of Shiga Toxin-Producing Escherichia coli in Spinach Using Metagenomic Sequencing. PLoS One. 2016; 11: e0167870 10.1371/journal.pone.0167870 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leonard SR, Mammel MK, Lacher DW, Elkins CA. Application of metagenomic sequencing to food safety: detection of Shiga toxin-producing Escherichia coli on fresh bagged spinach. Appl. Environ. Microbiol. 2015; 81: 8183–8191. 10.1128/AEM.02601-15 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lusk PT, Ramachandran P, Reed E, Kase JA, Ottesen A. Metagenomic description of preenrichment and postenrichment of recalled Chapati Atta flour using a shotgun sequencing approach. Genome Announc. 2018; 6 10.1128/genomeA.00305-18 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ottesen A, Ramachandran P, Chen Y, Brown E, Reed E, Strain E. Quasimetagenomic source tracking of Listeria monocytogenes from naturally contaminated ice cream. BMC Infect. Dis. 2020; 20: 83 10.1186/s12879-019-4747-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kovac J, Bakker Hd, Carroll LM, Wiedmann M. Precision food safety: a systems approach to food safety facilitated by genomics tools. TrAC Trends in Analytical Chemistry. 2017; 96: 52–61. 10.1016/j.trac.2017.06.001. [DOI] [Google Scholar]
- 38.Gigliucci F, von Meijenfeldt FAB, Knijn A, Michelacci V, Scavia G, Minelli F, et al. Metagenomic characterization of the human intestinal microbiota in fecal samples from STEC-infected patients. Front. Cell. Infect. Microbiol. 2018; 8: 25 10.3389/fcimb.2018.00025 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moss EL, Maghini DG, Bhatt AS. Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nat. Biotechnol. 2020. 10.1038/s41587-020-0422-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nicholls SM, Quick JC, Tang S, Loman NJ. Ultra-deep, long-read nanopore sequencing of mock microbial community standards. Gigascience. 2019; 8: giz043 10.1093/gigascience/giz043 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boykin LM, Sseruwagi P, Alicai T, Ateka E, Mohammed IU, Stanton JL, et al. Tree Lab: portable genomics for early detection of plant viruses and pests in Sub-Saharan Africa. Genes (Basel). 2019; 10: 632 10.3390/genes10090632 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grutzke J, Malorny B, Hammerl JA, Busch A, Tausch SH, Tomaso H, et al. Fishing in the soup—pathogen detection in food safety using metabarcoding and metagenomic sequencing. Front. Microbiol. 2019; 10: 1805 10.3389/fmicb.2019.01805 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bertrand D, Shaw J, Kalathiyappan M, Ng AHQ, Kumar MS, Li C, et al. Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nat. Biotechnol. 2019; 37: 937–944. 10.1038/s41587-019-0191-2 [DOI] [PubMed] [Google Scholar]
- 44.Gu G, Ottesen A, Bolten S, Luo Y, Rideout S, Nou X. Microbiome convergence following sanitizer treatment and identification of sanitizer resistant species from spinach and lettuce rinse water. Int. J. Food Microbiol. 2020; 318: 108458 10.1016/j.ijfoodmicro.2019.108458 [DOI] [PubMed] [Google Scholar]
- 45.Suttner B, Johnston ER, Orellana LH, Rodriguez R, Hatt JK, Carychao D, et al. Metagenomics as a public health risk assessment tool in a study of natural creek sediments influenced by agricultural and livestock runoff: potential and limitations. Appl Environ Microbiol. 2020; 86: e02525–19. 10.1128/AEM.02525-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loman NJ, Quick J, Simpson JT. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods. 2015; 12: 733–735. 10.1038/nmeth.3444 [DOI] [PubMed] [Google Scholar]
- 47.Juul S, Izquierdo F, Hurst A, Dai X, Wright A, Kulesha E, et al. Whats in my pot? Real-time species identification on the MinION. bioRxiv. 2015; 030742 10.1101/030742. [DOI] [Google Scholar]
- 48.Kim D, Song L, Breitwieser FP, Salzberg SL. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016; 26: 1721–1729. 10.1101/gr.210641.116 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019; 37: 540–546. 10.1038/s41587-019-0072-8 [DOI] [PubMed] [Google Scholar]
- 50.Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019; 20: 257 10.1186/s13059-019-1891-0 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018; 46: W537–W544. 10.1093/nar/gky379 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Darling ACE, Mau B, Blattner FR, Perna NT. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004; 14: 1394–1403. 10.1101/gr.2289704 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alcock BP, Raphenya AR, Lau TTY, Tsang KK, Bouchard M, Edalatmand A, et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020; 48: D517–D525. 10.1093/nar/gkz935 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Archer DL. The evolution of FDA's policy on Listeria monocytogenes in ready-to-eat foods in the United States. Curr Opin Food Sci. 2018; 20: 64–68. 10.1016/j.cofs.2018.03.007. [DOI] [Google Scholar]
- 55.[Anonymous] (2017) https://www.fda.gov/media/102633/download.
- 56.[Anonymous] (2012) https://www.fda.gov/media/83177/download.
- 57.Karp BE, Tate H, Plumblee JR, Dessai U, Whichard JM, Thacker EL, et al. National antimicrobial resistance monitoring system: two decades of advancing public health through integrated surveillance of antimicrobial resistance. Foodborne Pathog Dis. 2017; 14: 545–557. 10.1089/fpd.2017.2283 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cooper RM, Tsimring L, Hasty J. Inter-species population dynamics enhance microbial horizontal gene transfer and spread of antibiotic resistance. Elife. 2017; 6 10.7554/eLife.25950 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holzel CS, Tetens JL, Schwaiger K. Unraveling the role of vegetables in spreading antimicrobial-resistant bacteria: a need for quantitative risk assessment. Foodborne Pathog Dis. 2018; 15: 671–688. 10.1089/fpd.2018.2501 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thakur S, Gray GC. The mandate for a global "One Health" approach to antimicrobial resistance surveillance. Am J Trop Med Hyg. 2019; 2018/12/27: 227–228. 10.4269/ajtmh.18-0973 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
(DOCX)
(XLSX)
(DOCX)
(DOCX)
Each sample extracted EDL933_2 matching contigs is laid out in a horizontal track and homologous segments are indicated in the same color and connected across genomes. Respective scales show the sequence coordinates in base pairs. A colored similarity plot is shown for each genome, the height of which is proportional to the level of sequence identity in that region. Contigs boundaries are represented by a red line. A) EDL933_2 vs all E. coli O157 MAGs from the different EDL933_2 spiking levels. From level Water+Ecoli4 we could not recover a complete fragmented O157 MAG. B) EDL933_2 vs levels where we could recover a completely closed O157 MAG, including the pO157 plasmid for visualization purposes.
(DOCX)
Data Availability Statement
The metagenomic sequence data from this study and the nanopore data for the EDL933 strain used in this study are available in GenBank under bioproject number PRJNA639799.