

Abstract
More than forty loci contribute to genetic risk for Alzheimer’s disease (AD). These risk alleles are enriched in myeloid cell enhancers suggesting that microglia, the brain-resident macrophages, contribute to AD risk. We have previously identified SPI1/PU.1, a master regulator of myeloid cell development in the brain and periphery, as a genetic risk factor for AD. Higher expression of SPI1 is associated with increased risk for AD, while lower expression is protective. To investigate the molecular and cellular phenotypes associated with higher and lower expression of PU.1 in microglia, we used stable overexpression and knock-down of PU.1 in BV2, an immortalized mouse microglial cell line. Transcriptome analysis suggests that reduced PU.1 expression suppresses expression of homeostatic genes similar to the disease-associated microglia response to amyloid plaques in mouse models of AD. Moreover, PU.1 knock-down resulted in activation of protein translation, antioxidant action and cholesterol/lipid metabolism pathways with a concomitant decrease of pro-inflammatory gene expression. PU.1 overexpression upregulated and knock-down downregulated phagocytic uptake in BV2 cells independent of the nature of the engulfed material. However, cells with reduced PU.1 expression retained their ability to internalize myelin similar to control albeit with a delay, which aligns with their anti-inflammatory profile. Here we identified several microglial responses that are modulated by PU.1 expression levels and propose that risk association of PU.1 to AD is driven by increased pro-inflammatory response due to increased viability of cells under cytotoxic conditions. In contrast, low expression of PU.1 leads to increased cell death under cytotoxic conditions accompanied by reduced pro-inflammatory signaling that decreased A1 reactive astrocytes signature supporting the protective effect of SPI1 genotype in AD. These findings inform future in vivo validation studies and design of small molecule screens for therapeutic discovery in AD.
Keywords: PU.1, Alzheimer’s disease, phagocytosis, disease-associated microglia, anti-inflammatory microglia, amyloid β
Introduction
Pathogenesis of neurodegenerative conditions is influenced by several factors the most notable being age and genetics (Pimenova et al., 2017b). Sporadic AD is the most common form of dementia, characterized neuropathologically by deposition of amyloid plaques and neurofibrillary tangles in the brain. These pathological hallmarks are also accompanied by neuronal cell loss, neuroinflammation and lipidosis (De Strooper and Karran, 2016), which are propagated by microglia, the brain-resident macrophages. Under normal conditions microglia perform tissue surveillance and phagocytic uptake and clearance associated with maintenance of tissue homeostasis and immune tolerance. In aged or diseased brain microglia respond to tissue damage by orchestrating an inflammatory response that may modulate disease onset or progression. Meta-analysis of genome-wide association studies for AD suggests that genes expressed in microglia modulate risk for late onset AD (Jones et al., 2015; Kunkle et al., 2019). The disease-associated phenotype of microglia seems to be shared by aging, as well as neurological and non-neurological diseases with different etiology, e.g. AD and amyotrophic lateral sclerosis (Holtman et al., 2015; Keren-Shaul et al., 2017), atherosclerosis and obesity, characterized by lipid overload (Jaitin et al., 2019; Martinet et al., 2019). Thus, it is important to elucidate the functional effect of molecules driving the disease-associated phenotype of microglia in the brain.
The molecular signature of disease-associated microglia (DAM), described in the 5XFAD mouse model of AD, shows repression of microglial homeostatic genes that are expressed in microglia within a healthy brain and upregulation of genes with roles in phagocytosis and lipid metabolism (Keren-Shaul et al., 2017). The DAM response is dependent on TREM2 and APOE, two major AD risk genes, in mouse models of AD and metabolic disease (Krasemann et al., 2017; Pimenova et al., 2017a). This signaling pathway seems to be conserved in human microglia (Zhou et al., 2020), although the mouse DAM transcriptome profile may not be fully present in AD brain, as human microglia show a distinct AD-related gene signature (Jaitin et al., 2019; Mathys et al., 2019). This discrepancy may be due to differences in the timing and collection of the cells, since mouse microglia are extracted and analyzed during active stages of amyloidosis, while human brain tissue is post-mortem, representing the terminal stage of disease with failed microglial response. It is hypothesized that DAM are protective in AD because changes in expression of AD risk genes in the DAM signature correlate with reduced disease risk based on human genetics (Deczkowska et al., 2018). Efforts to describe functional consequences of changes in AD risk gene expression based on genetic variation in patient cohorts present the best way to decipher the significance of the DAM response.
We have previously reported that SPI1 is the most likely gene mediating the association of the CELF1 locus with AD risk (Huang et al., 2017). The mechanism behind the association is based on the differential expression of SPI1 mRNA depending on the genotype of the haplotype tagged by the rs1057233 SNP. Reduced PU.1 expression confers protection, while increased expression is associated with higher AD risk. SPI1/PU.1 is a myeloid lineage-determining transcription factor that together with MEF2C shapes the enhancer landscape specific to microglia in the brain (Lavin et al., 2014). Expression of PU.1 is transcriptionally regulated (Mak et al., 2011). In microglia and macrophages PU.1 is bound to myeloid-specific enhancers sustaining cell response to injury induced by stimulus-dependent transcription factors (Holtman et al., 2017). This hierarchical cooperation leads to robust modulation of gene expression contributing to cytokine production, lysosomal function, phagocytosis and lipid metabolism. SPI1/PU.1 is a master regulator of microglial development and homeostasis and the expression of several AD genetic risk genes, e.g. ABCA7, CD33, MS4A4A, MS4A6A, TREM2, APOE and CLU, and small changes in PU.1 expression can significantly affect cell function (Huang et al., 2017; Satoh et al., 2014). The role of PU.1 in microglial cell function has not been investigated before, and as a genetic risk factor with plausible mechanism of action based on difference in expression, it is important to investigate the impact of increased and reduced PU.1 expression in microglia in vitro and in vivo.
Here, we report the role of PU.1 in microglial cell function in vitro. Using BV2 immortalized mouse microglial cells, we generated lines with stable overexpression and knock-down of PU.1 mimicking the risk and protective genotypes of SPI1. While the role of PU.1 in microglial phagocytosis has been described before, this report is the first to show that increased PU.1 expression renders cells more resistant to cell death and increased inflammatory response, which may be detrimental for other cell types of the diseased brain. Interestingly, the effect of PU.1 overexpression on microglial function shows opposing effects to PU.1 knock-down, which supports the mechanism of action for SPI1 contribution to AD pathogenesis based on differential expression. PU.1 knock-down makes microglia more susceptible to cell death, but also less pro-inflammatory, which was beneficial in reducing A1 reactive astrocytes signature relevant to diseased brain milieu. Altogether, we report a comprehensive functional validation of the role of PU.1 in microglia, which will facilitate the interpretation of PU.1 effects in vivo and guide the development of phenotypic screens for drug discovery. On a broader scale these findings may be widely applicable to the treatment of neurological or metabolic disorders displaying gliosis, independent of the trigger, such as Aβ, tau or accumulation of other pathogenic proteins.
Methods
Reagents, antibodies, plasmids and cell lines
Tissue culture reagents were purchased from Thermo Fisher. pHrodo™ Red Zymosan Bioparticles™ Conjugate for Phagocytosis was from Thermo Fisher (P35364). pHrodo™ Red, succinimidyl ester (pHrodo™ Red, SE) was from Thermo Fisher (P36600). Staurosprorine was from Alfa Aesar (J62837-M). Rotenone was from Sigma-Aldrich (R8875-1G). Z-VAD-FMK was from Enzo Life Science (ALX-260-020-M001). Aβ42 was from CaliforniaPeptide (641-15). Lipopolysaccharides (LPS) were from Sigma-Aldrich (L5886-10MG). N-Acetyl-L-cysteine (NAC) was from Sigma-Aldrich (A7250-5G). Antibodies were from: PU.1 (Cell Signaling #2266), β-actin (Sigma-Aldrich #A5441), iNOS (Cell Signaling #13120), pro and cleaved caspase-3 (8G10, #9665), pro-caspase-8 (#4927), cleaved caspase-8 (D5B2, # 8592), pro- and cleaved caspsase-9 (C9, #9508), pro- and cleaved Lamin A/C (#4777), pro- and cleaved PARP (#9542), anti-mouse CD11b APC was from eBioscience (#17-0112-81). pcDNA3-FLAG-PU.1 was a gift from Christopher Vakoc (Roe et al., 2015) (Addgene plasmid 66974). pGFP-V-RS with either non-targeting shRNA (#TR30013) or PU.1-targeting shRNA (GI595749) was purchased from OriGene Technologies (#TG502008). BV2 mouse microglial cell line was kindly provided by Dr. Marc Diamond (UT Southwestern Medical Center). The BV2 cells were cultured in DMEM (Gibco #11965) supplemented with 5% heat-inactivated fetal bovine serum (FBS, Gibco #16140) and 100 U/ml penicillin-streptomycin (Pen/Strep, Gibco #15140). Jurkat human T cell leukemia cells were kindly provided by Dr. Benjamin Chen (MSSM). Jurkat cells were cultured in RPMI (Gibco #11875) supplemented with 10% FBS and 1:100 Pen/Strep. N2A mouse neuroblastoma cells were kindly provided by Dr. Eric Nestler (MSSM). N2A cells were cultured in 50% DMEM (Gibco #11965), 50% OPTI-MEM (Gibco #11058) supplemented with 5% FBS, and 1:100 Pen/Strep. Routine testing of cell lines using MycoAlert PLUS mycoplasma detection kit (Lonza) showed that all cells were negative for mycoplasma contamination.
Stable cell line generation
Lenti ORF particles, Sfpi1 (Myc-DDK-tagged), pLenti-C-Myc-DDK-P2A-Puro (MR203632L3V) and LentiORF control particles of pLenti-C-Myc-DDK-P2A-Puro (PS100092V) were purchased from OriGene Technologies. Mouse SPI1 MISSION shRNA Lentiviral pLKO.1 Particles (TRCN0000009500) and MISSION® pLKO.1-puro Non-Target shRNA Control Transduction Particles (SHC016V-1EA) were purchased from Sigma-Aldrich. BV2 cells were seeded in 24-well plates at 50.000 per well and transduced the next day with lentiviral particles at MOI 5 using spinfection protocol 2000 rpm for 2 hours at room temperature (RT). After 48 hours of incubation, cells were split into a 6-well plate and selected with 10 μg/ml puromycin for 5 days. Surviving cells were amplified and frozen to create BV2 cells with overexpression of mouse PU.1 (OE) and respective empty frame control (CTL) or cells with knock-down of PU.1 (KD) with the respective scrambled control (SCR). Changes in PU.1 expression were confirmed on the mRNA level using qPCR and protein level using western blotting of polyclonal selected lines.
iPSCs culture and differentiation
Human induced pluripotent stem cells (iPSCs) were maintained on Matrigel (BD Biosciences) in complete StemFlex medium (Thermo Fisher #A3349401) supplemented with Pen/Strep. iPSCs were passaged every week using ReLeSR dissociation reagent (StemCell Technologies) and used for hematopoietic stem cell differentiation with STEMdiff Hematopoietic kit (STEMCELL Technologies #05310) followed by differentiation to induced microglial-like cells (iMGLs) using a previously published protocol by (McQuade et al., 2018). F11349, F12455 and 75.11-IW1A12 (Karch et al., 2019) iPSC lines were used with confirmed normal karyotyping (WiCell). iMGLs maturation was confirmed with immunostaining for PU.1 and CD11b after 3–5 days of culture in mature microglial medium. Mature iMGLs were transfected with a set of 4 ON-TARGETplus human siRNAs (Horizon Discovery #LQ-010537-00-0002) at 40nM in 6-well plates (~300.000 cells per well in 3–4 ml) using jetPRIME reagent (Polyplus #114–15) for 48 hours. Cells were collected for qPCR analysis as described below using TaqMan Gene Expression Assays (Thermo Fisher) for a set of selected genes in Fig. 1c.
Figure 1. RNA sequencing analysis of BV2 microglial cells with transient modulation of Spi1/PU.1.
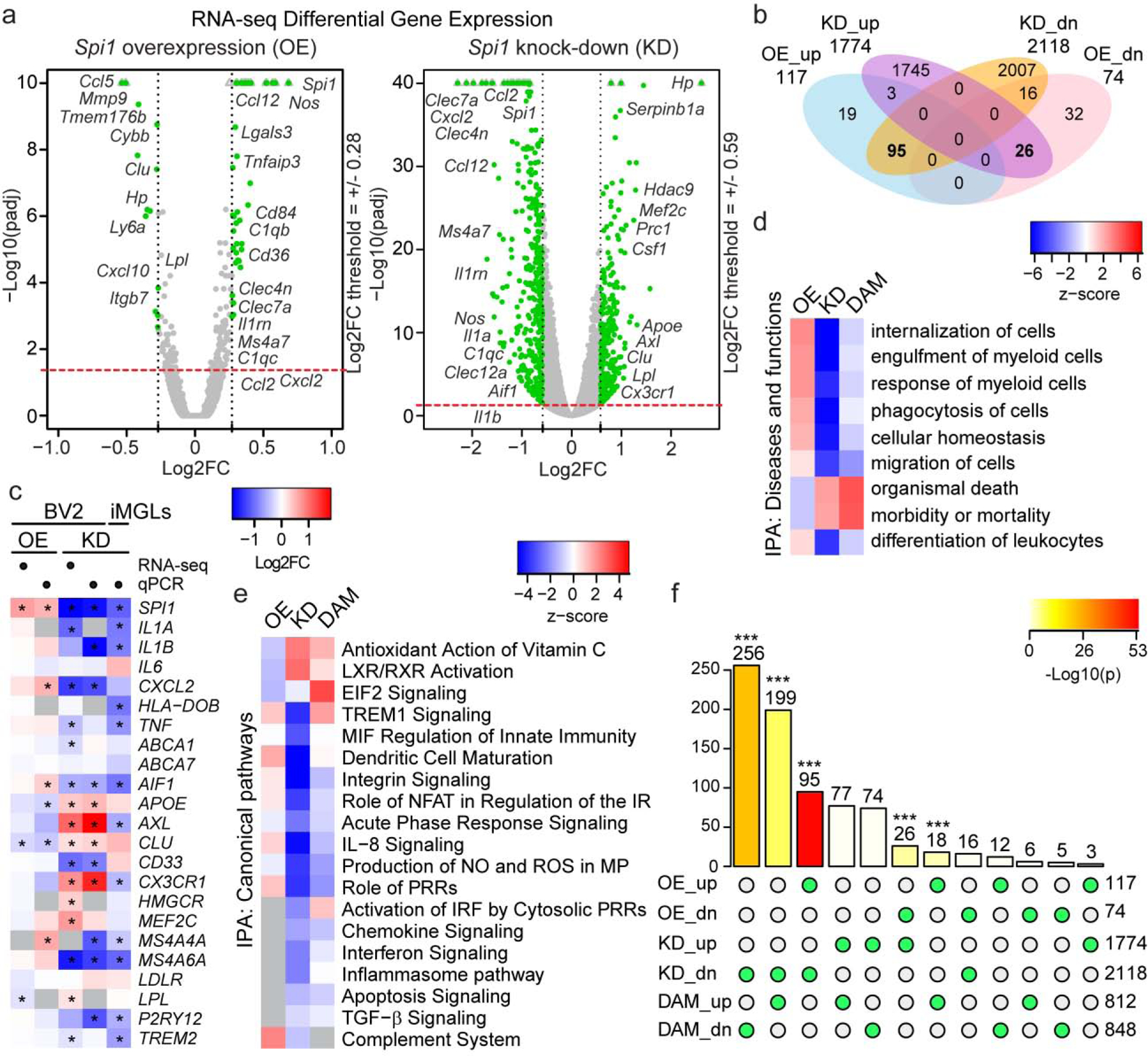
(a) Differential gene expression volcano plots and (b) Venn diagram revealed significant changes in BV2 cells transcriptome after transient knock-down of PU.1 affecting 3892 genes, while overexpression of PU.1 affected 191 genes (cutoff: padj < 0.05). (c) Heatmap of DEGs analyzed in BV2 cells with overexpression or knock-down of PU.1 using RNA sequencing or qPCR and in human induced microglial-like cells (iMGLs) with knock-down of PU.1 using qPCR. (d) Diseases and functions and (e) canonical pathways affected by differential expression of PU.1 in BV2 cells. Grey squares label pathways not found in select conditions. (f) Intersection of significantly up- or downregulated DEGs in BV2 cells with transient PU.1 overexpression and knock-down and disease-associated microglia based on Keren-Shaul et al., 2017. Number of genes overlap and its significance between conditions labeled with green circles below is shown on top of the bars; number of total significant DEGs in each condition is shown on the right (cutoff: padj < 0.05). * P < 0.05, *** P < 0.001.
Quantitative PCR
BV2 cells collected during passaging were used for mRNA extraction using the RNeasy mini kit (QIAgen #74106) including the DNase I treatment step with RNase-Free DNase set (QIAgen #79254) according to manufacturer’s instructions. mRNA quantity was measured using Nanodrop 8000 (Thermo Scientific) and reverse transcription reaction was performed with 1000 ng of RNA using High-Capacity RNA-to-cDNA kit (Thermo Fisher #4387406). 10 ng cDNA was used in the qPCR reaction with Power SYBR Green Master Mix (Applied Biosystems #4368706) run using QuantStudio 7 Flex Real-Time PCR System (Thermo Fisher Scientific). Primers were from PrimerBank (Wang and Seed, 2003) or designed using the Primer-BLAST program (NCBI) and are listed in Supplementary Table 1. Ct values were averaged from two technical replicates for each gene, Gapdh Ct values were used for normalization. Gene expression levels were quantified using the 2−ddCt method relative to control.
Western blotting
BV2 cells were lysed in RIPA buffer (Sigma-Aldrich #R0278) supplemented with Protease/Phosphatase Inhibitor Cocktail (Cell Signaling #5872) with one freeze-thaw cycle and 30 min incubation on ice. Lysates were cleared with 30 min centrifugation at 15,000 g. Protein concentration was measured using the BCA kit (Thermo Fisher #PI-23225) and equal quantities were used to prepare the samples for western blotting. Samples were resolved by electrophoresis with Bolt 4–12% Bis-Tris Plus Gels (Invitrogen) in Bolt MES SDS running buffer (Invitrogen, B0002) and transferred using iBlot 2 nitrocellulose transfer stacks (Invitrogen). Membranes were blocked for 1 hour and probed with antibodies 1:1000–4000 in 3% non-fat dry milk in PBS / 0.1% Tween-20 buffer overnight at +4°C. Secondary antibody staining 1:2000 was applied for 1 hour at RT and visualized with WesternBright ECL HRP Substrate Kit (Advansta, K-12045) and UVP ChemiDoc-ItTS2 Imager (UVP). Images were quantified using ImageJ (NIH).
Phagocytosis assays
Uptake of different bioparticles was measured with FACS in BV2 cells with transient PU.1 overexpression and knock-down or with IncuCyte live cell analysis systems in BV2 cells with stable PU.1 overexpression and knock-down generated as described above. For transient PU.1 modulation BV2 cells were seeded at 200,000 cells/well in 24-well plates and transfected the next day with 1 μg plasmids and 2 μl Lipofectamine 2000 prepared in 100 μl OPTI-MEM added to 500 μl of growth medium per well. The following day 1 ml growth medium was added and 48 hours after transfection cells were treated with bioparticles for uptake in 500 μl (zymosan and myelin 20 μg/well, apoptotic cells to BV2 cells 10:1). Early Jurkat apoptotic cells were prepared using 3-hour incubation with 1 μM staurosporine, late Jurkat and N2A apoptotic cells were prepared using 16-hour incubation with 1 μM staurosporine. Cells were collected, washed with PBS and stained with pHrodo dye 1:100 in PBS for 1 hour in the dark at RT. Cells were washed once with PBS and resuspended at 10,000,000 cells/ml in PBS to be added for uptake to plated BV2 cells. To inhibit phagocytosis cells were pre-treated with 2 μM Cytochalasin D for 30 min and during incubation with bioparticles. After 3-hour incubation cells were collected with trypsin (Gibco #25200) and analyzed on an LSR II flow cytometer (BD Biosciences). To distinguish apoptotic Jurkat or N2A cells from BV2 cells in FACS, samples for the phagocytosis assays were pre-stained with CD11b antibody 1:400 for 30 min and then used for sorting. Data were quantified using FCS Express 7 (De Novo Software) and GraphPad Prism 8 (GraphPad Software). GFP+/pHrodo + cells were used to quantify the phagocytic index: percentage of pHrodo+ cells in GFP+ gated population x geometric mean pHrodo intensity / 106; and represented as phagocytic activity. CD11b-APC+/GFP+/pHrodo+ cells were gated to quantify phagocytic index for apoptotic cell uptake. Stable BV2 cells with overexpression and knock-down of PU.1 were seeded at 50,000 cells/well in 24-well plates and treated with bioparticles in 2 ml the next day to start live imaging of uptake with IncuCyte for 3–4 days.
Viability assays
BV2 cells were seeded at 30,000 cells/well in 96-well plates and treated the following day with 30 nM staurosporine in 100 μl/well for 24 hours. Cells were then used for MTS/PMS proliferation assay (CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay) and the LDH cytotoxicity assay (CytoTox 96® Non-Radioactive Cytotoxicity Assay) according to the manufacturers’ instructions (Promega). For apoptosis and live-imaging analysis BV2 cells were seeded at 130,000 cells/well in 24-well plates and the next day were treated in 500 μl with 30 nM staurosporine for 24 hours or 5 μM rotenone for 2 hours that was then washed off and replaced with fresh medium. Cells were collected with trypsin and stained with PE Annexin V (BD Biosciences #559763) and DAPI or DRAQ7 (Cell Signaling #7406) according to manufacturers’ instructions for further analyses using an LSR II flow cytometer (BD Biosciences). To inhibit apoptosis, cells were pre-treated with 20 μM Z-VAD-FMK for 30 min before and/or during the incubation with staurosporine. For live-imaging analysis DRAQ7 1:300 was added into the treatment medium and cells were placed in an IncuCyte for 24-hour incubation.
Oxidative and inflammatory stress assays
For reactive oxygen species (ROS) and nitric oxide (NO) analyses BV2 cells were plated 24 hours before treatment at 130,000 cells/well in 24-well plates. For ROS cells were pre-loaded with Molecular Probes CellROX Green Reagent (Thermo Fisher #C10444) for 4 hours, which was washed off and replaced with treatment medium. 5 μM rotenone was incubated with cells for 2 hours during incubation with the Cell ROX reagent, then washed off and cells were incubated in growth medium for another 20 hours. 10 μM Aβ42 or 1 μg/ml LPS was added to the medium after CellROX pre-treatment for 20 hours. The following day cells were collected with trypsin, stained with DAPI or DRAQ7 and analyzed using an LSR II flow cytometer (BD Biosciences). For NO assay cells were treated with 1 μg/ml LPS for 4 hours, then washed and medium was replaced with growth medium for another 20 hours. To inhibit NO production cells were pre-treated with 10 mM NAC for 1 hour and during the incubation with LPS. After incubation we used the Griess Reagent Nitrite Measurement Kit (Cell Signaling #13547S) according to the manufacturers’ instructions to measure NO in the conditioned medium. For cytokine analyses BV2 cells were seeded at 750,000/well in 6-well plates, the following day cells were treated with 1 μg/ml LPS for 4 hours, then washed and incubated in 3 ml OPTI-MEM plus Pen/Strep for 24 hours. Conditioned medium was collected, cleared of cell debris with 10 min centrifugation at 3,000 rpm and used for the Proteome Profiler Mouse Cytokine Array Kit, Panel A (R&D Systems #ARY006) according to the manufacturers’ instructions. Primary astrocytes were prepared using cortices from 2 – 5 days old pups that were triturated into single cell suspension. Cells were grown till confluent for 1 – 2 weeks after isolation in DMEM plus 20% FBS and P/S and split at 350,000 cells/well in 6-well plates 5 days before treatment. For treatment of primary astrocytes, conditioned medium from BV2 cells treated with LPS was collected as described above and 2 ml were used to replace medium on astrocytes washed with PBS. Astrocytes were incubated with conditioned medium for 24 hours, then cells were collected in RLT buffer and used for RNA extraction as described above for qPCR or Custom TaqMan Gene Expression Array Cards (Thermo Fisher Scientific).
RNA sequencing and analyses
RNA sequencing was performed on BV2 cell lysates with transient PU.1 overexpression and knock-down as described above for phagocytosis. 48 hours after transfection cells were collected and pooled together from several wells for FAC sorting of GFP+ population using FACSARIA III (BD Biosciences). Sorted cells were used for RNA extraction as described above for qPCR. mRNA cells were submitted to the Genomics Core Facility at the Icahn School of Medicine at Mount Sinai. Samples passed quality control with Qubit and BioAnalyzer showing RIN = 10. Libraries were prepared using TruSeq RNA Sample Prep Kit v2 and sequenced using HiSeq4000 with 100 nt paired-end reads. Raw data was processed by the core and aligned to the mouse genome. Differentially expressed genes (DEGs) were identified using DEseq2 (Bioconductor). Nominal p value is reported for PU.1 overexpression samples in Supplementary Table S2 as correction for multiple comparisons filtered out all significant changes. FDR-corrected padj values are reported for PU.1 knock-down samples in Supplementary Table S3. Data were processed using R Studio for principal component analysis, volcano plots, Venn diagram, heatmaps (gplots). Pathways network analysis was performed using Ingenuity Pathway Analysis software (Qiagen Bioinformatics). Gene enrichment analysis was performed using the SuperExactTest package (Wang et al., 2015). RNA sequencing results were validated using qPCR on 75 genes with Pearson correlation analysis.
Statistics
Data were visualized in GraphPad Prism 8 (GraphPad Software). In each analysis three to five independent experiments were performed (n = 3 – 5 biological replicates). The researcher was not blinded to sample collection. Differences between multiple means of data were analyzed with one-way ANOVA with Bonferroni’s or Sidak’s post-hoc test for selected pairs. An unpaired t test was used for analyses of WB data. A paired t test was used for analyses of uptake half-life. * P<0.05, ** P<0.01, *** P<0.001. All data are represented as mean ± standard deviation (SD).
Results
Knock-down of PU.1 repressed homeostatic microglial gene expression and activated disease-associated microglial signature pathways
We have previously reported that the association of the so-called “CELF1 locus” to AD (Lambert et al., 2013) is likely mediated by a haplotype (tagged by rs1057233-G) associated with lower expression of SPI1 and delayed onset of AD (Huang et al., 2017). SPI1/PU.1 is a myeloid lineage-determining transcription factor expressed in monocytes, macrophages and their progenitors, while in the brain it is exclusively expressed in microglia, the brain-resident macrophages. To predict the effect of PU.1 gene expression levels on microglial function that could be relevant to AD pathogenesis and suggest the mechanism of action for the protective effect of lower PU.1 expression, we performed RNA sequencing in BV2 mouse microglial cells with transient overexpression and knock-down of Spi1/PU.1. BV2 cells were transfected with vector constructs carrying mouse PU.1 cDNA or shRNA using Lipofectamine RNAiMAX and FAC-sorted for GFP+ population to enrich for targeted cells. This set-up allowed us to assess the direct effect of temporary modulation of PU.1 on the microglial transcriptome, and potentially mimic use of a PU.1 inhibitor, which should be unrelated to compensation mechanisms that would otherwise take place in cell lines with stable overexpression and knock-down of PU.1. Principal component analysis revealed good separation of samples with PU.1 knock-down (KD_S1–3) versus respective scrambled treated control cells (KD_C1–3); however, PU.1 overexpression samples (OE_S1–3) were not so different from control samples (OE_C1–3) (Supplementary Fig. S1a). The result of this PCA for the overexpression condition reflects the small number of genes affected by PU.1 overexpression – 191 (Spi1 was upregulated by 60%), while there were 3892 genes differentially expressed after PU.1 knock-down (Spi1 was downregulated by 71%) (Fig. 1a–b, Supplementary Tables S2–3). We selected a panel of genes related to AD and the disease-associated microglial signature (DAM) based on (Keren-Shaul et al., 2017) for validation of RNA sequencing data with qPCR (Supplementary Table S1). Pearson correlation of 75 genes showed good overlap of DEGs from RNA sequencing with qPCR results in BV2 cells (Supplementary Fig. S1b, Supplementary Tables S2–3). In order to assess the relevance of the RNA sequencing findings we validated a selection of BV2 DEGs in human induced microglial-like cells (iMGLs) upon transient siRNA-mediated knock-down of PU.1. Changes in the expression of genes mediating immune response and lipid metabolism or direct targets of PU.1 in iMGLs overlapped with BV2 cells suggesting PU.1 affects similar aspects of microglial biology in human and mouse cells (Figure 1c). In addition, we validated the effect of PU.1 on Itgam expression at the protein level by immunostaining for CD11b, which showed a significant reduction of CD11b after PU.1 knock-down, but not overexpression consistent with the RNA sequencing data (Supplementary Fig. S1c–d). Next we used DEGs for Ingenuity Pathway Analysis (IPA) to dissect the functions that are affected by PU.1 expression in microglia (Fig. 1d). To describe how PU.1 affects microglia and predict its role in vivo we compared pathways affected by PU.1 expression and the disease-associated microglia (DAM) (Keren-Shaul et al., 2017) that surround amyloid β (Aβ) plaques in AD and have been reported to be the protective population of microglia in patients’ brains. The majority of genes associated with microglial core functions were repressed by PU.1 knock-down, e.g. phagocytosis, cell migration and immune response (Supplementary Table S4). We observed a stark distinction and opposite directionality between the functions affected by PU.1 overexpression versus knock-down. Pathway analysis showed activation of LXR/RXR and EIF2 signaling and increased antioxidant action of vitamin C in cells with PU.1 knock-down suggesting a sustained level of oxidative stress response, cholesterol/lipid metabolism and protein translation (Fig. 1e). These pathways are similarly activated in DAM and oppositely inhibited in PU.1 overexpression that is associated with increased risk for AD based on human genetics, suggesting these pathways may be central to microglial response to amyloid β in the brain. Interestingly, DAM and PU.1 overexpression both upregulated TREM1 pathways, while PU.1 knock-down showed decreased activation, which aligns with reduced uptake capacity in BV2 cells after PU.1 downregulation (Huang et al., 2017). Along with reduced phagocytosis we detect reduced activation of IRF by cytosolic pattern recognition receptors in BV2 cells with PU.1 knock-down, suggesting these cells exhibit lower activity and inflammatory response in contrast to DAM. Indeed, 199 up- and 256 downregulated genes in the DAM signature were significantly enriched in the PU.1-downregulated gene set (Fig. 1f), perhaps underlying the differences we observed in the pathways regulated by PU.1 knock-down and DAM (Fig. 1e). Only 18 genes were upregulated by PU.1 overexpression and DAM, which may underlie shared activation of TREM signaling in line with the increased phagocytic capacity of these cells. Naturally, intersection analysis detected a significant enrichment of genes reciprocally regulated by PU.1 in overexpression and knock-down conditions – 95 positively and 26 negatively regulated genes by PU.1 (Fig. 1f). To validate our findings on the functions and pathways affected by PU.1 we generated cell lines with lentiviral overexpression and knock-down of PU.1 using puromycin selection in BV2 cells. Analysis of mRNA and protein expression of PU.1 showed that we were able to obtain 2-fold overexpression of PU.1 (OE cell line) and 75% knock-down of PU.1 (KD cell line) at the protein level (Supplementary Fig. S2a–c) relative to their respective controls (CTL line for overexpression and SCR line for knock-down). These lines were used for several assays discussed below instead of transient modulation of PU.1 to circumvent low efficiency and cell death associated with lipofectamine transfection. Altogether, PU.1 regulates a plethora of functions in microglial cells that we have evaluated in vitro to begin to describe microglial phenotypes important in supporting the brain milieu that is affected in AD.
PU.1 overexpression increased and knock-down decreased microglial phagocytosis
Phagocytic capability of microglia has been shown to be affected by risk factors for AD, such as TREM2 and CD33 (Griciuc et al., 2013; Kleinberger et al., 2014). We have previously reported that PU.1 regulates zymosan phagocytosis (Huang et al., 2017) and others reported that PU.1 knock-down reduces Aβ42 uptake in human microglial cells (Smith et al., 2013). Here we have expanded the number of bioparticles used to test microglial phagocytic uptake of myelin, zymosan, early and late apoptotic cells – human Jurkat T lymphocyte and N2A mouse neuroblastoma cells, respectively (Supplementary Fig. S3f). BV2 cells with transient overexpression of PU.1 increased and knock-down of PU.1 decreased phagocytic uptake of all bioparticles independent of their nature as measured by FACS 3 hours after treatment (Supplementary Fig. S3a–e). Longitudinal experiments tracking internalization of pHrodo-labeled myelin and zymosan in BV2 cells with stable overexpression or knock-down of PU.1 showed similar results with increased PU.1 expression leading to increased uptake and decreased PU.1 expression inhibiting bioparticle uptake, based on the quantification of uptake half-life (Fig. 2d, h). Interestingly, live imaging of myelin and zymosan internalization showed different kinetics of uptake, while proliferation of PU.1 overexpression and knock-down cells was comparable to control cell lines (Fig. 2a, e). Myelin uptake was accelerated in PU.1 overexpressing cells and delayed in PU.1 knock-down cells, however, both lines with differential PU.1 expression show a similar capacity to internalize myelin over time illustrated by the plateau that is reached by both lines in 60 – 80 hours (Fig. 2b, f). PU.1 overexpressing cells showed increased zymosan internalization compared to control CTL cells, while PU.1 knock-down cells displayed limited capability to internalize zymosan in comparison to control SCR cells (Fig. 2c, g). Overall, these data align with previous reports and show that PU.1 is a positive regulator of phagocytic uptake in microglia independent of the nature of the bioparticles. However, PU.1 may differentially regulate the capacity of microglia to store and degrade internalized myelin and zymosan in BV2 microglial cells.
Figure 2. Stable PU.1 overexpression increases and knock-down decreases phagocytic uptake in BV2 microglial cells.
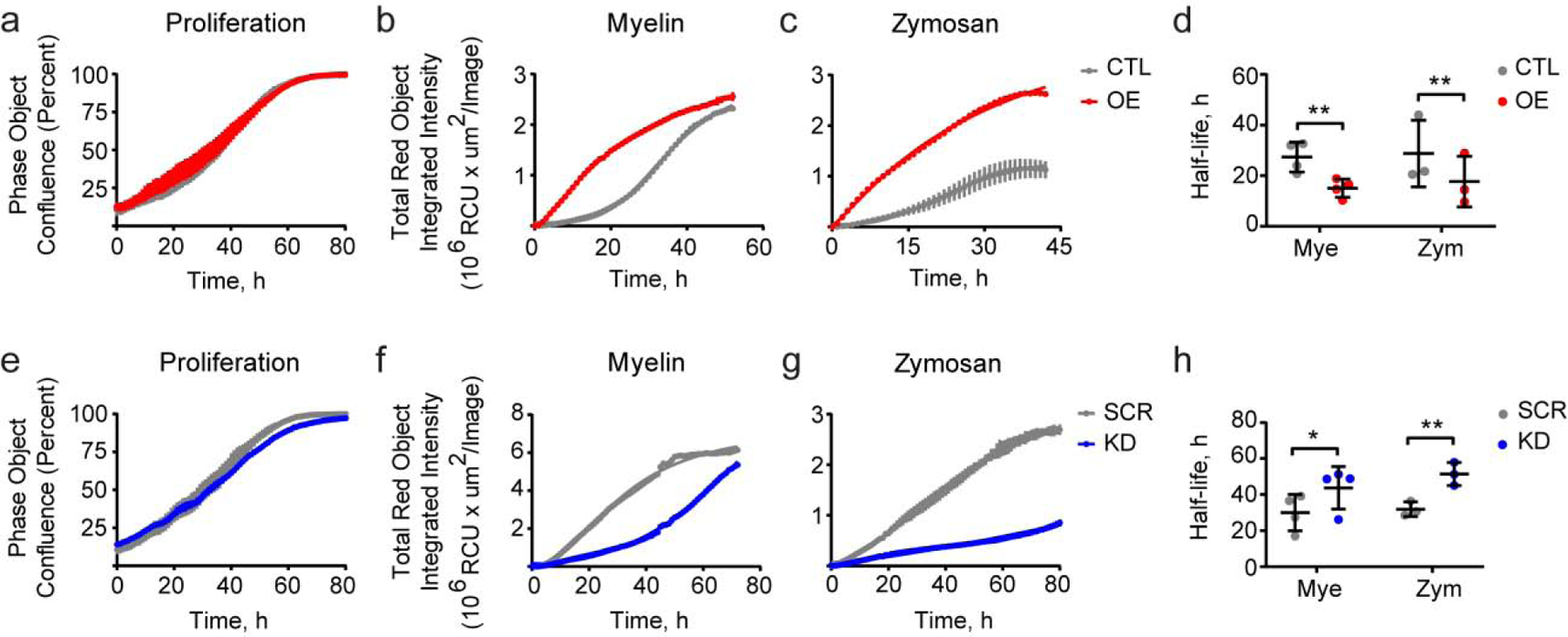
(a, e) Proliferation of cells with overexpression and knock-down of PU.1 is not affected as imaged by IncuCyte. (b, f) Uptake of myelin and (c, g) zymosan is increased in PU.1 overexpressing cells and decreased in PU.1 knock-down BV2 cells. (d, h) Half-life of myelin and zymosan uptake is reduced in cells with increased PU.1 expression (d) and increased in cells with PU.1 knock-down (h). Values shown are mean ± SD (a–c, e–g) representative of n = 4–5 independent experiments with two technical replicates each or (d, h) n = 4–5 independent experiments with two technical replicates. * P < 0.05, ** P < 0.01. (d, h) Paired t test.
PU.1 overexpression increased resistance and knock-down increased susceptibility of microglia to apoptosis
Myeloid cells have limited capacity to internalize debris and excessive uptake uncoupled from effective degradation can lead to cell death in vivo. To elucidate the contribution of PU.1 to cell death under stimulated conditions we challenged BV2 cells with staurosporine, which induces caspase-dependent cell death. PU.1 overexpression reduced staurosporine-induced cell death as measured by an MTS assay assessing cell proliferation potential, which was enhanced by increased PU.1 (Fig.3a) likely due to increased viability observed with an LDH assay assessing the rate of cell death after staurosporine treatment (Fig. 3a–b). Interestingly, the extent of cell death rescue by PU.1 overexpression was comparable to the effect of Z-VAD-FMK, a pan-caspase inhibitor, which prevents staurosporine-mediated cell death. On the contrary, knock-down of PU.1 decreased viability of BV2 cells after staurosporine treatment (Fig. 3c–d). The effect of PU.1 on the viability of BV2 cells was further investigated by measuring plasma membrane permeability and outer-leaflet phosphatidylserine exposure in BV2 cells after staurosporine treatment (Supplementary Fig. S4a–b). PU.1 overexpression reduced and PU.1 knock-down increased the population of Annexin V+/DRAQ7− cells describing viable early apoptotic cells positive for phosphatidylserine staining with Annexin V and negative for DRAQ7, which stains nuclei in dead and permeabilized cells (Fig. 3e–f). Populations of nonviable phosphatidylserine-negative late apoptotic (Annexin V+/DRAQ7 +) and necrotic (Annexin V−/DRAQ7+) cells seem to be unaffected by PU.1 expression when compared to Z-VAD-FMK, which reduced the number of cells in all three states. Live-imaging of the DRAQ7+ necrotic cells revealed that PU.1 overexpression delayed the onset of BV2 cell death after staurosporine treatment, while PU.1 knock-down cells died at the same rate as control cells (Supplementary Fig. S4c–d). To elucidate the mechanism of PU.1 regulation of staurosporine-mediated cell death in BV2 cells we evaluated activation of caspases by western blotting (Supplementary Fig. S5). PU.1 overexpression reduced cleavage of caspases-3, -8, -9, Lamin A/C, and PARP suggesting that the rescue of staurosporine-mediated cell death was prevented by reduced activation of the caspase cascade similar to the mechanism of action of the Z-VAD-FMK inhibitor (Fig. 3g–h). Interestingly, staurosporine treatment of BV2 cells with PU.1 overexpression reduced PU.1 expression levels, but had no effect on PU.1 in control CTL cells (Supplementary Fig. S4e). In fact, levels of PU.1 were equal under staurosporine treatment in overexpression and knock-down conditions, however, the latter was due to decreased PU.1 expression in control SCR cells, but not in PU.1 knock-down cells (Supplementary Fig. S4f). Knock-down of PU.1 also did not affect caspase activation only showing a trend towards increased activation of caspases (Fig. 3g–i). In conclusion, PU.1 overexpression renders microglial cells resistant to caspase-mediated apoptosis, while PU.1 knock-down cells become more susceptible to caspase-induced cell death.
Figure 3. Stable PU.1 overexpression decreases and knock-down increases apoptotic cell death in BV2 microglial cells.
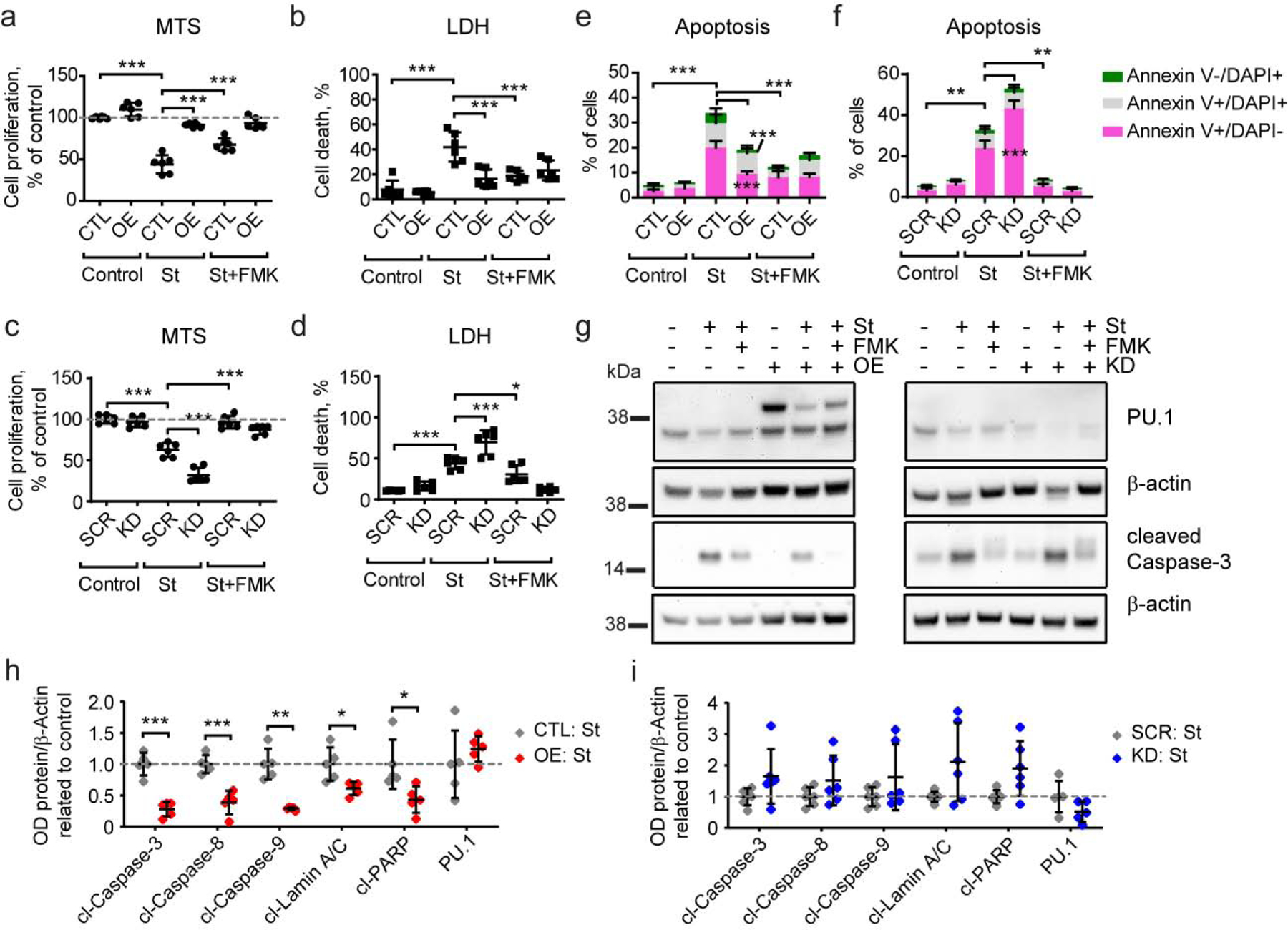
(a, b) PU.1 overexpression leads to increased resistance of BV2 cells to staurosporine treatment, while (c, d) PU.1 knock-down cells are more sensitive to staurosporine treatment measured by MTS (a, c) and LDH (b, d) assays. (e) PU.1 overexpression decreases the number of early apoptotic and necrotic cells after staurosporine treatment. (f) PU.1 knock-down increases the number of early apoptotic cells after staurosporine treatment. (g) Activation of caspase-3 was reduced in PU.1 overexpressing cells and unaffected in PU.1 knock-down cells after staurosporine treatment, representative western blot image. (h, i) Quantification of apoptosis-dependent activation of proteins based on western blot as shown in (g) and Supplementary Fig. S5. Values shown are mean ± SD of (a-d) n = 3 independent experiments with two technical replicates each or (g–i) n = 4–5 independent experiments with one technical replicate. * P < 0.05, ** P < 0.01, *** P < 0.001. (a–d) One-way ANOVA with Bonferroni’s post-hoc multiple comparisons, (h–i) unpaired t test with Welch’s correction.
PU.1 overexpression potentiates and knock-down mitigates microglial oxidative stress response
Since PU.1 regulated cell viability upon exposure to cytotoxic stimuli it is plausible that survival of microglial cells will result in differential inflammatory response transmitted by microglia in the brain milieu. To test the effect of PU.1 expression on microglial stress response we treated BV2 cells with rotenone, an inhibitor of complex I of the mitochondrial electron transport chain, which leads to an increase in mitochondrial production of reactive oxygen species (ROS) and cytochrome c release inducing apoptosis. Rotenone-treated BV2 cells showed an increase in ROS with increased number of cells responding to rotenone treatment and increased production of ROS per cell. PU.1 overexpression potentiated both parameters after rotenone treatment, which may due to high levels of ROS and ROS+ cells at baseline (Fig. 4a–b, Supplementary Fig. S6a), suggesting that PU.1 overexpression increases the capacity of microglial cells to tolerate oxidative stress. In contrast, knock-down of PU.1 decreased the number of ROS+ cells and ROS production per cell as measured by FACS, while baseline levels of ROS were unaffected (Fig. 4c–d, Supplementary Fig. S6b). Rotenone treatment of BV2 cells affected cell viability in a similar manner to staurosporine, with PU.1 overexpression showing increased survival upon rotenone treatment (Supplementary Fig. S6e) and a reduced number of early apoptotic cells (Fig. 4e, Supplementary Fig. S6c). This correlated with reduced activation of caspase-3 (Fig. 4g–h), suggesting PU.1 overexpressing cells show increased survival upon rotenone treatment. Thus, the effect of PU.1 overexpression on microglial survival and capacity for ROS production seems to underlie the increased production of ROS in these cells. On the contrary, viability of PU.1 knock-down cells was affected to the same extent as in control SCR cells showing similar levels of early apoptotic cells (Fig. 4f), rate of cell death (Supplementary Fig. S6f) and caspase-3 activation (Fig. 4g–h). Thus, PU.1 knock-down reduces oxidative stress in microglia independent of its effect on cell survival. Indeed, expression levels of PU.1 were unchanged under stimulated conditions compared to baseline, contrary to data after staurosporine treatment (Supplementary Fig. S6g). Next we tested the effect of Aβ42 and LPS on oxidative stress response in microglial cells as amyloid and inflammation trigger and exacerbate pathological changes in AD brain (Wendeln et al., 2018). Treatment of control CTL cells with Aβ42 or LPS did not produce a robust ROS response, nevertheless, PU.1 overexpression amplified production of ROS in cells exposed to these stimuli (Fig. 4i–j). Control SCR cells increased ROS production upon Aβ42 and LPS stimulation, which was repressed by knock-down of PU.1 (Fig. 4k–l), suggesting PU.1 regulates ROS production by microglia upon exposure to pro-inflammatory stimuli. The difference in response of respective control cells may be due to the transformation procedure rendering SCR cells more inflammatory prone than CTL cell line. In conclusion, PU.1 overexpression potentiates microglial oxidative stress through a direct effect on ROS production and increased survival after treatment with cytotoxic stimuli, while PU.1 knock-down represses the microglial oxidative stress response after rotenone, Aβ42 and LPS treatment.
Figure 4. Stable PU.1 overexpression increases and knock-down decreases rotenone-dependent cell death and production of reactive oxygen species in BV2 microglial cells.
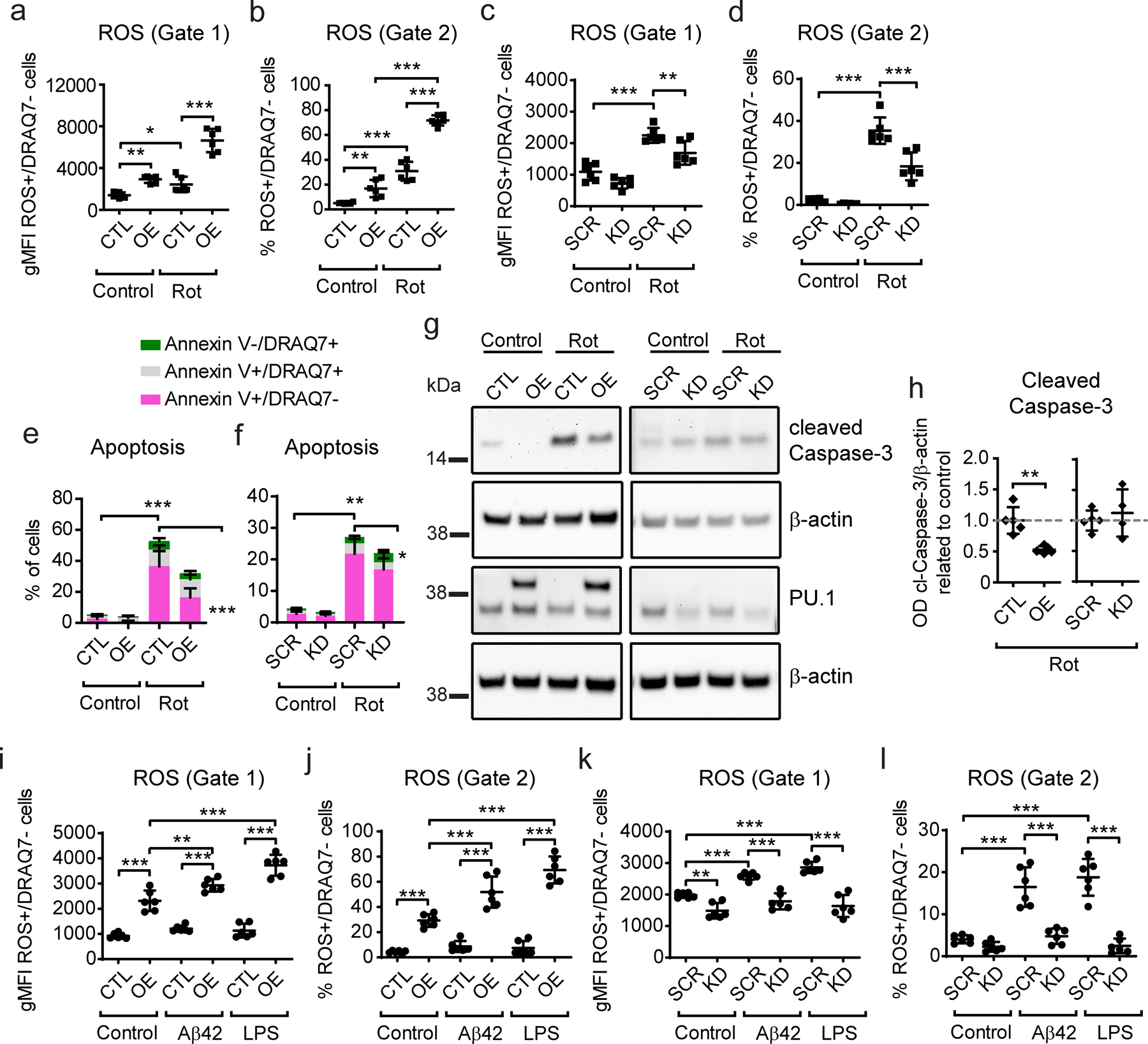
(a, c) Quantification of ROS intensity and (b, d) number of ROS+/DRAQ7− cells in PU.1 overexpressing and knock-down cells after rotenone treatment based on gate 1 for the whole cell population and gate 2 for the rotenone-reactive cells set in Supplementary Fig. S6a–b. (e, f) Analysis of the number of early, late apoptotic and necrotic cells with (e) PU.1 overexpression and (f) knock-down after rotenone treatment. (g) Representative western blot image of caspase-3 activation and PU.1 levels after rotenone treatment. (h) Quantification of cleaved caspase-3 levels based on western blot as shown in (g). (i, k) Quantification of ROS intensity and (j, l) number of ROS+/DRAQ7− cells in PU.1 overexpressing and knock-down cells after Aβ42 and LPS treatment. Values shown are mean ± SD of (a–f, i–l) n = 3 independent experiments with two technical replicates each or (h) n = 4–5 independent experiments with one technical replicate. * P < 0.05, ** P < 0.01, *** P < 0.001. (a–f, j–l) One-way ANOVA with Bonferroni’s post-hoc multiple comparisons, (h) unpaired t test with Welch’s correction.
PU.1 overexpression amplifies and knock-down abates microglial inflammatory response
Microglial inflammatory response is transmitted through different cytokines affecting astrocytes reactivity and neuronal viability, which contribute to AD pathology. To assess the magnitude of PU.1 effect on the regulation of microglial inflammatory response we analyzed nitric oxide (NO) and cytokine production in BV2 cells upon LPS stimulation. PU.1 overexpression further potentiated NO production after treatment with LPS compared to control cells, while PU.1 knock-down repressed NO production (Fig. 5a–b). This effect was specific to induction of oxidative stress as it could be partially reduced by N-acetyl cysteine (NAC) antioxidant pre-treatment and was due to PU.1-mediated regulation of inducible nitric oxide synthase (iNOS) expression which showed increased levels in PU.1 overexpressing cells and reduced levels in cells with PU.1 knock-down (Fig. 5c–d). Furthermore, analysis of cytokine production in BV2 cells suggests several molecules produced prior to treatment (Supplementary Fig. S7a–b), e.g. TNFα, CXCL2, CXCL10, CCL5, CCL12, IL-6, TREM1, etc. show increased secretion after LPS treatment. As we have previously seen with ROS and NO, PU.1 overexpression amplified cytokine secretion suggesting an increased pro-inflammatory response (Fig. 5e). Baseline production of several cytokines in PU.1 overexpressing cells was also increased, likely contributing to the amplified response after LPS treatment. Control SCR cells responded to LPS treatment with secretion of a larger number of cytokines, as previously observed for ROS, after Aβ42 and LPS stimulation (Fig. 5f). In contrast, PU.1 knock-down diminished LPS-mediated inflammatory response in BV2 microglial cells seemingly to baseline levels. Activated microglia propagate the inflammatory response to astrocytes, which become reactive and neurotoxic (Liddelow et al., 2017). Treatment of primary astrocytes with conditioned medium from LPS-stimulated microglial cells with PU.1 overexpression resulted in increased expression of the astrocytes reactive signature, while PU.1 knock-down negated induction of A1 reactive astrocytes after LPS treatment (Fig. 5g, Supplementary Fig. S7d). These data suggest that PU.1 regulates the inflammatory response of microglial cells to pathogenic stimuli and that increased PU.1 expression would amplify and propagate it, while decreased PU.1 expression will mitigate microglia-mediated pro-inflammatory responses.
Figure 5. Stable PU.1 overexpression exacerbates and knock-down represses inflammatory response to LPS in BV2 microglial cells.
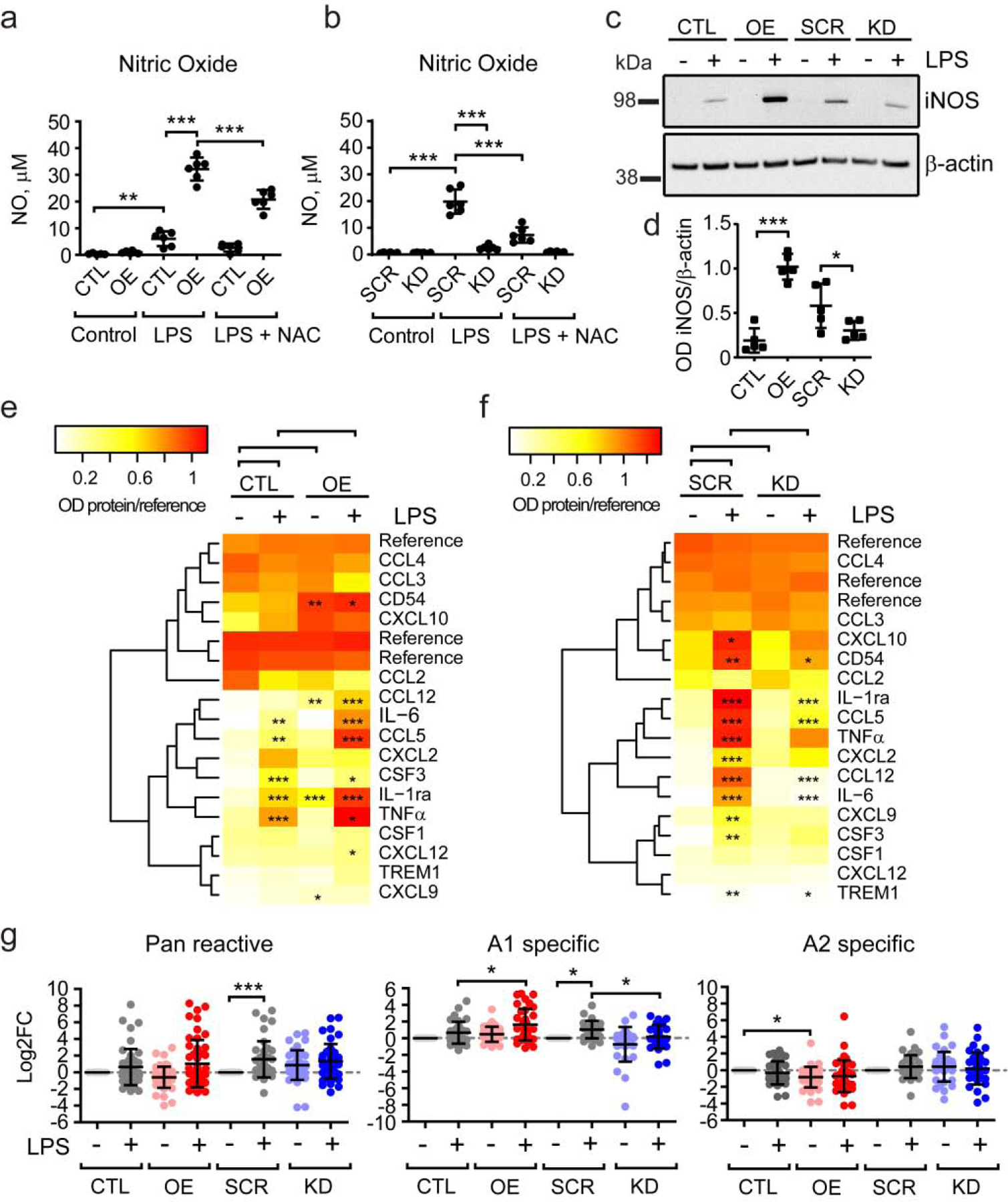
(a, b) Nitric oxide production is potentiated by PU.1 overexpression and repressed by PU.1 knock-down after LPS treatment. (c) Representative western blot image of iNOS levels in PU.1 overexpression and knock-down cells stimulated with LPS. (d) Quantification of iNOS expression in LPS-stimulated cells based on western blot as shown in (c). (e, f) Heatmap of protein expression levels in conditioned medium of (e) PU.1 overexpression and (f) knock-down cells treated with LPS. (g) Effect of microglial conditioned medium treatment as in (e, f) on astrocytes gene signature analyzed by qPCR. Values shown are mean ± SD of (a–b) n = 3 independent experiments with two technical replicates each or (d, g) n = 4–5 and (e–f) n = 3 independent experiments with one technical replicate. * P < 0.05, ** P < 0.01, *** P < 0.001. One-way ANOVA with (a, b, d, g) Bonferroni’s and (e, f) Sidak’s post-hoc multiple comparisons.
Discussion
The SPI1 locus is one of the few AD risk loci for which a mechanism of action based on the modulation of gene expression by common disease risk alleles has been delineated (Huang et al., 2017). Here we have generated and characterized BV2 microglial cell lines with high and low PU.1 expression to examine changes in and microglial functions, that may be relevant to AD pathogenesis. Using phagocytosis, apoptosis and oxidative stress assays we have revealed the detrimental and beneficial features of microglia underlying the risk and protective effects of PU.1 in AD. This phenotypic characterization of PU.1 effects in vitro will inform future in vivo investigations and small molecule phenotypic screening strategies for drug discovery.
Modulation of PU.1 significantly affected the microglial transcriptome with knock-down of PU.1 showing opposing effects to overexpression (Fig. 1), which we further compared to the previously described DAM signature to elaborate on the possible significance of these changes in vivo (Keren-Shaul et al., 2017). In comparison with BV2 cells with differential PU.1 expression the DAM signature had a larger number of genes enriched in the set of genes downregulated by PU.1 knock-down rather than overexpression (Fig. 1d). Indeed, modest repression of microglial functions and homeostatic genes in DAM aligned with a marked decrease of the same functions and pathways by PU.1 knock-down (Fig. 1c). However, a few pathways were activated with PU.1 knock-down and at the same time downregulated with PU.1 overexpression (Fig. 1d). We propose that the protective effect of reduced PU.1 expression in AD is mediated through pathways of LXR/RXR signaling, antioxidant action of vitamin C and/or EIF2 protein translation similar to the protective effect of DAM. LXR/RXR agonists have long been considered for treatment of AD in order to modulate lipid metabolism and inflammatory response. Indeed, genetic variants in several targets of LXR/RXR transcription factors have been associated with risk for AD, e.g. APOE, CLU, ABCA1, ABCA7, ABCG1, TREM2 (A-González and Castrillo, 2011; Pimenova et al., 2017b; Savage et al., 2015). SPI1 is a master regulator of this gene network, upregulation of which suggests clearance of lipids and debris may be preserved with PU.1 knock-down. Importantly, APOE signaling is required for microglia clustering and transcriptional response to amyloid plaques (Sala Frigerio et al., 2019), which based on our data will be sustained with PU.1 downregulation, similarly to how impaired degradation of myelin debris by Trem2−/− or Apoe−/− microglia in mouse model of demyelination is rescued with the agonist of LXR (Cantuti-Castelvetri et al., 2018; Nugent et al., 2020). Oxidative stress is part of AD pathology as Aβ42 neurotoxicity leads to induction of reactive oxygen species and other free radicals, which in turn may escalate production and deposition of Aβ and phosphorylation and polymerization of tau (Kim et al., 2015). Mutations in EIF2B1-5 genes, encoding subunits of eIF2b, responsible for protein translation, lead to leukoencephalopathy with vanishing white matter characterized by progressive degeneration of white matter due to myelination failure and impairments in oligodendrocyte function (Bugiani et al., 2010). Since impairments in these pathways contribute to AD progression, counteracting consequences of defects in these pathways will be beneficial in AD, supporting the protective effect of reduced PU.1 expression in microglia.
The phagocytic capacity of microglia is considered to be beneficial for AD pathology based on functional studies of TREM2 and CD33 AD risk-increasing mutations. TREM2 rare genetic variants associated with AD risk impair microglial lipid sensing and phagocytosis (Kleinberger et al., 2014; Yeh et al., 2016). The AD-associated risk allele of CD33 likely produces the longer more active spliced form of this inhibitory receptor (Pimenova et al., 2017b), which correlates with increased cell surface expression and reduced phagocytic capacity observed in human monocytes (Bradshaw et al., 2013). Moreover, activation of the DAM signature is dependent on TREM2 suggesting DAM have increased phagocytic clearance ability (Keren-Shaul et al., 2017). In BV2 microglial cells protective low expression of PU.1 led to decreased phagocytic uptake (Fig. 2), which is in line with previous data showing that siRNA knock-down of PU.1 in human microglia in vitro reduces expression of genes involved in phagocytic and antigen presentation pathways (Rustenhoven et al., 2018). This effect may be beneficial in AD considering how microglial uptake of Aβ or lipids may hinder effective debris clearance and result in microglial cell death contributing to seeding of the amyloid plaque core (Baik et al., 2016). This process may be similar to how macrophages contribute to atherosclerotic plaques when failure to efficiently eliminate lipids results in the formation of foam cells in cardiovascular disease (Moore and Tabas, 2011). Another possibility explaining the protective effect of a reduction in phagocytic uptake is that microglial cells with PU.1 knock-down may continue to maintain a barrier around the amyloid plaques, which has been shown to insulate toxic Aβ42 and improve cognition in mice independent of microglial phagocytic activity based on studies of Trem2−/−mice (Condello et al., 2015; Yuan et al., 2016). On the other hand, under normal conditions microglia sculpt neuronal brain connections and increased phagocytic activity by PU.1 overexpression may result in elevated removal of dendritic spines contributing to neuronal loss in disease based on studies of microglia depletion in mouse models (Elmore et al., 2018). Interestingly, PU.1 may modulate specificity of microglial phagocytosis towards pro- and anti-inflammatory bioparticles, e.g. amyloid β and zymosan versus myelin, as microglia with PU.1 knock-down specifically showed preserved capacity for storing internalized myelin albeit with a delayed uptake, which was not observed for zymosan (Fig. 2f–g). Demyelination and white matter abnormalities are observed in AD brains early in disease progression (Nasrabady et al., 2018; Raj et al., 2017) and dampening microglia functions in the early stages of disease can be beneficial as shown for Trem2−/− mouse models of AD (Jay et al., 2017). However, the impaired ability of Trem2−/− or Apoe−/− microglia to remove damaged myelin hinders subsequent remyelination (Cantoni et al., 2015; Cantuti-Castelvetri et al., 2018; Poliani et al., 2015; Yeh et al., 2016). Based on these observations and our data we propose that PU.1 knock-down may be beneficial in slowing overt neurodegeneration at the onset of disease due to decreased ability of microglia to seed amyloid plaques, possibly without compromising on clearance and maintenance of brain tissue homeostasis at later stages. Thus, it will be important to further test these possibilities in mouse models of AD and demyelination.
Deletion of PU.1 results in ablation of myeloid lineage cell types in mouse, e.g. macrophages and B cells, suggesting modulation of PU.1 levels in adult brain may affect microglial viability (Kierdorf et al., 2013; McKercher et al., 1996). While baseline cell viability was not affected by modulation of PU.1, likely because of in vitro cell culture conditions, PU.1 overexpression led to increased resistance and knock-down to increased susceptibility of microglial cells to staurosporine- and rotenone-induced cell death (Figs. 3a–d and 4e–f). Specifically, PU.1 overexpression reduced and knock-down increased caspase-activated apoptosis of microglia (Fig. 3e–i), which was corroborated by PU.1 regulation of “organismal death” and “morbidity and mortality” functions (Fig. 1c). In mouse models of AD genetic or pharmacological depletion of microglia prevented microglia association with amyloid plaques and neuronal loss, which led to improved learning and memory without affecting amyloid plaque load in mouse brain (Dagher et al., 2015; Rice et al., 2015; Spangenberg et al., 2016). Thus, depletion of microglia utilizing knock-down of PU.1 expression may be beneficial in AD pathogenesis. Moreover, microglial repopulation after depletion reversed age-related deficits in long-term potentiation as initial elimination of microglia increased neurogenesis and dendritic spine densities (Elmore et al., 2018). In mouse and human brain turnover of microglia depends on high proliferation rates spatially coupled to efficient apoptosis in order to maintain steady state cell numbers in each brain region (Askew et al., 2017; Füger et al., 2017). Proliferation of microglia depends on CSF1R signaling through PU.1 and C/EBPα (Gomez-Nicola et al., 2013), and we now show that microglial apoptosis is also modulated by PU.1 (Fig. 3), which in vivo leads to regeneration of white matter after death of pro-inflammatory microglia following demyelination (Lloyd et al., 2019). We propose that the protective effect of reduced PU.1 expression in AD may be due to increased turnover and repopulation of microglia initiated by increased susceptibility of microglia to apoptotic cell death under pathological conditions.
Inflammation is an integral part of the innate immune response to tissue damage and pathogen infection, so the impact of differential PU.1 expression can be further modulated by changes in microglial cell viability, thus prolonging, dampening or differentially steering the innate immune response. Indeed, we show here that PU.1 overexpression potentiated and knock-down repressed production of ROS, NO and inflammatory cytokines after treatment with rotenone, Aβ42 or LPS (Figs. 4 and 5). Specifically, PU.1 overexpression converted microglial cells to a pro-inflammatory phenotype in control conditions suggesting microglial cells are primed for exaggerated inflammatory response. This effect was propagated to primary astrocytes inducing a neurotoxic reactive gene signature, which was reduced by PU.1 knock-down in LPS-stimulated microglial cells (Fig. 5g), suggesting reduced PU.1 expression converts microglial cells to a neuroprotective phenotype (Joshi et al., 2019; Yun et al., 2018). The concept of microglial priming is related to the idea of trained immunity that is controlled by epigenetic mechanisms imprinted after first exposure to a pro-inflammatory stimulus (Haley et al., 2019). Our data suggest that PU.1 may contribute to this mechanism of innate immune memory in the brain (Wendeln et al., 2018). PU.1 knock-down dampened stimulus-dependent pro-inflammatory response, which is generally considered protective in neurodegenerative conditions. Indeed, co-expression network analysis of transcriptomic changes in mouse models of AD revealed distinct states of DAM with pro- and anti-inflammatory profiles (Rangaraju et al., 2018). Further analyses of single-cell RNA sequencing data identified interferon-related and neurodegeneration-related states of microglia activation that are present in aging and several models of neurodegeneration and inflammation in mice (Friedman et al., 2018; Sala Frigerio et al., 2019). The neurodegeneration-related state is controlled by APOE, and agonists of LXR promoted this anti-inflammatory state and microglial phagocytosis of Aβ42 in vitro (Rangaraju et al., 2018). Overall, downregulation of PU.1 expression seems to transform microglial cells into an anti-inflammatory state with activated LXR/RXR and reduced IRF signaling that is protective in AD (Fig. 1), albeit distinct from previously described DAM.
The most well studied genetic risk factors for AD, e.g. APOE, TREM2, CD33, describe specific aspects of changes in microglial biology under pathogenic conditions, which is defined by the gene function. As PU.1 is a master regulator of microglial function and several genetic risk factors for AD (APOE, CLU, MS4A4A/MS4A6A, PILRA), it may be possible to modulate several phenotypes at once by targeting a single gene for treatment of AD. In this report we investigated the impact of modulating PU.1 dosage on microglial cell phenotypes. PU.1 downregulation reduced phagocytosis, increased apoptotic cell death and inhibited immune response, which promoted the anti-inflammatory state of microglia that is likely protective in AD. These studies represent the first step toward investigating PU.1 inhibition/lower expression as a possible therapeutic target for AD treatment.
Supplementary Material
Highlights:
PU.1 knock-down abates homeostatic microglial signature and inflammatory response
PU.1 regulates staurosporine-mediated apoptotic cell death in BV2 cells
PU.1 regulates ROS production in response to rotenone, Aβ42 and LPS in BV2 cells
PU.1 regulates LPS-induced nitric oxide and cytokine release in BV2 cells
Acknowledgements
This study was funded by the JPB Foundation (A.M.G.), NIA grant RF1AG054011 (A.M.G.), the Paul A. Slavik Fund (A.M.G.), and the BrightFocus Foundation (A.A.P.). We thank the National Centralized Repository for Alzheimer’s Disease and Related Dementias (NCRAD) for 75.11-IW1A12 PBMCs used for iPSCs generation. The Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) and Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS) Studies were made possible through the support of the U.S Department of Health and Human Services (DHHS), the National Institute on Aging (NIA), the National Institute of Neurological Disorders and Stroke (NINDS) and the National Center for Advancing Translational Sciences (NCATS) grants: U54NS092089 and U01AG045390. NCARD thanks the staff and investigators of the study as well as the participants and their families, whose help and participation made their work possible. We thank Celeste Karch (Washington University in St. Louis) for iPSC lines F11349, F12455. The recruitment and clinical characterization of research participants at Washington University were supported by NIH P50 AG05681, P01 AG03991, and P01 AG026276.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
A.M.G. has consulted for Eisai, Biogen, Pfizer, AbbVie, Cognition Therapeutics and GSK. A.A.P., M.H., I.G., S.I.M., K.R.B. and E.M. report no conflicts of interest.
References
- A-González N, Castrillo A, 2011. Liver X receptors as regulators of macrophage inflammatory and metabolic pathways. Biochim. Biophys. Acta - Mol. Basis Dis 1812, 982–994. 10.1016/j.bbadis.2010.12.015 [DOI] [PubMed] [Google Scholar]
- Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, Richardson P, Tipton T, Chapman MA, Riecken K, Beccari S, Sierra A, Molnár Z, Cragg MS, Garaschuk O, Perry VH, Gomez-Nicola D, 2017. Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep 18, 391–405. 10.1016/j.celrep.2016.12.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baik SH, Kang S, Son SM, Mook-Jung I, 2016. Microglia contributes to plaque growth by cell death due to uptake of amyloid β in the brain of Alzheimer’s disease mouse model. Glia 10.1002/glia.23074 [DOI] [PubMed]
- Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A, Morris MC, Evans D. a, Johnson K, Sperling R. a, Schneider J. a, Bennett D. a, De Jager PL, 2013. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat. Neurosci 16, 848–50. 10.1038/nn.3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugiani M, Boor I, Powers JM, Scheper GC, Van Der Knaap MS, 2010. Leukoencephalopathy with vanishing white matter: A review. J. Neuropathol. Exp. Neurol 69, 987–996. 10.1097/NEN.0b013e3181f2eafa [DOI] [PubMed] [Google Scholar]
- Cantoni C, Bollman B, Licastro D, Xie M, Mikesell R, Schmidt R, Yuede CM, Galimberti D, Olivecrona G, Klein RS, Cross AH, Otero K, Piccio L, 2015. TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol 129, 429–447. 10.1007/s00401-015-1388-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti-Castelvetri L, Fitzner D, Bosch-Queralt M, Weil M-T, Su M, Sen P, Ruhwedel T, Mitkovski M, Trendelenburg G, Lütjohann D, Möbius W, Simons M, 2018. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 359, 684–688. 10.1126/science.aan4183 [DOI] [PubMed] [Google Scholar]
- Condello C, Yuan P, Schain A, Grutzendler J, 2015. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun 6, 1–14. 10.1038/ncomms7176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagher NN, Najafi AR, Kayala KMN, Elmore MRP, White TE, Medeiros R, West BL, Green KN, 2015. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J. Neuroinflammation 12, 139 10.1186/s12974-015-0366-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Karran E, 2016. The cellular phase of Alzheimer’s disease. Cell 164, 603–615. 10.1016/j.cell.2015.12.056 [DOI] [PubMed] [Google Scholar]
- Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I, 2018. Disease-associated microglia: A universal immune sensor of neurodegeneration. Cell 173, 1073–1081. 10.1016/j.cell.2018.05.003 [DOI] [PubMed] [Google Scholar]
- Elmore MRP, Hohsfield LA, Kramár EA, Soreq L, Lee RJ, Pham ST, Najafi AR, Spangenberg E, Wood MA, West BL, Green KN, 2018. Replacement of microglia in the aged brain reverses cognitive, synaptic, and neuronal deficits in mice 10.1111/acel.12832 [DOI] [PMC free article] [PubMed]
- Friedman BA, Srinivasan K, Ayalon G, Kaminker JS, Brug M.P. Van Der, Hansen DV, Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, 2018. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep 22, 832–847. 10.1016/j.celrep.2017.12.066 [DOI] [PubMed] [Google Scholar]
- Füger P, Hefendehl JK, Veeraraghavalu K, Wendeln AC, Schlosser C, Obermüller U, Wegenast-Braun BM, Neher JJ, Martus P, Kohsaka S, Thunemann M, Feil R, Sisodia SS, Skodras A, Jucker M, 2017. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat. Neurosci 20, 1371–1376. 10.1038/nn.4631 [DOI] [PubMed] [Google Scholar]
- Gomez-Nicola D, Fransen NL, Suzzi S, Perry VH, 2013. Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci 33, 2481–2493. 10.1523/JNEUROSCI.4440-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE, 2013. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643. 10.1016/j.neuron.2013.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley MJ, Brough D, Quintin J, Allan SM, 2019. Microglial priming as trained immunity in the brain. Neuroscience 405, 47–54. 10.1016/j.neuroscience.2017.12.039 [DOI] [PubMed] [Google Scholar]
- Holtman IR, Raj DD, Miller J. a, Schaafsma W, Yin Z, Brouwer N, Wes PD, Möller T, Orre M, Kamphuis W, Hol EM, Boddeke EWGM, Eggen BJL, 2015. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol. Commun 3 10.1186/s40478-015-0203-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtman IR, Skola D, Glass CK, 2017. Transcriptional control of microglia phenotypes in health and disease. J. Clin. Invest 127, 3220–3229. 10.1172/JCI90604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, Harari O, Bertelsen S, Fairfax BP, Czajkowski J, Chouraki V, Grenier-Boley B, Bellenguez C, Deming Y, McKenzie A, Raj T, Renton AE, Budde J, Smith A, Fitzpatrick A, Bis JC, DeStefano A, Adams HHH, Ikram MA, van der Lee S, Del-Aguila JL, Fernandez MV, Ibañez L, Sims R, Escott-Price V, Mayeux R, Haines JL, Farrer LA, Pericak-Vance MA, Lambert JC, van Duijn C, Launer L, Seshadri S, Williams J, Amouyel P, Schellenberg GD, Zhang B, Borecki I, Kauwe JSK, Cruchaga C, Hao K, Goate AM, 2017. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat. Neurosci 20(8), 1052–1061. 10.1038/nn.4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A, Keren-Shaul H, David E, Zmora N, Eldar SM, Lubezky N, Shibolet O, Hill DA, Lazar MA, Colonna M, Ginhoux F, Shapiro H, Elinav E, Amit I, 2019. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell 178, 686–698.e14. 10.1016/j.cell.2019.05.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Hirsch XAM, Broihier XML, Miller XCM, Neilson XLE, Ransohoff XRM, Lamb XBT, Landreth GE, 2017. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci 37, 637–647. 10.1523/JNEUROSCI.2110-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L, Lambert JC, Wang LS, Choi SH, Harold D, Vedernikov A, Escott-Price V, Stone T, Richards A, Bellenguez C, Ibrahim-Verbaas CA, Naj AC, Sims R, Gerrish A, Jun G, DeStefano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thornton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin CF, Schmidt H, Kunkle BW, Dunstan ML, Ruiz A, Bihoreau MT, Reitz C, Pasquier F, Hollingworth P, Hanon O, Fitzpatrick AL, Buxbaum JD, Campion D, Crane PK, Becker T, Gudnason V, Cruchaga C, Craig D, Amin N, Berr C, Lopez OL, De Jager PL, Deramecourt V, Johnston JA, Evans D, Lovestone S, Letteneur L, Kornhuber J, Tarraga L, Rubinsztein DC, Eiriksdottir G, Sleegers K, Goate AM, Fievet N, Huentelman MJ, Gill M, Emilsson V, Brown K, Kamboh MI, Keller L, Barberger-Gateau P, McGuinness B, Larson EB, Myers AJ, Dufouil C, Todd S, Wallon D, Love S, Kehoe P, Rogaeva E, Gallacher J, St George-Hyslop P, Clarimon J, Lleo A, Bayer A, Tsuang DW, Yu L, Tsolaki M, Bossu P, Spalletta G, Proitsi P, Collinge J, Sorbi S, Sanchez Garcia F, Fox N, Hardy J, Deniz Naranjo MC, Razquin C, Bosco P, Clarke R, Brayne C, Galimberti D, Mancuso M, Moebus S, Mecocci P, Del Zompo M, Maier W, Hampel H, Pilotto A, Bullido M, Panza F, Caffarra P, Nacmias B, Gilbert JR, Mayhaus M, Jessen F, Dichgans M, Lannfelt L, Hakonarson H, Pichler S, Carrasquillo MM, Ingelsson M, Beekly D, Alavarez V, Zou F, Valladares O, Younkin SG, Coto E, Hamilton-Nelson KL, Mateo I, Owen MJ, Faber KM, Jonsson PV, Combarros O, O’Donovan MC, Cantwell LB, Soininen H, Blacker D, Mead S, Mosley TH, Bennett DA, Harris TB, Fratiglioni L, Holmes C, De Bruijn RFAG, Passmore P, Montine TJ, Bettens K, Rotter JI, Brice A, Morgan K, Foroud TM, Kukull WA, Hannequin D, Powell JF, Nalls MA, Ritchie K, Lunetta KL, Kauwe JSK, Boerwinkle E, Riemenschneider M, Boada M, Hiltunen M, Martin ER, Pastor P, Schmidt R, Rujescu D, Dartigues JF, Mayeux R, Tzourio C, Hofman A, Nothen MM, Graff C, Psaty BM, Haines JL, Lathrop M, Pericak-Vance MA, Launer LJ, Farrer LA, Van Duijn CM, Van Broeckhoven C, Ramirez A, Schellenberg GD, Seshadri S, Amouyel P, Williams J, Holmans PA, MRC CFAS, 2015. Convergent genetic and expression data implicate immunity in Alzheimer’s disease. Alzheimer’s Dement 11, 658–671. 10.1016/j.jalz.2014.05.1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AU, Minhas PS, Liddelow SA, Haileselassie B, Andreasson KI, Ii GWD, Mochly-rosen D, 2019. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci 22, 1635–1648. 10.1038/s41593-019-0486-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, Kao AW, Karydas A, Onanuga K, Martinez R, Argouarch A, Wang C, Huang C, Sohn PD, Bowles KR, Spina S, Silva MC, Marsh JA, Hsu S, Pugh DA, Ghoshal N, Norton J, Huang Y, Lee SE, Seeley WW, Theofilas P, Grinberg LT, Moreno F, McIlroy K, Boeve BF, Cairns NJ, Crary JF, Haggarty SJ, Ichida JK, Kosik KS, Miller BL, Gan L, Goate AM, Temple S, Alquezar C, Bowles K, Butler D, Hernandez I, Hennes V, Kampmann M, 2019. A comprehensive resource for induced pluripotent stem cells from patients with primary tauopathies. Stem Cell Reports 13, 939–955. 10.1016/j.stemcr.2019.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I, 2017. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 1–15. 10.1016/j.cell.2017.05.018 [DOI] [PubMed]
- Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Gomez Perdiguero E, Wieghofer P, Heinrich A, Riemke P, Hölscher C, Müller DN, Luckow B, Brocker T, Debowski K, Fritz G, Opdenakker G, Diefenbach A, Biber K, Heikenwalder M, Geissmann F, Rosenbauer F, Prinz M, 2013. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci 16, 273–280. 10.1038/nn.3318 [DOI] [PubMed] [Google Scholar]
- Kim GH, Kim JE, Rhie SJ, Yoon S, 2015. The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol 24, 325–340. 10.5607/en.2015.24.4.325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin J-J, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C, 2014. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med 6, 243ra86 10.1126/scitranslmed.3009093 [DOI] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Ochando J, Haass C, Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, Fatimy R. El, Beckers L, Loughlin EO, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Mazaheri F, Hartmann K, Madi A, Ulrich JD, Glatzel M, Worthmann A, Heeren J, 2017. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e9. 10.1016/j.immuni.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, Bellenguez C, Frizatti A, Chouraki V, Martin ER, Sleegers K, Badarinarayan N, Jakobsdottir J, Hamilton-Nelson KL, Moreno-Grau S, Olaso R, Raybould R, Chen Y, Kuzma AB, Hiltunen M, Morgan T, Ahmad S, Vardarajan BN, Epelbaum J, Hoffmann P, Boada M, Beecham GW, Garnier JG, Harold D, Fitzpatrick AL, Valladares O, Moutet ML, Gerrish A, Smith AV, Qu L, Bacq D, Denning N, Jian X, Zhao Y, Del Zompo M, Fox NC, Choi SH, Mateo I, Hughes JT, Adams HH, Malamon J, Sanchez-Garcia F, Patel Y, Brody JA, Dombroski BA, Naranjo MCD, Daniilidou M, Eiriksdottir G, Mukherjee S, Wallon D, Uphill J, Aspelund T, Cantwell LB, Garzia F, Galimberti D, Hofer E, Butkiewicz M, Fin B, Scarpini E, Sarnowski C, Bush WS, Meslage S, Kornhuber J, White CC, Song Y, Barber RC, Engelborghs S, Sordon S, Voijnovic D, Adams PM, Vandenberghe R, Mayhaus M, Cupples LA, Albert MS, De Deyn PP, Gu W, Himali JJ, Beekly D, Squassina A, Hartmann AM, Orellana A, Blacker D, Rodriguez-Rodriguez E, Lovestone S, Garcia ME, Doody RS, Munoz-Fernadez C, Sussams R, Lin H, Fairchild TJ, Benito YA, Holmes C, Karamujić-Čomić H, Frosch MP, Thonberg H, Maier W, Roschupkin G, Ghetti B, Giedraitis V, Kawalia A, Li S, Huebinger RM, Kilander L, Moebus S, Hernández I, Kamboh MI, Brundin RM, Turton J, Yang Q, Katz MJ, Concari L, Lord J, Beiser AS, Keene CD, Helisalmi S, Kloszewska I, Kukull WA, Koivisto AM, Lynch A, Tarraga L, Larson EB, Haapasalo A, Lawlor B, Mosley TH, Lipton RB, Solfrizzi V, Gill M, Longstreth WT, Montine TJ, Frisardi V, Diez-Fairen M, Rivadeneira F, Petersen RC, Deramecourt V, Alvarez I, Salani F, Ciaramella A, Boerwinkle E, Reiman EM, Fievet N, Rotter JI, Reisch JS, Hanon O, Cupidi C, Andre Uitterlinden AG, Royall DR, Dufouil C, Maletta RG, de Rojas I, Sano M, Brice A, Cecchetti R, George-Hyslop PS, Ritchie K, Tsolaki M, Tsuang DW, Dubois B, Craig D, Wu CK, Soininen H, Avramidou D, Albin RL, Fratiglioni L, Germanou A, Apostolova LG, Keller L, Koutroumani M, Arnold SE, Panza F, Gkatzima O, Asthana S, Hannequin D, Whitehead P, Atwood CS, Caffarra P, Hampel H, Quintela I, Carracedo Á, Lannfelt L, Rubinsztein DC, Barnes LL, Pasquier F, Frölich L, Barral S, McGuinness B, Beach TG, Johnston JA, Becker JT, Passmore P, Bigio EH, Schott JM, Bird TD, Warren JD, Boeve BF, Lupton MK, Bowen JD, Proitsi P, Boxer A, Powell JF, Burke JR, Kauwe JSK, Burns JM, Mancuso M, Buxbaum JD, Bonuccelli U, Cairns NJ, McQuillin A, Cao C, Livingston G, Carlson CS, Bass NJ, Carlsson CM, Hardy J, Carney RM, Bras J, Carrasquillo MM, Guerreiro R, Allen M, Chui HC, Fisher E, Masullo C, Crocco EA, DeCarli C, Bisceglio G, Dick M, Ma L, Duara R, Graff-Radford NR, Evans DA, Hodges A, Faber KM, Scherer M, Fallon KB, Riemenschneider M, Fardo DW, Heun R, Farlow MR, Kölsch H, Ferris S, Leber M, Foroud TM, Heuser I, Galasko DR, Giegling I, Gearing M, Hüll M, Geschwind DH, Gilbert JR, Morris J, Green RC, Mayo K, Growdon JH, Feulner T, Hamilton RL, Harrell LE, Drichel D, Honig LS, Cushion TD, Huentelman MJ, Hollingworth P, Hulette CM, Hyman BT, Marshall R, Jarvik GP, Meggy A, Abner E, Menzies GE, Jin LW, Leonenko G, Real LM, Jun GR, Baldwin CT, Grozeva D, Karydas A, Russo G, Kaye JA, Kim R, Jessen F, Kowall NW, Vellas B, Kramer JH, Vardy E, LaFerla FM, Jöckel KH, Lah JJ, Dichgans M, Leverenz JB, Mann D, Levey AI, Pickering-Brown S, Lieberman AP, Klopp N, Lunetta KL, Wichmann HE, Lyketsos CG, Morgan K, Marson DC, Brown K, Martiniuk F, Medway C, Mash DC, Nöthen MM, Masliah E, Hooper NM, McCormick WC, Daniele A, McCurry SM, Bayer A, McDavid AN, Gallacher J, McKee AC, van den Bussche H, Mesulam M, Brayne C, Miller BL, Riedel-Heller S, Miller CA, Miller JW, Al-Chalabi A, Morris JC, Shaw CE, Myers AJ, Wiltfang J, O’Bryant S, Olichney JM, Alvarez V, Parisi JE, Singleton AB, Paulson HL, Collinge J, Perry WR, Mead S, Peskind E, Cribbs DH, Rossor M, Pierce A, Ryan NS, Poon WW, Nacmias B, Potter H, Sorbi S, Quinn JF, Sacchinelli E, Raj A, Spalletta G, Raskind M, Caltagirone C, Bossù P, Orfei MD, Reisberg B, Clarke R, Reitz C, Smith AD, Ringman JM, Warden D, Roberson ED, Wilcock G, Rogaeva E, Bruni AC, Rosen HJ, Gallo M, Rosenberg RN, Ben-Shlomo Y, Sager MA, Mecocci P, Saykin AJ, Pastor P, Cuccaro ML, Vance JM, Schneider JA, Schneider LS, Slifer S, Seeley WW, Smith AG, Sonnen JA, Spina S, Stern RA, Swerdlow RH, Tang M, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Van Eldik LJ, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Wilhelmsen KC, Williamson J, Wingo TS, Woltjer RL, Wright CB, Yu CE, Yu L, Saba Y, Pilotto A, Bullido MJ, Peters O, Crane PK, Bennett D, Bosco P, Coto E, Boccardi V, De Jager PL, Lleo A, Warner N, Lopez OL, Ingelsson M, Deloukas P, Cruchaga C, Graff C, Gwilliam R, Fornage M, Goate AM, Sanchez-Juan P, Kehoe PG, Amin N, Ertekin-Taner N, Berr C, Debette S, Love S, Launer LJ, Younkin SG, Dartigues JF, Corcoran C, Ikram MA, Dickson DW, Nicolas G, Campion D, Tschanz JA, Schmidt H, Hakonarson H, Clarimon J, Munger R, Schmidt R, Farrer LA, Van Broeckhoven C, O’Donovan C, M., DeStefano AL, Jones L, Haines JL, Deleuze JF, Owen MJ, Gudnason V, Mayeux R, Escott-Price V, Psaty BM, Ramirez A, Wang LS, Ruiz A, van Duijn CM, Holmans PA, Seshadri S, Williams J, Amouyel P, Schellenberg GD, Lambert JC, Pericak-Vance MA, 2019. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet 51, 414–430. 10.1038/s41588-019-0358-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin CF, Gerrish A, Schmidt H, Kunkle B, Dunstan ML, Ruiz A, Bihoreau MT, Choi SH, Reitz C, Pasquier F, Cruchaga C, Craig D, Amin N, Berr C, Lopez OL, De Jager PL, Deramecourt V, Johnston JA, Evans D, Lovestone S, Letenneur L, Morón FJ, Rubinsztein DC, Eiriksdottir G, Sleegers K, Goate AM, Fiévet N, Huentelman MW, Gill M, Brown K, Kamboh MI, Keller L, Barberger-Gateau P, McGuiness B, Larson EB, Green R, Myers AJ, Dufouil C, Todd S, Wallon D, Love S, Rogaeva E, Gallacher J, St George-Hyslop P, Clarimon J, Lleo A, Bayer A, Tsuang DW, Yu L, Tsolaki M, Bossù P, Spalletta G, Proitsi P, Collinge J, Sorbi S, Sanchez-Garcia F, Fox NC, Hardy J, Deniz Naranjo MC, Bosco P, Clarke R, Brayne C, Galimberti D, Mancuso M, Matthews F, Moebus S, Mecocci P, Del Zompo M, Maier W, Hampel H, Pilotto A, Bullido M, Panza F, Caffarra P, Nacmias B, Gilbert JR, Mayhaus M, Lannefelt L, Hakonarson H, Pichler S, Carrasquillo MM, Ingelsson M, Beekly D, Alvarez V, Zou F, Valladares O, Younkin SG, Coto E, Hamilton-Nelson KL, Gu W, Razquin C, Pastor P, Mateo I, Owen MJ, Faber KM, Jonsson PV, Combarros O, O’Donovan MC, Cantwell LB, Soininen H, Blacker D, Mead S, Mosley TH, Bennett DA, Harris TB, Fratiglioni L, Holmes C, de Bruijn RF, Passmore P, Montine TJ, Bettens K, Rotter JI, Brice A, Morgan K, Foroud TM, Kukull WA, Hannequin D, Powell JF, Nalls MA, Ritchie K, Lunetta KL, Kauwe JS, Boerwinkle E, Riemenschneider M, Boada M, Hiltuenen M, Martin ER, Schmidt R, Rujescu D, Wang LS, Dartigues JF, Mayeux R, Tzourio C, Hofman A, Nöthen MM, Graff C, Psaty BM, Jones L, Haines JL, Holmans PA, Lathrop M, Pericak-Vance MA, Launer LJ, Farrer LA, van Duijn CM, Van Broeckhoven C, Moskvina V, Seshadri S, Williams J, Schellenberg GD, Amouyel P, 2013. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet 45, 1452–8. 10.1038/ng.2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I, 2014. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326. 10.1016/j.cell.2014.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung W-S, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA, 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd AF, Davies CL, Holloway RK, Labrak Y, Ireland G, Carradori D, Dillenburg A, Borger E, Soong D, Richardson JC, Kuhlmann T, Williams A, Pollard JW, des Rieux A, Priller J, Miron VE, 2019. Central nervous system regeneration is driven by microglia necroptosis and repopulation. Nat. Neurosci 22, 1046–1052. 10.1038/s41593-019-0418-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak KS, Funnell APW, Pearson RCM, Crossley M, 2011. PU.1 and haematopoietic cell fate: dosage matters. Int. J. Cell Biol 2011 10.1155/2011/808524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinet W, Coornaert I, Puylaert P, De Meyer GRY, 2019. Macrophage death as a pharmacological target in atherosclerosis. Front. Pharmacol 10, 1–18. 10.3389/fphar.2019.00306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai LH, 2019. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. 10.1038/s41586-019-1195-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, Klemsz M, Feeney AJ, Wu GE, Paige CJ, Maki RA, 1996. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J 15, 5647–58. 10.1002/j.1460-2075.1996.tb00949.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade A, Coburn M, Tu CH, Hasselmann J, Davtyan H, Blurton-Jones M, 2018. Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener 13, 1–13. 10.1186/s13024-018-0297-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, Tabas I, 2011. Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355. 10.1016/j.cell.2011.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrabady SE, Rizvi B, Goldman JE, Brickman AM, 2018. White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol. Commun 6, 1–10. 10.1016/j.neuron.2014.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D, Lucas A, Baskaran S, Haddick PCG, Lenser M, Earr TK, Shi J, Dugas JC, Andreone BJ, Logan T, Solanoy HO, Chen H, Srivastava A, Poda SB, Sanchez PE, Watts RJ, Sandmann T, Astarita G, Lewcock JW, Monroe KM, Di Paolo G, 2020. TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105, 837–854.e9. 10.1016/j.neuron.2019.12.007 [DOI] [PubMed] [Google Scholar]
- Pimenova AA, Marcora E, Goate AM, 2017a. A tale of two genes: microglial Apoe and Trem2. Immunity 47, 398–400. 10.1016/j.immuni.2017.08.015 [DOI] [PubMed] [Google Scholar]
- Pimenova AA, Raj T, Goate AM, 2017b. Untangling genetic risk for Alzheimer’s disease. Biol. Psychiatry 1–11. 10.1016/j.biopsych.2017.05.014 [DOI] [PMC free article] [PubMed]
- Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, Colonna M, 2015. TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Invest 125, 2161–2170. 10.1172/JCI77983DS1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj D, Yin Z, Breur M, Doorduin J, Holtman IR, Olah M, Mantingh-Otter IJ, Van Dam D, De Deyn PP, den Dunnen W, Eggen BJL, Amor S, Boddeke E, 2017. Increased white matter inflammation in aging- and Alzheimer’s disease brain. Front. Mol. Neurosci 10, 1–18. 10.3389/fnmol.2017.00206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju S, Dammer EB, Raza SA, Rathakrishnan P, Xiao H, Gao T, Duong DM, Pennington MW, Lah JJ, Seyfried NT, Levey AI, 2018. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener 13, 1–25. 10.1186/s13024-018-0254-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice RA, Spangenberg EE, Yamate-Morgan H, Lee RJ, Arora RPS, Hernandez MX, Tenner AJ, West BL, Green KN, 2015. Elimination of microglia improves functional outcomes following extensive neuronal loss in the hippocampus. J. Neurosci 35, 9977–89. 10.1523/JNEUROSCI.0336-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe J, Mercan F, Pappin DJ, Christopher R, Roe J, Mercan F, Rivera K, Pappin DJ, Vakoc CR, 2015. BET bromodomain inhibition suppresses the function of hematopoietic transcription factors in acute myeloid leukemia. Mol. Cell 58, 1028–1039. 10.1016/j.molcel.2015.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustenhoven J, Smith AM, Smyth LC, Jansson D, Scotter EL, Swanson MEV, Aalderink M, Coppieters N, Narayan P, Handley R, Overall C, Park TIH, Schweder P, Heppner P, Curtis MA, Faull RLM, Dragunow M, 2018. PU.1 regulates Alzheimer’s disease-associated genes in primary human microglia. Mol. Neurodegener 13, 44 10.1186/s13024-018-0277-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, Mancuso R, Chen WT, Woodbury ME, Srivastava G, Möller T, Hudry E, Das S, Saido T, Karran E, Hyman B, Perry VH, Fiers M, De Strooper B, 2019. The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep 27, 1293–1306.e6. 10.1016/j.celrep.2019.03.099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh J-I, Asahina N, Kitano S, Kino Y, 2014. A comprehensive profile of ChIP-seq-based PU.1/Spi1 target genes in microglia. Gene Regul. Syst. Bio 8, 127–39. 10.4137/GRSB.S19711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage JC, Jay T, Goduni E, Quigley C, Mariani MM, Malm T, Ransohoff RM, Lamb XBT, Landreth GE, 2015. Nuclear receptors license phagocytosis by Trem2+ myeloid cells in mouse models of Alzheimer’s disease. J. Neurosci 35, 6532–6543. 10.1523/JNEUROSCI.4586-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Gibbons HM, Oldfield RL, Bergin PM, Mee EW, Faull RLM, Dragunow M, 2013. The transcription factor PU.1 is critical for viability and function of human brain microglia. Glia 61, 929–942. 10.1002/glia.22486 [DOI] [PubMed] [Google Scholar]
- Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MRP, Blurton-Jones M, West BL, Green KN, 2016. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain 139, 1265–1281. 10.1093/brain/aww016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Zhao Y, Zhang B, 2015. Efficient test and visualization of multi-set intersections. Sci. Rep 1–12. 10.1038/srep16923 [DOI] [PMC free article] [PubMed]
- Wang X, Seed B, 2003. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res 31, e154 10.1093/nar/gng154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendeln A-C, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, Wagner J, Häsler LM, Wild K, Skodras A, Blank T, Staszewski O, Datta M, Centeno TP, Capece V, Islam MR, Kerimoglu C, Staufenbiel M, Schultze JL, Beyer M, Prinz M, Jucker M, Fischer A, Neher JJ, 2018. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338. 10.1038/s41586-018-0023-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M, 2016. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of Amyloid-beta by microglia. Neuron 91, 328–340. 10.1016/j.neuron.2016.06.015 [DOI] [PubMed] [Google Scholar]
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J, 2016. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 92, 252–264. 10.1016/j.neuron.2016.09.016 [DOI] [PubMed] [Google Scholar]
- Yun SP, Kam T, Panicker N, Kim Sangmin, Oh Y, Park J, Kwon S, Park YJ, Karuppagounder SS, Park H, Kim Sangjune, Oh N, Kim NA, Lee Saebom, Brahmachari S, Mao X, Lee JH, Kumar M, An D, Kang S, Lee Y, Lee KC, Na DH, Kim D, Lee SH, Roschke VV, Liddelow SA, Mari Z, Barres BA, Dawson VL, Lee Seulki, 2018. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med 24 10.1038/s41591-018-0051-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, Cella M, Ulland TK, Zaitsev K, Miyashita A, Ikeuchi T, Sainouchi M, Kakita A, Bennett DA, Schneider JA, Nichols MR, Beausoleil SA, Ulrich JD, Holtzman DM, Artyomov MN, Colonna M, 2020. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med 26, 131–142. 10.1038/s41591-019-0695-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.