

Abstract
As the importance of effective vaccines and the role of protein therapeutics in the drug industry continue to expand, alternative strategies to characterize protein complexes are needed. Mass spectrometry (MS) in conjunction with enzymatic digestion or chemical probes has been widely used for mapping binding epitopes at the molecular level. However, advances in instrumentation and application of activation methods capable of accessing higher energy dissociation pathways have recently allowed direct analysis of protein complexes. Here we demonstrate a workflow utilizing native MS and ultraviolet photodissociation (UVPD) to map the antigenic determinants of a model antibody–antigen complex involving hemagglutinin (HA), the primary immunogenic antigen of the influenza virus, and the D1 H1–17/H3–14 antibody which has been shown to confer potent protection to lethal infection in mice despite lacking neutralization activity. Comparison of sequence coverages upon UV photoactivation of HA and of the HA·antibody complex indicates the elimination of some sequence ions that originate from backbone cleavages exclusively along the putative epitope regions of HA in the presence of the antibody. Mapping the number of sequence ions covering the HA antigen versus the HA·antibody complex highlights regions with suppressed backbone cleavage and allows elucidation of unknown epitopes. Moreover, examining the observed fragment ion types generated by UVPD demonstrates a loss in diversity exclusively along the antigenic determinants upon MS/MS of the antibody–antigen complex. UVPD-MS shows promise as a method to rapidly map epitope regions along antibody–antigen complexes as novel antibodies are discovered or developed.
Graphical Abstract

The design of immunotherapeutic drugs and vaccines relies on identification of the epitopes to which antibodies bind. The location within the intact antibody of corresponding paratopes comprising predominantly but not exclusively loops within the variable domain, i.e. the complementarity determining regions (CDRs), is very important for understanding antibody function and also for generating improved variants having higher antigen affinity or specificity.1,2 Although structural biology approaches, namely X-ray crystallography, NMR spectroscopy, and cryo-electron microscopy, provide high resolution information on the residues at the antibody●antigen interface, alternative epitope/paratope mapping pipelines offer certain advantages including requiring lower quantities of proteins and allowing more rapid analysis.3 Over the past three decades, mass spectrometry (MS) has emerged as a rapid and sensitive technique for determining the higher order structure of antibodies and identifying residues comprising the binding epitope and paratope.4 Traditionally MS-based approaches to map antigenic epitopes involve formation of the complexes in solution followed by enzymatic digestion to preserve structural information prior to mass spectrometric read-out.4 Proteolytic digestion of the antigen can occur before or after formation of the complex with the antibody, termed respectively epitope extraction and epitope excision, followed by washing of unbound peptides and MS analysis of epitope peptides.4,5 More modern MS-based epitope and paratope mapping methods rely on hydrogen/deuterium exchange (HDX),6–9 carboxyl footprinting,10,11 or fast photochemical oxidation of proteins (FPOP)12,13 to compare the uptake of unbound and bound antigens and detect regions protected upon antibody binding. Chemical cross-linking of immune complexes has also been demonstrated for identifying antigenic determinants.14
With the advent of native MS, intact antibody–antigen complexes that have not been subjected to proteolytic digestion can now be interrogated directly.15,16 The native MS approach utilizes electrospray ionization of analytes in solutions of high ionic strength to maintain noncovalent interactions and transfer intact proteins into the gas phase with architectures reminiscent of their solution structures.17–19 While the absence of solvent certainly impacts structure to some extent, there is growing evidence that charged protein complexes maintain a large portion of the folded tertiary and quaternary structures adopted in solution.20–22 An early experiment to address this issue involved electrospray ionization of the tobacco mosaic virus and subsequent capture of the sprayed protein by soft landing.20 Visualization by transmission electron microscopy suggested the virus was still structurally intact, further demonstrated by its ability to infect tobacco plants after transition through the gas phase. More recently, ion mobility spectrometry (IMS) experiments have provided convincing evidence that protein structures are partially retained based on gas-phase measurements of collision cross sections (CCS) that can be directly compared to solution-phase values.21,22 Such analysis has demonstrated similarity between solution-phase CCS values for the trp RNA-binding protein, TRAP, as well as GroEL-GroES complexes with those measured in the absence of bulk water.21,22 As such, the stoichiometry and higher order structures of antibodies and antibody–antigen complexes are now routinely detected with MS.23–26 Notable improvements in instrumentation have focused on extending the observable mass range to allow detection of high MW complexes such as those involved in complement initiation by the classical pathway, specifically the interaction of hexameric immunoglobulin G (IgG) with C1q.27,28 Most recently charge detection MS was utilized to probe virus-like particles conjugated to antibodies.29 Additionally, utilizing native MS streamlines sample handling for epitope extraction workflows by allowing analysis of antigen digest and antibody mixtures.30,31 In this latter method, collisionally activated separation of epitope peptides from the antibody–peptide complexes and subsequent sequencing of the peptides is carried out within the mass spectrometer.31 Ion mobility mass spectrometry (IM-MS) has also demonstrated utility in defining antibody heterogeneity and detecting immune complexes.26,32–34 Specifically, collision-induced unfolding (CIU) footprints can be used to distinguish antibodies with divergent higher order structures34 or those bound to the same antigen along different epitopes.33
While collisional activation, including collision-induced dissociation (CID) and higher-energy collisional dissociation (HCD), is most commonly used to sequence resulting epitope or paratope peptides, studies aimed at detection of protected regions following antibody binding in the gas phase have opted for alternative MS/MS approaches.35 In particular, electron-based activation methods, including electron transfer dissociation (ETD) and electron capture dissociation (ECD), afford higher sequence coverage of epitope/paratope regions, allowing utilization of middle-down or even top-down analysis to determine sites of chemical modification after HDX of antibody–antigen complexes or to characterize the amino acids along CDRs.36,37 Moreover, native MS and ECD of an antigen bound to the Fab region of an antibody yielded sequence ions exclusively from flexible regions of the complexes,38 a trend also commonly observed for ETD or ECD of protein complexes.39,40 Similarly, UVPD41 has afforded unsurpassed sequence coverage of CDRs for monoclonal antibody mixtures, but the feasibility of using UVPD for epitope mapping has not been reported.
Previous UVPD-MS workflows include modification of the CDR of a monoclonal antibody with a chromogenic, cysteine-selective tag and subsequent liquid chromatography (LC) MS/MS analysis with 351 nm UVPD42 and middle-down digestion of monoclonal antibody mixtures followed by identification using 193 nm photoactivation.43 A recent study demonstrated the utility of both ECD and 157 nm UVPD for determining heavy and light chain antibody pairing and successful sequencing of the CDR-H3.44 Native MS/UVPD studies of other types of proteins and protein complexes have tracked suppression or enhancement of backbone cleavages as a means to characterize changes in noncovalent interactions and conformation, including loop movements and structural variations stemming from single point mutations.41 Owing to the production of a wide array of fragment ions which afford high sequence coverages for increasingly large proteins45–47 in combination with the apparent sensitivity of UVPD for profiling secondary and tertiary structures,41,48 application of this frontier strategy for characterization of antibody–antigen complexes was warranted.
Here we explore the epitope mapping capabilities of 193 nm UVPD-MS for a recently identified antibody that recognizes influenza A hemagglutinin (HA). Despite widespread vaccination efforts, seasonal outbreaks of influenza A virus remain a major threat to public health and affect millions of people worldwide every year.49 Along with the glycoside hydrolase neuraminidase (NA), HA resides on the surface of virus particles. It recognizes sialic acid moieties presented along the surfaces of target cells and facilitates fusion of the viral envelope with the host endosomal membrane.50 Functioning as a homotrimer, each HA protomer consists of two domains: HA1 and HA2.51 Apart from residues along the receptor binding site, HA1 is subject to intense evolutionary pressure causing antigenic drift, while HA2 remains relatively conserved.52–54 Vaccines must be updated continually to match predicted seasonal strains of influenza but are still ineffective against pandemic outbreaks resulting from more significant antigenic shift. Efforts to develop a prophylactic offering universal protection across influenza strains and subtypes rely on elucidation of the HA epitopes recognized by broadly protective antibodies.55–57 As illustrated in the present study, comparison of sequence coverages of the HA1 antigen upon UVPD of unbound HA1 and antibody-bound HA1 suggests the presence of the antibody curbs fragmentation specifically along the putative epitope regions of the antigen, resulting in an observed suppression in sequence coverage. Moreover, charting the sequence coverage per residue based on UVPD reveals attenuation of sequence ions specifically along the two epitope regions and provides a more general approach for elucidation of an unknown epitope.
EXPERIMENTAL SECTION
Sample Preparation.
The D1 H1–17/H3–14 IgG monoclonal antibody and the monomeric HA1 domain (residues 57–267) of the corresponding HA protomer from H1N1 A/California/04/2009 were expressed and purified as described in the Supporting Information (expressed protein sequences of the antibody and antigen are shown in Figure S1). For glycan removal, 20 μg of the antigen was diluted in 50 mM sodium phosphate (pH 7.5) and incubated for 48 h at room temperature with 1000 units of PNGase F (New England BioLabs Inc., Ipswich, MA). The antibody was then added to the antigen solution at a 1:2 or 1:20 antibody:antigen ratio, and the resulting solution was flash frozen in an effort to minimize deglycosylation of the antibody by PNGase F. Samples were desalted and exchanged into 20 mM ammonium acetate (pH 6.8) at 5–10 μM using 50 kDa molecular weight cutoff filters (MilliporeSigma, Burlington, MA) for MS analysis.
Mass Spectrometry and Data Analysis.
MS experiments were performed on a Thermo Scientific Q Exactive UHMR (Bremen, Germany) modified as previously described58 to allow photodissociation in the HCD cell through incorporation of a 193 nm ArF Coherent Excistar excimer laser (Santa Cruz, CA). Protein solutions were subjected to electrospray ionization using source voltages of 1.0–1.2 kV and a source temperature of 200 °C. Solutions were introduced via an offline nano-ESI source using borosilicate emitters fabricated in-house and coated with Au/Pd. Details for implementation of the online size-exclusion chromatography experiment are given in the Supporting Information. In-source trapping (IST) provided low energy collisional activation to promote desolvation of the antibody or antibody–antigen complex with an optimal value of −100 V. All other ion optics were tuned to optimize transmission of the species of interest: the antigen (lower m/z region) or the antibody–antigen complex (higher m/z region). ESI mass spectra represent 60 transients collected at a resolving power of 12500 at m/z 200. For MS/MS spectra, a single charge state was isolated using a width of 10–15 m/z and activated in the HCD cell. A resolving power of 140 K at m/z 200 was used while collecting 500 transients for each MS/MS spectrum. Collision energies of 200–300 eV/q were used for HCD spectra while UVPD spectra represent a single laser pulse at 3 mJ. Automated gain control (AGC) was turned off during collection of all MS and MS/MS data, and instead the ion population was controlled by adjusting the ion time (IT). Specifically, IT values were set at 20 and 350 ms for MS and MS/MS spectra, respectively. Lowering the nitrogen bath gas pressure of the HCD cell from a corresponding pressure in the ultrahigh vacuum (UHV) region of 1 × 10−9 mbar to 1 × 10−10 mbar aided in detection of fragments after photoactivation. However, for collisional activation a minimum UHV pressure of 4 × 10−10 mbar was necessary for effective HCD to occur.
Lower resolution ESI-MS was used to assign average masses of the antigen, antibody, and antibody–antigen complex species using UniDec.59 High resolution MS/MS spectra were decharged and deisotoped using the Xtract algorithm from Thermo Scientific (S/N ratio of 3, fit factor 44%, remainder 25%) to create lists of monoisotopic fragment ion masses that could be assigned as sequence ions using ProSight Lite v1.4. Identifications of HCD (b, y) and UVPD (a, a+1, b, c, x, x+1, y, y−1, z) sequence ions were made within ±10 ppm. All MS/MS spectra were collected in triplicate and only sequence ions identified in all three spectra are reported in the lists of matched ions shown in Tables S1–S3. For HCD or UVPD of the antibody–antigen complex in which sequence ions could arise from the antibody or antigen, there were no overlapping masses in the assigned fragment ions. Sequence coverage maps were made using ProSight Lite in which sequence ion types are color coded as a/x-type green, b/y-type blue, and c/z-type red. All cysteine residues in the antigen and antibody sequences were assumed to be disulfide bound, and identifications accounted for the loss of a hydrogen at each cysteine (−1.0078 Da). A custom script in R was used to calculate the number of sequence ions observed per residue for HCD or UVPD of the antigen and antibody–antigen complex. Briefly, for each amino acid the number of N-terminal product ions (a, b, c) resulting from cleavage of the backbone C-terminal to a given residue and C-terminal product ions (x, y, z) arising from backbone fragmentation N-terminal to that position were summed. Values are reported as a percentage of the total possible number of cleavages adjacent to a given residue (i.e., two for HCD and nine for UVPD, except residues at the N- and C-terminus). A crystal structure of the HA1 domain of an HA protomer bound to the antigen binding fragment (Fab) region of an IgG antibody (PDB ID: 6E4X)60 is shown in Figure S2. Although not the same sequence as the D1 H1–17/H3–14 antibody, both IgGs are known to bind along the HA1 domain near the trimeric interface.60,61 The two expected epitope regions are highlighted and, based on the HA protomer sequence, include residues 90–109 and 213–233, herein referred to respectively as epitope regions 1 and 2. This corresponds to amino acids T35-R55 and F163-Y183 in the HA1 sequence expressed for this study.
RESULTS AND DISCUSSION
Deglycosylation of HA1 Antigen for Improved MS Analysis.
Previous analysis of the repertoire of monoclonal antibodies that comprise the serological response to influenza vaccination led to the identification of a set of antibodies showing broad binding to HA from divergent influenza virus strains.61 In the present study, one such antibody (termed D1 H1–17/H3–14), shown to bind to the interface region of HA trimer via negative-stain EM, was expressed recombinantly to target the HA1 domain of HA from an H1N1 strain of influenza A virus responsible for a pandemic in 2009.62 Without the HA2 domain to stabilize trimer formation, HA1 exists in solution as a 25.4 kDa monomer. ESI-MS of HA1 under native conditions yields low charge states (9+ – 11+) of eight proteoforms corresponding to the attachment of up to seven variations of sugars at an asparagine residue along the sequence (Figures 1A and 1B). The locations of glycan modifications along the HA1 domain are known to vary by HA strain with the glycosylation sites likely evolving under selective pressure to provide steric blocking to antigenic sites and impede interaction with neutralizing antibodies.63
Figure 1.
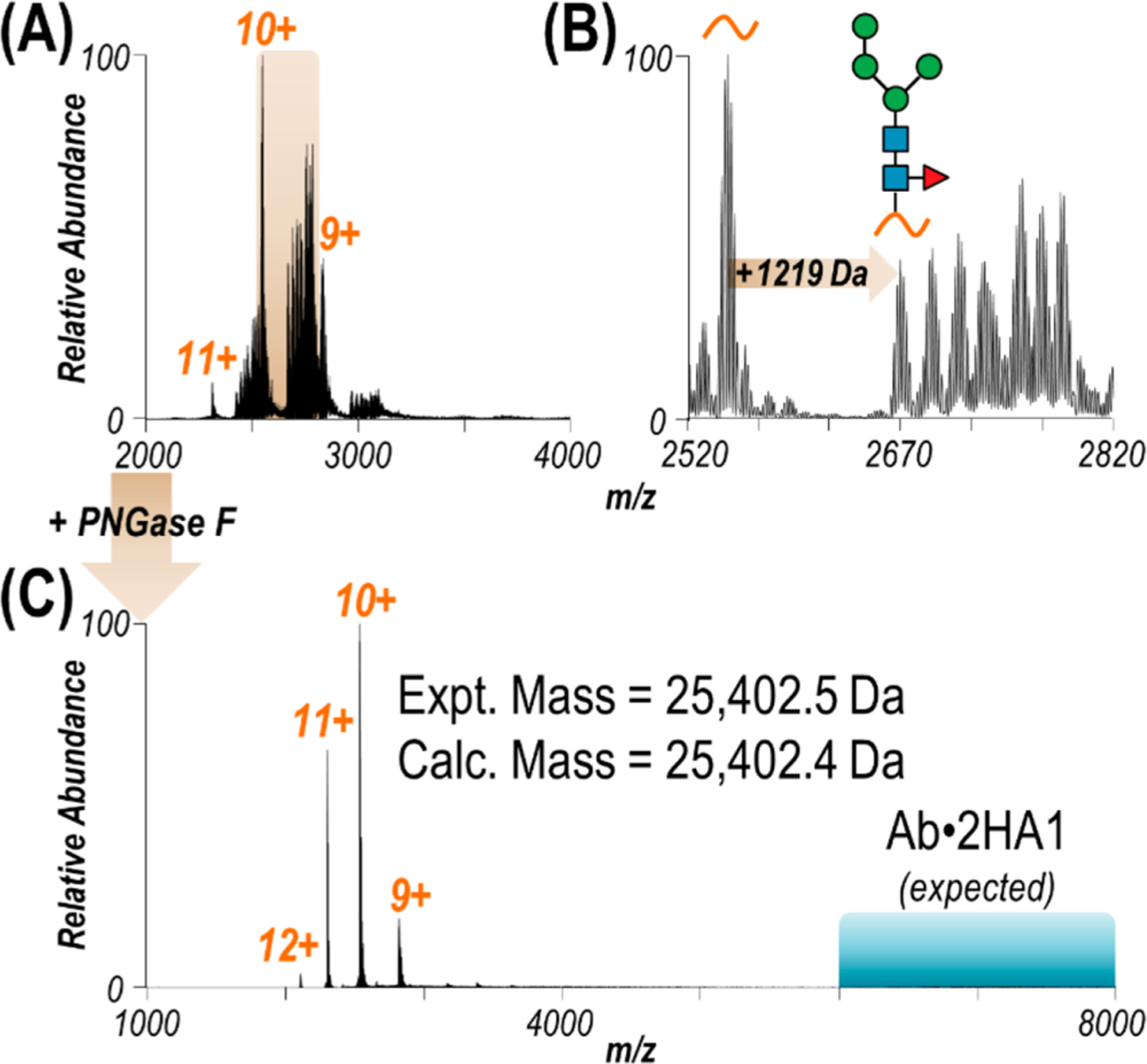
(A) ESI mass spectrum of HA1 sprayed in 20 mM ammonium acetate. (B) Expanded view of the region of the spectrum spanning m/z 2520–2820 revealing several proteoforms of the 10+ charge state resulting from glycosylation. Based on a mass shift of +1219 Da, one possible glycan structure is shown, with up to six additional sugars accounting for all observed glycoforms. (C) ESI mass spectrum of HA1 after glycan removal with PNGase F. Observed as a monomer, the experimental mass of deglycosylated HA1 matches the calculated mass accounting for two disulfide bonds. The m/z region in which antibody·HA1 complexes would be expected is highlighted in turquoise.
Although understanding HA glycosylation patterns can offer insight into viral mechanisms, the various glycoforms result in charge states overlapping with the unmodified protein of interest and hinder MS/MS analysis.64,65 The amidase PNGase F was used to cleave N-linked glycosylations from HA1.15 After the deglycosylation reaction, a substoichiometric amount of antibody was used to bind and isolate the antigen. The result is a simplified mass spectrum of the unmodified antigen with an intact mass matching the expected sequence, accounting for two disulfide bonds (Figure 1C). Owing to the lower concentration at which the antibody was present in solution compared to the antigen, the antibody–antigen complex is not observed under these conditions (m/z region of expected antibody–antigen complexes highlighted in turquoise in Figure 1C). The 10+ charge state of unmodified HA1 was selectively isolated and activated with HCD or UVPD (Figure S3A). Deconvolution of the MS/MS spectra allowed fragment ions to be assigned as backbone cleavage sites along the sequence of HA1 (Figure S3B). Sequence coverage maps shown in Figure S3C demonstrate that UVPD affords coverage of over twice as much of the protein sequence compared to HCD (60% and 27%, respectively). As expected, the presence of two disulfide bonds along the N-terminal region hampers production of informative fragments between those cysteine residues using HCD and to a lesser extent UVPD.66,67
Given our workflow requires exposing the antibody to PNGase F as it is added to the deglycosylation reaction mixture to form the antibody–antigen complex and recover HA1, it is likely the amidase will also competitively cleave the G0F glycans present along the constant region (Fc) of the antibody. Glycosylation of the heavy chains alters the affinity of Fc receptors but has little impact on antigen binding.68 As such, the antibody was reacted with PNGase F separately, mirroring the conditions used when the antigen was present, to determine the extent of deglycosylation. The most abundant species in Figure S4A corresponds to removal of the glycan from each of the heavy chains. Nevertheless, the presence of the singly and doubly glycosylated antibody forms in the spectrum suggests the short reaction time in the presence of PNGase F prevented complete glycan removal. The 25+ charge state of the antibody with no glycans was subsequently isolated and activated using HCD or UVPD (Figures S4B and S4C). Owing to the presence of one intermolecular and seven intramolecular disulfide bonds that preserve the overall structure of the antibody, the majority of fragment ions identified as portions of the sequences of the light or heavy chain are observed in the region encompassing m/z 500–4000. By matching fragment ions from the deconvoluted MS/MS spectra, the resulting sequence coverages were 6% each for the light and heavy chains using HCD, and 45% and 37%, respectively, for the light and heavy chains using UVPD (Figure S5). While the cysteine bridges significantly hinder analysis with HCD as evidenced by the low sequence coverages, the majority of observed light and heavy chain sequence ions upon UVPD occur along regions not restricted by disulfide bonds. Overall, UVPD affords moderate to high sequence coverage of each of the separate components that form the antibody–antigen complex.
Formation and MS Characterization of the Antibody–Antigen Complex.
Size-exclusion chromatography (SEC) MS was used to confirm the expected 1:2 antibody:antigen stoichiometry after incubation of HA1 and the D1 H1–17/H3–14 antibody (Figure 2). The SEC trace shown in Figure 2A demonstrates one major species present in solution (RT 2.58 min) with a small amount of a second species (RT 3.56 min). Corresponding mass spectra confirm these species respectively as the antibody–antigen complex (Figure 2B) and unbound HA1 (Figure 2C). An inset of Figure 2B for the region encompassing m/z 6500–10000 shows the antibody–antigen complex was formed between the antibody retaining either 0, 1, or 2 G0F glycans (due to exposure of the antibody to PNGase F) and two HA1 subunits. In the mass spectrum of the complex, a 1:1 antibody:antigen species is also observed as well as unbound HA1 (Figure 2B). Extracted ion chromatograms of the m/z values corresponding to these three species highlight that unbound HA1 (m/z 3175 (7+ charge state)) elutes in two separate peaks while the antibody–antigen complexes (1:1 and 1:2, m/z 8185 (24+ charge state) and 13157 (13+ charge state)) elute simultaneously (Figure 2A). As such, the unbound HA1 and 1:1 antibody:antigen species coeluting with the antibody–antigen complex (Figure 2A, RT 2.58 min) are likely due to disassembly of the 1:2 Ab:HA1 complexes as a result of in-source trapping (IST, a type of nonselective front-end collisional activation), a method typically used to desolvate large biomolecules in mass spectrometry workflows.69 The use of higher IST parameters can cause disassembly of desired protein complexes. To demonstrate this outcome, mass spectra of the complex were collected using various IST values (−10 V to −250 V) for comparison of the relative abundances of the three major species: unbound HA1, the Ab·2HA1 complex, and the Ab·HA1 complex (Figure S6). Specifically, at low IST values (i.e., −10 V) the complex is poorly desolvated and the quality of the MS1 spectrum is subpar. As the applied voltage is increased, the adducts are collisionally removed resulting in a cleaner spectrum of the antibody–antigen complex. However, at the same time the abundances of ejected unbound HA1 and the corresponding 1:1 antibody:antigen complex increase. While an IST value of −150 V was used during SEC-MS, an optimized value of −100 V was selected based on the trend in Figure S6, representing a justifiable compromise between removal of adducts and decomposition of intact Ab·2HA1 complexes. For the later eluting peak in the SEC trace (Figure 2A, RT 3.56 min, corresponding to free HA1), its narrower charge state distribution (7+ to 9+) compared to the HA1 ejected from the complex in Figure 2B (6+ to 11+) suggests the former HA1 species was unbound in solution and ionized separately from the complex.
Figure 2.
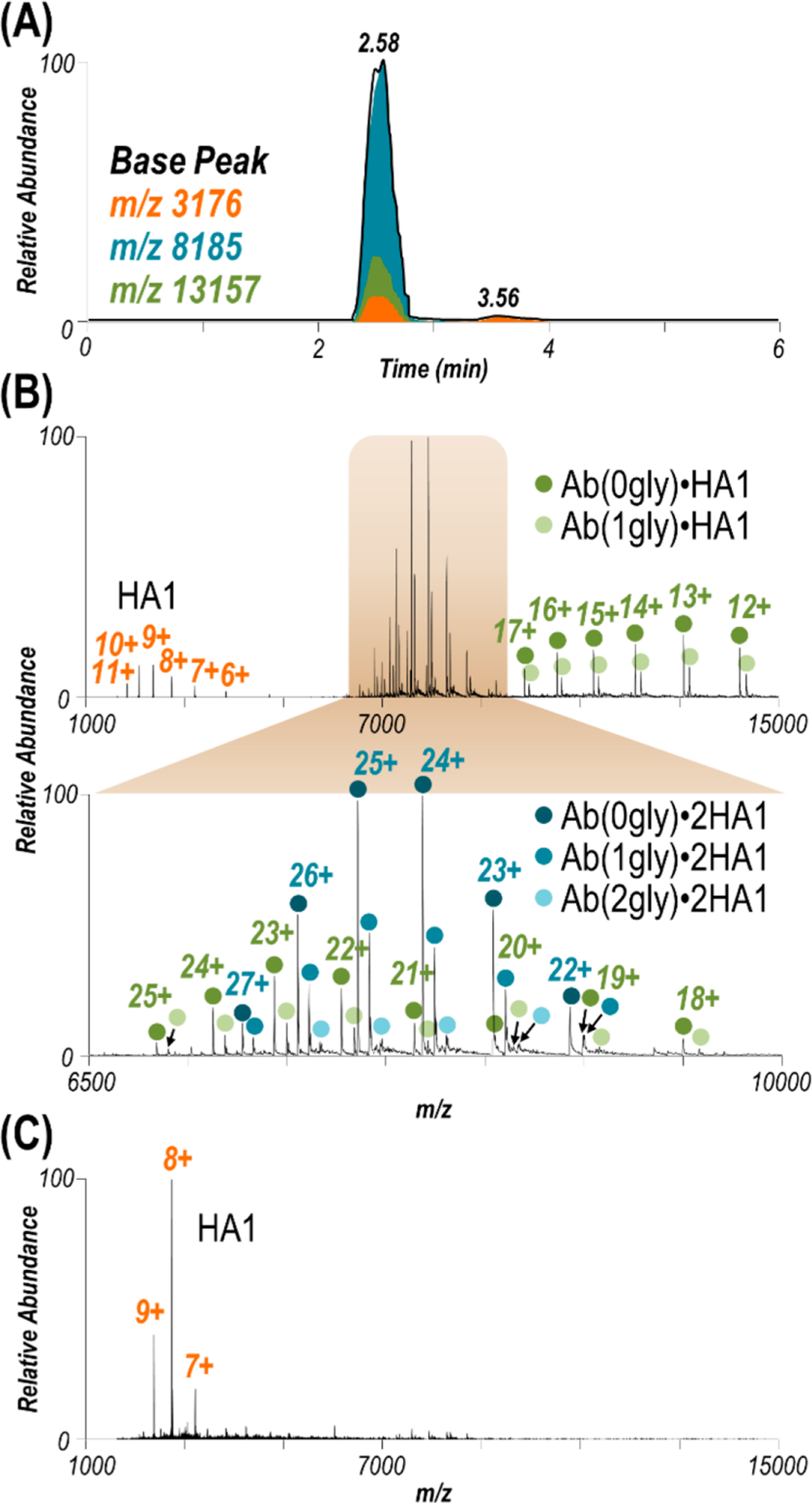
(A) SEC LC trace for HA1 with the antibody at a 1:2 antibody:antigen ratio. Extracted ion chromatograms of the m/z values corresponding to HA1 (orange), Ab·HA1 (green), and Ab·2HA1 (turquoise). (B) ESI mass spectrum collected at retention time 2.58 min along with an expanded view of the range spanning m/z 6500–10000. Observed species include HA1, Ab·HA1, and Ab·2HA1 with 0–2 glycans (gly) attached to the antibody (glycoforms are denoted with colored circles for the complex). In-source trapping (−150 V) necessary to desolvate the Ab·2HA1 complex causes ejection of one HA1, resulting in the observed Ab·HA1 species and unbound HA1. Mass spectra of the complexes obtained using various in-source trapping energies are shown in Figure S6. (C) ESI mass spectrum collected at retention time 3.56 min suggesting a low abundance of unbound HA1 is in solution.
Using these optimized conditions, the 1:2 antibody:antigen mixture was infused by nano-ESI and the 29+ charge state of the antibody–antigen complex (with no G0F glycans bound to antibody) was activated with HCD or UVPD (Figure S7). Ejection of one HA1 monomer to yield the 1:1 antibody:antigen complex was a dominant pathway following activation with HCD and to a lesser extent for UVPD (Figures S7B and S7C). In addition to production of intact protein subunits (unbound HA1 and Ab·HA1), both activation methods yielded a vast array of fragment ions that were deconvoluted and assigned as portions of the sequences of HA1 or the light and heavy chains of the antibody (Figure S8). Briefly, sequence coverages were 20% (HCD) and 49% (UVPD) for HA1, 9% (HCD) and 46% (UVPD) for the antibody light chain, and 6% (HCD) and 37% (UVPD) for the antibody heavy chain. Activation of the Ab·2HA1 complex compared to activation of unbound HA1 resulted in lower sequence coverage of HA1 using either HCD or UVPD (Figure S9), whereas the observed coverage of either chain of the antibody was not significantly impeded by the presence of the antigen (Figures S5 and S8).
UVPD-MS for Epitope Mapping.
Past studies leveraging UVPD-MS to probe protein–ligand and protein–protein complexes have relied on comparison of backbone cleavage propensities upon photoactivation of protein states of interest.41 In essence, observed enhancement of backbone cleavages for a specific protein region upon UVPD correlates with an increase in flexibility or reduction in noncovalent interactions, while suppression of backbone cleavages suggests the formation of new or strengthened stabilizing interactions that hamper the separation/release of fragment ions and inhibits their detection.41,70 As such, during UVPD experiments a decrease in the overall sequence coverage of HA1 upon complexation with the antibody is expected. Nevertheless, to be useful in mapping antibody–antigen complex interactions, the reduction in backbone cleavages must occur specifically along the epitope regions of the antigen. The D1 H1–17/H3–14 antibody is known to bind a conformational epitope spanning two regions near the trimeric interface along the HA1 domain of H1 strains.61 Although existence of epitopes at the contact surface between HA subunits is not intuitive, this interface is exposed postfusion as well as by molecular breathing of the HA trimer.60,71,72 A published crystal structure of the Fab fragment of the S5 V2–29 antibody that binds similarly near the HA trimer interface (PDB ID: 6E4X) visualizes these two putative epitope regions for the HA1 domain of H3N2 A/Texas/50/2014 corresponding to residues 90–109 and 213–233 in the H1 strain used in this study (or residues 35–55 and 163–183 in the expressed HA1 sequence) (Figure S2).60 These two regions are highlighted in green and red along the HCD and UVPD sequence coverage maps of unbound HA1 and the antibody–antigen complex in Figure S9. Although coverage is visibly curbed along these regions upon HCD and UVPD of the complex compared to the monomeric antigen, sequence coverage was plotted by protein region to better assess the corresponding changes in identified sequence ions for the rest of the protein.
Bar graphs of sequence coverages (i.e., the number of backbone cleavages as a percent of the total number of protein residues) are shown for HCD and UVPD of unbound HA1 versus the Ab·2HA1 complex for four different regions: entire protein, epitope region 1, epitope region 2, and the rest of the protein excluding the epitope regions (Figure 3). Both HCD and UVPD resulted in statistically significant lower sequence coverages along the entire protein for activation of the Ab·2HA1 complex. Along the two epitope regions, HCD resulted in a significant suppression in sequence coverage for only one of the regions while UVPD did for both. Comparison of coverage for the rest of the protein sequence excluding the two epitope regions for HCD showed significant suppression but for UVPD remained the same for activation of unbound HA1 or the Ab·2HA1 complex. These results suggest the presence of the antibody prevents production/detection of fragments originating from backbone cleavages of the epitope regions of the antigen during UVPD, resulting in the observed suppression in sequence coverage. An analogous change in sequence coverage of the antibody was not observed upon HCD nor UVPD likely owing to the overall lower sequence coverage, extensive disulfide bonding, and size difference between the antibody and antigen (e.g. the antibody accounts for 75% of the mass of the Ab·2HA1 complex).
Figure 3.
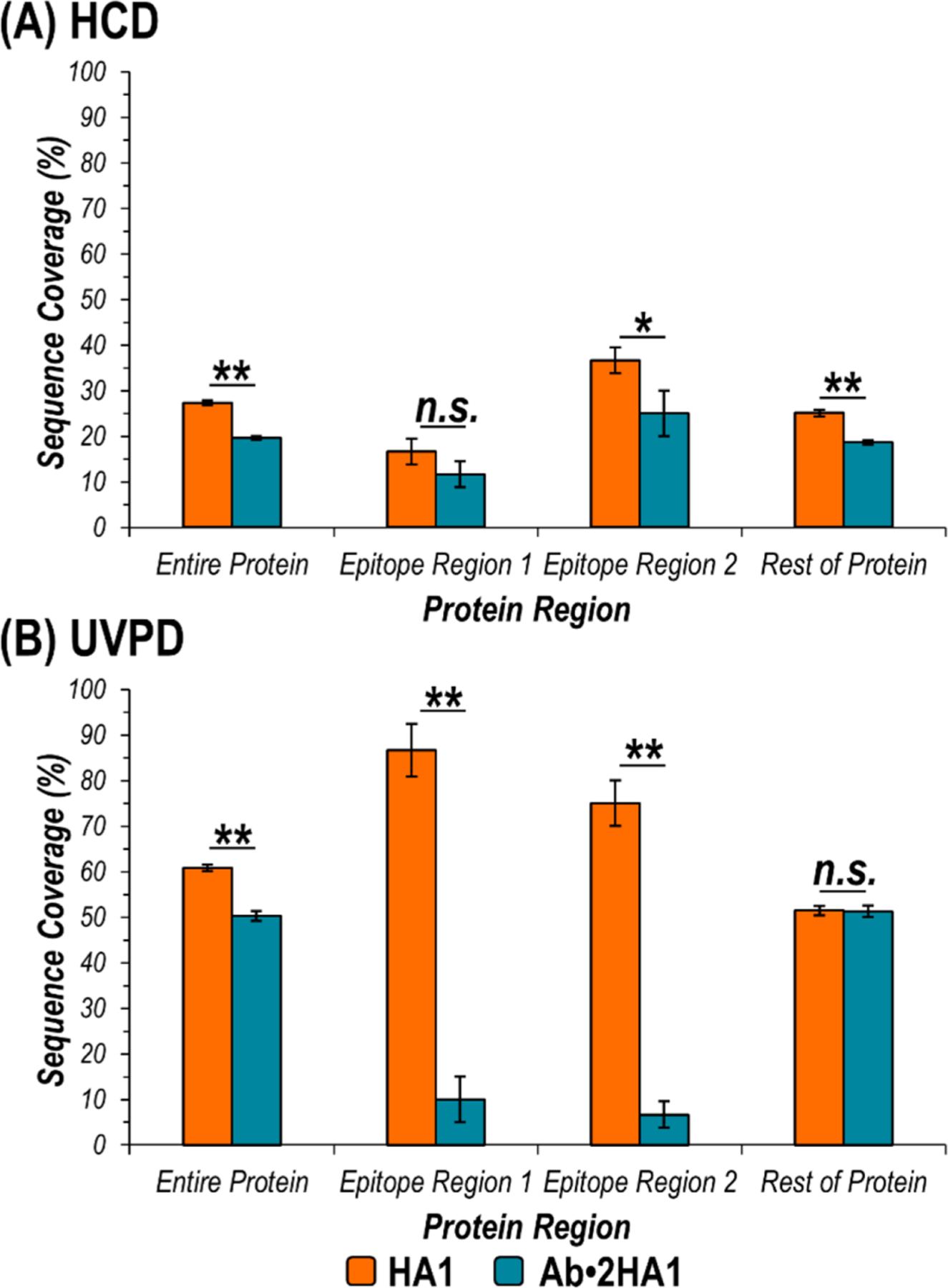
Graphs displaying sequence coverage of various regions of HA1 afforded by activation of HA1 (10+) (orange) or the Ab·2HA1 (29+) complex (turquoise) using (A) HCD and (B) UVPD. In the expressed HA1 sequence, epitope region 1 encompasses residues T35-R55, while epitope region 2 includes amino acids F163-Y183 (corresponding to residues 90–109 and 213–233 in the HA protomer). The rest of the protein is defined as the entire expressed HA1 protein sequence excluding the two epitope regions. Asterisks indicate statically significant differences in sequence coverage for the two precursors at a 95% or 99% confidence level (*p-value < 0.05, **p-value < 0.01, n.s. = not significant).
The distributions of fragment ion types for each of these four regions of HA1 were also plotted as a percentage of the total number of sequence ions identified upon HCD or UVPD of unbound HA1 or the Ab·2HA1 complex (Figure S10). While HCD typically yields only two ion types (b- and y-type from cleavage of the labile C–N amide backbone bond), UVPD produces up to nine different ion types from cleavage of different backbone bonds between pairs of amino acids (a, a+1, x, and x+1 from cleavage of Cα–C bonds, b, y, and y−1 from C–N amide bonds, and c and z from N–Cα bonds).41 For HCD generally more b-type ions (containing the N-terminus) were observed for activation of the Ab·2HA1 complex compared to the antigen alone, except along epitope region 1 spanning residues 35–55 (Figure S10A). For UVPD only b- and y-type fragment ions were observed in the epitope regions upon activation of the Ab·2HA1 complex, while the entire array of expected ion types (a/x, b/y, c/z) were produced for the free antigen (Figure S10B). Moreover, the diversity in the ion type distributions upon UVPD was maintained for the rest of the protein excluding the two epitope regions.
While the complex mechanisms underlying UVPD of intact proteins have yet to be fully unraveled, the hypothesis that both direct fragmentation from excited states and dissociation from vibrationally excited ground states after internal conversion are at play is generally accepted.73 The latter leads to fragmentation pathways similar to those that occur during collisional activation and produces b- and y-type ions.74 The former describes direct cleavage of the backbone by excitation of an electron into an excited state orbital, typically yielding the other ion types observed upon UV photoactivation: a/x and c/z.73 These considerations align with observation of exclusively b- and y-type ions upon UVPD of the Ab·2HA1 complex along epitope regions that produced the entire array of possible sequence ions in the absence of the antibody. In particular, upon formation of the Ab·2HA1 complex, it seems that the network of noncovalent interactions between the antibody and antigen suppresses direct dissociation pathways (e.g. formation and release of a/x or c/z-type ions) especially at residues involved in the protein–protein interface. However, the fragmentation pathways that proceed via redistribution of vibrational energy and disruption of weak noncovalent bonds result in cleavage of backbone bonds in the epitope region and concomitant release of b/y ions.
Approach for Elucidation of an Unknown Epitope using UVPD-MS.
While the epitope regions for the model antibody–antigen system in the present study are reputed,61 a similar UVPD-MS approach could be used to resolve disputed sites or even elucidate unknown antigenic determinants. Examining the number of observed sequence ions bracketing each individual residue of the antigen after UVPD of the Ab·2HA1 complex compared to the unbound antigen highlights possible epitope regions. Stretches of amino acids exhibiting suppressed or completely curbed backbone cleavage for the complexes relative to the free Ab or HA1 subunits suggest involvement in the interface. Such analysis is demonstrated in Figure S11 for the Ab·2HA1 complex. The number of observed HA1 sequence ions upon activation of the antibody–antigen complex or unbound antigen is graphed as a percentage of the total possible number of fragment ions per residue (e.g. two for HCD (b/y) and nine for UVPD (a, a+1, b, c, x, x+1, y, y−1, z)). Subtraction of the corresponding values for the Ab·2HA1 complex and monomeric antigen yields a difference plot and aids in visualization of changes (Figure 4). In short, for residues with values that lie below the x-axis, fewer sequence ions originated from backbone cleavage adjacent to that amino acid upon activation of the antibody–antigen complex compared to the unbound antigen. Conversely the production of more sequence ions arising from backbone cleavage adjacent to a given residue when comparing the Ab·2HA1 complex compared to HA1 is indicated by a value above the x-axis. For HCD there is no distinct pattern or specific region in which fewer sequence ions were observed (Figure 4A). On the contrary, for UVPD there are two main stretches of amino acids resulting in fewer sequence ions upon formation of the Ab·2HA1 complex: residues 35–59 and residues 163–188 of the expressed HA1 sequence (Figure 4B). These regions largely align with the presumed antigenic determinants for the specific antibody–antigen complex used in this study (T35-R55 and F163-Y183 or residues 90–109 and 213–233 of the HA protomer).61
Figure 4.
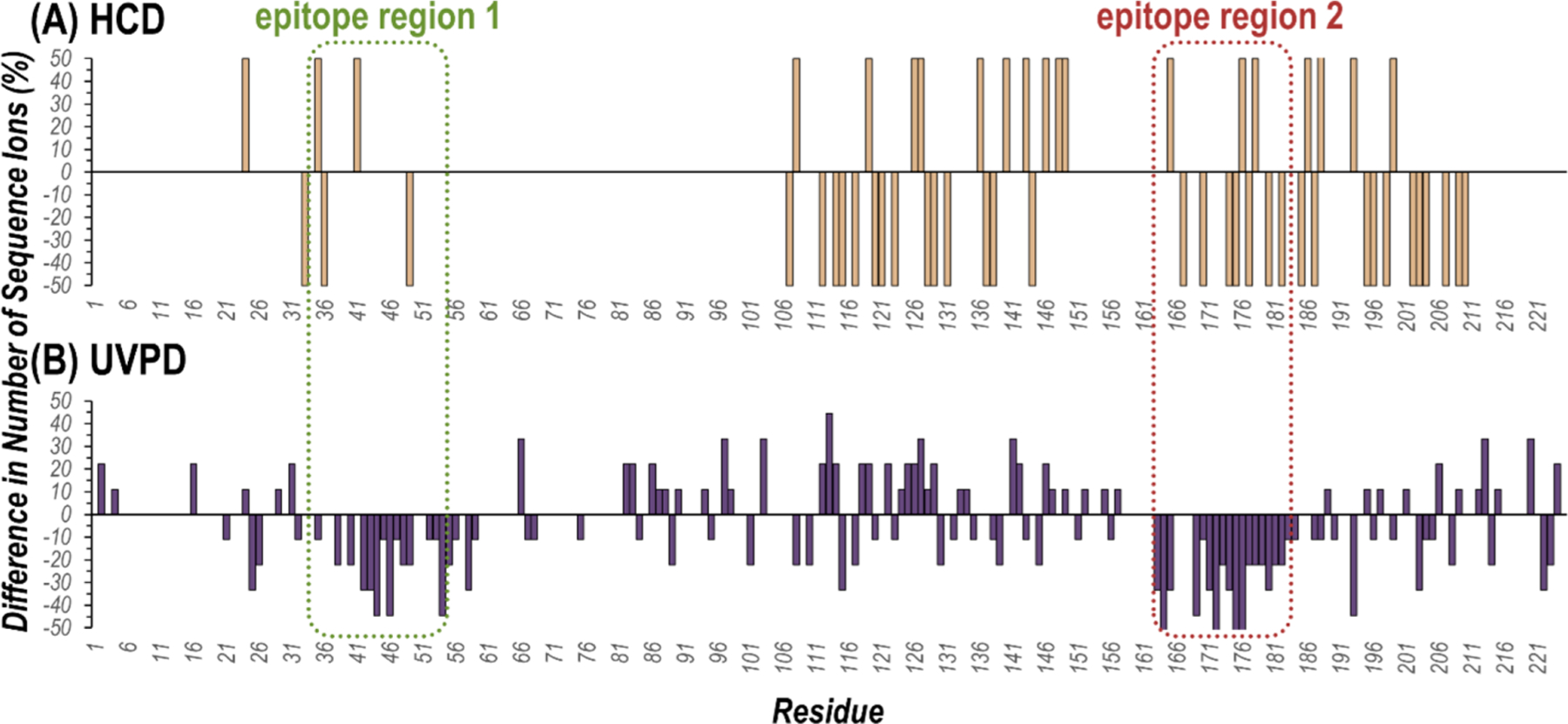
Difference plots representing the changes in the number of observed sequence ions produced by backbone cleavages that bracket each residue upon activation of the Ab·2HA1 antibody–antigen complex (29+) compared to unbound HA1 (10+) using (A) HCD and (B) UVPD. Values greater than zero mean more sequence ions were observed from cleavages adjacent to a given residue when activating the complex compared to the unbound antigen, while values less than zero mean fewer sequence ions were observed. The two known epitope regions of HA1 (residues 90–109 and 213–233 in the HA protomer sequence or 35–55 and 163–183 in the expressed HA1 sequence) are outlined with dashed lines. Plots of the number of sequence ions per residue as a percentage of the possible number of backbone cleavages are shown in Figure S11.
Moreover difference plot values can be represented as heat maps by residue number or visualized along a crystal structure of HA1 bound to the Fab region of an antibody known to interact similarly with HA as D1 H1–17/H3–14 (PDB ID: 6E4X)60 (Figure 5). Suppression (blue) or enhancement (red) in the number of observed sequence ions generated upon backbone cleavages bracketing each residue is shown for activation of the antibody–antigen complex compared to the antigen alone using HCD and UVPD. Residues demarcated in blue (Figure 5A) or shaded in blue (Figure 5B) signal likely involvement in the antibody–antigen complex interface. Conversely an increase in the number of sequence ions produced adjacent to a given residue after formation of the antibody–antigen complex (colored red) can result from increased flexibility/fewer stabilizing interactions in that region or simply the redistribution of energy to dissociation pathways that were inaccessible in the unbound antigen. For the UVPD data, the majority of amino acids exhibiting suppressed cleavage (colored blue) are located along the interface with the Fab region of the antibody in the crystal structure (e.g., the epitope regions), whereas for the HCD data there is both suppression and enhancement of fragmentation along those regions (Figure 5B, 5C). When a structure of the antibody–antigen complex is lacking, mapping differences in the number of UVPD cleavages adjacent to each residue along a high-resolution structure of an unbound antigen could help correlate observed changes in photodissociation with structural features of the protein and aid in localization of the epitopes. Lastly, plotting the observed fragment ion types per residue makes apparent a loss in diversity of observed sequence ions for activation of the antibody–antigen complex compared to the unbound antigen (Figure S12). In particular, the full array of UVPD ion types (a/x, b/y, c/z) covers the residues involved in the epitope regions upon activation of HA1, yet only b- and y-type ions are identified for those same regions upon UVPD of the antibody–antigen complex. Comparison of the number and type of sequence ions arising from backbone cleavages adjacent to individual residues upon UVPD of unbound antigens and antibody–antigen complexes offers an alternative approach for probing unidentified epitopes.
Figure 5.
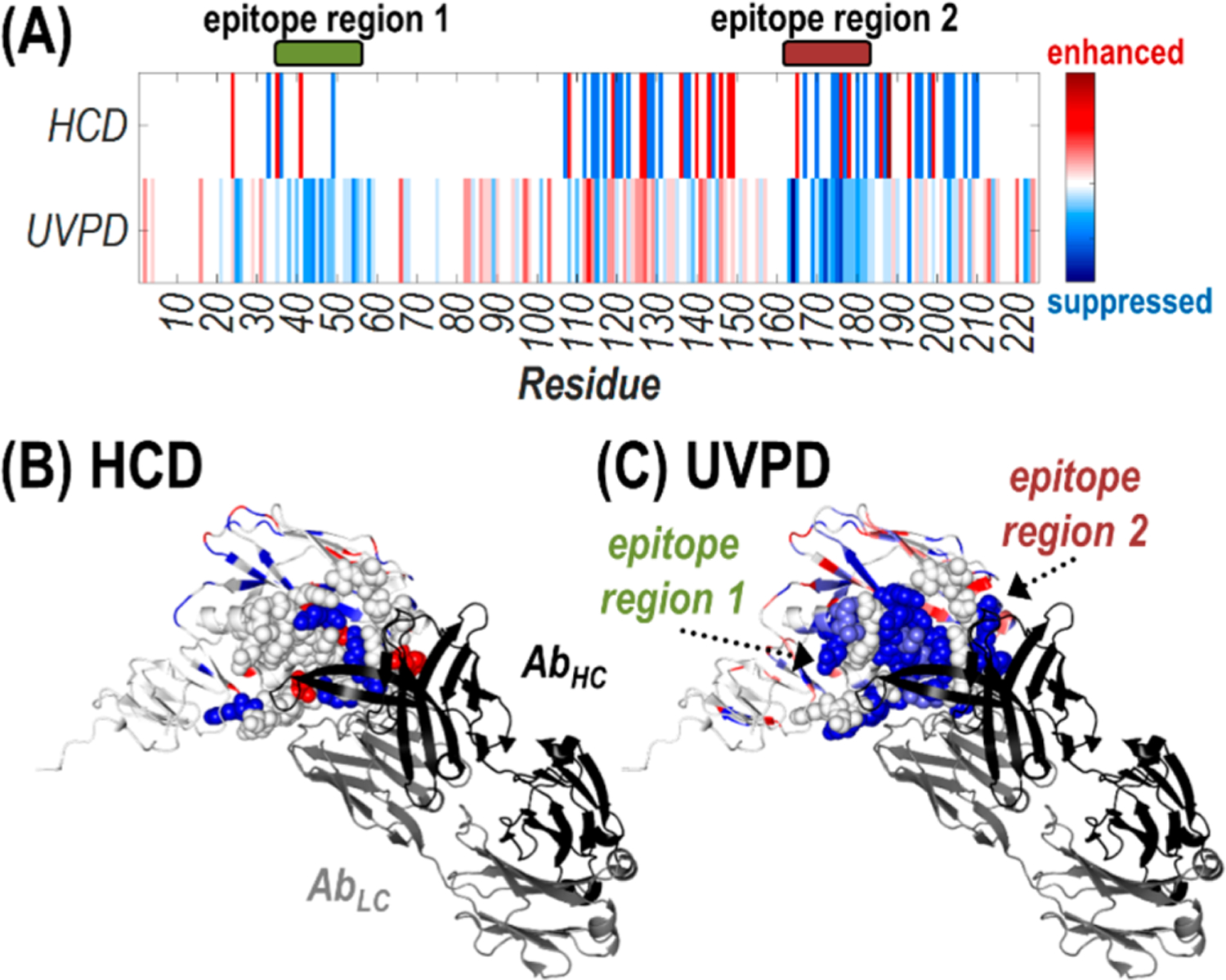
Heat maps of the suppression (blue) or enhancement (red) in the number of observed sequence ions generated upon backbone cleavages bracketing each residue for activation of the Ab·2HA1 complex (29+) compared to the antigen alone (10+) using HCD and UVPD shown (A) for the HA1 sequence or (B, C) along the crystal structure of the HA1 domain of an HA protomer (H3N2 A/Texas/50/2012) bound to the antigen binding fragment (Fab) region of the S5 V2–29 IgG monoclonal antibody (PDB ID: 6E4X). Residues encompassing the two epitope regions are shown as spheres, while the light chain (gray) and heavy chain (black) of the antibody are labeled in (B).
CONCLUSION
Native MS in conjunction with 193 nm UVPD was utilized to probe the antigenic determinants of an antibody–antigen complex. Plotting sequence coverage revealed suppression of UVPD along the two expected epitope regions and provides an approach for elucidation of an unknown antigenic determinant. Moreover, comparing the sequence ion types produced upon UVPD of the antigen in the presence and absence of the antibody highlighted a loss in diversity of fragment ion types covering the epitope regions for the complex. This observation merits future investigation in which utilizing different laser wavelengths or laser energies could provide insight into the time scales and extent to which individual processes govern UVPD. Additional information about the factors that influence the lack of fragmentation of specific regions of antibody–antigen complexes might be obtained by employing supplemental activation prior to and/or after UVPD. When integrated with electron activation methods, supplemental activation has proven effective for disruption of noncovalent interactions that prevent the separation and release of fragment ions.75
Further experiments are underway to apply this UVPD-MS workflow to a wider variety of antibody–antigen complexes to determine if there is an ideal size regime or limit for the antigen as well as if targets that exist as higher order oligomers can be probed. Additionally, MS analysis can be hampered by the presence of post-translational modifications along the antigen, as demonstrated by extensive glycosylation of HA1. Nevertheless, leveraging native MS along with structurally sensitive MS/MS techniques such as ECD or UVPD could further minimize sample handling and provide a new era of MS-based epitope mapping to aid in the discovery of novel therapeutics for use in passive immunotherapy or the identification of conserved epitopes for improved vaccine design.
Supplementary Material
ACKNOWLEDGMENTS
Funding from the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) (R01GM121714), the NIH (P01AI089618 to G.G. and P20GM113132 to J.L.), the Clayton Foundation, and the Robert A. Welch Foundation (F-1155) is acknowledged.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c02237.
Detailed descriptions of the protein expression and purification and online size-exclusion chromatography experiment; Figure S1, expressed protein sequences of the antibody and antigen; Figure S2, crystal structure of HA1 bound to the Fab fragment of an IgG antibody; Figure S3, MS/MS spectra and sequence coverage maps of the antigen; Figure S4, ESI-MS and MS/MS spectra of the antibody; Figure S5, sequence coverage maps of the antibody; Figure S6, ESI-MS for optimization of the in-source trapping voltage; Figure S7, ESI-MS and MS/MS spectra of the antibody–antigen complex; Figure S8, sequence coverage maps for each component of the antibody–antigen complex; Figure S9, comparison of sequence coverage of the antigen before and after complexation with the antibody; Figure S10, sequence ion type distributions for the four distinct regions of the antigen; Figure S11, number of sequence ions produced adjacent to each residue; Figure S12, distributions of ion types produced adjacent to each residue; Table S1, identified fragment ions from MS/MS spectra of the antigen; Table S2, identified fragment ions from MS/MS spectra of the antibody; Table S3, identified fragment ions from MS/MS spectra of the antibody–antigen complex. (PDF)
The authors declare no competing financial interest.
Contributor Information
M. Rachel Mehaffey, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States.
Jiwon Lee, Thayer School of Engineering, Hanover, New Hampshire 03755, United States; Department of Chemical Engineering, University of Texas at Austin, Austin, Texas 78712, United States.
Jiwon Jung, Department of Biomedical Engineering, University of Texas at Austin, Austin, Texas 78712, United States.
Michael B. Lanzillotti, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States
Edwin E. Escobar, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States
Keith R. Morgenstern, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States
George Georgiou, Department of Chemical Engineering, Department of Biomedical Engineering, Institute for Cellular and Molecular Biology, Center for Systems and Synthetic Biology, and Department of Molecular Biosciences, University of Texas at Austin, Austin, Texas 78712, United States.
Jennifer S. Brodbelt, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States.
REFERENCES
- (1).Mahler M; Fritzler MJ Ann. N. Y. Acad. Sci 2010, 1183, 267–287. [DOI] [PubMed] [Google Scholar]
- (2).Regenmortel MHVV J. Mol. Recognit 2014, 27, 627–639. [DOI] [PubMed] [Google Scholar]
- (3).Abbott WM; Damschroder MM; Lowe DC Immunology 2014, 142, 526–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Opuni KFM; Al-Majdoub M; Yefremova Y; El-Kased RF; Koy C; Glocker MO Mass Spectrom. Rev 2018, 37, 229–241. [DOI] [PubMed] [Google Scholar]
- (5).Stefanescu R; Iacob RE; Damoc EN; Marquardt A; Amstalden E; Manea M; Perdivara I; Maftei M; Paraschiv G; Przybylski M Eur. J. Mass Spectrom 2007, 13, 69–75. [DOI] [PubMed] [Google Scholar]
- (6).Wei H; Mo J; Tao L; Russell RJ; Tymiak AA; Chen G; Iacob RE; Engen JR Drug Discovery Today 2014, 19, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Puchades C; Kükrer B; Diefenbach O; Sneekes-Vriese E; Juraszek J; Koudstaal W; Apetri A Sci. Rep 2019, 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhu S; Liuni P; Ettorre L; Chen T; Szeto J; Carpick B; James DA; Wilson DJ Biochemistry 2019, 58, 646–656. [DOI] [PubMed] [Google Scholar]
- (9).Huang RY-C; Krystek SR; Felix N; Graziano RF; Srinivasan M; Pashine A; Chen G mAbs 2018, 10, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wecksler AT; Kalo MS; Deperalta G J. Am. Soc. Mass Spectrom 2015, 26, 2077–2080. [DOI] [PubMed] [Google Scholar]
- (11).Kaur P; Tomechko SE; Kiselar J; Shi W; Deperalta G; Wecksler AT; Gokulrangan G; Ling V; Chance MR mAbs 2015, 7, 540–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhang Y; Rempel DL; Zhang H; Gross ML J. Am. Soc. Mass Spectrom 2015, 26, 526–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhang Y; Wecksler AT; Molina P; Deperalta G; Gross ML J. Am. Soc. Mass Spectrom 2017, 28, 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Pimenova T; Nazabal A; Roschitzki B; Seebacher J; Rinner O; Zenobi RJ Mass Spectrom 2008, 43, 185–195. [DOI] [PubMed] [Google Scholar]
- (15).Rosati S; Yang Y; Barendregt A; Heck AJR Nat. Protoc 2014, 9, 967–976. [DOI] [PubMed] [Google Scholar]
- (16).Thompson NJ; Rosati S; Heck AJR Methods 2014, 65, 11–17. [DOI] [PubMed] [Google Scholar]
- (17).Leney AC; Heck AJR J. Am. Soc. Mass Spectrom 2017, 28, 5–13. [DOI] [PubMed] [Google Scholar]
- (18).Eschweiler JD; Kerr R; Rabuck-Gibbons J; Ruotolo BT Annu. Rev. Anal. Chem 2017, 10, 25–44. [DOI] [PubMed] [Google Scholar]
- (19).Kaur U; Johnson DT; Chea EE; Deredge DJ; Espino JA; Jones LM Anal. Chem 2019, 91, 142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Siuzdak G; Bothner B; Yeager M; Brugidou C; Fauquet CM; Hoey K; Change C-M Chem. Biol 1996, 3, 45–48. [DOI] [PubMed] [Google Scholar]
- (21).Ruotolo BT; Giles K; Campuzano I; Sandercock AM; Bateman RH; Robinson CV Science 2005, 310, 1658–1661. [DOI] [PubMed] [Google Scholar]
- (22).Duijn E. van; Barendregt A; Synowsky S; Versluis C; Heck AJR J. Am. Chem. Soc 2009, 131, 1452–1459. [DOI] [PubMed] [Google Scholar]
- (23).Tito MA; Miller J; Walker N; Griffin KF; Williamson ED; Despeyroux-Hill D; Titball RW; Robinson CV Biophys. J 2001, 81, 3503–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Haberger M; Leiss M; Heidenreich A-K; Pester O; Hafenmair G; Hook M; Bonnington L; Wegele H; Haindl M; Reusch D; Bulau P mAbs 2016, 8, 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Yang Y; Wang G; Song T; Lebrilla CB; Heck AJR mAbs 2017, 9, 638–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Tian Y; Ruotolo BT Analyst 2018, 143, 2459–2468. [DOI] [PubMed] [Google Scholar]
- (27).Dyachenko A; Wang G; Belov M; Makarov A; de Jong RN; van den Bremer ETJ; Parren PWHI; Heck AJR Anal. Chem 2015, 87, 6095–6102. [DOI] [PubMed] [Google Scholar]
- (28).Wang G; de Jong RN; van den Bremer ETJ; Beurskens FJ; Labrijn AF; Ugurlar D; Gros P; Schuurman J; Parren PWHI; Heck AJR Mol. Cell 2016, 63, 135–145. [DOI] [PubMed] [Google Scholar]
- (29).Bond KM; Aanei IL; Francis MB; Jarrold MF Anal. Chem 2020, 92, 1285–1291. [DOI] [PubMed] [Google Scholar]
- (30).Lu X; DeFelippis MR; Huang L Anal. Biochem 2009, 395, 100–107. [DOI] [PubMed] [Google Scholar]
- (31).Danquah BD; Röwer C; Opuni KFM; El-Kased R; Frommholz D; Illges H; Koy C; Glocker MO Mol. Cell. Proteomics 2019, 18, 1543–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Atmanene C; Wagner-Rousset E; Malissard M; Chol B; Robert A; Corvaïa N; Dorsselaer AV; Beck A; Sanglier-Cianférani S Anal. Chem 2009, 81, 6364–6373. [DOI] [PubMed] [Google Scholar]
- (33).Huang Y; Salinas ND; Chen E; Tolia NH; Gross ML J. Am. Soc. Mass Spectrom 2017, 28, 2515–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ehkirch A; Goyon A; Hernandez-Alba O; Rouviere F; D’Atri V; Dreyfus C; Haeuw J-F; Diemer H; Beck A; Heinisch S; Guillarme D; Cianferani S Anal. Chem 2018, 90, 13929–13937. [DOI] [PubMed] [Google Scholar]
- (35).Allison TM; Bechara C Biochem. Soc. Trans 2019, 47, 317–327. [DOI] [PubMed] [Google Scholar]
- (36).Pan J; Zhang S; Chou A; Hardie DB; Borchers CH Anal. Chem 2015, 87, 5884–5890. [DOI] [PubMed] [Google Scholar]
- (37).Pan J; Zhang S; Chou A; Borchers CH Chem. Sci 2016, 7, 1480–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zhang Y; Cui W; Wecksler AT; Zhang H; Molina P; Deperalta G; Gross ML J. Am. Soc. Mass Spectrom 2016, 27, 1139–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lermyte F; Sobott F Proteomics 2015, 15, 2813–2822. [DOI] [PubMed] [Google Scholar]
- (40).Li H; Nguyen HH; Ogorzalek Loo RR; Campuzano IDG; Loo JA Nat. Chem 2018, 10, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Brodbelt JS; Morrison LJ; Santos I Ultraviolet Photodissociation Mass Spectrometry for Analysis of Biological Molecules. Chem. Rev 2020, 120, 3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Cotham VC; Wine Y; Brodbelt JS Anal. Chem 2013, 85, 5577–5585. [DOI] [PubMed] [Google Scholar]
- (43).Cotham VC; Horton AP; Lee J; Georgiou G; Brodbelt JS Anal. Chem 2017, 89, 6498–6504. [DOI] [PubMed] [Google Scholar]
- (44).Shaw JB; Liu W; Vasil’ev YV; Bracken CC; Malhan N; Guthals A; Beckman JS; Voinov VG Anal. Chem 2020, 92, 766–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Shaw JB; Li W; Holden DD; Zhang Y; Griep-Raming J; Fellers RT; Early BP; Thomas PM; Kelleher NL; Brodbelt JS J. Am. Chem. Soc 2013, 135, 12646–12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).O’Brien JP; Li W; Zhang Y; Brodbelt JS J. Am. Chem. Soc 2014, 136, 12920–12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Greisch J-F; Tamara S; Scheltema RA; Maxwell HWR; Fagerlund RD; Fineran PC; Tetter S; Hilvert D; Heck AJR Chem. Sci 2019, 10, 7163–7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Zhou M; Liu W; Shaw JB Anal. Chem 2020, 92, 1788–1795. [DOI] [PubMed] [Google Scholar]
- (49).Krammer F; Palese P Nat. Rev. Drug Discovery 2015, 14, 167–182. [DOI] [PubMed] [Google Scholar]
- (50).Gamblin SJ; Skehel JJ J. Biol. Chem 2010, 285, 28403–28409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Wu NC; Wilson IA Nat. Struct. Mol. Biol 2018, 25, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Air GM Proc. Natl. Acad. Sci. U. S. A 1981, 78, 7639–7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Brandenburg B; Koudstaal W; Goudsmit J; Klaren V; Tang C; Bujny MV; Korse HJWM; Kwaks T; Otterstrom JJ; Juraszek J; Oijen A. M. van; Vogels R; Friesen RHE PLoS One 2013, 8, No. e80034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Sparrow E; Friede M; Sheikh M; Torvaldsen S; Newall AT Vaccine 2016, 34, 5442–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Laursen NS; Friesen RHE; Zhu X; Jongeneelen M; Blokland S; Vermond J; Eijgen A. van; Tang C; Diepen H. van; Obmolova G; Kolfschoten M. van der N.; Zuijdgeest D; Straetemans R; Hoffman RMB; Nieusma T; Pallesen J; Turner HL; Bernard SM; Ward AB; Luo J; Poon LLM; Tretiakova AP; Wilson JM; Limberis MP; Vogels R; Brandenburg B; Kolkman JA; Wilson IA Science 2018, 362, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Sautto GA; Kirchenbaum GA; Ross TM Virol. J 2018, 15, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Zhang Y; Xu C; Zhang H; Liu GD; Xue C; Cao Y Targeting Hemagglutinin: Approaches for Broad Protection against the Influenza A Virus. Viruses 2019, 11, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Mehaffey MR; Sanders JD; Holden DD; Nilsson CL; Brodbelt JS Anal. Chem 2018, 90, 9904–9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Marty MT; Baldwin AJ; Marklund EG; Hochberg GKA; Benesch JLP; Robinson CV Anal. Chem 2015, 87, 4370–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Watanabe A; McCarthy KR; Kuraoka M; Schmidt AG; Adachi Y; Onodera T; Tonouchi K; Caradonna TM; Bajic G; Song S; McGee CE; Sempowski GD; Feng F; Urick P; Kepler TB; Takahashi Y; Harrison SC; Kelsoe G Cell 2019, 177, 1124–1135 (e16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Lee J; Boutz DR; Chromikova V; Joyce MG; Vollmers C; Leung K; Horton AP; DeKosky BJ; Lee C-H; Lavinder JJ; Murrin EM; Chrysostomou C; Hoi KH; Tsybovsky Y; Thomas PV; Druz A; Zhang B; Zhang Y; Wang L; Kong W-P; Park D; Popova LI; Dekker CL; Davis MM; Carter CE; Ross TM; Ellington AD; Wilson PC; Marcotte EM; Mascola JR; Ippolito GC; Krammer F; Quake SR; Kwong PD; Georgiou G Nat. Med 2016, 22, 1456–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Garten RJ; Davis CT; Russell CA; Shu B; Lindstrom S; Balish A; Sessions WM; Xu X; Skepner E; Deyde V; Okomo-Adhiambo M; Gubareva L; Barnes J; Smith CB; Emery SL; Hillman MJ; Rivailler P; Smagala J; Graaf M. de; Burke DF; Fouchier RAM; Pappas C; Alpuche-Aranda CM; López-Gatell H; Olivera H; López I; Myers CA; Faix D; Blair PJ; Yu C; Keene KM; Dotson PD; Boxrud D; Sambol AR; Abid SH; George KS; Bannerman T; Moore AL; Stringer DJ; Blevins P; Demmler-Harrison GJ; Ginsberg M; Kriner P; Waterman S; Smole S; Guevara HF; Belongia EA; Clark PA; Beatrice ST; Donis R; Katz J; Finelli L; Bridges CB; Shaw M; Jernigan DB; Uyeki TM; Smith DJ; Klimov AI; Cox NJ Science 2009, 325, 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Tate MD; Job ER; Deng Y-M; Gunalan V; Maurer-Stroh S; Reading PC Viruses 2014, 6, 1294–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Wang L; Qin Y; Ilchenko S; Bohon J; Shi W; Cho MW; Takamoto K; Chance MR Biochemistry 2010, 49, 9032–9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Wohlschlager T; Scheffler K; Forstenlehner IC; Skala W; Senn S; Damoc E; Holzmann J; Huber CG Nat. Commun 2018, 9, 1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Lioe H; O’Hair RAJ J. Am. Soc. Mass Spectrom 2007, 18, 1109–1123. [DOI] [PubMed] [Google Scholar]
- (67).Quick MM; Crittenden CM; Rosenberg JA; Brodbelt JS Anal. Chem 2018, 90, 8523–8530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Jennewein MF; Alter G Trends Immunol 2017, 38, 358–372. [DOI] [PubMed] [Google Scholar]
- (69).Fort KL; Waterbeemd M. van de; Boll D; Reinhardt-Szyba M; Belov ME; Sasaki E; Zschoche R; Hilvert D; Makarov AA; Heck AJR Analyst 2018, 143, 100–105. [DOI] [PubMed] [Google Scholar]
- (70).Cammarata MB; Brodbelt JS Chem. Sci 2015, 6, 1324–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Bajic G; Maron MJ; Adachi Y; Onodera T; McCarthy KR; McGee CE; Sempowski GD; Takahashi Y; Kelsoe G; Kuraoka M; Schmidt AG Cell Host Microbe 2019, 25, 827–835 (e6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Bangaru S; Lang S; Schotsaert M; Vanderven HA; Zhu X; Kose N; Bombardi R; Finn JA; Kent SJ; Gilchuk P; Gilchuk I; Turner HL; García-Sastre A; Li S; Ward AB; Wilson IA; Crowe JE Cell 2019, 177, 1136–1152 (e18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).R. Julian R J. Am. Soc. Mass Spectrom 2017, 28, 1823–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Paizs B; Suhai S Mass Spectrom. Rev 2005, 24, 508–548. [DOI] [PubMed] [Google Scholar]
- (75).Riley NM; Coon JJ Anal. Chem 2018, 90, 40–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.